
Submitted:
19 September 2025
Posted:
19 September 2025
You are already at the latest version
Abstract
The phenotypic resistance of 56 E. faecalis isolates from Eurasian griffon vultures was subjected to surveillance testing with the microdilution method using a standardized panel of antimicrobials. Isolates were also characterized by MLST. Additionally, the genome of 19 isolates with phenotypic resistance to linezolid, ciprofloxacin, chloramphenicol and/or high-level resistance to gentamicin were sequenced to determine their antimicrobial resistance (ARGs) and virulence-associated genes and to identify mobile genetic elements (MGEs). Most isolates (82.1%) exhibited non-wild-type phenotypes to six antimicrobial agents, and multidrug resistance (MDR) was detected in 34% of the isolates. Most MDR isolates (53%) belonged to ST16, ST116 and ST35. ARGs were localized on the chromosome as well as on different MGEs previously reported in humans, foods and livestock isolates, suggesting the exchange of antimicrobial-resistant bacteria and/or ARGs to vultures consequence of an anthropogenic pollution. Twenty-two virulence-associated genes encoding cell surface and secreted factors were identified, some of them located on MGEs that also carried ARGs. The significant proportion of E. faecalis isolates recovered from vultures exhibiting MDR phenotypes and harbored MGEs carrying ARGs and virulence-associated genes, is a cause for concern, since vultures may act as spreaders of these genes to the environment, domestic animals and humans.
Keywords:
ultures
; microbiota
; Enterococcus faecalis
; antimicrobial resistance
; virulence genes
; mobile genetic elements
; genotyping
1. Introduction
Antimicrobial resistance (AMR) is a complex problem that affects humans, animals and the environment. It is considered the greatest public and animal health threat of the 21st century [1] and requires a coordinated approach across different sectors to mitigate its impact [2]. Wildlife serves as a link between environmental, human and animal domains through direct and indirect contact, faecal contamination and shared environmental resources [3]. Although the role of wildlife has been highlighted with the application of the “One Health” concept to AMR, studies characterizing AMR in wild animals are not as exhaustive as those carried out with humans and livestock [4]. These studies show that antibiotic-resistant bacteria (ARBs) can be isolated from a multitude of wild animals across different geographical areas, including wild birds, mammals and reptiles [5,6,7,8]. Wildlife is often referred to as sentinels, reservoirs and bridging hosts, facilitating the persistence of AMR in their habitats [4,9]. In addition, because many wild animal species are highly mobile, they can play a crucial role in the transmission of ARBs over long distances [10]. Consequently, the role of wildlife in the dissemination of AMR might be underestimated [4,9], and further research in this area is needed to properly assess the public health implications of AMR in wild animals.
The Eurasian griffon vulture (Gyps fulvus) is the most widespread vulture across Europe, Asia and Africa (http://www.birdlife.org). These vultures are obligate scavengers, feeding primarily on carcasses of dead livestock left at supplementary feed stations but also on carcasses from other unmanaged sources, such as wild species and human waste [11,12]. Although vultures are wild animals that do not receive antibiotics, they can be exposed to active antibiotics ingested when feeding on livestock carcasses [13], or they can be contaminated or colonized by ARBs through environmental contamination resulting from human activities [14]. Previous studies have isolated different ARBs, such as Escherichia coli, Salmonella spp., Campylobacter spp. and Enterococcus spp., from the faecal microbiota of vultures [14,15,16]. In addition, the diet of vultures has been identified as a frequent source of AMR genes (ARGs) in their gut microbiome [13]. Therefore, owing to their feeding habits, migratory behaviour, and capacity to adapt to different environments, these wild birds may serve as reservoirs of ARBs present in their microbiomes with the potential to spread resistance genes over long distances and into new areas [5].
Enterococci are among the indicator bacteria used to study the extent of AMR in populations owing to their ability to spread resistance and virulence genes [9], as demonstrated in the surveillance program implemented in the European Union [17]. These bacteria are common among the commensal microbiota of domesticated and wild animals and are also opportunistic pathogens associated with significant morbidity and mortality in humans and animals [18]. In a previous study, analysis of cloacal and pharyngeal samples of Eurasian griffon vultures identified Enterococcus faecalis as one of the most common bacterial species [19]. Strains of E. faecalis isolated from faecal samples of vultures have been found to carry clinically important resistance determinants and virulence traits [14,16]. Both ARGs and virulence genes can spread between bacterial species or genera via horizontal gene transfer (HGT) through different mobile genetic elements (MGEs), such as conjugative transposons or plasmids [20]. From the perspective of the “One Health” concept, the study of AMR is a field of growing interest in microbial ecology [21]. In addition, there is no information available on the presence of MGEs among E. faecalis in the vulture microbiota. Thus, the purpose of this study was (a) to determine the phenotypic resistance of cloacal and pharyngeal E. faecalis isolates from Eurasian griffon vultures to various antimicrobials used in animal health and human medicine and (b) to characterize ARGs, virulence genes and MGEs in the genome of ARBs to four critical antimicrobials: linezolid, chloramphenicol, ciprofloxacin and high-level resistance (HLR) to gentamicin.
2. Materials and Methods
2.1. Enterococcus faecalis Isolates and Susceptibility Testing
The study focused on 56 E. faecalis isolates recovered from cloacal (n=35) or pharyngeal (n=21) samples of 45 Eurasian griffon (Gyps fulvus) vultures [19]. The E. faecalis isolates were subjected to surveillance testing with the microdilution method [22] using a standardized panel of antimicrobials (Sensititre EU Surveillance Enterococcus EUVENC Antimicrobial Susceptibility Testing Plates). In brief, inocula of the isolates were prepared in Muller–Hinton broth, adjusted to a 0.5 McFarland standard, and further diluted 1/220 in sterile distilled water. They were then deposited into each well of the microdilution plates, which were subsequently incubated at 37°C for 24 h. E. faecalis ATCC 29212 and Staphylococcus aureus ATCC 29213 were used as control strains. The epidemiological cut-off (ECOFF) values (ECVs) used for the interpretation of the minimal inhibitory concentrations (MICs) of the isolates were in accordance with the guidelines of the European Committee for Antimicrobial Susceptibility Testing (https://www.eucast.org) and the European Decision 2020/1729 [23]. Based on the ECV, the isolates were classified into wild-type (WT, without phenotypically detectable resistance) and non-WT (NWT, with phenotypically detectable resistance) categories (Table 1). HLR to gentamicin was defined as strains presenting MICs > 500 μg/mL, in accordance with the recommendations of the Clinical and Laboratory Standards Institute [24]. Isolates with phenotypically detectable resistance to three or more antimicrobials were classified as multidrug resistant (MDR). To assess the contribution of putative active efflux, the MICs of gentamicin, erythromycin and ciprofloxacin were determined in the presence or absence of the inhibitor reserpine (final concentration, 20 µg/ml; Sigma–Aldrich). The experiments were repeated three times. An efflux mechanism was inferred to be present when the antibiotic MICs in the presence of reserpine were at least 4-fold lower than the corresponding MICs in the absence of this compound [25].
2.2. Multilocus Sequence Typing (MLST)
The 56 isolates were characterized using primers and conditions for PCR amplification of seven housekeeping gene fragments (gdh, gyd, pstS, gki, aroE, xpt and yqiL) included on the website of the E. faecalis MLST database (https://pubmlst.org/organisms/enterococcus-faecalis). The MLST alleles and resulting sequence types (STs) were assigned through the submission of the amplified sequences or respective allelic profiles to the E. faecalis MLST database. Genetic diversity (GD) was calculated as the ratio of the total number of STs to the total number of isolates [26]. The MLST profiles of E. faecalis available for download from the MLST website (https://pubmlst.org/organisms/enterococcus-faecalis) were used to generate a minimum-spanning tree with Phyloviz V2.0 and the goeBURST algorithm [27]. Clonal complexes (CCs) were defined as groups of isolates that differed in no more than two of the seven loci analysed, consisting of double-locus variants (DLVs) of a founder isolate.
