
Submitted:
21 June 2025
Posted:
23 June 2025
You are already at the latest version
Abstract
Short-chain fatty acids (SCFAs) are microbial metabolites, also known as postbiotics, produced by the gut microbiota, essential in maintaining gut health. Research on the antiproliferative effects of SCFAs against gastric cancer cells and their interactions with conventional cancer therapies is limited. This study investigates the antiproliferative effects of SCFA salts—magnesium acetate (A), sodium propionate (P), and sodium butyrate (B)—and their combinations with dexamethasone (Dex) in AGS gastric adenocarcinoma cells. Our results showed that SCFA salts were potent against AGS cells, and combining A, P, and B (APB) also led to significant growth inhibition of the cells, with an IC50 of 568.33 μg/mL. Furthermore, combining APB with Dex enhanced the antiproliferative effect of the former, showing strong synergy against AGS cells with a combination index value of 0.76. Flow cytometry analysis confirmed that both APB (p < 0.0001) and APB+Dex (p < 0.0001) induced substantial apoptosis in AGS cells compared to the negative control, with minimal necrosis, suggesting a primarily apoptotic mode of cell death. Additionally, APB (p < 0.0001) alone significantly elevated reactive oxygen species (ROS) levels, with Dex moderating this increase in the APB+Dex group (p < 0.0001), compared to the untreated control. Proteomic analysis using LC-MS identified several protein groups in the AGS cells after treatment with APB, Dex, and their combination APB+Dex. Proteomics revealed that APB suppressed key regulators of the cell cycle checkpoint (e.g., CHEK1, log2FC = –1.31), chromatin remodelling (e.g., CREBBP, –1.14), and DNA repair enzymes (e.g., BRCA1, –1.40), indicating a disruption of proliferation and genome maintenance machinery in AGS gastric adenocarcinoma cells. In contrast, Dex induced transcriptional reprogramming via modulation of CDKN1A (p21, 1.01), alongside activation of cholesterol biosynthesis through HMGCR (3.33), and enhancement of ferroptosis-related responses, notably through suppression of the antioxidant transporter SLC7A11 (–2.60). The APB+Dex treatment synergistically targeted multiple tumour-promoting mechanisms, including the impairment of redox homeostasis through SLC7A11 suppression, and inhibition of the haemostasis, platelet activation network and NF-κB signalling pathway via downregulation of NFKB1 (–1.34), exemplified by increased expression of SERPINE1 (1.99) within the “response to elevated platelet cytosolic Ca²⁺” pathway. These findings underscored a multifaceted anticancer mechanism by APB+Dex that may collectively impair cell proliferation, survival signalling, immune modulation, and tumour microenvironment support.
Keywords:
Gastric cancer
; Gastric Adenocarcinoma
; Postbiotics
; Gut microbiome
; Dexamethasone
; Apoptosis
; Proteomics
1. Introduction
Cancer continues to be one of the most significant global health challenges, with gastric cancer ranking as the fifth most common malignancy and the third leading cause of cancer-related mortality worldwide [1, 2]. Despite significant advancements in treatment options such as chemotherapy, radiotherapy, and immunotherapy, the prognosis for patients, particularly those with advanced or metastatic gastric cancer, remains poor [3]. Conventional therapies often come with severe side effects, suboptimal effectiveness, and the eventual development of drug resistance, highlighting the urgent need for more effective and less toxic therapeutic strategies [4]. Recent advances in cancer therapy have increasingly focused on harnessing the potential of postbiotics derived from gut microbial communities, particularly short-chain fatty acids (SCFAs). These metabolites, including acetate, propionate, and butyrate, are produced during the fermentation of dietary fibers and have demonstrated anti-inflammatory, immunomodulatory, and anticancer properties [5-7]. In gastric cancer, SCFAs have shown potential in modulating tumour cell metabolism, enhancing apoptosis, and inhibiting proliferation [8]. This makes SCFAs attractive candidates for combination therapies aimed at enhancing the efficacy of conventional cancer treatments while mitigating their side effects.
Sodium butyrate (B) has emerged as a potent anticancer agent due to its ability to induce apoptosis, arrest cell cycles, and alter gene expression by inhibiting histone deacetylases (HDACs) [9]. Research has shown that B can sensitise cancer cells to chemotherapeutic drugs such as 5-Fluorouracil (5-FU), oxaliplatin, and cisplatin, increasing apoptosis while decreasing drug resistance in colorectal, cervical, and gastric cancers [9, 10]. For example, combining B and cisplatin in gastric cancer has enhanced apoptosis and reduced cell migration and invasion [10]. Sodium propionate (P), though less studied than B, has also shown promising anticancer effects, particularly in enhancing apoptosis through modulation of critical signalling pathways such as Wnt/β-catenin and NF-Κb [11, 12]. Dexamethasone (Dex), a glucocorticoid commonly used in cancer treatment for its immunomodulatory and anti-inflammatory effects, has been shown to enhance the action of chemotherapeutic agents [13]. However, its use in treating solid tumours such as gastric cancer remains limited. Combining Dex with SCFAs could offer a novel approach by simultaneously targeting cancer cell survival mechanisms, modulating the tumour microenvironment, and enhancing immune responses.
The concept of synergistic interactions between SCFAs and chemotherapeutic agents is gaining traction [10]. Synergy occurs when the combined effect of two or more agents exceeds the sum of their individual effects, leading to enhanced therapeutic outcomes [10]. For instance, the pairing of B with cisplatin enhanced apoptosis and decreased tumour volume in cervical cancer models [10]. Likewise, when combined with 5-FU or oxaliplatin, B has been shown to potentiate the effects of these drugs, improving their efficacy and reducing drug resistance in colorectal cancer [10]. Given the diverse mechanisms by which SCFAs modulate cancer cell behaviour, their combination with conventional therapies like Dex could pave the way for more effective treatments in gastric cancer. To build upon existing research, the current study employs a combination index model to assess synergy between SCFAs and Dex, utilising flow cytometry, reactive oxygen species (ROS) analysis, and proteomics to investigate their molecular interactions. Unlike previous studies that primarily focus on individual SCFAs or their general anticancer properties, this research aims to elucidate their combined effects with Dex, providing a more comprehensive mechanistic understanding. Additionally, our previous findings demonstrated that B exhibited the most potent antiproliferative effects among SCFAs and showed synergy with Dex in AGS gastric adenocarcinoma cells [14]. The current study extends that work by exploring the role of multiple SCFA salts in combination with Dex, analysing their effects on cancer cell metabolism, oxidative stress, and proteomic alterations. These insights may contribute to developing SCFA-based adjuvant therapies for gastric cancer.
2. Materials and Methods
2.1. Chemicals and Drug Preparation
All solvents utilized in this study were of analytical grade and sourced from Sigma Aldrich (Castle Hill, NSW, Australia). A, P, B and Dex standards were also obtained from Sigma Aldrich (Castle Hill, NSW, Australia). All reagents used in the proteomics analyses were purchased from Thermo Fisher Scientific (Waltham, Massachusetts, USA) unless stated otherwise.
2.2. Cell Culture
AGS gastric adenocarcinoma (CRL-1739, ATCC), HS738.St/Int stomach intestinal (CRL-7869, ATCC) and murine RAW264.7 macrophage (CRK-TIB-71, ATCC) cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, Virginia, USA). AGS cells were cultured in ATCC-formulated F-12K medium (Kaighn's Modification of Ham's F-12) containing 2 mM L-glutamine, 1500 mg/L sodium bicarbonate, and 10% foetal bovine serum (FBS; Bio-Strategy PTY, Campbellfield, VIC, Australia), with the addition of 1% penicillin and streptomycin (Sigma-Aldrich, Castle Hill, NSW, Australia). HS738.St/Int cells were grown in ATCC-formulated DMEM (Dulbecco’s Modified Eagle Medium), which included 4.5 g/L glucose, L-glutamine, sodium pyruvate, and 10% FBS, also supplemented with 1% penicillin and streptomycin. Both cell lines were maintained at 37 °C in a 5% CO2 environment, with cell maintenance carried out every 48–72 h to allow for confluent monolayers.
Murine RAW264.7 macrophage cells were cultured in DMEM (Lonza Australia, Mount Waverley, VIC, Australia) supplemented with 5% FBS and 1% penicillin and streptomycin and incubated at 37 °C in a humidified atmosphere with 5% CO2.
2.3. Cell Viability Assays
The viability of AGS cells following treatment with six-point dose responses in 1:2 dilutions starting from 3000 µg/mL of the postbiotics combinations (AP (1:1), AB (1:1), PB (1:1), APB (1:1:1)) and 200 µg/mL Dex were assessed using the Alamar Blue assay, as described in previous studies [15, 16]. Briefly, 100 µL of cells were seeded at a density of 105 cells/mL in 96-well plates. After 24 h, the cells were treated with the test compounds and incubated for 72 h. At the end of the incubation, the culture medium was removed, and 100 µL of a 0.1 mg/mL Alamar Blue solution (prepared from a 1 mg/mL resazurin stock in PBS, diluted 1:10 with serum-free media) was added to each well. Fluorescence was measured using a microplate spectrophotometer (BMG CLARIOstar, Mornington, VIC, Australia), with excitation at 555 nm and emission at 595 nm. All compounds were tested in triplicate, with the negative control set to 100% cell viability.
2.4. Synergy
Dex was combined with the postbiotics combination APB in 1:1 (3000 μg/mL of APB + 100 μg/mL of Dex) for combination index (CI) analyses. The potential interactions between Dex and APB were analysed using the CI model, and the CompuSyn version 2.0 (Biosoft, CA, USA) was used for the CI calculations based on the median-effect equation, which was derived from mass action law [16]. In the current study, the combination APB+Dex was studied with a six-point dose-response curve using the CI model.
2.5. Flow Cytometry Analyses of Apoptotic Profiles
The effect of APB (3000 µg/mL), Dex (200 µg/mL), and APB+Dex (3100 µg/mL), on apoptosis in AGS gastric adenocarcinoma cells was assessed using an annexin V/7-AAD kit (#ab214663, Abcam) as previously described [16, 17]. Briefly, AGS cells (1 × 10⁶ cells/10 mL) were cultured in T75 flasks at 37 °C with 5% CO₂ for 24 h. Cells were treated with APB (3000 µg/mL), Dex (200 µg/mL), and APB+Dex, with serum-containing media as the control. After 24 h, cells were harvested, stained with annexin V-CF Blue and 7-AAD, and analyzed using a Novocyte 3000 flow cytometer (ACEA Biosciences). Data were processed with NovoExpress software (v1.5.0) and visualized using GraphPad Prism (v9.0). Apoptotic profiles were determined by gating live, early apoptotic, late apoptotic, and necrotic cells based on annexin V and 7-AAD fluorescence.
2.6. ROS Production Analysis
The effect of APB, Dex and APB+Dex on oxidative stress in the AGS gastric adenocarcinoma cancer cells was evaluated using the H2DCFDA (2′,7′-dichlorofluorescein diacetate) Cellular Reactive Oxygen Species (ROS) Detection Assay Kit (#ab113851; Abcam, Melbourne, VIC, Australia) [15]. In brief, AGS cells (2.5 × 105 cells/mL) were seeded in a 96-well plate, allowed to adhere overnight, and then treated with 20 μM H2DCFDA for 45 min to assess the ROS levels. After the dye solution was removed, the cells were washed with 1X buffer. The cells were then exposed to 750, 1500, and 3000 µg/mL of APB; 50, 100, and 200 µg/mL of Dex; 3000:100 μg/mL, 1500:50 μg/mL, and 750:25 μg/mL of APB+Dex; and 250 μM of tert-butyl hydroperoxide (TBHP) and incubated at 37 °C for 4 h. The fluorescence was measured immediately at an excitation/emission of 485/535 nm using a microplate spectrophotometer (BMG CLARIOstar, Mornington, VIC, Australia). The fold increase in ROS production was calculated relative to the negative control (cells treated with buffer as per the manufacturer’s instructions).
2.7. Liquid Chromatography-Mass Spectrometry, Label Free Quantification Bottom-Up Proteomics Analysis
Cell Culture, Treatment, and Protein Extraction
The AGS adenocarcinoma cells were initially placed in T75 flasks at a concentration of 1.0 × 106 cells/mL and were allowed to incubate overnight at 37 °C in the presence of 5% CO2. After removing the media, it was replaced with fresh F-12K medium supplemented with 10% FBS, and the cultured flasks were treated, as well as specific doses of the APB (3000 µg/mL), Dex (200 µg/mL), and APB+Dex (3100 µg/mL). Treatments were done in triplicate and incubated for 24 h at 37 °C in a humidified atmosphere with 5% CO2. Following the incubation, each flask of cells was subjected to 0.25% w/v trypsin treatment for 3 min at 37 °C, and the cell culture media were collected. To neutralise the trypsin, an equal volume of media containing F-12K medium (containing 10% FBS) was added before mixing with the previously collected media. The cells were then spun in a centrifuge at 500× g for 5 min at RT. The cell pellets were subsequently washed twice with ice-cold PBS and spun again at 500× g for 5 min.
Sample Preparation
The AGS gastric adenocarcinoma cells treated with APB, Dex and APB+Dex were homogenized in 300 µL lysis buffer (4 % w/v) sodium deoxycholate, 50 mM ammonium bicarbonate, pH 7.6) and sonicated using a Sonifiers SFX tip probe (Branson, Danbury, Connecticut, USA) for eight cycles (30 sec on and 30 sec off) at a frequency of 30 % whilst on ice. The debris was removed through centrifugation (Eppendorf, Hamburg, Germany) at 9000 g for 5 min, the resulting supernatant was aliquoted, and 4X volume of ice-cold acetone was added to denature and precipitate proteins. Samples were incubated at -30 °C overnight before centrifugation at 10000 g for 20 min, the supernatant was removed, and the protein pellets were air dried for 3 h. Protein pellets were reconstituted with 100 µl of denaturing buffer (6 M urea, 2 M thiourea, 100 mM HEPES buffer, pH 7.5) and then reduced with dithiothreitol (10 mM final concentration, room temperature, 1 h incubation) and alkylated with iodoacetamide (25 mM final concentration, room temperature, 30 min incubation in darkness) before being diluted 6-fold (50 mM ammonium bicarbonate, pH 7.6). Protein quantification was determined using a Qubit 4 (Invitrogen, Carlsbad, California, USA) according to the manufacturer’s instructions. Samples were aliquoted to a final concentration of 2 ug, and trypsin (4 µL, 12 ng/µL, Promega, Madison, Wisconsin, USA) was added to each sample. Digestion occurred at 37 °C and samples were incubated overnight with shaking at 600 rpm. After digestion, samples were then acidified to 0.1 % TFA, concentrated and desalted using C18 Zip-Tips (Millipore, Bedford, Massachusetts, USA) per the manufacturer’s instructions. The solvent was removed from the desalted peptides through vacuum using a Concentrator plus (Eppendorf, Hamburg, Germany), and samples were then resuspended in 20 µL of loading buffer (3% (v/v), acetonitrile, 0.1 % (v/v) formic acid), briefly sonicated and centrifuged at 16000 g for 10 min. Samples were run in triplicates.
Liquid Chromatography-Mass Spectrometry Data Independent Analysis
Samples were separated by nano-LC using an Ultimate 3000 HPLC and autosampler system (Dionex, Amsterdam, Netherlands) coupled to an in-house built fritless nano 75 μm × 45 cm column packed with ReproSil Pur 120 Å, 1.9 μm, C18 stationary phase (Dr Maisch GmbH, Germany). LC mobile phase buffers were comprised of buffer A: 0.1% (v/v) formic acid in HPLC grade water and buffer B: 80% (v/v) acetonitrile, 0.1% (v/v) formic acid in HPLC water. Peptides were eluted using a linear gradient of 5 % buffer B to 40 % buffer B over 60 mins and then an increase to 60 % buffer B over 5 mins and then 98 % buffer B over 3 mins before an isocratic wash at 98 % buffer B for 7 min at a flow rate of 300 nL/min. The LC was coupled to a Q Exactive HF-X Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Column voltage was set to 2400 V, the heated capillary set to 300 °C, and no sheath and auxiliary gas flow. Positive ions were generated by electrospray, and the orbitrap operated in data-independent acquisition mode. A survey scan of 350-1650 m/z was kept constant, acquired with a resolution at 60,000, and an accumulation target value of 3,000,000 ions followed by 20 narrow isolations windows covering 350-1650 m/z with varying widths of 26-589 m/z, resolution set to 30,000, and an accumulation gain control value of 3,000,000 ions. Stepped normalised higher energy collisional dissociation (HCD) collision energy was set to 22.5, 25 and 27.5 %.
Data Processing
The raw data was analysed with SpectronautTM software v. 19.0.240604.62635 (Biognosys, Schlieren, Switzerland) using the directDIATM analysis and were searched against the 2023 Human FASTA file and the Human Gene Annotation file downloaded from the UniProt website (https://www.uniprot.org, 20,595 protein sequence entries and 19,756 gene sequence entries). The search library parameters were the default parameters of the BGS Factory Setting for the directDIATM analysis process; this included the following variable modifications: enzymes/ cleavage rules: trypsin, peptide length 7-52, missed cleavages: 2, max variable modifications: 5, fixed modifications: carbamidomethyl (C), variable modifications: acetyl (protein N-term), oxidation (M), PSM FDR: 0.01, peptide FDR: 0.01, protein group FDR: 0.01, directDIA workflow: directDIA+ (deep), all tolerance parameters (ion trap, orbitrap, ToF) calibration search: dynamic, MS1 correction factor: 1, MS2 correction factor: 1. XIC extraction: dynamic, MZ extraction strategy: maximum intensity, single hit definition: by stripped sequence, precursor filtering: identified (Qvalue), quantity type: area, differential abundance testing: unpaired t-tests. The raw and processed data have been deposited to the ProteomeXchange Consortium via the PRoteomics IDEntifications (PRIDE) repository with the dataset identifier PXD061822.
