
Submitted:
10 October 2025
Posted:
14 October 2025
You are already at the latest version
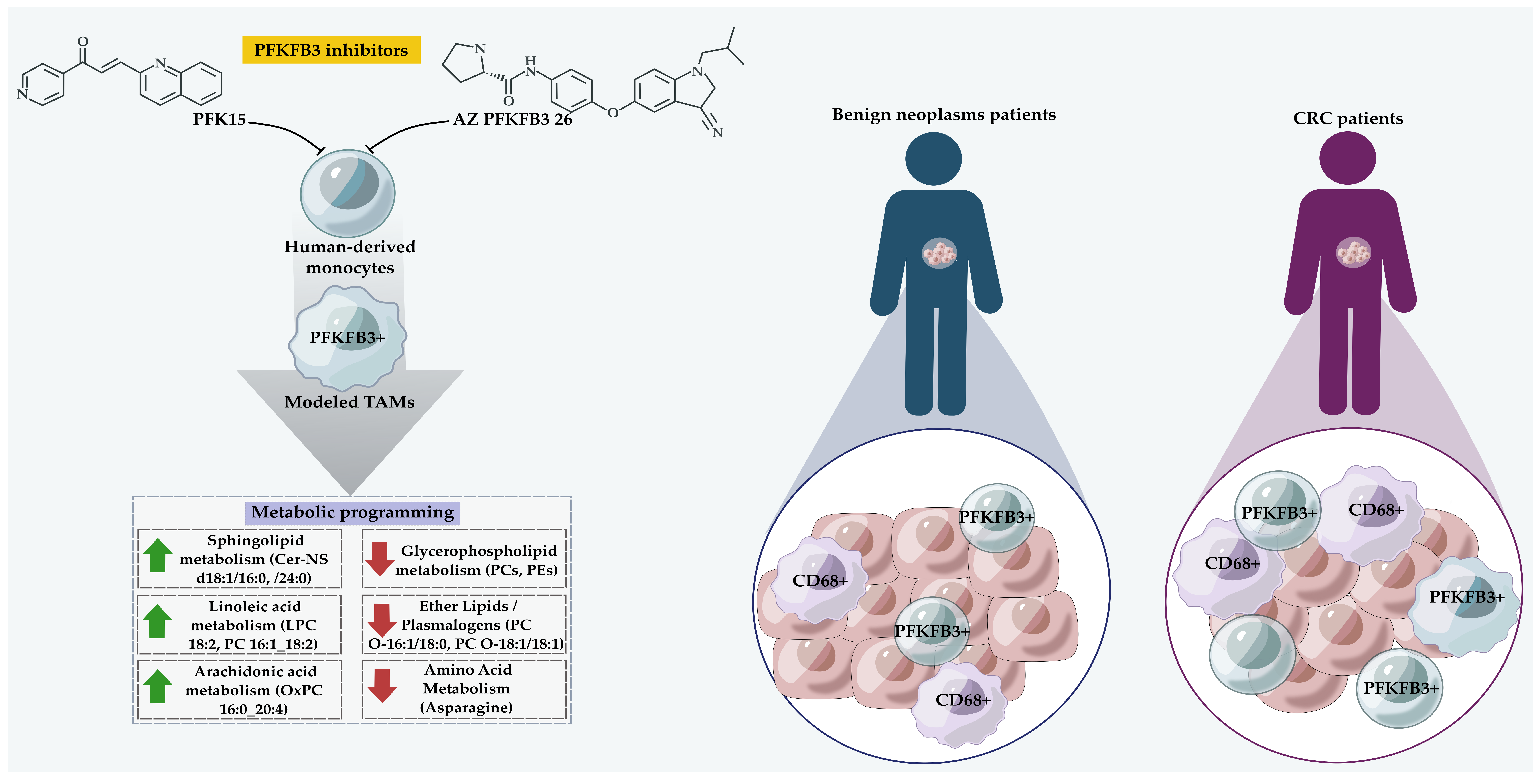
Abstract
Colorectal cancer is the third most common cancer worldwide, making lymph node re-covery critical for treatment decisions and prognosis. Within the colorectal tumor mi-croenvironment, the metabolic programming of tumor-associated macrophages (TAMs) can drive both pro- and anti-tumor responses, yet the specific glycolytic pathways gov-erning their pro-metastatic conversion present promising therapeutic targets. This study investigated the role of glycolysis activating enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) in mediating TAM metabolic polarization, and its potential as a therapeutic target. PFKFB3 expression was found to be predominant in tumor-associated macrophages in CRC human tumor sam-ples. Lipidomic analysis performed by HPLC-MS/MS revealed that PFKFB3 inhibition altered glycerophospholipid metabolism (p=6.13E-10), and shifted TAMs toward sphingolipid-mediated immunosuppressive metabolism. PFKFB3 activity was associated with a specific reduction in asparagine availability, potentially pointing to a targeted reprogramming of amino acid metabolism supporting distinct macrophage functions under conditions of intra-tumoral metabolic stress. These findings highlight PFKFB3 as an essential metabolic regulator of TAMs pro-tumoral activity in CRC, particularly in colon cancer.
Keywords:
colorectal cancer
; tumor-associated macrophages
; PFKFB3
; lipid metabolism
; amino acid metabolism
1. Introduction
Tumor-associated macrophages (TAMs) are potent innate immune cells that can support all stages of solid tumor initiation and progression, including proliferation benefits of cancer cells, cancer cell migration and immune escape, tumor angiogenesis, extravasation of cancer cells into blood and lymphatic circulation, finding favorable accommodation in a metastatic niche in distant organs or in sentinel lymph nodes [1,2]. The effector mediators produced by TAMs include reactive oxide species (ROS), cytokines, growth factors, chitinase-like proteins, enzymes, and components of the extracellular matrix [2,3,4,5]. Resident TAMs have intrinsic ability to fight with transformed cells, but once cancer cells overcome this barrier, they start to program both resident macrophages and incoming monocyte-derived macrophages to develop tumor-associated TAM phenotype. Selective scavenging of these mediators also enables TAMs to modulate tumor growth control. [6]. However, if in the majority of solid cancers TAMs have dominating pro-tumoral activity, in colorectal cancer (CRC) high levels of CD68+ cells correlate with good prognosis [7].
Currently, accumulating data indicate that metabolism of TAMs is principal for their pro-tumor activity [2,8]. If in the acute inflammation macrophages utilize glycolysis for the rapid anti-bacterial response, while homeostatic resident macrophages use fatty acid oxidation allowing slow and economical use of their energy resources, TAMs have mixed metabolism [2,9]. Such mixed metabolism allows TAMs to have sufficient energy for the low-grade production of pro-tumoral mediators for a long time period. There is an urgent need to understand which factors allow TAMs to combine glycolysis with fatty acid oxidation, and how cancer cells can promote low grade glycolysis in tumor-supporting tolerogenic TAMs.
We have recently identified that expression of the stimulator of glycolysis PFKFB3 is elevated in monocytes of patients with colon but no rectal cancer. PFKFB3 is a gene that encodes the protein 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3. This enzyme is needed to convert glucose-derived metabolite fructose-6-phosphate into fructose-2,6-biphosphate, which later is needed for glycolysis [10]. The role of PFKFB3 in the glycolytic pathway in cancer cells is well established, and has been under consideration for the therapeutic targeting in various cancer types [10,11]. However, its role in metabolic programing of TAMs is only emerging. PFKFB3 was also found in monocyte-derived peritumoral macrophages in hepatocellular carcinoma (HCC), where PFKFB3 modulated the metabolic switch and the increased NF-κB-dependent PD-L1 expression [12]. The levels of PFKFB3+CD68+ macrophage infiltration in peritumoral tissues negatively correlated with overall survival in patients with HCC. We followed the fate of PFKFB3+ monocytes in colon cancer, and using spatial transcriptomics demonstrated their massive infiltration and differentiation into PFKFB3+CD68+ TAMs in the tumor tissue in colon cancer patients [13]. PFKFB3 correlated with unfavorable prognosis specifically in colon cancer. However, whether such elevated expression of PFKFB3 is an adaptation of macrophages to the increased energy requirements, or cancer cells have ability to actively induce PFKFB3 expression in TAMs, thus providing TAMs with the ability to use glycolysis in the tolerogenic environment, remains unknown. It was also unclear which functions of TAMs depend on PFKFB3.
In our study we demonstrate that PFKFB3 is predominantly expressed in tumor-associated macrophages in CRC tumor microenvironment. For the first time by using mass spectrometry, we identified that inhibition of glycolysis activating enzyme PFKFB3 re-programs lipid metabolism in primary human monocyte-derived tumor-associated macrophages.
