Submitted:
28 August 2023
Posted:
31 August 2023
Read the latest preprint version here
Abstract
Cellular organisms possess intricate DNA damage repair and tolerance pathways to manage various DNA lesions arising from endogenous or exogenous sources. Dysregulation of these pathways is associated with cancer development and progression. Synthetic lethality (SL), a promising cancer therapy concept, involves exploiting the simultaneous functional loss of two genes for selective cell death. PARP inhibitors (PARPis) have demonstrated success in BRCA-deficient tumors. Cisplatin (CPT), a widely used chemotherapy agent, forms DNA adducts and crosslinks, rendering it effective against various cancers but less so for prostate cancer (PCa) due to resistance and toxicity. Here, we explore the therapeutic potential of TLK1, a kinase upregulated in androgen-insensitive PCa cells, as a target for enhancing CPT-based therapy. TLK1 phosphorylates key homologous recombination repair (HRR) proteins RAD54L and RAD54B, critical for HRR alongside RAD51. The combination of CPT with TLK1 inhibitor J54 exhibits SL in androgen-insensitive PCa cells. The formation of double-strand break intermediates during inter-strand crosslink processing necessitates HRR for effective repair. Therefore, targeting TLK1 with J54 enhances the SL of CPT by impeding HRR, leading to increased sensitivity in PCa cells. These findings suggest a promising approach for improving CPT-based therapies in PCa, particularly in androgen-insensitive cases. By elucidating the role of TLK1 in CPT resistance, this study provides valuable insights into potential therapeutic targets to overcome PCa resistance to CPT chemotherapy. Further investigations into TLK1 inhibition in combination with other DNA-damaging agents may pave the way for more effective and targeted treatments for PCa and other cancers that exhibit resistance to traditional chemotherapy agents.
Keywords:
PCa
; Synthetic lethality
; TLK1 signaling
; Homologous recombination repair
; TLK1 inhibitor J54
; CPT-based PCa therapy
Introduction
All cellular organisms, including humans, are equipped with multiple highly conserved DNA damage repair and tolerance pathways to contend with different types of DNA lesions formed spontaneously or upon treatment with DNA damaging agents, including radio- and chemo-therapeutics [1]. For example, bulky and/or helix-distorting lesions are repaired by nucleotide excision repair (NER). On the other hand, non-bulky and non-helix-distorting base lesions, as well as single strand breaks (SSBs), are repaired by base excision repair (BER). Double strand breaks (DSBs) can be repaired by homologous recombination repair (HRR), which is generally error-free, and nonhomologous end-joining (NHEJ), which generally creates insertion/deletion at the damaged sites. DNA lesions can also be tolerated by the so-called translesion synthesis (TLS), which is catalyzed by specialized and generally error-prone DNA polymerases. Activation of DNA damage checkpoint (DDC), which temporarily arrests the cell cycle before DNA lesions are repaired, is also an important mechanism for the cell to contend with DNA damage [2]. By maintaining genome stability, most genes involved in DNA damage repair, tolerance and DDC serve as bona fide cancer suppressors.
Synthetic lethality (SL), where functional loss of two genes together, but not alone, results in cell death, is a conceptually new strategy for cancer therapies [3]. The first successful SL treatment strategy is using PARP inhibitors (PARPis) to treat tumors with functional loss of BRCA1 or BRAC2, two cancer suppressors involved in HRR of DSBs. PARPis prevent PARP1 and PARP2 from repairing SSBs, leading to stalled and collapsed DNA replication forks. Subsequently, SSBs are converted to DSBs that HRR-deficient cells cannot repair effectively, leading to cell death. Recently, PARPis have been shown to be effective for killing PCa cells with functional loss of BRCA1, BRCA2 or other proteins directly or indirectly involved in HRR, and moderately extend survival of mCRPC patients having the functional loss [4,5,6].
Cisplatin (CPT) reacts with DNA, producing single purine adducts, and intra- and inter-strand crosslinks (ICLs) [7,8,9,10]. The single purine adducts and intra-strand crosslinks can be repaired by NER, or tolerated by TLS. The repair of inter-strand crosslinks is more complicated, requiring the participations of Fanconi anemia (FA) proteins, and subsets of proteins involved in NER, TLS, HRR, NHEJ and BER (Figure 1B) [11,12,21,22][M1] . Therefore, deficiency in any of the DNA damage repair, tolerance and DDC mechanisms may cause CPT sensitivity, and combined deficiencies of two or more of these mechanisms will synergistically enhance the sensitivity.
