
Submitted:
03 February 2025
Posted:
04 February 2025
You are already at the latest version
Abstract
Mitophagy plays a critical role in maintaining mitochondrial quality and cellular homeostasis. But the specific contribution of mitophagy-related E3 ubiquitin ligases to prognosis remains largely unexplored. In this study, we identified a novel mitophagy-related E3 ubiquitin ligases prognostic signature using least absolute shrinkage and selector operator (LASSO) and multivariate Cox regression analyses in breast cancer. Based on median risk scores, patients were divided into high-risk and low-risk groups. Functional enrichment analyses were conducted to explore the biological differences between the two groups. Immune infiltration, drug sensitivity, and mitochondrial-related phenotypes were also analyzed to evaluate the clinical implications of the model. A four-gene signature (ARIH1, SIAH2, UBR5, and WWP2) was identified and Kaplan-Meier analysis demonstrated that the high-risk group had significantly worse overall survival (OS). High-risk patients exhibited disrupted mitochondrial metabolism and immune dysregulation with upregulated immune checkpoint molecules. Additionally, high-risk group exhibited higher sensitivity to several drugs targeting at Akt/PI3K/mTORC1 signaling axis. Accompanying mitochondrial metabolic dysregulation, mtDNA stress was elevated, contributing to the activation of senescence-associated secretory phenotype (SASP) in the high-risk group. In conclusion, the identified signature provides a robust tool for risk stratification and offers insights into the interplay between mitophagy, immune modulation, and therapeutic responses for breast cancer.
Keywords:
breast cancer
; prognostic signature
; mitophagy
; E3 ubiquitin ligase
; metabolic
; immune
1. Introduction
Breast cancer is the most common cancer worldwide, with its occurrence being influenced by various risk factors, including genetic and hereditary predisposition [1]. Current therapeutic strategies primarily involve endocrine therapy, anti-HER2 targeting, and chemotherapy. Despite these advancements, breast cancer mortality continues to rise [2,3]. The genomic landscape of diverse type of breast cancer, along with the tumor immune microenvironment (TIME), often leads to different immune infiltration and functionality, which eventually leads to the high recurrence risk and decreased probability of disease-free survival [4]. Overall, better prognostic signatures are needed that can accurately identify risk stratification and deliver personalized treatment.
Mitochondrial autophagy, commonly referred to as mitophagy, is a selective form of autophagy that plays a critical role in maintaining mitochondrial and cellular homeostasis [5]. However, the functions and mechanisms of mitophagy in breast cancer development and progression appear to be complex, as mitophagy acts as both a tumor suppressor and a tumor promoter depending on the specific context, microenvironment, or cancer stage [6]. Therefore, a deeper understanding of mitophagy-related molecules and their regulatory mechanisms is urgently needed to facilitate the development of more effective therapeutic strategies.
Existing studies have shown that mitophagy can reduce the production of reactive oxygen species (ROS) in tumor cells [7]. In tumor cells with BRCA1 or ULK1 depletion, mitophagy deficiency leads to intracellular ROS accumulation, which triggers NLRP3 inflammasome activation and promotes breast cancer cell growth and metastasis [8]. ROS accumulation also promotes the activation of HIF-1α, which drives the metabolic reprogramming of tumor cells from oxidative phosphorylation to aerobic glycolysis, thereby ensuring energy production during cancer development [9]. Moreover, in the tumor microenvironment (TME), mitophagy plays multifaceted roles, including mediating the metabolic reprogramming of cancer-associated fibroblasts [10], regulating immune cell function [11], and influencing oxidative stress [7].
The mechanism of mitophagy can be broadly categorized into ubiquitin-dependent and ubiquitin-independent pathways [12]. The ubiquitin-dependent pathway is primarily characterized by PINK1/Parkin-mediated mitophagy [13]. In this process, PINK1 accumulates on the outer mitochondrial membrane and phosphorylates ubiquitin, which subsequently recruits Parkin to the mitochondria. This recruitment activates Parkin's E3 ligase activity, leading to the generation of additional ubiquitin chains. This positive feedback loop facilitates the recruitment of autophagy receptor proteins that recognize ubiquitin chains and interact with LC3 family proteins, ultimately initiating the autophagosome biogenesis machinery [14]. However, the mRNA and protein levels of Parkin are frequently downregulated in breast cancer [15,16]. Considering the significance of PINK1/Parkin-mediated mitophagy, it is worthwhile to find out the other E3 ubiquitin ligases which affect the survival and investigate the mechanism for these potential biomarkers.
In this study, RNA sequencing and corresponding clinical data of breast cancer were obtained from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases for analysis. A mitophagy-related E3 ligase prognostic model comprising ARIH1, SIAH2, UBR5, and WWP2 was developed to predict patient prognosis. Based on the risk model scores, breast cancer patients were stratified into two groups. Notably, patients in the high-risk group exhibited poorer overall survival (OS). Our analysis further revealed significant differences in energy metabolism and the immune microenvironment between the two groups. The high-risk group is more sensitive to multiple drugs such as MK-2206/Rapamycin/Pictilisib. Data analysis indicates that the process of vesicles derived from the inner mitochondrial membrane (VDIMs), responsible for regulating mitochondrial quality, is notably inhibited in the high-risk group. Additionally, SASP driven by mtDNA stress is more pronounced in high-risk group. In summary, our mitophagy-related E3 ligase risk model shows potential as a prognostic biomarker for guiding personalized treatment strategies in breast cancer patients. The flowchart of this study is shown in Figure 1.
2. Results
2.1. Construction of Mitophagy-related E3 Ubiquitin Ligases Prognostic Model
Mitochondrial metabolism plays a crucial role in supporting tumor anabolism [17]. Mitophagy is a critical process for maintaining mitochondrial quality control [18]. Classical mitophagy pathways include PINK1/PARKIN-mediated mitophagy and receptor-mediated mitophagy [14]. Receptor-mediated mitophagy has been demonstrated to play a pivotal role in tumorigenesis and contributes to chemotherapy resistance [19,20,21]. PARKIN expression is frequently downregulated, mutated, or functionally absent in many tumors, particularly in breast cancer, limiting its role in this context [15,16]. Therefore, elucidating the underlying biological mechanisms of other E3 ubiquitin ligases involved in mitophagy, could provide novel perspectives and strategies for breast cancer treatment.
To further investigate mitophagy-related E3 ubiquitin ligases, we retrieved data from GeneCards using the keyword "mitophagy". Based on the "Description" category, 31 E3 ubiquitin ligases were identified. Combined with transcriptomic data and corresponding clinical data of BRCA obtained from TCGA database, statistical analysis of the expression levels of these ligases revealed that most exhibited significant differences (Figure S1A). Subsequently, we utilized the online analysis platform UALCAN (https://ualcan.path.uab.edu) to evaluate the OS of these mitophagy-related E3 ubiquitin ligases in BRCA patients (Figure S2, Figure 2C).
The survival analysis indicated that the higher expression of ARIH1, SIAH1, and UBR5 was associated with shorter OS in breast cancer patients, whereas the higher expression of SIAH2 correlated with prolonged survival. Univariate analysis (Figure S1B) revealed that SIAH2 had a protective effect on prognosis (hazard ratio (HR): 0.76, 95% confidence interval (CI): 0.64−0.89, p= 0.00059), whereas UBR5 was associated with poorer overall survival (OS) in BRCA patients (HR: 1.3, 95% CI: 1.0−1.6, p= 0.038).
The gene number was further narrowed down to 4 according to LASSO-Cox regression analysis (Figure 2A, B). Finally, a prognostic model for BRCA patients, referred to as ASUW, was constructed based on four mitophagy-related E3 ubiquitin ligases: ARIH1, SIAH2, UBR5, and WWP2. Multivariate regression analysis of these four ligases (Figure 2D) revealed that UBR5 was associated with poor prognosis (HR: 1.41, 95% CI: 1.11–1.81, p = 0.006), while SIAH2 had a favorable effect on prognosis (HR: 0.72, 95% CI: 0.61–0.85, p < 0.001). The risk score formula was established as follows: Risk score = ∑ (Coefᵢ × xᵢ), where xᵢ represents the normalized expression level of the target gene i and Coefᵢ is the corresponding regression coefficient. These genes and corresponding coefficients were shown in Table 1.
Figure 2.
LASSO regression and clinical analyses of prognostic model in breast cancer. (A, B) Results of LASSO regression analysis. (C) Kaplan-Meier survival curves of 4 mitophagy-related E3 ubiquitin ligases (ARIH1, SIAH2, UBR5, WWP2) in BRCA. (D) Multivariate Cox regression analysis of the four selected E3 ubiquitin ligases (ARIH1, SIAH2, UBR5, and WWP2). HRs with 95% CI are displayed. (E) Multivariate Cox regression analysis integrating the risk score with clinical characteristics. HRs with 95% CI are shown. T tumor, N lymph node, M metastasis.
Figure 2.
LASSO regression and clinical analyses of prognostic model in breast cancer. (A, B) Results of LASSO regression analysis. (C) Kaplan-Meier survival curves of 4 mitophagy-related E3 ubiquitin ligases (ARIH1, SIAH2, UBR5, WWP2) in BRCA. (D) Multivariate Cox regression analysis of the four selected E3 ubiquitin ligases (ARIH1, SIAH2, UBR5, and WWP2). HRs with 95% CI are displayed. (E) Multivariate Cox regression analysis integrating the risk score with clinical characteristics. HRs with 95% CI are shown. T tumor, N lymph node, M metastasis.
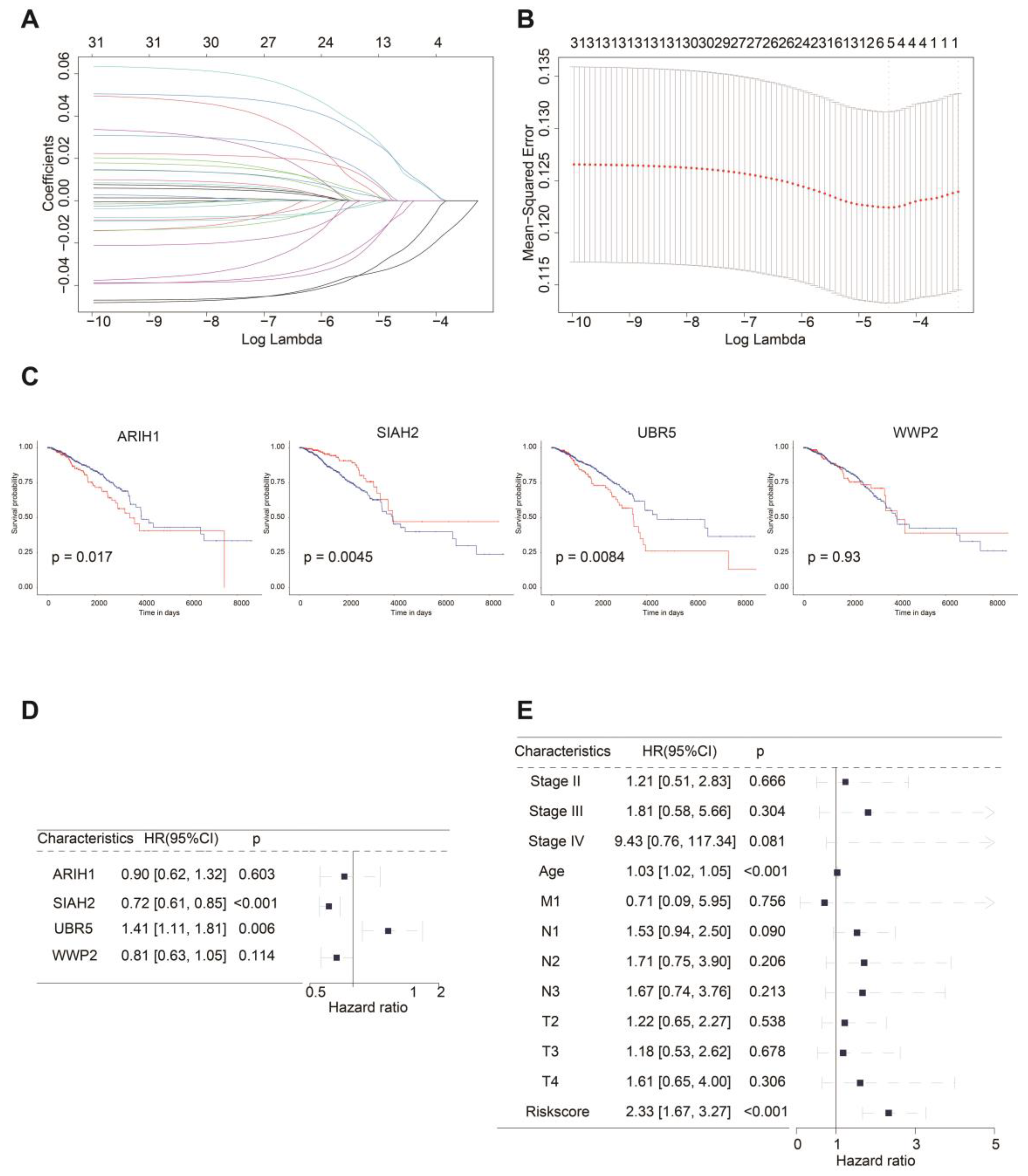
Assessment of the effectiveness of risk scores and clinical characteristics (age, stage, T-typing, N-typing, M-typing) on prognosis by multivariate Cox regression was conducted to determine whether risk scores could independently predict patient survival (Figure 2E). The results demonstrated that the risk score served as an independent prognostic factor (HR: 2.33, 95% CI: 1.67–3.27, p < 0.001).