2. Results
2.1. PFKFB3 is expressed in tumor microinvironment in colorecal cancer as well as in benign neoplasms
Analysis of PFKFB3 protein expression was performed in malignant and benign lesions, as well as across CRC anatomical sites. Based on our previous findings that demonstrated elevated PFKFB3 levels in monocytes from colon cancer (CC) relative to rectal cancer (RC) patients [13], the current study extends these observations to tissue-level characterization. High number of PFKFB3+ cells were observed in tumor tissues, and these cells were single infiltrating cells. Notably, benign intestinal neoplasms exhibited even higher stromal PFKFB3 expression compared to CRC tissues (78.22 ± 35.54 vs. 30.19 ± 25.33, p < 0.00001) (Figure 1).
This difference was particularly pronounced when comparing benign lesions to RC (3.2-fold increase; 78.22 ± 35.54 vs. 24.72 ± 21.53, p < 0.00001) and to CC (2.3-fold increase; 78.22 ± 35.54 vs. 34.50 ± 27.37, p < 0.00001) (Table 1).
Within the CRC cohort, the CC subgroup maintained significantly higher PFKFB3 expression than RC (1.4-fold increase; 34.50 ± 27.37 vs. 24.72 ± 21.53, p = 0.045), corroborating prior monocyte data and underscoring a specific role for PFKFB3 in colon cancer pathogenesis [13]. Furthermore, tumor compartments exhibited a 3.8-fold higher PFKFB3 expression relative to matched stromal regions (65.94 ± 50.59 vs. 17.27 ± 17.50, p < 0.00001).
We next analyzed, whether PFKFB3 is pronominally expressed in TAMs, but not in cancer cells. We focused our attention on the stromal compartment and confirmed that TAMs significantly express PFKFB3 (Figure 2).
This observation emphasizes the critical role of stromal cells, particularly TAMs, in the metabolic reprogramming of the tumor niche. Our findings support the importance of investigating cell-specific contributions to PFKFB3-mediated pathways in CRC development. Specifically, the role of PFKFB3 expression in TAMs may be critical for regulating immunosuppression and metabolic adaptation within the TME.
2.2. PFKFB3 inhibition shifts tumor-associated macrophage metabolism toward immunosuppressive sphingolipid and pro-inflammatory arachidonic acid pathways
To gain deeper insight into the biochemical reprogramming induced by PFKFB3 inhibition, we next sought to determine whether these changes were underpinned by specific metabolic reprogramming. To systematically investigate the global lipidomic shifts induced by PFKFB3 blockade, we performed pathway enrichment analysis on lipids that exhibited statistically significant alterations following treatment with PFK15 or AZ PFKFB3 26. Using MetaboAnalyst and the KEGG database, we mapped these lipid changes to biological pathways, ranking them by both statistical significance and overall metabolic impact (Table 3, 4).

Table 4.
KEGG-derived metabolic pathways enriched for lipids exhibiting statistically significant differential abundance between control and AZ PFKFB3 26-treated macrophages.
Table 4.
KEGG-derived metabolic pathways enriched for lipids exhibiting statistically significant differential abundance between control and AZ PFKFB3 26-treated macrophages.
| Name metabolic pathways | The total number of metabolites involved | The number of metabolites matching the experimental data | P | -LOG(P) | Effect on pathway |
| Glycerophospholipid metabolism | 36 | 7 | 6.13E-10 | 9.2127 | 0.26922 |
| Glycosylphosphatidylinositol (GPI)-anchor biosynthesis | 14 | 2 | 0.003979 | 2.4002 | 0.1875 |
| Glycerolipid metabolism | 16 | 2 | 0.005207 | 2.2834 | 0.13636 |
| Sphingolipid metabolism | 21 | 2 | 0.008937 | 2.0488 | 0.1875 |
| Phosphatidylinositol signaling system | 28 | 2 | 0.015655 | 1.8053 | 0.12766 |
| Linoleic acid metabolism | 5 | 1 | 0.035028 | 1.4556 | 0.25 |
| alpha-Linolenic acid metabolism | 13 | 1 | 0.08876 | 1.0518 | 0.125 |
| Ether lipid metabolism | 20 | 1 | 0.13353 | 0.87443 | 0.05263 |
| Inositol phosphate metabolism | 30 | 1 | 0.19402 | 0.71216 | 0.05 |
| Arachidonic acid metabolism | 36 | 1 | 0.22844 | 0.64122 | 0.02778 |
This approach revealed that PFKFB3 inhibition profoundly redirects TAM metabolism, particularly toward immunosuppressive sphingolipid and pro-inflammatory arachidonic acid pathways, suggesting a complex metabolic reprograming that may paradoxically sustain pro-tumorigenic functions and reduce monocyte recruitment. The following section presents a detailed analysis of these metabolic pathways and their potential implications for the TME (Figure 3).
(left panel) Control vs. PFK15 Treatment: Perturbations were observed in glycerophospholipid metabolism (1), glycosylphosphatidylinositol (GPI)-anchor biosynthesis (2), sphingolipid metabolism (3), linoleic acid metabolism (4), alpha-linolenic acid metabolism (5), glycerolipid metabolism (6), ether lipid metabolism (7), phosphatidylinositol signaling system (8), inositol phosphate metabolism (9), and arachidonic acid metabolism (10).(right panel) Control vs. AZ PFKFB3 26 Treatment: Similar analysis comparing control macrophages with those treated with PFKB3_26 identified alterations within the same pathways: glycerophospholipid metabolism (1), glycosylphosphatidylinositol (GPI)-anchor biosynthesis (2), sphingolipid metabolism (3), linoleic acid metabolism (4), alpha-linolenic acid metabolism (5), glycerolipid metabolism (6), ether lipid metabolism (7), phosphatidylinositol signaling system (8), inositol phosphate metabolism (9), and arachidonic acid metabolism (10). Node color represents significance level, while node radius correlates with pathway impact. Pathway impact is calculated as the sum of impact values for corresponding metabolites, normalized to the sum of impact values for all metabolites in each pathway.
Two hundred lipids were identified in both positive and negative ion modes, primarily belonging to triglyceride, ceramide, sphingomyelin, cardiolipin, fatty acid, phosphatidylinositol, various phosphatidylcholine and phosphatidylethanolamine classes, including their oxidized derivatives. Figure 4 present comparative lipid levels across macrophage groups.
Lipid metabolism potentially influences the pro- or anti-inflammatory activities of macrophages by fulfilling their energy demands and regulating membrane fluidity (Table 5).
Various lipids, including free fatty acids (FFAs), neutral lipids, glycerophospholipids (GLs), and sphingolipid, constitute cell membranes and lipoproteins, as well as play critical roles in diverse biological processes such as inflammation and cell differentiation [23]. Pathway enrichment analysis using KEGG and MetaboAnalyst highlighted glycerophospholipid metabolism as the most significantly affected pathway (p = 6.13E-10) in both inhibitor-treated groups, with notable involvement of glycosylphosphatidylinositol (GPI)-anchor biosynthesis, sphingolipid metabolism, and linoleic acid metabolism. The impact of PFK15 and PFKB3_26 diverged in specific pathways: PFK15 exhibited stronger associations with ether lipid metabolism and arachidonic acid metabolism, while PFKB3_26 showed greater influence on glycerolipid metabolism and phosphatidylinositol signaling. These findings suggest that both inhibitors disrupt membrane lipid homeostasis and inflammatory signaling networks.
The observed lipidomic shifts imply that PFK15 and PFKB3_26 modulate macrophage polarization by altering metabolic fluxes, particularly in pathways linked to pro-inflammatory (e.g., arachidonic acid derivatives) and immunosuppressive (e.g., sphingolipids) mediators. The consistent downregulation of glycerophospholipids, critical for membrane structure and signaling, may impair macrophage functionality in the TME. Furthermore, the differential effects on linoleic and alpha-linolenic acid metabolism suggest context-dependent roles for these lipids in CRC progression.
To further characterize the systemic metabolic impact of PFKFB3 inhibition beyond lipid remodeling, we extended our analysis to the amino acid repertoire in macrophages. Utilizing high-performance liquid chromatography coupled with tandem mass spectrometry in multiple reaction monitoring mode (HPLC-MS/MS-MRM), we quantitatively profiled the levels of 20 amino acids.
Notably, control samples occupied an intermediate position in the principal component space, situated between the two separate clusters formed by PFK15 and AZ PFKFB3 26-treated groups. This clear separation indicates that PFKFB3 inhibition induces a profound and specific reprogramming of amino acid metabolism, suggesting a metabolic rewiring that extends beyond carbon flux through glycolysis to encompass nitrogen metabolism and protein synthesis precursors.
This observed clustering, despite the absence of statistically significant differences in individual amino acids by Mann-Whitney U test, suggests subtle but coordinated shifts in the amino acid profile that are associated with inhibitor treatment. The clear separation of groups along the second principal component (PC2) prompted an investigation into the variables driving this distribution. Variable loading analysis (Figure 6) identified asparagine as the dominant contributor to the separation along PC2, with its levels decreasing progressively along this axis in the inhibitor-treated groups.