2.3. Whole-Genome Sequencing (WGS)
A total of 19 E. faecalis isolates were selected for WGS based on phenotypically detectable resistance to four antimicrobials (linezolid, ciprofloxacin, chloramphenicol and/or HLR to gentamicin) (Table S1). The genomic DNA of 13 isolates was sent to STAB-VIDA (Caparica, Portugal) for WGS. Genomic DNA of the isolates was extracted using the MagMax core kit (Applied Biosystems) and the KingFisher Flex System automated extraction instrument (Thermo Fisher Scientific) according to the manufacturer’s protocol. The concentration of genomic DNA was quantified and verified using the Qubit® dsDNA BR Assay Kit (Thermo Fisher Scientific). The degree of genomic DNA degradation was evaluated through agarose gel electrophoresis. The sequencing libraries were prepared using the KAPA HyperPrep Library Preparation Kit (Roche) following the manufacturer’s recommended protocol and sequenced using the Illumina NovaSeq platform with the TrueSeq Library Prep Kit (paired end 150 bp). Quality control of the raw data generated was performed using FastQC v0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc). Trimming and de novo assembly were carried out with CLC Genomics Workbench v12.0.3 (Qiagen). All assemblies were performed with an automatic word size, a similarity fraction of 0.95, a length fraction of 0.95 and a minimum contig size of 500 bp. In addition, genomic DNA from six other isolates was sent to Secugen (Madrid, Spain) for WGS. Total DNA extraction and purification were performed with the MagAttract HMW DNA kit (Qiagen). Subsequently, the quality and concentration of DNA were assessed using NanoDrop (Thermo Fisher Scientific) and Qubit (Invitrogen; Thermo Fisher Scientific) devices. Genomic libraries were prepared in accordance with the 1D native barcoding genomic DNA protocol using SKQ-LSK114 and SQK-NBD114.96 kits (Oxford Nanopore Technologies). Sequencing was performed using MinION equipment on a FLO-MIN114 vR10.4.1 flow cell at a speed of 400 bp/s (5 kHz). Long-read assemblies were carried out with Flye v2.9.1 [28] using default parameters.
Raw Illumina and nanopore sequence data were deposited under project PRJNA1277192 (https://www.ebi.ac.uk/ena).
2.4. Bioinformatic Analysis of Whole-Genome Sequences
The assembled genomes were subjected to in silico screening for ARGs and virulence genes using the genomic tools ResFinder v4.0 and VirulenceFinder v2.0, respectively (http://www.genomicepidemiology.org), with the following cut-off values: a minimum of 90% coverage and 80% identity. ARGs were also screened against the Comprehensive Antibiotic Resistance Database (https://card.mcmaster.ca/analyze/rgi) using the default criteria (perfect and strict hits only). LRE-Finder (http://www.genomicepidemiology.org) was used to detect 23S rRNA mutations and optrA, cfr, cfr(B), and poxtA genes encoding linezolid resistance in enterococci from whole-genome sequences [29]. Mobile Element Finder was used to identify the MGEs in the genomes (http://www.genomicepidemiology.org), while plasmid replicon genes, incompatibility groups and associated contigs were determined using PlasmidFinder (http://www.genomicepidemiology.org) and MOBsuite v3.0.3 [30].
2.5. Phylogenetic Analysis
Mapping and whole-genome and core-genome alignment of the 19 E. faecalis genomes were performed using Snippy v4.6.0 software. A maximum likelihood tree based on the concatenated alignment of high-quality single-nucleotide polymorphisms (SNPs) was constructed with FastTree v2.1.10 [31] using the GTR+CAT models of nucleotide evolution, and the phylogenetic tree was visualized with iTOL v6.9.1.
3. Results
3.1. Phenotypic Antimicrobial Resistance
The percentages of isolates showing the NWT phenotype to the 12 antimicrobials tested are presented in Table 1. No significant differences were found between enterococci from cloacal and pharyngeal swabs for any of the 12 antimicrobials (data not shown). Most (82.1%) of the enterococcal isolates exhibited NWT phenotypes to six antimicrobial agents (Table 2). The most prevalent NWT phenotype was to tetracycline (82.1%), followed by erythromycin (58.9%). The NWT rates for ciprofloxacin, gentamicin, chloramphenicol and linezolid ranged between 25.0% and 3.5%, whereas no NWT phenotypes were found for the remaining six antibiotics (vancomycin, teicoplanin, daptomycin, quinupristin/dalfopristin, tigecycline and ampicillin). HLR to gentamicin was detected in 13 isolates.
The antimicrobial resistance profiles are presented in Table 2. MDR was detected in more than 30% of the isolates (Table 2). MDR profiles were generally characterized by phenotypic resistance to gentamicin, tetracycline and erythromycin. The most common phenotypic resistance profile was erythromycin-tetracycline (ERY-TET, 25.0% of the isolates), followed by tetracycline (TET, 23.2% of the isolates).
3.2. Molecular Characterization of the Isolates
Overall, significant genetic heterogeneity (GD 0.55) was observed among the 56 E. faecalis isolates, which were classified into 31 STs after MLST analysis (Table 3). The most frequent genotypes were ST300, ST16 and ST648, represented by nine, five and four isolates, respectively. The other two STs included three isolates each (ST4 and ST116). Additionally, six STs were represented by two strains each (ST35, ST40, ST82, ST256, ST287 and ST706). The remaining STs were represented by single isolates.
The 19 MDR isolates also exhibited significant genetic diversity (GD 0.68) and were classified into 13 STs, all of them previously documented in the E. faecalis MLST database from different sources (Table 3). Three STs (ST16, ST116 and ST35) were detected in 52.6% of the MDR isolates (Table 3). Except for ST4, ST16 and ST82, the remaining 10 STs included only MDR isolates.
The goeBURST analyses, using all STs available on the E. faecalis MLST database website, enabled the classification of the 31 STs identified in this study into 10 CCs and one singleton based on the DLV criterion (Table 2 and Table 3). CC16 was the largest CC identified, comprising 18 STs, followed by CC863, which contained three STs. The isolates with the MDR phenotype included four CCs (CC16, CC57, CC863 and CC1688) (Table 3). CC16 included most STs containing NWT isolates to ciprofloxacin, linezolid and chloramphenicol and exhibiting HLR to gentamicin isolates (Table 4).
Pairwise SNP differences among the 19 vulture E. faecalis with NWT phenotype to ciprofloxacin, linezolid and chloramphenicol and/or with HLR to gentamicin are shown in Table S2. The minimum and maximum SNP differences were 0 and 17168, respectively. The phylogenetic tree generated on the basis of whole-genome SNP (WG-SNP) revealed three small clades for the sequence types ST116 (3 isolates), ST16 (5 isolates) and ST35 (2 isolates), whereas the other STs were genetically diverse (Figure 1).
3.3. Antimicrobial Resistance Genes and Mobile Gene Elements (Plasmids, Transposons, and Integrative and Conjugative Elements)
Table 4 shows the presence of ARGs detected in the genomes of the 19 isolates exhibiting an NWT phenotype to linezolid, ciprofloxacin and chloramphenicol and/or with HLR to gentamicin. Twelve NWT isolates to chloramphenicol carried cat genes encoding resistance to this antibiotic, cat(pC223) (58.3% isolates) and cat(pC221) (41.7%), whereas the two isolates showing phenotypic resistance to chloramphenicol and linezolid (3126-2A and 3137-2D) were found to contain genes encoding resistance to phenicols (fexA) and phenicols/oxazolidinones (optrA). The aac(6′)-Ie-aph(2”)-Ia gene conferring resistance to aminoglycosides was detected in 12 isolates with the HLR phenotype to gentamicin. Seven isolates with an NWT phenotype to ciprofloxacin carried chromosomal point mutations in the quinolone resistance-determining regions (QRDRs) of the DNA gyrase subunit A (gyrA) and topoisomerase IV subunit A (parC) genes. Three amino acid mutations were detected in the gyrA gene, two at codon 83 (Ser to Tyr [n = 1] and Ser to Ile [n = 2]) and another at codon 87 (Glu to Gly [n =4]), whereas a single amino acid mutation was found at parC codon 80 (Ser to Ile [n = 7]). This amino acid change at codon 80 in the parC gene was also detected in two WT isolates.
Genes conferring resistance to other antimicrobials included in the commercial panel used for phenotypic resistance screening were also identified. The tetM gene and tetM/tetL genes were found in 31.5% and 68.4% of the isolates with an NWT phenotype to tetracycline, respectively (Table 4). The ermB gene was identified in 94.7% of the isolates with the NWT phenotype to erythromycin.
In addition, genes conferring resistance to other antimicrobials not included in the commercial panel used for phenotypic resistance screening were also identified (Table S1). Thus, the ant(6)-Ia and str genes, which confer resistance to streptomycin, were detected in 11 and four isolates, respectively, whereas the aph(3’)-III gene, which confers resistance to kanamycin, was detected in 13 isolates. The presence of the dfrG gene, which confers resistance to trimethoprim, was identified in the genomes of 16 isolates. The lnu (lnuA, LnuB and lnuG) genes and lsa (lsaA and lsaE) genes conferring resistance to lincomycin were also frequently detected.
