
Submitted:
17 February 2025
Posted:
17 February 2025
You are already at the latest version
Abstract
The rapid expansion of pomegranate (Punica granatum L.) cultivation in central and southern Italy has revealed emerging phytosanitary challenges, including "pomegranate wood decay syndrome”, characterised by cortical cankers, wood browning, and progressive plant decline. This study investigates the fungal microbiota associated with symptomatic pomegranate wood using a combined approach of traditional fungal isolation and ITS2 metabarcoding analysis. Samples from two orchards in Lazio were examined, revealing a complex fungal community with a high prevalence of Neofusicoccum parvum and species of the Diaporthe complex. Pathogenicity tests confirmed the role of N. parvum in causing significant wood browning, while other isolates showed variable virulence. Statistical analyses validated the pathogenicity of select isolates, with Diaporthe eres (Nitschke) consistently demonstrating potential pathogenic activity across all trials. Metabarcoding identified 289 taxa highlighting a richer fungal diversity in symptomatic wood compared to asymptomatic sections. Notably, Coniella granati, previously implicated in pomegranate decline, was absent in the studied orchards. The findings reveal that pomegranate wood decay is a complex syndrome driven by fungal pathogens and environmental stressors, such as low temperatures. This study highlights the value of integrative approaches for understanding and managing fungal-associated wood diseases in pomegranate orchards.
Keywords:
wood decay
; Diaporthe
; Pathogenicity test
; Metabarcoding
; ITS 2
1. Introduction
The interest in the cultivation of pomegranate (Punica granatum L.) in Italy has led the research to increase the knowledge about this crop, whose fruit is considered a functional product of great benefit from the organoleptic and nutraceutical point of view as well as for the possibility of pharmaceutical applications given the content of active molecules that reduce the risk of vascular diseases, coronary artery problems and canker mortality [1,2]. Among the bioactive compounds present in pomegranate juice, there are phenols and tannins such as punicalin, punicalagin, and ellagic acid; moreover, the varieties with red arils contain a large number of anthocyanins including cyanidin, delphinidin, pelargonidin, and the related glycosides with a strong antioxidant capacity [3,4,5].
Some young pomegranate plants in central and southern Italy, the major cultivated areas together with the islands, have experienced phytosanitary problems that, in some cases, caused the loss of fruit production and plants [6]. This disease, called “Pomegranate canker” or, more generically “Pomegranate decay”, shows progressively worsening conditions of the plant: the first symptoms are leaf chlorosis, and later, the plant shows the formation of cankers on the stem and branches with wood browning below the symptoms. The main fungi isolated from the symptomatic portions of pomegranate wood were: Coniella granati, already been reported in Italy, Iran, Israel, Turkey, Greece and Spain [6,7,8,9,10,11] and N. parvum (Pennycook & Samuels) Crous, Slippers and Phillips, already reported in Greece [12], in Florida [13] and in Italy where it was recently reported in an orchard in Lazio [14]. N. parvum was also reported on other plants of agricultural interest including the grapevine, kiwifruit, and avocado [15,16,17]. This fungus enters through wounds (e.g., pruning cuts or mechanical damage due to abiotic agents such as frost or hail). The infection causes browning of the bark on the trunk and branches and the discolouration of the wood underlying the affected areas, resulting in the general decay of the plant and subsequent die-off of the shoots. The fungus often causes significant cankers that can affect the whole plant [14].
The wood decay often represents a disease complex due to the characteristic mixed infections [18] and the simultaneous presence of several fungal species complicates the diagnosis. The traditional phytosanitary approach requires an initial culturing step, which excludes the detection of the biotrophic, endophytic species and may limit the detection of slow-growing fungi. As trunk pathogens cause mixed infections, it is important to characterize the composition of the species complex. In the last years, due to the growing power and reducing cost of the next generation sequencing technologies, the DNA metabarcoding analysis has been used extensively for the characterization of microbial communities [19,20,21,22,23]. The internal transcribed spacer (ITS) regions of ribosomal DNA (rDNA) are the most predominant DNA barcode sequences used for fungal metabarcoding since they can be easily amplified and sequenced and they are highly represented in GenBank and other databases [24,25]. The Illumina MiSeq NGS technology is the most used in metabarcoding studies [26], due to its high diversity coverage, high accuracy, low cost and availability of several consolidated data processing pipelines; however, due to the limited sequence lengths, it allows the sequencing of one of the ITS subregions. The choice of using either ITS1 or ITS2 for the best taxonomic resolution is still debated [27,28]. ITS2 shows some advantageous features: it is less variable in length, has no intron regions, is better represented in public databases, and has a longer portion of the sequence providing taxonomic information [29]. This work aims to study and characterise the fungal flora present in diseased pomegranate wood through traditional phytosanitary analysis combined with an ITS metabarcoding analysis and to verify the disease involvement of the more frequently isolated fungi through a pathogenicity test. To the best of our knowledge, this is the first study combining culturing and DNA-metabarcoding approaches to assess fungal pathogen diversity associated with pomegranate wood decay.
2. Materials and Methods
2.1. Collection of Samples
Two pomegranate Italian orchards, both in the Lazio region, were monitored: Sermoneta (41.556459, 12.979830) and Pavona (41.742874, 12.603806). The soil of the Sermoneta orchard was classified predominantly as Haplic Luvisols (>75%), with Luvic Phaeozems (10–25%), while the Pavona orchard soil was classified as Haplic Phaeozems (25–50%), Luvic Phaeozems (10–25%), and Cambic Endoleptic Phaeozems (<10%) [30]. Furthermore, the Lazio region experiences colder winter climatic conditions than southern Italy, where pomegranate cultivation is more successful [31]. Fifteen symptomatic plants obtained from both the orchards from 2017 to 2019 were transferred to the laboratory of the Research Centre for Plant Protection and Certification (CREA-DC, Roma) and analysed by isolation and morphological and molecular identification of the fungal flora. Two plants, showing the symptoms of pomegranate decay (identified as Plant 1 and Plant 2, Figure 1), from the Pavona site (2018) were analysed by conventional approach (isolation and identification) and by the metabarcoding NGS tool. Plant 1 presented some cankers on the stem and some branches, subcortical browning of the wood, and yellowing; Plant 2 showed an extended canker on the whole trunk, with extended subcortical browning (Figure 1).
The plants (stem and branches) were divided into sections (Figure 2) representative of different asymptomatic and symptomatic wood tissues, from the trunks and the branches.
2.2. Fungal Isolation and Identification
Longitudinal and transversal sections of tissues sampled as described above were obtained aseptically; small portions between the healthy and symptomatic tissue were explanted from the browned wood and placed on the Potato Dextrose Agar (PDA) culture medium amended with 100 µg/mL of streptomycin and ampicillin sodium salt antibiotic and incubated at 22°C until the development of the fungal colonies. The fungal identification was carried out based on the morphological features of the colonies and of the fruiting structures. The morphological identification was confirmed by the sequencing of the ITS region amplified with ITS4/ITS5 primers [32] performed at BioFab Research laboratory (Rome, Italy). The sequences were blasted in the GenBank database by using the nBLAST search. The isolated and identified species were preserved in the fungal collection of CREA-DC (Rome).
2.3. DNA Extraction
The DNA from the fungal mycelium was extracted with the rapid protocol of Cenis [33] starting from 100 mg of mycelium modified for plate-grown mycelium [34]. Pomegranate wood was ground in liquid nitrogen; an aliquot of ground wood (about 100 mg) was used for DNA extraction performed with the DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. The quantity and quality of purified DNA extracts were determined using a Qubit 2.0 fluorometer (Life Technologies, Carlsbad, CA, USA) and DS-11 FX spectrophotometer (DeNovix Inc., Wilmington, DE, USA) respectively.
2.4. Metabarcoding Analysis
A metabarcoding analysis was carried out on Plant 1 and Plant 2 from the Pavona pomegranate orchard to confirm the data derived from the isolation and identification of the fungal flora, and to have more information about the fungal communities in the asymptomatic and symptomatic wood tissues. The different samples were combined in 8 experimental units (Table 1), 7 from Plant 1 and 1 from Plant 2.
DNA samples were amplified by PCR targeting the ITS2 region of the fungal rDNA using ITS3 and ITS4 primers [32]. These primers were modified by the 5′ end Illumina overhang adapter sequences (34 and 35 nucleotides) and by different 5′-end identifier (index) sequences of 6 nucleotides generating 8 oligo pairs of unique tags (Table S1). PCR was conducted for the 8 samples with each of the 8 newly designed primer pairs to test for variability in amplification success. Each sample was assigned a different forward/reverse index combination for sample-specific labelling..
The amplification reaction was performed following the protocol proposed by Miller, et al. [35]. The reaction mix contained 10 ng template DNA, 1× BioTaq PCR NH4-based reaction buffer, 4 mM MgCl2, 0.8 mM dNTP mix (Bioline, London, UK), 0.8 mg/mL bovine serum albumin (BSA, Invitrogen, Carlsbad, CA, USA) 2.5 U BioTaqTM DNA polymerase (Bioline, London, UK), and 0.5 μM of each forward and reverse primers in a 25 μL reaction volume. For each sample, three PCRs were carried out at three different annealing temperatures:combining PCR products using a range of annealing temperatures reduces primer bond distortion and decreases the stochasticity of individual PCRs [19]. The following cycling conditions were used for the amplification: initial denaturation at 94°C for 30 sec, followed by 30 cycles of denaturation at 94°C for 1 min, annealing at 50/53/56°C for 1 min, extension at 72°C for 30 sec, and final extension at 72°C for 2 min. The PCR products were visualised in 1% agarose gel electrophoresis using GelRed™ dye (Biotium, Hayward, CA, USA). Purification of the amplicons was performed using the Isolate II PCR and Gel Kit (Bioline, London, UK). The cleaned PCR products were then quantified using a Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and mixed in equimolar amounts (75 ng for each sample). A single sample, consisting of the pooled amplification products was sent to the BioFab Research laboratory (Rome, Italy). The sequencing was performed on the Illumina platform (Miseq) with a 2×300 paired-ends run and a reading depth of 100000 reads. The reads forward and reverse have been merged and filtered by quality (Q> 30). Subsequently, a demultiplexing of the sequenced sample was carried out obtaining the results of the 8 separate experimental units. Bioinformatic processing of the sequences was conducted using the USEARCH pipeline and UPARSE-OTU algorithm [36].