This indicates that modulation of PFKFB3 activity is associated with a specific reduction in asparagine availability, potentially pointing to a targeted reprogramming of amino acid metabolism that may support distinct macrophage functions under conditions of metabolic stress.
In summary, this comprehensive metabolic analysis demonstrates that PFKFB3 inhibition induces profound and multifaceted reprogramming of TAM metabolism, extending beyond glycolytic suppression to encompass significant alterations in lipid and amino acid metabolic pathways. The observed shifts reveal a paradoxical dual effect: while effectively reducing monocyte recruitment and suppressing certain pro-inflammatory mediators, PFKFB3 inhibition simultaneously promotes immunosuppressive metabolic reprograming. Specifically, both inhibitors consistently disrupted glycerophospholipid metabolism, critical for membrane integrity, while differentially modulating sphingolipid and inflammatory pathways. PFK15 showed stronger association with arachidonic acid metabolism while AZ PFKFB3 26 exhibited greater impact on phosphatidylinositol signaling. The distinct clustering patterns in amino acid profiles, driven primarily by reduced asparagine availability, further indicate that PFKFB3 blockade triggers coordinated nitrogen metabolic reprograming that may support macrophage adaptation to metabolic stress. These findings collectively suggest that PFKFB3 serves as a central metabolic integrator in TAMs, and its inhibition creates a complex metabolic reconfiguration that may sustain certain pro-tumorigenic functions despite impairing others. The observed metabolic flexibility suggests that targeting the broader metabolic network, rather than inhibiting a single pathway, will be more effective in overcoming potential compensatory mechanisms.
3. Discussion
This study provides compelling evidence that CRC cells actively orchestrate the metabolic reprogramming of TAMs by inducing the expression of the glycolytic regulator PFKFB3. Our findings reveal a complex interplay between tumor-derived signals and macrophage metabolism, which culminates in the promotion of a pro-tumoral phenotype of TAMs characterized by a distinct lipidomic profile. The significantly higher expression of PFKFB3 in the stroma of benign neoplasms compared to malignant CRC tissues suggests a dynamic regulation of this metabolic enzyme during cancer progression, potentially reflecting an early host an-ti-tumor response that becomes suppressed or reprogrammed in advanced disease. Our observation that colon cancer tissues exhibit higher PFKFB3 expression than rectal cancer tissues further underscore the metabolic heterogeneity within CRC subtypes and aligns with our previous findings of elevated PFKFB3 in monocytes of colon cancer patients, pointing to a subtype-specific role for this metabolic pathway in disease pathogenesis [13].
Most intriguing is the lipidomic changes in macrophages after PFKFB3 inhibition, which revealed a profound metabolic plasticity in TAMs. Significant alteration in glycerophospholipid metabolism suggests substantial membrane remodeling, which could impact numerous cellular functions including phagocytosis, cytokine secretion, cell signaling, in M2 macrophage polarization and anti-inflammatory processes [24,25]. The shift toward sphingolipid metabolism is particularly noteworthy given the established role of sphingolipids in promoting immunosuppressive and M2-like macrophage polarization [25,26]. Simultaneously, the observed changes in linoleic and arachidonic acid metabolism indicate modulation of inflammatory signaling pathways, creating a complex metabolic landscape where PFKFB3 inhibition may simultaneously impair some pro-tumoral functions while potentially reinforcing others through alternative metabolic routes. This metabolic flexibility may represent an adaptive mechanism that allows TAMs to maintain immunosuppressive functions even under glycolytic restriction, explaining why PFKFB3 inhibition alone may be insufficient to completely reverse the immunosuppressive TME.
The distinct clustering patterns in amino acid profiles following PFKFB3 inhibition, driven primarily by reduced asparagine availability, further expanded our understanding of the metabolic reprograming beyond carbon metabolism to include nitrogen metabolism. This coordinated reprogramming suggests that PFKFB3 serves as a metabolic integrator coordinated multiple metabolic pathways to support tumor-promoting functions of TAMs.
Our findings strongly support that PFKFB3 inhibition can be a promising therapeutic strategy to disrupt tumor–macrophage crosstalk in CRC, particularly in colon cancer. However, the metabolic plasticity revealed by our lipidomic and amino acid analyses suggests that compensatory mechanisms may limit the efficacy of single-agent targeting. Future studies should explore rational combinations of PFKFB3 inhibitors with TAM-targeting immunotherapeutic agents or other metabolic inhibitors to achieve more effective reprogramming of the TME.
In summary, our work established PFKFB3 as a key metabolic node in the reprogramming of TAMs in colorectal cancer, inducing their recruitment, functional polarization, and metabolic switch.
4. Materials and Methods
4.1. Patients
The study enrolled patients (N=111) with morphologically confirmed colorectal adenocarcinoma treated at the Department of Abdominal Oncology, Cancer Research Institute of Tomsk National Research Medical Center (Tomsk, Russia) between 2019 and 2021, along with healthy donors. Conducted in accordance with the Declaration of Helsinki (1964, revised 1975 and 1983), the study was approved by the Local Ethics Committee of the Tomsk Cancer Research Institute (protocol number 7 dated 25 August 2020), with all participants providing written informed consent. Patients were stratified into two groups based on tumor type and treatment approach: colon cancer patients (T2-4N0-3M0, stages I–III) and rectal cancer patients (T2-4N0-3M0, stages I–III). Colon cancers involved various segments, including the cecum, ascending, transverse, descending, and sigmoid colon.
Rectal cancer patients underwent surgery, followed by adjuvant chemotherapy (using the same regimens) for up to six months. Rectal cancers included tumors of the rectum and rectosigmoid junction.
For comparative immunohistochemical analysis of the tumor microenvironment, a control group of patients with benign intestinal neoplasms (N=50) was additionally enrolled. This group consisted of individuals with histologically verified benign lesions, including tubular adenomas, tubulovillous adenomas, and hyperplastic polyps, which were identified and resected during standard diagnostic or screening colonoscopies. Patients with benign neoplasms were selected to provide a representative baseline for stromal and immune cell characteristics in non-malignant intestinal tissue, allowing for a direct comparison with the malignant cohorts.
Healthy volunteers, matched for age and sex, served as controls. Eligibility criteria for healthy participants included age 48–70 years, absence of acute inflammatory or severe chronic conditions (e.g., diabetes, hepatitis, HIV, myocarditis), no immunomodulatory drug use within 30 days prior to enrollment, capacity to provide informed consent, and no personal history of malignancy.
4.2. Immunohistochemical (IHC) analysis
Quantitative assessment of PFKFB3-positive TAMs in TME and benign intestinal tumors (n=50) was performed. A comparative analysis of TAM presence (using the general marker CD68), along with evaluation of M2-polarized TAM markers (CD206, CD163), blood vessel density (CD31), and lymphatic vessel density (LYVE1) was performed. Standard IHC staining procedures were employed on FFPE tissue sections using the following primary antibodies: rabbit monoclonal anti-PFKFB3 (Abcam, ab181861, dilution 1:50), mouse monoclonal anti-CD68 (Novus biologicals, NBP2 445-39, clone KP1, dilution 1:100), mouse polyclonal anti-CD31 (Thermo fisher scientific, MA5-13188, dilution 1:100), rabbit monoclonal anti-CD163 (Abcam, ab182422, clone EPR19518, dilution 1:250), goat anti-CD206 (R&D systems, AF2534, dilution 1:20), and goat anti-LYVE1 (R&D systems, AF2089, dilution 1:40). For mouse and rabbit antibodies, the Bond Oracle IHC system (TA9145, Leica Biosystems) was used for visualization with DAB chromogen. Nuclear counterstaining was performed with hematoxylin. Tumor tissue sections were scanned using a Leica Aperio AT2 slide scanner and ScanScope software. QuPath software (freely available from https://qupath.github.io) was utilized for image analysis and marker quantification. Regions of interest were selected and analyzed using cell detection and intensity classification. "Cell: Mean DAB OD" was employed to analyze both membrane and cytoplasmic staining. Intensity thresholds were set to further classify cells as negative, weak, moderate, or strong for PFKFB3 staining based on the mean nuclear DAB optical density.