One isolate (841-1C) with an NWT phenotype to ciprofloxacin and erythromycin and with HLR to gentamicin but without resistance genes or chromosomal point mutations in the gyrA and parC genes was further analysed using an antibiotic/reserpine (A/R) MIC test. The isolate had lower MICs for these antibiotics after exposure to reserpine than the corresponding MICs in the absence of this compound (MICs of 16 µg/ml vs 1024 µg/ml, 0.25 µg/ml vs 16 µg/ml and <1 µg/ml vs >128 µg/ml for gentamicin, ciprofloxacin and erythromycin, respectively). The efrA and erfB genes, which encode efflux pumps for different antimicrobials, were detected in the genome of this isolate (data not shown).
Four plasmid replicon types harbouring ARGs were detected in the 19 isolates (Table 4; Table S1, Figure 1). The most prevalent was repUS43, which was detected in 16 isolates (89.5%). This replicon was found to be chromosomally integrated and co-located on the same contig with tetM (10 genomes) or tetM/temL (3 genomes; 820-1A, 828-1B and 832-1A) genes in 13 E. faecalis isolates exhibiting an NWT to tetracycline. In addition, cat genes, together with tetM/temL, were bound to repUS43 in three genomes (822-2A, 824-1A and 825-1C). The tetL gene was also associated with the rep22-type plasmid, whereas the str, aph(3’)-III, ant(6)-Ia, ermB and cat genes were associated with rep7a (Table 4; Table S1, Figure 1). The repUS40 replicon was associated with the optrA and fexA genes in both isolates exhibiting NWT phenotypes to linezolid and chloramphenicol.
In addition, other MGEs, such as transposons (Tns) and composite transposons (ComTn), were also associated with ARGs. Tn6009 was detected in 57.9% of the isolates. This MGE was associated with repUS43 and tetracycline resistance genes. The lnuG gene was found to be embedded in Tn6260 in five genomes, whereas ComTn, cn_43171_ISS1N, was linked to tetracycline (tetM, tetL) and macrolide (ermB) resistance genes in one genome.
3.4. Virulence Factors
A total of 22 virulence factors were found among the 19 genomes (Figure 1). All isolates harboured genes encoding sex pheromone-associated proteins (cad, cCF10, camE, cOB1), protection against oxidative stress (tpx), cell wall adhesion (efaAfs), biofilm-associated pili (ebpA, ebpB, ebpC) and the cell wall anchor surface protein sortase A (srtA). Genes associated with the cytolysin toxin (cylA, cylB, cylL and cylM) were identified in seven isolates (12.5%). Other virulence genes identified in most of the 19 isolates were elrA (Rgg-like regulator gene associated with macrophage persistence; 94.7% of the isolates), agg (aggregation substance; 57.9% of the isolates), frsB (quorum-sensing regulator; 63.2% of the isolates), gelE (gelatinase toxin; 63.2% of the isolates), ace (collagen adhesion precursor; 78.9% of the isolates), hylA and hylB (hyaluronidase genes; 73.7% and 47.4% of the isolates, respectively). The espfs gene (enterococcal surface protein) was identified in 5.3% of the isolates. Genes associated with the cytolysin toxin (cylA, cylB, cylL and cylM) were found only in the ST16, ST82, ST1291 and ST1768 isolates.
Some virulence genes were associated with MGEs (Figure 1). Thus, the cCF10 and cad genes bound to repUS43 in three and one isolates, respectively. In addition, the agg gene bound to replicons rep9b and rep9c, in two isolates each, and the tpx gene to transposon Tn6260, in five isolates.
4. Discussion
The impact of wild animals as reservoirs of AMR and ARGs, that can later be disseminated among different hosts, and the environment, has received particular attention from the “One Health” perspective in recent years [3]. Thus, the present study investigated the prevalence of AMR in a collection of 56 E. faecalis isolates from the pharynx and cloaca of Eurasian griffon vultures. These birds were chosen because of their wide geographic distribution and their scavenging habits, and because they live in close proximity to humans. In addition, enterococci are widely considered key microbiota indicators for tracing the spread and evolution of MDR bacteria in environments and wildlife [9]; furthermore, E. faecalis is a common inhabitant of the cloacal and pharyngeal microbiota of Eurasian griffon vultures [19].
Different studies have shown high rates of AMR and MDR in enterococci from various wild bird species, including vultures [14,32,33]. In this study, a high percentage of E. faecalis isolates (82.1%) exhibited NWT phenotypes to six antimicrobials, indicating the widespread presence of AMR in isolates of E. faecalis in this vulture population. The highest resistance rates were detected for erythromycin and tetracycline (Table 1), and one-third of the isolates were phenotypically MDR (Table 2). Accordingly, the most common antimicrobial resistance profile observed was resistance to erythromycin and tetracycline (Table 2), which is in line with previous studies on E. faecalis collected from different animal and human sources [8,34]. Of special concern is the detection of isolates with an NWT phenotype to linezolid, chloramphenicol and ciprofloxacin and with HLR to gentamicin (Table 1), antimicrobials considered clinically important in human medicine [21]. Furthermore, over half (63%; 12 out 19 isolates) of the MRD isolates exhibited an NWT phenotype to at least two of these antimicrobials (Table 2). E. faecalis strains with a resistant phenotype to any of these four antimicrobial agents have been reported in various wild bird species, including vultures [6,16,32].
Human activities and food-animal production can contribute to the development and spread of AMR in the environment, which has a negative impact on wildlife. Thus, vultures living in close contact with anthropogenically impacted areas or high densities of livestock may be colonized by ARB strains, which are likely selected by antimicrobial agents used in humans and domestic animals [35]. In this way, the rates of E. faecalis with an NWT phenotype to antimicrobials authorized only for human use [21], such as linezolid, could be linked to the exposure of these vultures to antimicrobials via contaminated water [36]. The feeding habits of Eurasian griffon vultures can also contribute to the acquisition of ARBs. These animals feed almost exclusively on carrion, primarily mammals from intensive farming that have died from disease or accidents and are discarded at predictable sites that represent an abundant food source [37]. Consequently, the concentration of vultures at these feeding sites may facilitate their exposure to active antimicrobial residues that are present in the carcasses of medicated livestock [13]. Erythromycin and tetracycline are widely used in veterinary medicine in Spain (https://www.aemps.gob.es/), and the direct feeding of livestock carcasses treated with these antimicrobials could explain the high rates of E. faecalis isolates with the NWT phenotype to both antimicrobials (Table 2). Once ARBs are introduced into wildlife, they can persist for extended periods even in the complete absence of selection pressure from antimicrobial agents [38]. These bacteria can also spread to other species and environments, which could explain the relatively high rates of E. faecalis isolates with an NWT phenotype to chloramphenicol (Table 1), despite the ban on its use in humans, pets and non-food-producing animals in the mid-1990s in the EU [https://www.aemps.gob.es/, 39].
MLST analysis identified 31 STs with high genetic heterogeneity (GD 0.55) among the 56 E. faecalis isolates. The genetic diversity was similar between non-MRD and MRD E. faecalis isolates (GD 0.56 and GD 0.68, respectively). Despite the similar genetic heterogeneity of the MRD and non-MRD isolates, most STs identified in MRD isolates (10 out of 13) were detected exclusively in these isolates. Similarly, 19 of the 21 STs identified in the non-MRS isolates were detected in only these isolates (Table 3). Moreover, ST16 and ST116 accounted for almost 40% of the MRD isolates, whereas only one non-MRD isolate belonged to either of these two genotypes. These results suggest a different genetic background for the E. faecalis populations of MRD and non-MRD isolates. Additionally, SNP differences among the 19 E. faecalis isolates from vultures with an NWT to ciprofloxacin, linezolid and chloramphenicol and/or with HLR to gentamicin also indicated high genetic heterogeneity. Except for isolates 3126-2A and 3137-2D, both belonging to ST116, all other 17 isolates exhibited SNP differences greater than 100 SNP, which is above the proposed cut-off value for considering isolates as clonal [40]. Thus, the genetic diversity observed by MLST suggests that the high rates of antimicrobial resistance detected in the E. faecalis isolates are more likely a consequence of exposure to multiple strains rather than the clonal spread of resistant isolates. All the STs identified have been detected previously in animals, humans, foods and the environment according to the information available on the E. faecalis MLST database website (https://pubmlst.org/organisms/enterococcus-faecalis). These data suggest that the detection of NWT phenotypes to different antimicrobial agents in the E. faecalis vulture isolates is likely a consequence of anthropogenic ARB contamination of the ecosystems inhabited by vultures [41].