To compare the expression levels of ARIH1, SIAH2, UBR5, and WWP2 across different molecular subtypes and stages of BRCA, we analyzed their expression differences using the UALCAN online database (Figure S3). Significant differences were identified for ARIH1, SIAH2, UBR5, and WWP2 in breast cancer stages: ARIH1 showed significant differences in Normal-vs-Stages 1, 2, 3, and 4 (p < 0.05) (Figure S3A); SIAH2 in Normal-vs-Stages 1, 2, 3, and 4 (p < 0.05) (Figure S3C); UBR5 in Normal-vs-Stages 1, 2, and 3 (p < 0.05) (Figure S3E); and WWP2 in Normal-vs-Stages 2 and 3 (p < 0.05) (Figure S3G). Similarly, differences in molecular subtypes were observed: ARIH1 showed significant differences in Normal-vs-Luminal and Normal-vs-TNBC (p < 0.05) (Figure S3B); SIAH2 in Normal-vs-Luminal, Normal-vs-TNBC, Luminal-vs-HER2 Positive, Luminal-vs-TNBC, and HER2 Positive-vs-TNBC (p < 0.05) (Figure S3D); UBR5 in Normal-vs-Luminal, Normal-vs-TNBC, and Luminal-vs-HER2 Positive (p < 0.05) (Figure S3F); and WWP2 in Normal-vs-Luminal, Luminal-vs-TNBC, and HER2 Positive-vs-TNBC (p < 0.05) (Figure S3H).
2.2. ASUW Prognostic Model Effectively Predicted Poor Breast Cancer Outcomes
To assess the prognostic value of the four mitophagy-related E3 ubiquitin ligases signature, patients in the TCGA-BRCA cohort were classified into high-risk and low-risk groups based on the median risk score. Kaplan-Meier curves revealed that patients in the high-risk group had significantly shorter OS compared to those in the low-risk group (p < 0.0001; Figure 3A). The distribution of risk scores and survival status across the two risk subgroups was illustrated in Figure 3C, showing a higher number of deaths in the high-risk group. The heatmap of the genes of 4 mitophagy-related E3 ubiquitin ligases was also shown in Figure 3C. To further evaluate the predictive accuracy of the ASUW prognostic model, time-dependent receiver operating characteristic (ROC) curve analysis was performed. The area under the curves (AUCs) for 5-year, 10-year, and 15-year survival predictions in the TCGA-BRCA cohort were 0.648, 0.654, and 0.552, respectively (Figure 3E), showing the model's potential for survival prediction.
The efficiency of the prognostic risk model was further validated using the GSE25066 dataset. The same risk formula was applied to the validation cohort from the GEO dataset (GSE25066), and patients were also stratified into high-risk and low-risk groups based on the median risk score. Consistent with the TCGA-BRCA training cohort, Kaplan-Meier survival analysis demonstrated that patients in the high-risk group had significantly worse overall survival (OS) compared to those in the low-risk group (p = 0.0031; Figure 3B). Additionally, higher risk score was correlated with poorer survival outcomes (Figure 3D). Notably, the expression patterns of the four mitophagy-related E3 ubiquitin ligases in the GSE25066 validation cohort closely mirrored those observed in the TCGA-BRCA training cohort. In GSE25066 dataset, the AUCs of the ROC curve for 1-year, 3-year, and 5-year survival were 0.690, 0.665, and 0.634, respectively (Figure 3F). These findings underscored the reliability of the ASUW model as an effective prognostic tool for BRCA patients.
2.3. Functional Enrichment Analyses of DEGs between Two Groups
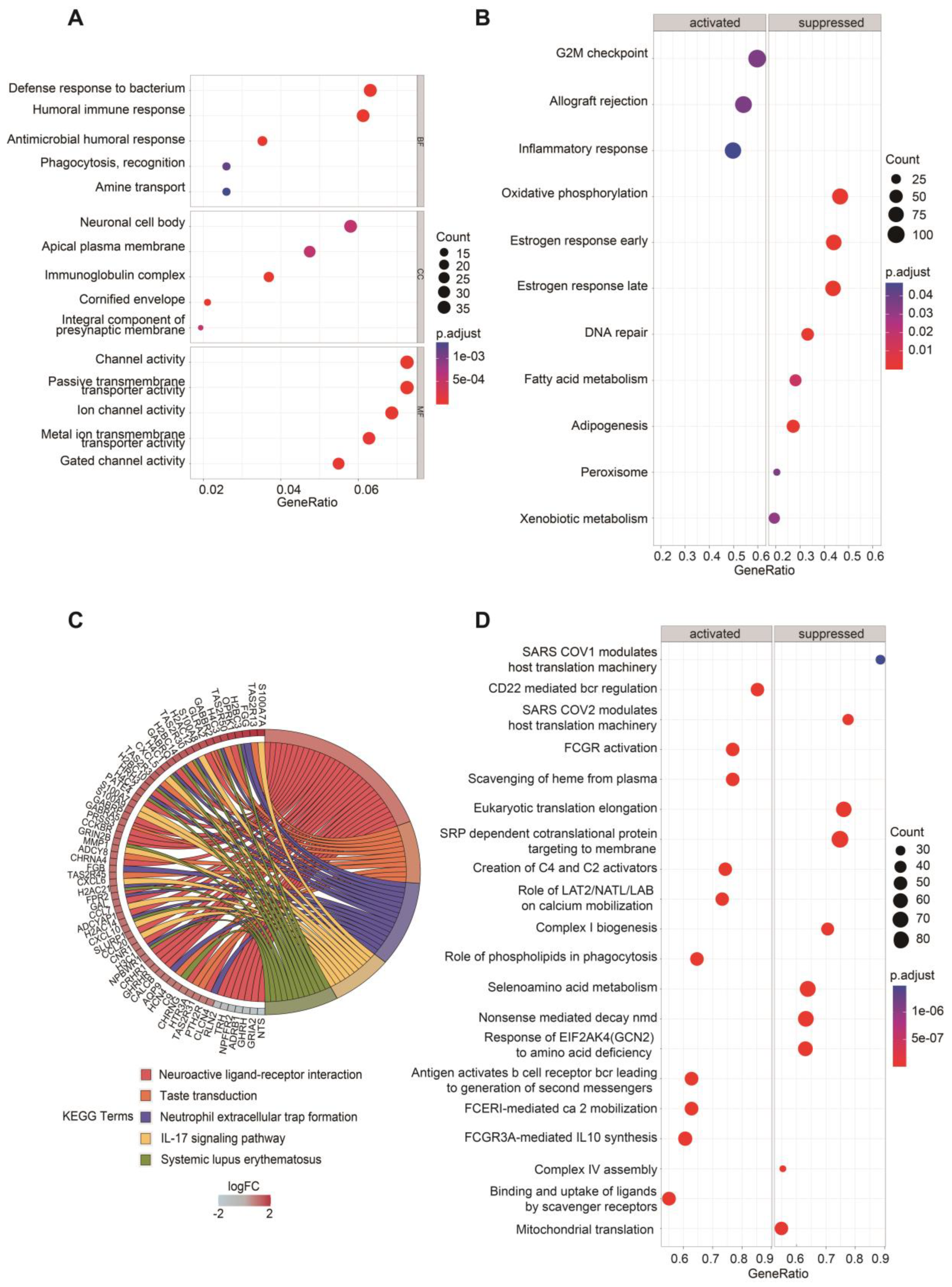
Given the significantly different OS observed between the two groups, we sought to further investigate the biological differences between the high-risk and low-risk groups. Differentially expressed genes (DEGs) were identified using the DESeq2 package, and their distribution was visualized through volcano plots to compare high-risk and low-risk groups (Figure S4A, B). Functional enrichment analyses were subsequently performed to explore the biological significance of these DEGs in the two groups. Gene Ontology (GO) enrichment analysis revealed that DEGs annotated to biological processes (BP) were primarily associated with defense response to bacterium, humoral immune response, antimicrobial humoral response, phagocytosis, recognition, and amine transport. DEGs annotated to cellular component (CC) categories were predominantly enriched in neuronal cell body, apical plasma membrane, immunoglobulin complex, cornified envelope, and integral component of presynaptic membrane. For molecular function (MF), the enriched terms included channel activity, passive transmembrane transporter activity, ion channel activity, metal ion transmembrane transporter activity, and gated channel activity (Figure 4A).
Additionally, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis highlighted the top five enriched pathways as neuroactive ligand-receptor interaction, taste transduction, neutrophil extracellular trap formation, IL-17 signaling pathway and systemic lupus erythematosus. The genes enriched in these pathways were visualized in the left half of the circle plot, showing their involvement in these key pathways (Figure 4C).
For the HALLMARK gene set collection defined by the Molecular Signatures Database (MSigDB), Gene Set Enrichment Analysis (GSEA) was conducted to further compare the biological differences between the two groups (Figure 4B). The results revealed that the activated pathways in the high-risk group included G2M checkpoint, allograft rejection, and inflammatory response. In contrast, metabolism-related gene sets such as oxidative phosphorylation, fatty acid metabolism, adipogenesis, peroxisome, and xenobiotic metabolism were significantly suppressed in the high-risk group. Additionally, DNA repair, estrogen response early and estrogen response late were also inhibited in the high-risk group.
To gain deeper insights into the metabolic differences between two groups, the Reactome gene set collection from MSigDB was utilized (Figure 4D). The top 10 activated and suppressed pathways, ranked by the Normalized Enrichment Score (NES), were identified. Notably, mitochondrial-related pathways, including mitochondrial translation, complex I biogenesis and complex IV assembly were significantly suppressed in the high-risk group. What’s more, immune-related pathways, such as antigen activates B cell receptor leading to generation of second messengers and FCGR3A-mediated IL-10 synthesis” were activated in the high-risk group.
Figure 4.
