
Submitted:
16 April 2025
Posted:
16 April 2025
You are already at the latest version
Abstract
We combined atomistic simulations and experiments to assess the photocatalytic potential of the rutile phase of TiO2 combined with phenyl-modified carbon nitride (PhCN). Density Functional Tight Binding calculations are employed to investigate the electronic properties, band alignment, and adsorption behavior of TiO2/PhCN heterostructures. The results show a favorable adhesion and band alignment indicating strong potential for photocatalytic applications. XRD measurements, optical characterization, and photocatalytic degradation experiments provide insight on the beneficial integration of the organic and inorganic components, identifying the PhCN/rutile heterostructure as a promising green photocatalyst.
Keywords:
photocatalysis
; gC3N4
; DFTB
; atomistic simulations
1. Introduction
Graphitic carbon nitride () has emerged as an auspicious material for photocatalytic applications, owing to its exceptional structural and optoelectronic properties[1,2]. Its two-dimensional layered framework comprises tri-s-triazine (or heptazine) units linked by nitrogen bridges, forming a robust polymeric network stabilized by van der Waals forces. This distinctive configuration imparts several key advantages, including excellent thermal and chemical stability, biocompatibility, and eco-friendliness. The -conjugated planar structure enhances charge carrier mobility. At the same time, its moderate bandgap ( eV) enables efficient absorption of visible light, making it a good candidate for solar energy conversion and other light-driven applications[3]. Building on these peculiar properties, has shown great potential over the years in diverse fields such as photocatalytic water splitting, reduction, environmental remediation, organic synthesis, and energy harvesting[3]. Despite its potential, the practical application is limited by its high electron-hole recombination rates and restricted absorption within the visible spectrum. To overcome these challenges, significant research efforts have been devoted to couple with other semiconductors to develop hybrid systems[4,5,6]. Among these, /titanium dioxide () stands out as a particularly effective combination, offering improved photocatalytic performance and expanded light absorption capabilities[1,7]. is a well-established photocatalyst known for its excellent photocatalytic activity, high chemical stability, and non-toxic nature. However, its wide bandgap (3.2 eV for the anatase phase) limits its light absorption primarily to the ultraviolet (UV) region, which accounts for only about of the solar spectrum. When combined with graphitic carbon nitride, the resulting heterostructure effectively integrates the complementary properties of both materials. provides a stable platform for charge separation and transport. At the same time, broadens the light absorption range into the visible spectrum. This synergistic interaction in the heterojunction promotes efficient separation of photogenerated electron-hole pairs, significantly enhancing photocatalytic performance[8]. Furthermore, the realization of phenyl-modified carbon nitride (PhCN)[9,10,11] has represented a significant breakthrough: incorporating phenyl groups extends the -conjugation within the framework, effectively reducing its bandgap and shifting its light absorption deeper into the visible spectrum. In addition, PhCN significantly improves light-harvesting efficiency and enhanced charge transport properties[9]. Upon light excitation, electrons transition from the highest occupied to lowest unoccupied molecular orbitals (HOMO and LUMO) of PhCN are transferred to the conduction band leading to higher rates of pollutant degradation under visible light. Under visible-light irradiation, the degradation efficiency of Rhodamine B solutions increased from with / to with the PhCN/ hybrid system. This substantial improvement highlights the critical role of phenyl modification in optimizing photocatalytic activity[12].
In this work, we further improve the hybrid system: replacing anatase with rutile phase as a more sustainable and cost-effective alternative. Rutile offers significant advantages in terms of simpler and greener synthesis methods. For instance, replacing ethanol with water as a solvent during the synthesis process aligns with the principles of green chemistry[13,14,15]. This eco-friendly approach minimizes the environmental impact and reduces production costs, making the PhCN/ hybrid system a more practical and sustainable solution. Here, we explore the effectiveness of this strategy through a combined computational and experimental approach. Density Functional Tight Binding (DFTB) calculations are employed to investigate the electronic properties, band alignment, and adsorption energetics of PhCN/ heterostructures. The results reveal that (i) the adhesion properties of PhCN with the rutile phase are comparable to those observed with anatase and pristine , and (ii) the band alignment indicates strong potential for photocatalytic applications. All computational findings are paralleled with experimental Raman, X-ray diffraction (XRD), diffuse reflectance spectroscopy, and photocatalytic degradation experiments. Specifically, Raman and XRD characterizations confirm the effective formation of the anatase and rutile structures, as well as the successful integration of and PhCN, with adhesion observed in both phases. Optical characterization further demonstrates interactions between the organic and inorganic components, highlighting the charge transfer process from the organic moiety, which acts as a sensitizer, to . Finally, photocatalytic degradation experiments validate the predicted charge transfer behavior and photocatalytic efficiency, identifying PhCN/rutile as a promising green photocatalyst.
2. Materials and Methods
2.1. Computational Methods
This study adopts DFTB calculations to investigate the electronic properties and band alignment of PhCN/ heterostructures. DFTB is a semi-empirical quantum mechanical method derived from Density Functional Theory (DFT) that simplifies the Kohn-Sham equations by employing a minimal localized basis set and precomputed integrals [16,17,18]. This approach retains the essential physics of DFT while significantly reducing computational cost, enabling the simulation of large systems with hundreds of atoms that would be computationally prohibitive using standard DFT methods. Such efficiency is crucial for modeling complex interfaces between extended surfaces and sizeable organic molecules such as triazine and heptazine. While DFT offers high accuracy for electronic structure calculations, its computational demands scale poorly with system size. DFTB provides a balance between accuracy and efficiency and has been successfully applied to modeling both gas-phase molecules [19] and condensed matter under inert and reactive conditions [20,21,22,23], including extreme pressures and temperatures [24,25].
The DFTB total energy is derived from an expansion of the Kohn-Sham energy to second or third-order in charge fluctuations, yielding the expression:
Here, is the band structure energy, is the charge fluctuation term, and is the repulsive energy. The band structure energy is calculated as a sum over occupied electronic states from the DFTB Hamiltonian. The Hamiltonian matrix elements are determined from pre-tabulated Slater-Koster tables derived from reference calculations with a minimal basis set. The onsite matrix elements are the free-atom orbital energies, and the off-site terms are computed using a two-center approximation, where both wavefunctions and electron density are subjected to confining potentials. The repulsive energy, instead, accounts for ion-ion repulsions as well as Hartree and exchange-correlation double-counting terms. This term is typically expressed as an empirical function with parameters fitted to reproduce high-level quantum mechanical or experimental reference data. In addition, a dispersion correction can be included, such as those commonly used in DFT calculations [26,27]. To model in our simulations, we employed a combination of publicly available DFTB parameter sets: mio-1-1 [16,28] and tiorg-0-1 [29]. These parameter sets have been developed for organic molecules containing O, N, C, H, and S, as well as for bulk titanium oxide, titanium oxide surfaces, and the interactions of titanium oxide with organic molecules within the DFTB framework.
Reference DFT simulations were performed using the Vienna Ab initio Simulation Package (VASP)[30,31,32], employing the projector augmented wave (PAW) method [33,34] and the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional [35]. Partial occupancies of the electronic states were set using fourth-order Methfessel-Paxton smearing [36] with a width of 0.05 eV. Converged energies for the bulk systems were achieved with a plane-wave energy cutoff of 600 eV and a self-consistent field (SCF) convergence criterion of eV. The force convergence tolerance was set to eV/Å for each atom in all directions. DFTB calculations were performed using the DFTB+ code[17], employing orbital-resolved self-consistent charge (SCC) calculations[16]. The same convergence criteria and electron thermal smearing as in the DFT calculations were adopted. In DFTB, the total energy expression with SCC is derived by assuming spherically symmetric charge densities and expanding the Kohn-Sham total DFT energy expression to second[16] or third order[18] in charge fluctuations; in this work, we rely exclusively on the second-order charge fluctuation model. A -point Monkhorst-Pack mesh sampling of the Brillouin zone [37] was used: for bulk anatase, a mesh was considered, while for bulk rutile, we adopted a mesh. For the slabs, a mesh was used for anatase-derived slabs and a mesh for rutile slabs.
Slab generation and assessment of simulation parameters
A series of model systems were constructed for both the anatase and rutile phases of . Two crystallographic orientations were considered for each phase: the (100) and (110) surfaces. These surfaces were selected due to their distinct atomic arrangements and surface energies, which can significantly influence adsorption behavior and electronic interactions with organic molecules [38,39]. The bulk structures of anatase and rutile were first optimized using DFT calculations to obtain accurate lattice parameters. A comparison between DFT and DFTB calculated lattice constants is reported in Table 1.