Given the importance of understanding AMR mechanisms, the genomes of a subset of 19 selected isolates with an NWT phenotype to the clinically important antimicrobials linezolid, chloramphenicol and ciprofloxacin and with HLR to gentamicin were sequenced, and an in-depth analysis of their genetic mechanisms of resistance was performed. Several ARGs encoding resistance to chloramphenicol and gentamicin were identified on the chromosomes of the E. faecalis isolates (Table 4). Notably, the aac(6’)-Ie-aph(2″)-Ia gene was detected in most isolates (92.3%) with an HLR phenotype to gentamicin, which is consistent with [42] that described this resistance gene as the most frequently modifying enzyme underlying HLR to aminoglycosides in enterococci. The NWT phenotype to chloramphenicol was associated with the cat(pC221) and cat(pC223) genes (Table 4), which are catA variants that have been detected in a wide variety of bacteria [39]. Both isolates with an NWT phenotype to chloramphenicol and linezolid carried the optrA and fexA genes in their genomes (Table 4), which are the main mechanisms of resistance of Enterococcus to oxazolidinone and phenicol antimicrobials [43]. Seven of the eight isolates with an NWT phenotype to ciprofloxacin and MICs of ≥16 µg/ml exhibited amino acid changes in the GyrA (S83I or S83Y, and E87G) and ParC (S80I) proteins, which have been previously reported in ciprofloxacin-resistant E. faecalis isolates [44,45].
ARGs to other antimicrobials included in the commercial panel that exhibited NWT phenotypes were also identified on the chromosome of the E. faecalis isolates. Thus, the NWT phenotype to erythromycin and tetracycline was associated with the detection of the emrB (94.7% isolates) and tetM/tetL genes, in accordance with previous studies, which indicated that these genes are the most prevalent among erythromycin- and tetracycline-resistant enterococci [6,32]. We also identified other genes associated with resistance to antimicrobials not included in the commercial panel, such as the aph(3’)-III gene, which confers resistance to kanamycin and amikacin, and the ant(6)-Ia and str genes, which are associated with resistance to streptomycin [46,47]. However, phenotypic resistance to these antimicrobials was not tested in this study. The efrA, erfB and lsa genes, encoding efflux pumps for different antimicrobials in E. faecalis isolates [48], were detected in the genome of one isolate with an NWT phenotype to ciprofloxacin and erythromycin and with HLR to gentamicin but without resistance genes or chromosomal point mutations in the gyrA and parC genes. The decrease in the MIC values for these antimicrobials by six- to seven-fold in the presence of reserpine versus in the absence of this compound suggests that antibiotic efflux pumps are involved in resistance to these antimicrobials [49]. Although the efflux pump system has not been involved in erythromycin resistance in enterococci, it has been reported in other gram-positive bacteria, such as streptococci [25].
Enterococci readily acquire ARGs through MGEs such as transposons or plasmids, which play a key role in the acquisition and dissemination of ARGs via HGT [50]. In addition to chromosomal ARGs, MGEs, including plasmid replicons and transposons, were identified among the E. faecalis vulture isolates, suggesting the potential of these isolates to acquire and transfer AMR. Notably, resistance genes to chloramphenicol, linezolid, tetracycline, erythromycin, streptomycin and kanamycin more frequently co-existed in different replicons and transposons (Figure 1; Table S1). Chloramphenicol resistance genes were detected in rep7a, repUS43 and rpUS40 replicons. Tetracycline resistance genes (tetM, tetL) were detected in transposons cn_43171_ISS1N and Tn6009 and in the plasmid replicons repUS43 and rep22, whereas a macrolide resistance gene (ermB) was also detected in cn_43171_ISS1N and rep7a (Table 4). The association between the tetM and tetL genes and the repUS43 replicon, as well as with Tn6009, has been previously reported [45,51]. Tn6260 was associated with the lnuG gene (Table 4), which is involved in resistance to lincosamide, in agreement with previous studies [52,53]. The detection of ARGs for different antimicrobials in the same MGE has also been reported [51]. In line with the observations of these authors, we found that different MGEs carried the same ARG and that different ARGs were carried by the same MGE, which could increase their dissemination potential. Thus, genes conferring resistance to different classes of antimicrobial agents were found in six isolates co-located with a plasmid replicon on the same contig (31.6%; Table S1): three isolates carried the tetM, tetL and cat(pC223) genes located in tandem close to the plasmid replicon repUS43; two isolates carried the fexA and optrA genes on the repUS40 replicon; and one isolate carried the ant(6)-Ia, aph(3’)-III, ermB and cat(pC221) genes on the same contig as the rep7a replicon. Other resistance genes located on MGEs not reported previously in E. faecalis were also detected in this study. Notably, rep22 carried the tetL gene, which has been reported in other Gram-positive bacteria, such as Macrococcus caseolyticus [54], while the optrA and fexA genes located on the repUS40 plasmid replicon have been reported in Enterococcus hirae and Enterococcus casseliflavus [53,55]. In this study, ComTra with ISS1N on both sides was linked to the tetM, tetL and ermB genes in one isolate. This MGE has been reported in Listeria [56] but not in Enterococcus or in association with resistance genes. Thus, it is the first description of these three MGEs (rep22, repUS40 and ComTra) carrying antimicrobial resistance genes in E. faecalis. The acquisition of ARGs carried by MGEs can lead to the establishment of multidrug resistance [57]. In this context, the detection of different MGEs in the 19 sequenced isolates could explain why one-third of the 56 isolates were MDR (Table S1; Figure 1). According to the available data (https://www.ncbi.nlm.nih.gov), all replicons and transposons identified in the present study have been reported previously in E. faecalis and other enterococcal species isolated from humans, foods and livestock. Hence, the detection of E. faecalis isolates in vultures with NWT phenotypes to several antimicrobials could result from the exchange of ARBs and/or ARGs from these sources to vultures. This finding also supports the MLST data, suggesting that AMR in these vultures is likely a consequence of anthropogenic pollution.
Most enterococci are not virulent and are considered relatively harmless, with little potential for human infection. However, they have also been identified as nosocomial opportunistic pathogens with increased resistance to antimicrobial-approved agents [58]. The 19 isolates investigated in this study harboured 22 different virulence-related genes (Figure 1), which could facilitate colonization and cause infections [59]. In particular, several isolates carried genes (cylA, cylB, cylL and cylM) associated with cytolysin production, which is related to increased severity of infection in humans [59]. These genes are more prevalent among ST16 isolates [42], the second most frequently detected genotype in our study. In addition, vulture isolates harboured genes encoding sex pheromone-associated proteins, protection against oxidative stress, cell wall adhesion, biofilm formation and macrophage persistence (Figure 1), which are also involved in enterococcal pathogenicity [59]. Some virulence genes were found on the same MGEs, such as the replicon repUS43 and the transposon Tn6260, which simultaneously carried ARGs. The co-presence of ARGs and virulence-related genes on the same MGE could facilitate the ability of E. faecalis vulture isolates to survive under antibiotic pressure and increase their pathogenic potential [58]. Consequently, they could represent a risk factor for domestic animals and humans.
5. Conclusions
Overall, the results of this study show that a significant proportion of E. faecalis strains recovered from cloacal or pharyngeal samples of vultures were phenotypically MDR and harboured MGEs (plasmid replicons, transposons and composite transposons) that carried AMR and virulence-associated genes. These findings are cause for concern, since vultures may act as spreaders of virulence and antimicrobial resistance genes to the environment and even to other hosts.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Phenotypic, genomic antimicrobial resistance and mobile genetic elements associated with antimicrobial resistance genes in 19 E. faecalis isolates investigated in this study; Table S2: Pairwise SNP differences between E. faecalis isolates with a non-wide type to ciprofloxacin, linezolid, chloramphenicol and/or high-level resistance to gentamycin.
Author Contributions
Conceptualization, L.C. and L.D.; methodology, A.B., A.C., J.M.C. and M.A; software, C.S.; validation, J.F.F.G. and A.I.V,; formal analysis, A.I.V.; investigation, A.I.V.; resources, A.I.V.; data curation, C.S.; writing—original draft preparation, A.I.V.; writing—review and editing, J.F.F.G.; visualization, L.C. and L.D.; supervision, A.I.V.; project administration, A.I.V.; funding acquisition, A.I.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Santander-Universidad Complutense de Madrid, grant number PR12/24-31585.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The genomic sequencing data generated throughout this research have been deposited under project PRJNA1277192, publicly available in the National Center for Biotechnology Information (NCBI) Database.