Enrichment analyses of DEGs of two groups. (A) GO enrichment analysis of DEGs between the high- and low-risk groups based on the ASUW model. The size of the dots indicates the ratio of genes enriched, while the colors represent the adjusted p-value. (B) GSEA of “Hallmark gene sets” between high-risk and low-risk groups. Significant pathways were categorized as activated (left panel) or suppressed (right panel). The size of the dots represents the ratio of enriched genes, while the color indicates the adjusted p-value. (C) KEGG enrichment analysis of DEGs. A chord diagram demonstrates the associations between the 5 top enriched KEGG pathways and the corresponding genes. The colors of the bands correspond to log (Foldchange, FC) values, showing upregulated (red) and downregulated (blue) genes in the high-risk group. (D) GSEA of “Reactome gene sets” between high-risk and low-risk groups. Significant pathways were categorized as activated (left panel) or suppressed (right panel). The size of the dots represents the ratio of enriched genes, while the color indicates the adjusted p-value.
Figure 4.
Enrichment analyses of DEGs of two groups. (A) GO enrichment analysis of DEGs between the high- and low-risk groups based on the ASUW model. The size of the dots indicates the ratio of genes enriched, while the colors represent the adjusted p-value. (B) GSEA of “Hallmark gene sets” between high-risk and low-risk groups. Significant pathways were categorized as activated (left panel) or suppressed (right panel). The size of the dots represents the ratio of enriched genes, while the color indicates the adjusted p-value. (C) KEGG enrichment analysis of DEGs. A chord diagram demonstrates the associations between the 5 top enriched KEGG pathways and the corresponding genes. The colors of the bands correspond to log (Foldchange, FC) values, showing upregulated (red) and downregulated (blue) genes in the high-risk group. (D) GSEA of “Reactome gene sets” between high-risk and low-risk groups. Significant pathways were categorized as activated (left panel) or suppressed (right panel). The size of the dots represents the ratio of enriched genes, while the color indicates the adjusted p-value.
These findings suggest that risk score is closely associated with alterations in metabolism and the status of TME, highlighting the interplay between mitochondrial function, immune responses, and cancer progression.
2.4. TIME Comparisons between Two Groups Upon ASUW Prognostic Model
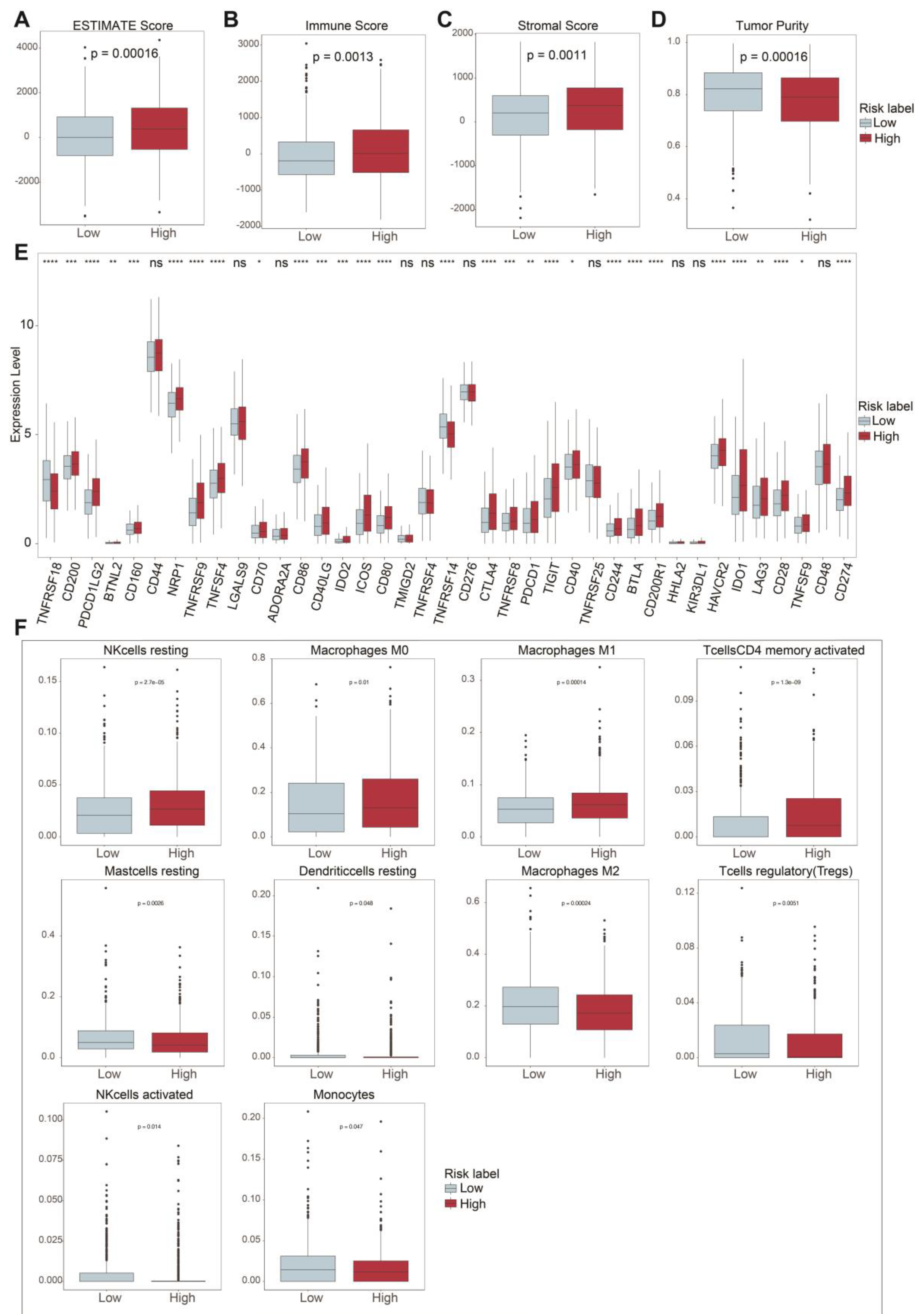
Mitophagy dysregulation has been shown to play a crucial role in shaping an immunosuppressive tumor microenvironment, thereby facilitating cancer initiation and progression. To further explore the correlation between the risk score and immune microenvironment, we used ESTIMATE to analyze differences in TIME. Patients in the high-risk group exhibited higher ESTIMATE Score (p= 0.00016; Figure 5A). Consistently, the high-risk group also showed elevated Immune Scores (p = 0.0013; Figure 5B) and Stromal Scores (p = 0.0011; Figure 5C) compared to the low-risk group. Additionally, tumor purity was significantly lower in the high-risk group (p = 0.00016; Figure 5D). These findings suggest that the high-risk group was characterized by a higher level of immune infiltration.
The overexpression of immune checkpoint molecules is frequently linked to immune escape mechanisms [4], and targeting these checkpoints through immunotherapy has proven to be an effective strategy for BRCA treatment [22]. We further examined the expression of several immune checkpoint related genes between the high-risk and low-risk groups. Immune checkpoint genes [23,24,25,26,27,28,29,30,31,32], including PD-L1 (CD274), CTLA-4, TIGIT, HAVCR2 (TIM3), BTLA, CD200, CD200R1, IDO1, IDO2 and CD160, were significantly upregulated in the high-risk group (Figure 5E). Among these, PD-L1, CTLA-4, TIM3, LAG3, and TIGIT are key immune checkpoint molecules that play essential roles in regulating immune responses and have been utilized in clinical therapies [33,34,35,36,37].
Using CIBERSORT, we analyzed the proportions of immune cells, as depicted in Fig S5A. The analysis revealed the distribution of 22 immune cell types in breast cancer patients. Additionally, there was a certain degree of correlation among these immune cells, as shown in Figure S5B. To further investigate the immune microenvironment, we compared the differences in immune cell proportions between the high-risk and low-risk groups. Out of the 22 immune cell types, 10 exhibited significant changes. The proportions of T cells CD4 memory activated, resting NK cells, Macrophages M0, and Macrophages M1 were upregulated in the high-risk group, while the proportions of regulatory T cells (Tregs), activated NK cells, Monocytes, Macrophages M2, resting Dendritic cells, and resting Mast cells were downregulated (Figure 5F). These results highlighted distinct immune cell dynamics between the two risk groups, suggesting a potential role of immune cell composition in shaping the tumor microenvironment and influencing prognosis.
Figure 5.
Immune-related analysis between two groups. (A) Total ESTIMATE score indicates tumor purity and immune-stromal components differences (p = 0.00016). (B) Immune score indicates immune infiltration differences (p = 0.0013). (C) Stromal score indicates stromal content differences (p = 0.0011). (D) Tumor purity score indicates tumor purity differences (p = 0.00016). (E) Comparison of immune checkpoint-related gene expression between the two risk groups. (F) Proportions of immune cell types with significant differences between high- and low-risk groups. Statistical significance is indicated as: ns (not significant), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Figure 5.
Immune-related analysis between two groups. (A) Total ESTIMATE score indicates tumor purity and immune-stromal components differences (p = 0.00016). (B) Immune score indicates immune infiltration differences (p = 0.0013). (C) Stromal score indicates stromal content differences (p = 0.0011). (D) Tumor purity score indicates tumor purity differences (p = 0.00016). (E) Comparison of immune checkpoint-related gene expression between the two risk groups. (F) Proportions of immune cell types with significant differences between high- and low-risk groups. Statistical significance is indicated as: ns (not significant), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
2.5. Prediction of Sensitive Drugs for BRCA upon ASUW Prognostic Model
Mitochondria are essential for tumor anabolism, providing critical building blocks, regulating redox and calcium homeostasis, contributing to transcriptional control, and orchestrating cell death [38,39]. Consequently, mitochondrial dysfunction plays a pivotal role in determining the efficacy of innovative anticancer therapies. To further explore the clinical relevance of the ASUW prognostic model for BRCA treatment, the ability of the risk score to predict responses of 198 drugs was analyzed using the "Oncopredict" tool and the GDSC2 database. Drug sensitivity, measured by half-maximal inhibitory concentration (IC50) values, was compared between high- and low-risk groups. The findings revealed that patients in the high-risk group exhibited increased sensitivity to MK-2206, Pictilisib, Rapamycin, Sorafenib, GSK1904529A, Uprosertib, LGK974, Elephantin, AZD5363, Ipatasertib, and AT13148, but decreased sensitivity to BI-2536 (Figure 6). Notably, MK-2206, Pictilisib, Rapamycin, Uprosertib, AZD5363, and Ipatasertib are drugs targeting the PI3K/Akt/mTOR signaling pathway. Detailed information of these 12 drugs was provided in Table 2. These results highlight the potential clinical utility of ASUW prognostic model in guiding personalized therapeutic strategies for BRCA patients.