2.8. Statistical Analysis
Data collection and management were conducted using Microsoft Excel and GraphPad Prism (version 9.0, San Diego, CA, USA) for statistical analysis and visualisation. All data collection and analyses were performed in triplicate, with results presented as the mean ± standard deviation. Statistical significance between the mean values was assessed at p < 0.05 using a two-way ANOVA. For multiple comparisons, Tukey and Dunnett's tests were applied within GraphPad Prism software. Additionally, the IC50 value, which indicates the concentration of a drug needed to inhibit cell growth by 50%, was nonlinearly regressed using GraphPad Prism software.
3. Results and Discussion
3.1. Antiproliferative Activity of Postbiotic Combinations, Standard Immunotherapy and Standard Chemotherapy
In our recent study, we showed that SCFA salts- A, P and B exhibited a significant inhibitory effect (p < 0.05) against the AGS gastric adenocarcinoma cells [14]. At 3000 μg/ml, B demonstrated an inhibition value of 100.36 ± 1.23%, followed by P at 95.15 ± 1.99 % and A at 81.47 ± 20.28% against AGS cells following a 72-h treatment [14]. Additionally, the combination of Dex and B showed strong synergy at a 2:8 ratio (40 μg/mL Dex + 2400 μg/mL B) with significantly greater inhibitory activity (p < 0.05) compared to the mono treatments [14]. In the current study, the effect of different combinations of A, P and B were investigated for their antiproliferative activity against AGS cells. Furthermore, the combination of APB+Dex was studied against the AGS gastric adenocarcinoma cells. Table 1 presents the cell growth inhibition (%) of AGS cells when treated with various combinations of the SCFA salts (AP, AB, PB, APB), Dex, and APB+Dex following a 72-treatment using the Alamar Blue assay.
This study is among the first to investigate the antiproliferative effects of the combined A and P on AGS gastric adenocarcinoma cells. The AP combination showed modest growth inhibition at higher concentrations (93.75 μg/mL - 3000 μg/mL), with the highest inhibition of 59.79 ± 4.32% at 3000 μg/mL. The IC50 for AP was found to be relatively high at 1141.13 ± 362.00 μg/mL, indicating weaker potency. The AB combination showed 99.49 ± 0.85% inhibition at 3000 μg/mL and a potent IC50 of 446.53 ± 19.55 μg/mL, suggesting strong anti-proliferative activity against the AGS cells. The PB combination displayed high cell inhibition, reaching 99.22 ± 1.42% at 3000 μg/mL, with an IC50 of 421.23 ± 15.31 μg/mL, making it comparable to AB (p<0.05). The APB combination displayed inhibitory action similar to AB and PB (p< 0.05), with 95.65 ± 7.90% inhibition at 3000 μg/mL and an IC50 of 568.33 ± 82.56 μg/mL.
The cell viability of Hs 738.St/Int normal intestinal cells remained relatively high across all tested concentrations for all three combinations (AP, AB, and PB). At the highest concentration (3000 μg/mL), the viability was 82.39% (AP), 61.08% (AB), and 58.86% (PB). As the concentration decreased, the cell viability generally increased, with the highest viability observed at 93.75 μg/mL, where AP (175.62%), AB (149.29%), and PB (138.92%) compared to the negative control. These values suggest that the normal cells might have recovered or even proliferated slightly at lower concentrations in response to treatment. This is likely due to the fact that SCFAs, particularly butyrate, are known to serve as a primary energy source for intestinal epithelial cells, promoting their growth and maintenance of barrier function [18]. This cell viability study suggested that the SCFA, even at their highest tested concentration of 3000 μg/mL showed less than 50% toxicity against the normal Hs 738.St/Int cells, indicating their therapeutic potential against gastric adenocarcinoma.
SCFAs have been investigated in many studies for their potential to enhance the effectiveness of chemotherapy, radiotherapy, and immunotherapy [19, 20]. Previous studies have shown that B, when combined with cisplatin or Dex, can induce apoptosis in various tumour cells, including gastric and cervical cancers, both in vitro and in vivo [14, 19]. Here, we sought to study the synergistic potential of the SCFAs (APB) and Dex. We previously reported Dex with potent inhibition of AGS cells by 80.46% at 200 μg/mL in a dose-dependent manner with an IC50 value of 86.60 ± 11.85 μg/mL [14]. In the current study, the APB+Dex combination (3000 μg/mL of APB + 100 μg/mL of Dex) showed 103.17 ± 3.06% inhibition against AGS cells, with an IC50 of 643.30 ± 58.26 μg/mL (Table 2).
The combination of APB and Dex was also tested on normal Hs 738.St/Int human cells, revealing a dose-dependent effect on cell viability. At the highest concentration of APB+Dex (3100 μg/mL), cell viability significantly decreased to 68.57 ± 11.74%, indicating cytotoxicity at this dose. However, at lower concentrations (1550 and 775 μg/mL), cell viability increased to 85.91 ± 10.59% and 110.88 ± 14.35%, respectively, suggesting recovery of cellular activity. Interestingly, at lower concentrations (387.5, 193.75, and 96.875 μg/mL), the combination appeared to stimulate cell proliferation, with viability reaching 115.29 ± 16.81%, 134.46 ± 24.72%, and 159.21 ± 14.16%, respectively compared to the negative control. Notably, our previous findings demonstrated that B and Dex, when tested individually, exhibited a favourable safety profile, as they did not significantly reduce the viability of normal intestinal cells [14]. B even promoted cell proliferation, likely due to its role as a primary energy source for intestinal epithelial cells. In contrast, the APB+DEX combination exhibited cytotoxic effects at higher concentrations and proliferation at lower doses in the Hs 738.St/Int cells. This suggested that while B and Dex are well tolerated in normal cells, combining APB with Dex alters the cellular response. This may be attributed to mechanisms such as nutrient utilisation or stress-induced compensatory responses in normal intestinal cells [21]. Even at the highest tested concentration of 3100 μg/mL, the APB+Dex combination showed less than 50% cytotoxicity against the normal Hs 738.St/Int human cells, supporting its favourable safety profile at lower doses. These results underscored the potential therapeutic utility of APB+DEX while highlighting the importance of dose optimisation to minimise toxicity in healthy tissues.
3.2. Synergistic Potential of APB with Dex against the AGS gastric Adenocarcinoma Cells
The potential synergistic effects of AP, AB, PB, and APB+Dex on AGS cells were analysed using the CI model, as shown in Table 3. The interactions were categorized into three distinct groups: synergistic effects (CI < 1), additive effects (CI = 1), and antagonistic effects (CI > 1). The AP, AB and BP combinations showed CI values of 3.53, 1.14 and 1.41, respectively, indicating antagonistic interactions. The results indicated that the APB+Dex yielded a CI value of 0.76, signifying a strong synergistic interaction. This was particularly evident at the IC50, IC75, IC90, and IC95 levels, where the CI values consistently remained below 1, reinforcing the effectiveness of this combination in inhibiting the growth of AGS cells. Interestingly, our previous study demonstrated a similar strong synergistic effect between B and Dex in AGS cells, with a B+Dex ratio of 2:8 (2,400 μg/mL B + 40 μg/mL Dex) yielding CI values <1 compared to their mono treatments [14]. The B+Dex combination achieved 80.32–103.78% inhibition of AGS cell growth while also improving the viability of normal Hs 738.St/Int intestinal cells, highlighting its favourable safety profile [14]. The findings from the current study align with our previous results, further supporting the potential of combining Dex with SCFA-based compounds to enhance antiproliferative efficacy while minimising toxicity.
3.3. Flow Cytometric Analyses of Apoptotic Profiles of Mono and Combination Therapies
Apoptosis, a type of programmed cell death, plays a crucial role in preventing cancer by eliminating malignant cells from the body [22]. Due to the importance of apoptosis in cancer suppression, many new anticancer therapies aim to target this biological process [22]. Annexin V and 7-AAD were employed to identify apoptosis and necrosis, respectively, using flow cytometry. Annexin V binds to phosphatidylserine, which is exposed on the outer membrane of cells during apoptosis, while 7-AAD, with a high affinity for guanine-cytosine residues, intercalates into double-stranded DNA to indicate necrosis. The effects of the mono treatments APB, Dex, and the combination APB+Dex were investigated using flow cytometric analysis, compared to the negative control (Figure 1). The results are categorised into live cells, early apoptotic cells, late apoptotic cells and necrotic cells.
Dex (200 μg/mL) alone showed a relatively balanced effect, with a moderate proportion of live cells (33.11%), early apoptotic cells (10.17%), and a notable proportion of late apoptotic cells (52.08%). The necrotic cell population remained low (4.64%) in the Dex treatment (P < 0.0001), although slightly higher than the combination treatments (Figure 1). Nevertheless, apoptosis was still the dominant cell death mechanism in the Dex-treated cells. APB (3000 μg/mL) treatment led to a high proportion of apoptotic cells, including late apoptotic cells (71.91%) and early apoptotic cells (11.81%), indicating significant induction (P < 0.0001) of apoptosis compared to the negative control. The proportion of necrotic cells was low (3.32%), suggesting that APB primarily induced apoptosis rather than necrosis and a small proportion of live cells (12.96%) was observed (Figure 1). Overall, APB induced a substantial amount of apoptosis, predominantly in the late stage, while maintaining a low percentage of necrosis, underscoring its effectiveness in triggering programmed cell death. When combined with Dex, the apoptotic effect was enhanced, particularly in the early stage, and the proportion of live cells decreased, indicating a synergistic interaction between APB and Dex. This APB+Dex combination promoted apoptosis in the AGS gastric adenocarcinoma cells more effectively (p < 0.0001) than either agent alone, with minimal necrosis observed. These findings aligned with our previous study on the B+Dex combination, where the synergy between Dex and B also significantly increased apoptosis while keeping necrosis at a low level [14]. Notably, the B+Dex combination (40:2400 μg/mL) resulted in a significant increase in early apoptotic cells (48.97%; p < 0.0001) compared to Dex alone, similar to the enhancement of early apoptosis observed in the APB+Dex combination. However, in contrast to the APB+Dex combination, which showed a predominant late apoptotic response, the B+Dex combination favoured early apoptosis. This difference may not necessarily indicate distinct mechanisms of action but could suggest that APB+Dex induces apoptosis more rapidly, causing a greater proportion of cells to progress to the late apoptotic stage by the time of measurement.
3.4. ROS production in the AGS cells after Treatment with Different Concentrations of APB, Dex and APB+Dex
Oxidative stress, marked by elevated levels of ROS, is a critical factor in the development and progression of cancer [23]. Therefore, enhancing or reducing ROS levels in cancer cells can be a promising strategy for anticancer therapy. This study evaluated the effects of APB, its combination with Dex, and individual treatments on ROS production in AGS gastric adenocarcinoma cells at different concentrations. TBHP, used as a positive control for ROS induction, resulted in significantly higher ROS levels (11.94-fold) compared to the negative control, which showed baseline ROS levels (0.91), confirming the sensitivity of the assay to detect oxidative stress changes in the cells (Figure 2). At the highest concentration (3000 μg/mL), APB alone significantly increased ROS production (6.14-fold), suggesting a strong pro-oxidative response. When combined with Dex, the ROS levels were moderately reduced to 5.64-fold (p<0.0001), indicating that Dex may mitigate APB-induced oxidative stress. At the lower concentrations, a consistent pattern emerged; at 1500 μg/mL, APB alone led to a ROS increase of 4.50-fold, while its combination with (APB+Dex) reduced ROS to 3.98-fold, reinforcing the potential antioxidative role of Dex. The APB+Dex combination at this concentration (1500 μg/mL of APB + 50 μg/mL of Dex) maintained elevated ROS levels at 4.21-fold (Figure 2). At 750 μg/mL, APB alone induced a ROS increase of 3.35-fold, while the combination APB+Dex brought ROS down further to 3.08-fold, suggesting a consistent reduction in oxidative stress by Dex. Lower concentrations of Dex alone (200 μg/mL, 100 μg/mL, 50 μg/mL) consistently showed very low ROS production (0.30-0.31), indicating its strong antioxidative properties (Figure 2).
These findings align with our previous study, which similarly demonstrated that Dex significantly reduced ROS production in AGS cells at all tested concentrations (p ≤ 0.01), indicating its potent antioxidative effects [14]. In contrast, B exhibited strong pro-oxidative properties, significantly increasing ROS levels compared to the negative control (p < 0.0001). Notably, the Dex+B combination displayed a unique ROS modulation pattern, with ROS levels significantly higher than Dex alone but lower than B alone. This suggested that Dex counteracted some of the oxidative stress induced by B, a pattern that parallels the current study’s findings with APB and Dex.
While B and APB both demonstrated pro-oxidative effects, APB (p < 0.0001) induced a stronger ROS response at high concentrations (3000 μg/mL). Dex exhibited a protective antioxidative effect in both studies, reducing ROS when combined with pro-oxidative agents. This consistent ROS-modulating effect of Dex across different SCFA-based combinations suggested its potential role in balancing oxidative stress, which may be crucial for optimising therapeutic efficacy while minimising oxidative damage. Overall, these findings display the importance of ROS regulation in gastric cancer therapy and highlight the potential benefits of combining Dex with pro-oxidative agents to fine-tune oxidative stress responses in AGS gastric cancer cells.
3.5. Proteomics Study of the AGS Cells Treated with the Synergistic Combination vs Mono Treatments
While studies have evaluated the anticancer potential of SCFAs on various cancer cell lines, the molecular mechanisms underlying SCFA combinations remain largely unexplored [24]. In the current study, proteomic analysis was conducted for the SCFA combination APB, Dex, and their combinations APB+Dex). The mono and combination treatments were compared to the untreated control to decipher their mechanisms of action against AGS cells as explained in the following sections.
3.5.1. Enrichment Analyses of Differentially Expressed Proteins (DEPS) In Apb-Treated Ags Cells Compared to the Untreated Control Cells
The volcano plot (Figure 3A) comparing APB-treated AGS gastric cancer cells to untreated control highlights several key proteins with significant differential expression that may underpin APB's antiproliferative effects. On the upregulated side, CLU (Clusterin), also known as apolipoprotein J, stands out as one of the most highly induced genes (log2FC > 3), often associated with stress response, apoptosis, and inhibition of cell growth in certain contexts, including gastric cancer [25]. CLU has been shown to play a protective role in the gastric epithelium by regulating cellular responses to injury and limiting abnormal proliferation during the emergence of spasmolytic polypeptide-expressing metaplasia, a preneoplastic condition [26]. Similarly, SERPINB2 also known as plasminogen activator inhibitor type 2 (PAI-2), and HIP1 (Huntingtin-interacting protein 1-related) are notably upregulated—each implicated in cell death, and or inhibition of metastasis, respectively [27, 28] SERPINB2, has been shown to inhibit tumour metastasis by being present on tumour cell-derived microparticles, thereby reducing the invasive potential of cancer cells [27]. HIP1R functions as a tumour suppressor in gastric cancer by promoting apoptosis and inhibiting proliferation, migration, and invasion of cancer cells. It achieves this, in part, by modulating the Akt signaling pathway, which is crucial for cell survival and growth [28]. DNAJB1, a heat shock protein, is also elevated and may play a role in proteostasis under therapeutic stress, promoting apoptosis in cancer cells [29].
Conversely, several proteins critical for proliferation and tumour maintenance are downregulated (Figure 3A). Notably, TYMS (Thymidylate Synthase), a key enzyme involved in DNA synthesis and repair, is strongly suppressed [30]. TYMS is frequently overexpressed in various types of cancer and is a well-known contributor to chemotherapy resistance. [30]. Its overexpression has been associated with increased genomic instability, a hallmark of cancer progression [31, 32]. Therefore, its downregulation in response to APB treatment suggested a potential reduction in genomic instability, contributing to a less favourable environment for tumour growth and survival. EPCAM, involved in cell adhesion and widely recognised as a gastric cancer stem cell marker, is also significantly downregulated, suggesting a loss of proliferative and invasive potential [33]. Other suppressed genes such as CLDN7, ID1, and PLA2G2A further reflect diminished epithelial integrity and proliferative signalling, all aligning with anticancer responses [34-36].
The graphical summary (Figure 3B) ties multiple dysregulated proteins and pathways to gastric adenocarcinoma biology. Cell cycle regulation, a critical hallmark of cancer, was notably impacted upon APB treatment through CCND1 (Cyclin D1) and CDKN1A (p21), both of which are central to the transition from G1 to S phase [37, 38]. Dysregulation of these proteins is frequently observed in gastric and other gastrointestinal malignancies, where CCND1 is often overexpressed to promote proliferation, while CDKN1A can act as a tumour suppressor or be inactivated [37, 38]. In this network, their interplay suggests a mechanistic shift in cell cycle dynamics, directly influencing gastric tumour growth. The Figure 3B also highlights tumour-related processes, with connections to terms like gastrointestinal tumour, gastrointestinal tract cancer, and connective tissue tumour, emphasizing the relevance to gastric adenocarcinoma. The involvement of proteins such as TP73 and HGF was detected in this study upon APB treatment — both known contributors to gastric cancer progression, TP73 in apoptosis resistance and HGF in invasive growth[39, 40]. The proliferation and colony formation cluster, featuring FOXM1, HGF, and RAEL6, underscored increased cellular turnover, a key trait in GIT tumour aggressiveness[40, 41]. Additionally, chromatin remodelling components like KDM5B and let-7 (miRNA) hinted at epigenetic reprogramming often seen in GIT cancers, affecting gene silencing and oncogene activation[42, 43]. Lastly, signal transduction pathways like Ribonucleotide Reductase Signalling are essential for DNA synthesis and repair, processes often hijacked in gastric cancer to sustain rapid cell division[44]. Altogether, the dysregulated genes and pathways in this network mirrored known molecular disruptions in gastrointestinal cancers and suggested that APB treatment exerted therapeutic effects by restoring balance across these oncogenic circuits.
The bar chart (Figure 3C) represents the canonical pathways and was filtered to include enriched terms with a –log Benjamini–Hochberg (BH) p-value greater than 3 and an absolute z-score of ≥ 2. Notably, Chromatin organisation and Ribonucleotide Reductase Signalling are the most significantly dysregulated pathways with a strong negative z-score, indicating suppression. Other inhibited pathways included regulation of endogenous retroelements, and cell cycle checkpoints, suggesting that APB disrupted key proliferative and repair mechanisms, reinforcing its antiproliferative activity.