Overall, the DFTB lattice constants show good agreement with the DFT results and experimental data [40], confirming the reliability of our computational approach. Furthermore, the calculated band gaps ( eV for anatase and eV for rutile) are consistent with experimental values of eV and eV, respectively [41]. Using these optimized bulk structures, slab models were generated for the (100) and (110) surfaces of both anatase and rutile phases. The slabs were constructed by cleaving the bulk crystals along the respective crystallographic planes, resulting in surfaces with specific atomic terminations. We chose the ones with the lowest energy among all possible terminations. Each slab consisted of three atomic layers, which provides a balance between capturing the representative properties of the bulk material and computational efficiency. A vacuum layer of 15 Å was included perpendicular to the surface to prevent interactions between periodic images in the slab model. To accommodate the adsorption of the organic molecules considered in this study without introducing significant strain or artificial interactions, supercells were constructed by replicating the slabs in the in-plane directions. Specifically, a supercell was used for anatase slabs and a supercell for rutile slabs. As a further check for the validity of our parameter sets, we computed the formation energy for the obtained surfaces. We compared them with values available in the literature, as presented in Table 2. A complete collection of the atomistic views for he structures here investigated (both bulk and slab systems) is shown in Supporting Information (SI) in Fig. S1.
Our findings compare very well with the range of values reported in the literature for anatase surfaces, while the formation energies are somewhat overestimated for rutile. However, we remark that the formation energies are highly dependent upon the choice of simulation parameters and functional, thus the observed offset is not a significant issue here. These observations further support the validity of the adopted computational methodology.
2.2. Experimental Methods
Phenyl-triazine, melamine, Rhodamin B (RhB 95%), and absolute ethanol were purchased from Merck/Sigma-Aldrich (Darmstadt, Germany) and Carlo Erba (France), respectively.
Preparation of
Graphitic Carbon Nitride () was synthesized through the thermal polymerization of melamine. Precisely, 1 of melamine was placed in a crucible and heated in a furnace at 550 °C for 4 hours under static air conditions.
Preparation of Phenyl-Modified Carbon Nitride
The phenyl-modified carbon nitride was synthesized by placing 1 of 6-phenyl-1,3,5-triazine-2,4-diamine powder in a quartz tube, then positioned in a tubular furnace. The material was subjected to a controlled heating process, with the temperature gradually increased to 400 °C over 1 hour.
Preparation of PhCN/ in Ethanol
A total of 100 of hCN was dispersed in 20 of ethanol and stirred at room temperature for 30 minutes to ensure uniform dispersion. Subsequently, 0.5 of titanium tetrachloride (TiCl4) was added dropwise to the solution, stirring continuously for an additional 2 hours. The resulting mixture was transferred to an autoclave and subjected to hydrothermal treatment at 180 °C. Following the reaction, the product was filtered, washed with ethanol to remove impurities, and dried. Finally, the dried sample was ground into a fine powder for subsequent characterization.
Preparation of PhCN/ in Water
In this case, we used wet-chemical and sol-gel synthesis methods. Initially, 100 of PhCN was dispersed in 20 of deionized water and stirred at room temperature for 30 minutes to form a homogeneous suspension. Then, 0.5 of titanium tetrachloride (TiCl4) was added dropwise to the mixture, followed by continuous stirring for another 2 hours to promote interaction between the components. The resulting product was separated by filtration, thoroughly washed with deionized water to remove unreacted residues, and dried. Finally, the dried sample was ground into a fine powder to obtain the final material.
Preparation of / in Water
The same methods (wet-chemical, sol-gel) used for synthesizing PhCN/ in water were applied.
Characterization
XRD analysis was conducted at room temperature using a Rigaku Miniflex II diffractometer equipped with Cu K radiation (Å) in a Bragg–Brentano geometry. Diffuse reflectance spectroscopy was used to measure the samples absorption properties, employing a UV–Vis-NIR JASCO FP-8550ST spectrometer (Jasco, Easton, MD, USA) equipped with a PbS solid-state photodetector. The measurements were carried out in a reflection configuration, with the diffuse reflectance compared against a BaSO4 reference. Absorption features were determined using the Kubelka–Munk equation. The Raman spectra were recorded using a Sol Instruments MS750 series monochromator-spectrograph (Sol Instruments, Augsburg, Germany). An excitation wavelength of 785 nm was employed for samples, while 1064 nm was used for the hybrid systems, with a spectral resolution of approximately 1.
Photodegradation of Rhodamine B
The photocatalytic performance was tested by measuring the degradation of Rhodamine B (RhB) in an aqueous solution under visible light. A Philips 13 W white LED light source (100 optical power) was used for irradiation. To ensure equilibrium between the catalyst and the dye, 40 of the catalyst was mixed with 40 mL of a 10 mg/L RhB solution and stirred in the dark for 30 minutes. Afterward, the mixture was exposed to visible light. During the reaction, 1.5 samples were taken every 60 minutes. These samples were centrifuged to separate the catalyst, and the remaining RhB concentration was measured using a Jasco V-750 spectrophotometer with a spectral bandwidth of 2 in the 200–800 range monitoring the maximum at 554.
3. Results and Discussion
3.1. Computational Results: Stability and Energetics of the Heterostructure
As mentioned in the Methods section, the preparation of typically involves the thermal condensation of nitrogen-rich organic precursors; triazine and heptazine are recognized as such precursors[44,45] and serve as fundamental building blocks of graphitic carbon nitride. Given the large spatial dimensions of a typical sample, the computational workload would be prohibitive even for DFTB calculations. Therefore, we opted to study the band alignment properties of triazine and heptazine with . In fact, the extended electronic properties of polymeric systems such as are often dominated by the characteristics of their essential components. In the case of , triazine and heptazine units largely determine the frontier orbital distribution and, hence, the band edges that govern photocatalytic and charge-transfer processes. Previous theoretical studies have shown that the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO) in are primarily localized on these units[46,47]. In addition, recent studies have shown that doping carbon nitride-based materials with phenyl rings can decrease the band gap and increase the separation rate of electron-hole pairs[48]. For this reason, we decided to compare triazine and heptazine with their phenyl-functionalized counterparts (Ph-triazine and Ph-heptazine). The molecules investigated in this work are shown in Fig. S2 (SI).
The organic molecules were initially fully relaxed in vacuum conditions and then placed atop the constructed slabs, with their principal planes parallel to the surface and randomly oriented in the in-plane directions. The resulting organic molecule–substrate complexes were fully optimized according to the aforementioned convergence criteria. Figure 1 illustrates two different views of heptazine and phenyl-heptazine deposited on the (100) surface of anatase . We generated a total of 16 heterostructures by varying the deposited molecule, the surface orientation, and the phase of the substrate. The computed adsorption energies for these heterostructures are reported in Table 3.
The adsorption energies indicate that adsorption on the (110) facet is stronger than on the (100) facet, suggesting that chemisorption may occur on the former. At the same time, the interaction on the latter is predominantly a physisorption phenomenon, dominated by van der Waals forces and weak electrostatic interactions. (110) surface orientation has a higher density of unsaturated surface atoms and undercoordinated sites[49], which enhance interactions with adsorbed molecules through stronger chemical bonding or increased van der Waals forces. In comparing the role of the substrate, we observe that, across all molecules and surface orientations, the adsorption energies are more negative on anatase surfaces than on rutile surfaces. Anatase typically exhibits higher photocatalytic activity and more reactive surface sites due to its electronic structure and surface energy[50]. Specifically, the anatase phase presents more undercoordinated Ti atoms and oxygen vacancies, which can form stronger bonds with adsorbates. Heptazine molecules exhibit slightly more negative adsorption energies for most surfaces than triazine, especially on the anatase (110) surface. This trend may be related to the more extensive conjugated ring system of heptazine compared to triazine, which provides a greater area for interaction with the surface. The extended interaction area enhances orbital interactions between the delocalized electrons of the organic molecule and the orbitals of Ti atoms. Finally, phenyl-functionalized molecules exhibit more negative adsorption energies on the (110) surfaces than their pristine counterparts. The addition of a phenyl group increases the molecular size and introduces additional electrons, which can enhance stacking interactions with the surface and increase van der Waals forces, especially on surfaces with higher atomic density such as the (110) facet.
These results show that phenyl functionalization enhances triazine and heptazine adhesion properties. Furthermore, moving from anatase to rutile, both heptazine and triazine—though with slightly reduced strength—still adhere to the surface. In all cases, the preferred adsorption configuration remains the face-on orientation.