Conflicts of Interest
The authors declare no conflicts of interest
Abbreviations
The following abbreviations are used in this manuscript:
| AMR | Antimicrobial resistance |
| ARBs | Antibiotic-resistant bacteria |
| ARGs | AMR genes |
| HGT | Horizontal gene transfer |
| MGEs | Mobile genetic elements |
| HLR | High-level resistance |
| ECOFF | Epidemiological cut-off |
| ECVs | Epidemiological cut-off F values |
| MICs | Minimal inhibitory concentrations |
| WT | Wild-type |
| NWT | non-WT |
| MDR | Multidrug resistant |
| MLST | Multilocus sequence typing |
| STs | Sequence types |
| GD | Genetic diversity |
| CCs | Clonal complexes |
| DLVs | Double-locus variants |
| WGS | Whole-genome sequencing |
| SNPs | Single-nucleotide polymorphisms |
| TET | Tetracycline |
| ERY-TET | Erythromycin- tetracycline |
| A/R | Antibiotic/reserpine |
| Tns | Transposons |
| ComTn | Composite transposons |
References
- Antimicrobial Resistance Collaborators (AMR Col). Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Antimicrobial resistance: global report on surveillance. Geneva, Switzerland, World Health Organization 2014.
- Dias, D. , Fonseca, C., Caetano, T., Mendo, S. Oh, deer! How worried should we be about the diversity and abundance of the faecal resistome of red deer? Sci. Total Environ. 2022, 825, 153831. [Google Scholar] [CrossRef] [PubMed]
- Dolejska, M. , Guenther, I. Wildlife is overlooked in the epidemiology of medically important antibiotic-resistant bacteria. Antimicrob. Agents Chemother. 2019, 63, e0116719. [Google Scholar] [CrossRef]
- Allen, H.K. , Donato, J., Wang, H.H., Cloud-Hansen, K.A., Davies, J., Handelsman, J. Call of the wild: antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Santos, T. , Silva, N., Igrejas, G., Rodrigues, P., Micael, J., Rodrigues, T., Resendes, R., Gonçalves, A., Marinho, C., Gonçalves, D., Cunha, R., Poeta, P. Dissemination of antibiotic resistant Enterococcus spp. and Escherichia coli from wild birds of Azores Archipelago. Anaerobe 2013, 24, 25–31. [Google Scholar] [PubMed]
- Vittecoq, M. , Godreuil, S., Prugnolle, F., Durand, P., Brazier, L., Renaud, N., Arnal, A., Aberkane, S., Jean-Pierre, H., Gauthier-Clerc, M., Thomas, F., Renaud, F. Antimicrobial resistance in wildlife. J. Appl. Ecol. 2016, 53, 519–529. [Google Scholar] [CrossRef]
- Stępień-Pyśniak, D. , Hauschild, T., Nowaczek, A., Marek, A., Dec, M. Wild birds as a potential source of known and novel multilocus sequence types of antibiotic-resistant Enterococcus faecalis. J. Wildl. Dis 2018, 54, 219–228. [Google Scholar] [CrossRef]
- Radhouani, H. , Silva, N., Poeta, P., Torres, C., Correia, S., Igrejas, G. Potential impact of antimicrobial resistance in wildlife, environment and human health. Front. Microbiol. 2014, 5, 1–12. [Google Scholar] [CrossRef]
- Benavides, J.A. , Salgado-Caxito, M., Torres, C., Godreuil, S. Public health implications of antimicrobial resistance in wildlife at the One Health Interface. Med. Sci. Forum 2024, 25, 1–7. [Google Scholar]
- Slotta-Bachmayr, L., Bögel, R., Camiña, C.A. The Eurasian Griffon vulture in Europe and the Mediterranean. Status Report & Action Plan. EGVWG; Salzburg, Austria, 2005.
- Pirastru, M. , Mereu,, P., Manca, L., Bebber, D., Naitana, S., Leoni, G.G. Anthropogenic drivers leading to population decline and genetic preservation of the Eurasian Griffon vulture (Gyps fulvus). Life (Basel) 2021, 11, 1038. [Google Scholar]
- Blanco, G. , Bautista, L.M. Avian Scavengers as Bioindicators of Antibiotic Resistance Due to Livestock Farming Intensification. Int. J. Environ. Res. Public. Health 2020, 2020 17, 3620. [Google Scholar] [CrossRef]
- Tallon, A.K. , Smith, R.K., Rush, S., Naveda-Rodriguez, A., Brooks, J.P. The role of New World vultures as carriers of environmental antimicrobial resistance. BMC Microbiol. 2024, 24, 487. [Google Scholar] [CrossRef]
- Blanco, G. Supplementary feeding as a source of multiresistant Salmonella in endangered Egyptian vultures. Transbound. Emerg. Dis. 2018, 65, 806–816. [Google Scholar] [CrossRef]
- González-Martín, M.R. , Suárez-Pérez, A., Álamo-Peña, A., Valverde Tercedor, C., Corbera, J.A., Tejedor-Junco, M.T. Antimicrobial susceptibility of enterococci isolated from nestlings of wild birds feeding in supplementary feeding stations: the case of the Canarian Egyptian vulture. Pathogens 2024, 13, 855. [Google Scholar] [PubMed]
- European Food Safety Authority, European Centre for Disease Prevention and Control (EFSA). The European Union summary report on antimicrobial resistance in zoonotic and indicator bacteria from humans, animals and food in 2011. EFSA J. 2013, 11, 3196. [Google Scholar] [CrossRef]
- Ellerbroek, L. , Mac, K.N., Peters, J., Hultquist, L. Hazard potential from antibiotic-resistant commensals like Enterococci. J. Vet. Med. B Infect. Dis. Vet. Public Health 2004, 51, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Vela, A.I. , Casas-Díaz, E., Fernández-Garayzábal, J.F., Serrano, E., Agustí, S., Porrero, M.C., Sánchez del Rey, V., Marco, I., Lavín, S., Domínguez, L. Estimation of cultivable bacterial diversity in the cloacae and pharynx in Eurasian Griffon vultures (Gyps fulvus). Microb. Ecol. 2015, 69, 597–607. [Google Scholar]
- Partridge, S.R. , Kwong, S.M., Firth, N., Jensen, S.O. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 2018, 31, e00088–17. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). WHO List of Medically Important Antimicrobials: A risk management tool for mitigating antimicrobial resistance due to non-human use. Geneva, Switzerland, 2024.
- Clinical and Laboratory Standards Institute (CLSI). Performance standards for antimicrobial disk and dilution susceptibility tests for bacteria isolated from animals, fourth edition. CLSI Approved Standard M31-A4; Clinical and Laboratory Standards Institute: Wayne, PA, 2013. [Google Scholar]
- European Union. Commission Implementing Decision (EU) 2020/1729 of 17 November 2020 on the monitoring and reporting of antimicrobial resistance in zoonotic and commensal bacteria and repealing Implementing Decision 2013/652/EU (2020/1729/EU). Off. J. Eur. Union 2020, 8–21.