The paired-end (PE) sequences were merged using the -fastq_mergepairs command. Next, PE reads were quality-filtered (maximum e-value: 1.0), trimmed 340 bp, dereplicated, and sorted by abundance (singletons removed). Chimaera detection and OTU (operational taxonomic unit) clustering at 97% sequence identity followed. The original sequences were then mapped to OTUs at the 97% identity threshold, generating separate OTU tables for prokaryotic and fungal communities.
The taxonomic affiliation of each OTU was assigned using the -syntax algorithm against the UNITE_all_eukaria database [37] with an 80% confidence threshold. Sequencing depth was normalized across libraries, and the resulting OTU tables were used for downstream analyses.
2.5. Pathogenicity Test
Artificial inoculations were performed using the 5 fungal species isolated with higher incidence and belonging to the most represented Genus in metabarcoding analysis (N. parvum, isolate ER 2123; D. eres (Nitschke), isolate ER 2111; D. phaseolorum (Cooke & Ellis) Sacc., isolates ER 2126 and ER 2126; D. rudis (Sacc.) Sacc., isolate ER 2127; D. foeniculina (Udayanga et al., 2014), isolates ER 2125 and ER 2128. N. parvum ER 2123 was used as a positive control since it is reported as a pathogen of pomegranate wood [14].
The pathogenicity was evaluated through a test carried out on cut twigs and plants. The twigs were prepared in sections approximately 20 cm long, harbouring at least two buds between which were performed the inoculation. The inoculation area was previously surface sterilised with denatured ethyl alcohol.
Three inoculation procedures were performed.
First procedure: Detached pomegranate twigs at least one year old as described by Palavouzis, et al. [12]. The test was carried out with 5 mm diameter mycelium plugs taken from the margin of fungal colonies, about one week old, grown in purity on PDA medium. A 5 mm diameter disc of the bark was previously removed from the twigs with a cork borer to expose the cambium to the mycelium applied with a PDA plug. Then the twigs were placed in the dark in a humid chamber at room temperature to promote the infection (Figure 3). After 3 days, when presumably the mycelium colonised the wood, the humid chambers were opened. The test was completed in 7 days post inoculation (DPI).
Second procedure: detached twigs of at least four years old placed in water were inoculated wounding the bark with a razor blade in a basipetal orientation exposing the youngest wood; then the strip of tissue was pulled back to place the PDA with the mycelium on the exposed xylem. The inoculation area was covered with a cotton disc soaked with 3 ml of sterile tap water, wrapped with tinfoil strips, and stuck with paper tape as shown in Figure 4 [38]. They were placed in water at 25°C with photoperiod 13 h of light (13/11) and weekly checked to change the water. After seven days tinfoil strips were removed. The test was completed in 90 DPI.
Third procedure. In vivo, plants were inoculated as described in the second procedure and placed outdoors in the nursery. After 30 days, when presumably the mycelium colonized the wood, the tinfoil strips and the paper tape were removed; The test was completed in 90 days.
At the end of these tests, the first layers of the bark were removed with a razor blade to show and measure the length of the browned area in the longitudinal direction of the twig (Figure 5).
The tests were conducted in randomized blocks with four repetitions for procedures 1 and 2, and five repetitions for procedure 3. This was applied to each fungal isolate, as well as to the negative control (sterile PDA plugs) and the positive control.
Moreover, control isolations were carried out from the symptomatic areas to verify the Kock’s postulates.
2.5. Statistical Analysis
The data from the pathogenicity test were analysed using R software version 4.2.1 to perform a One-Way ANOVA, assessing significant differences in browning length (cm) among treatments to evaluate the virulence of the fungus compared to negative controls.
Before the ANOVA, normality was checked using shapiro.test function, while homoscedasticity and non-additivity were verified with bartlett.test and leveneTest functions, respectively, considering p > 0.05 as the threshold for homogeneity. The ANOVA was performed using the aov function, followed by Tukey’s HSD post hoc test with Tukey HSD function to identify significant differences between treatment groups (p = 0.05). The ANOVA results were considered significant at p < 0.05 and highly significant at p < 0.01.
3. Results
3.1. Phytosanitary Analysis of Pomegranate Plants Affected by Decay
Plants exhibiting wood decay symptoms, including reduced growth, leaf yellowing, and reddening, were sampled. Their trunks and twigs displayed cortical cankers, some located above the collar and others extending along the entire trunk. In the early stages of the disease, before canker formation, the bark appeared darker, and the underlying cambium showed distinct darkening and damage.
Laboratory dissections revealed browned wood, particularly beneath the cankers, with necrosis spreading in both basipetal and acropetal directions. In cases of multiple cankers, browning could extend throughout the entire trunk.
Fungal isolations from 325 symptomatic tissues yielded 188 colonies. Morphological and DNA sequence analyses identified 11 distinct species (Table 2). N. parvum was detected at both the Pavona and Sermoneta sites but only in early autumn. Diaporthe spp. and Gliocladium spp. were more frequently isolated and exhibited a high incidence.
3.2. Metabarcoding Analysis
A fragment of the expected size (340 bp) was obtained from ITS2 amplification in all the samples investigated.
The dataset comprises 114,486 reads distributed across eight samples, with a total of 41 operational taxonomic units (OTUs) identified. Notably, all OTUs and samples contain nonzero counts with no missing data or singleton OTUs.
In terms of OTU abundance and distribution, none of the OTUs have a total count of one, meaning that all identified taxa are represented by multiple reads across samples. A total of 328 counts were recorded, with 44.5% (146 counts) being zero and 55.5% (182 counts) showing at least ten occurrences. This suggests a relatively balanced distribution of microbial taxa, although a portion of the OTUs remains rare across samples.
Regarding the core microbiome and ubiquity of OTUs, 13 OTUs (31.7%) are present in all samples, defining a core microbiome that is consistently detected across the dataset; 23 OTUs (56.1%) are detected in at least 50% of the samples, indicating a moderate level of species turnover across different sampling points.
The sample size and sequencing depth vary, with the smallest sample containing 3,749 reads and the largest 19,253 reads. The median sample size is 15,599 reads, while the mean is 14,310 reads, suggesting an overall well-balanced sequencing depth across samples. The lower quartile (14,171 reads) and upper quartile (19,157 reads) indicate that most samples fall within a reasonable range, minimising the risk of uneven sequencing depth affecting downstream analyses.
Based on Figure 6, the most frequently occurring taxon across samples was Diaporthe, which shows consistently high relative abundances, ranging from 17.10% to 86.60%. This genus dominates several samples, particularly in samples 1, 2, 5, and 6, where its relative abundance exceeds 60%.
Another notable taxon is Punica, which exhibits the highest relative abundance in sample 4 (91.30%) and sample 3 (54.50%), which were asymptomatic sections. In contrast, taxa such as Vishniacozyma, Phaeoacremonium, and Fusarium are present in lower proportions but show some variability across samples, with Phaeoacremonium reaching 10.70% in sample 7.
Regarding species richness, sample 7 (symptomatic section) appears to have the highest diversity, as it contains multiple taxa with substantial relative abundances, including Diaporthe (41.80%), Phaeoacremonium (10.70%), Fusarium (5.44%), and Clonostachys (4.03%). This indicates a more heterogeneous microbial community in this sample.
Conversely, samples 3 and 4 (asymptomatic sections) exhibited the lowest species richness, as it is overwhelmingly dominated by Punica (54.50% and 91.30%, respectively) and Diaporthe (17.10% and 4.29%, respectively), with other taxa either absent or present in very low amounts.
Overall, the results highlight a strong predominance of Diaporthe, variations in microbial community structure across samples, and a marked difference in species richness, with sample 7 exhibiting the highest diversity and sample 4 the lowest.
The results confirmed the correlation between isolated fungal and fungal genera detected with metabarcoding analysis.
3.3. Pathogenicity Test
The artificial inoculation tests on a twig were able to reproduce the symptoms of browning of the wood by all tested fungi, which had been obtained from diseased wood of pomegranate plants affected by decay, and to highlight their greater or lesser aggressiveness. The positive control N. parvum (ER 2123 tube) caused the expected symptoms in the three tests to confirm that the inoculation method works properly (Tables S2–S4, Figure S1). The ANOVA results highlighted highly significant differences in terms of the length of browning between the treatments in all three tests (Table 3, Table 4 and Table 5).
D. eres (ER 2111 tube) is the only isolate that shows constant pathogenicity in the three analyses (Figure 9). The other isolates, D. phaseolorum (ER 2126 tube), D. foeniculina (ER 2128 and ER 2125 tubes) and D. rudis (ER 2127 tube) were less aggressive and showed no statistically significant differences compared to the control. Despite this, their presence on symptomatic wood is not completely negligible and although with less aggression the mycelium manages to develop through the tissues of pomegranate wood.
Figure 10, Figure 11 and Figure 12 show the average lengths (cm) of browning caused by the 5 inoculated isolates.
The Tukey’s HSD test, performed on the data of the inoculations on detached twigs placed in humid chamber, shows that: D. phaseolorum (ER 2126 tube) and D. eres (ER 2111 Tube) shows statistically significant differences compared to the control. Among D. foeniculina (tubes ER 2128 and ER 2125) and D. rudis (ER 2127), there are no significant differences compared to the control, so they have not pathogenicity activity.
The Tukey’s HSD test, performed on the data of the inoculations on detached twigs in water, shows that: D. foeniculina (ER 2125 tube) and D. eres (ER 2111 Tube) showed statistically significant differences compared to the control. Among D. phaseolorum (ER 2126 tube) D. foeniculina (ER 2128 tube) and D. rudis (ER 2127 tube), there are no significant differences compared to the control, so they have not pathogenicity activity.
The Tukey’s HSD test, performed on the data of the inoculations on pomegranate plants, showed that D. eres (ER 2111 Tube), D. foeniculina (ER 2125 tube), and D. rudis (ER 2127 tube) showed statistically significant differences compared to the control (Figure 13). Among D. phaseolorum (ER 2126 tube) and D. foeniculina (ER 2128 tube), there were no significant differences compared to the control, so they had no pathogenicity activity.