2.6. mtDNA Stress, SASP, VDIM Analyses for Two Groups Upon ASUW Prognostic Model
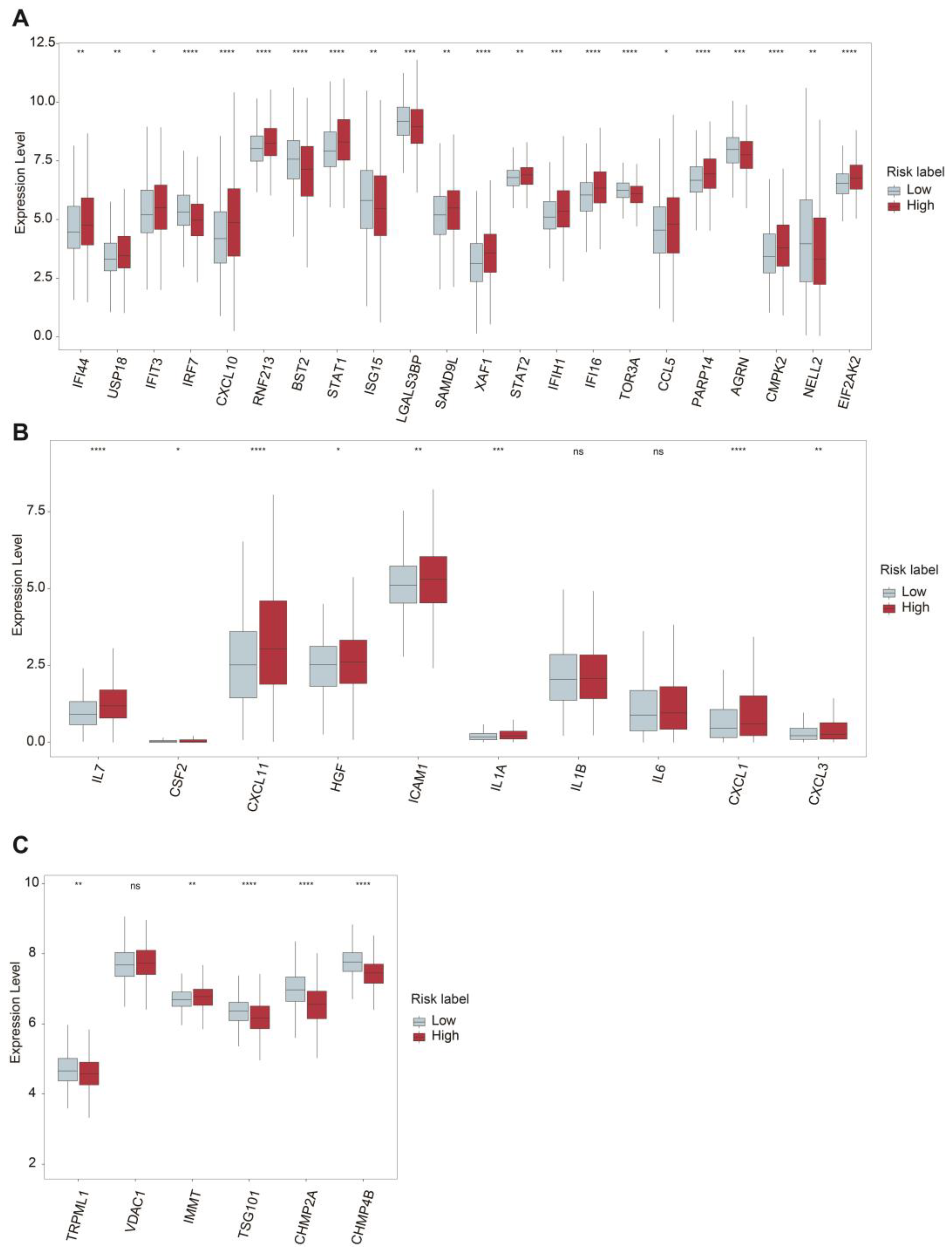
Mitochondrial dysfunction is a hallmark of aging and cellular senescence, significantly influencing the development of senescence-associated secretory phenotype (SASP), which actively reshape the tumor microenvironment and facilitate immune evasion by tumors [57,58]. In our analysis, we compared the expression of mtDNA-stress signature genes between the high-risk and low-risk groups. Among these genes, IFI44, USP18, IFIT3, CXCL10, RNF213, STAT1, SAMD9L, XAF1, STAT2, IFIH1, IFI16, CCL5, PARP14, CMPK2, and EIF2AK2 were significantly upregulated in the high-risk group, while IRF7, BST2, STAT1, ISG15, LGALS3BP, TOR3A, AGRN, and NELL2 were down regulated in the low-risk group (Figure 7A).
Additionally, the expression levels of several SASP-associated genes were examined. Genes such as IL7, CSF2, CXCL11, HGF, ICAM1, IL1A, CXCL1, and CXCL3 were significantly upregulated in the high-risk group, whereas IL1β and IL6 did not show significant differences between the two groups (Figure 7B). These findings highlighted the potential role of SASPs in modulating the TME and promoting tumor progression in the high-risk group.
The maintenance of mitochondrial homeostasis relies on the coordinated activity of various quality control mechanisms [59,60]. Among these, mitophagy is a critical process responsible for the removal of damaged mitochondria. Recently, a newly discovered pathway involving vesicles derived from the inner mitochondrial membrane (VDIMs) has been identified especially as a steady-state mechanism for mitochondrial quality control. The formation of VDIMs is primarily dependent on key molecules, including TRPML1, VDAC1, IMMT, TSG101, CHMP2A, and CHMP4B, which participate in the four main stages of VDIM formation [61].
Our analysis revealed that TRPML1, TSG101, CHMP2A, and CHMP4B were significantly downregulated in the high-risk group. While the expression level of IMMT was increased in the high-risk group. Interestingly, the expression level of VDAC1 showed no significant changes between the two groups (Figure 7C). These results suggest a potential impairment in VDIMs-related mitochondrial quality control mechanisms in the high-risk group, contributing to mitochondrial dysfunction and its associated pathological effects.
Figure 7.
Comparative expression analysis of mtDNA Stress, SASP, and VDIM-Related Genes between two groups. (A) Differential expression levels of mtDNA stress-related genes between high- and low-risk groups. (B) Differential expression levels of SASP-related genes. (C) Differential expression levels of critical proteins involved in the VDIM process. Statistical significance is indicated as: ns (not significant), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Figure 7.
Comparative expression analysis of mtDNA Stress, SASP, and VDIM-Related Genes between two groups. (A) Differential expression levels of mtDNA stress-related genes between high- and low-risk groups. (B) Differential expression levels of SASP-related genes. (C) Differential expression levels of critical proteins involved in the VDIM process. Statistical significance is indicated as: ns (not significant), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
3. Discussion
Aberrant regulation of mitophagy has been linked to a variety of diseases, including breast cancer progression. In breast cancer, mitophagy-related signaling molecules demonstrate abnormal expression patterns and play critical roles in biological processes [6]. Mitochondrial function is closely associated with metabolic adaptation, cell proliferation and senescence, metastasis [20,62,63]. Recent studies have revealed that mitophagy actively reshapes the immune microenvironment, contributing to an immunosuppressive tumor microenvironment that diminishes the efficacy of immunotherapy [64,65,66]. A deeper understanding of the intricate relationship between tumors and mitophagy offers opportunities to propose novel therapeutic strategies. While previous studies have sought to identify mitophagy-related biomarkers at a broad level [67,68], the specific role of mitophagy-related E3 ubiquitin ligases in predicting breast cancer prognosis remains largely unexplored.
This study identified a novel mitophagy-related E3 ubiquitin ligase signature that can effectively predict prognosis in BRCA patients. The risk score derived from this model was significantly associated with metabolism dysfunction, immune microenvironment, and drug sensitivity, highlighting its potential as a prognostic biomarker and therapeutic target.
After constructing the prognostic model consisting of ARIH1, SIAH2, UBR5, and WWP2, TCGA-BRCA patients were stratified into two groups based on the median risk score. The expression level of 4 E3 ligases were all significantly changed compared to normal tissue especially in luminal type of BRCA. Notably, ARIH1 is a key E3 ubiquitin ligase involved in mitophagy via a Parkin-independent pathway, particularly in response to mitochondrial damage, ultimately influencing chemotherapy sensitivity [69].
Currently, no direct studies have confirmed whether SIAH2, UBR5, or WWP2 promote or inhibit mitophagy. However, knocking out SIAH2 has been shown to prevent the degradation of mitochondrial NCX3, regulate mitochondrial fission and fusion dynamics, and restore mitochondrial function in hypoxic neurons [70]. SIAH1, a member of the same family as SIAH2, has been reported to promote mitophagy through the PINK1-synphilin-1-SIAH1 signaling axis, contributing to the progression of Parkinson's disease [71]. Conversely, SIAH3, which lacks catalytic activity, has been shown to inactivate PINK1 and accumulate on damaged mitochondria, thereby inhibiting mitophagy [72]. In colorectal carcinoma, UBR5 knockdown inhibits cancer cell proliferation by reducing the expression of PYK2, which subsequently downregulates oxidative phosphorylation and suppresses metabolic reprogramming [73]. Meanwhile, studies have suggested that WWP2 may play a role in the degradation of mitophagy receptors, such as NDP52, OPTN, and SQSTM [74]. However, direct evidence linking WWP2 to regulation of mitophagy through this pathway is currently lacking. Considering these findings, further research is urgently needed to elucidate the roles of ARIH1, SIAH2, UBR5, and WWP2 in modulating mitophagy, mitochondrial homeostasis, and drug resistance in cancer.
As the Kaplan-Meier survival analysis demonstrated a significant association between a high-risk group and poor prognosis, we further examined the biological differences between the high- and low-risk groups. GSEA results revealed that pathways such as the G2M checkpoint and inflammatory response were upregulated in the high-risk group. These pathways are well-known contributors to poor survival, reduced drug responsiveness, and a more immunosuppressive tumor microenvironment [75,76]. Additionally, estrogen response (early and late) pathways were downregulated in the high-risk group. Given the established association between estrogen receptor positivity and lower recurrence rates [22], the downregulated estrogen response further explains the poor prognosis observed in the high-risk group.
GO enrichment analysis highlighted both immune dysregulation and apparent ion channel activity dysregulation in the high-risk group. Worth noting, ion channel activity is critical for maintaining lysosomal and mitochondrial homeostasis [77,78]. The proper functioning of mitochondria largely relies on electrochemical proton gradient. A key component of this gradient, the inner mitochondrial membrane potential, is tightly regulated by ion transport across mitochondrial membranes [79]. These results corresponded that mitophagy have connection with ion channel activity.
As we observed that pathways related to defense response to bacterium, antimicrobial humoral response, and phagocytosis recognition were all upregulated in the high-risk group according to GO enrichment results, we hypothesized that microbes hijacking tumor cells may act as drivers of tumor cell seeding. Supporting this notion, the upregulation of neutrophil extracellular trap formation and IL-17 signaling pathways, as revealed by KEGG enrichment analysis, aligns with findings from previous studies [80].
Recently, accumulating evidence has shown that human tumors harbor substantial amounts of viable commensal microbiota [81,82,83]. For instance, a recent study demonstrated that a gut microbial metabolite activates mitophagy to regulate microglia-mediated neuroinflammation and mitigate the progression of herpes simplex encephalitis [84]. Additionally, Escherichia coli Nissle 1917 was found to inhibit mitophagy and ameliorate mitochondrial damage by promoting IL-22 expression in polycystic ovary syndrome mice [85]. These findings suggest that microbiota can influence the immune environment through mitophagy, raising the intriguing possibility of a connection between tumor-resident intracellular microbiota and mitophagy. Further investigation into this relationship could provide valuable insights, potentially uncovering drug targets such as E3 ubiquitin ligases that regulate mitophagy, thereby offering novel therapeutic strategies for cancer treatment.
The enrichment analysis indicated immune dysregulation in the high-risk group, which was further supported by ESTIMATE analysis showing higher immune infiltration accompanied by an immunosuppressive status in the high-risk group. Dysregulation of mitophagy has been demonstrated to play a significant role in establishing an immunosuppressive microenvironment, thereby fostering cancer initiation and progression [86]. Studies have revealed that mitophagy shapes the tumor microenvironment by regulating the PD-1/PD-L1 signaling pathway. Impaired mitophagy weakens antitumor immune responses and facilitates immune evasion, as observed in hepatocellular carcinoma. Parkin, an E3 ubiquitin ligase, has been shown to induce the ubiquitination and degradation of PD-1, ultimately modulating antitumor immunity through the PD-1/PD-L1 pathway [87].