- Clinical and Laboratory Standards Institute (CLSI). Performance standards for antimicrobial susceptibility testing. CLSI Approved Standard M100-S15; Clinical and Laboratory Standards Institute: Wayne, PA, 2018. [Google Scholar]
- Bast, D.J. , Low, D.E., Duncan, C.L., Kilburn, L., Mandell, L.A., Davidson, R.J., de Azavedo, J.C.S. Fluoroquinolone resistance in clinical isolates of Streptococcus pneumoniae: Contributions of type II topoisomerase mutations and efflux to levels of resistance. Antimicrob. Agents Chemother. 2000, 44, 3049–3054. [Google Scholar] [CrossRef]
- Martinez, G. , Harel, J., Lacouture, S., Gottschalk, M. Genetic diversity of Streptococcus suis serotypes 2 and 1/2 isolates recovered from carrier pigs in closed herds. Can. J. Vet. Res. 2002, 66, 240–248. [Google Scholar]
- Nascimento, M. , Sousa, A., Ramirez, M., Francisco, A.P., Carriço, J.A., Vaz, C. PHYLOViZ 2.0: Providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 2017, 33, 128–129. [Google Scholar] [CrossRef]
- Kolmogorov, M. , Yuan, J., Lin, Y., Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Hasman, H. , Clausen, P.T.L.C., Kaya, H., Hansen, F., Knudsen, J.D., Wang, M., Holzknecht, B.J., Samulioniene, J., Roeder, B., Frimodt-Møller, N., Lund, O., Hammerum, A.M. LRE-Finder, a web tool for detection of the 23S rRNA mutations, and the optrA, cfr, cfr(B) and poxtA genes, encoding linezolid resistance in Enterococci from whole genome sequences. J. Antimicrob. Chemother. 2019, 74, 1473–1476. [Google Scholar]
- Robertson, J. , Nash, J.H.E. MOB-suite: Software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genomics 2018, 4, e000206. [Google Scholar] [CrossRef]
- Price, M.N. , Dehal, P.S., Arkin, A.P. FastTree 2 –Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Cagnoli, G. , Bertelloni, F., Interrante, P., Ceccherelli, R., Marzoni, M., Ebani, V.V. Antimicrobial-resistant Enterococcus spp. in wild avifauna from central Italy. Antibiotics 2022, 11, 852. [Google Scholar] [CrossRef]
- Freitas, A.A.R. , Faria, A.R., Mendes, L.T., Merquior, V.L.C., Neves, D.M., Pires, J.R., Teixeira, L.M. The gut microbiota of wild birds undergoing rehabilitation as a reservoir of multidrug resistant enterococci in a metropolitan area in Brazil. Braz. J. Microbiol. 2024, 55, 3849–3861. [Google Scholar] [CrossRef]
- Thu, W.P. , Sinwat, N., Bitrus, A.A., Angkittitrakul, S., Prathan, R., Chuanchuen, R. Prevalence, antimicrobial resistance, virulence gene, and class 1 integrons of Enterococcus faecium and Enterococcus faecalis from pigs, pork and humans in Thai-Laos border provinces. J. Glob. Antimicrob. Resist. 2019, 18, 130–138. [Google Scholar]
- Literak, I. , Dolejska, M., Janoszowska, D., Hrusakova, J., Meissner, W., Rzyska, H., Bzoma, S., Cizek, A. Antibiotic-resistant Escherichia coli bacteria, including strains with genes encoding the extended-spectrum beta-lactamase and QnrS, in water-birds on the Baltic Sea coast of Poland. Appl. Environ. Microbiol. 2010, 76, 8126–8134. [Google Scholar] [PubMed]
- Guenther, S. , Grobbel, M., Heidemanns, K., Schlegel, M., Ulrich, R.G., Ewers, C., Wieler, L.H. First insights into antimicrobial resistance among faecal Escherichia coli isolates from small wild mammals in rural areas. Sci. Total Environ. 2010, 408, 3519–3522. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Avizanda, G. , Blanco, B., Deault, T.L., Markandya, A., Virani, M.Z., Donázar, J.A. Supplementary feeding and endangered avian scavengers: Benefits, caveats and controversies. Front. Ecol. Environ. 2016, 14, 191–199. [Google Scholar] [CrossRef]
- Sunde, M. , Ramstad, S.N., Rudi, K., Porcellato, D., Ravi, A., Ludvigsen, J., das Neves, C.G., Tryland, M., Ropstad, E., Slettemeås, J.S., Telke, A.A. Plasmid-associated antimicrobial resistance and virulence genes in Escherichia coli in a high Arctic reindeer subspecies. J. Glob. Antimicrob. Resist. 2021, 26, 317–322. [Google Scholar]
- Schwarz, S. , Kehrenberg, C., Doublet, B., Cloeckaert, A. Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol. Rev. 2004, 28, 519–542. [Google Scholar] [CrossRef]
- van Engelen, E. , Mars, J., Dijkman, R. Molecular characterisation of Mycoplasma bovis isolates from consecutive episodes of respiratory disease on Dutch veal farms. Vet. Microbiol. 2024, 298, 56–57. [Google Scholar] [CrossRef]
- Martinez, J.L. Environmental pollution by antibiotics and by antibiotic resistance determinants. Environ. Pollut. 2009, 157, 2893–2902. [Google Scholar] [CrossRef]
- Farman, M. , Yasir, M., Al-Hindi, R.R., Farraj, S.A., Jiman-Fatani, A.A., Alawi, M., Azhar, E. Genomic analysis of multi-drug-resistant clinical Enterococcus faecalis isolates for antimicrobial resistance genes and virulence factors from the western region of Saudi Arabia. Antimicrob. Resist. Infect. Control 2019, 8, 55. [Google Scholar] [PubMed]
- Wang, Y. , Lv, Y., Cai, J., Schwarz, S., Cui, L., Hu, Z., Zhang, R., Li, J., Zhao, Q., He, T., Wang, D., Wang, Z., Shen, Y., Li, Y., Feßler, A.T., Wu, C., Yu, H., Deng, X., Xia, X., She, J. A novel gene, optrA, that confers transferable resistance to oxazolidinones and phenicols and its presence in Enterococcus faecalis and Enterococcus faecium of human and animal origin. J. Antimicrob. Chemother. 2015, 70, 2182–2190. [Google Scholar] [PubMed]
- Petersen, A. , Jensen, L.B. Analysis of gyrA and parC mutations in enterococci from environmental samples with reduced susceptibility to ciprofloxacin. FEMS Microbiol. Lett. 2004, 231, 73–76. [Google Scholar] [CrossRef]
- Amuasi, G.R. , Dsani, E., Owusu-Nyantakyi, C., Owusu, F.A., Mohktar, Q., Nilsson, P., Adu, B., Hendriksen, R.S., Egyir, B. Enterococcus species: Insights into antimicrobial resistance and whole-genome features of isolates recovered from livestock and raw meat in Ghana. Front. Microbiol. 2023, 14, 1254896. [Google Scholar]
- Shaw, K.J. , Rather, P.N., Hare, R.S., Miller, G.H. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 1993, 57, 138–163. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M. , Arias, C.A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 2016, 4, VMBF–0016. [Google Scholar] [CrossRef]
- Lee, E.W. , Huda, N., Kuroda, T., Mizushima, T., Tsuchiya, T. EfrAB, an ABC multidrug efflux pump in Enterococcus faecalis. Antimicrob. Agents Chemother. 2003, 47, 3733–3738. [Google Scholar]
- Shiadeh, S.M.J. , Azimi, L., Azimi, T., Pourmohammad, A., Goudarzi, M., Chaboki, B.G., Hashemi, A. Upregulation of efrAB efflux pump among Enterococcus faecalis ST480, ST847 in Iran. Acta Microbiol. Immunol. Hung. 2020, 67, 187–192. [Google Scholar] [CrossRef]
- Iweriebor, B.C. , Obi, L.C., Okoh, A.I. Virulence and antimicrobial resistance factors of Enterococcus spp. isolated from fecal samples from piggery farms in Eastern Cape, South Africa ecological and evolutionary microbiology. BMC Microbiol. 2015, 15, 136. [Google Scholar] [CrossRef]
- Fatoba, D.O. , Amoako, D.G., Akebe, A.L.K., Ismail, A., Essack, S.Y. Genomic analysis of antibiotic-resistant Enterococcus spp. reveals novel enterococci strains and the spread of plasmid-borne tet(M), Tet(L) and erm(B) genes from chicken litter to agricultural soil in South Africa. J. Environ. Manag. 2022, 302, 114101. [Google Scholar]
- Zhu, X.Q. , Wang, X.M., Li, H., Shang, Y.H., Pan, Y.S., Wu, C.M., Wang, Y., Du, X.D., Shen, J.Z. Novel lnu(G) gene conferring resistance to lincomycin by nucleotidylation, located on Tn6260 from Enterococcus faecalis E531. J. Antimicrob. Chemother. 2017, 72, 993–999. [Google Scholar]
- Abdullahi, I.N. , Lozano, C., Zarazaga, M., Latorre-Fernández, J., Hallstrøm, S., Rasmussen, A., Stegger, M., Torres, C. Genomic characterization and phylogenetic analysis of linezolid-resistant Enterococcus from the nostrils of healthy hosts identifies zoonotic transmission. Curr. Microbiol. 2024, 81, 225. [Google Scholar]
- Zhang, Y. , Min, S., Sun, Y., Ye, J., Zhou, Z., Li, H. Characteristics of population structure, antimicrobial resistance, virulence factors, and morphology of methicillin-resistant Macrococcus caseolyticus in global clades. BMC Microbiol. 2022, 22, 266. [Google Scholar] [CrossRef]
- Biggel, M. , Nüesch-Inderbinen, M., Jans, C., Stevens, M.J.A., Stephan, R. Genetic context of optrA and poxtA in florfenicol-resistant enterococci isolated from flowing surface water in Switzerland. Antimicrob. Agents Chemother. 2021, 65, e0108321. [Google Scholar] [CrossRef]
- Rivu, S. , Shourav, A.H., Ahmed, S. Whole genome sequencing reveals circulation of potentially virulent Listeria innocua strains with novel genomic features in cattle farm environments in Dhaka, Bangladesh. Infect. Genet. Evol. 2024, 126, 105692. [Google Scholar] [CrossRef]
- Nikaido, H. Multidrug resistance in bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef] [PubMed]
- Ramos, S. , Silva, V., Dapkevicius, M.d.L.E., Igrejas, G., Poeta, P. Enterococci, from harmless bacteria to a pathogen. Microorganisms 2020, 8, 1118. [Google Scholar] [PubMed]
- Jett, B.D. , Huycke, M.M., Gilmore, M.S. Virulence of Enterococci. Clin. Microbiol. Rev. 1994, 7, 462–478. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Phylogenetic tree generated from whole-genome SNP (WG-SNP) analysis of 19 non-wide type vulture enterococci with resistance to ciprofloxacin, linezolid and chloramphenicol and/or high-level resistance to gentamicin. The figure shows the in silico identification of virulence-associated genes, antimicrobial resistance genes and mobile genetic elements (MGEs). The colour of the symbols and leaves link ARGs and virulence genes to the corresponding MGE. Except for isolates 822-2A, 824-1A, 825-1C, 828-1B and 835-1B, repUS43 and Tn6009 were found co-located with tetracycline resistance genes. Abbreviations: Amg, Aminoglycoside; Mac-Linc-Strep, Macrolide-Lincosamide-Streptogramin; Phe, Phenicols; Oxa-Phe, Oxazolidinone-Phenicols; Tet, Tetracyclines; Tri; Trimethoprim; Qui, Quinolones; REP, replicon type; Tn, Transposon; ComTn, Composite Transposon.