4. Discussion
The rapid expansion of pomegranate cultivation in central and southern Italy, primarily using foreign varieties, has revealed unexpected phytosanitary issues. This is notable for a plant traditionally considered hardy and relatively free from phytosanitary concerns, apart from well-documented issues affecting the fruit. Over the past three years, the first cases of plant decline and mortality have been reported, with losses reaching 30–40% in 2015 and escalating to 90% in subsequent years. In Puglia, in 2016, Pollastro, et al. [6] identified C. granati, a pathogen already known in various pomegranate-growing regions worldwide, as the causative agent of fruit rot. However, in China, Iran, and Spain, C. granati has also been associated with cankers and pomegranate tree mortality, while in Greece and Turkey, it has been reported as a cause of stem cankers [39]. In central Italy, particularly in Lazio, this issue was initially unknown at the start of our investigation. However, following reports and subsequent inspections and analyses described in this study, two potential fungal pathogens were identified: N. parvum and Diaporthe spp., which are likely to act synergistically. Initially, N. parvum was considered the only causal agent [14] and was, therefore, the primary focus of further investigations.
The metabarcoding analysis had limitations due to the availability of ITS2 sequences in the UNITE database, which, at present, does not allow for taxonomic resolution sufficient to classify all DNA in the sample at the species level. As a result, a proportion of fungal DNA could only be identified at the division or, at best, genus level. Additionally, a significant proportion of DNA (on average 29.3%) remained unassigned, potentially concealing other pathogenic fungi. The metabarcoding analysis, even within its limits, allowed us to highlight in the two plants analysed that N. parvum and C. granati were not present in the diseased wood where it was not isolated, nor even in the healthy wood, excluding the possibility that it was present as an endophyte.
This result was significant as it validated the test performed, confirming the presence of a substantially richer fungal community in symptomatic tissue, which exhibited clear signs such as cankers and wood browning. In contrast, the fungal DNA detected in asymptomatic samples was likely associated with the endophytic fungal flora naturally present in pomegranate wood.
Different diseases are already known as the Esca of Grapevine [40] and Esca-like symptoms of Actinidia [41] caused by fungi complex. Today, there are other diseases, such as “Kiwifruit Vine Decline Syndrome” (KVDS), caused by different microorganisms and abiotic disorders. The causes of the decline are still discussed; currently are involved and reported pathogenic species such as Phytopythium vexans [42,43], Phytophthora spp [44], Cylindrocarpon [45] and, anaerobic bacteria Clostridium [46].
Similarly to other complex syndromes, abiotic stress could represent a trigger factor for the disease. The pomegranate, mainly the ’Wonderful’ cultivar, is widely cultivated in warm climates and originates from regions such as Iran and surrounding regions, from where it spreads to other parts of the world [47], where temperature conditions are more similar to those of southern Italy rather than central regions like Lazio. This difference is attributed to the latitudinal gradient, as well as variations in altitude and proximity to the sea, which influence mean winter temperatures [31] For this reason, climatic conditions play a central role as a trigger for wood decay.
Few studies have dealt with the taxonomic resolution obtained using both the ITS1 and the ITS2 barcodes on the same dataset [27,48,49,50,51]. They have been carried out on both Ascomycota [52,53] and Basidiomycota [54]. The choice of primers can introduce taxonomic bias, as they may produce a higher number of mismatches in certain taxa [53,55,56]. Some studies also reported that the two spacers are prone to preferential amplification at different levels [48,49,50,52,53]. Basidiomycetes have on average longer amplicon sequences for the ITS2, and since the shorter sequences are preferentially sequenced with high-throughput sequencing (HTS), the use of ITS2 would favour the detection of ascomycetes [53]. On the other hand, ITS1 often contains an intron that extends its sequence at the 5′-end [57], thereby promoting an over-representation of those sequences that lack the intron [49]. Because ITS2 is more frequently represented in public databases, has a higher number of available sequences, and offers a better taxonomic resolution, it has been proposed as the better choice for parallel sequencing [52]. In some cases, however, no substantial differences between ITS1 and ITS2 were recovered [27,54]. Finally, there are numerous studies that consider a single spacer, either the ITS1 or ITS2 [35,53,58,59].
As fungal metabarcoding studies have used different HTS platforms [60], different bioinformatic pipelines have been proposed [61,62]. These have been developed based on experience gained from data analyses of prokaryote datasets [63]. However, no standard procedure has been established so far for fungal sequence data. The analyses seem strongly dependent on the working hypotheses of each study and on the type of sequence at hand. As the majority of studies target fungal communities to uncover unknown diversity, an important and ongoing problem is the definition of those sequences lacking an assigned taxonomy [52]. For this reason, many sequences remain identified as “unassigned”. In addition, many fungi have not yet been sequenced and cannot offer reference sequences for ongoing studies [63].
The molecular markers used in this investigation have a broad level of fungal species identification, so the failure to detect Coniella granati among the identified species allows us to speculate that other species are implicated in pomegranate wood decay syndrome. In fact, the metabarcoding results suggested a relatively stable microbial community, with a substantial proportion of OTUs present in multiple samples. C. granati, in the two plants object of our study, it can be said with certainty that it was not present and, therefore, was not involved with the cankers and deaths found in Pavona and Sermoneta. This is confirmed both by the isolations carried out on diseased plants and by the metagenomic analysis where the presence of Schizoparmaceae was not found.
The presence of a well-defined core microbiome (31.7% of OTUs in all samples) indicates a consistent microbial signature across different conditions. The data also suggest good sequencing depth, with minimal bias from zero or low-count OTUs.
5. Conclusions
To conclude, pomegranate wood decay syndrome is probably a complex syndrome, where the triggering factor could be climatic conditions such as low temperatures stress, for which plants become susceptible to various wood pathogens, including N. parvum and other fungi of the Diaporthe complex. The wood deterioration caused by C. granati may represent a disease with its own epidemiology and progression, capable of damaging pomegranate plants even in the absence of abiotic stress.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Bar plot showing the average browning lengths (cm) caused by inoculations with the positive control Neofusicoccum parvum (ER 2123) across three inoculation methods. The letters above the bars indicate significant statistical differences between treatments (Tukey’s HSD, p < 0.05). Different letters indicate significant differences based on One-Way ANOVA results at p < 0.05. Error bars represent the standard error of the mean (4 replicates); Table S1: Sequences of primer pairs used for the 8 experimental units (sample) showing, separately, the adapter sequences for the Illumina system, the indexed tag and the sequences of the universal ITS3 and ITS4 (original primer) are shown separately; Table S2: ANOVA results conducted on length (cm) of browning caused by the positive control, N. parvum (ER 2123) inoculated on the pomegranate detached twigs in humid chamber in 2019; Table S3: ANOVA results conducted on length (cm) of browning caused by the positive control, N. parvum (ER 2123) inoculated on the pomegranate detached twigs in water in 2019; Table S4: ANOVA results conducted on length (cm) of browning caused by the positive control, N. parvum (ER 2123) inoculated on the pomegranate plants in 2020.
Author Contributions
Conceptualization, V.B., M.T.V. and R.M.; methodology, V.B., M.T.V. and R.M.; formal analysis, V.B., M.T.V. and R.M.; investigation, V.B., M.T.V. and R.M.; resources, V.B., M.T.V. and R.M.; data curation, V.B., M.T.V. and R.M.; writing—original draft preparation, V.B., M.T.V. and R.M.; writing—review and editing, V.B., M.T.V. and R.M.; visualization, V.B., M.T.V. and R.M.; supervision, V.B., M.T.V. and R.M.. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially carried out within the framework of The Ministry of University and Research (MUR) initiative “Department of Excellence” (Law 232/2016) DAFNE Project 2023-27 “Digital, Intelligent, Green and Sustainable” (acronym: D.I.Ver.So). https://www.unitus.it/ricerca/dipartimenti-di-eccellenza/
Data Availability Statement
Data is contained within the article.
Acknowledgments
This study is dedicated to our friend, Luca Riccioni, for his contributions and memory. We are grateful to him that making this work possible
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Cristofori, V.; Caruso, D.; Latini, G.; Dell’Agli, M.; Cammilli, C.; Rugini, E.; Bignami, C.; Muleo, R. Fruit quality of italian pomegranate (punica granatum l.) autochthonous varieties. European Food Research and Technology 2011, 232, 397–403. [Google Scholar] [CrossRef]
- Hajleh, M.N.A.; Al-Dujaili, E.A.S. In Anti-cancer activity of pomegranate and its biophenols; general review, 2016.