Chromatin Organisation
The inhibition of the chromatin organisation pathway following APB treatment represented a pivotal mechanism contributing to its antiproliferative activity in AGS gastric adenocarcinoma cells. Chromatin remodelling is essential for regulating gene expression, DNA repair, and cell cycle progression—all processes frequently dysregulated in cancer[45] (Table S2, SI). Among the significantly downregulated genes were the DNA methyltransferase (DNMT3A) and the histone acetyltransferase CREBBP, and NSD2 (a histone methyltransferase) (log2FC = -1.08, -1.14 and -1.73, respectively) indicated impaired epigenetic silencing machinery, potentially reversing oncogenic transcriptional programs[46, 47].
Breast Cancer Metastasis Suppressor 1 (BRMS1) is a protein involved in chromatin remodelling and gene expression regulation, particularly in the context of metastasis [48]. It is known to suppress metastasis in various cancers. In gastric cancer, its expression is often reduced; however, overexpression of BRMS1 (log2FC = 0.89) (Table S2, SI) has been shown to inhibit tumour growth and metastasis in vivo [49]. Meanwhile, the suppression of chromatin remodelling complex components such as SMARCA4, SMARCB1, and PBRM1 (log2FC = -0.90, -1.09 and -1.97, respectively) further supported widespread disruption of nucleosome dynamics (Table S2, SI) [50-52]. Collectively, these alterations indicated a destabilisation of chromatin architecture, resulting in reduced tumour cell proliferation and viability. This chromatin-targeting effect underscored APB’s potential as an epigenetic modulator in gastric cancer therapy.
Ribonucleotide Reductase Signalling Pathway
APB treatment significantly suppresses the Ribonucleotide Reductase (RNR) signalling pathway, which plays a central role in maintaining the deoxyribonucleotide (dNTP) pool necessary for DNA replication and repair in proliferating and quiescent cells [53]. RNR is composed of the catalytic subunit RRM1 and regulatory subunits RRM2 or RRM2B, forming a complex tightly regulated during the cell cycle and stress responses [53]. In cancer, particularly gastric cancer, high RRM2 expression is commonly observed and is associated with enhanced proliferation, angiogenesis, and chemoresistance via pathways such as NF-κB (NFKB1, log2FC = –1.34), AKT-mTOR (AKT1S1, log2FC = –0.67), and EGFR (log2FC = –0.78) (Table S2, SI) [54-56]. In the current study, APB treatment resulted in downregulation of RRM1 (log2FC = –1.1), indicating a disruption of nucleotide biosynthesis that likely contributes to cell cycle arrest and impaired tumour DNA synthesis [57]. This inhibition cascaded through related oncogenic pathways. For example, CHEK1, a DNA damage checkpoint kinase that can upregulate RRM2 via E2F1, was also suppressed (log2FC = –1.31), reinforcing the downregulation of RNR activity [58]. Similarly, key regulators in RNR expression and function—such as NFKB1 (–1.34), AKT1S1 (–0.67), and CDK6 (log2FC = –1.63)—were also downregulated, reflecting a coordinated silencing of survival and proliferative signalling (Table S2, SI) [54, 59, 60]. Intriguingly, CDKN1A (p21) was upregulated (log2FC = 0.95), suggesting cell cycle arrest at the G1/S checkpoint, further halting DNA synthesis and aligning with the suppressed RNR activity [38]. Together, these results suggested that APB imposes a multi-level inhibition of dNTP production and cell cycle progression, strongly implicating the RNR pathway as a targetable vulnerability in gastric cancer.
In the broader context of gastric tumour biology, APB-induced downregulation of the SMARCD family members—SMARCD1 (log2FC = –0.795), SMARCD2 (–0.677), and SMARCD3 (–1.021)—suggested disrupted chromatin remodelling, a process vital for transcriptional control in cancer cells (Table S2, SI) [61]. The downregulation of MAPK13 (log2FC = –0.863), a mitogen-activated protein kinase implicated in inflammatory and stress responses, further indicated attenuation of signalling pathways that support tumour survival [62]. Additionally, key members of the poly (adenosine diphosphate-ribose) polymerase (PARP) family—PARP9 (–1.017), PARP12 (–1.190), and PARP14 (–1.208)—are significantly suppressed (Table S2, SI) [63]. While these genes are commonly upregulated in gastric cancer and contribute to tumour progression, stress adaptation, and cell survival, their inhibition may sensitise tumour cells to damage and promote therapeutic response. Notably, PHF10 (–1.773), a component of the chromatin remodelling complex, was also strongly downregulated, reinforcing the global collapse of transcriptional machinery following APB treatment [64].
Regulation of endogenous retroelements
APB treatment appeared to modulate the regulation of endogenous retroelements, which are typically silenced in normal cells but often become aberrantly activated in cancer through epigenetic dysregulation. Transposable elements—including LINEs (Long Interspersed Nuclear Elements) and SINEs (Short Interspersed Nuclear Elements) can influence oncogenic transcription through enhancer activity or transcription factor binding [65]. In the context of gastric cancer, APB downregulated several proteins associated with retroelement-linked transcriptional regulation. These include CDK6 (log2FC = –1.63), a key G1/S checkpoint regulator often influenced by retroelement-driven chromatin changes, and PHF10 (log2FC = –1.77), a subunit of the ATP-dependent PBAF chromatin remodeling complex, essential for its interaction with chromatin[60, 64]. Additionally, WEE1 (log2FC = –1.12), ERBB2 (log2FC = –1.07), and EGFR (log2FC = –0.78)—each implicated in DNA damage responses and retroelement-associated promoter activation—are suppressed (Table S2, SI) [66, 67]. The anti-apoptotic proteins BIRC5 (log2FC = –0.659) and mTOR complex regulator MLST8 (log2FC = –0.66) were also downregulated, further supporting reduced cell survival and metabolic activity [68, 69]. Finally, the transcription factor subunit NFYC (log2FC = –0.61) was suppressed, indicating disrupted regulatory control over retroelement-responsive gene networks (Table S2, SI) [70].
Cell Cycle Checkpoint
Disruption of the cell cycle checkpoint pathway is a defining mechanism through which APB exerted its antiproliferative activity in AGS gastric adenocarcinoma cells. Normally, cell cycle checkpoints ensure genomic integrity by coordinating critical transitions such as G1/S and G2/M, with failure in these processes being a hallmark of cancer progression. In the current study, APB treatment led to strong downregulation of multiple key genes involved in checkpoint regulation and mitotic fidelity. These include UBE2C (log2FC = –1.75), NSD2 (log2FC = –1.73), ZWINT (log2FC = –1.63), BRCA1 (BReast CAncer gene 1; log2FC = –1.40), and CHEK1 (–1.31)—all of which are typically overexpressed in gastric cancer and promote cell proliferation, chromosomal segregation, and DNA damage repair (Table S2, SI) [46, 71-76]. Their suppression reflected a broad collapse in checkpoint surveillance and genome maintenance functions, potentially leading to mitotic catastrophe and apoptosis in rapidly dividing tumour cells. Additional downregulated regulators such as WEE1 (–1.12), PLK1 (–1.07), MAD2L1 (–0.96), CENPE (–0.81), and AURKB (–0.77) further highlighted the inhibition of mitotic entry and spindle checkpoint integrity (Table S2, SI) [37, 66, 77-80]. Collectively, these proteins play essential roles in ensuring proper kinetochore function, centrosome duplication, and chromosome segregation. Notably, many of these factors are normally upregulated in gastric cancer and contribute to tumour aggressiveness, poor prognosis, and therapy resistance.
Figure 3D presents a disease and disorder association analysis following APB treatment in AGS gastric cancer cells. Notably, the strongest associations (highest significance, blue bars with negative z-scores) are linked to squamous cell tumour and squamous-cell carcinoma, suggesting that APB induces transcriptional changes opposing gene expression profiles typically observed in these malignancies. This is particularly relevant given the epithelial nature of gastric cancer and its overlap with squamous differentiation in some subtypes [81]. Other significantly downregulated disease signatures included malignant intracranial tumours, cerebellar neoplasms, and medulloblastoma, indicating a broad anticancer transcriptional shift. The data imply that APB enforced a general suppression of proliferative and neoplastic transcriptional programs
3.5.2. Enriched Pathways of DEPs in Dex Treated AGS Gastric Adenocarcinoma Cells Compared to Control
Dex, a potent synthetic glucocorticoid, has been shown to significantly affect various pathways and biological processes (Figure 4). The volcano plot (Figure 4A) comparing Dex-treated AGS gastric cancer cells to untreated control revealed a distinct pattern of gene expression changes, highlighting Dex’s broad regulatory effects. A substantial number of genes were significantly downregulated (blue), including key cell cycle and DNA repair regulators such as RAD51AP1, MCM5, TPX2, and HELls, indicating suppression of proliferative and genomic maintenance pathways. Conversely, a cluster of genes was significantly upregulated (red), including CLU (Clusterin), HERPUD1, MT2A, CEBPB, and NDFIP1, many of which are linked to stress response, apoptosis, or metabolic regulation. The presence of strongly upregulated chaperones and antioxidant-related genes, along with suppression of proliferation-associated factors, suggested that Dex induces a stress-adaptive, anti-proliferative transcriptional program in the AGS gastric adenocarcinoma cells.
The graphical summary (Figure 4B) of differentially expressed proteins (DEPs) in AGS cells treated with Dex revealed several key biological processes and regulatory networks disrupted by the treatment. The central themes identified included inhibition of cell proliferation, suppression of tumour-promoting transcriptional networks, and modulation of cytokine-responsive gene expression. Core regulators such as CCND1, E2F3, FOXM1, SOX9, and GATA6 are significantly affected, many of which are commonly overexpressed in gastric cancer and are associated with tumour growth, cell cycle progression, and epithelial transformation [82]. The interplay of these proteins with cytokine mediators (e.g., IL1B) further suggested a strong immunomodulatory component of Dex’s action.
Figure 4C presents enriched canonical pathways in AGS gastric cancer cells following Dex treatment, ranked by statistical significance (–log BH P-value). It highlighted both positively and negatively regulated processes, indicating Dex's dual impact on metabolic and cell cycle-related signalling. The most significantly upregulated pathways include activation of protein expression by SREBF (SREBP, Sterol Regulatory Element-Binding Proteins) and multiple branches of cholesterol biosynthesis, suggesting that Dex strongly enhanced lipid metabolic programs. Given that cancer cells often rewire lipid metabolism to support membrane synthesis and rapid proliferation, this shift may reflect either a compensatory adaptation or Dex-driven metabolic stress. Conversely, several key proliferative pathways showed strong inhibition, most notably cell cycle checkpoints, activation of the pre-replicative complex, and cell cycle control of chromosomal replication. These pathways are critical for ensuring DNA replication fidelity and progression through S-phase and mitosis—core processes often dysregulated in gastric and other cancers.
Activation of SREBF-Mediated Cholesterol Biosynthesis in Dex-Treated AGS Cells
Dex treatment in AGS gastric adenocarcinoma cells resulted in a robust activation of the SREBF (SREBP)-mediated transcriptional programme, as evidenced by the significant upregulation of key proteins involved in the cholesterol biosynthesis pathway (Figure 4C). SREBPs are master regulators of lipid homeostasis, primarily controlling the expression of enzymes required for cholesterol and fatty acid synthesis [83]. Dex enhanced the nuclear activity of SREBP1/2, driving the expression of canonical downstream targets including HMGCR (log2FC = 3.327), CYP51A1 (2.695), SQLE (2.064), FDFT1 (1.889), SC5D (1.737), and MSMO1 (1.761) (Table S2, SI) [83-85]. These genes encoding these proteins span multiple stages of the mevalonate pathway and both branches of cholesterol synthesis—via desmosterol (Bloch pathway) and lathosterol (Kandutsch–Russell pathway)—highlighting widespread pathway engagement.
Increased cholesterol synthesis intermediates could sensitise cells to ferroptosis or oxidative stress, both being anticancer mechanisms [86]. Dex treatment resulted in the downregulation of key SREBP co-factors—including SP1 (log2FC = –0.594), CREBBP (–0.645), and NFYB (–0.669)—all of which are typically upregulated in cancer (Table S2, SI) [87, 88]. These co-factors are essential for full transcriptional activation, and their suppression could limit the tumour-promoting effects of the cholesterol biosynthesis programme [89]. Moreover, repression of RXRA (–0.718), a nuclear receptor involved in lipid signalling and inflammatory crosstalk, may further modulate this axis toward a tumour-suppressive phenotype [90]. Altogether, these findings suggested that Dex-induced activation of cholesterol biosynthesis may not reflect metabolic support for tumour growth but rather a form of stress-induced reprogramming. Enhanced cholesterol biosynthesis may reflect a cellular stress response or a mechanism promoting membrane synthesis in proliferative cells.
Proteins related to the cell cycle
Dex treatment also led to the dysregulation of key proteins involved in apoptosis, DNA repair, cell cycle progression, and autophagy [91]. Among these, Survivin BIRC5 (log2FC = -1.36), an inhibitor of apoptosis protein, was downregulated, which aligned with studies indicating that Dex promotes apoptotic pathways by reducing survival signals in cancer cells [68]. Similarly, the suppression of cell division cycle 20 (CDC20; log2FC = -1.56) and ubiquitin-conjugating enzyme E2 C (UBE2C; log2FC = -1.58)—key players in cell cycle progression—pointed to Dex-induced mitotic arrest, a mechanism often exploited to curb cancer cell proliferation. Upregulation of CDKN1A (log2FC = 1.01) and downregulation of polo-like kinase 1 (PLK1; log2FC = -0.94) reflected Dex's dual role in promoting cell cycle arrest and limiting mitotic progression (Table S2, SI) [92-94]. CDKN1A, also known as p21, is a cyclin-dependent kinase inhibitor that enforces G1/S checkpoint control, preventing cells from proceeding through the cell cycle. This effect, coupled with PLK1 suppression, a key regulator of mitotic entry, suggested that Dex enforced a halt in cell proliferation of AGS cells in our study. The p21 was reported to be a negative regulator of p53 stability [38]. Furthermore, the downregulation of BRCA1 (log2FC = -1.93) and TP53 (log2FC = -0.75), critical mediators of the DNA damage response—indicated a potential attenuation of repair mechanisms, which might sensitise cancer cells to chemotherapy or further DNA damage. Notably, BRCA1 was also downregulated by APB treatment (log2FC = –1.40). The significant upregulation of P62, known as SQSTM1 (log2FC = 2.46), correlated with its role as a mediator in autophagy and cellular stress pathways (Table S2, SI) [95]. This could indicate an adaptive response of AGS cells to Dex-induced stress, possibly balancing autophagy with cell death mechanisms. Additionally, the marked upregulation of P35527 (log2FC = 4.41) and downregulation of C4BPB (log2FC = -2.17) and CD320 (log2FC = -2.11) further emphasised the impact of Dex on the cellular microenvironment, particularly influencing immune-modulatory and nutrient uptake pathways (Table S2, SI) [96, 97]
Ferroptosis
Ferroptosis is a form of programmed cell death distinct from apoptosis, necrosis, or autophagy, characterised by the accumulation of lipid peroxidation products and lethal ROS derived from iron-dependent reactions [98]. This process is tightly regulated and plays a significant role in various biological contexts, including cancer, neurodegeneration, and ischemia-reperfusion injuries [98]. The upregulation of HMGCR (log2FC = 3.32), HMOX1 (log2FC = 0.83), ACSL4 (log2FC = 0.74), FTH1 (log2FC = -0.75), GCLC (log2FC = 0.85), GCLM (log2FC = 0.87), TXNRD1 (log2FC = 0.98) in combination with the downregulation of SLC11A2 (log2FC = -0.65), TFRC (log2FC = -0.64), and OLR1 (log2FC = -2.51) indicated induction of ferroptosis in the AGS cells upon Dex treatment (Table S2, SI) [99]. This observation aligned with previous investigations conducted by us and others [14, 100].
3.5.3. Enriched Pathways Using DEPs of APB+Dex Combination Treated AGS Cells vs Mono Treatments
Based on the promising antiproliferative and apoptotic effects of the APB+Dex combination against AGS gastric adenocarcinoma cells, we analysed the proteins linked to these activities. We compared the proteomic profiles of combination therapy with those of the individual treatments and the untreated control.
The volcano plot comparing APB+Dex versus APB and Dex mono treatments alone highlighted key DEPs that reveal the molecular effects of adding dexamethasone to APB therapy in AGS gastric cancer cells (Figure 5A). Several proteins, including INSIG1, PCBD1, and COL4A2, were significantly upregulated (log2FC > 0.58, Q ≤ 0.05), suggesting enhanced extracellular matrix organisation, cholesterol homeostasis, and metabolic regulation, possibly linked to Dex’s modulatory effects [101-103]. Conversely, a large number of proteins involved in cell proliferation, oxidative stress, and immune response—such as AGTRAP, COX17, TAF15, ELF1, NFE2L1, OSGIN1, and CCND1—were significantly downregulated, indicating suppression of proliferative and inflammatory pathways upon combination treatment [37, 104-108]. The marked repression of CCND1, a critical cell cycle regulator, and EGR1, an early growth response gene, supported a Dex-mediated antiproliferative shift [109]. Overall, this comparison demonstrated that Dex addition to APB intensified the suppression of tumour-promoting pathways while modestly activating stress-adaptive and metabolic programs, underscoring its potential to enhance therapeutic efficacy. A summary of other dysregulated proteins upon the combination treatment is listed in Table 4.