Figure 1.
Phylogenetic tree generated from whole-genome SNP (WG-SNP) analysis of 19 non-wide type vulture enterococci with resistance to ciprofloxacin, linezolid and chloramphenicol and/or high-level resistance to gentamicin. The figure shows the in silico identification of virulence-associated genes, antimicrobial resistance genes and mobile genetic elements (MGEs). The colour of the symbols and leaves link ARGs and virulence genes to the corresponding MGE. Except for isolates 822-2A, 824-1A, 825-1C, 828-1B and 835-1B, repUS43 and Tn6009 were found co-located with tetracycline resistance genes. Abbreviations: Amg, Aminoglycoside; Mac-Linc-Strep, Macrolide-Lincosamide-Streptogramin; Phe, Phenicols; Oxa-Phe, Oxazolidinone-Phenicols; Tet, Tetracyclines; Tri; Trimethoprim; Qui, Quinolones; REP, replicon type; Tn, Transposon; ComTn, Composite Transposon.
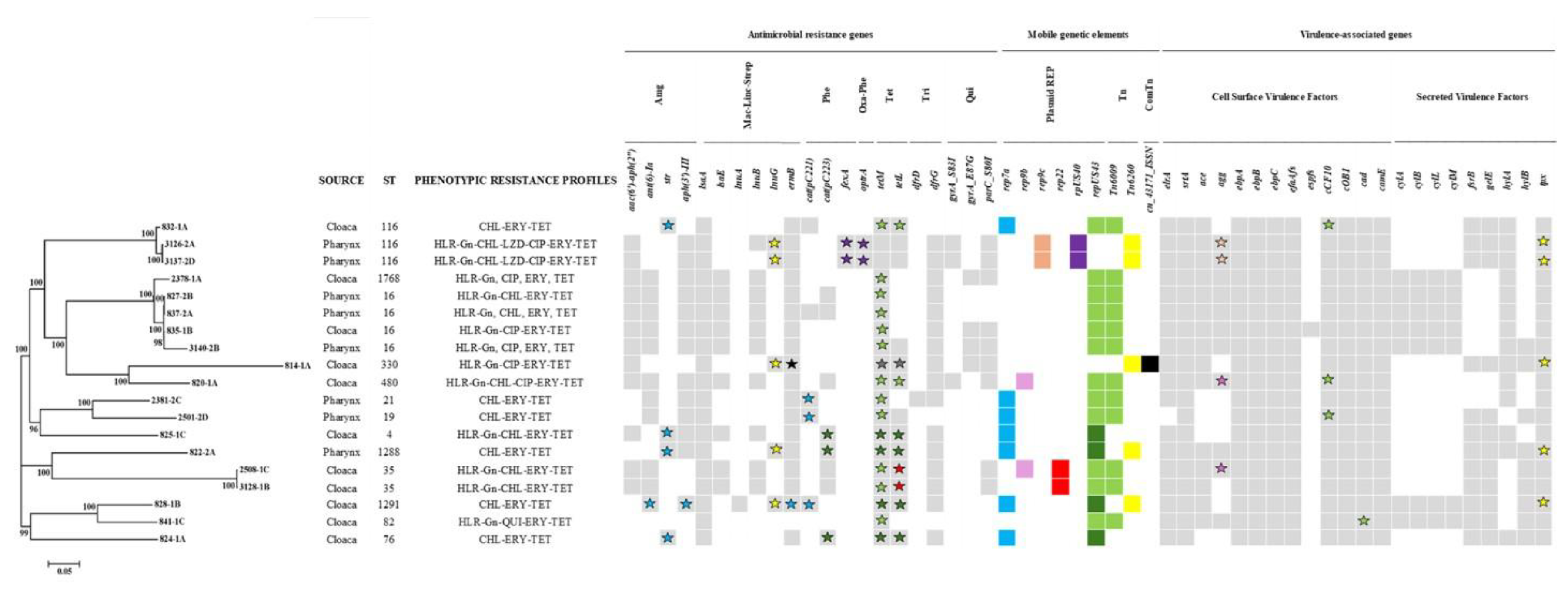

Table 1.
Minimal inhibitory concentrations for 12 antimicrobial agents of E. faecalis isolates from Eurasian griffon vultures.
Table 1.
Minimal inhibitory concentrations for 12 antimicrobial agents of E. faecalis isolates from Eurasian griffon vultures.
| Class | Antimicrobial | No. of isolates with MIC of (μg/ml) | % non-WT isolates | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.03 | 0.06 | 0.12 | 0.25 | 0.50 | 1 | 2 | 4 | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 1024 | ≥2056 | |||
| β-Lactam | AMP | <2 | 38 | 16 | > | 0 | |||||||||||||
| Quinolone | CIP | < | 3 | 35 | 9 | 1 | 2> | 6 | 14.3 | ||||||||||
| Macrolide | ERY | <10 | 8 | 5 | 2 | > | 31 | 58.9 | |||||||||||
| Aminoglycoside | GEN | <31 | 11 | 1 | 1a> | 12a | 25.0 | ||||||||||||
| Liopeptide | DAP | <1 | 4 | 39 | 12 | > | 0 | ||||||||||||
| Tetracycline | TET | <10 | 1 | 1 | 5 | 37> | 2 | 82.1 | |||||||||||
| TGC | < | 16 | 32 | 8 | > | 0 | |||||||||||||
| Phenicol | CHL | <2 | 38 | 1 | 1 | 9 | 5> | 25.0 | |||||||||||
| Oxazolidinone | LZD | < | 8 | 45 | 1 | 2 | > | 3.6 | |||||||||||
| Streptogramin | SYN | <3 | 1 | 1 | 10 | 34 | 7 | > | 0 | ||||||||||
| Glycopeptide | TEI | <56 | > | 0 | |||||||||||||||
| VAN | <29 | 20 | 7 | > | 0 | ||||||||||||||
|, lines indicate epidemiological cut-off values (ECOFF). The grey zone indicates the number of bacteria with decreased susceptibility above ECOFF (https://mic.eucast.org). <, minimum value of concentration used; >, maximum value of concentration used. aNumber of HLR-Gn isolates. Abbreviations: non-WT, non-wild type; AMP, ampicillin; CIP, ciprofloxacin; ERY, erythromycin; GEN, gentamicin; DAP, daptomycin; TET, tetracycline; TGC, tigecycline; CHL, chloramphenicol; LZD, linezolid; SYN, quinupristin/dalfopristin; TEI, teicoplanin; VAN, vancomycin.
Table 2.