3.2. Computational Results: Band Alignment
To calculate the band alignment, we performed a detailed analysis of the electronic density of states (DOS) for the fully relaxed heterostructures. The total electronic density of states (DOS) was projected onto the atomic species to separate the contributions from the substrate and the adsorbed organic molecules to identify the specific electronic states near the Fermi level (). In determining the top of the valence band (VBM) and the bottom of the conduction band (CBM) for , as well as the HOMO and LUMO of the adsorbed molecules, a threshold criterion based on the normalized DOS was applied. Specifically, energy levels were defined as the energies at which the normalized DOS above and below . This threshold ensured that only the relevant electronic states near the band gap are considered, isolating the significant electronic states contributing to interface charge transfer processes. As illustrated in Figure 2, this approach allows for a clear visualization of how the energy levels of the substrate align with those of the adsorbed molecules, providing insights into the potential for efficient charge separation and transfer. The complete set of projected DOS and band alignment results is provided in the SI.
The results of our calculations are rationalized by quantifying the energy difference between the CBM and the LUMO and the difference between the CBM and the HOMO . These values are reported in Table 4.
Consistent with the observations in Figure 2, all heterostructures exhibit a type-II (staggered gap) band alignment, as proven by : the LUMO is higher in energy than the CBM. This alignment promotes efficient charge separation and transfers across the interface, essential for enhancing device performance[51,52]. In particular, the phenyl-functionalized versions of the organic molecules consistently exhibit smaller values compared to their pristine counterparts. As already pointed out rationalizing the adsorption energies, adding a phenyl group extends the conjugation of the molecule, lowering its LUMO energy level. This shift brings the LUMO closer to the CBM, enhancing electron transfer. The stronger adsorption of phenyl-functionalized molecules suggests improved electronic interaction and orbital overlap, increasing charge transfer efficiency. Similarly, smaller values are observed for heptazine when comparing triazine with heptazine. The more extensive conjugated system of heptazine lowers its LUMO energy level relative to triazine, enhancing electronic interactions with [53]. Anatase tends to show lower values compared to rutile; the electronic structure of anatase has its CBM at a higher energy level relative to rutile, reducing the energy gap with the molecule LUMO and promoting electron transfer. This trend correlates with the interaction energies, as anatase is characterized by stronger adsorption, likely due to a higher ratio of undercoordinated Ti atoms and reactive sites than rutile. Finally, considering the surface orientation, it is observed that the (110) orientation is characterized by lower values, particularly for phenyl-functionalized molecules adsorbed on anatase. Combining surface reactivity and molecular design leads to enhanced photocatalytic properties[54]. A reduced energy gap between the CBM and the LUMO facilitates efficient electron transfer from the molecule to the substrate upon photoexcitation[55,56].
helps in quantifying the potential for direct electron transfer from the HOMO of the molecule to the CBM of upon photoexcitation. This quantity provides a crucial insight into an additional charge transfer pathway, complementing the values, which primarily focus on transitions involving the LUMO. Table 4 indicates that, in almost all cases, functionalization with the phenyl group leads to an increase in . In contrast, pristine triazine and heptazine molecules generally exhibit smaller values, suggesting that, for phenyl-functionalized molecules, an indirect charge transfer from the LUMO of the molecule to the CBM of is favored. Hence, in pristine triazine or heptazine molecules, a direct transfer from the HOMO to the CBM of appears to be more likely. These two electron transfer mechanisms are schematically shown in Figure 2. The analysis of the band alignment suggests that, in both anatase and rutile, the addition of organic molecules extends absorption into the visible region. In fact, the reduction of the LUMO and HOMO difference, effectively decreases the band gap compared to that of . Furthermore, incorporating a phenyl group further reduces the HOMO-LUMO gap, resulting in a further red shift of the absorption spectrum toward the visible range. Our analysis also confirms that the staggered band alignment is achieved for both anatase and rutile, enabling efficient charge transfer from the organic molecule to .
Incorporating the phenyl group significantly reduces the value of , suggesting differing charge transfer mechanisms. In systems without the phenyl group, direct electron transfer from the HOMO of the molecule to the conduction band of is expected. In contrast, in phenyl-functionalized systems, an indirect transfer mechanism is likely, where the electron first transitions to the LUMO of the molecule after photoexcitation and subsequently transfers to the conduction band of .
3.3. Experimental Results: Raman and XRD Measurements
These theoretical predictions have been validated through a series of measurements (i) assessing the effective formation of the hybrid systems with the organic polymers on surfaces using Raman spectroscopy and XRD analysis and (ii) analyzing the kinetics behavior of the excited systems using time-resolved measurements of the hybrid systems to determine whether charge transfer occurs between the polymers and polymorphs upon photoexcitation. Furthermore, we evaluated whether the addition of the phenyl group alters the charge transfer mechanism, shifting it from a direct transfer (from the HOMO of the molecule to the conduction band of ) to an indirect mechanism, where the electron transitions to the LUMO of the molecule before transferring to the conduction band.
To synthesize in the anatase phase, a hydrothermal-assisted method using ethanol as a solvent was employed (see the experimental methods section)[12]. However, the same procedure cannot be directly applied to produce the rutile phase. The synthesis of rutile typically requires thermal treatments at elevated temperatures, often above 600 °C, to promote the transition from anatase to rutile or to directly form rutile, depending on the precursor materials. These high temperatures are incompatible with hybrid structures, as the polymer component degrades above 400–450 °C. An alternative approach involves modifying the hydrothermal method used for anatase synthesis by replacing ethanol with water and using TiCl4 as the titanium precursor[57]. In previous studies, this method was followed by additional hydrothermal treatment to achieve rutile [57,58]. However, this step leads to the oxidation and destruction of the organic component in the autoclave. To address this, we extended the duration of the initial solution-based process and omitted the autoclave step. Titanium was hydrolyzed by OH groups in water and then slowly crystallized into . The resulting larger crystallite dimensions favored the formation of the rutile phase over the anatase phase[59,60].
We realized a total of three sets of samples:
- two PhCN/ hybrids synthesized either with ethanol or water, thus in the anatase and rutile phase, respectively (PhCN/anatase, PhCN/rutile) to verify the charge transfer mechanism and assess the potential of the hybrid structure for visible, solar-driven applications
- / synthesized with water, thus in the rutile phase (/rutile) to explore the specific role of the phenyl group
To assess the effectiveness of this modified approach, we synthesized pristine anatase and rutile phases by following both methods and analyzed their crystalline form, ensuring the viability of the process before applying it to hybrid structures. The Raman spectra of polymorphs (Figure 3a and c) reveal that for the sample synthesized via the hydrothermal method, the main peaks occur at 144, 398, 518, and 640, corresponding to the Eg (144 and 398), A1g, and Eg vibrational modes of anatase crystallites, respectively (Figure 3c). Conversely, the Raman spectrum of the water-assisted-synthesized sample revealed peaks at 210 (B1g), 448 (Eg), and 613 (A1g), characteristic of the rutile phase (Figure 3a)[60,61], proving the efficacy of the alternative synthesis approach. In the case of the hybrid compounds, exciting the sample with an infrared laser (1064) allowed a clear observation of the leading bands of the two polymorphs, overcoming the interference caused by the heptazine luminescence (Figure 3b and d). This confirmed the effective adhesion of the organic molecules on surfaces.
As for the structural characterization, the XRD pattern of the hydrothermal compound (PhCN/anatase) reveals all the characteristic peaks of the anatase phase, along with a minor peak at 25°, attributed to the organic component (Figure 3g). The XRD patterns of the water synthesized samples (/rutile and PhCN/rutile) exhibit similar features, with a prominent peak at approximately 27° (Figure 3e and f), corresponding to the (001) reflection (slightly varying between 27.5° and 27.7° due to interplanar distance changes), and a broader peak at around 15°, that we attribute to the (210) reflection from the separation distance of heptazine chains[10]. These organic peaks overlap with the dominant peaks of the rutile phase, with its main peak also appearing at 27°. In the /rutile compound, the presence of a small amount of anatase is indicated by a shoulder at 25° (Figure 3e). These patterns provide clear evidence of the adhesion of the organic molecules on both phases. The detection of organic-specific peaks in conjunction with diffraction patterns highlights the integration of the two components.
3.4. Experimental Results: Absorption and Emission Spectra
Optical characterization provides valuable insights into the structural differences and interactions between the organic and inorganic parts. , in both its anatase and rutile phases, strongly absorbs light in the UV range, with a sharp increase in absorption for wavelengths shorter than 410 nm, corresponding to its bandgap ( eV for anatase and eV for rutile). In contrast, exhibits optical absorption below 450 (bandgap eV).