The graphical summary of differentially expressed proteins in the APB+Dex versus APB-only and Dex-only treatment highlighted significant transcriptional reprogramming and immune modulation driven by the addition of Dex (Figure 5B). Central to this network are key regulators such as CREBBP, TP53, NFKB1, and MYD88, which coordinate critical processes like transcriptional activation, immune signalling, and inflammation [110-113]. Downregulation of transcription factors (ELK1, REL, ATF4) and transcriptional co-activators like CREBBP suggested a global reduction in RNA and DNA transcription, potentially curbing tumour-promoting gene expression [114]. Notably, suppression of the TNF-NFKB1-MYD88 axis implied attenuation of the I-kappaB kinase/NF-kappaB cascade, a pathway often hyperactivated in gastric and other cancers to promote survival and inflammation [115]. Moreover, the network reflected a decrease in pro-inflammatory cytokine signalling through downregulation of IFNG, TLR3, and related mediators, consistent with Dex’s immunosuppressive effects [116]. This may contribute to the observed suppression of transcription and transactivation processes across multiple genes. Additionally, reduced activity of growth and proliferation-associated proteins (HGF, FLCN, CD40LG) aligned with predictions of impaired cell growth and development, as visualized in Figure 5B by links to phenotypes like "Growth Failure" and "Short Stature" [117].
It should provide a concise and precise description of the experimental results, their interpretation, as well as the experimental conclusions that can be drawn.
Proteins Related to homeostasis and Tumour Microenvironment Regulation
APB+Dex treatment in AGS gastric cancer cells significantly altered the expression of proteins involved in haemostasis, extracellular matrix remodelling, and oxidative stress regulation, as reflected in the top canonical pathways enriched in the analysis (Figure 5C). Most notably, the “Response to elevated platelet cytosolic Ca²⁺” and “LXR/RXR Activation” pathways—ranked as the most significantly upregulated—underscored a coordinated activation of calcium and lipid-mediated signalling that impacts coagulation, immune regulation, and vascular interaction. These processes are central to tumour progression and metastasis but may also act as targets for tumour suppression when tightly modulated.
The upregulation of key coagulation and fibrinolysis-related proteins—SERPINE1 (log2FC = 1.993), F5 (1.765), F13A1 (1.63), FGA, FGB, FGG (1.458, 1.338, 1.172), and FN1 (1.404)—pointed to enhanced clotting and extracellular matrix (ECM) stabilization (Table S2, SI) [131]. These changes, tied to “Formation of Fibrin Clot (Clotting Cascade)” and “Cell surface interactions at the vascular wall”, suggested a shift toward a more adhesive and structurally reinforced microenvironment that may hinder tumour cell dissemination. In parallel, key tumour-supportive proteins were notably suppressed upon APB+Dex treatment. SLC7A11 (log2FC = –2.60), a cystine/glutamate transporter critical for glutathione biosynthesis and resistance to ferroptosis, was strongly downregulated, suggesting APB+Dex disrupted redox homeostasis, potentially sensitising cancer cells to oxidative damage [132, 133]. Similarly, FSTL3 (log2FC = –1.62), a glycoprotein implicated in extracellular matrix regulation and associated with metastasis in gastric cancer, was also reduced. Its repression may contribute to decreased tumour cell proliferation and invasion by limiting cell–matrix interactions [134, 135]. Together, these haemostasis- and matrix-related changes illustrated how APB+Dex may reprogram the tumour microenvironment, impair survival mechanisms, and reinforce cytotoxic and anti-metastatic effects in gastric cancer.
Proteins related to Transcription Regulation and Cell Growth Modulation
Pleckstrin homology domain-containing family G member 2 (PLEKHG2) is a key regulator of Rho GTPases, which are pivotal in cytoskeletal remodelling, cell migration, and intracellular signal transduction. These functions make it critical in maintaining cellular architecture and response to environmental stimuli [136]. PLEKHG2 was reported to be upregulated in gastric cancer [137]. The downregulation of PLEKHG1 (log2FC=-3.72) suggested the efficacy of APB+Dex in targeting pathways critical to gastric cancer progression. By disrupting PLEKHG1-mediated functions, APB+Dex might induce structural and signalling deficits, contributing to its antiproliferative activity. APB+Dex combination treatment also resulted in the downregulation of Tensin-4 (log2FC=-1.35) (Table S2, SI). Tensin-4 (TNS4, also known as Cten) is an oncogene involved in regulating cell adhesion, migration, and signalling pathways and has been associated with cancer progression [138]. Interestingly, we previously reported that mono treatment with B resulted in the upregulation of both TNS4 and PLEKHM1 [14]. The downregulation of structural and signalling proteins such as PLEKHG1 (log2FC = –3.72) and TNS4 (log2FC = –1.35) following APB+Dex treatment in AGS gastric adenocarcinoma cells underscored a broader transcriptional repression of oncogenic regulators linked to cytoskeletal dynamics, adhesion, and migration (Table S2, SI).
Insulin Induced Gene 1 (INSIG1) is a key regulator of intracellular signalling, particularly in sterol biosynthesis and metabolic homeostasis [101]. It modulates the activity of sterol regulatory element-binding proteins (SREBPs), which are critical for lipid metabolism [101]. A previous study suggested that reduced expression of the INSIG1 gene may be involved in gastric cancer development or progression [139]. In AGS cells treated with APB+Dex, the upregulation of INSIG1 (log2FC = 2.66) and SRBP1 (log2FC = 1.19) could indicate a reversal of this cancer-associated silencing (Table S2, SI) [139]. Given its potential tumour-suppressive role, this restoration may indicate a reactivation of normal cellular regulatory processes, contributing to an antiproliferative effect [139]. A list of dysregulated genes by the combination treatment is illustrated in Table 4 and Figure 5.
Proteins related to amino acid transport across the plasma membrane
Solute carrier (SLC) proteins are membrane transport proteins that move nutrients, ions, drugs, and neurotransmitters across cell membranes [140]. APB+Dex treatment significantly downregulated key amino acid transporters across the plasma membrane, disrupting essential metabolic pathways in gastric cancer cells. Among these, SLC7A11 showed the most pronounced decrease (Log2FC = –2.60), impacting its crucial role in cystine/glutamate exchange and redox balance [133]. This transporter is typically upregulated in gastric cancer to combat oxidative stress and prevent ferroptosis, underscoring its role in tumour survival [133]. Similarly, the downregulation of SLC7A1 (Log2FC = – 1.26) indicated the impaired uptake of cationic amino acids like arginine, essential for cancer cell proliferation and survival [133].
The neutral amino acid transporters SLC1A4 (Log2FC = – 1.20), SLC7A5 (Log2FC = – 0.71), and SLC43A2 (Log2FC = – 1.20) were also significantly suppressed after APB+Dex treatment. SLC1A4 and SLC43A2 facilitate neutral amino acid uptake, essential for metabolic adaptations in proliferative cancer cells, while SLC7A5 supports the import of large neutral amino acids such as leucine, fuelling tumour growth and is associated with poor prognosis in gastric cancer (Table S2, SI) [140]. Together, these changes reflected a comprehensive disruption of amino acid transport and metabolic reprogramming critical for tumour growth and oxidative stress resistance. The coordinated suppression of these transporters highlighted their potential as therapeutic targets in gastric cancer treatment, especially under metabolic stress induced by APB+Dex therapy.Table 4: List of the relevant dysregulated proteins in APB+Dex treated AGS gastric adenocarcinoma cells
The disease association analysis for APB+Dex treatment (Figure 5D) in AGS gastric cancer cells highlighted a strong inverse correlation with gene expression signatures typically found in a variety of solid tumours, particularly hepatocellular carcinoma, non-hematological solid tumours, and extracranial or non-melanoma solid tumours—as evidenced by prominent negative z-scores (blue bars). These findings suggested that APB+Dex potentially downregulated gene programs that are characteristically upregulated in these cancers, including gastric malignancies. Since gastric cancer shares oncogenic pathways and molecular hallmarks with these solid tumour types, such as dysregulated proliferation, angiogenesis, and extracellular matrix remodelling, the observed transcriptional reversal strongly supported the potential antitumor efficacy of APB+Dex in gastric adenocarcinoma.
4. Conclusions
In conclusion, this study highlighted the potent antiproliferative effects of SCFA salts A, P, and B, both individually and in combination with Dex on AGS gastric adenocarcinoma cells. B exhibited significant growth inhibition, and the combination of SCFAs (APB) with Dex demonstrated strong synergistic effects, further enhancing antiproliferative activity. The strong synergistic interaction observed with Dex indicated that APB combinations could be promising adjuncts in gastric cancer treatment, potentially improving patient outcomes by enhancing drug efficacy while possibly reducing the required dosages and associated side effects. Flow cytometric analysis revealed increased apoptosis, with minimal necrosis, indicating that APB and their combination with Dex primarily induced apoptotic cell death. APB treatment elevated ROS levels, with Dex moderating this increase in the combination therapy. Proteomic analysis using LC-MS provided insights into the molecular mechanisms underlying these effects, identifying key proteins involved in apoptosis, autophagy, and immune modulation. The combination APB+Dex targeted critical pathways, including cell cycle regulation and redox balance, potentially sensitising gastric cancer cells to oxidative stress and ferroptosis. These findings suggested that APB+Dex combinations hold promise as an adjunct therapy in gastric cancer treatment, offering enhanced anticancer activity and providing a basis for further investigation into their clinical applications. Future studies on appropriate animal and organoid models are required to validate these findings further.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, R.A.E and D.J.B; methodology, R.A.E and M.F.; software, R.A.E, M.F and M.A.A.; validation, R.A.E; formal analysis, R.A.E. and M.F.; investigation, R.A.E.; data curation, R.A.E; writing—original draft preparation, R.A.E and M.F.; writing—review and editing, R.A.E, M.F., M.A.A and D.J.B; visualization, R.A.E and M.F.; supervision, D.C., C.G.L and D.J.B; project administration, R.A.E and D.J.B; funding acquisition, R.A.E and D.J.B. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research received no external funding. We acknowledge the support of Western Sydney University, Australia, through the PhD Research Training Program Scholarship (RE) and the Research Support Program Fellowship (DJB) to conduct this research.
Informed Consent Statement
Any research article describing a study involving humans should contain this statement. Please add “Informed consent was obtained from all subjects involved in the study.” OR “Patient consent was waived due to REASON (please provide a detailed justification).” OR “Not applicable.” for studies not involving humans. You might also choose to exclude this statement if the study did not involve humans.Written informed consent for publication must be obtained from participating patients who can be identified (including by the patients themselves). Please state “Written informed consent has been obtained from the patient(s) to publish this paper” if applicable.
Acknowledgments
The authors acknowledge and pay respect to the Traditional Owners of the land on which we research, teach, and collaborate—the Darug People (Darug Nation: Western Sydney University Westmead and Penrith Campuses), the Bidigal People (Dharawal Nation: Western Sydney University Campbelltown Campus), and the Gadigal People (Eora Nation: The University of Sydney). The authors would also like to acknowledge the University of Sydney Mass Spectrometry Facility for providing access to its instrumentation and assistance with the MS analyses.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Burz, Claudia, Vlad Pop, Ciprian Silaghi, Iulia Lupan, and Gabriel Samasca. "Prognosis and Treatment of Gastric Cancer: A 2024 Update." Cancers 16, no. 9 (2024): 1708. [CrossRef]
- Rawla, Prashanth, and Adam Barsouk. "Epidemiology of Gastric Cancer: Global Trends, Risk Factors and Prevention." Gastroenterology Review/Przegląd Gastroenterologiczny 14, no. 1 (2019): 26-38.
- Joshi, Smita S, and Brian D Badgwell. "Current Treatment and Recent Progress in Gastric Cancer." CA: a cancer journal for clinicians 71, no. 3 (2021): 264-79. [CrossRef]
- Hani, Umme, Riyaz Ali M Osmani, Sabina Yasmin, BH Jaswanth Gowda, Hissana Ather, Mohammad Yousuf Ansari, Ayesha Siddiqua, Mohammed Ghazwani, Adel Al Fatease, and Ali H Alamri. "Novel Drug Delivery Systems as an Emerging Platform for Stomach Cancer Therapy." Pharmaceutics 14, no. 8 (2022): 1576. [CrossRef]
- Jaye, Kayla, Chun Guang Li, Dennis Chang, and Deep Jyoti Bhuyan. "The Role of Key Gut Microbial Metabolites in the Development and Treatment of Cancer." Gut Microbes 14, no. 1 (2022): 2038865.
- Eladwy, Radwa A, Hang Thi Vu, Ravi Shah, Chun Guang Li, Dennis Chang, and Deep Jyoti Bhuyan. "The Fight against the Carcinogenic Epstein-Barr Virus: Gut Microbiota, Natural Medicines, and Beyond." International Journal of Molecular Sciences 24, no. 2 (2023): 1716.
- Chambers, Laura M, Emily L Esakov Rhoades, Rashmi Bharti, Chad Braley, Surabhi Tewari, Lexie Trestan, Zahraa Alali, Defne Bayik, Justin D Lathia, and Naseer Sangwan. "Disruption of the Gut Microbiota Confers Cisplatin Resistance in Epithelial Ovarian Cancer." Cancer research 82, no. 24 (2022): 4654-69.
- Facchin, Sonia, Luisa Bertin, Erica Bonazzi, Greta Lorenzon, Caterina De Barba, Brigida Barberio, Fabiana Zingone, Daria Maniero, Marco Scarpa, and Cesare Ruffolo. "Short-Chain Fatty Acids and Human Health: From Metabolic Pathways to Current Therapeutic Implications." Life 14, no. 5 (2024): 559. [CrossRef]
- Sun, Jinzhe, Shiqian Chen, Dan Zang, Hetian Sun, Yan Sun, and Jun Chen. "Butyrate as a Promising Therapeutic Target in Cancer: From Pathogenesis to Clinic." International Journal of Oncology 64, no. 4 (2024): 44. [CrossRef]
- Son, Mi-Young, and Hyun-Soo Cho. "Anticancer Effects of Gut Microbiota-Derived Short-Chain Fatty Acids in Cancers." Journal of Microbiology and Biotechnology 33, no. 7 (2023): 849. [CrossRef]
- Filippone, Alessia, Giovanna Casili, Sarah Adriana Scuderi, Deborah Mannino, Marika Lanza, Michela Campolo, Irene Paterniti, Anna Paola Capra, Cristina Colarossi, and Annalisa Bonasera. "Sodium Propionate Contributes to Tumor Cell Growth Inhibition through Ppar-Γ Signaling." Cancers 15, no. 1 (2022): 217. [CrossRef]
- Mirzaei, Rasoul, Azam Afaghi, Sajad Babakhani, Masoud Reza Sohrabi, Seyed Reza Hosseini-Fard, Kiandokht Babolhavaeji, Shabnam Khani Ali Akbari, Rasoul Yousefimashouf, and Sajad Karampoor. "Role of Microbiota-Derived Short-Chain Fatty Acids in Cancer Development and Prevention." Biomedicine & Pharmacotherapy 139 (2021): 111619.
- Twycross, Robert. "The Risks and Benefits of Corticosteroids in Advanced Cancer." Drug Safety 11 (1994): 163-78.
- Eladwy, Radwa A, Muhammad A Alsherbiny, Dennis Chang, Mohamed Fares, Chun-Guang Li, and Deep Jyoti Bhuyan. "The Postbiotic Sodium Butyrate Synergizes the Antiproliferative Effects of Dexamethasone against the Ags Gastric Adenocarcinoma Cells." Frontiers in Nutrition 11 (2024): 1372982.
- Dissanayake, Indeewarie Hemamali, Muhammad A Alsherbiny, Dennis Chang, Chun Guang Li, and Deep Jyoti Bhuyan. "Antiproliferative Effects of Australian Native Plums against the Mcf7 Breast Adenocarcinoma Cells and Uplc-Qtof-Im-Ms-Driven Identification of Key Metabolites." Food Bioscience 54 (2023): 102864.
- Alsherbiny, Muhammad A, Deep J Bhuyan, Mitchell N Low, Dennis Chang, and Chun Guang Li. "Synergistic Interactions of Cannabidiol with Chemotherapeutic Drugs in Mcf7 Cells: Mode of Interaction and Proteomics Analysis of Mechanisms." International journal of molecular sciences 22, no. 18 (2021): 10103.
- Jaye, Kayla, Muhammad A Alsherbiny, Dennis Chang, Chun-Guang Li, and Deep Jyoti Bhuyan. "Mechanistic Insights into the Anti-Proliferative Action of Gut Microbial Metabolites against Breast Adenocarcinoma Cells." International journal of molecular sciences 24, no. 20 (2023): 15053.
- Donohoe, Dallas R, Nikhil Garge, Xinxin Zhang, Wei Sun, Thomas M O'Connell, Maureen K Bunger, and Scott J Bultman. "The Microbiome and Butyrate Regulate Energy Metabolism and Autophagy in the Mammalian Colon." Cell metabolism 13, no. 5 (2011): 517-26.
- Li, Yangbo, Pengzhan He, Yinghui Liu, Mingming Qi, and Weiguo Dong. "Combining Sodium Butyrate with Cisplatin Increases the Apoptosis of Gastric Cancer in Vivo and in Vitro Via the Mitochondrial Apoptosis Pathway." Frontiers in Pharmacology 12 (2021): 708093.
- Li, Yuanqing, Yaxuan Huang, Haili Liang, Wen Wang, Bo Li, Ting Liu, Yuqi Huang, Zhe Zhang, Yutao Qin, and Xiaoying Zhou. "The Roles and Applications of Short-Chain Fatty Acids Derived from Microbial Fermentation of Dietary Fibers in Human Cancer." Frontiers in Nutrition 10 (2023): 1243390. [CrossRef]
- Den Besten, Gijs, Karen Van Eunen, Albert K Groen, Koen Venema, Dirk-Jan Reijngoud, and Barbara M Bakker. "The Role of Short-Chain Fatty Acids in the Interplay between Diet, Gut Microbiota, and Host Energy Metabolism." Journal of lipid research 54, no. 9 (2013): 2325-40.
- Elmore, Susan. "Apoptosis: A Review of Programmed Cell Death." Toxicologic pathology 35, no. 4 (2007): 495-516. [CrossRef]
- Hayes, John D, Albena T Dinkova-Kostova, and Kenneth D Tew. "Oxidative Stress in Cancer." Cancer cell 38, no. 2 (2020): 167-97.
- Sudaarsan, Aruna Senthil Kumar, and Asit Ranjan Ghosh. "Appraisal of Postbiotics in Cancer Therapy." Frontiers in Pharmacology 15 (2024): 1436021.