Moreover, defective mitophagy results in the accumulation of damaged mitochondria, leading to increased reactive oxygen species production and the release of mitochondrial DNA. Mitochondrial homeostasis is maintained by the removal of damaged cristae via vesicles derived from the inner mitochondrial membrane (VDIMs) at steady status. The significant downregulation of TRPML1, TSG101, CHMP2A, and CHMP4B in the high-risk group suggests impaired VDIM-mediated mitochondrial quality control, potentially contributing to mtDNA stress and the activation of senescence-associated secretory phenotypes. These changes promote immunosuppressive signaling and recruit tumor-promoting immune cells, such as myeloid-derived suppressor cells and regulatory T cells (Tregs) [86]. Intriguingly, a recent study developed a mitochondrial localized in situ self-assembly system that shifts mitophagy from a pro-survival to a pro-death mechanism, transforming the immunosuppressive tumor microenvironment into an immunostimulatory one [88]. Overall, dysregulated mitophagy disrupts the balance of the immune and metabolic microenvironment, thereby creating conditions that support cancer development and progression.
Notably, the enhanced drug sensitivity to many PI3K/Akt/mTOR inhibitors in the high-risk group suggests that these patients may benefit from targeted therapies, offering a potential strategy for improving treatment outcomes [89]. Recent studies have highlighted the interplay between the PI3K/Akt/mTOR signaling pathway and mitophagy in maintaining mitochondrial homeostasis. For example, inhibition of SGLT1 was found to increase mitophagy and restore mitochondrial function by suppressing the PI3K/Akt pathway in radiation-induced intestinal injury, underscoring the critical role of this pathway in modulating mitophagy [90]. Similarly, metformin treatment, while alleviating ultraviolet radiation-induced cellular damage, regulated the PI3K/Akt/mTOR pathway, company with reduced mitochondrial oxidative stress and decreased mitophagy [91]. These findings suggest that dysregulation of the PI3K/Akt/mTOR signaling pathway not only affects mitochondrial quality control via mitophagy but also exacerbates oxidative stress, thereby establishing a connection to finding therapeutic strategies for conditions involving mitochondrial dysfunction. However, the specific role of mitophagy-related E3 ubiquitin ligases in regulating the crosstalk between PI3K/Akt/mTOR signaling and mitophagy remains an area that needs further investigation.
While this study leveraged comprehensive transcriptomic datasets to construct and validate the prognostic model, further experimental validation is necessary to confirm the functional roles of the identified E3 ubiquitin ligases. Additionally, incorporating multi-omics data, such as proteomics and metabolomics, could provide deeper insights into the underlying mechanisms.
In conclusion, this study sheds light on the intricate relationship between mitophagy, tumor microenvironment, and breast cancer prognosis. The identified mitophagy-related E3 ligase signature holds promise as a prognostic biomarker and therapeutic target, paving the way for personalized treatment strategies in breast cancer.
4. Materials and Methods
4.1 Data collection and preparation of BRCA
The gene expression profiles and clinical data for breast cancer were obtained from the TCGA database via UCSC Xena. After excluding samples lacking corresponding clinical information, 111 normal samples and 1,052 tumor samples with both clinical and expression data were included for further analysis.
For external validation, the dataset GSE25066 was downloaded from the Gene Expression Omnibus (GEO). GSE25066 was generated using the GPL96 platform (Affymetrix Human Genome U133A Array) and comprises 508 tumor samples of breast cancer patients. Probe identifiers were mapped to gene symbols according to the platform annotation for GSE25066 dataset. Patients from the TCGA dataset were used for the training cohort, while the GSE25066 dataset was used for external validation.
4.2 Mitophagy-related gene collection
A total of 5,177 mitophagy-related genes were identified from GeneCards. Based on their descriptions, 31 mitophagy-related E3 ubiquitin ligases were selected for further analysis.
4.3 Univariate Cox regression analysis
Univariate Cox regression analysis was performed to evaluate the prognostic significance of individual mitophagy-related E3 ubiquitin ligases. The hazard ratio and corresponding 95% confidence interval were calculated for each gene to evaluate its association with overall survival in BRCA.
4.4. Construction of a prognostic model
The 31 mitophagy-related E3 ubiquitin ligases were further refined using least absolute shrinkage and selection operator regression analysis, conducted with the R package “glmnet”, to identify key genes for constructing a multivariate Cox regression model. Regression coefficients (Coefᵢ) for each selected gene included in the prognostic model were determined through multivariate Cox regression analysis. Subsequently, the risk score for each patient in the TCGA dataset was calculated based on the formula derived from the multivariate Cox model. Patients were stratified into low-risk and high-risk groups using the median risk score as the cut-off value. The same regression coefficient was used to calculate the risk score of patients in the GSE25066 dataset, patients were divided into low-risk and high-risk groups according to the median risk score.
4.5. Overall survival analysis
Survival curves were generated using the Kaplan–Meier method, and the log-rank test was applied to assess survival differences between the two groups. Statistical significance was defined as a p-value < 0.05. Additionally, the R package “survivalROC” was utilized to generate time-dependent receiver operating characteristic curves, allowing for the evaluation of the prognostic model's predictive performance. The area under the curve values were calculated to quantify the accuracy of the model in predicting survival probabilities over specific time intervals. The R package “ggrisk” was used to generate the risk score distribution, survival status and heatmap. This risk factor association plot provided an integrated view of the relationship between risk scores, survival status, and gene expression, enabling a comprehensive assessment of the prognostic model.
4.6 DEG identification
Differentially expressed genes (DEGs) between two groups were identified through the R package “Deseq2”. |Log (2) fold change|>1 and adjusted P < 0.05 were the criteria for defining DEGs. R package “pheatmap” and “ggplot2” were employed to generate volcano map and heatmap to visualize the DEGs.
4.7 Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses
To better understand the underlying differences between two groups, enrichment analyses, including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG), were conducted using the R packages “clusterProfiler”, “org.Hs.eg.db”, “enrichplot”, and “ggplot2”. A significance threshold of p < 0.05 was applied. The GO analysis categorized the enriched terms into three domains: biological process (BP), cellular component (CC), and molecular function (MF), providing insights into the biological roles and mechanisms associated with the DEGs. These analyses were instrumental in uncovering the functional annotations and pathway associations of the identified DEGs.
4.8 Gene set enrichment analysis (GSEA)
The R packages “clusterProfiler” was used to perform GSEA to evaluate the functional pathway differences between the two groups. Enrichment analysis was conducted using the Hallmark and Reactome gene sets from the Molecular Signatures Database. Pathways with Benjamini & Hochberg (BH) adjusted p-values < 0.05 were considered significantly enriched. The results were visualized using the R packages “enrichplot”, providing a presentation for the pathway significantly enriched.
4.9 ESTIMATE analysis
The ESTIMATE algorithm was employed to evaluate the tumor microenvironment's stromal and immune components by the R package “estimate”. This method quantifies stromal and immune cell infiltration within tumor samples based on gene expression profiles, providing four key scores: stromal score, immune score, tumor purity and ESTIMATE score, which collectively reflect tumor purity. Gene expression profiles were input into the ESTIMATE algorithm to calculate the stromal and immune scores for each sample. These scores were used to compare the tumor microenvironment characteristics between the high-risk and low-risk patient groups. The results were visualized using the R packages “ggplot2” to highlight differences in tumor microenvironment features between the groups.
4.10 Evaluation of drug sensitivity
The R package “oncoPredict” is a powerful tool for predicting clinical drug response in patients using gene expression profiles. It constructs statistical models based on gene expression profiles and drug sensitivity data from cell lines in Sanger’s Genomics of Drug Sensitivity in Cancer (GDSC) database [92]. Using the “oncoPredict” package, we estimated the half-maximal inhibitory concentration (IC50) values for 198 drugs in both the high-risk and low-risk patient groups, enabling a comprehensive comparison of drug sensitivity between the groups.
4.11 Statistical analysis
Statistical analyses were conducted using R Studio (version 4.2.3). For comparisons between two groups with continuous variables, the independent t-test was employed to assess the statistical significance of variables with a normal distribution. For non-normally distributed variables, the Wilcoxon rank-sum test was used. A p-value < 0.05 was considered statistically significant.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Kangjing Bian, Feng Zhang and Lei Huang; Formal analysis, Kangjing Bian; Funding acquisition, Lei Huang; Writing – original draft, Kangjing Bian; Writing – review & editing, Kangjing Bian, Chihyu Yang, Feng Zhang and Lei Huang. All the authors revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (grant numbers 82372764) to L.H. and the Fundamental Research Funds for the Central Universities.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
The datasets of this article were generated from TCGA database and GEO database.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| SASP | senescence-associated secretory phenotype |
| TME | tumor microenvironment |
| BRCA | breast cancer |
| TIME | tumor immune microenvironment |
| TCGA | The Cancer Genome Atlas |
| GEO | Gene Expression Omnibus |
| OS | overall survival |