4.3. Immunofluorescent staining and confocal microscopy
FFPE tissue sections from ten colon cancer patients were utilized for immunohistochemical analysis. Antigen retrieval was performed using the PT Link module (Dako, Denmark) in T/E buffer (pH 9.0). For immunofluorescence (IF) staining, FFPE tumor sections were deparaffinized in xylol, and subsequently blocked with 3% BSA in PBS for 45 minutes. Sections were then incubated with primary antibodies for 1.5 hours, washed, and incubated with appropriate secondary antibodies for 45 minutes. The following primary antibodies were used: rabbit monoclonal anti-PFKFB3 (1:50, #ab181861, Abcam, USA), mouse monoclonal anti-CD68 (1:100, clone KP1, #NBP2-44539, Novus Biologicals). Secondary antibodies included donkey Cy3-conjugated anti-rabbit antibody (1:400, #711-165-152, Dianova, Germany), donkey AlexaFluor488-conjugated anti-mouse antibody (1:400, #715-545-150, Dianova, Germany), and. Slides were mounted with Fluoroshield Mounting Medium with DAPI (#ab104135, Abcam, USA). Confocal microscopy was performed using a Carl Zeiss LSM 780 NLO laser scanning spectral confocal microscope (Carl Zeiss, Germany) equipped with a 40x objective. Images were acquired and analyzed using ZEN software (RRID: SCR_018163) in sequential scan mode for all four-color images
4.4. Isolation of monocytes and model of primary human TAMs
Monocytes were isolated from human buffy coats using magnetic-activated cell sorting (MACS) with CD14-positive selection as previously described [14]. The resulting cell population contained 95-98% monocytes, as confirmed by flow cytometry analysis of CD14 surface expression. Monocytes from four healthy donors were cultured at a concentration of 2 × 106 cells/mL at 37°C for 6 days in X-VIVO medium. All macrophages were stimulated with M-CSF (1 ng/mL) and dexamethasone (10−8 M). For M1 polarization, IFNγ (100 ng/mL) was added; for M2 polarization, IL-4 (10 ng/mL) was added; and for tumor-associated macrophage (TAM) differentiation, 80% X-VIVO and 20% tumor supernatant from the colorectal cancer cell line Caco2 were used. PFKFB3 inhibition was performed using PFK15, 207 nM (Tocris, UK) and AZ PFKFB3 26, 0.023 μM (Tocris, UK) after monocyte-to macrophage differentiation. After 6 days of culture, cells were harvested and used for HPLC-MS/MS analysis.
4.5. Macrophage lipidome analysis
The macrophage lipidome was characterized using HPLC-MS/MS analysis of lipid extracts. Monocyte samples were mixed with 480 μL of chloroform-methanol (2:1, v/v) at 4°C, followed by 10-minute sonication. After adding 150 μL of water, the solution was centrifuged at 13,000 g for 5 minutes at room temperature. The lower organic layer containing lipids was collected, dried under nitrogen stream, and reconstituted in 200 μL of isopropanol-acetonitrile (1:1, v/v). Lipid extracts were analyzed using a Dionex UltiMate 3000 liquid chromatograph (Thermo Scientific, Germany) coupled to a Maxis Impact qTOF mass spectrometer with an ESI ion source (Bruker Daltonics, Germany). Reverse-phase chromatography was performed on a Zorbax C18 column (150 × 2.1 mm, 5 μm; Agilent, USA) with a 20-minute linear gradient from 30% to 90% eluent B (acetonitrile/isopropanol/water, 90/8/2 v/v/v, containing 0.1% formic acid and 10 mmol/L ammonium formate). Eluent A consisted of acetonitrile/water (60/40, v/v) with 0.1% formic acid and 10 mmol/L ammonium formate. The flow rate was maintained at 35 μL/min with a 1 μL injection volume. Mass spectra were acquired in positive and negative ion modes (m/z 100-1700) with the following parameters: capillary voltage 4.1 kV, nebulizer gas pressure 0.7 bar, drying gas flow rate 6 L/min, and drying gas temperature 200°C. Lipid identification was performed using tandem mass spectrometry in data-dependent acquisition mode with a 5 Da isolation window.
Chromatographic data were preprocessed using Proteowizard 3.0.9987 (msConvert) to generate MzXML (full scan) and MS2 (tandem spectra) files, followed by peak detection and feature table generation in MZmine. Lipids were identified using LipidMatch [26] with a mass accuracy of 0.01 Da, fragment ion matching within 5 Da mass windows, and retention time tolerances of 0.5 minutes. Statistical analysis was performed using custom R scripts in RStudio, comparing lipid levels between control macrophages and those treated with PFK15 or PFKB3_26 (pairwise comparisons, p<0.05). Principal component analysis (PCA) was employed to assess global lipid profile differences.
4.6. Amino acid profile analysis
The amino acid profile analysis was performed using HPLC-MS/MS on a system comprising an Agilent 6460 triple quadrupole mass spectrometer equipped with an electrospray ionization source coupled to an Agilent 1260 II liquid chromatograph with a binary high-pressure pump, column thermostat, and 122-vial autosampler. Sample preparation followed this protocol: macrophage samples were mixed with 480 μL of chloroform-methanol (2:1, v/v) at 4°C, sonicated for 10 minutes, then supplemented with 150 μL of water and vortexed for 5 minutes. After centrifugation at 13,000 g for 5 minutes at room temperature, 150 μL of the upper aqueous-methanolic layer was collected, evaporated under nitrogen stream for 30 minutes at 60°C, and reconstituted in 200 μL of 0.1 M hydrochloric acid in butanol. The mixture was vortexed for 3 minutes, centrifuged at 13,000 g for 15 seconds, then incubated at 60°C for 15 minutes to complete derivatization. Following another centrifugation (13,000 g, 15 seconds), samples were dried under nitrogen (30 minutes, 60°C), redissolved in 300 μL of acetonitrile/water (1:1, v/v), vortexed for 5 minutes, and centrifuged (13,000 g, 10 minutes). Finally, 200 μL aliquots were transferred to vials with inserts for analysis.
Statistical analysis was performed using custom R scripts in RStudio. Amino acid levels in macrophages from colorectal cancer patients were compared pairwise between control, PFK15-treated, and PFKB3_26-treated groups using Student's t-test with a significance threshold of p<0.05. Principal component analysis was employed to evaluate global differences in amino acid profiles.
4.7. Statistical analysis
Statistical analysis was performed using STATISTICA 8.0 for Windows (STATISTICA, RRID: SCR_014213) and GraphPad Prism 8.4.2 (GraphPad Prism, RRID: SCR_002798). The Manna-Whitney test and t-test for independent groups were implemented.
5. Conclusions
In conclusion, our study demonstrates that CRC cells strongly induce PFKFB3 expression in TAMs, thereby promoting their metabolic and functional reprogramming. We found that PFKFB3 expression is significantly elevated in the colorectal tumors, particularly in colon cancer, and is associated with increased infiltration of pro-tumoral macrophages. Lipidomic profiling revealed that PFKFB3 inhibition profoundly alters glycerophospholipid metabolism and shifts macrophage lipidomes toward immunosuppressive sphingolipid and pro-inflammatory arachidonic acid pathways. Additionally, amino acid metabolic profiling indicated coordinated changes in nitrogen metabolism, particularly a reduction in asparagine availability. These findings establish PFKFB3 as a central metabolic regulator in TAMs, governing both their recruitment and pro-tumoral polarization within the colorectal tumor microenvironment. Targeting PFKFB3 may therefore represent a promising therapeutic strategy to disrupt tumor–macrophage crosstalk and impede cancer progression, although the metabolic plasticity of TAMs suggests that combination therapies addressing multiple pathways may be necessary to fully overcome tumor-induced immunosuppression.
Author Contributions
Conceptualization, K.J.G., L.I.V.; methodology, S.E.O., S.T.S, S.K.D, C.V.V., S.N.L., T.A.O.; formal analysis, S.E.O., S.T.S., N.A.V., F.V.E., T.A.S., K.D.N., D.A.Y., A.S.G.; project administration, K.J.G., L.I.V; writing - original draft, K.J.G., S.E.O.
Funding
This research was funded by Russian Science Foundation, grant number 24-25-00455.
Institutional Review Board Statement
The study was conducted in accordance with the Declarationof Helsinki, and approved by the Institutional Ethics Committee of Cancer Research Institute, TomskNRMC (protocol code 7 and date of 25 August 2020).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
All data generated and analyzed during this study are included in this published article.