- Martín-García, Desirée, Marilina García-Aranda, and Maximino Redondo. "Therapeutic Potential of Clusterin Inhibition in Human Cancer." Cells 13, no. 8 (2024): 665. [CrossRef]
- Vange, Pål, Torunn Bruland, Bjørn Munkvold, Elin Synnøve Røyset, Martin Gleave, and Ingunn Bakke. "Subtle Protective Roles of Clusterin in Gastric Metaplasia after Acute Oxyntic Atrophy." Cellular and Molecular Gastroenterology and Hepatology 7, no. 1 (2019): 246-50. e1. [CrossRef]
- Zhu, Jinliang, Xin Wang, Huiyuan Guan, Qiong Xiao, Zhonghua Wu, Jinxin Shi, Fei Zhang, Peng Gao, Yongxi Song, and Zhenning Wang. "Hip1r Acts as a Tumor Suppressor in Gastric Cancer by Promoting Cancer Cell Apoptosis and Inhibiting Migration and Invasion through Modulating Akt." Journal of Clinical Laboratory Analysis 34, no. 9 (2020): e23425.
- Schroder, Wayne A, Lee D Major, Thuy T Le, Joy Gardner, Matthew J Sweet, Sabina Janciauskiene, and Andreas Suhrbier. "Tumor Cell-Expressed Serpinb2 Is Present on Microparticles and Inhibits Metastasis." Cancer medicine 3, no. 3 (2014): 500-13. [CrossRef]
- Kim, Hye-Youn, and Suntaek Hong. "Multi-Faceted Roles of Dnajb Protein in Cancer Metastasis and Clinical Implications." International journal of molecular sciences 23, no. 23 (2022): 14970.
- Liu, Jun, John C Schmitz, Xiukun Lin, Ningwen Tai, Wu Yan, Michael Farrell, Michelle Bailly, Tian-min Chen, and Edward Chu. "Thymidylate Synthase as a Translational Regulator of Cellular Gene Expression." Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1587, no. 2-3 (2002): 174-82. [CrossRef]
- Voeller, Donna, Lambratu Rahman, and Maria Zajac-Kaye. "Elevated Levels of Thymidylate Synthase Linked to Neoplastic Transformation of Mammalian Cells." Cell Cycle 3, no. 8 (2004): 1003-05. [CrossRef]
- Guijarro, Maria V, Akbar Nawab, Peter Dib, Sandra Burkett, Xiaoping Luo, Michael Feely, Elham Nasri, Robert P Seifert, Frederic J Kaye, and Maria Zajac-Kaye. "Tyms Promotes Genomic Instability and Tumor Progression in Ink4a/Arf Null Background." Oncogene 42, no. 23 (2023): 1926-39. [CrossRef]
- Liu, Yiyang, Yufei Wang, Sheng Sun, Zeyu Chen, Shuai Xiang, Zeyang Ding, Zhao Huang, and Bixiang Zhang. "Understanding the Versatile Roles and Applications of Epcam in Cancers: From Bench to Bedside." Experimental hematology & oncology 11, no. 1 (2022): 97. [CrossRef]
- Sachdeva, Rohit, Megan Wu, Sandra Smiljanic, Oleksandra Kaskun, Kimia Ghannad-Zadeh, Angela Celebre, Keren Isaev, A Sorana Morrissy, Jennifer Guan, and Jiefei Tong. "Id1 Is Critical for Tumorigenesis and Regulates Chemoresistance in Glioblastoma." Cancer research 79, no. 16 (2019): 4057-71.
- Wang, Xin, Shen Li, Chen Liu, Jiawei Zhao, Gangfeng Ren, Feng Zhang, Xuyang Liu, Shuang Cao, Yuming Xu, and Zongping Xia. "High Expression of Pla2g2a in Fibroblasts Plays a Crucial Role in the Early Progression of Carotid Atherosclerosis." Journal of Translational Medicine 22, no. 1 (2024): 967.
- Hashimoto, Itaru, and Takashi Oshima. "Claudins and Gastric Cancer: An Overview." Cancers 14, no. 2 (2022): 290.
- Nie, Min, Yadong Wang, Zenong Yu, Xinyu Li, Yexuan Deng, Ying Wang, Dongjun Yang, Qixiang Li, Xiangwei Zeng, and Junyi Ju. "Aurkb Promotes Gastric Cancer Progression Via Activation of Ccnd1 Expression." Aging (Albany NY) 12, no. 2 (2020): 1304. [CrossRef]
- Broude, Eugenia V, Zoya N Demidenko, Claire Vivo, Mari E Swift, Brian M Davis, Mikhail V Blagosklonny, and Igor B Roninson. "P21 (Cdkn1a) Is a Negative Regulator of P53 Stability." Cell Cycle 6, no. 12 (2007): 1467-70.
- Bao, Chenhui, and Lin Guo. "Tp73-As1 Promotes Gastric Cancer Proliferation and Invasion by Regulation Mir-27b-3p/Tmed5 Axis." Journal of Cancer 13, no. 4 (2022): 1324.
- Koh, Sung Ae, and Kyung Hee Lee. "Function of Hepatocyte Growth Factor in Gastric Cancer Proliferation and Invasion." Yeungnam University Journal of Medicine 37, no. 2 (2020): 73-78.
- Zeng, Jiping, Lixiang Wang, Qiao Li, Wenjuan Li, Magnus Björkholm, Jihui Jia, and Dawei Xu. "Foxm1 Is up-Regulated in Gastric Cancer and Its Inhibition Leads to Cellular Senescence, Partially Dependent on P27kip1." The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland 218, no. 4 (2009): 419-27.
- Zhao, Long-Fei, Feng-Yu Qi, Jin-Ge Zhang, Jing-Ru Pang, Hong-Mei Ren, Dan-Dan Shen, Li-Juan Zhao, Lin Qi, Hong-Min Liu, and Yi-Chao Zheng. "Identification of the Upstream Regulators of Kdm5b in Gastric Cancer." Life Sciences 298 (2022): 120458. [CrossRef]
- Ma, Yuxi, Na Shen, Max S Wicha, and Ming Luo. "The Roles of the Let-7 Family of Micrornas in the Regulation of Cancer Stemness." Cells 10, no. 9 (2021): 2415. [CrossRef]
- Morikawa, Teppei, Rumi Hino, Hiroshi Uozaki, Daichi Maeda, Tetsuo Ushiku, Aya Shinozaki, Takashi Sakatani, and Masashi Fukayama. "Expression of Ribonucleotide Reductase M2 Subunit in Gastric Cancer and Effects of Rrm2 Inhibition in Vitro." Human pathology 41, no. 12 (2010): 1742-48. [CrossRef]
- Mjelle, Robin, Siv Anita Hegre, Per Arne Aas, Geir Slupphaug, Finn Drabløs, Pål Sætrom, and Hans E Krokan. "Cell Cycle Regulation of Human DNA Repair and Chromatin Remodeling Genes." DNA repair 30 (2015): 53-67. [CrossRef]
- Topchu, Iuliia, Rajendra P Pangeni, Igor Bychkov, Sven A Miller, Evgeny Izumchenko, Jindan Yu, Erica Golemis, John Karanicolas, and Yanis Boumber. "The Role of Nsd1, Nsd2, and Nsd3 Histone Methyltransferases in Solid Tumors." Cellular and Molecular Life Sciences 79, no. 6 (2022): 285. [CrossRef]
- Kataoka, Isao, Sayaka Funata, Kiyotaka Nagahama, Kazunobu Isogaya, Hirohisa Takeuchi, Nobutsugu Abe, and Junji Shibahara. "Dnmt3a Overexpression Is Associated with Aggressive Behavior and Enteroblastic Differentiation of Gastric Adenocarcinoma." Annals of Diagnostic Pathology 44 (2020): 151456.
- Meehan, William J, Rajeev S Samant, James E Hopper, Michael J Carrozza, Lalita A Shevde, Jerry L Workman, Kristin A Eckert, Michael F Verderame, and Danny R Welch. "Breast Cancer Metastasis Suppressor 1 (Brms1) Forms Complexes with Retinoblastoma-Binding Protein 1 (Rbp1) and the Msin3 Histone Deacetylase Complex and Represses Transcription." Journal of Biological Chemistry 279, no. 2 (2004): 1562-69. [CrossRef]
- Guo, Xiu-Li, Ya-Jie Wang, Pei-Lin Cui, Yan-Bin Wang, Pi-Xia Liang, Ya-Nan Zhang, and You-Qing Xu. "Effect of Brms1 Expression on Proliferation, Migration and Adhesion of Mouse Forestomach Carcinoma." Asian Pacific Journal of Tropical Medicine 8, no. 9 (2015): 724-30. [CrossRef]
- Bhat, Vipul, Manisha Koneru, Kristen Knapp, Upasana Joneja, Jamin Morrison, and Young K Hong. "Identification and Treatment of Smarca4 Deficient Poorly Differentiated Gastric Carcinoma." The American Surgeon™ 89, no. 11 (2023): 4987-89.
- Kohashi, Kenichi, and Yoshinao Oda. "Oncogenic Roles of Smarcb 1/Ini 1 and Its Deficient Tumors." Cancer science 108, no. 4 (2017): 547-52.
- Zhou, Zhiyi, Dandan Huang, Shudong Yang, Jiabei Liang, Xuan Wang, and Qiu Rao. "Clinicopathological Significance, Related Molecular Changes and Tumor Immune Response Analysis of the Abnormal Swi/Snf Complex Subunit Pbrm1 in Gastric Adenocarcinoma." Pathology and Oncology Research 28 (2022): 1610479. [CrossRef]
- Foskolou, Iosifina P, Christian Jorgensen, Katarzyna B Leszczynska, Monica M Olcina, Hanna Tarhonskaya, Bauke Haisma, Vincenzo D’Angiolella, William K Myers, Carmen Domene, and Emily Flashman. "Ribonucleotide Reductase Requires Subunit Switching in Hypoxia to Maintain DNA Replication." Molecular cell 66, no. 2 (2017): 206-20. e9. [CrossRef]
- O’Reilly, Lorraine A, Tracy L Putoczki, Lisa A Mielke, Jun T Low, Ann Lin, Adele Preaudet, Marco J Herold, Kelvin Yaprianto, Lin Tai, and Andrew Kueh. "Loss of Nf-Κb1 Causes Gastric Cancer with Aberrant Inflammation and Expression of Immune Checkpoint Regulators in a Stat-1-Dependent Manner." Immunity 48, no. 3 (2018): 570-83. e8.
- Riquelme, Ismael, Oscar Tapia, Jaime A Espinoza, Pamela Leal, Kurt Buchegger, Alejandra Sandoval, Carolina Bizama, Juan Carlos Araya, Richard M Peek, and Juan Carlos Roa. "The Gene Expression Status of the Pi3k/Akt/Mtor Pathway in Gastric Cancer Tissues and Cell Lines." Pathology & Oncology Research 22 (2016): 797-805.
- Weng, Xiaoling, Hong Zhang, Junyi Ye, Mengyuan Kan, Fatao Liu, Ting Wang, Jiaying Deng, Yanfang Tan, Lin He, and Yun Liu. "Hypermethylated Epidermal Growth Factor Receptor (Egfr) Promoter Is Associated with Gastric Cancer." Scientific reports 5, no. 1 (2015): 10154. [CrossRef]
- Zhao, Li-Ping, Cong Xue, Jian-Wei Zhang, Zhi-Huang Hu, Yuan-Yuan Zhao, Jing Zhang, Yan Huang, Hong-Yun Zhao, and Li Zhang. "Expression of Rrm1 and Its Association with Resistancy to Gemcitabine-Based Chemotherapy in Advanced Nasopharyngeal Carcinoma." Chinese journal of cancer 31, no. 10 (2012): 476.
- Jiang, Kailong, Minjie Deng, Wenjing Du, Tao Liu, Jia Li, and Yubo Zhou. "Functions and Inhibitors of Chk1 in Cancer Therapy." Medicine in Drug Discovery (2024): 100185.
- Qi, Zhaolai, Ting Zhang, Lei Song, Hongyong Fu, Haifeng Luo, Jie Wu, Shuyun Zhao, Tianhua Zhang, Lianying Guo, and Lingling Jin. "Pras40 Hyperexpression Promotes Hepatocarcinogenesis." EBioMedicine 51 (2020). [CrossRef]
- Ooi, Akishi, Takeru Oyama, Ritsuko Nakamura, Ryousuke Tajiri, Hiroko Ikeda, Sachio Fushida, and Yoh Dobashi. "Gene Amplification of Ccne1, Ccnd1, and Cdk6 in Gastric Cancers Detected by Multiplex Ligation-Dependent Probe Amplification and Fluorescence in Situ Hybridization." Human pathology 61 (2017): 58-67.
- Park, Sun Yi, Ji-Ho Park, Jung Wook Yang, Eun-Jung Jung, Young-Tae Ju, Chi-Young Jeong, Ju-Yeon Kim, Taejin Park, Tae-Han Kim, and Miyeong Park. "Smarcd3 Overexpression Promotes Epithelial–Mesenchymal Transition in Gastric Cancer." Cancers 16, no. 12 (2024): 2282. [CrossRef]
- Kudaravalli, Sriya, Petra den Hollander, and Sendurai A Mani. "Role of P38 Map Kinase in Cancer Stem Cells and Metastasis." Oncogene 41, no. 23 (2022): 3177-85. [CrossRef]
- Chen, Hongjie, Yangchan Hu, Zirui Zhuang, Dingyi Wang, Zu Ye, Ji Jing, and Xiangdong Cheng. "Advancements and Obstacles of Parp Inhibitors in Gastric Cancer." Cancers 15, no. 21 (2023): 5114.
- Fan, Zhiyuan, Wenjing Yan, Jianfang Li, Min Yan, Bingya Liu, Zhongyin Yang, and Beiqin Yu. "Phf10 Inhibits Gastric Epithelium Differentiation and Induces Gastric Cancer Carcinogenesis." Cancer Gene Therapy 31, no. 10 (2024): 1511-24.
- Grundy, Erin E, Noor Diab, and Katherine B Chiappinelli. "Transposable Element Regulation and Expression in Cancer." The FEBS journal 289, no. 5 (2022): 1160-79.
- Kim, Hye-Young, Yunhee Cho, HyeokGu Kang, Ye-Seal Yim, Seok-Jun Kim, Jaewhan Song, and Kyung-Hee Chun. "Targeting the Wee1 Kinase as a Molecular Targeted Therapy for Gastric Cancer." Oncotarget 7, no. 31 (2016): 49902.
- Hino, Kaori, Tomohiro Nishina, Takeshi Kajiwara, Hideaki Bando, Maho Nakamura, Shigenori Kadowaki, Keiko Minashi, Satoshi Yuki, Takashi Ohta, and Hiroki Hara. "Association of Erbb2 Copy Number and Gene Coalterations with Trastuzumab Efficacy and Resistance in Human Epidermal Growth Factor Receptor 2–Positive Esophagogastric and Gastric Cancer." JCO Precision Oncology 6 (2022): e2200135.
- Xing, Kailin, Bingxin Gu, Ping Zhang, and Xianghua Wu. "Dexamethasone Enhances Programmed Cell Death 1 (Pd-1) Expression During T Cell Activation: An Insight into the Optimum Application of Glucocorticoids in Anti-Cancer Therapy." BMC immunology 16 (2015): 1-9. [CrossRef]
- Kakumoto, Kyoko, Jun-ichiro Ikeda, Masato Okada, Eiichi Morii, and Chitose Oneyama. "Mlst8 Promotes Mtor-Mediated Tumor Progression." PLoS One 10, no. 4 (2015): e0119015.
- Gallo, Alberto, Mirko Ronzio, Eugenia Bezzecchi, Roberto Mantovani, and Diletta Dolfini. "Nf-Y Subunits Overexpression in Gastric Adenocarcinomas (Stad)." Scientific reports 11, no. 1 (2021): 23764.
- Liu, Pei-Feng, Chun-Feng Chen, Chih-Wen Shu, Hui-Min Chang, Cheng-Hsin Lee, Huei-Han Liou, Luo-Ping Ger, Chun-Lin Chen, and Bor-Hwang Kang. "Ube2c Is a Potential Biomarker for Tumorigenesis and Prognosis in Tongue Squamous Cell Carcinoma." Diagnostics 10, no. 9 (2020): 674. [CrossRef]
- Wang, Ying, Feifei Huang, Ming Liu, and Quan Zhao. "Ube2c Mrna Expression Controlled by Mir-300 and Hur Determines Its Oncogenic Role in Gastric Cancer." Biochemical and Biophysical Research Communications 534 (2021): 597-603.
- Chen, Peng, Zhiwei He, Jie Wang, Jian Xu, Xueyi Jiang, Yankun Chen, Xinyuan Liu, and Jianxin Jiang. "Hypoxia-Induced Zwint Mediates Pancreatic Cancer Proliferation by Interacting with P53/P21." Frontiers in cell and developmental biology 9 (2021): 682131.
- Marks, Jeffrey R. "Refining the Role of Brca1 in Combating Oxidative Stress." Breast Cancer Research 15 (2013): 1-2. [CrossRef]
- Zhang, Youwei, and Tony Hunter. "Roles of Chk1 in Cell Biology and Cancer Therapy." International journal of cancer 134, no. 5 (2014): 1013-23. [CrossRef]
- Yin, Yuping, Qian Shen, Peng Zhang, Ruikang Tao, Weilong Chang, Ruidong Li, Gengchen Xie, Weizhen Liu, Lihong Zhang, and Prabodh Kapoor. "Chk1 Inhibition Potentiates the Therapeutic Efficacy of Parp Inhibitor Bmn673 in Gastric Cancer." American journal of cancer research 7, no. 3 (2017): 473.
- Matheson, Christopher J, Donald S Backos, and Philip Reigan. "Targeting Wee1 Kinase in Cancer." Trends in pharmacological sciences 37, no. 10 (2016): 872-81.
- Iliaki, Styliani, Rudi Beyaert, and Inna S Afonina. "Polo-Like Kinase 1 (Plk1) Signaling in Cancer and Beyond." Biochemical Pharmacology 193 (2021): 114747.