| VDIMs | vesicles derived from the inner mitochondrial membrane |
| GSEA | Gene Set Enrichment Analysis |
| LASSO | least absolute shrinkage and selector operator |
| ROC | receiver operating characteristic |
| AUC | area under curve |
| DEG | differentially expressed gene |
| GO | Gene Ontology |
| BP | Biological processes |
| CC | Cellular Component |
| MF | Molecular Function |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GSEA | Gene set enrichment analysis |
| MSigDB | Molecular Signatures Database |
| HR | hazard ratio |
| CI | confidence interval |
| KM | Kaplan-Meier |
References
- Hong, R.; Xu, B. Breast cancer: an up-to-date review and future perspectives. Cancer Commun (Lond) 2022, 42, 913–936. [Google Scholar] [CrossRef]
- Heater, N.K.; Warrior, S.; Lu, J. Current and future immunotherapy for breast cancer. J Hematol Oncol 2024, 17, 131. [Google Scholar] [CrossRef]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef] [PubMed]
- Onkar, S.S.; Carleton, N.M.; Lucas, P.C.; Bruno, T.C.; Lee, A.V.; Vignali, D.A.A.; Oesterreich, S. The Great Immune Escape: Understanding the Divergent Immune Response in Breast Cancer Subtypes. Cancer Discov 2023, 13, 23–40. [Google Scholar] [CrossRef]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct Target Ther 2023, 8, 304. [Google Scholar] [CrossRef]
- Chen, C.; Xiang, A.; Lin, X.; Guo, J.; Liu, J.; Hu, S.; Rui, T.; Ye, Q. Mitophagy: insights into its signaling molecules, biological functions, and therapeutic potential in breast cancer. Cell Death Discov 2024, 10, 457. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Lei, J.H.; Bao, J.; Wang, H.; Hao, W.; Li, L.; Peng, C.; Masuda, T.; Miao, K.; Xu, J.; et al. BRCA1 Deficiency Impairs Mitophagy and Promotes Inflammasome Activation and Mammary Tumor Metastasis. Adv Sci (Weinh) 2020, 7, 1903616. [Google Scholar] [CrossRef]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.N.; Drake, L.E.; Sachleben, J.R.; Asara, J.M.; Locasale, J.W.; Karczmar, G.S.; et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep 2015, 16, 1145–1163. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Zhang, H.L.; Huang, J.H.; Cai, R.Z.; Wang, Y.; Chen, Y.H.; Hu, B.X.; Ye, Z.P.; Li, Z.L.; Mai, J.; et al. MAPK1/3 kinase-dependent ULK1 degradation attenuates mitophagy and promotes breast cancer bone metastasis. Autophagy 2021, 17, 3011–3029. [Google Scholar] [CrossRef]
- Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Lin, Z.; Lamb, R.; Hulit, J.; Howell, A.; Sotgia, F.; Rubin, E.; Lisanti, M.P. Ethanol exposure induces the cancer-associated fibroblast phenotype and lethal tumor metabolism: implications for breast cancer prevention. Cell Cycle 2013, 12, 289–301. [Google Scholar] [CrossRef]
- Zheng, B.; Wang, Y.; Zhou, B.; Qian, F.; Liu, D.; Ye, D.; Zhou, X.; Fang, L. Urolithin A inhibits breast cancer progression via activating TFEB-mediated mitophagy in tumor macrophages. J Adv Res 2024. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, Z.; Zhang, S.; Zhang, T.; Liu, Y.; Zhang, L. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics 2023, 13, 736–766. [Google Scholar] [CrossRef]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef]
- Song, C.; Pan, S.; Zhang, J.; Li, N.; Geng, Q. Mitophagy: A novel perspective for insighting into cancer and cancer treatment. Cell Prolif 2022, 55, e13327. [Google Scholar] [CrossRef] [PubMed]
- Wahabi, K.; Perwez, A.; Kamarudheen, S.; Bhat, Z.I.; Mehta, A.; Rizvi, M.M.A. Parkin gene mutations are not common, but its epigenetic inactivation is a frequent event and predicts poor survival in advanced breast cancer patients. BMC Cancer 2019, 19, 820. [Google Scholar] [CrossRef]
- Villa, E.; Marchetti, S.; Ricci, J.E. No Parkin Zone: Mitophagy without Parkin. Trends Cell Biol 2018, 28, 882–895. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab 2020, 32, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.N. Mitochondrial fidelity and metabolic agility control immune cell fate and function. J Clin Invest 2018, 128, 3651–3661. [Google Scholar] [CrossRef]
- Hou, H.; Er, P.; Cheng, J.; Chen, X.; Ding, X.; Wang, Y.; Chen, X.; Yuan, Z.; Pang, Q.; Wang, P.; et al. High expression of FUNDC1 predicts poor prognostic outcomes and is a promising target to improve chemoradiotherapy effects in patients with cervical cancer. Cancer Med 2017, 6, 1871–1881. [Google Scholar] [CrossRef]
- Yan, C.; Luo, L.; Guo, C.Y.; Goto, S.; Urata, Y.; Shao, J.H.; Li, T.S. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett 2017, 388, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Glytsou, C.; Chen, X.; Zacharioudakis, E.; Al-Santli, W.; Zhou, H.; Nadorp, B.; Lee, S.; Lasry, A.; Sun, Z.; Papaioannou, D.; et al. Mitophagy Promotes Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Discov 2023, 13, 1656–1677. [Google Scholar] [CrossRef]
- Hulahan, T.S.; Angel, P.M. From ductal carcinoma in situ to invasive breast cancer: the prognostic value of the extracellular microenvironment. J Exp Clin Cancer Res 2024, 43, 329. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Freeman, G.J. The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol Rev 2009, 229, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Nollen, E.A.A.; Rohrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov 2019, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Wu, Y.H.; Song, Y.; Yu, B. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J Hematol Oncol 2021, 14, 68. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, W.; Kadu, S.; Johnson, O.; Hasanali, Z.S.; Kelly, A.; Shestov, A.; Pajarillo, R.; Greenblatt, E.; Holmes, M.; et al. Exploiting the CD200-CD200R immune checkpoint axis in multiple myeloma to enhance CAR T-cell therapy. Blood 2024, 143, 139–151. [Google Scholar] [CrossRef]
- Erin, N.; Podnos, A.; Tanriover, G.; Duymus, O.; Cote, E.; Khatri, I.; Gorczynski, R.M. Bidirectional effect of CD200 on breast cancer development and metastasis, with ultimate outcome determined by tumor aggressiveness and a cancer-induced inflammatory response. Oncogene 2015, 34, 3860–3870. [Google Scholar] [CrossRef]
- Guruprasad, P.; Carturan, A.; Zhang, Y.; Cho, J.H.; Kumashie, K.G.; Patel, R.P.; Kim, K.H.; Lee, J.S.; Lee, Y.; Kim, J.H.; et al. The BTLA-HVEM axis restricts CAR T cell efficacy in cancer. Nat Immunol 2024, 25, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- de Mingo Pulido, A.; Hanggi, K.; Celias, D.P.; Gardner, A.; Li, J.; Batista-Bittencourt, B.; Mohamed, E.; Trillo-Tinoco, J.; Osunmakinde, O.; Pena, R.; et al. The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity 2021, 54, 1154–1167 e1157. [Google Scholar] [CrossRef]
- Monteran, L.; Ershaid, N.; Scharff, Y.; Zoabi, Y.; Sanalla, T.; Ding, Y.; Pavlovsky, A.; Zait, Y.; Langer, M.; Caller, T.; et al. Combining TIGIT Blockade with MDSC Inhibition Hinders Breast Cancer Bone Metastasis by Activating Antitumor Immunity. Cancer Discov 2024, 14, 1252–1275. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, J.; Zhang, Q.; Ouyang, Q.; Zhang, Y.; Liu, Q.; Sun, T.; Ye, F.; Zhang, B.; Xia, S.; et al. The anti-PD-L1/CTLA-4 bispecific antibody KN046 in combination with nab-paclitaxel in first-line treatment of metastatic triple-negative breast cancer: a multicenter phase II trial. Nat Commun 2024, 15, 1015. [Google Scholar] [CrossRef]
- Jin, M.; Fang, J.; Peng, J.; Wang, X.; Xing, P.; Jia, K.; Hu, J.; Wang, D.; Ding, Y.; Wang, X.; et al. PD-1/PD-L1 immune checkpoint blockade in breast cancer: research insights and sensitization strategies. Mol Cancer 2024, 23, 266. [Google Scholar] [CrossRef]
- Cai, L.; Li, Y.; Tan, J.; Xu, L.; Li, Y. Targeting LAG-3, TIM-3, and TIGIT for cancer immunotherapy. J Hematol Oncol 2023, 16, 101. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3--potential mechanisms of action. Nat Rev Immunol 2015, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wang, S.; Bindeman, W. Clinical applications of PD-L1 bioassays for cancer immunotherapy. J Hematol Oncol 2017, 10, 110. [Google Scholar] [CrossRef]
- Nagaraju, G.P.; Malla, R.R.; Basha, R.; Motofei, I.G. Contemporary clinical trials in pancreatic cancer immunotherapy targeting PD-1 and PD-L1. Semin Cancer Biol 2022, 86, 616–621. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther 2024, 9, 124. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.L.; Cui, Q.; Wang, J.Q.; Ashby, C.R., Jr.; Chen, Y.; Shen, Z.X.; Chen, Z.S. The AKT inhibitor, MK-2206, attenuates ABCG2-mediated drug resistance in lung and colon cancer cells. Front Pharmacol 2023, 14, 1235285. [Google Scholar] [CrossRef] [PubMed]
- Chien, A.J.; Tripathy, D.; Albain, K.S.; Symmans, W.F.; Rugo, H.S.; Melisko, M.E.; Wallace, A.M.; Schwab, R.; Helsten, T.; Forero-Torres, A.; et al. MK-2206 and Standard Neoadjuvant Chemotherapy Improves Response in Patients With Human Epidermal Growth Factor Receptor 2-Positive and/or Hormone Receptor-Negative Breast Cancers in the I-SPY 2 Trial. J Clin Oncol 2020, 38, 1059–1069. [Google Scholar] [CrossRef]
- Soria, J.C.; Adjei, A.A.; Bahleda, R.; Besse, B.; Ferte, C.; Planchard, D.; Zhou, J.; Ware, J.; Morrissey, K.; Shankar, G.; et al. A phase IB dose-escalation study of the safety and pharmacokinetics of pictilisib in combination with either paclitaxel and carboplatin (with or without bevacizumab) or pemetrexed and cisplatin (with or without bevacizumab) in patients with advanced non-small cell lung cancer. Eur J Cancer 2017, 86, 186–196. [Google Scholar] [CrossRef]
- Cheng, W.J.; Lin, S.Y.; Chuang, K.H.; Chen, M.; Ho, H.O.; Chen, L.C.; Hsieh, C.M.; Sheu, M.T. Combined Docetaxel/Pictilisib-Loaded mPEGylated Nanocarriers with Dual HER2 Targeting Antibodies for Synergistic Chemotherapy of Breast Cancer. Int J Nanomedicine 2022, 17, 5353–5374. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 2012, 366, 520–529. [Google Scholar] [CrossRef]
- Xu, X.; Li, Y.; Wu, Y.; Wang, M.; Lu, Y.; Fang, Z.; Wang, H.; Li, Y. Increased ATF2 expression predicts poor prognosis and inhibits sorafenib-induced ferroptosis in gastric cancer. Redox Biol 2023, 59, 102564. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther 2020, 5, 87. [Google Scholar] [CrossRef]
- Schoffski, P.; Blay, J.Y.; De Greve, J.; Brain, E.; Machiels, J.P.; Soria, J.C.; Sleijfer, S.; Wolter, P.; Ray-Coquard, I.; Fontaine, C.; et al. Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network Of Core Institutes (NOCI). Eur J Cancer 2010, 46, 2206–2215. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Hou, P.; Lv, Y.; Li, G.; Cao, G.; Wang, H.; Lin, W. BRCA1 orchestrates the response to BI-2536 and its combination with alisertib in MYC-driven small cell lung cancer. Cell Death Dis 2024, 15, 551. [Google Scholar] [CrossRef]