At the same time, PhCN extends absorption across the entire visible range due to the presence of phenyl groups in its structure (Fig. S3). This aligns with the previous theoretical predictions, where we showed that adding a phenyl group results in an overall decrease in the HOMO-LUMO gap, leading to a red shift in the absorption spectrum. The composites with organic structures show an expanded working range between 400 and 600 (Figure 4a). However, the high-energy portion of the spectra shows minimal differences between anatase and rutile phases. As anticipated above, the two hybrid systems exhibit distinct absorption mechanisms. In the PhCN/rutile system, the photoinduced electron transfer (Figure 2b) occurs, where excited electrons in the LUMO state of PhCN are transferred to the conduction band of . Conversely, the /rutile complex operates through a direct optical electron transfer (Figure 2a), where photons promote electrons directly from the HOMO ground state of to the conduction band of [1,12]. This direct transfer mechanism results in a red-shifted optical absorption compared to the simple sum of the absorption features of the individual components, regardless of the polymorph used (see Figure 4a). The observed behavior confirms our calculations, where we showed that adding a phenyl group results in an increase of (see Table 4) indicating that an indirect electron transfer mechanism in the case of PhCN/rutile is favorable, while it might follow a direct pathway in /rutile. The luminescence of the hybrid samples reveals the difference induced by the phenyl group. The excitation/emission characteristics of the photoluminescence of the five samples are reported in Figure 4b and c. While the inorganic parts do not seem to contribute significantly, the presence of the phenyl groups in the heptazine mesh generates a redshift of the emission. In carbon nitrides systems, the photoluminescence (PL) arises from recombination between and lone pairs electrons (LP), and LP, and and energy levels[48,62]. These recombinations create a broad emission spectrum centered around 530 in the PhCN (Figure 4b). In contrast, in the emission is dominated by transitions from to LP levels, generating a blue-shifted spectrum (Figure 4c) and, mainly, a strongly reduced PL efficiency due to competitive thermal recombination from the levels.
3.5. Experimental Results: Photocatalytic Efficiency
The formation of active heterostructure can be directly tested by photocatalytic properties (Figure 5a). While it was already proved that PhCN with anatase forms an efficient photocatalyst activated with visible light, the use of as the organic part in the hybrid compound with has a reduced activity if the incident light has wavelength higher than 450 [12].
In the PhCN/rutile sample, the photocatalytic activity is slightly reduced with respect to the anatase sample but still noteworthy, indicating an effective charge transfer from PhCN to rutile. On the contrary, the absence of the phenyl group in the organic part strongly reduces the connection between and rutile structure, as indicated by the lower photocatalytic activity. Time-resolved photoluminescence (TRPL) measurements help in quantifying these aspects: the information from TRPL data is connected to the probability of recombination from the excited states via the relation
where is the recombination probability from the excited state with and as the probabilities for radiative and non-radiative recombination, respectively. and are the relative time lifetime of the radiative and non-radiative paths and as the overall decay time is obtained through fitting the measured signal (see Figure 5b and c), therefore a decrease of the experimental luminescence decays is a clear indication of the formation of non-radiative pathways. In Table 5, we report the weighted average of the time decay constant, together with the efficiency of the non-radiative charge transfer mechanism referred to PhCN according to
While it seems that there is almost no charge transfer from the (which aligns with the low absorption in the visible range of the polymer), the charge transfer efficiency for the PhCN hybrids is about 40% for both the phases. The increased efficiency in the anatase-based hybrid system is most probably related to the more active role of the anatase with respect to the rutile in the photocatalytic process. However, the high efficiency of photocatalysis for the rutile heterostructure opens new possibilities for environmentally friendly and cost-efficient solutions.
4. Conclusions
In this study, we investigated the photocatalytic properties of phenyl-modified carbon nitride in combination with the rutile phase of with a combined computational and experimental approach. DFTB calculations proved the effective interaction of PhCN with rutile, favorable band alignment, and enhanced charge transfer efficiency. These results indicate efficient charge separation and reduced recombination, supporting its potential for photocatalytic applications. Furthermore, phenyl functionalization improves visible light absorption by reducing the energy gap between the HOMO and the lowest unoccupied molecular orbital LUMO. Raman spectroscopy, XRD, and photocatalytic degradation measurements confirmed the computational predictions. These analyses showed the successful integration of PhCN onto both anatase and rutile , demonstrating effective charge transfer interactions. Photoluminescence and time-resolved spectroscopy revealed approximately 40% charge transfer efficiency in both phases, aligning with computational findings. Additionally, photocatalytic experiments confirmed significant activity under visible light, supporting the practical viability of PhCN/rutile as a green and sustainable photocatalyst, emphasizing its role as an effective alternative to anatase-based systems.
Author Contributions
Conceptualization, L.C., C.M. and P.C.R.; methodology, R.D. S.A.G., and S.P.; validation, R.D. S.A.G., and S.P.; formal analysis, R.D., S.P.; investigation, R.D. S.A.G., and S.P.; writing—original draft preparation, R.D., C.M. and S.P.; writing—review and editing, R.D., C.M. and L.C.; visualization, X.X.; supervision, L.C., C.M. and P.C.R.; project administration, P.C.R.; funding acquisition, L.C., C.M. and P.C.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by “e.INS - Ecosystem of Innovation for Next Generation Sardinia" – Project code ECS00000038, SPOKE 7 "Low carbon technologies for efficient energy systems", under the National Recovery and Resilience Plan (NRRP), Ministero dell’Università e della Ricerca (MUR); funded by the European Union—NextGenerationEU.
Data Availability Statement
The simulation cells for all the structures investigated in this paper and the experimental data are available at https://github.com/rdettori/PhCN_TiO2.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bledowski, M.; Wang, L.; Ramakrishnan, A.; Khavryuchenko, O.V.; Khavryuchenko, V.D.; Ricci, P.C.; Strunk, J.; Cremer, T.; Kolbeck, C.; Beranek, R. Visible-light photocurrent response of TiO2–polyheptazine hybrids: evidence for interfacial charge-transfer absorption. Physical Chemistry Chemical Physics 2011, 13, 21511. [Google Scholar] [CrossRef]
- Basivi, P.K.; Selvaraj, Y.; Perumal, S.; Bojarajan, A.K.; Lin, X.; Girirajan, M.; Kim, C.W.; Sangaraju, S. Graphitic carbon nitride (g–C3N4)–Based Z-scheme photocatalysts: Innovations for energy and environmental applications. Materials Today Sustainability 2025, 29, 101069. [Google Scholar] [CrossRef]
- Zhao, Z.; Sun, Y.; Dong, F. Graphitic carbon nitride based nanocomposites: a review. Nanoscale 2015, 7, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.J.; Tan, L.L.; Ng, Y.H.; Yong, S.T.; Chai, S.P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability? Chemical Reviews 2016, 116, 7159–7329. [Google Scholar] [CrossRef]
- Pei, J.; Li, H.; Yu, D.; Zhang, D. g-C3N4-Based Heterojunction for Enhanced Photocatalytic Performance: A Review of Fabrications, Applications, and Perspectives. Catalysts 2024, 14, 825. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Antonietti, M. Polymeric Graphitic Carbon Nitride as a Heterogeneous Organocatalyst: From Photochemistry to Multipurpose Catalysis to Sustainable Chemistry. Angewandte Chemie International Edition 2011, 51, 68–89. [Google Scholar] [CrossRef]
- Porcu, S.; Secci, F.; Ricci, P.C. Advances in Hybrid Composites for Photocatalytic Applications: A Review. Molecules 2022, 27, 6828. [Google Scholar] [CrossRef]
- Yu, J.; Wang, S.; Low, J.; Xiao, W. Enhanced photocatalytic performance of direct Z-scheme g-C3N4–TiO2 photocatalysts for the decomposition of formaldehyde in air. Physical Chemistry Chemical Physics 2013, 15, 16883. [Google Scholar] [CrossRef]
- Li, Y.; Jin, R.; Li, G.; Liu, X.; Yu, M.; Xing, Y.; Shi, Z. Preparation of phenyl group functionalized g-C3N4 nanosheets with extended electron delocalization for enhanced visible-light photocatalytic activity. New Journal of Chemistry 2018, 42, 6756–6762. [Google Scholar] [CrossRef]
- Porcu, S.; Roppolo, I.; Salaun, M.; Sarais, G.; Barbarossa, S.; Casula, M.F.; Carbonaro, C.M.; Ricci, P.C. Come to light: Detailed analysis of thermally treated Phenyl modified Carbon Nitride Polymorphs for bright phosphors in lighting applications. Applied Surface Science 2020, 504, 144330. [Google Scholar] [CrossRef]
- Chen, T.; Chen, C.; Liu, Q.; Zhang, Z.; Fang, X. A one-step process for preparing a phenyl-modified g-C3N4 green phosphor with a high quantum yield. RSC Advances 2017, 7, 51702–51710. [Google Scholar] [CrossRef]
- Porcu, S.; Castellino, M.; Roppolo, I.; Carbonaro, C.M.; Palmas, S.; Mais, L.; Casula, M.F.; Neretina, S.; Hughes, R.A.; Secci, F.; et al. Highly efficient visible light phenyl modified carbon nitride/TiO2 photocatalyst for environmental applications. Applied Surface Science 2020, 531, 147394. [Google Scholar] [CrossRef]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, 2000. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Green Chemistry | US EPA — epa.gov. https://www.epa.gov/greenchemistry.