CPT has been most frequently used for adjuvant or neoadjuvant treatment of the majority of solid tumors[13]. Impressively, CPT treatment is highly efficient against testicular germ cell cancer, leading to a durable complete remission in > 80% of patients [14]. While CPT-based therapy has not been the treatment of choice for PCa, partly due to significant renal toxicity (which can be faithfully recapitulated in mice [15]), recent studies indicated that lower dosing in combination with inhibitors of DNA damage repair could be quite effective [10]. However, as multiple DNA damage repair, tolerance and DDC mechanisms are implicated in CPT resistance (Figure 1) [8,16], the genes that can be targeted to improve CPT-based therapies of PCa, remain largely unexplored. CPT is highly effective against several forms of cancer, most notably testicular tumors, and it is also commonly used to treat breast, ovarian, bladder, lung, and head and neck cancer. But prostate cancer (PCa) is resistant to CPT chemotherapy due to poor targeting and the development of resistance. Some strategies for targeting CPT by piggybacking carrier nanoparticles onto PSMA[17], as well as recent clinical studies that indicated that lower dosing in combination with DNA damage repair inhibitors (or for cases with defective DNA repair genes) have shown to be quite effective [18,19,20] However, as multiple DNA damage repair, tolerance and DDC mechanisms are implicated in CPT resistance [8,16], it is not obvious which genes need to be targeted to improve CPT-based therapies for the majority of PCa cases.
TLK1 is a serine/threonine kinase [21] that is frequently upregulated in PCa cells upon anti-androgen therapies [22,23]. By phosphorylating its substrates, TLK1 plays roles in resistance to a number of DNA damaging agents, including CPT[24,25,26,27]. Our recent work showed that TLK1 specifically catalyzes the phosphorylation of the HRR proteins RAD54L and RAD54B [28], which at least in part explains the role for TLK1 in CPT resistance, as together these two paralogs carry out some fundamental functions in HRR along with RAD51 [29]. Since the processing of ICLs typically results in the formation of DSB intermediates that require the HRR pathway to complete lesions’ repair, it is expected that the combination of CPT with inhibitors of HRR will result in synthetic lethality. In this work we tested the effect of a specific inhibitor of TLK1 in combination with CPT as a therapeutic strategy for androgen-insensitive PCa cells.
Results
Combining CPT with J54 enhances its growth inhibitory potential on CRPC cells. For our work, we selected two CRPC cell lines that utilize different mechanisms of androgen-insensitive growth (PC3 and C4-2B), both of which are known to be rather resistant to CPT. In Figure 1, we show the viability results, using MTT, of the two cell lines culture over 48h with cisplatin alone, or in combination with the TLK inhibitor, J54. It is noticeable that at up to a concentration of 0.3 μg/ml (already quite high for therapeutic index) both cell lines were quite insensitive to CPT, and in fact, C4-2B grew even slightly better than untreated cells (possibly an effect of the induction of DNA repair enzymes by CPT that may help in the complex process of DNA replication). At higher CPT concentration, both cell lines display a progressive growth inhibition, as expected. The situation, however, was quite different when the experiment was carried out in combination with 5 μM J54. In this case, both cell lines displayed a much stronger, dose-dependent reduction in viability, and the peculiar growth stimulation observed at low doses of CPT for C4-2B cells was completely eliminated. Note that J54 alone (0 CPT) had no growth inhibitory effect on the cells, as previously reported [30].
CPT treatment induces pRA54-T700. The fundamental hypothesis of our work is that the repair process of ICL-lesions induced by CPT results in the formation of transient DSBs that require repair via HRR. Hence, interfering with the mechanism of HRR by preventing the critical phosphorylation of RAD54-T700 mediated specifically by TLK1, will enhance the toxic effects of CPT.
We have recently generated a highly specific pRAD-T700 antiserum that can specifically monitor the activity of TLK1 during HRR[28], However, this antiserum has not been tested yet on DNA damaging agents other than IR. We now observed that CPT is potent inducer of RAD54 phosphorylation, while the TLK1 inhibitor J54 partly suppresses this. This was independently confirmed in both CRPC PCa cell lines (Figure 2). Since this was observed also for the recovery period after IR [28], it is tempting to speculate that phosphorylation of RAD54-T700 is either a general phenomenon that occurs with other DNA damaging agents, or (more likely) that ICLs getting converted to DSBs are partly repaired via canonical HRR requiring pRAD54-T700 modification.