- Fanali, C.; Belluomo, M.G.; Cirilli, M.; Cristofori, V.; Zecchini, M.; Cacciola, F.; Russo, M.; Muleo, R.; Dugo, L. Antioxidant activity evaluation and hplc-photodiode array/ms polyphenols analysis of pomegranate juice from selected italian cultivars: A comparative study. Electrophoresis 2016, 37, 1947–1955. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.K.; Gupta, S.K.; Jacob, M.R.; Khan, S.I.; Ferreira, D. Antioxidant, antimalarial and antimicrobial activities of tannin-rich fractions, ellagitannins and phenolic acids from punica granatum l. Planta medica 2007, 73, 461–467. [Google Scholar] [CrossRef]
- Giménez-Bastida, J.A.; Ávila-Gálvez, M.Á.; Espín, J.C.; González-Sarrías, A. Evidence for health properties of pomegranate juices and extracts beyond nutrition: A critical systematic review of human studies. Trends in Food Science & Technology 2021, 114, 410–423. [Google Scholar] [CrossRef]
- Pollastro, S.; Dongiovanni, C.; Gerin, D.; Pollastro, P.; Fumarola, G.; De Miccolis Angelini, R.M.; Faretra, F. First report of coniella granati as a causal agent of pomegranate crown rot in southern italy. Plant Disease 2016, 100, 1498. [Google Scholar] [CrossRef]
- Mirabolfathy, M.; Groenewald, J.Z.; Crous, P.W. First report of pilidiella granati causing dieback and fruit rot of pomegranate (punica granatum) in iran. Plant Dis 2012, 96, 461. [Google Scholar] [CrossRef]
- Levy, E.; Elkind, G.; Ben-Arie, R.; Ben-Ze’ev, I.S. First report of coniella granati causing pomegranate fruit rot in israel. Phytoparasitica 2011, 39, 403–405. [Google Scholar] [CrossRef]
- Çeliker, N.M.; Uysal, A.; Çetinel, B.; Poyraz, D. Crown rot on pomegranate caused by coniella granati in turkey. Australasian Plant Disease Notes 2012, 7, 161–162. [Google Scholar] [CrossRef]
- Tziros, G.T.; Tzavella-Klonari, K. Pomegranate fruit rot caused by coniella granati confirmed in greece. Plant Pathology 2008, 57, 783. [Google Scholar] [CrossRef]
- Palou, L.; Guardado, A.; Montesinos-Herrero, C. First report of penicillium spp. And pilidiella granati causing postharvest fruit rot of pomegranate in spain. New Disease Reports 2010, 22, 21. [Google Scholar] [CrossRef]
- Palavouzis, S.C.; Tzamos, S.; Paplomatas, E.; Thomidis, T. First report of neofusicoccum parvum causing shoot blight of pomegranate in northern greece. New Disease Reports 2015, 32, 10. [Google Scholar] [CrossRef]
- KC, A.N.; Vallad, G.E. First report of neofusicoccum parvum causing shoot blight and stem cankers on pomegranate in florida. Plant Disease 2016, 100, 1783. [Google Scholar] [CrossRef]
- Riccioni, L.; Valente, M.T.; Giambattista, G.d. First report of neofusicoccum parvum causing shoot blight and plant decay on pomegranate in tarquinia, italy. Journal of Plant Pathology 2017, 99, 294. [Google Scholar]
- Molina-Gayosso, E.; Silva-Rojas, H.V.; García-Morales, S.; Avila-Quezada, G. First report of black spots on avocado fruit caused by neofusicoccum parvum in mexico. Plant Dis 2012, 96, 287. [Google Scholar] [CrossRef]
- Phillips, A.J.L.; Alves, A.; Abdollahzadeh, J.; Slippers, B.; Wingfield, M.J.; Groenewald, J.Z.; Crous, P.W. The botryosphaeriaceae: Genera and species known from culture. Studies in Mycology 2013, 76, 51–167. [Google Scholar] [CrossRef]
- Úrbez-Torres, J.R.; Gubler, W.D. Susceptibility of grapevine pruning wounds to infection by lasiodiplodia theobromae and neofusicoccum parvum. Plant Pathology 2011, 60, 261–270. [Google Scholar] [CrossRef]
- Lamichhane, J.R.; Venturi, V. Synergisms between microbial pathogens in plant disease complexes: A growing trend. Frontiers in Plant Science 2015, 6. [Google Scholar] [CrossRef]
- Schmidt, P.-A.; Bálint, M.; Greshake, B.; Bandow, C.; Römbke, J.; Schmitt, I. Illumina metabarcoding of a soil fungal community. Soil Biology and Biochemistry 2013, 65, 128–132. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Li Destri Nicosia, M.G.; Cacciola, S.O.; Droby, S.; Schena, L. Metabarcoding analysis of fungal diversity in the phyllosphere and carposphere of olive (olea europaea). PLoS ONE 2015, 10, e0131069. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Cacciola, S.O.; Mosca, S.; Zappia, R.; Schena, L. Analysis of the fungal diversity in citrus leaves with greasy spot disease symptoms. Microbial ecology 2017, 73, 739–749. [Google Scholar] [CrossRef]
- Morales-Cruz, A.; Figueroa-Balderas, R.; García, J.F.; Tran, E.; Rolshausen, P.E.; Baumgartner, K.; Cantu, D. Profiling grapevine trunk pathogens in planta: A case for community-targeted DNA metabarcoding. BMC Microbiology 2018, 18, 214. [Google Scholar] [CrossRef] [PubMed]
- Del Frari, G.; Gobbi, A.; Aggerbeck, M.R.; Oliveira, H.; Hansen, L.H.; Ferreira, R.B. Characterization of the wood mycobiome of vitis vinifera in a vineyard affected by esca. Spatial distribution of fungal communities and their putative relation with leaf symptoms. Frontiers in Plant Science 2019, 10. [Google Scholar] [CrossRef]
- Kõljalg, U.; Larsson, K.-H.; Abarenkov, K.; Nilsson, R.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; et al. Unite: A database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytologist 2005, 166, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Meiklejohn, K.A.; Damaso, N.; Robertson, J.M. Assessment of bold and genbank – their accuracy and reliability for the identification of biological materials. PLoS ONE 2019, 14, e0217084. [Google Scholar] [CrossRef] [PubMed]
- van der Loos, L.M.; Nijland, R. Biases in bulk: DNA metabarcoding of marine communities and the methodology involved. Molecular ecology 2021, 30, 3270–3288. [Google Scholar] [CrossRef]
- Blaalid, R.; Kumar, S.; Nilsson, R.H.; Abarenkov, K.; Kirk, P.M.; Kauserud, H. Its1 versus its2 as DNA metabarcodes for fungi. Molecular Ecology Resources 2013, 13, 218–224. [Google Scholar] [CrossRef]
- Mbareche, H.; Veillette, M.; Bilodeau, G.; Duchaine, C. Comparison of the performance of its1 and its2 as barcodes in amplicon-based sequencing of bioaerosols. PeerJ 2020, 8, e8523. [Google Scholar] [CrossRef]
- Yang, R.-H.; Su, J.-H.; Shang, J.-J.; Wu, Y.-Y.; Li, Y.; Bao, D.-P.; Yao, Y.-J. Evaluation of the ribosomal DNA internal transcribed spacer (its), specifically its1 and its2, for the analysis of fungal diversity by deep sequencing. PLoS ONE 2018, 13, e0206428. [Google Scholar] [CrossRef]
- Napoli, R.; Paolanti, M.; Di Ferdinando, S. Atlante dei suoli del lazio. ARSIAL Regione Lazio. ISBN 978-88-904841-2-4 2019.
- Fratianni, S.; Acquaotta, F. The climate of italy. In Landscapes and landforms of italy, Soldati, M.; Marchetti, M., Eds. Springer International Publishing: Cham, 2017; pp 29-38.
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. 38 - amplification and direct sequencing of fungal ribosomal rna genes for phylogenetics. In Pcr protocols, Innis, M.A.; Gelfand, D.H.; Sninsky, J.J.; White, T.J., Eds. Academic Press: San Diego, 1990; pp 315-322.
- Cenis, J.L. Rapid extraction of fungal DNA for pcr amplification. Nucleic Acids Research 1992, 20, 2380. [Google Scholar] [CrossRef]
- Aragona, M.; Minio, A.; Ferrarini, A.; Valente, M.T.; Bagnaresi, P.; Orrù, L.; Tononi, P.; Zamperin, G.; Infantino, A.; Valè, G.; et al. De novo genome assembly of the soil-borne fungus and tomato pathogen pyrenochaeta lycopersici. BMC Genomics 2014, 15, 313. [Google Scholar] [CrossRef]
- Miller, K.E.; Hopkins, K.; Inward, D.J.G.; Vogler, A.P. Metabarcoding of fungal communities associated with bark beetles. Ecology and Evolution 2016, 6, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Uparse: Highly accurate otu sequences from microbial amplicon reads. Nature Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Abarenkov, K.Z., Allan; Piirmann, Timo; Pöhönen, Raivo; Ivanov, Filipp; Nilsson, R. Henrik; Kõljalg, Urmas. Unite usearch/utax release for eukaryotes. Version 04.04.2024. UNITE Community. 2024.
- Manetti, G.; Brunetti, A.; Lumia, V.; Sciarroni, L.; Marangi, P.; Cristella, N.; Faggioli, F.; Reverberi, M.; Scortichini, M.; Pilotti, M. Identification and characterization of neofusicoccum stellenboschiana in branch and twig dieback-affected olive trees in italy and comparative pathogenicity with n. Mediterraneum. Journal of Fungi 2023, 9, 292. [Google Scholar] [CrossRef] [PubMed]
- Moncini, L.; Pachetti, G.; Pollastro, S. Il cancro del fusto minaccia il melograno italiano. 2017.
- Graniti, A.; Surico, G.; Mugnai, L. Esca of grapevine: A disease complex or a complex of diseases? Phytopathologia Mediterranea 2000, 39, 16–20. [Google Scholar]
- Hennion, B.; Lecomte, P.; Larignon, P.; Baudry, A.; Durpaire, M.P.; Mouillon, M.; Tailleur, J.L.; Dupuy, O. First observations of a wood decay (esca-loke disease) on kiwifruit in france. Acta Horticulturae 2003, 409–413. [Google Scholar] [CrossRef]
- Savian, F.; Ginaldi, F.; Musetti, R.; Sandrin, N.; Tarquini, G.; Pagliari, L.; Firrao, G.; Martini, M.; Ermacora, P. Studies on the aetiology of kiwifruit decline: Interaction between soil-borne pathogens and waterlogging. Plant and Soil 2020, 456, 113–128. [Google Scholar] [CrossRef]
- Prencipe, S.; Savian, F.; Nari, L.; Ermacora, P.; Spadaro, D.; Martini, M. First report of phytopythium vexans causing decline syndrome of actinidia deliciosa ‘hayward’ in italy. Plant Disease 2020, 104, 2032. [Google Scholar] [CrossRef]
- Donati, I.; Cellini, A.; Sangiorgio, D.; Caldera, E.; Sorrenti, G.; Spinelli, F. Pathogens associated to kiwifruit vine decline in italy. Agriculture 2020, 10, 119. [Google Scholar] [CrossRef]
- Tacconi, G.; Paltrinieri, S.; Mejia, J.F.; Fuentealba, S.P.; Bertaccini, A.; Tosi, L.; Giacopini, A.; Mazzucchi, U.; Favaron, F.; Sella, L.; et al. Vine decline in kiwifruit: Climate change and effect on waterlogging and phytophthora in north italy. 2015, 93–97.
- Spigaglia, P.; Barbanti, F.; Marocchi, F.; Mastroleo, M.; Baretta, M.; Ferrante, P.; Caboni, E.; Lucioli, S.; Scortichini, M. Clostridium bifermentans and c. Subterminale are associated with kiwifruit vine decline, known as moria, in italy. Plant Pathology 2020, 69, 765–774. [Google Scholar] [CrossRef]
- Chandra, R.; Babu, K.D.; Jadhav, V.T.; Silva, J.T.d. In Origin, history and domestication of pomegranate, 2010.