- Kanaji, Shingo, Hiroaki Saito, Shunichi Tsujitani, Sachiko Matsumoto, Shigeru Tatebe, Akira Kondo, Mitsuhiko Ozaki, Hisao Ito, and Masahide Ikeguchi. "Expression of Polo-Like Kinase 1 (Plk1) Protein Predicts the Survival of Patients with Gastric Carcinoma." Oncology 70, no. 2 (2006): 126-33.
- Li, Qian, Dongdong Tong, Xintao Jing, Peihan Ma, Fang Li, Qiuyu Jiang, Jinyuan Zhang, Hua Wen, Manli Cui, and Chen Huang. "Mad2l1 Is Transcriptionally Regulated by Tead4 and Promotes Cell Proliferation and Migration in Colorectal Cancer." Cancer Gene Therapy 30, no. 5 (2023): 727-37. [CrossRef]
- Seidlitz, Therese, Bon-Kyoung Koo, and Daniel E Stange. "Gastric Organoids—an in Vitro Model System for the Study of Gastric Development and Road to Personalized Medicine." Cell Death & Differentiation 28, no. 1 (2021): 68-83. [CrossRef]
- Liao, Guo-Bin, Xin-Zhe Li, Shuo Zeng, Cheng Liu, Shi-Ming Yang, Li Yang, Chang-Jiang Hu, and Jian-Ying Bai. "Regulation of the Master Regulator Foxm1 in Cancer." Cell Communication and Signaling 16, no. 1 (2018): 57. [CrossRef]
- Zhang, Yan, Xiangming Fang, Fen Shuang, and Guoxun Chen. "Dexamethasone Potentiates the Insulin-Induced Srebp-1c Expression in Primary Rat Hepatocytes." Food Science and Human Wellness 12, no. 5 (2023): 1519-25.
- Nakamura, Tomoko, Akira Iwase, B Bayasula, Yoshinari Nagatomo, Mika Kondo, Tatsuo Nakahara, Sachiko Takikawa, Maki Goto, Tomomi Kotani, and Tohru Kiyono. "Cyp51a1 Induced by Growth Differentiation Factor 9 and Follicle-Stimulating Hormone in Granulosa Cells Is a Possible Predictor for Unfertilization." Reproductive sciences 22, no. 3 (2015): 377-84.
- Ershov, Pavel, Leonid Kaluzhskiy, Yuri Mezentsev, Evgeniy Yablokov, Oksana Gnedenko, and Alexis Ivanov. "Enzymes in the Cholesterol Synthesis Pathway: Interactomics in the Cancer Context." Biomedicines 9, no. 8 (2021): 895.
- Yang, Yongfei, Meiying Luo, Kexin Zhang, Jun Zhang, Tongtong Gao, Douglas O’ Connell, Fengping Yao, Changwen Mu, Bingyu Cai, and Yuxue Shang. "Nedd4 Ubiquitylates Vdac2/3 to Suppress Erastin-Induced Ferroptosis in Melanoma." Nature communications 11, no. 1 (2020): 433.
- Liu, Pulin, Naifei Xing, Zhikai Xiahou, Jingwei Yan, Zhiheng Lin, and Junlong Zhang. "Unraveling the Intricacies of Glioblastoma Progression and Recurrence: Insights into the Role of Nfyb and Oxidative Phosphorylation at the Single-Cell Level." Frontiers in Immunology 15 (2024): 1368685.
- Beishline, Kate, and Jane Azizkhan-Clifford. "Sp1 and the ‘Hallmarks of Cancer’." The FEBS journal 282, no. 2 (2015): 224-58.
- He, Ying, Shasha Qi, Lu Chen, Jinyu Zhu, Linda Liang, Xudong Chen, Hao Zhang, Lvjia Zhuo, Shujuan Zhao, and Shuiping Liu. "The Roles and Mechanisms of Srebp1 in Cancer Development and Drug Response." Genes & Diseases 11, no. 4 (2024): 100987.
- Guarrera, Luca, Mami Kurosaki, Silvio-Ken Garattini, Maurizio Gianni’, Gianpiero Fasola, Luca Rossit, Michele Prisciandaro, Maria Di Bartolomeo, Marco Bolis, and Paola Rizzo. "Anti-Tumor Activity of All-Trans Retinoic Acid in Gastric-Cancer: Gene-Networks and Molecular Mechanisms." Journal of Experimental & Clinical Cancer Research 42, no. 1 (2023): 298. [CrossRef]
- Lauder, I, AM Zaitoun, and WA Aherne. "The Effects of Dexamethasone on the Cell Kinetics of a Murine Malignant Lymphoma." The Journal of Pathology 129, no. 1 (1979): 1-8. [CrossRef]
- Goya, Luis, Anita C Maiyar, Ying Ge, and Gary L Firestone. "Glucocorticoids Induce a G1/G0 Cell Cycle Arrest of Con8 Rat Mammary Tumor Cells That Is Synchronously Reversed by Steroid Withdrawal or Addition of Transforming Growth Factor-Alpha." Molecular endocrinology 7, no. 9 (1993): 1121-32.
- He, Wenning, and Jun Meng. "Cdc20: A Novel Therapeutic Target in Cancer." American Journal of Translational Research 15, no. 2 (2023): 678.
- Jalali, Pooya, Amir Samii, Malihe Rezaee, Arvin Shahmoradi, Fatemeh Pashizeh, and Zahra Salehi. "Ube2c: A Pan-Cancer Diagnostic and Prognostic Biomarker Revealed through Bioinformatics Analysis." Cancer Reports 7, no. 4 (2024): e2032.
- Mylka, Viacheslav, Julie Deckers, Dariusz Ratman, Lode De Cauwer, Jonathan Thommis, Riet De Rycke, Francis Impens, Claude Libert, Jan Tavernier, and Wim Vanden Berghe. "The Autophagy Receptor Sqstm1/P62 Mediates Anti-Inflammatory Actions of the Selective Nr3c1/Glucocorticoid Receptor Modulator Compound a (Cpda) in Macrophages." Autophagy 14, no. 12 (2018): 2049-64. [CrossRef]
- Zou, Juan, Yaokun Chen, Zeqi Ji, Danyi Liu, Xin Chen, Mengjia Chen, Kexun Chen, Haojia Lin, Yexi Chen, and Zhiyang Li. "Identification of C4bpa as Biomarker Associated with Immune Infiltration and Prognosis in Breast Cancer." Translational Cancer Research 13, no. 1 (2024): 25. [CrossRef]
- Quadros, Edward V, Yasumi Nakayama, and Jeffrey M Sequeira. "Saporin Conjugated Monoclonal Antibody to the Transcobalamin Receptor Tcblr/Cd320 Is Effective in Targeting and Destroying Cancer Cells." Journal of cancer therapy 4, no. 6 (2013): 1074.
- Yan, Hong-fa, Ting Zou, Qing-zhang Tuo, Shuo Xu, Hua Li, Abdel Ali Belaidi, and Peng Lei. "Ferroptosis: Mechanisms and Links with Diseases." Signal transduction and targeted therapy 6, no. 1 (2021): 49.
- Tang, Daolin, Xin Chen, Rui Kang, and Guido Kroemer. "Ferroptosis: Molecular Mechanisms and Health Implications." Cell research 31, no. 2 (2021): 107-25. [CrossRef]
- Li, Huifang, Shuxia Jiang, Chun Yang, Shu Yang, Bin He, Wenqiang Ma, and Ruqian Zhao. "Long-Term Dexamethasone Exposure Down-Regulates Hepatic Tfr1 and Reduces Liver Iron Concentration in Rats." Nutrients 9, no. 6 (2017): 617. [CrossRef]
- Dong, Xiao-Ying, and Sheng-Qiu Tang. "Insulin-Induced Gene: A New Regulator in Lipid Metabolism." Peptides 31, no. 11 (2010): 2145-50.
- Ferrè, Silvia, Jeroen HF de Baaij, Patrick Ferreira, Roger Germann, Johannis BC de Klerk, Marla Lavrijsen, Femke van Zeeland, Hanka Venselaar, Leo AJ Kluijtmans, and Joost GJ Hoenderop. "Mutations in Pcbd1 Cause Hypomagnesemia and Renal Magnesium Wasting." Journal of the American Society of Nephrology 25, no. 3 (2014): 574-86.
- Zeng, Xi, Hao-Ying Wang, Yu-Ping Wang, Su-Yang Bai, Ke Pu, Ya Zheng, Qing-Hong Guo, Quan-Lin Guan, Rui Ji, and Yong-Ning Zhou. "Col4a Family: Potential Prognostic Biomarkers and Therapeutic Targets for Gastric Cancer." Translational Cancer Research 9, no. 9 (2020): 5218. [CrossRef]
- Liu, Shanshan, Wei Zhao, Xuemei Li, La Zhang, Yu Gao, Qiling Peng, Chengyou Du, and Ning Jiang. "Agtrap Is a Prognostic Biomarker Correlated with Immune Infiltration in Hepatocellular Carcinoma." Frontiers in oncology 11 (2021): 713017.
- Liu, Yan, Jihai Zhu, Jun Liu, Xueman Ma, Jun Zhao, and Zhanhai Su. "Knockdown of Gastrin Promotes Apoptosis of Gastric Cancer Cells by Decreasing Ros Generation." BioMed Research International 2021, no. 1 (2021): 5590037. [CrossRef]
- Tang, Li, Chengming Guo, Xu Li, Bo Zhang, and Liuye Huang. "Taf15 Promotes Cell Proliferation, Migration and Invasion of Gastric Cancer Via Activation of the Raf1/Mek/Erk Signalling Pathway." Scientific reports 13, no. 1 (2023): 5846. [CrossRef]
- Wang, Yu, Zhicheng Zhao, GuiMei Li, Qin Jin, and Shu Zhang. "Human Gastric Cancer Decellularized Scaffold Promotes Epithelial-Mesenchymal Transition of Gastric Cancer Cells in Vitro with the Involvement of Elf1." Human Gastric Cancer Decellularized Scaffold Promotes Epithelial-Mesenchymal Transition of Gastric Cancer Cells in Vitro with the Involvement of Elf1.
- Liu, Xingzhu, Chang Xu, Wanglong Xiao, and Nianlong Yan. "Unravelling the Role of Nfe2l1 in Stress Responses and Related Diseases." Redox biology 65 (2023): 102819. [CrossRef]
- Wang, Bin, Hanfei Guo, Hongquan Yu, Yong Chen, Haiyang Xu, and Gang Zhao. "The Role of the Transcription Factor Egr1 in Cancer." Frontiers in oncology 11 (2021): 642547.
- Graziano, Francesco, Nicholas W Fischer, Irene Bagaloni, Maria Di Bartolomeo, Sara Lonardi, Bruno Vincenzi, Giuseppe Perrone, Lorenzo Fornaro, Elena Ongaro, and Giuseppe Aprile. "Tp53 Mutation Analysis in Gastric Cancer and Clinical Outcomes of Patients with Metastatic Disease Treated with Ramucirumab/Paclitaxel or Standard Chemotherapy." Cancers 12, no. 8 (2020): 2049.
- Kim, Min Sung, Nam Jin Yoo, and Sug Hyung Lee. "Expressional and Mutational Analysis of Crebbp Gene in Gastric and Colorectal Cancers with Microsatellite Instability." Pathology & Oncology Research 20 (2014): 221-22.
- Chaithongyot, Supattra, Phatcharida Jantaree, Olga Sokolova, and Michael Naumann. "Nf-Κb in Gastric Cancer Development and Therapy." Biomedicines 9, no. 8 (2021): 870.
- Du, Qiupeng, Chenchen Zhu, Qingqing Shang, Haiyan Mao, Xiaoyun Li, Yingchun Huang, and Na Du. "A Study on the Correlation of Myd88 Expression with Gastric Cancer." International Journal of Clinical and Experimental Pathology 11, no. 10 (2018): 4836.
- Talukdar, Priyanka Dey, and Urmi Chatterji. "Transcriptional Co-Activators: Emerging Roles in Signaling Pathways and Potential Therapeutic Targets for Diseases." Signal transduction and targeted therapy 8, no. 1 (2023): 427.
- Guo, Qing, Yizi Jin, Xinyu Chen, Xiaomin Ye, Xin Shen, Mingxi Lin, Cheng Zeng, Teng Zhou, and Jian Zhang. "Nf-Κb in Biology and Targeted Therapy: New Insights and Translational Implications." Signal transduction and targeted therapy 9, no. 1 (2024): 53. [CrossRef]
- Ivashkiv, Lionel B. "Ifnγ: Signalling, Epigenetics and Roles in Immunity, Metabolism, Disease and Cancer Immunotherapy." Nature Reviews Immunology 18, no. 9 (2018): 545-58.
- Ahn, So Yeong, Jin Kim, Min A Kim, Jiwoon Choi, and Woo Ho Kim. "Increased Hgf Expression Induces Resistance to C-Met Tyrosine Kinase Inhibitors in Gastric Cancer." Anticancer Research 37, no. 3 (2017): 1127-38. [CrossRef]
- Liu, Xinmei, Shasha Huang, Yun Guan, and Qing Zhang. "Long Noncoding Rna Oser1-As1 Promotes the Malignant Properties of Non-Small Cell Lung Cancer by Sponging Microrna-433-3p and Thereby Increasing Smad2 Expression." Oncology reports 44, no. 2 (2020): 599-610.
- Chu, Hang Yin, Zihao Chen, Luyao Wang, Zong-Kang Zhang, Xinhuan Tan, Shuangshuang Liu, Bao-Ting Zhang, Aiping Lu, Yuanyuan Yu, and Ge Zhang. "Dickkopf-1: A Promising Target for Cancer Immunotherapy." Frontiers in Immunology 12 (2021): 658097.
- Ebrahimnezhad, Mohammad, Mohammad Natami, Ghazaleh Hafezi Bakhtiari, Peyman Tabnak, Niloufar Ebrahimnezhad, Bahman Yousefi, and Maryam Majidinia. "Foxo1, a Tiny Protein with Intricate Interactions: Promising Therapeutic Candidate in Lung Cancer." Biomedicine & Pharmacotherapy 169 (2023): 115900. [CrossRef]
- Lin, Ching-Yih, Ying-En Lee, Yu-Feng Tian, Ding-Ping Sun, Ming-Jen Sheu, Chen-Yi Lin, Chien-Feng Li, Sung-Wei Lee, Li-Ching Lin, and I-Wei Chang. "High Expression of Epha4 Predicted Lesser Degree of Tumor Regression after Neoadjuvant Chemoradiotherapy in Rectal Cancer." Journal of Cancer 8, no. 6 (2017): 1089.
- Greuber, Emileigh K, Pameeka Smith-Pearson, Jun Wang, and Ann Marie Pendergast. "Role of Abl Family Kinases in Cancer: From Leukaemia to Solid Tumours." Nature Reviews Cancer 13, no. 8 (2013): 559-71.
- Kim, Eric S, and Ravi Salgia. "Met Pathway as a Therapeutic Target." Journal of Thoracic Oncology 4, no. 4 (2009): 444-47.
- Pelaz, Sara G, and Arantxa Tabernero. "Src: Coordinating Metabolism in Cancer." Oncogene 41, no. 45 (2022): 4917-28. [CrossRef]
- Zhang, Ying, and Zhaoyong Liu. "Stat1 in Cancer: Friend or Foe?" Discovery medicine 24, no. 130 (2017): 19-29.
- Jiménez-Martínez, Marta, Konstantinos Stamatakis, and Manuel Fresno. "The Dual-Specificity Phosphatase 10 (Dusp10): Its Role in Cancer, Inflammation, and Immunity." International journal of molecular sciences 20, no. 7 (2019): 1626.
- Zhang, Keyi, Xiao Ni, Xiaoling Ma, Rui Sun, Jiangnan Qiu, and Chengyan Luo. "Linc01012 Upregulation Promotes Cervical Cancer Proliferation and Migration Via Downregulation of Cdkn2d." Oncology Letters 25, no. 3 (2023): 1-9. [CrossRef]
- Silva-Rodríguez, Paula, Daniel Fernández-Díaz, Manuel Bande, María Pardo, Lourdes Loidi, and María José Blanco-Teijeiro. "Gnaq and Gna11 Genes: A Comprehensive Review on Oncogenesis, Prognosis and Therapeutic Opportunities in Uveal Melanoma." Cancers 14, no. 13 (2022): 3066.
- Vera-Puente, Olga, Carlos Rodriguez-Antolin, Ana Salgado-Figueroa, Patrycja Michalska, Olga Pernia, Brett M Reid, RocÍo Rosas, Alvaro Garcia-Guede, Silvia SacristÁn, and Julia Jimenez. "Mafg Is a Potential Therapeutic Target to Restore Chemosensitivity in Cisplatin-Resistant Cancer Cells by Increasing Reactive Oxygen Species." Translational Research 200 (2018): 1-17.
- Marni, Rakshmitha, Anindita Chakraborty, and RamaRao Malla. "Oncogenic Tetraspanins: Implications for Metastasis, Drug Resistance, Cancer Stem Cell Maintenance and Diagnosis of Leading Cancers in Females." Gene Reports 27 (2022): 101548. [CrossRef]
- Mast, Alan E, Alisa S Wolberg, David Gailani, Michael R Garvin, Christiane Alvarez, J Izaak Miller, Bruce Aronow, and Daniel Jacobson. "Sars-Cov-2 Suppresses Anticoagulant and Fibrinolytic Gene Expression in the Lung." Elife 10 (2021): e64330.
- Shan, Qingqing, Chi Zhang, Yangke Li, Qunying Li, Yifan Zhang, Xue Li, Junqing Shi, and Fengying Hu. "Slc7a11, a Potential Immunotherapeutic Target in Lung Adenocarcinoma." Scientific reports 13, no. 1 (2023): 18302. [CrossRef]
- Jiang, Yun, Jingyi Cui, Ming Cui, and Rongrong Jing. "Slc7a11 Promotes the Progression of Gastric Cancer and Regulates Ferroptosis through Pi3k/Akt Pathway." Pathology-Research and Practice 248 (2023): 154646.
- Sun, Fengfeng, Peng Sun, Xiaofeng Yang, Liangliang Hu, Jianguo Gao, and Tao Tian. "Inhibition of Fstl3 Abates the Proliferation and Metastasis of Renal Cell Carcinoma Via the Gsk-3β/Β-Catenin Signaling Pathway." Aging (Albany NY) 13, no. 18 (2021): 22528.