Antimicrobial resistance profiles of E. faecalis isolates from Eurasian griffon vultures.
| Antimicrobial resistance profile | Nº isolates showing the antimicrobial resistance profile (%) | ST (CC)3 |
|---|---|---|
| WT isolates | 10 (17.9) | ST4 (2; CC16); ST40 (1; CC16); ST441 (1; CC16); ST648 (4; CC16); ST1600 (1; CC860); ST1875 (1; CC863) |
| TET | 13 (23.2) 1 | ST9 (1; CC16), ST40 (1; CC16); ST59 (1; CC16); ST82 (1; CC16); ST200 (1; CC16); ST256 (2; CC16); ST268 (1; CC16); ST631 (1; CC631); ST699 (1; CC1567); ST706 (2; CC376); ST860 (1; CC860) |
| ERY-TET | 14 (25.0)1 | ST7 (1; CC16); ST16 (1; CC16); ST287 (2; CC287); ST300 (8; CC863); ST1287 (1; 1861) |
| GEN-ERY-TET2 | 2 (3.6) | ST16 (2; CC16) |
| CHL-ERY-TET2 | 5 (8.9) | ST19 (1; CC16); ST21 (1; CC16); ST76 (1; CC1688); ST116 (1; CC16); ST1288 (1; CC863) |
| GEN-CIP-TET2 | 1 (1.8) | ST330 (1; CC16); |
| GEN-CIP-ERY-TET2 | 4 (7.1) | ST16 (2; CC16); ST82 (1; CC16); ST1768 (1; CC16) |
| GEN-CHL-ERY-TET 2 | 4 (7.1) | ST4 (1; CC16); ST35 (2; CC57); ST1291 (1; Singleton) |
| GEN-CIP- CHL-ERY-TET2 | 1 (1.8) | ST480 (1; CC16) |
| GEN-CIP-CHL-LZD-ERY-TET2 | 2 (3.6) | ST116 (2; CC16) |
| Total NWT isolates | 46 (82.1) | |
| Total MDR isolates | 19 (33.9) |
1Most frequent profiles are in bolds.2 Multidrug resistance phenotypes are underlined. 3Number of isolates and CCs in brackets. Abbreviations: WT, wild type, NWT, non-wild type; MDR, multidrug resistant; CIP, ciprofloxacin; ERY, erythromycin; GEN, gentamicin; TET, tetracycline; CHL, chloramphenicol; LZD, linezolid.
Table 3.
Multilocus sequence types of the 56 E. faecalis isolates from Eurasian griffon vultures and their relationship to data available on E. faecalis the MLST database.
Table 3.
Multilocus sequence types of the 56 E. faecalis isolates from Eurasian griffon vultures and their relationship to data available on E. faecalis the MLST database.
| ST | CC (DVL)a |
Nº vulture isolates | Nº isolates of E. faecalis in the MLST database | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MDR | non-MDR | TOTAL | Animalsb | Human | Foods | Environment | Unknown | TOTAL | ||
| 4 | 16 | 1 | 2 | 3 | 12 | 1 | 1 | 14 | ||
| 7 | 16 | 1 | 1 | 1 | 1 | |||||
| 9 | 16 | 1 | 1 | 22 | 2 | 24 | ||||
| 16 | 16 | 4 | 1 | 5 | 68 | 1 | 11 | 80 | ||
| 19 | 16 | 1 | 1 | 7 | 4 | 7 | 3 | 21 | ||
| 21 | 16 | 1 | 1 | 41 | 23 | 22 | 4 | 90 | ||
| 35 | 57 | 2 | 2 | 2 | 2 | |||||
| 40 | 16 | 2 | 2 | 15 | 42 | 8 | 3 | 6 | 74 | |
| 59 | 16 | 1 | 1 | 6 | 4 | 1 | 11 | |||
| 76 | 1688 | 1 | 1 | 1 | 1 | 3 | 5 | |||
| 82 | 16 | 1 | 1 | 2 | 13 | 1 | 25 | 2 | 41 | |
| 116 | 16 | 3 | 3 | 9 | 8 | 1 | 1 | 1 | 20 | |
| 200 | 16 | 1 | 1 | 1 | 1 | |||||
| 256 | 16 | 2 | 2 | 6 | 1 | 3 | 3 | 13 | ||
| 268 | 16 | 1 | 1 | 1 | 1 | |||||
| 287 | 287 | 2 | 2 | 4 | 1 | 5 | ||||
| 300 | 863 | 9 | 9 | 4 | 1 | 5 | ||||
| 330 | 16 | 1 | 1 | 3 | 3 | 3 | 9 | |||
| 441 | 16 | 1 | 1 | 1 | 1 | |||||
| 480 | 16 | 1 | 1 | 11 | 4 | 1 | 16 | |||
| 631 | 631 | 1 | 1 | 10 | 1 | 11 | ||||
| 648 | 16 | 4 | 4 | 4 | 1 | 5 | ||||
| 699 | 1567 | 1 | 1 | 1 | 1 | |||||
| 706 | 376 | 2 | 2 | 1 | 1 | |||||
| 860 | 860 | 1 | 1 | 1 | 1 | |||||
| 1287 | 1861 | 1 | 1 | 2 | 1 | 3 | ||||
| 1288 | 863 | 1 | 1 | 5 | 5 | |||||
| 1291 | singleton | 1 | 1 | 1 | 1 | |||||
| 1600 | 860 | 1 | 1 | 1 | 1 | |||||
| 1768 | 16 | 1 | 1 | 1 | 1 | |||||
| 1875 | 863 | 1 | 1 | 1 | 1 | |||||
a CC (DVL), clonal complexes using the double-locus variants (DLV) criterion. b Chicken, pets, livestock and wildlife animals. Abbreviations: MDR; multidrug resistant.
Table 4.
Location of the antimicrobial resistance genes in the genome of 19 E. faecalis isolated from Eurasian griffon vultures.
Table 4.
Location of the antimicrobial resistance genes in the genome of 19 E. faecalis isolated from Eurasian griffon vultures.
| Antimicrobial | Nº Isolates | ST (Nº isolates) |
Chromosome | MGEs | |||
|---|---|---|---|---|---|---|---|
| QRDRs (Nº isolates) |
ARGs (Nº isolates) |
REP (Nº isolates/ARG) |
Tn (Nº isolates/ARG) | ||||
| Included in the commercial panel | HLR-Gn | 13 | ST4 (1); ST16 (4); ST82 (1); ST35 (2); ST116 (2); ST330 (1); ST480 (1); ST1768 (1) | aac(6’)-aph(2’’) (12) | |||
| CIP | 8 | ST16 (2); ST82 (1); ST116 (2); ST330 (1); ST480 (1); ST1768 (1) | gyrA_E87G/parC_S80I (4) gyrA_E87G/parC_S80I (3) |
||||
| CIP (WT) | 2 | ST35 (2) | parC_S80I (2) | ||||
| CHL | 14 | ST480 (1); ST116 (3); ST4 (1); ST16 (2); ST1291 (1); ST35 (2); ST1288 (1); ST76 (1); ST21 (1); ST19 (1) |
cat(pC221) (1) cat(pC223) (2) |
rep7a (3/cat(pC221)) repUS43 (3/cat(pC223)) rpUS40 (2/fexA) |
|||
| LZD | 2 | ST116 (2) | rpUS40 (2/optrA) | ||||
| TET | 19 | ST4 (1); ST16 (4); ST19 (1); ST21 (1); ST35 (2); ST76 (1); ST82 (1); ST116 (2); ST330 (1); ST480 (1); ST1288 (1); ST1291(1); ST1768 (1) |
tetM (2) tetL (4) |
repUS43 (10/tetM) repUS43 (7/tetM&tetL) rep22 (2/tetL) |
Tn6009 (10/ tetM) Tn6009 (2/tetM&tetL) cn_43171_ISS1N (1/tetM&teL) |
||
| ERY | 19 | ST4 (1); ST16 (4); ST19 (1); ST21 (1); ST35 (2); ST76 (1); ST82 (1); ST116 (2); ST330 (1); ST480 (1); ST1288 (1); ST1291(1); ST1768 (1) | ermB (16) | rep7a (1/ ermB) | cn_43171_ISS1N (1/ermB) | ||
| No included in the commercial panel | STR | - | ant(6)-Ia (10) |
rep7a (1/ant(6)-Ia) rep7a (4/str) |
|||
| KAN | - | aph(3’)-III (12) | rep7a (1/ aph(3’)-III) | ||||
| TRM | - |
dfrD (1) dfrG (16) |
|||||
| LIN | - |
lsaA (19) lsaE (9) lnuA(1) lnuB (10) lnuG (5) |
Tn6260 (5/lnuG) | ||||
Abbreviations: ST. sequence type; HLR-Gn, high level resistance to gentamicin AMR, Antimicrobial resistance; REP, replicon type; Tn, Transposon; ComTn, Composite Transposon; CIP, ciprofloxacin; ERY, erythromycin; TET, tetracycline; CHL, chloramphenicol; LZD, linezolid; STR, streptomycin; KAN, Kanamycin; TRM, Trimethoprim; LIN, Lincomicin QRDRs, quinolone resistance determinant regions; MEG, mobile genetic elements; ARGs, antimicrobial resistance genes; REP, replicon; Tn, transposons.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