- Mello, A.; Lumini, E.; Napoli, C.; Bianciotto, V.; Bonfante, P. Arbuscular mycorrhizal fungal diversity in the tuber melanosporum brûlé. Fungal Biology 2015, 119, 518–527. [Google Scholar] [CrossRef]
- Bazzicalupo, A.L.; Bálint, M.; Schmitt, I. Comparison of its1 and its2 rdna in 454 sequencing of hyperdiverse fungal communities. Fungal Ecology 2013, 6, 102–109. [Google Scholar] [CrossRef]
- Monard, C.; Gantner, S.; Stenlid, J. Utilizing its1 and its2 to study environmental fungal diversity using pyrosequencing. FEMS Microbiology Ecology 2013, 84, 165–175. [Google Scholar] [CrossRef]
- Orgiazzi, A.; Bianciotto, V.; Bonfante, P.; Daghino, S.; Ghignone, S.; Lazzari, A.; Lumini, E.; Mello, A.; Napoli, C.; Perotto, S.; et al. 454 pyrosequencing analysis of fungal assemblages from geographically distant, disparate soils reveals spatial patterning and a core mycobiome. Diversity 2013, 5, 73–98. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Ryberg, M.; Abarenkov, K.; Sjökvist, E.; Kristiansson, E. The its region as a target for characterization of fungal communities using emerging sequencing technologies. FEMS Microbiology Letters 2009, 296, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Bellemain, E.; Davey, M.L.; Kauserud, H.; Epp, L.S.; Boessenkool, S.; Coissac, E.; Geml, J.; Edwards, M.; Willerslev, E.; Gussarova, G.; et al. Fungal palaeodiversity revealed using high-throughput metabarcoding of ancient DNA from arctic permafrost. Environ Microbiol 2013, 15, 1176–1189. [Google Scholar] [CrossRef]
- Badotti, F.; de Oliveira, F.S.; Garcia, C.F.; Vaz, A.B.M.; Fonseca, P.L.C.; Nahum, L.A.; Oliveira, G.; Góes-Neto, A. Effectiveness of its and sub-regions as DNA barcode markers for the identification of basidiomycota (fungi). BMC Microbiology 2017, 17, 42. [Google Scholar] [CrossRef]
- Tedersoo, L.; Anslan, S.; Bahram, M.; Põlme, S.; Riit, T.; Liiv, I.; Kõljalg, U.; Kisand, V.; Nilsson, H.; Hildebrand, F.; et al. Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 2015, 10. [Google Scholar] [CrossRef]
- Tedersoo, L.; Lindahl, B. Fungal identification biases in microbiome projects. Environmental Microbiology Reports 2016, 8, 774–779. [Google Scholar] [CrossRef]
- Martin, K.J.; Rygiewicz, P.T. Fungal-specific pcr primers developed for analysis of the its region of environmental DNA extracts. BMC Microbiology 2005, 5, 28. [Google Scholar] [CrossRef]
- Langarica-Fuentes, A.; Zafar, U.; Heyworth, A.; Brown, T.; Fox, G.; Robson, G.D. Fungal succession in an in-vessel composting system characterized using 454 pyrosequencing. FEMS Microbiology Ecology 2014, 88, 296–308. [Google Scholar] [CrossRef]
- U’Ren, J.M.; Riddle, J.M.; Monacell, J.T.; Carbone, I.; Miadlikowska, J.; Arnold, A.E. Tissue storage and primer selection influence pyrosequencing-based inferences of diversity and community composition of endolichenic and endophytic fungi. Molecular Ecology Resources 2014, 14, 1032–1048. [Google Scholar] [CrossRef]
- Cuadros-Orellana, S.; Leite, L.R.; Smith, A.; Medeiros, J.D.; Badotti, F.; Fonseca, P.L.; Vaz, A.B.; Oliveira, G.; Góes-Neto, A. Assessment of fungal diversity in the environment using metagenomics: A decade in review. Fungal Genomics & Biology 2013, 3, 1. [Google Scholar]
- Bálint, M.; Schmidt, P.-A.; Sharma, R.; Thines, M.; Schmitt, I. An illumina metabarcoding pipeline for fungi. Ecology and Evolution 2014, 4, 2642–2653. [Google Scholar] [CrossRef] [PubMed]
- Gweon, H.S.; Oliver, A.; Taylor, J.; Booth, T.; Gibbs, M.; Read, D.S.; Griffiths, R.I.; Schonrogge, K. Pipits: An automated pipeline for analyses of fungal internal transcribed spacer sequences from the illumina sequencing platform. Methods in ecology and evolution 2015, 6, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Hibbett, D.; Abarenkov, K.; Kõljalg, U.; Öpik, M.; Chai, B.; Cole, J.; Wang, Q.; Crous, P.; Robert, V.; Helgason, T.; et al. Sequence-based classification and identification of fungi. Mycologia 2016, 108, 1049–1068. [Google Scholar] [CrossRef]
Figure 1.
Sub-cortical browning on the sampled Plant 2 in Pavona (RM).

Figure 2.
Sampling points of Plant1 (left) and Plant2 (right), showing the cankers and browning wood, with the identification legend of each sampled section, later described in Table 1. The circled sections were used for the metabarcoding analysis.
Figure 2.
Sampling points of Plant1 (left) and Plant2 (right), showing the cankers and browning wood, with the identification legend of each sampled section, later described in Table 1. The circled sections were used for the metabarcoding analysis.
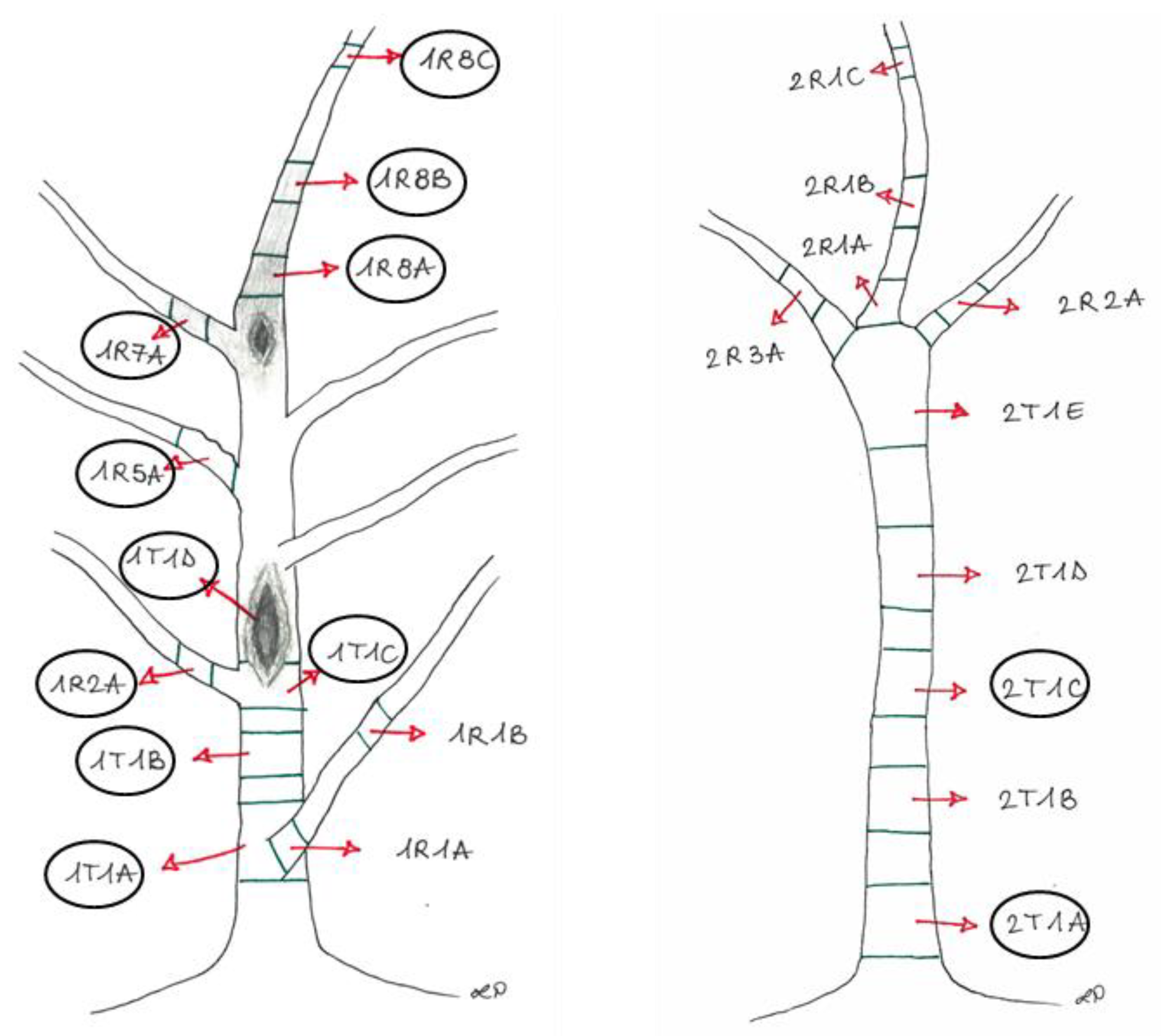
Figure 3.
Pomegranate twigs inoculated and placed in the humid chamber.

Figure 4.
Pomegranate twigs (4 years old) inoculated and placed in water.

Figure 5.
Pomegranate branches without the first layers of the bark to discover the browned area and measure its length in the longitudinal direction (highlighted in red).
Figure 5.
Pomegranate branches without the first layers of the bark to discover the browned area and measure its length in the longitudinal direction (highlighted in red).

Figure 6.
Fungal relative abundance at genus level in the different sections of the 8 experimental units. A description of each sample is provided in Table 1.
Figure 6.
Fungal relative abundance at genus level in the different sections of the 8 experimental units. A description of each sample is provided in Table 1.