- Yuan, L.; Liu, J.; Bao, L.; Qu, H.; Xiang, J.; Sun, P. Upregulation of the ferroptosis-related STEAP3 gene is a specific predictor of poor triple-negative breast cancer patient outcomes. Front Oncol 2023, 13, 1032364. [Google Scholar] [CrossRef]
- Barnes, E.M.E.; Xu, Y.; Benito, A.; Herendi, L.; Siskos, A.P.; Aboagye, E.O.; Nijhuis, A.; Keun, H.C. Lactic acidosis induces resistance to the pan-Akt inhibitor uprosertib in colon cancer cells. Br J Cancer 2020, 122, 1298–1308. [Google Scholar] [CrossRef]
- Thacker, G.; Henry, S.; Nandi, A.; Debnath, R.; Singh, S.; Nayak, A.; Susnik, B.; Boone, M.M.; Zhang, Q.; Kesmodel, S.B.; et al. Immature natural killer cells promote progression of triple-negative breast cancer. Sci Transl Med 2023, 15, eabl4414. [Google Scholar] [CrossRef]
- Fu, Y.; Dong, W.; Xu, Y.; Li, L.; Yu, X.; Pang, Y.; Chan, L.; Deng, Y.; Qian, C. Targeting mitochondrial dynamics by AZD5363 in triple-negative breast cancer MDA-MB-231 cell-derived spheres. Naunyn Schmiedebergs Arch Pharmacol 2023, 396, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Turner, N.C.; Barrios, C.H.; Isakoff, S.J.; Kim, S.B.; Sablin, M.P.; Saji, S.; Savas, P.; Vidal, G.A.; Oliveira, M.; et al. First-Line Ipatasertib, Atezolizumab, and Taxane Triplet for Metastatic Triple-Negative Breast Cancer: Clinical and Biomarker Results. Clin Cancer Res 2024, 30, 767–778. [Google Scholar] [CrossRef]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Rath, N.; Munro, J.; Cutiongco, M.F.; Jagiello, A.; Gadegaard, N.; McGarry, L.; Unbekandt, M.; Michalopoulou, E.; Kamphorst, J.J.; Sumpton, D.; et al. Rho Kinase Inhibition by AT13148 Blocks Pancreatic Ductal Adenocarcinoma Invasion and Tumor Growth. Cancer Res 2018, 78, 3321–3336. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Luo, Y.; Yuan, Z.; Tian, Y.; Jin, T.; Xu, F. Cellular senescence and SASP in tumor progression and therapeutic opportunities. Mol Cancer 2024, 23, 181. [Google Scholar] [CrossRef]
- Victorelli, S.; Salmonowicz, H.; Chapman, J.; Martini, H.; Vizioli, M.G.; Riley, J.S.; Cloix, C.; Hall-Younger, E.; Machado Espindola-Netto, J.; Jurk, D.; et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature 2023, 622, 627–636. [Google Scholar] [CrossRef]
- Ban-Ishihara, R.; Ishihara, T.; Sasaki, N.; Mihara, K.; Ishihara, N. Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. Proc Natl Acad Sci U S A 2013, 110, 11863–11868. [Google Scholar] [CrossRef] [PubMed]
- Saunders, T.L.; Windley, S.P.; Gervinskas, G.; Balka, K.R.; Rowe, C.; Lane, R.; Tailler, M.; Nguyen, T.N.; Ramm, G.; Lazarou, M.; et al. Exposure of the inner mitochondrial membrane triggers apoptotic mitophagy. Cell Death Differ 2024, 31, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Prashar, A.; Bussi, C.; Fearns, A.; Capurro, M.I.; Gao, X.; Sesaki, H.; Gutierrez, M.G.; Jones, N.L. Lysosomes drive the piecemeal removal of mitochondrial inner membrane. Nature 2024, 632, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Faitg, J.; Auwerx, J.; Ferrucci, L.; D'Amico, D. Mitophagy in human health, ageing and disease. Nat Metab 2023, 5, 2047–2061. [Google Scholar] [CrossRef] [PubMed]
- Alcala, S.; Sancho, P.; Martinelli, P.; Navarro, D.; Pedrero, C.; Martin-Hijano, L.; Valle, S.; Earl, J.; Rodriguez-Serrano, M.; Ruiz-Canas, L.; et al. ISG15 and ISGylation is required for pancreatic cancer stem cell mitophagy and metabolic plasticity. Nat Commun 2020, 11, 2682. [Google Scholar] [CrossRef]
- Xie, X.Q.; Yang, Y.; Wang, Q.; Liu, H.F.; Fang, X.Y.; Li, C.L.; Jiang, Y.Z.; Wang, S.; Zhao, H.Y.; Miao, J.Y.; et al. Targeting ATAD3A-PINK1-mitophagy axis overcomes chemoimmunotherapy resistance by redirecting PD-L1 to mitochondria. Cell Res 2023, 33, 215–228. [Google Scholar] [CrossRef]
- Newman, L.E.; Shadel, G.S. Mitochondrial DNA Release in Innate Immune Signaling. Annu Rev Biochem 2023, 92, 299–332. [Google Scholar] [CrossRef]
- Zhu, W.; Rao, J.; Zhang, L.H.; Xue, K.M.; Li, L.; Li, J.J.; Chen, Q.Z.; Fu, R. OMA1 competitively binds to HSPA9 to promote mitophagy and activate the cGAS-STING pathway to mediate GBM immune escape. J Immunother Cancer 2024, 12. [Google Scholar] [CrossRef]
- Tang, Y.; Guo, H.; Chen, L.; Wang, X.; Chen, Q.; Gou, L.; Liu, X.; Wang, X. Development and validation of a prognostic model for mitophagy-related genes in colon adenocarcinoma: A study based on TCGA and GEO databases. PLoS One 2023, 18, e0284089. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, Z.; Wang, L.; Yang, Q.; Huang, J.; Huang, J. A Mitophagy-Related Gene Signature for Subtype Identification and Prognosis Prediction of Hepatocellular Carcinoma. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Proics, E.; Rubio-Patino, C.; Obba, S.; Zunino, B.; Bossowski, J.P.; Rozier, R.M.; Chiche, J.; Mondragon, L.; Riley, J.S.; et al. Parkin-Independent Mitophagy Controls Chemotherapeutic Response in Cancer Cells. Cell Rep 2017, 20, 2846–2859. [Google Scholar] [CrossRef]
- Sisalli, M.J.; Ianniello, G.; Savoia, C.; Cuomo, O.; Annunziato, L.; Scorziello, A. Knocking-out the Siah2 E3 ubiquitin ligase prevents mitochondrial NCX3 degradation, regulates mitochondrial fission and fusion, and restores mitochondrial function in hypoxic neurons. Cell Commun Signal 2020, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Szargel, R.; Shani, V.; Abd Elghani, F.; Mekies, L.N.; Liani, E.; Rott, R.; Engelender, S. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum Mol Genet 2016, 25, 3476–3490. [Google Scholar] [CrossRef]
- Abd Elghani, F.; Safory, H.; Hamza, H.; Savyon, M.; Farhoud, M.; Toren-Hershoviz, M.; Vitic, Z.; Ebanks, K.; Shani, V.; Bisharat, S.; et al. SIAH proteins regulate the degradation and intra-mitochondrial aggregation of PINK1: Implications for mitochondrial pathology in Parkinson's disease. Aging Cell 2022, 21, e13731. [Google Scholar] [CrossRef] [PubMed]
- Qin, R.; Huang, Y.; Yao, Y.; Wang, L.; Zhang, Z.; Huang, W.; Su, Y.; Zhang, Y.; Guan, A.; Wang, H. The role and molecular mechanism of metabolic reprogramming of colorectal cancer by UBR5 through PYK2 regulation of OXPHOS expression study. J Biochem Mol Toxicol 2023, 37, e23376. [Google Scholar] [CrossRef]
- Jiang, H.; Chiang, C.Y.; Chen, Z.; Nathan, S.; D'Agostino, G.; Paulo, J.A.; Song, G.; Zhu, H.; Gabelli, S.B.; Cole, P.A. Enzymatic analysis of WWP2 E3 ubiquitin ligase using protein microarrays identifies autophagy-related substrates. J Biol Chem 2022, 298, 101854. [Google Scholar] [CrossRef]
- Oshi, M.; Takahashi, H.; Tokumaru, Y.; Yan, L.; Rashid, O.M.; Matsuyama, R.; Endo, I.; Takabe, K. G2M Cell Cycle Pathway Score as a Prognostic Biomarker of Metastasis in Estrogen Receptor (ER)-Positive Breast Cancer. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Diakos, C.I.; Charles, K.A.; McMillan, D.C.; Clarke, S.J. Cancer-related inflammation and treatment effectiveness. Lancet Oncol 2014, 15, e493–503. [Google Scholar] [CrossRef]
- Cui, M.; Yamano, K.; Yamamoto, K.; Yamamoto-Imoto, H.; Minami, S.; Yamamoto, T.; Matsui, S.; Kaminishi, T.; Shima, T.; Ogura, M.; et al. HKDC1, a target of TFEB, is essential to maintain both mitochondrial and lysosomal homeostasis, preventing cellular senescence. Proc Natl Acad Sci U S A 2024, 121, e2306454120. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, H.; Liu, Y.; Zhang, S.; Su, P.; Wang, L.; Li, Y.; Liang, Y.; Wang, X.; Zhao, W.; et al. Hypoxia-induced GPCPD1 depalmitoylation triggers mitophagy via regulating PRKN-mediated ubiquitination of VDAC1. Autophagy 2023, 19, 2443–2463. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Szewczyk, A. Mitochondrial Ion Channels. Annu Rev Biophys 2023, 52, 229–254. [Google Scholar] [CrossRef]
- Fu, A.; Yao, B.; Dong, T.; Chen, Y.; Yao, J.; Liu, Y.; Li, H.; Bai, H.; Liu, X.; Zhang, Y.; et al. Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell 2022, 185, 1356–1372 e1326. [Google Scholar] [CrossRef]
- Villani, A.; Fontana, A.; Panebianco, C.; Ferro, C.; Copetti, M.; Pavlovic, R.; Drago, D.; Fiorentini, C.; Terracciano, F.; Bazzocchi, F.; et al. A powerful machine learning approach to identify interactions of differentially abundant gut microbial subsets in patients with metastatic and non-metastatic pancreatic cancer. Gut Microbes 2024, 16, 2375483. [Google Scholar] [CrossRef]
- Banerjee, S.; Tian, T.; Wei, Z.; Shih, N.; Feldman, M.D.; Peck, K.N.; DeMichele, A.M.; Alwine, J.C.; Robertson, E.S. Distinct Microbial Signatures Associated With Different Breast Cancer Types. Front Microbiol 2018, 9, 951. [Google Scholar] [CrossRef]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef]
- Li, F.; Wang, Y.; Zheng, K. Microglial mitophagy integrates the microbiota-gut-brain axis to restrain neuroinflammation during neurotropic herpesvirus infection. Autophagy 2023, 19, 734–736. [Google Scholar] [CrossRef]
- Luo, M.; Chen, Y.; Pan, X.; Chen, H.; Fan, L.; Wen, Y. E. coli Nissle 1917 ameliorates mitochondrial injury of granulosa cells in polycystic ovary syndrome through promoting gut immune factor IL-22 via gut microbiota and microbial metabolism. Front Immunol 2023, 14, 1137089. [Google Scholar] [CrossRef]
- Chen, F.; Xue, Y.; Zhang, W.; Zhou, H.; Zhou, Z.; Chen, T.; YinWang, E.; Li, H.; Ye, Z.; Gao, J.; et al. The role of mitochondria in tumor metastasis and advances in mitochondria-targeted cancer therapy. Cancer Metastasis Rev 2024, 43, 1419–1443. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.; Sun, X.; Li, J.; Pu, M.; Zhang, J. Parkin modulates the hepatocellular carcinoma microenvironment by regulating PD-1/PD-L1 signalling. J Adv Res 2025. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Q.; Cao, H.; Wang, Z.; Wang, D.; Liu, J.; Gao, T.; Ren, C.; Liu, J. Mitochondrial Localized In Situ Self-Assembly Reprogramming Tumor Immune and Metabolic Microenvironment for Enhanced Cancer Therapy. Adv Mater 2024, 36, e2311043. [Google Scholar] [CrossRef]
- Yu, L.; Wei, J.; Liu, P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin Cancer Biol 2022, 85, 69–94. [Google Scholar] [CrossRef]
- Jiao, W.; Cheng, Y.; Liu, C.; Feng, J.; Lin, J.; Shen, Y. SGLT1 inhibition alleviates radiation-induced intestinal damage through promoting mitochondrial homeostasis. Free Radic Biol Med 2024, 224, 831–845. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, H.; Yang, Y.; Zhang, S.; Wang, J.; Zhang, D.; Yu, H. Metformin Attenuates UVA-Induced Skin Photoaging by Suppressing Mitophagy and the PI3K/AKT/mTOR Pathway. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinform 2021, 22. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Workflow diagram. The flowchart graph of this research
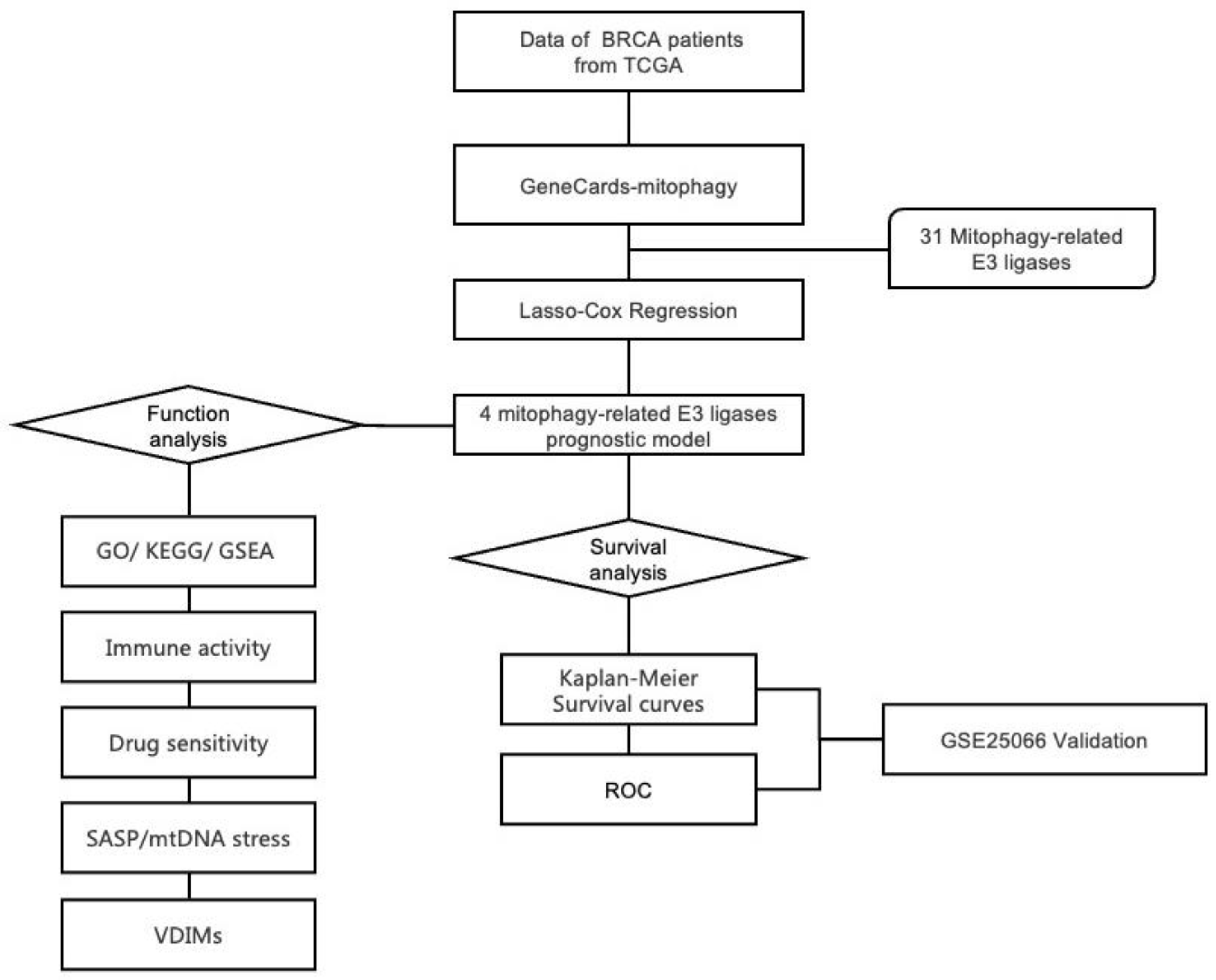
Figure 3.