Acknowledgments
The study was conducted in accordance with the Declaration of Helsinki and the Core Facility “Clinical Mass-spectrometry” of the National Medical Research Center for Obstetrics Gynecology and Perinatology Named after Academician V.I. Kulakov of the Ministry of Healthcare of Russian Federation, and approved by the Institutional Ethics Committee of Cancer Research Institute, Tomsk NRMC (protocol number 7 and date of 25 August 2020).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of Tumor Associated Macrophages in Tumor Angiogenesis and Lymphangiogenesis. Front Physiol 2014, 5 MAR. [Google Scholar] [CrossRef]
- Kzhyshkowska, J.; Shen, J.; Larionova, I. Targeting of TAMs: Can We Be More Clever than Cancer Cells? Cellular & Molecular Immunology 2024 21:12 2024, 21, 1376–1409. [Google Scholar] [CrossRef]
- Kzhyshkowska, J.; Yin, S.; Liu, T.; Riabov, V.; Mitrofanova, I. Role of Chitinase-like Proteins in Cancer. Biol Chem 2016, 397, 231–247. [Google Scholar] [CrossRef]
- Larionova, I.; Kazakova, E.; Gerashchenko, T.; Kzhyshkowska, J. New Angiogenic Regulators Produced by TAMs: Perspective for Targeting Tumor Angiogenesis. 2021, 13, 3253. [Google Scholar] [CrossRef]
- Li, X.; Lian, J.; Lu, H. The Role of SPP1+TAMs in Cancer: Impact on Patient Prognosis and Future Therapeutic Targets. Int J Cancer 2025. [Google Scholar] [CrossRef] [PubMed]
- Kazakova, E.; Iamshchikov, P.; Larionova, I.; Kzhyshkowska, J. Macrophage Scavenger Receptors: Tumor Support and Tumor Inhibition. Front Oncol 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front Oncol 2020, 10. [Google Scholar] [CrossRef]
- Karimova, A.F.; Khalitova, A.R.; Suezov, R.; Markov, N.; Mukhamedshina, Y.; Rizvanov, A.A.; Huber, M.; Simon, H.U.; Brichkina, A. Immunometabolism of Tumor-Associated Macrophages: A Therapeutic Perspective. Eur J Cancer 2025, 220. [Google Scholar] [CrossRef]
- Larionova, I.; Kazakova, E.; Patysheva, M.; Kzhyshkowska, J. Transcriptional, Epigenetic and Metabolic Programming of Tumor-Associated Macrophages. Cancers (Basel) 2020, 12, 1–40. [Google Scholar] [CrossRef]
- Kotowski, K.; Rosik, J.; Machaj, F.; Supplitt, S.; Wiczew, D.; Jabłońska, K.; Wiechec, E.; Ghavami, S.; Dzięgiel, P. Role of Pfkfb3 and Pfkfb4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets. Cancers (Basel) 2021, 13, 1–29. [Google Scholar] [CrossRef]
- Casillo, S.M.; Gatesman, T.A.; Chilukuri, A.; Varadharajan, S.; Johnson, B.J.; David Premkumar, D.R.; Jane, E.P.; Plute, T.J.; Koncar, R.F.; Stanton, A.C.J.; et al. An ERK5-PFKFB3 Axis Regulates Glycolysis and Represents a Therapeutic Vulnerability in Pediatric Diffuse Midline Glioma. Cell Rep 2024, 43. [Google Scholar] [CrossRef]
- Chen, D.P.; Ning, W.R.; Jiang, Z.Z.; Peng, Z.P.; Zhu, L.Y.; Zhuang, S.M.; Kuang, D.M.; Zheng, L.; Wu, Y. Glycolytic Activation of Peritumoral Monocytes Fosters Immune Privilege via the PFKFB3-PD-L1 Axis in Human Hepatocellular Carcinoma. J Hepatol 2019, 71, 333–343. [Google Scholar] [CrossRef]
- Larionova, I.; Patysheva, M.; Iamshchikov, P.; Kazakova, E.; Kazakova, A.; Rakina, M.; Grigoryeva, E.; Tarasova, A.; Afanasiev, S.; Bezgodova, N.; et al. PFKFB3 Overexpression in Monocytes of Patients with Colon but Not Rectal Cancer Programs Pro-Tumor Macrophages and Is Indicative for Higher Risk of Tumor Relapse. Front Immunol 2023, 13, 1080501. [Google Scholar] [CrossRef]
- Larionova, I.; Kiselev, A.; Kazakova, E.; Liu, T.; Patysheva, M.; Iamshchikov, P.; Liu, Q.; Mossel, D.M.; Riabov, V.; Rakina, M.; et al. Tumor-Associated Macrophages Respond to Chemotherapy by Detrimental Transcriptional Reprogramming and Suppressing Stabilin-1 Mediated Clearance of EGF. Front Immunol 2023, 14, 1000497. [Google Scholar] [CrossRef] [PubMed]
- Saito, R. de F.; Andrade, L.N. de S.; Bustos, S.O.; Chammas, R. Phosphatidylcholine-Derived Lipid Mediators: The Crosstalk Between Cancer Cells and Immune Cells. Front Immunol 2022, 13, 768606. [Google Scholar] [CrossRef] [PubMed]
- Rabold, K.; Aschenbrenner, A.; Thiele, C.; Boahen, C.K.; Schiltmans, A.; Smit, J.W.A.; Schultze, J.L.; Netea, M.G.; Adema, G.J.; Netea-Maier, R.T. Enhanced Lipid Biosynthesis in Human Tumor-Induced Macrophages Contributes to Their Protumoral Characteristics. J Immunother Cancer 2020, 8, e000638. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Weir, J.D.; Hartung, R. The Composition of Linoleic Acid and Conjugated Linoleic Acid Has Potent Synergistic Effects on the Growth and Death of RAW264.7 Macrophages: The Role in Anti-Inflammatory Effects. Int Immunopharmacol 2024, 141. [Google Scholar] [CrossRef] [PubMed]
- Schumann, T.; Adhikary, T.; Wortmann, A.; Finkernagel, F.; Lieber, S.; Schnitzer, E.; Legrand, N.; Schober, Y.; Nockher, W.A.; Toth, P.M.; et al. Deregulation of PPARβ/δ Target Genes in Tumor-Associated Macrophages by Fatty Acid Ligands in the Ovarian Cancer Microenvironment. Oncotarget 2015, 6, 13416–13433. [Google Scholar] [CrossRef]
- Henry, W.S.; Müller, S.; Yang, J.-S.; Innes-Gold, S.; Das, S.; Reinhardt, F.; Sigmund, K.; Phadnis, V. V.; Wan, Z.; Eaton, E.; et al. Ether Lipids Influence Cancer Cell Fate by Modulating Iron Uptake. 2024. [Google Scholar] [CrossRef]
- Zhang, J.G.; Zhang, X.M.; Wu, X.; Zhou, C.K.; Liu, Z.Z.; Luo, X.Y.; Zhang, L.; Chen, W.; Yang, Y.J. Covalent Organic Frameworks-Delivered Reuterin Drives Trained Immunity in Tumor-Associated Macrophages to Enhance Melanoma Immunotherapy via Glycerophospholipid Metabolism. Advanced Science 2025, e04784. [Google Scholar] [CrossRef]
- Cheng, Y.; Bai, F.; Ren, X.; Sun, R.; Guo, X.; Liu, W.; Wang, B.; Yang, Y.; Zhang, X.; Xu, Y.; et al. Phosphoinositide-Binding Protein TIPE1 Promotes Alternative Activation of Macrophages and Tumor Progression via PIP3/Akt/TGFb Axis. Cancer Res 2022, 82, 1603–1616. [Google Scholar] [CrossRef]
- Xu, M.; Wang, X.; Li, Y.; Geng, X.; Jia, X.; Zhang, L.; Yang, H. Arachidonic Acid Metabolism Controls Macrophage Alternative Activation Through Regulating Oxidative Phosphorylation in PPARγ Dependent Manner. Front Immunol 2021, 12, 618501. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.; Wang, F.; Wang, Z.; Lu, Y.; Xu, Y.; Wang, K.; Shen, H.; Yang, P.; Li, S.; et al. Quantitative Profiling of Glycerophospholipids during Mouse and Human Macrophage Differentiation Using Targeted Mass Spectrometry. Sci Rep 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.; Wang, F.; Wang, Z.; Lu, Y.; Xu, Y.; Wang, K.; Shen, H.; Yang, P.; Li, S.; et al. Quantitative Profiling of Glycerophospholipids during Mouse and Human Macrophage Differentiation Using Targeted Mass Spectrometry. Sci Rep 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.P.; Casas, J.; Balboa, M.A.; Balsinde, J. Bioactive Lipid Signaling and Lipidomics in Macrophage Polarization: Impact on Inflammation and Immune Regulation. Front Immunol 2025, 16, 1550500. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, W.Y.; Zhou, Q.D.; York, A.G.; Williams, K.J.; Scumpia, P.O.; Kronenberger, E.B.; Hoi, X.P.; Su, B.; Chi, X.; Bui, V.L.; et al. Toll-Like Receptors Induce Signal-Specific Reprogramming of the Macrophage Lipidome. Cell Metab 2020, 32, 128–143.e5. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
PFKFB3+ cells are present in colon cancer tissue as well as in the tissue of benign neoplasms. Immunohistochemical analysis of all samples demonstrated the presence of PFKFB3+ cells within the TME. The representative images from three patients are provided.
Figure 1.
PFKFB3+ cells are present in colon cancer tissue as well as in the tissue of benign neoplasms. Immunohistochemical analysis of all samples demonstrated the presence of PFKFB3+ cells within the TME. The representative images from three patients are provided.