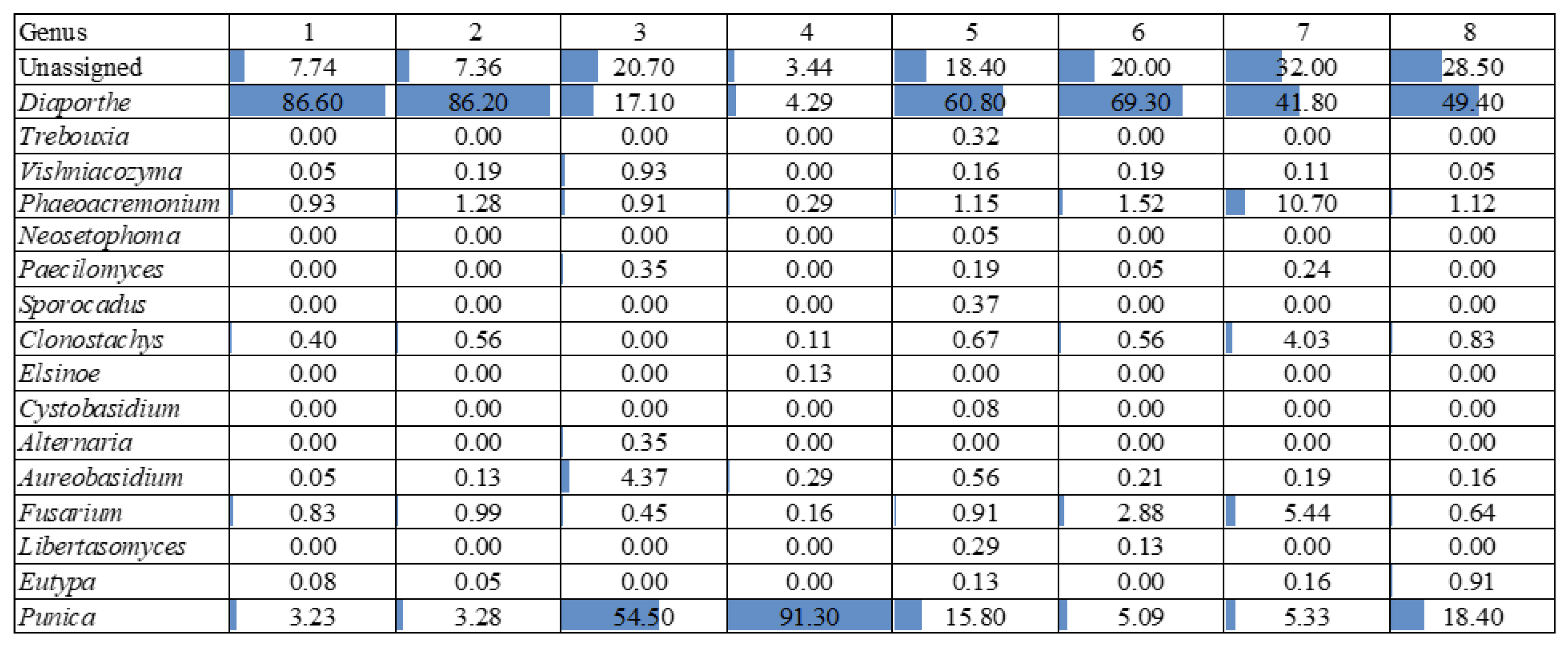
Figure 7.
Fungal abundance at the family level in different sections of the tested plants. A description of each sample is provided in Table 1.
Figure 7.
Fungal abundance at the family level in different sections of the tested plants. A description of each sample is provided in Table 1.
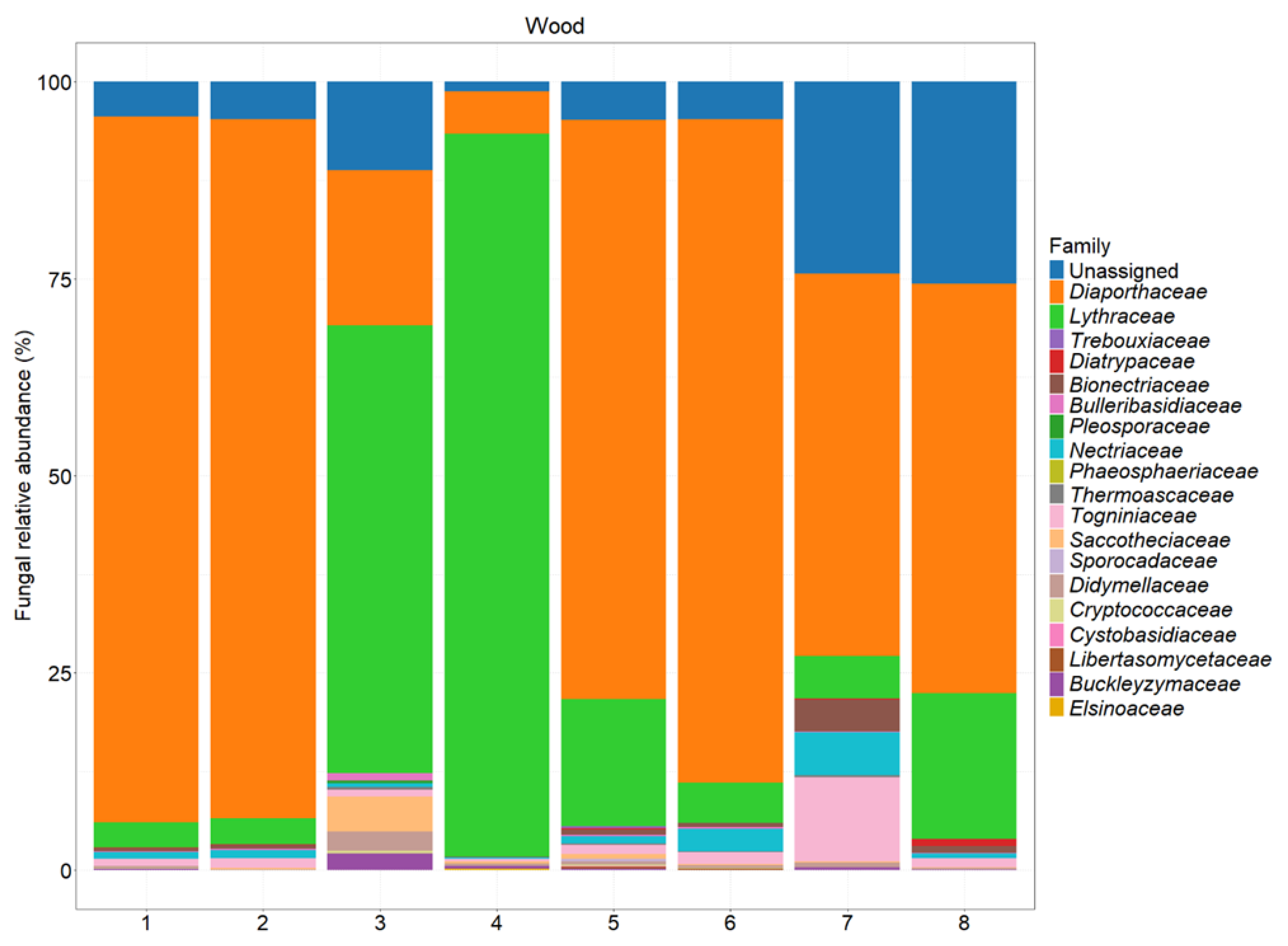
Figure 8.
Fungal abundance at the family level in different sections of the tested plants. A description of each sample is provided in Table 1.
Figure 8.
Fungal abundance at the family level in different sections of the tested plants. A description of each sample is provided in Table 1.
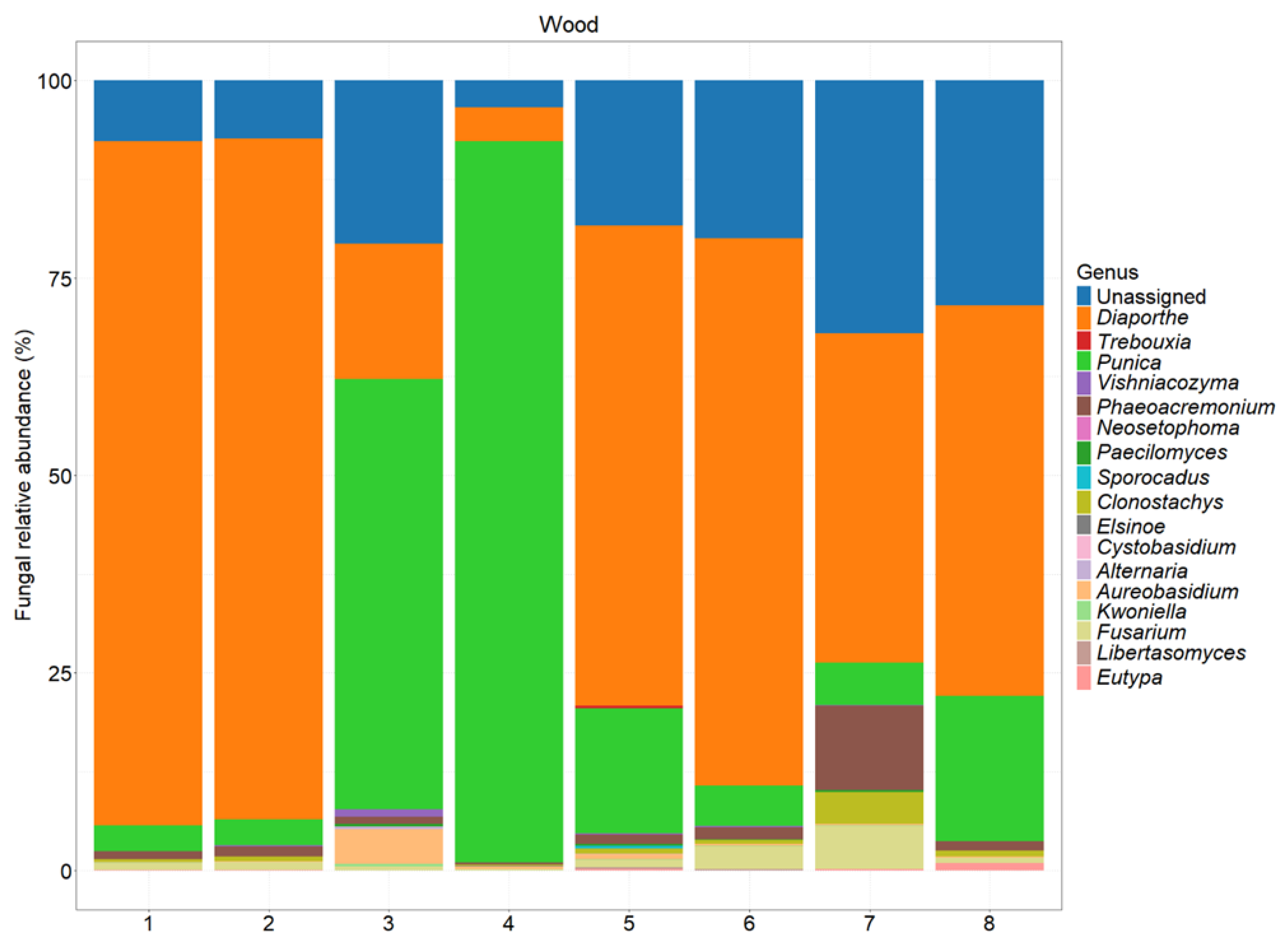
Figure 9.