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Physical Review B 1998, 58, 7260–7268. [Google Scholar] [CrossRef]
- Aradi, B.; Hourahine, B.; Frauenheim, T. DFTB+, a sparse matrix-based implementation of the DFTB method. J. Phys. Chem. A 2007, 111, 5678–5684. [Google Scholar] [CrossRef]
- Gaus, M.; Cui, Q.; Elstner, M. DFTB3: Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB). J. Chem. Theory Comput. 2011, 7, 931. [Google Scholar] [CrossRef] [PubMed]
- Kranz, J.J.; Kubillus, M.; Ramakrishnan, R.; von Lilienfeld, O.A.; Elstner, M. Generalized density-functional tight-binding repulsive potentials from unsupervised machine learning. J. Chem. Theory Comput. 2018, 14, 2341–2352. [Google Scholar] [CrossRef]
- Manaa, M.R.; Fried, L.E.; Melius, C.F.; Elstner, M.; Frauenheim, T. Decomposition of HMX at Extreme Conditions: A Molecular Dynamics Simulation. J. Phys. Chem. A 2002, 106, 9024. [Google Scholar] [CrossRef]
- Goyal, P.; Qian, H.J.; Irle, S.; Lu, X.; Roston, D.; Mori, T.; Elstner, M.; Cui, Q. Molecular Simulation of Water and Hydration Effects in Different Environments: Challenges and Developments for DFTB Based Models. The Journal of Physical Chemistry B 2014, 118, 11007–11027. [Google Scholar] [CrossRef]
- Goldman, N.; Fried, L.E.; Lindsey, R.K.; Pham, C.H.; Dettori, R. Enhancing the accuracy of density functional tight binding models through ChIMES many-body interaction potentials. The Journal of Chemical Physics 2023, 158. [Google Scholar] [CrossRef]
- Dettori, R.; Goldman, N. Creation of an Fe3P Schreibersite Density Functional Tight Binding Model for Astrobiological Simulations. The Journal of Physical Chemistry A 2025, 129, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Goldman, N.; Srinivasan, S.G.; Hamel, S.; Fried, L.E.; Gaus, M.; Elstner, M. Determination of a density functional tight binding model with an extended basis set and three-body repulsion for carbon under extreme pressures and temperatures. J. Phys. Chem. C 2013, 117, 7885–7894. [Google Scholar] [CrossRef]
- Srinivasan, S.G.; Goldman, N.; Tamblyn, I.; Hamel, S.; Gaus, M. Determination of a density functional tight binding model with an extended basis set and three-body repulsion for hydrogen under extreme thermodynamic conditions. J. Phys. Chem. A 2014, 118, 5520–5528. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. The Journal of Chemical Physics 2010, 132, 154104. [Google Scholar] [CrossRef]
- Niehaus, T.; Elstner, M.; Frauenheim, T.; Suhai, S. Application of an approximate density-functional method to sulfur containing compounds. Journal of Molecular Structure: THEOCHEM 2001, 541, 185–194. [Google Scholar] [CrossRef]
- Dolgonos, G.; Aradi, B.; Moreira, N.H.; Frauenheim, T. An Improved Self-Consistent-Charge Density-Functional Tight-Binding (SCC-DFTB) Set of Parameters for Simulation of Bulk and Molecular Systems Involving Titanium. Journal of Chemical Theory and Computation 2009, 6, 266–278. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Lazzeri, M.; Vittadini, A.; Selloni, A. Structure and energetics of stoichiometric TiO2 anatase surfaces. Physical Review B 2001, 63. [Google Scholar] [CrossRef]
- Gong, X.Q.; Selloni, A.; Batzill, M.; Diebold, U. Steps on anatase TiO2(101). Nature Materials 2006, 5, 665–670. [Google Scholar] [CrossRef]
- Howard, C.J.; Sabine, T.M.; Dickson, F. Structural and thermal parameters for rutile and anatase. Acta Crystallographica Section B Structural Science 1991, 47, 462–468. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, P.; Liu, J.; Yu, J. New understanding of the difference of photocatalytic activity among anatase, rutile and brookite TiO2. Physical Chemistry Chemical Physics 2014, 16, 20382–20386. [Google Scholar] [CrossRef]
- Martsinovich, N.; Troisi, A. How TiO2 crystallographic surfaces influence charge injection rates from a chemisorbed dye sensitiser. Physical Chemistry Chemical Physics 2012, 14, 13392. [Google Scholar] [CrossRef]
- Perron, H.; Domain, C.; Roques, J.; Drot, R.; Simoni, E.; Catalette, H. Optimisation of accurate rutile TiO2 (110), (100), (101) and (001) surface models from periodic DFT calculations. Theoretical Chemistry Accounts 2007, 117, 565–574. [Google Scholar] [CrossRef]
- Dong, F.; Zhao, Z.; Xiong, T.; Ni, Z.; Zhang, W.; Sun, Y.; Ho, W.K. In situ construction of g-C3N4/g-C3N4 metal-free heterojunction for enhanced visible-light photocatalysis. ACS Appl. Mater. Interfaces 2013, 5, 11392–11401. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J.M.; Domen, K.; Antonietti, M. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 2009, 8, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.; Chen, W.; Li, Y.; Zhao, M.; Shi, J.; Tang, Y.; Dai, X. Theoretical exploration of the structural, electronic and optical properties of g-C3N4/C3N heterostructures. Physical Chemistry Chemical Physics 2023, 25, 4081–4092. [Google Scholar] [CrossRef] [PubMed]
- Merschjann, C.; Tyborski, T.; Orthmann, S.; Yang, F.; Schwarzburg, K.; Lublow, M.; Lux-Steiner, M.C.; Schedel-Niedrig, T. Photophysics of polymeric carbon nitride: An optical quasimonomer. Physical Review B 2013, 87. [Google Scholar] [CrossRef]
- Cui, Q.; Xu, J.; Wang, X.; Li, L.; Antonietti, M.; Shalom, M. Phenyl-Modified Carbon Nitride Quantum Dots with Distinct Photoluminescence Behavior. Angewandte Chemie International Edition 2016, 55, 3672–3676. [Google Scholar] [CrossRef]
- Kowalkińska, M.; Dudziak, S.; Karczewski, J.; Ryl, J.; Trykowski, G.; Zielińska-Jurek, A. Facet effect of TiO2 nanostructures from TiOF2 and their photocatalytic activity. Chemical Engineering Journal 2021, 404, 126493. [Google Scholar] [CrossRef]
- Diebold, U. The surface science of titanium dioxide. Surface Science Reports 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Maeda, K.; Domen, K. New Non-Oxide Photocatalysts Designed for Overall Water Splitting under Visible Light. The Journal of Physical Chemistry C 2007, 111, 7851–7861. [Google Scholar] [CrossRef]
- Kamat, P.V. Meeting the Clean Energy Demand: Nanostructure Architectures for Solar Energy Conversion. The Journal of Physical Chemistry C 2007, 111, 2834–2860. [Google Scholar] [CrossRef]
- FUJISHIMA, A.; ZHANG, X.; TRYK, D. TiO2 photocatalysis and related surface phenomena. Surface Science Reports 2008, 63, 515–582. [Google Scholar] [CrossRef]
- Tachibana, Y.; Vayssieres, L.; Durrant, J.R. Artificial photosynthesis for solar water-splitting. Nature Photonics 2012, 6, 511–518. [Google Scholar] [CrossRef]
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Martin, D.J.; Reardon, P.J.T.; Moniz, S.J.A.; Tang, J. Visible Light-Driven Pure Water Splitting by a Nature-Inspired Organic Semiconductor-Based System. Journal of the American Chemical Society 2014, 136, 12568–12571. [Google Scholar] [CrossRef]
- Wategaonkar, S.; Pawar, R.; Parale, V.; Nade, D.; Sargar, B.; Mane, R. Synthesis of rutile TiO2 nanostructures by single step hydrothermal route and its characterization. Materials Today: Proceedings 2020, 23, 444–451. [Google Scholar] [CrossRef]
- Beyer, J.; Mamakhel, A.; Søndergaard-Pedersen, F.; Yu, J.; Iversen, B.B. Continuous flow hydrothermal synthesis of phase pure rutile TiO2 nanoparticles with a rod-like morphology. Nanoscale 2020, 12, 2695–2702. [Google Scholar] [CrossRef]
- Zhu, S.C.; Xie, S.H.; Liu, Z.P. Nature of Rutile Nuclei in Anatase-to-Rutile Phase Transition. Journal of the American Chemical Society 2015, 137, 11532–11539. [Google Scholar] [CrossRef]
- Ricci, P.C.; Carbonaro, C.M.; Stagi, L.; Salis, M.; Casu, A.; Enzo, S.; Delogu, F. Anatase-to-Rutile Phase Transition in TiO2 Nanoparticles Irradiated by Visible Light. The Journal of Physical Chemistry C 2013, 117, 7850–7857. [Google Scholar] [CrossRef]
- Balachandran, U.; Eror, N. Raman spectra of titanium dioxide. Journal of Solid State Chemistry 1982, 42, 276–282. [Google Scholar] [CrossRef]
- Zhang, Y.; Pan, Q.; Chai, G.; Liang, M.; Dong, G.; Zhang, Q.; Qiu, J. Synthesis and luminescence mechanism of multicolor-emitting g-C3N4 nanopowders by low temperature thermal condensation of melamine. Scientific Reports 2013, 3. [Google Scholar] [CrossRef]
Figure 1.