The combination of CPT+J54 results in dose-dependent regression of tumor xenografts. To establish if adding J54 to CPT has an actual therapeutic effect from a synthetic lethal combination with a genotoxic agent, we investigated the effect on the growth of a subcutaneous PC3-Luc tumor model in NOD-SCID mice. After inoculation of 1M cells on both dorsal flanks, the tumors were allowed to form to a size of ~150 mm3, after which the treatment was started. We used a fixed dose of 5 mg/kg J54 and 3 different dosages of CPT (0, 1, 3, and 6 mg/kg). The progression or regression of the tumors was followed in each mouse with calipers, and the average tumor size over time is plotted in Figure 3 bottom left panel. Notably, all the treatments were at least cytostatic compared to controls, while the combination treatments with 1 mg/kg and 3 mg/kg CPT after 40 days resulted in an actual regression of the tumors. In fact, when the experiment was concluded, and the tumors recovered from the group treated with 3 mg/kg CPT+J54, the remnants were hardly measurable with calipers or by weight. The 6 mg/kg CPT (+/- J54) dose was also highly effective at cytoreduction, but the treatment could be continued for only 6 cycles (bi-weekly) before the toxicity was such that it required euthanasia of the mice before the conclusion of the experiment, and even before the visualization with the IVIS.
At experiment midpoint and a day before sacrificing the mice (Figure 3) the animals were injected with luciferin and the actual tumor cells mass was visualized with an IVIS-Spectrum/CT machine (Perkin Elmer). While the IVIS was run unassisted with auto-acquisition, which allows for the detection of the full range of cells, from very few to very large numbers, resulting in the production of tumors images from all the animals, the details are much better appreciated in the heat-map scale (or from the individual radiance quantitations). For instance, the most remarkable result is seen in the CPT 3 mg/kg+J54 group. While the presence of PC3-Luc cells tumors are seen also in this group, the heat-map scale is drastically difference: it goes from 10,000 to 50,000. In contrast, in the control (for example) the scale goes from 0.2 to 1.2X1011, meaning up to 6 logs radiance units lower in the combo than in the tumors from the control group. For a more precise comparison between the control group and the 3mg/kg combo, we measured the average Flux of the all the tumors in each group. For the control, the average Flux was 4.61e10, while for the combo it was 31604. Another interesting observation was that J54 alone had some effect on tumor growth on some of the animals, for example very noticeable in the 3d mouse from the left. Although the growth curve measured with calipers is displayed as the average of all the tumors in a group, we did notice overall a partial cytostatic effect with J54 alone, which became an actual tumor regression toward the late part of experiment. Another important observation is that J54, not only had no apparent toxic effect and did not affect the weight and water/food consumption of the mice, but actually showed a partial improvement of the visual health conditions of the mice treated with the higher 3 mg/kg CPT dose. Rather than losing weight, a possible sign of cachexia from tumor burden and treatment toxicity, the animals in the combo groups showed stable weight and appeared to be more active and frequently foraging.