On the left (4 branches), is the result of the inoculation of D. foeniculina (ER 2128 tube); on the right (4 branches), is the result of the inoculation of D. eres (ER 2111 tube), using procedure 1.
Figure 9.
On the left (4 branches), is the result of the inoculation of D. foeniculina (ER 2128 tube); on the right (4 branches), is the result of the inoculation of D. eres (ER 2111 tube), using procedure 1.

Figure 10.
Bar plot showing the average lengths (cm) of browning caused by inoculations on the detached twigs placed in a humid chamber are reported. The letters above the bars indicate significant statistical differences between treatments (Tukey’s HSD, p < 0.05). Different letters indicate significant differences based on One-Way ANOVA results at p < 0.05. Error bars represent the standard error of the mean (4 replicates).
Figure 10.
Bar plot showing the average lengths (cm) of browning caused by inoculations on the detached twigs placed in a humid chamber are reported. The letters above the bars indicate significant statistical differences between treatments (Tukey’s HSD, p < 0.05). Different letters indicate significant differences based on One-Way ANOVA results at p < 0.05. Error bars represent the standard error of the mean (4 replicates).
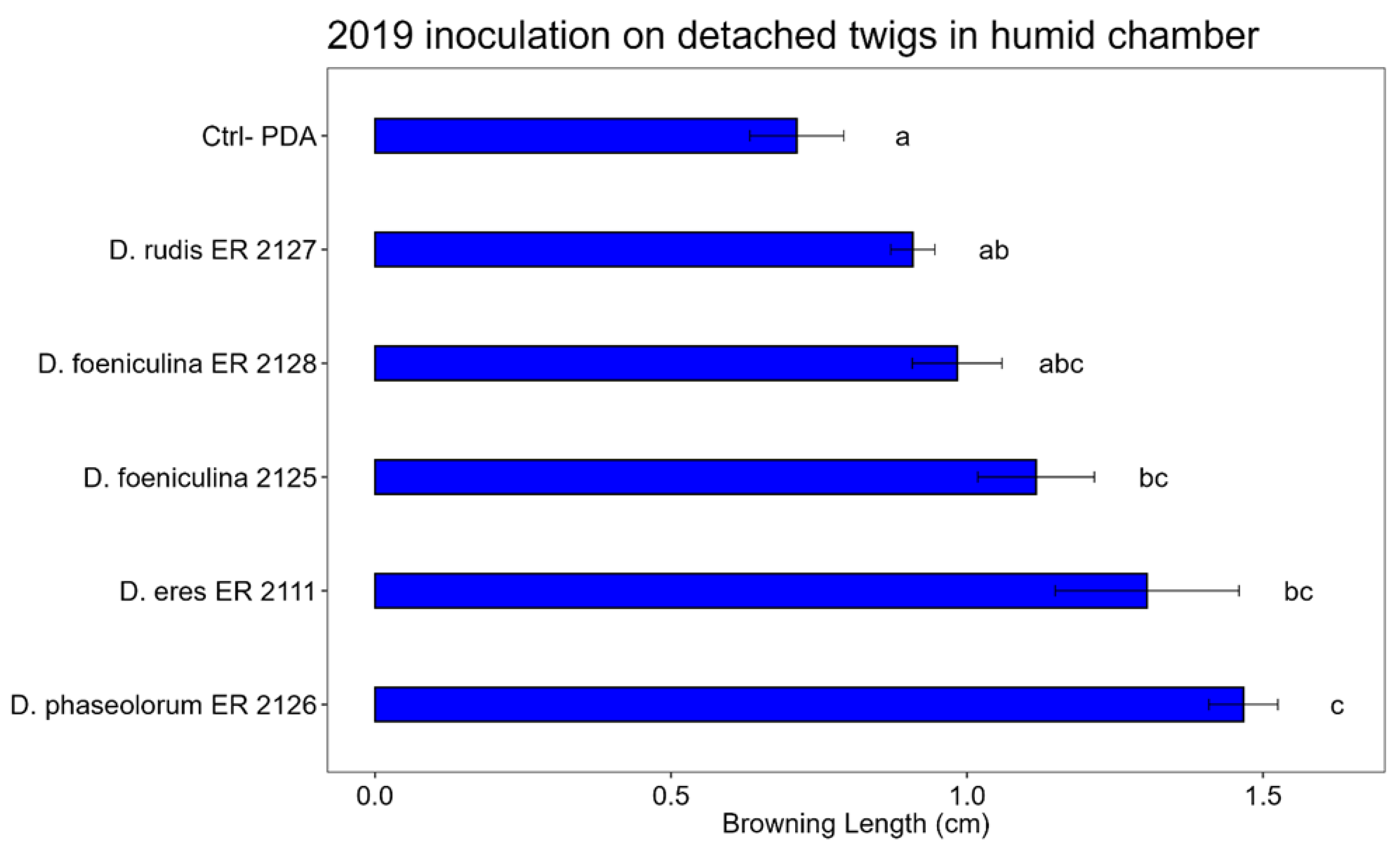
Figure 11.
Bar plot showing the average lengths (cm) of browning caused by inoculations on the detached twigs in water a is reported. The letters above the bars indicate significant statistical differences between treatments (Tukey’s HSD, p < 0.05). Different letters indicate significant differences based on One-Way ANOVA results at p < 0.05. Error bars represent the standard error of the mean (4 replicates).
Figure 11.
Bar plot showing the average lengths (cm) of browning caused by inoculations on the detached twigs in water a is reported. The letters above the bars indicate significant statistical differences between treatments (Tukey’s HSD, p < 0.05). Different letters indicate significant differences based on One-Way ANOVA results at p < 0.05. Error bars represent the standard error of the mean (4 replicates).
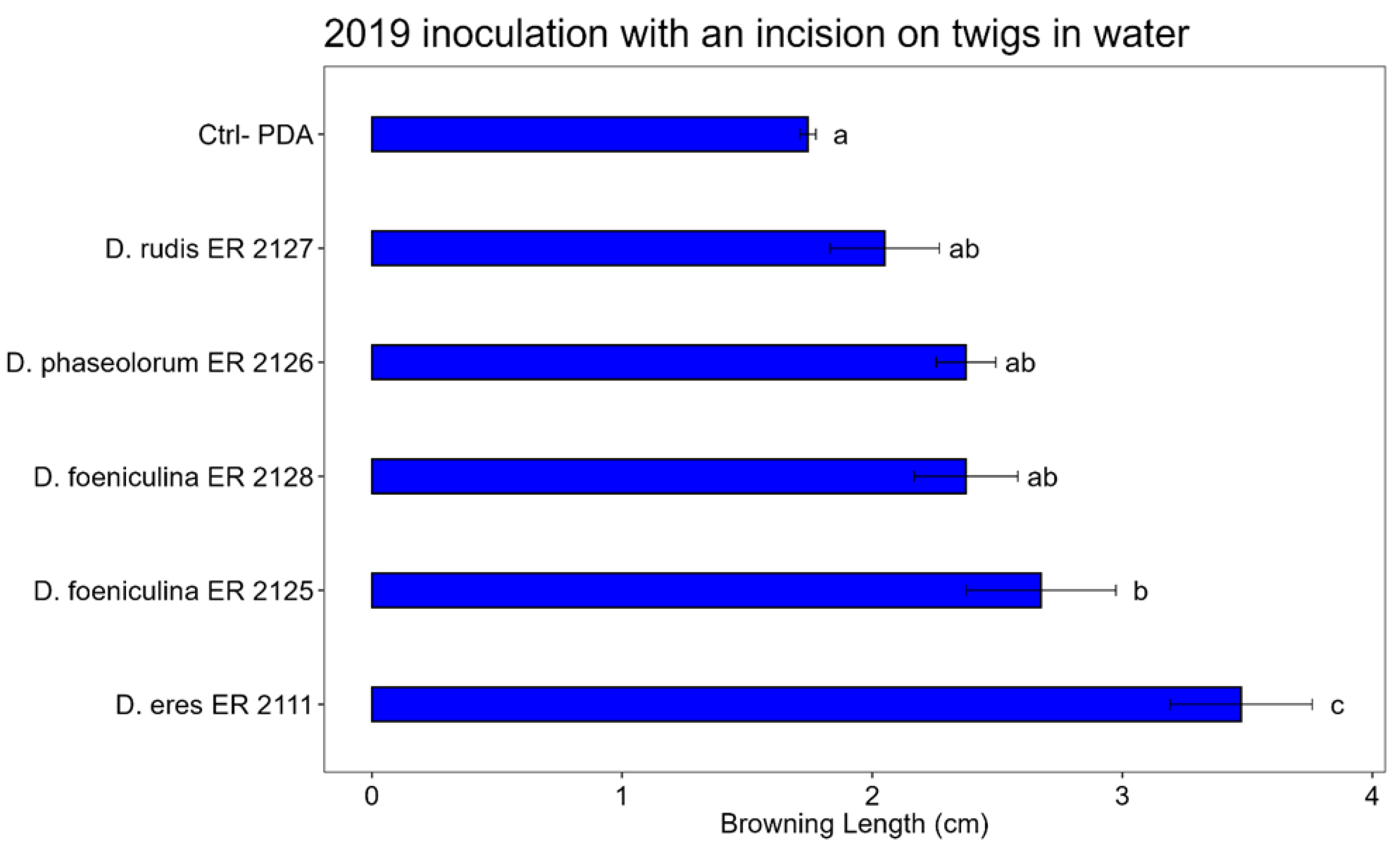
Figure 12.