(a) Top of the heptazine molecule on the (100) anatase slab, (b) top view of Ph-heptazine on the same facet.
Figure 1.
(a) Top of the heptazine molecule on the (100) anatase slab, (b) top view of Ph-heptazine on the same facet.
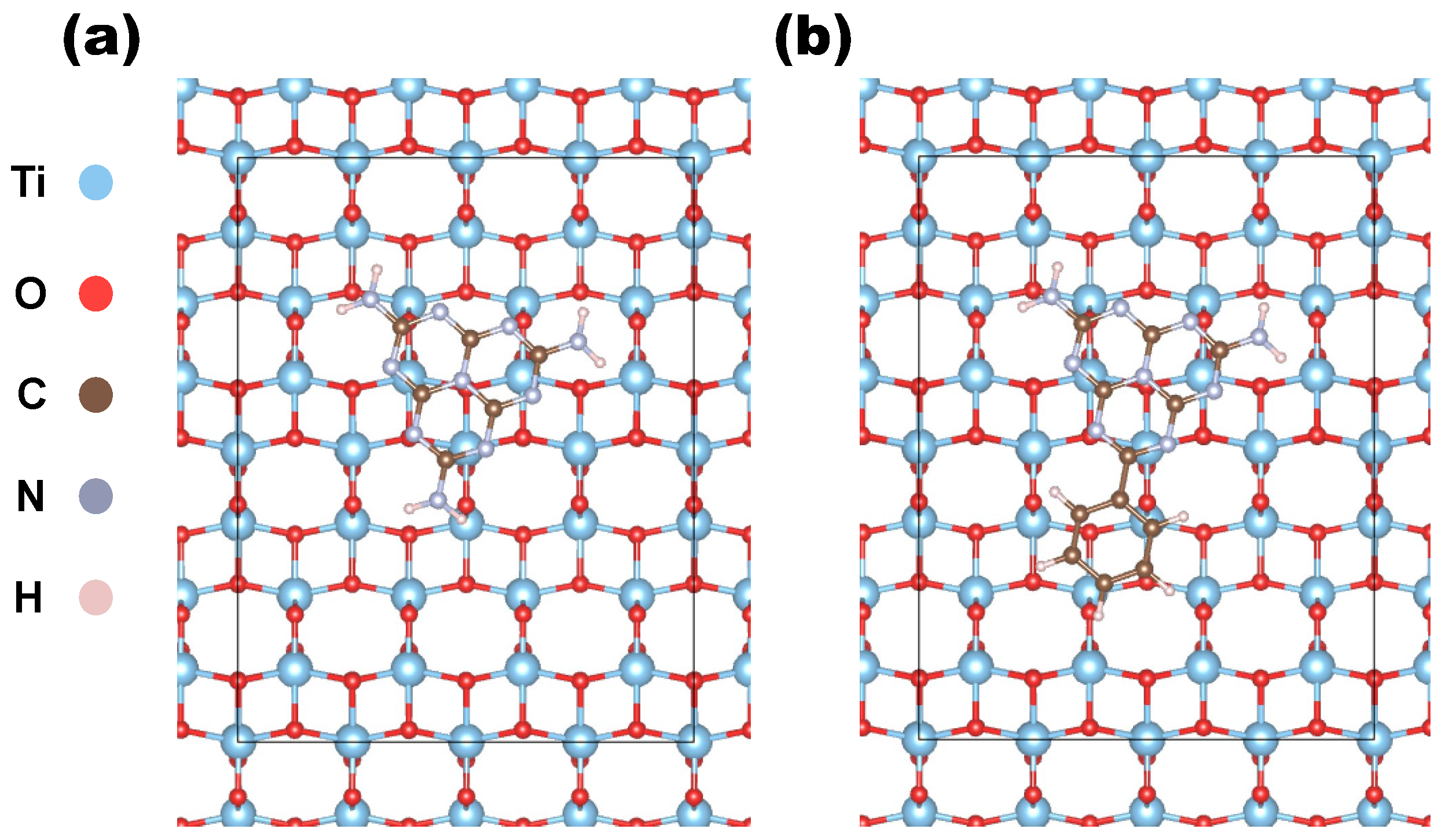
Figure 2.
Schematic representation of the band alignment along with the proposed electron transfer mechanism for (a) triazine on (100)-anatase (b) Ph-triazine on the same substrate.
Figure 2.
Schematic representation of the band alignment along with the proposed electron transfer mechanism for (a) triazine on (100)-anatase (b) Ph-triazine on the same substrate.
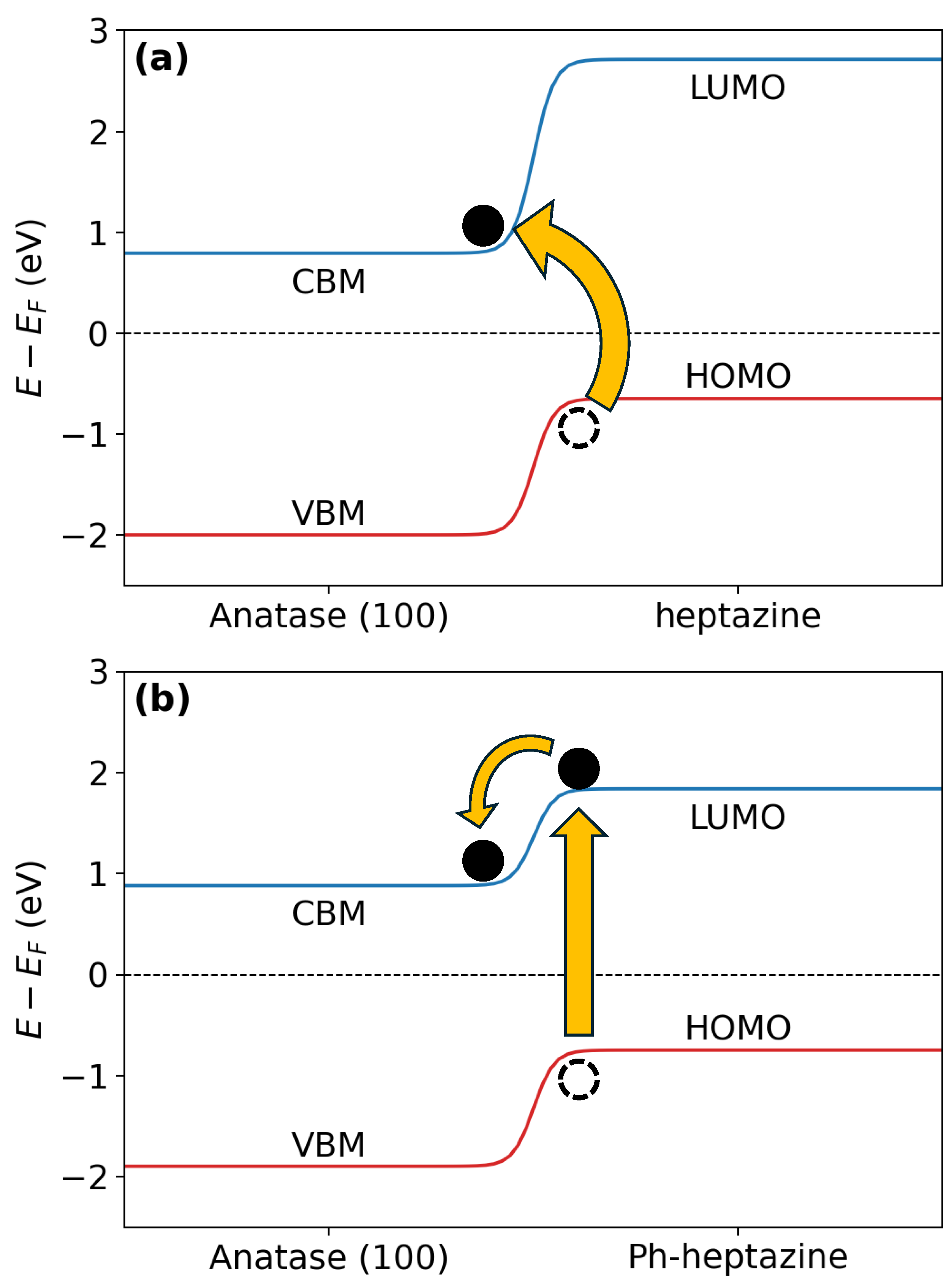
Figure 3.