- Liu, Yuan-Jie, Jie-Pin Li, Ying Zhang, Meng-Jun Nie, Yong-Hua Zhang, Shen-Lin Liu, and Xi Zou. "Fstl3 Is a Prognostic Biomarker in Gastric Cancer and Is Correlated with M2 Macrophage Infiltration." OncoTargets and therapy (2021): 4099-117.
- Nakano, Shun, Masashi Nishikawa, Tomoyo Kobayashi, Eka Wahyuni Harlin, Takuya Ito, Katsuya Sato, Tsuyoshi Sugiyama, Hisashi Yamakawa, Takahiro Nagase, and Hiroshi Ueda. "The Rho Guanine Nucleotide Exchange Factor Plekhg1 Is Activated by Interaction with and Phosphorylation by Src Family Kinase Member Fyn." Journal of Biological Chemistry 298, no. 2 (2022).
- Zhou, Xu-Dong, Ya-Wei Qu, Li Wang, Fu-Hua Jia, Peng Chen, Yin-Pu Wang, and Hai-Feng Liu. "Identification of Potential Hub Genes of Gastric Cancer." Medicine 101, no. 41 (2022): e30741. [CrossRef]
- Raposo, Teresa P, Susanti Susanti, and Mohammad Ilyas. "Investigating Tns4 in the Colorectal Tumor Microenvironment Using 3d Spheroid Models of Invasion." Advanced Biosystems 4, no. 6 (2020): 2000031. [CrossRef]
- Kaneda, Atsushi, Michio Kaminishi, Yukihiro Nakanishi, Takashi Sugimura, and Toshikazu Ushijima. "Reduced Expression of the Insulin-Induced Protein 1 and P41 Arp2/3 Complex Genes in Human Gastric Cancers." International journal of cancer 100, no. 1 (2002): 57-62.
- Jakobsen, Sebastian, and Carsten Uhd Nielsen. "Exploring Amino Acid Transporters as Therapeutic Targets for Cancer: An Examination of Inhibitor Structures, Selectivity Issues, and Discovery Approaches." Pharmaceutics 16, no. 2 (2024): 197.
Figure 1.
Flow cytometric assessment of the apoptotic profiles of the AGS gastric adenocarcinoma cells after 24 h of treatment A) The live, early apoptotic, late apoptotic, and necrotic cell percentages after 24 h treatment with APB (3000 µg/mL), APB+Dex (3100 µg/mL), Dex (200 µg/mL), and control (n = 4). (b) Represented are the density plots of each drug treatment that is most representative of the average data from the flow cytometric analyses, with Q3-1 = necrotic cells, Q3-2 = late-stage apoptotic cells, Q3-3 = live cells, and Q3-4 = early-stage apoptotic cells.
Figure 1.
Flow cytometric assessment of the apoptotic profiles of the AGS gastric adenocarcinoma cells after 24 h of treatment A) The live, early apoptotic, late apoptotic, and necrotic cell percentages after 24 h treatment with APB (3000 µg/mL), APB+Dex (3100 µg/mL), Dex (200 µg/mL), and control (n = 4). (b) Represented are the density plots of each drug treatment that is most representative of the average data from the flow cytometric analyses, with Q3-1 = necrotic cells, Q3-2 = late-stage apoptotic cells, Q3-3 = live cells, and Q3-4 = early-stage apoptotic cells.
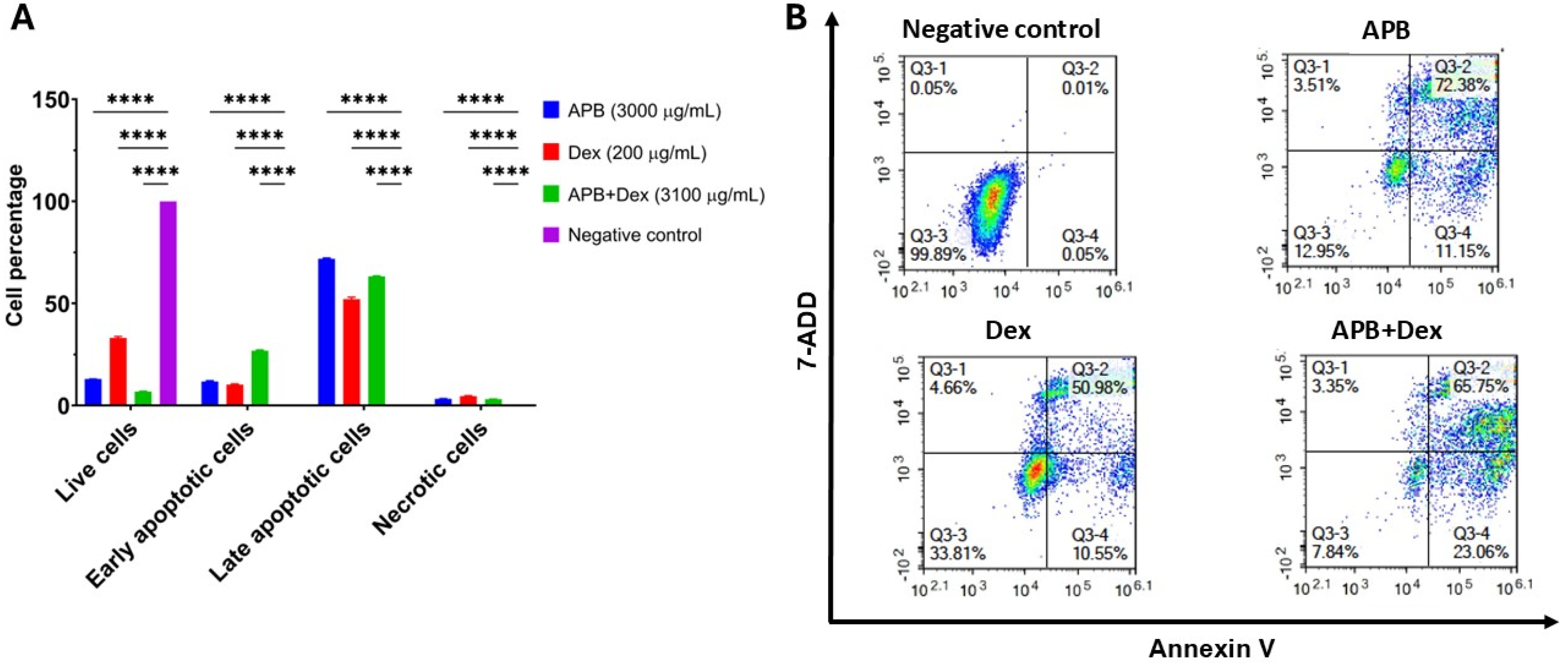
Figure 2.
Depicts the fold change in reactive oxygen species (ROS) generation following treatment with various concentrations: 3000 μg/mL, 1500 μg/mL, and 750 μg/mL of APB, and 3000:100 μg/mL, 1500:50 μg/mL, and 750:25 μg/mL of APB:Dex and, 200 μg/mL, 100 μg/mL, and 50 μg/mL of Dex. Additionally, tert-Butyl hydroperoxide (TBHP) (22.5 μg/mL or 250 μM) is included for comparative purposes. The values are expressed as mean ± SD. **indicates value of p ≤0.01, ****indicates p < 0.0001 compared to the negative control.
Figure 2.
Depicts the fold change in reactive oxygen species (ROS) generation following treatment with various concentrations: 3000 μg/mL, 1500 μg/mL, and 750 μg/mL of APB, and 3000:100 μg/mL, 1500:50 μg/mL, and 750:25 μg/mL of APB:Dex and, 200 μg/mL, 100 μg/mL, and 50 μg/mL of Dex. Additionally, tert-Butyl hydroperoxide (TBHP) (22.5 μg/mL or 250 μM) is included for comparative purposes. The values are expressed as mean ± SD. **indicates value of p ≤0.01, ****indicates p < 0.0001 compared to the negative control.
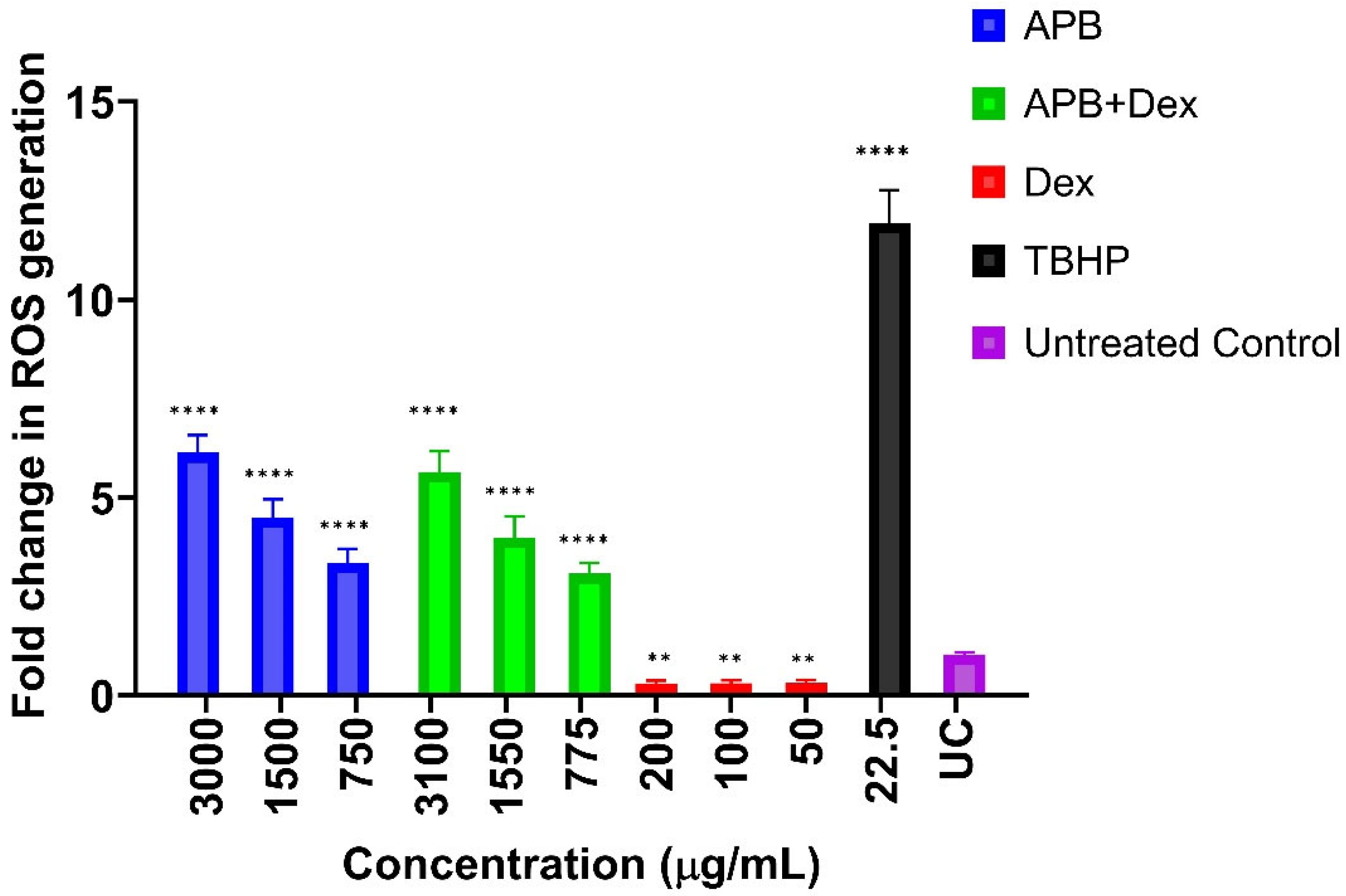
Figure 3.
A) Volcano plot showing significantly regulated proteins (Absolute Log2FC ≥0.58 and Q≤0.05) in APB-treated cells compared to the negative control cells and its significantly enriched pathways (Q ≤0.05), B) Graphical summary of top predictions in an IPA Interpret report presented in the form of a simple network. C) IPA enriched canonical pathways that identify the most significant signalling and metabolic pathways in the dataset and predicts whether each pathway is activated (red) or inhibited (blue). The chart was filtered to include enriched term with -log BH-P value >1.3 and Abs z score >= 1. D) IPA enriched Machine learning Disease-related pathways (chart was filtered to include enriched term with -log BH-P value >1.3 and Abs z score >= 1).
Figure 3.
A) Volcano plot showing significantly regulated proteins (Absolute Log2FC ≥0.58 and Q≤0.05) in APB-treated cells compared to the negative control cells and its significantly enriched pathways (Q ≤0.05), B) Graphical summary of top predictions in an IPA Interpret report presented in the form of a simple network. C) IPA enriched canonical pathways that identify the most significant signalling and metabolic pathways in the dataset and predicts whether each pathway is activated (red) or inhibited (blue). The chart was filtered to include enriched term with -log BH-P value >1.3 and Abs z score >= 1. D) IPA enriched Machine learning Disease-related pathways (chart was filtered to include enriched term with -log BH-P value >1.3 and Abs z score >= 1).
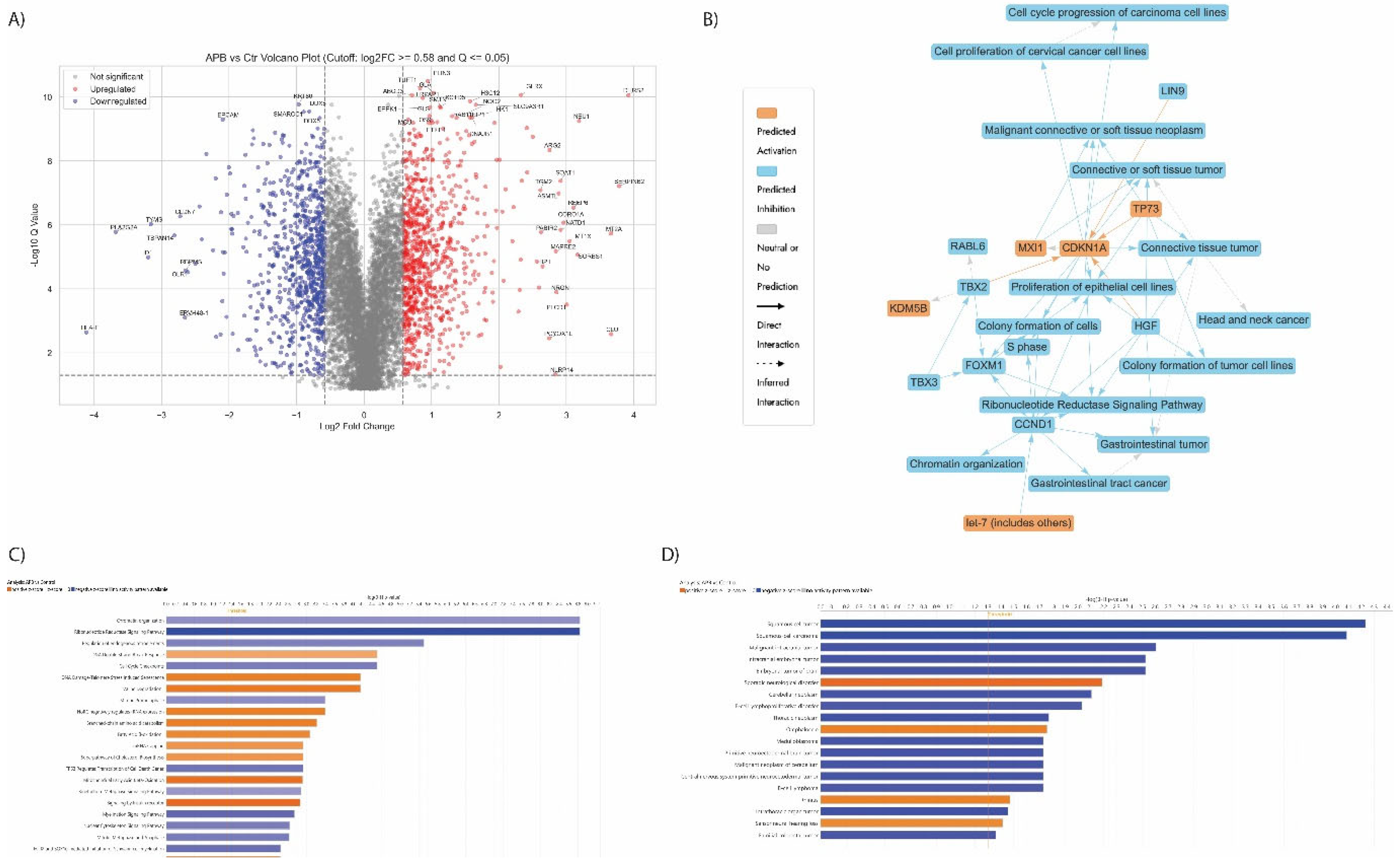
Figure 4.
A) Volcano plot showing significantly regulated proteins (Absolute Log2FC ≥0.58 and Q≤0.05) in Dex-treated cells compared to the negative control cells and its significantly enriched pathways (Q ≤0.05), B) Graphical summary of top predictions in an IPA Interpret report presented in the form of a simple network. C) IPA enriched canonical pathways that identify the most significant signalling and metabolic pathways in the dataset and predicts whether each pathway is activated (red) or inhibited (blue). Chart was filtered to include enriched term with -log BH-P value >1.3 and Abs z score >=
Figure 4.