Bar plot showing the average browning length (cm) caused by inoculations on pomegranate plants in 2020. The letters above the bars indicate significant statistical differences between treatments (Tukey’s HSD, p < 0.05). Different letters indicate significant differences based on One-Way ANOVA results at p < 0.05. Error bars represent the standard error of the mean (4 replicates).
Figure 12.
Bar plot showing the average browning length (cm) caused by inoculations on pomegranate plants in 2020. The letters above the bars indicate significant statistical differences between treatments (Tukey’s HSD, p < 0.05). Different letters indicate significant differences based on One-Way ANOVA results at p < 0.05. Error bars represent the standard error of the mean (4 replicates).
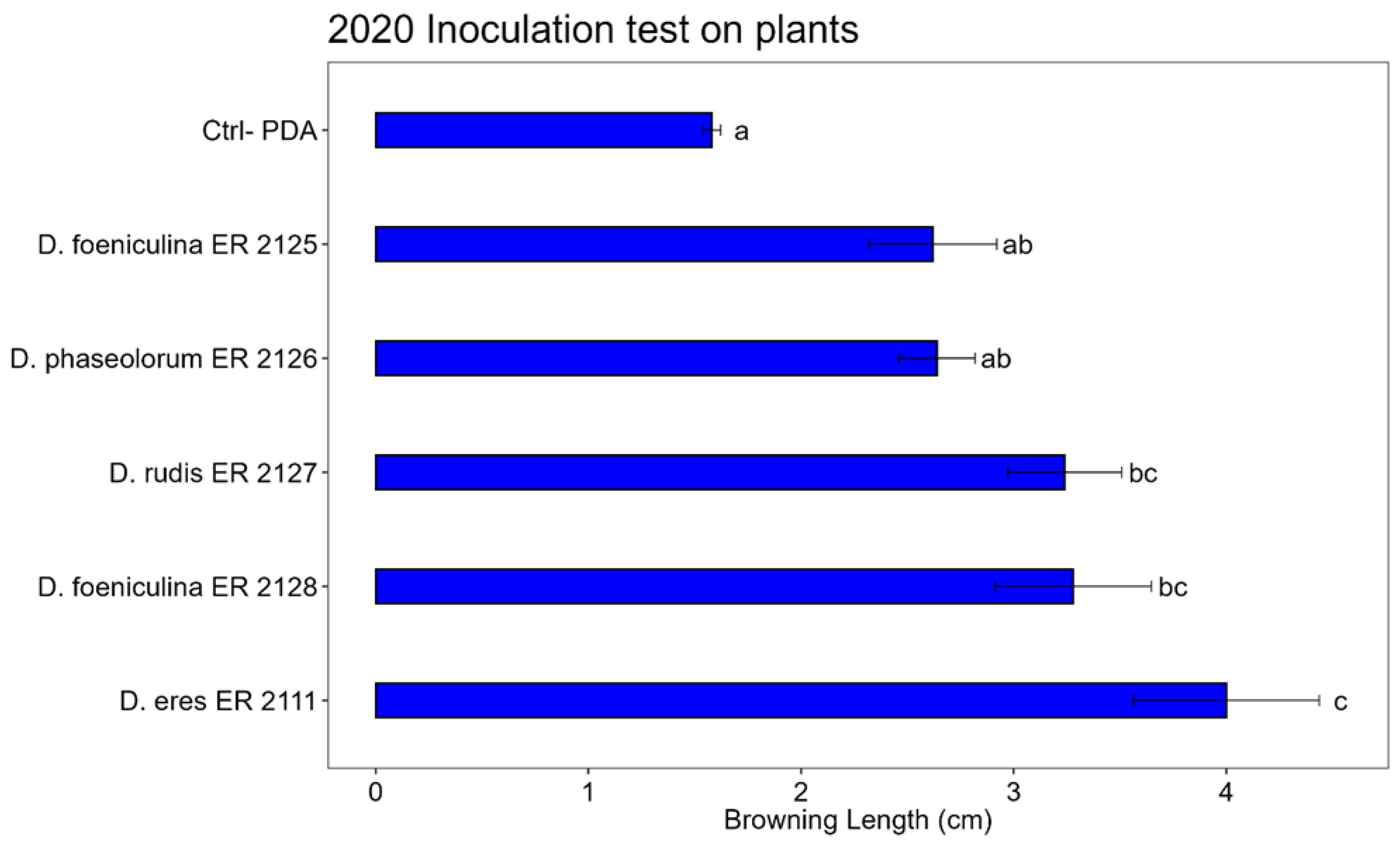
Figure 13.
(a) Pomegranate plants immediately after inoculation. (b) Pomegranate trunk with the outer bark layers removed, revealing a browned area at the end of the trial with D. eres. (c) Negative control: pomegranate trunk with the outer bark layers removed, inoculated with sterile PDA, showing healthy tissue.
Figure 13.
(a) Pomegranate plants immediately after inoculation. (b) Pomegranate trunk with the outer bark layers removed, revealing a browned area at the end of the trial with D. eres. (c) Negative control: pomegranate trunk with the outer bark layers removed, inoculated with sterile PDA, showing healthy tissue.


Table 1.
Description of the experimental units reported in Figure 2.
Table 1.
Description of the experimental units reported in Figure 2.
| Experimental Units | Section | Phenotypic Features |
|---|---|---|
| 1 | 1R8A+1R8B | Brown tissues above the canker from the apical portion (Plant 1) |
| 2 | 1R7A | Brown tissue from the lateral branch close to the canker (Plant 1) |
| 3 | 1R2A+1R8C | Asymptomatic tissues from branches (Plant 1) |
| 4 | 1R5A | Asymptomatic tissue (showing bark roughness) from the lateral branch (Plant 1) |
| 5 | 1T1A | Brown tissue from the basal area of the trunk, corresponding to the collar (Plant 1) |
| 6 | 1T1B | Brown tissue below the canker from the trunk (Plant 1) |
| 7 | 1T1C+1T1D | Brown tissues into the canker area (Plant 1) |
| 8 | 2T1A+2T1C | Brown tissues from the trunk (Plant 2) |
Table 2.
List of fungal species isolated from symptomatic pomegranate wood in the two sampling sites, with sampling time, representative strains and GenBank numbers. *Samples used for metabarcoding analysis, from all the 8 Experimental Units.
Table 2.
List of fungal species isolated from symptomatic pomegranate wood in the two sampling sites, with sampling time, representative strains and GenBank numbers. *Samples used for metabarcoding analysis, from all the 8 Experimental Units.
| Strain CREA-DC Collection Code | Species | Isolation incidence (% of the total fungal colonies) | Pavona | Sermoneta | GenBank Accession Number | |||
|---|---|---|---|---|---|---|---|---|
| July 2017 | October 2017 | March 2018* | September 2018 | February 2019 | ||||
| ER2123 | N. parvum | 27.6 | Absent | Present | Absent | Present | Absent | MW020287 |
| ER2111 | D. eres | 20.7 | Present | Absent | Present | Absent | Absent | MW020285 |
| ER2125, ER2128 | D. foeniculina | 17.2 | Absent | Present | Present | Absent | Absent | MW020288, MW032268 |
| ER2126 | D. phaseolorum | 3.4 | Absent | Present | Absent | Absent | Absent | MW020289 |
| ER2127 | D. rudis | 3.4 | Absent | Present | Present | Absent | Absent | MW032267 |
| ER2145 | Cytospora acaciae | 3.4 | Absent | Absent | Absent | Present | Absent | / |
| / | Eutypa lata | 0.5 | Absent | Absent | Present | Absent | Absent | / |
| ER2130 | Gliocladium spp. | 24.1 | Absent | Present | Present | Present | Present | / |
| / | Phoma glomerata | 5.3 | Absent | Absent | Present | Absent | Present | / |
| / | Neopestalotiopsis clavispora | 1 | Absent | Absent | Present | Absent | Absent | / |
| / | Fusarium spp. | 18.6 | Absent | Absent | Present | Absent | Absent | / |
Table 3.
ANOVA results conducted on length (cm) of browning caused by the inoculated fungi on the pomegranate detached twigs in the humid chamber in 2019.
Table 3.
ANOVA results conducted on length (cm) of browning caused by the inoculated fungi on the pomegranate detached twigs in the humid chamber in 2019.
| EFFECT | SS | DF | MS | F | ProbF |
|---|---|---|---|---|---|
| Isolate | 1.49215 | 5 | 0.29843 | 13.64701997 | 3.93174E-05 |
| Residual | 0.328016667 | 15 | 0.021867778 | ||
| Total | 1.94685 | 23 | 0.084645652 |
Table 4.
ANOVA results conducted on length (cm) of browning caused by the inoculated fungi on the pomegranate detached twigs in water in 2019.
Table 4.
ANOVA results conducted on length (cm) of browning caused by the inoculated fungi on the pomegranate detached twigs in water in 2019.
| EFFECT | SS | DF | MS | F | ProbF |
|---|---|---|---|---|---|
| Isolate | 8.419153439 | 5 | 1.683831 | 14.04785 | 4.26E-06 |
| Residual | 2.517142857 | 21 | 0.119864 | ||
| Total | 10.9362963 | 26 | 0.420627 |
Table 5.
ANOVA results conducted on length (cm) of browning caused by the inoculated fungi on the pomegranate plants in 2020.
Table 5.
ANOVA results conducted on length (cm) of browning caused by the inoculated fungi on the pomegranate plants in 2020.
| EFFECT | SS | DF | MS | F | ProbF |
|---|---|---|---|---|---|
| Isolate | 16.79066667 | 5 | 3.358133 | 9.631358 | 3.84E-05 |
| Residual | 8.368 | 24 | 0.348667 | ||
| Total | 25.15866667 | 29 | 0.86754 | ||
| C.V. (%): 20,4082959922632 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