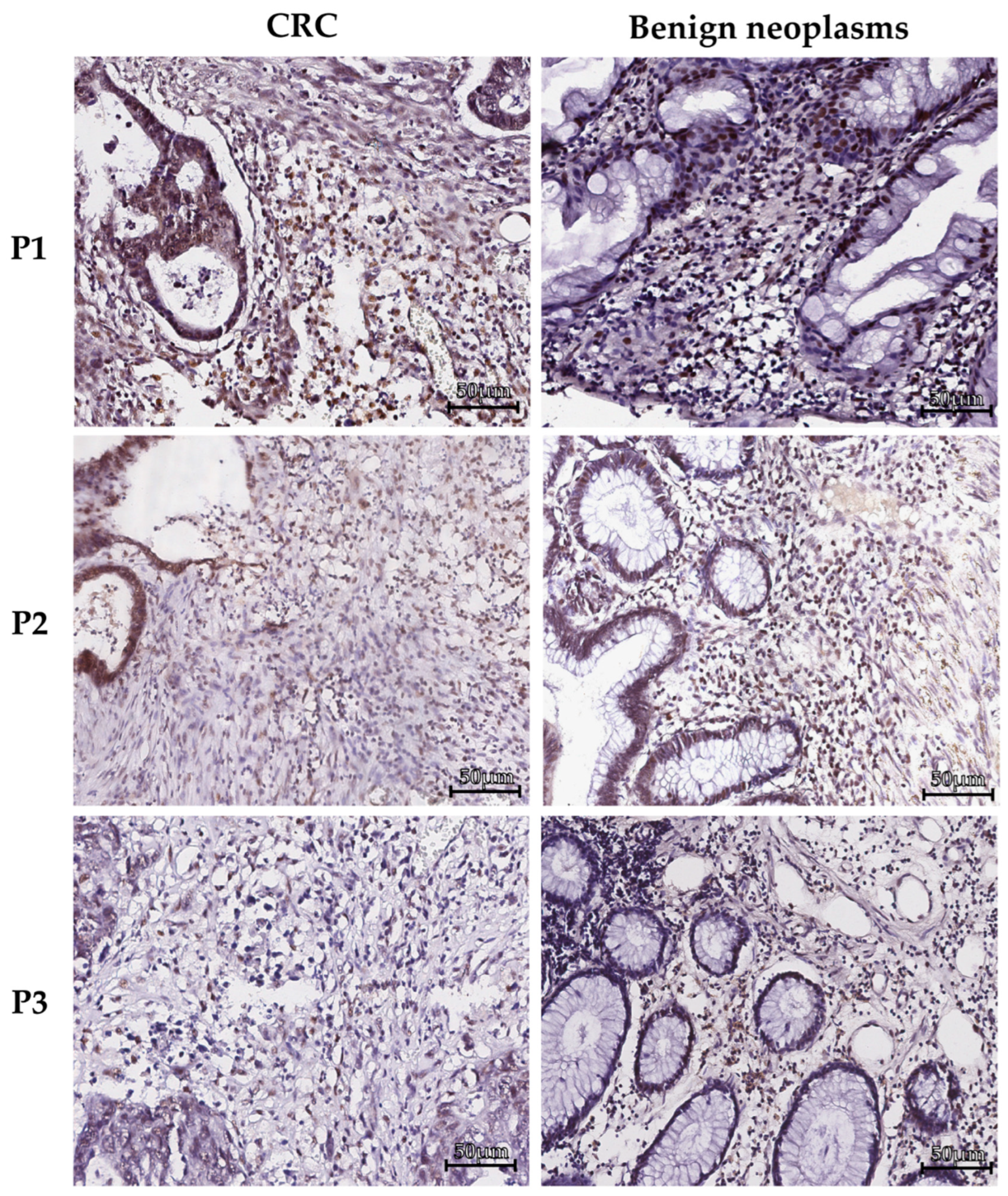
Figure 2.
PFKFB3 is preferential expressed in tumor-associated macrophages in human colon cancer tissues. Analysis performed by immunofluorescent staining and confocal microscopy. Examination of all samples demonstrated the presence of CD68+PFKFB3+ TAMs within the TME. Representative images from three patients are provided. The scale bar indicates 50 µm for the main image, allowing for detailed visualization of cellular localization and expression.
Figure 2.
PFKFB3 is preferential expressed in tumor-associated macrophages in human colon cancer tissues. Analysis performed by immunofluorescent staining and confocal microscopy. Examination of all samples demonstrated the presence of CD68+PFKFB3+ TAMs within the TME. Representative images from three patients are provided. The scale bar indicates 50 µm for the main image, allowing for detailed visualization of cellular localization and expression.
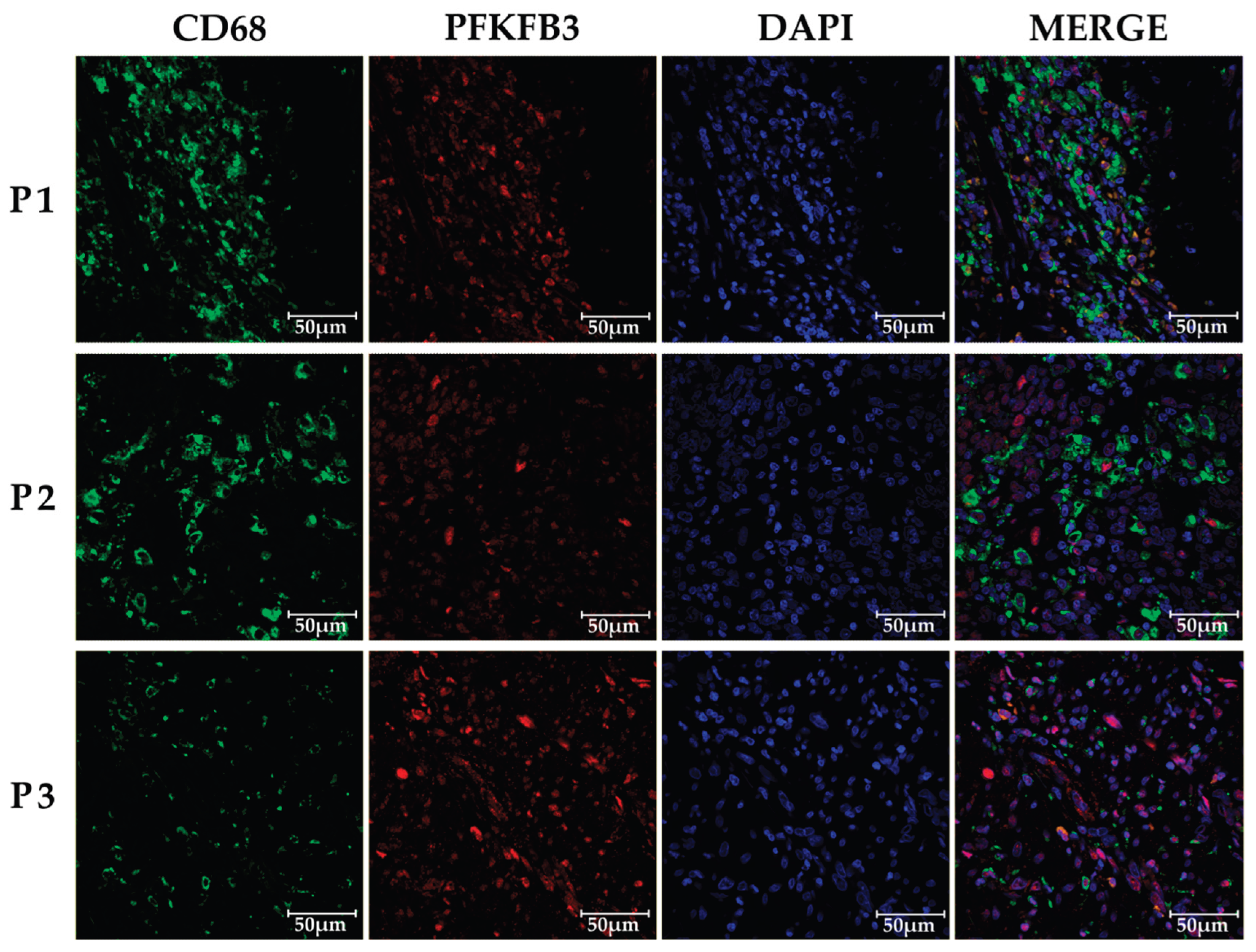
Figure 3.
Analysis of metabolic pathways enriched for lipids exhibiting statistically significant differential abundance in colorectal cancer-associated macrophages revealed the following:.
Figure 3.
Analysis of metabolic pathways enriched for lipids exhibiting statistically significant differential abundance in colorectal cancer-associated macrophages revealed the following:.