Raman spectra of (a) rutile, (b) PhCN, (c) anatase and (d) . XRD patterns for (e) /rutile, (f) PhCN/rutile, and (g) PhCN/anatase.
Figure 3.
Raman spectra of (a) rutile, (b) PhCN, (c) anatase and (d) . XRD patterns for (e) /rutile, (f) PhCN/rutile, and (g) PhCN/anatase.
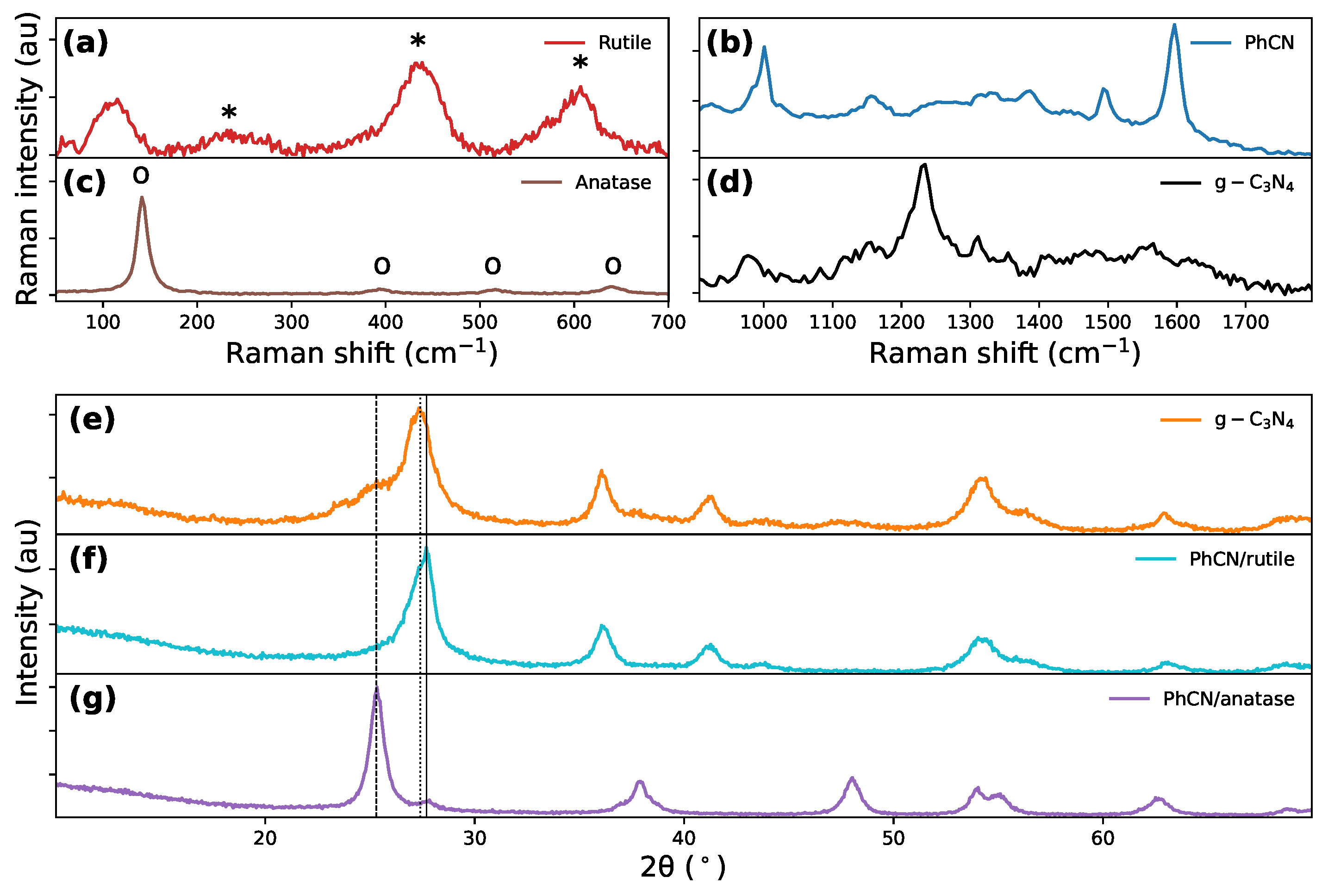
Figure 4.
(a) Absorption spectra of -PhCN hybrid systems. Emission spectra comparison for (b) PhCN, PhCN/rutile, PhCN/anatase, and (c) and /rutile.
Figure 4.
(a) Absorption spectra of -PhCN hybrid systems. Emission spectra comparison for (b) PhCN, PhCN/rutile, PhCN/anatase, and (c) and /rutile.
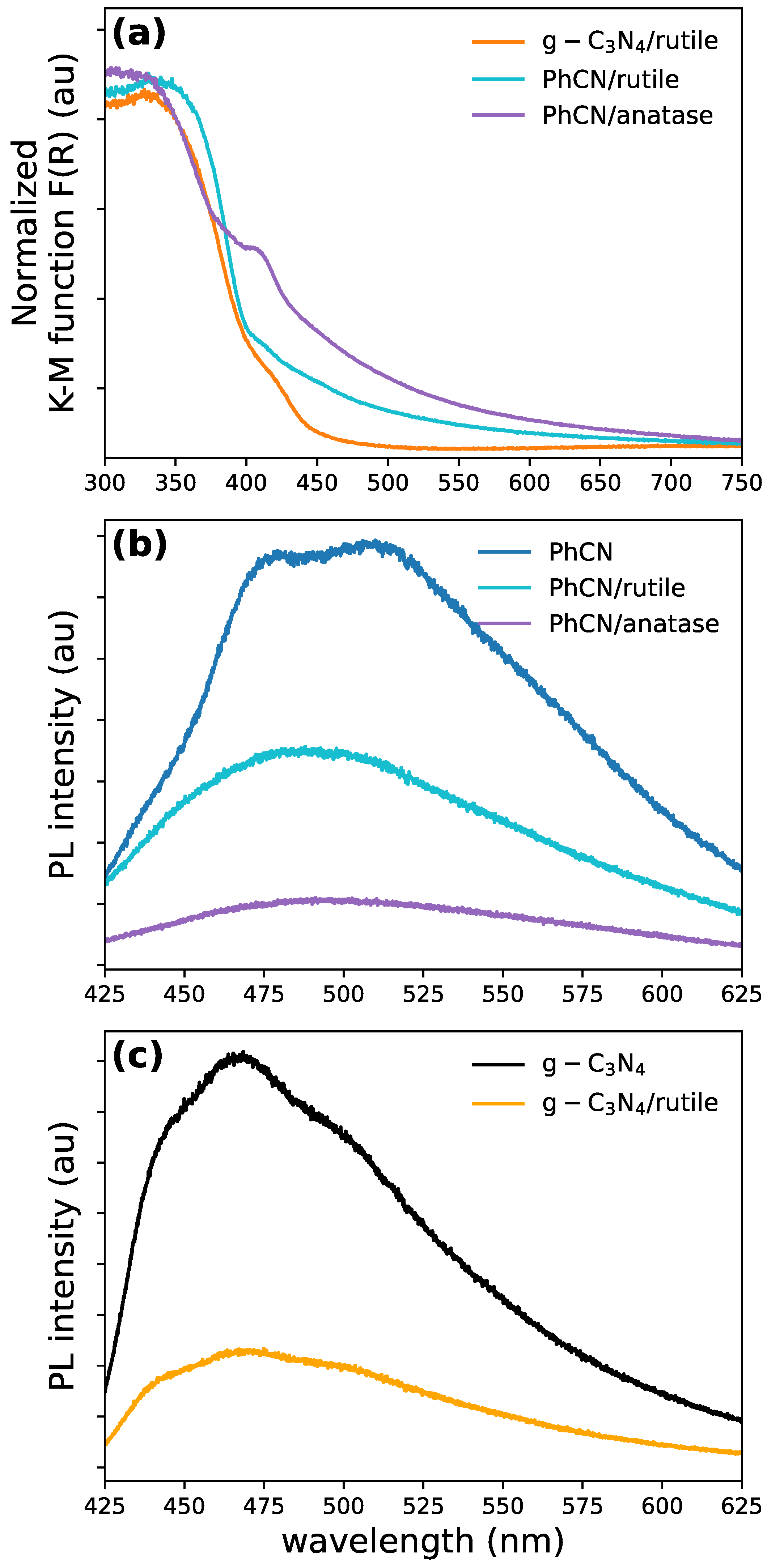
Figure 5.