A) Volcano plot showing significantly regulated proteins (Absolute Log2FC ≥0.58 and Q≤0.05) in Dex-treated cells compared to the negative control cells and its significantly enriched pathways (Q ≤0.05), B) Graphical summary of top predictions in an IPA Interpret report presented in the form of a simple network. C) IPA enriched canonical pathways that identify the most significant signalling and metabolic pathways in the dataset and predicts whether each pathway is activated (red) or inhibited (blue). Chart was filtered to include enriched term with -log BH-P value >1.3 and Abs z score >=
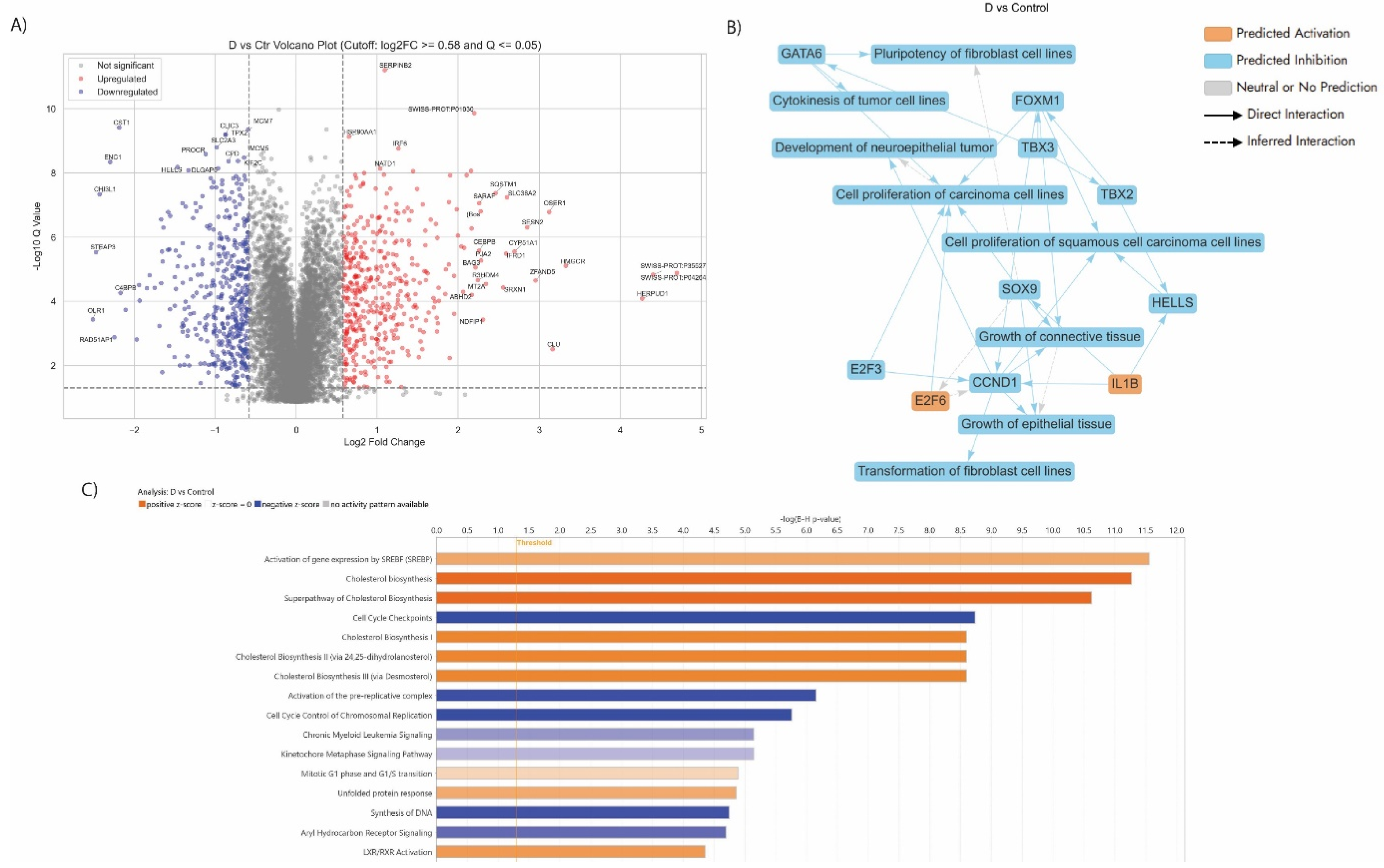
Figure 5.
Volcano plot showing significantly regulated proteins (Absolute Log2FC ≥0.58 and Q≤0.05) in APB+Dex treated AGS gastric adenocarcinoma cells compared to its monotreatment. (APB and Dex) B) Graphical summary of top predictions in an IPA Interpret report presented in the form of a simple network. C) IPA enriched canonical pathways that identify the most significant signalling and metabolic pathways in the dataset and predicts whether each pathway is activated (red) or inhibited (blue). Chart was filtered to include enriched term with -log BH-P value >1.3 and Abs z score
Figure 5.
Volcano plot showing significantly regulated proteins (Absolute Log2FC ≥0.58 and Q≤0.05) in APB+Dex treated AGS gastric adenocarcinoma cells compared to its monotreatment. (APB and Dex) B) Graphical summary of top predictions in an IPA Interpret report presented in the form of a simple network. C) IPA enriched canonical pathways that identify the most significant signalling and metabolic pathways in the dataset and predicts whether each pathway is activated (red) or inhibited (blue). Chart was filtered to include enriched term with -log BH-P value >1.3 and Abs z score
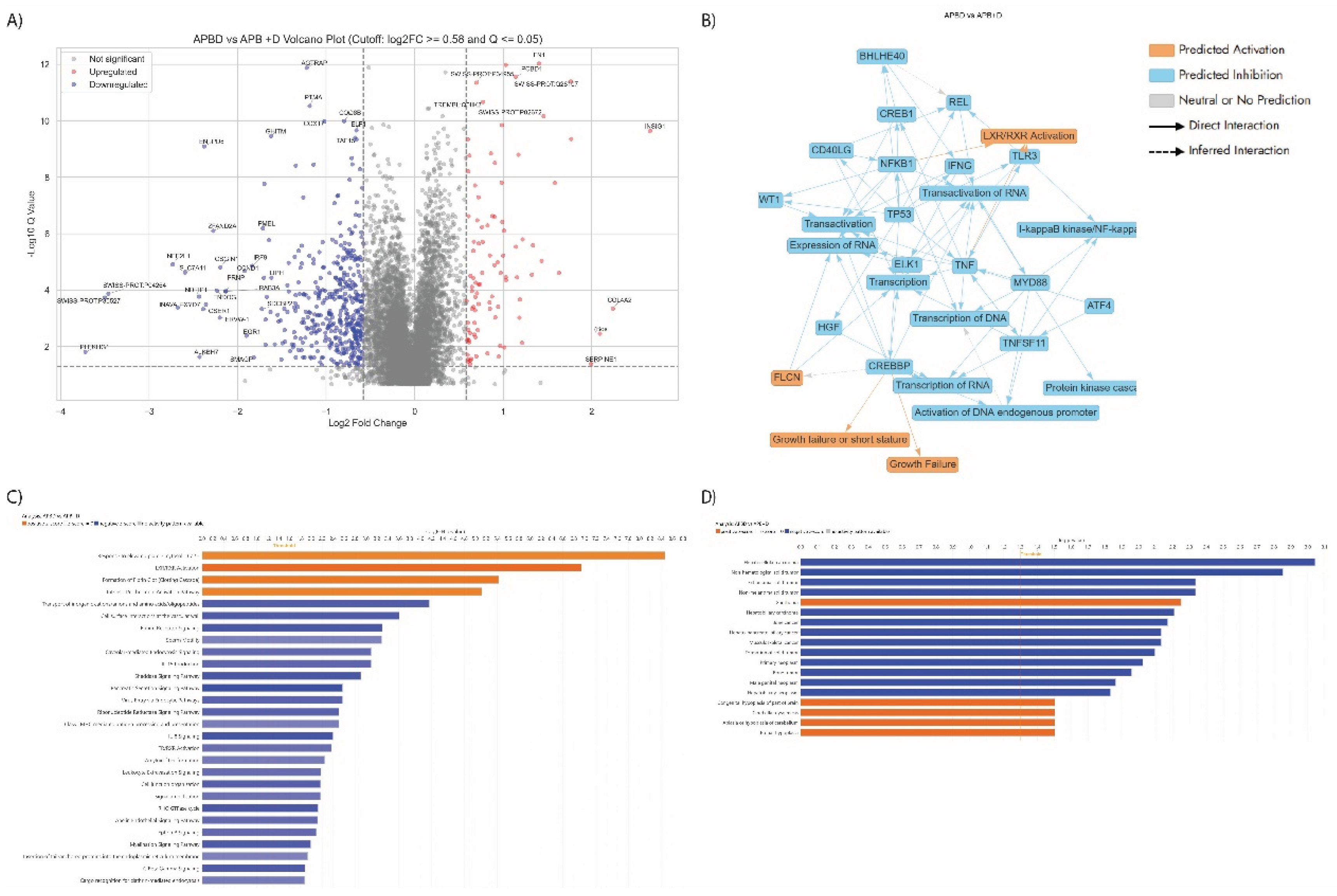

Table 1.
Cell growth inhibition (%) against the AGS gastric adenocarcinoma and cell viability (%) of the Hs 738.St/Int normal intestine cell lines at different concentrations of AP combination (magnesium acetate A: sodium propionate P), AB (magnesium acetate A: sodium butyrate B), PB (sodium propionate P: sodium butyrate B), for 72 h using the Alamar Blue assay (n = 3).
Table 1.
Cell growth inhibition (%) against the AGS gastric adenocarcinoma and cell viability (%) of the Hs 738.St/Int normal intestine cell lines at different concentrations of AP combination (magnesium acetate A: sodium propionate P), AB (magnesium acetate A: sodium butyrate B), PB (sodium propionate P: sodium butyrate B), for 72 h using the Alamar Blue assay (n = 3).
| Conc. μg/mL 1:1 |
Cell Growth Inhibition (%) of AGS cells |
Cell viability (%) of HS738.St/Int |
||||
| PB | AP | AB | PB | AP | AB | |
| 1500+1500 | 99.22 ± 1.42 a w | 59.79 ± 4.32 ax | 99.49 ± 0.85 ay | 58.86 ± 9.52 ax | 82.39 ± 12.30 ax | 61.08 ± 13.45 az |
| 750+750 | 91.14 ± 1.78 a w | 37.67 ± 9.68 bx | 90.08 ± 1.86 ax | 78.04 ± 4.98 aw | 93.48 ± 12.89 ax | 84.38 ± 13.01 ax |
| 375+375 | 75.93 ± 1.92 b,w | 29.31 ± 6.50 bx | 76.62 ± 2.81 by | 81.09 ± 10.51 aw | 107.71 ± 33.81 ay | 102.51 ± 17.25 ay |
| 187.5 +187.5 | 51.36 ± 7.50 cw | 23.87 ± 6.76 bw | 49.95 ± 7.31 cx | 100.62 ± 16.88 aw | 143.38 ± 26.78 ax | 103.77 ± 10.35 ax |
| 93.75 + 93.75 | 25.11 ± 8.21 dw | 19.20 ± 7.46 bx | 24.28 ± 7.26 dy | 127.45 ± 26.75 ax | 149.37 ± 9.05 ay | 122.20 ± 16.51 az |
| 46.875 + 46.875 | 9.79 ± 7.25 ew | 11.98 ± 12.23 bx | 12.01 ± 8.06 ey | 138.92 ± 9.91 ax | 175.62 ± 4.32 ay | 149.29 ± 21.90 az |
| IC50 | 421.23 ± 15.31 | 1141.13 ± 362.00 | 446.53 ± 19.55 | NA | NA | NA |
“NA” indicates the IC50 value could not be calculated as the highest tested concentration did not inhibit the normal HS738.St/Int cells by 50%.a,b,c,d,e The different superscript values in the same column for each cell line indicate a statistically significant difference (p < 0.05) compared to the highest concentration (3,000 μg/mL) within the same treatment group. w,x,y,z The different subscript values in the same row for each cell line indicate a statistically significant difference (p < 0.05) between the treatment groups.
Table 2.
Cell growth inhibition (%) against AGS gastric adenocarcinoma at different concentrations of the APB combination, Dex, and the APB+Dex combination for 72 h using the Alamar Blue assay (n = 3). The table also presents the cell viability (%) of Hs 738.St/Int normal intestinal cell lines for the APB+Dex combination.
Table 2.
Cell growth inhibition (%) against AGS gastric adenocarcinoma at different concentrations of the APB combination, Dex, and the APB+Dex combination for 72 h using the Alamar Blue assay (n = 3). The table also presents the cell viability (%) of Hs 738.St/Int normal intestinal cell lines for the APB+Dex combination.
| Conc. μg/mL |
Cell Growth Inhibition (%) of AGS cells |
Conc. μg/mL |
Cell Growth Inhibition (%) of AGS cells |
Conc. μg/mL APB+Dex |
Cell Growth Inhibition (%) of AGS cells |
Cell viability (%) of HS738.St/Int |
|---|---|---|---|---|---|---|
| APB | Dex | APB+Dex | ||||
| 3000 | 95.65 ± 7.90 ax | 200 | 80.46 ± 8.08 ax | 3000 + 100 | 103.17 ± 3.06 ax | 68.57 ± 11.74 ax |
| 1500 | 86.25 ± 8.42 ax | 100 | 39.38 ± 24.51bx | 1500 + 50 | 87.81 ± 5.57 bx | 85.91 ± 10.59 ax |
| 750 | 65.54 ± 4.91 bx | 50 | - | 750 + 25 | 62.42 ± 5.34 cx | 110.88 ± 14.35 ay |
| 375 | 40.28 ± 8.05 cx | 25 | - | 375+ 12.5 | 43.49 ± 10.93 dx | 115.29 ± 16.81 ay |
| 187.5 | 22.20 ± 8.55 dx | 12.5 | - | 187.5 + 6.25 | 28.70 ± 11.91 ex | 134.46 ± 24.72 ay |
| 93.75 | 19.20 ± 10.38 dx | 6.25 | - | 93.75 + 3.13 | 16.46 ± 9.71 fx | 159.21 ± 14.16 ay |
| IC50 | 568.33 ± 82.56 | IC50 | 86.60 ±11.85 | IC50 | 643.30 ± 58.26 | NA |
“NA” indicates the IC50 value could not be calculated as the highest tested concentration did not inhibit the HS738.St/Int cells by 50%.a,b,c,d,e,f The different superscript values in the same column for each cell line indicate a statistically significant difference (p < 0.05) compared to the highest concentration (3,000 μg/mL). w,x,y The different superscript values in the same row for the AGS gastric adenocarcinoma cell line indicate a statistically significant difference (p < 0.05) between the treatment groups.
Table 3.
Drug interaction analysis of Dex and APB combinations in AGS gastric adenocarcinoma cells. The bold numbers (CI values < 1) indicate synergistic interactions between Dex and APB.
Table 3.
Drug interaction analysis of Dex and APB combinations in AGS gastric adenocarcinoma cells. The bold numbers (CI values < 1) indicate synergistic interactions between Dex and APB.
| Combination Index (CI) values at: | ||||
|---|---|---|---|---|
| Combinations | IC50 | IC75 | IC90 | IC95 |
| AP 1:1 (1500 μg/mL A + 1500 μg/mL P) | 3.53 | 7.28 | 17.13 | 31.66 |
| AB 1:1 (1500 μg/mL A + 1500 μg/mL B) | 1.14 | 1.37 | 1.80 | 2.19 |
| BP 1:1 (1500 μg/mL B + 1500 μg/mL P) | 1.41 | 1.68 | 2.11 | 2.51 |
| APB + Dex (3000 μg/mL APB + 100 μg/mL Dex) | 0.76 | 0.48 | 0.31 | 0.23 |
IC = inhibitory concentration.
Table 4.
List of the dysregulated proteins in APB+Dex treated AGS gastric adenocarcinoma cells
| GENE | PROTEIN | LOG2FC | ROLE | REF |
|---|---|---|---|---|
| OSER1 | Oxidative stress-responsive serine-rich protein 1 | -2.39 | Noted for its role in the negative regulation of intracellular signal transduction. |
[118] |
| DKK1 | Dickkopf-related protein 1 | -1.54 | Overexpressed in cancer, affecting Wnt signaling pathways. Overexpression correlates with poor survival in gastric cancer. |
[119] |
| FOXO1 | Forkhead box protein O1 | -1.21 | A tumour suppressor transcription factor linked to cancer. | [120] |
| EPHA4 | Ephrin type-A receptor 4 | -1.07 | A receptor tyrosine kinase promoting cancer progression. |
[121] |
|
ABL1 ABL2 |
Tyrosine kinase ABL1 and ABL2 | -0.79 -0.76 |
Proto-oncogenes involved in cell differentiation, division, and adhesion and is linked various cancers, especially leukemia. Altered signalling associated with gastric cancer. |
[122] |
| MET | Hepatocyte growth factor receptor | -0.78 | A proto-oncogene involved in several cancers, including gastric cancer. Overexpression and mutations linked to poor prognosis in gastric cancer. |
[123] |
| SRC | Proto-oncogene tyrosine-protein kinase Src | -0.68 | Associated with numerous cancers through oncogenic signalling. Promotes gastric cancer progression through activation of oncogenic pathways. |
[124] |
| STAT1 | Signal transducer and activator of transcription 1-alpha/beta | 0.85 | Influences cancer progression and immune responses |
[125] |
| DUSP10 | Dual specificity protein phosphatase 10 | 1.33 | Regulates pathways connected to cancer development. | [126] |
| CDKN2D | Cyclin-dependent kinase 4 inhibitor D | 1.18 | A cyclin-dependent kinase inhibitor linked to multiple cancers. |
[127] |
| EPCAM | Epithelial cell adhesion molecule | -1.04 | Cell adhesion and signaling; upregulated in gastric tumors for proliferation and metastasis. | [33] |
| GNAQ | Guanine nucleotide-binding protein G(q) subunit alpha | -0.64 | Oncogene in G-protein signaling; implicated in tumor progression. |
[128] |
| GNAS | Guanine nucleotide-binding protein G(s) subunit alpha isoforms XLas | -0.73 | Oncogenic signaling driver; mutated in some gastric cancers. |
[128] |
| MAFG | Transcription factor MafG | -0.65 | Transcription factor in oxidative stress response; linked to oncogenesis. | [129] |
| TSPAN1, TSPAN6, TSPAN8, TSPAN14, TSPAN15, TSPAN31 | Tetraspanin | -0.78 to -1.49 |
Roles in cell signaling, adhesion, and metastasis. |
[130] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