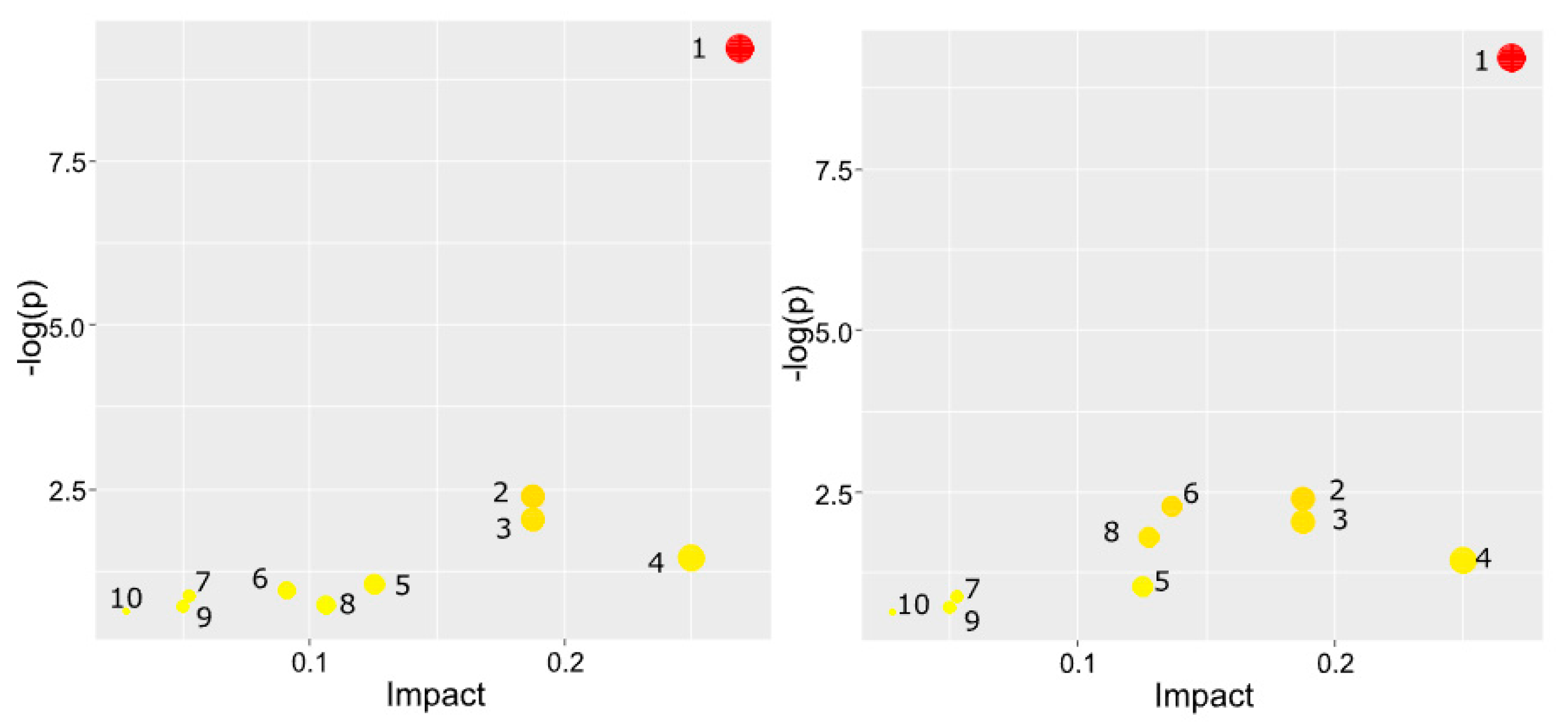
Figure 4.
Lipidomic profiles of colorectal cancer-associated macrophages were compared following treatment with PFK15 and PFKB3_26. Figure displays lipids exhibiting statistically significant differential abundance as determined by pairwise Mann-Whitney U tests comparing non-treated control (NS), PFK15-treated, and PFKB3_26-treated groups. Data are presented as median values with error bars representing standard deviation. Asterisks denote statistical significance (p < 0.05).
Figure 4.
Lipidomic profiles of colorectal cancer-associated macrophages were compared following treatment with PFK15 and PFKB3_26. Figure displays lipids exhibiting statistically significant differential abundance as determined by pairwise Mann-Whitney U tests comparing non-treated control (NS), PFK15-treated, and PFKB3_26-treated groups. Data are presented as median values with error bars representing standard deviation. Asterisks denote statistical significance (p < 0.05).
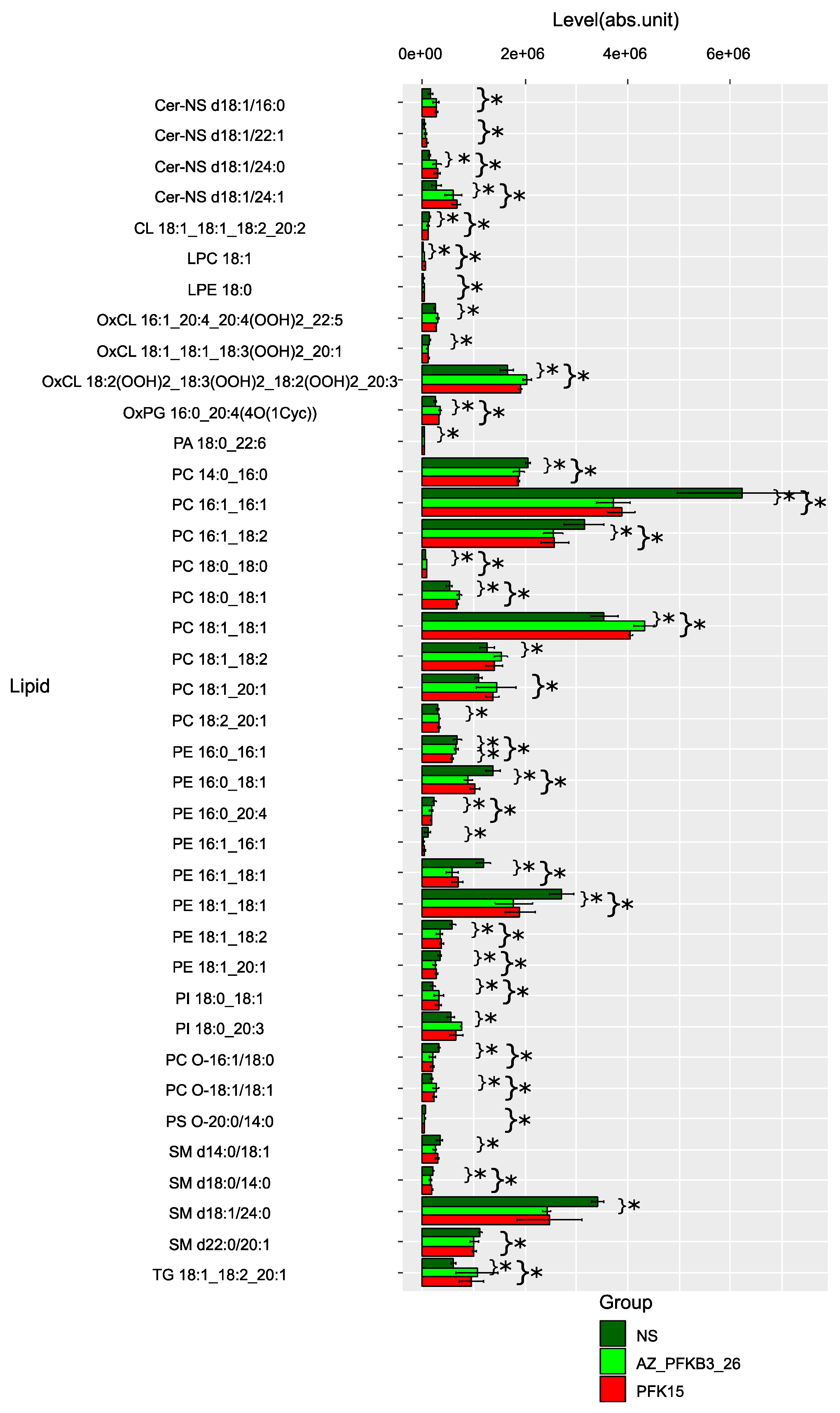
Figure 6.
Contribution of amino acids to the second principal component. Upper figure demonstrates Loadings of each amino acid on the PC2 axis. Down figure demonstrates Percentage contribution of each amino acid to the variance explained by PC2.
Figure 6.
Contribution of amino acids to the second principal component. Upper figure demonstrates Loadings of each amino acid on the PC2 axis. Down figure demonstrates Percentage contribution of each amino acid to the variance explained by PC2.