Validation of the ASUW prognostic model in TCGA-BRCA and GSE25066 databases. (A, B) Kaplan-Meier survival curve for high-risk and low-risk groups in the TCGA-BRCA cohort and GSE25066 cohort, based on the ASUW risk regression model. (C, D) The distribution of the risk scores, scatter plots showed whether the samples were alive and heatmap for the four E3 ubiquitin ligases in the TCGA-BRCA cohort and GSE25066 cohort. Top: Risk scores for each patient, classified by high-risk (red) and low-risk (blue) groups. Middle: Survival time and status (alive or dead). Bottom: Heatmap of expression levels of the four E3 ubiquitin ligases in two groups. (E) ROC curve analysis for 5-, 10-, and 15-year survival predictions in the TCGA-BRCA cohort. (F) ROC curve analysis for 1-, 3-, and 5-year survival predictions in the GSE25066 cohort.
Figure 3.
Validation of the ASUW prognostic model in TCGA-BRCA and GSE25066 databases. (A, B) Kaplan-Meier survival curve for high-risk and low-risk groups in the TCGA-BRCA cohort and GSE25066 cohort, based on the ASUW risk regression model. (C, D) The distribution of the risk scores, scatter plots showed whether the samples were alive and heatmap for the four E3 ubiquitin ligases in the TCGA-BRCA cohort and GSE25066 cohort. Top: Risk scores for each patient, classified by high-risk (red) and low-risk (blue) groups. Middle: Survival time and status (alive or dead). Bottom: Heatmap of expression levels of the four E3 ubiquitin ligases in two groups. (E) ROC curve analysis for 5-, 10-, and 15-year survival predictions in the TCGA-BRCA cohort. (F) ROC curve analysis for 1-, 3-, and 5-year survival predictions in the GSE25066 cohort.
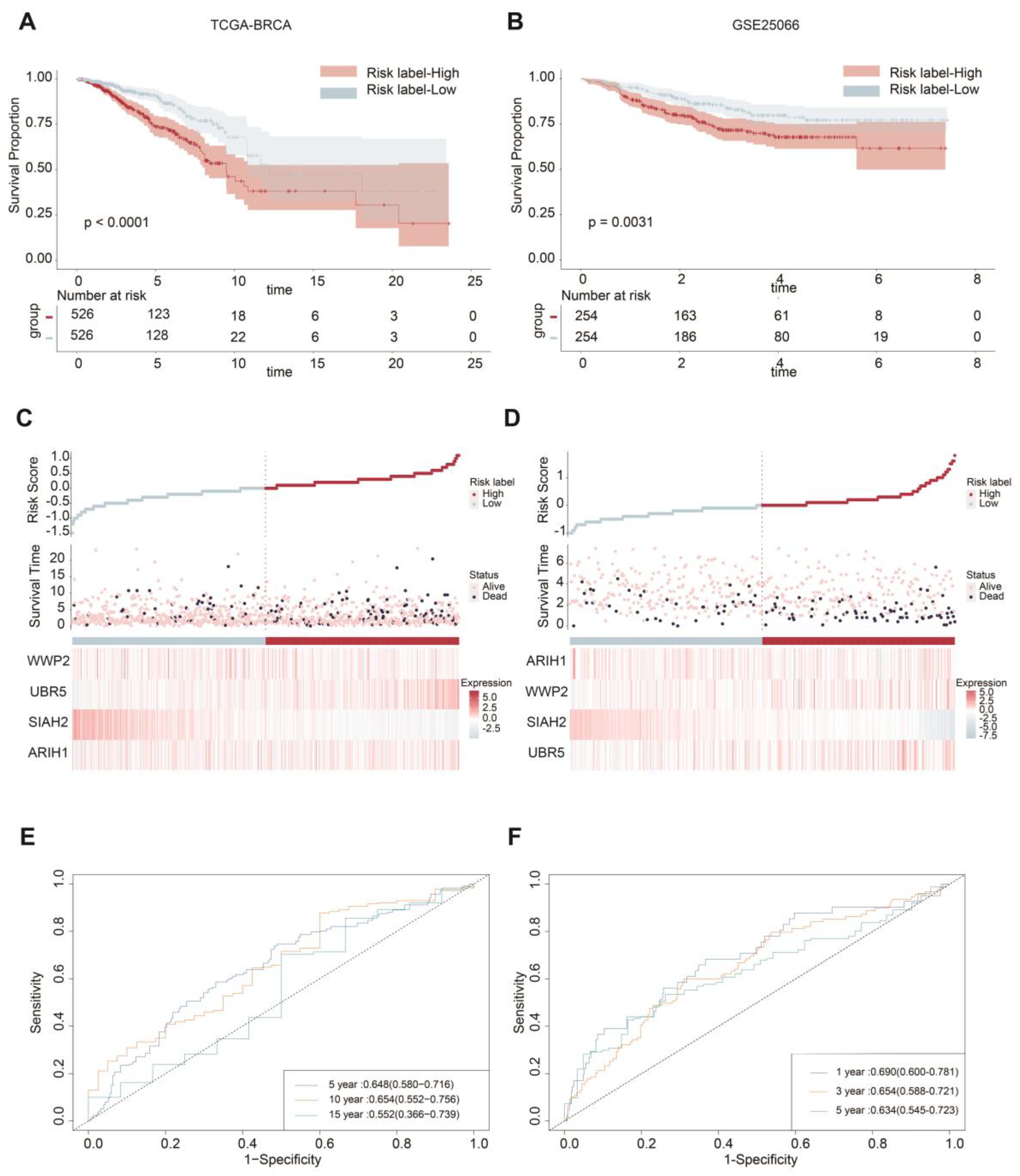
Figure 6.
Predicted drug sensitivity differences between two groups. The boxplots depict the predicted drug sensitivity (IC50) of 12 drugs between high-risk (red) and low-risk (blue) groups as predicted by the OncoPredict analysis. Drugs exhibiting statistically significant differences in sensitivity (p < 0.05) were shown. The high-risk group displayed higher sensitivity to drugs including MK-2206, Pictilisib, Rapamycin, Sorafenib, GSK1904529A, Uprosertib, LGK974, Elephantin, AZD5363, Ipatasertib, and AT13148. Conversely, the low-risk group demonstrated greater sensitivity to BI-2536. Statistical significance is indicated as: ns (not significant), *p < 0.05, **p < 0.01.
Figure 6.
Predicted drug sensitivity differences between two groups. The boxplots depict the predicted drug sensitivity (IC50) of 12 drugs between high-risk (red) and low-risk (blue) groups as predicted by the OncoPredict analysis. Drugs exhibiting statistically significant differences in sensitivity (p < 0.05) were shown. The high-risk group displayed higher sensitivity to drugs including MK-2206, Pictilisib, Rapamycin, Sorafenib, GSK1904529A, Uprosertib, LGK974, Elephantin, AZD5363, Ipatasertib, and AT13148. Conversely, the low-risk group demonstrated greater sensitivity to BI-2536. Statistical significance is indicated as: ns (not significant), *p < 0.05, **p < 0.01.

Table 1.
The information of 4 Mitophagy-Related E3 Ubiquitin Ligases.
| Gene symbol | Gene ID | Full name | Classes | Risk coefficient |
|---|---|---|---|---|
| ARIH1 | 25820 | Ariadne RBR E3 Ubiquitin Protein Ligase 1 | RBR (RING-IBR-RING) | -0.100073572 |
| SIAH2 | 6478 | Siah E3 Ubiquitin Protein Ligase 2 | RING (Really interesting new gene) | -0.333969422 |
| UBR5 | 51366 | Ubiquitin Protein Ligase E3 Component N-Recognin 5 | HECT (Homologous to the E6AP carboxyl terminus) | 0.34620336 |
| WWP2 | 11060 | WW Domain Containing E3 Ubiquitin Protein Ligase 2 | HECT (Homologous to the E6AP carboxyl terminus) | -0.204703988 |
Table 2.
Detailed information of 12 sensitivity drugs.
| Drug Name | Target | Application | Reference | IC50 (High vs Low) |
|---|---|---|---|---|
| MK-2206 | Akt | Lung cancer, Breast cancer, Colorectal cancer |
[40,41] | down |
| Pictilisib | PI3K | Breast cancer, Lung cancer | [42,43] | down |
| Rapamycin | mTORC1 | Breast cancer, Kidney cancer | [44,45] | down |
| Sorafenib | VEGFR, PDGFR, RAF kinases | Hepatocellular carcinoma, gastric cancer | [46,47] | down |
| BI-2536 | PLK1 | Breast cancer, Lung cancer | [48,49] | up |
| GSK1904529A | IGF-1R | Breast cancer | [50] | down |
| Uprosertib | Akt | Colorectal cancer | [51] | down |
| LGK974 | Porcupine | Colorectal cancer | [52] | down |
| Elephantin | Unclear | Further investigation is required | Unclear | down |
| AZD5363 | Akt | Breast cancer | [53] | down |
| Ipatasertib | Akt | Breast cancer, Prostate cancer | [54,55] | down |
| AT13148 | AGC family kinases | Pancreatic Ductal Adenocarcinoma | [56] | down |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