(a) Photocatalytic efficiency for RhB degradation. PL decay profile for (a) PhCN and PhCN hybrids, (b) and /rutile structure.
Figure 5.
(a) Photocatalytic efficiency for RhB degradation. PL decay profile for (a) PhCN and PhCN hybrids, (b) and /rutile structure.
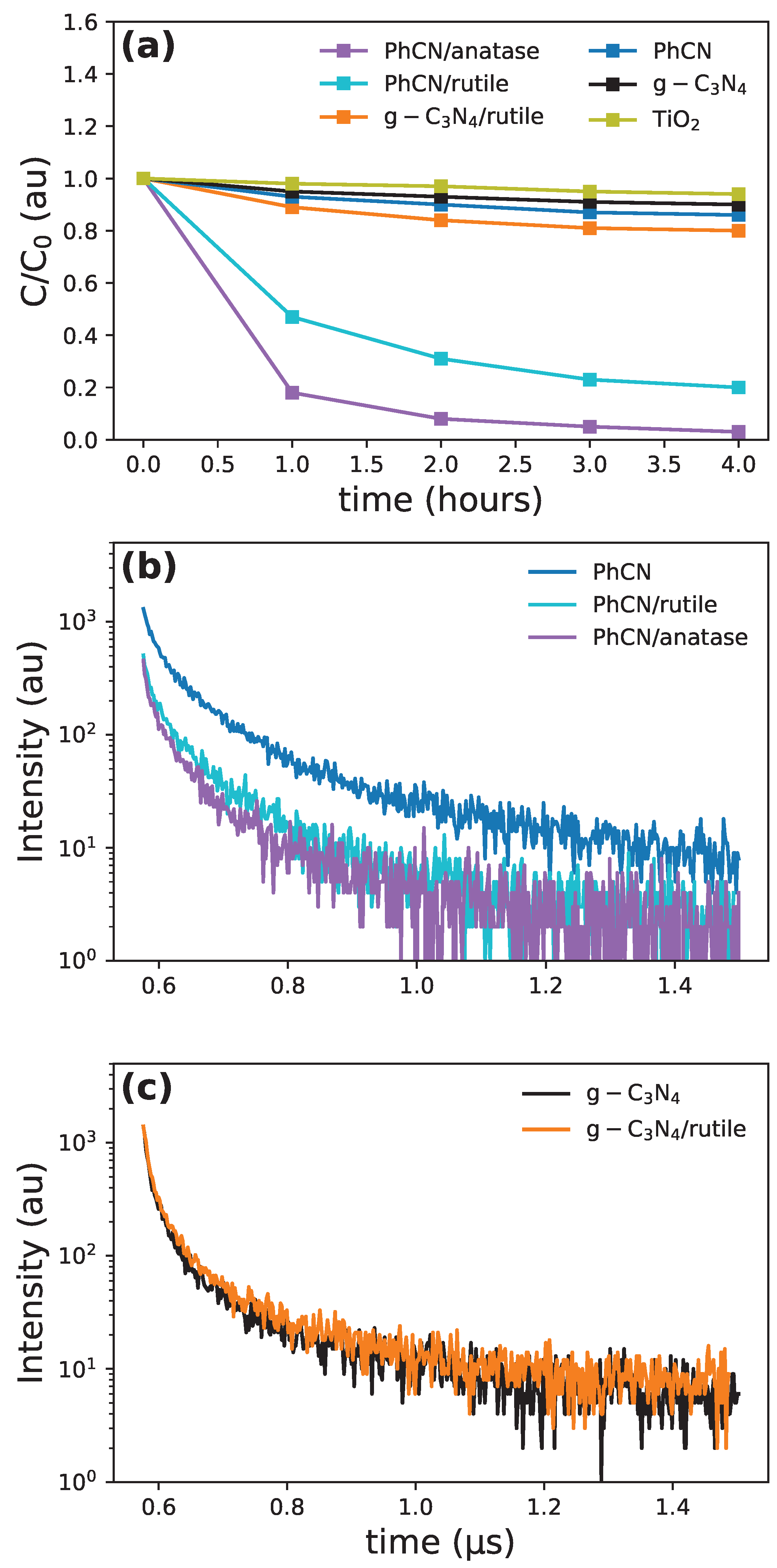

Table 1.
Cell parameters of bulk rutile and anatase with both DFT and DFTB methods. The third column represents the relative deviation of the DFTB value compared to the full ab initio one.
Table 1.
Cell parameters of bulk rutile and anatase with both DFT and DFTB methods. The third column represents the relative deviation of the DFTB value compared to the full ab initio one.
| DFT (Å) | DFTB (Å) | |||
| rutile | a | 4.648 | 4.619 | 0.63% |
| c | 2.971 | 2.991 | -0.68% | |
| anatase | a | 3.805 | 3.758 | 1.26% |
| c | 9.747 | 9.605 | 1.45% |
Table 2.
DFTB calculated formation energies for (100) and (110) facets of both anatase and rutile. The comparison is made with the range of values available in the literature, although they were obtained with different functionals and pseudopotentials.
Table 2.
DFTB calculated formation energies for (100) and (110) facets of both anatase and rutile. The comparison is made with the range of values available in the literature, although they were obtained with different functionals and pseudopotentials.
| anatase (J/ m 2) | rutile (J/ m 2) | ||
| DFTB | 100 | 0.85 | 1.09 |
| 110 | 1.39 | 0.88 | |
| Different XC[42] | 100 | 0.53-0.90 | 0.67-0.77 |
| PBE-D4+U[43] | 110 | 0.95-1.32 | 0.48-0.54 |
Table 3.
Absorption energies for the molecules here investigated.
| (eV) | triazine | Ph-triazine | heptazine | Ph-heptazine | |
| anatase | 100 | -0.59 | -0.76 | -0.64 | -0.63 |
| 110 | -0.82 | -0.99 | -0.96 | -1.21 | |
| rutile | 100 | -0.27 | -0.18 | -0.50 | -0.42 |
| 110 | -0.54 | -0.86 | -0.58 | -0.95 |
Table 4.
Energy differences between the electronic levels of the molecule and the substrate for all the cases investigated in this work. All energies are reported in eV. A complete collection of the energy levels for the systems investigated here is reported in Tab.SIII
Table 4.
Energy differences between the electronic levels of the molecule and the substrate for all the cases investigated in this work. All energies are reported in eV. A complete collection of the energy levels for the systems investigated here is reported in Tab.SIII
| Substrate | Molecule | |||
| 100 | Anatase | triazine | 2.34 | 1.89 |
| ph-triazine | 1.05 | 2.16 | ||
| Rutile | triazine | 4.34 | 0.75 | |
| ph-triazine | 2.36 | 1.07 | ||
| 110 | Anatase | triazine | 2.99 | 1.66 |
| ph-triazine | 1.11 | 2.23 | ||
| Rutile | triazine | 2.99 | 1.66 | |
| ph-triazine | 1.60 | 1.23 | ||
| 100 | Anatase | heptazine | 1.92 | 1.44 |
| ph-heptazine | 0.96 | 1.63 | ||
| Rutile | heptazine | 2.94 | 0.57 | |
| ph-heptazine | 1.81 | 0.76 | ||
| 110 | Anatase | heptazine | 1.62 | 1.68 |
| ph-heptazine | 0.61 | 1.87 | ||
| Rutile | heptazine | 2.90 | 0.60 | |
| ph-heptazine | 1.36 | 0.97 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