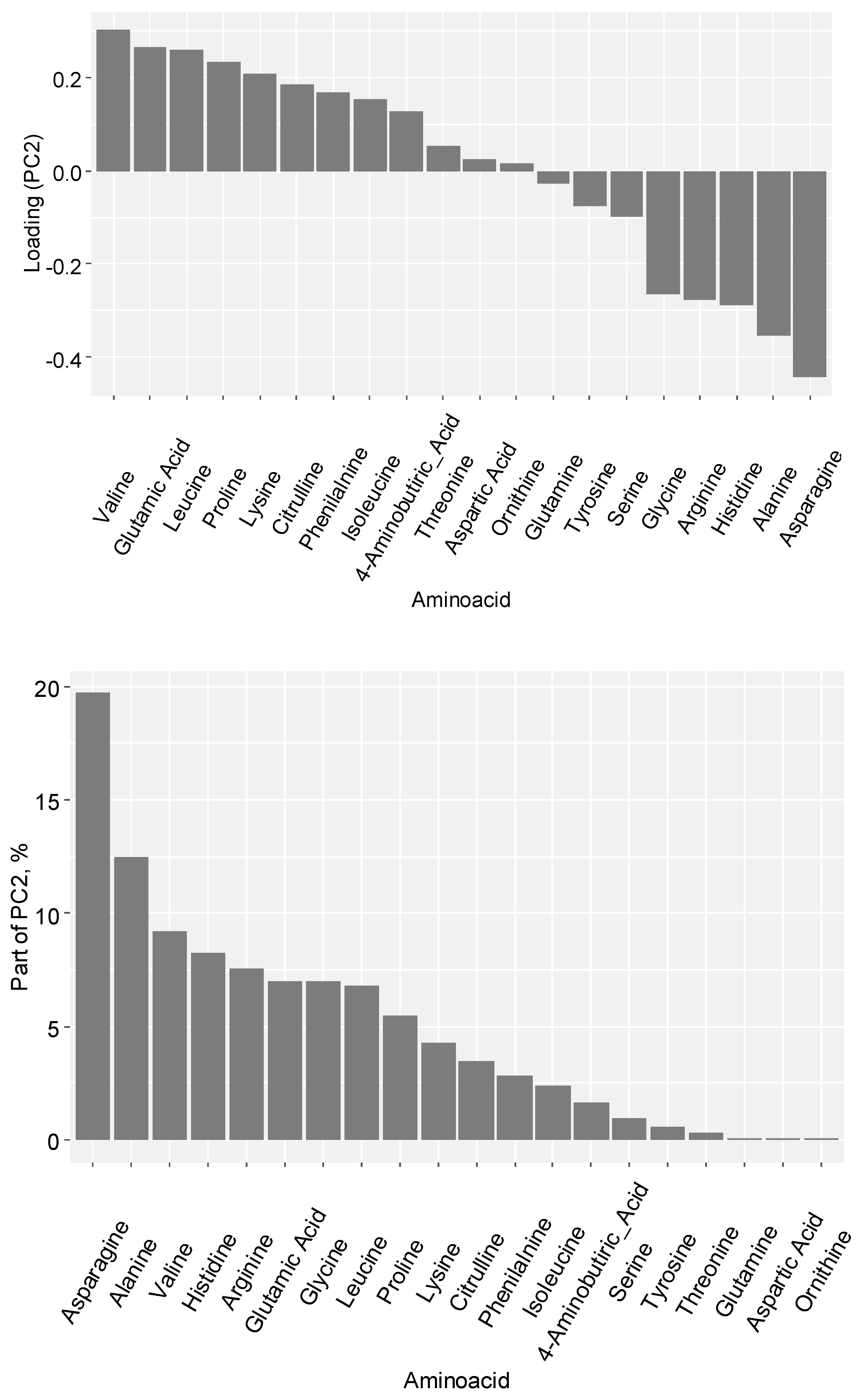
Table 1.
Comparative analysis of PFKFB3 expression in the tumor stroma of colorectal cancer patients and benign intestinal neoplasms.
Table 1.
Comparative analysis of PFKFB3 expression in the tumor stroma of colorectal cancer patients and benign intestinal neoplasms.
| Benign intestinal neoplasms (1) | Colorectal cancer (2) | Rectal cancer (3) | Colon cancer (4) |
|---|---|---|---|
| 78,22±35,54 (n=50) p1,2<0,00001 |
30,19±25,33 (n=111) |
24,72±21,53 (n=49) p1,3<0,00001 p3,4<0,045 |
34,50±27,37 (n=62) p1,2<0,00001 |
Table 3.
KEGG-derived metabolic pathways enriched for lipids exhibiting statistically significant differential abundance between control and PFK15-treated macrophages.
Table 3.
KEGG-derived metabolic pathways enriched for lipids exhibiting statistically significant differential abundance between control and PFK15-treated macrophages.
| Name metabolic pathways | The total number of metabolites involved | The number of metabolites matching the experimental data | P | LOG(P) | Effect on pathway |
| Glycerophospholipid metabolism | 36 | 7 | 6.13E-10 | 9.2127 | 0.26922 |
| Glycosylphosphatidylinositol (GPI)-anchor biosynthesis | 14 | 2 | 0.003979 | 2.4002 | 0.1875 |
| Sphingolipid metabolism | 21 | 2 | 0.008937 | 2.0488 | 0.1875 |
| Linoleic acid metabolism | 5 | 1 | 0.035028 | 1.4556 | 0.25 |
| alpha-Linolenic acid metabolism | 13 | 1 | 0.08876 | 1.0518 | 0.125 |
| Glycerolipid metabolism | 16 | 1 | 0.1082 | 0.96578 | 0.09091 |
| Ether lipid metabolism | 20 | 1 | 0.13353 | 0.87443 | 0.05263 |
| Phosphatidylinositol signaling system | 28 | 1 | 0.18224 | 0.73936 | 0.10638 |
| Inositol phosphate metabolism | 30 | 1 | 0.19402 | 0.71216 | 0.05 |
| Arachidonic acid metabolism | 36 | 1 | 0.22844 | 0.64122 | 0.02778 |
Table 5.
Effects of PFKFB3 inhibitors on macrophage lipid metabolism and functional polarization in colorectal cancer.
Table 5.
Effects of PFKFB3 inhibitors on macrophage lipid metabolism and functional polarization in colorectal cancer.
| Inhibitor | Lipid Class | Specific Lipids | Regulation | Pathway | Effects of PFKFB3 inhibitors on TAMs |
|---|---|---|---|---|---|
| PFK15 | Glycerophospholipids | Phosphatidylcholines, PCs (PC 14:0_16:0, PC 16:1_16:1, PC 16:1_18:2, PC 18:0_18:0, PC 18:0_18:1, PC 18:1_18:1, PC 18:1_18:2, PC 18:1_20:1, PC 18:2_20:1, PC O-16:1/18:0, PC O-18:1/18:1) | ↓ | Glycerophospholipid metabolism | Membrane destabilization Impairing of phagocytosis, monocyte migration and production of pro-inflammatory citokine production (Tumor-suppressive) [15] |
| Sphingolipids | Ceramides, Cer (Cer-NS d18:1/16:0, Cer-NS d18:1/22:1, Cer-NS d18:1/24:0, Cer-NS d18:1/24:1) | ↑ | Sphingolipid metabolism | •Anti-inflammatory skewing •M2-like polarization (Tumor-promotive) [16] |
|
| Fatty Acids | Linoleic acid derivatives, PCs (LPC 18:2, PC 16:1_18:2, PC 18:1_18:2, PC 18:2_20:1, PE 18:1_18:2, OxCL 18:1_18:1_18:3(OOH)2_20:1, OxCL 18:2(OOH)2_18:3(OOH)2_18:2(OOH)2_20:3) | ↑ | Linoleic acid metabolism | •Increasing of pro-inflammatory mediators [17,18] |
|
| Ether Lipids | Plasmalogens (PC O-16:1/18:0, PC O-18:1/18:1, PS O-20:0/14:0) | ↓ | Ether lipid metabolism | •Diminished antioxidant capacity [19] |
|
| AZ PFKFB3 26 | Glycerophospholipids | Phosphatidylethanolamines (PE 16:0_16:1, PE 16:0_18:1, PE 16:0_20:4, PE 16:1_16:1, PE 16:1_18:1, PE 18:1_18:1, PE 18:1_18:2, PE 18:1_20:1) | ↓ | Glycerophospholipid metabolism | • Supression of M1 activation • Tumor-promotive effects [20] |
| Phosphoinositides | PIP2, PIP3 | ↓ | PI signaling system of PI3K/AKT signaling pathway | Reduction of inflammatory responses [21] | |
| Oxidized Lipids | Oxidized phosphatidylcholines (OxPC 16:0_20:4(4O(1Oye)), OxPC 18:1_18:3(OOH)2_20:1, OxPC 18:2(OOH)2_18:3(OOH)2_18:2(OOH)2_20:3) | ↑ | Arachidonic acid metabolism | Suppression of lipid peroxidation and reduction of pro-inflammatory mediators, which may lead to attenuation of oxidative stress and modulation of the inflammatory response [22]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