The strong tumor regression in mice treated with CPT+J54 is hallmarked by apoptosis. To establish more firmly the mechanism of tumor regression (or at least cytostasis) in the mice treated with CPT, J54, or combination, we focused on the group of mice treated with the 3 mg/kg CPT, where the effects were more evident, but the toxicity was tolerable (the mice did not lose much weight and appeared healthy and active).
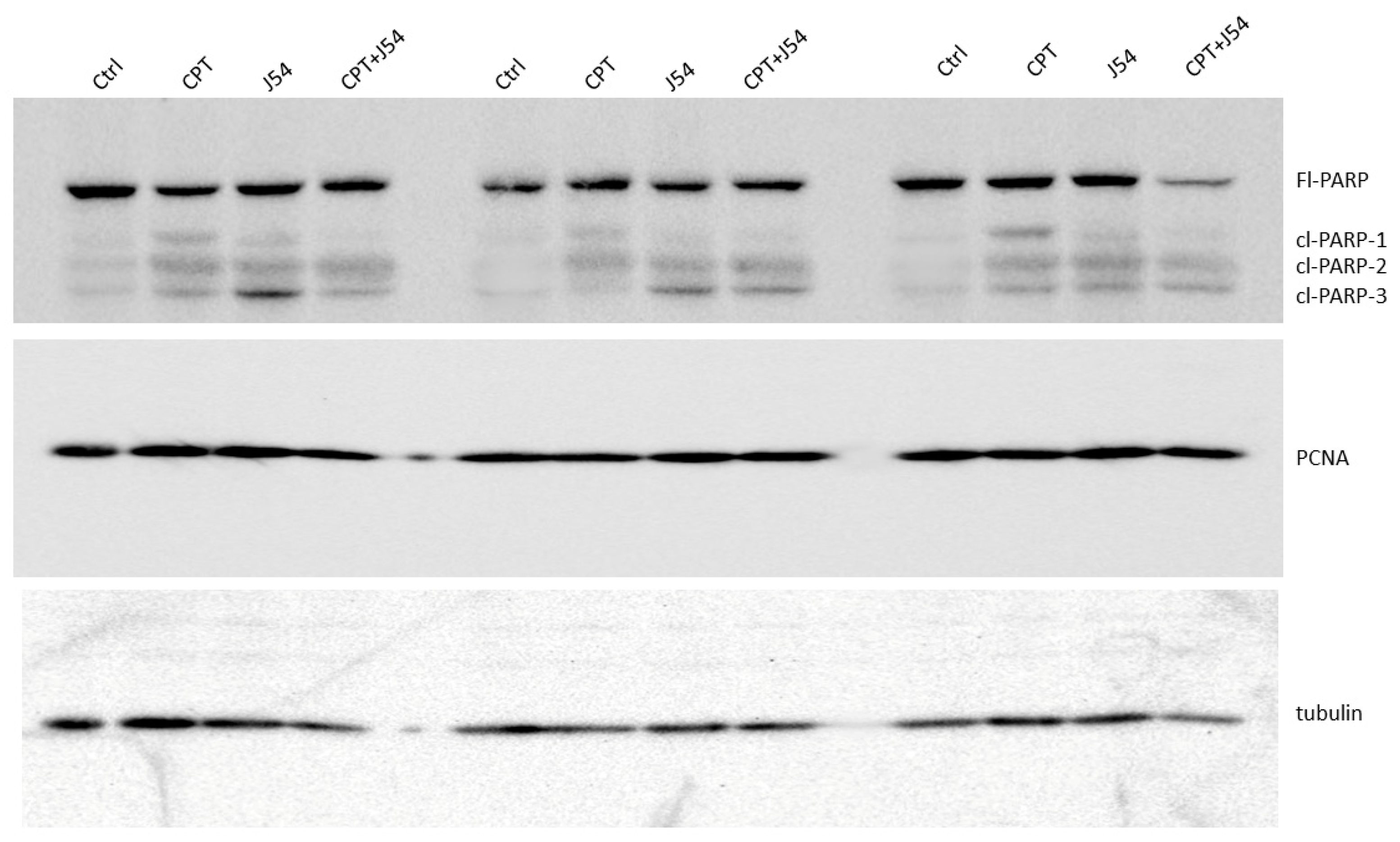
Immediately after necropsy, all the tumors were weighed, and one from three random mice in each group (from the 3 mg/kg dose) were prepared for western blot analysis of PARP/cleaved PARP (for determination of the intratumoral extent of apoptosis) and PCNA (for determination of the fraction of still proliferating cells), as illustrated in Figure 4, it is immediately clear that all the mice treated with CPT alone displayed reduced level of full-length (Fl) PARP and an array of its cleavage products, with an abundant ~90 kDa product (cl-PARP1) that is considered its classic apoptotic signature [31]; but smaller processed product were also generated as indicated in Figure 4. In the CPT+J54 combination treatment, and most prominently in the right-most tumor, Fl-PARP was strongly reduced, and there is a prominent formation of cl-PARP2 and cl-PARP-3 and entirely bypassing cl-PARP1 suggesting much more advanced stage of apoptosis and loss of PARP integrity (PARP degradation is mediated by a caspase cascade [32]). It should be noted that J54 treatment alone also resulted in some cleaved PARP products, consistent with our previous report that its related compound (Thioridazine also acting as inhibitor of TLK1), while not toxic for PC3 cells in vitro, resulted in some tumor regression with distinctive apoptotic signatures when administered in xenografts due to the partly hypoxic tumor micro-environment that tends to generate DNA lesions and activation of the DDR [33].
We should mention that we did not carry out other confirmatory assays of apoptosis, like TUNEL/IHC, due to the fact that several residual tumors, and particularly in the most significant 3mg/kg CPT+J54 group, were very small, and we had to make a choice between preparation for WBs or PEFF sections.
We also investigated the status of pRAD54-T700 in these tumor extracts. Apart from some biological variability in the three tumors from each group, it appears that J54 (or CPT+J54) actually increased pRAD54 signal, rather than its expected decrease (SI-Figure S3). While this may seem like a contradiction of our study, the fact is that the phosphorylation of pRAD54-T700 primarily alters its nucleoplasmic relocalization from the cytoplasm where it predominantly resides in the absence of DNA damage (DSBs); and we observed that the nuclear fraction remains unaltered by the addition of J54 likely because of the absence of a nuclear phosphatase [28]. Indeed, our fractionation studies revealed that after IR, pRAD54-T700 almost exclusively relocalizes to the nuclei, based on fractionation experiments. We further demonstrated that even treatment with J54, which as an inhibitor of TLK1, results in a reduction in the total cell extract pRAD54 signal, the pT700-nuclear fraction remains unaltered by the addition of J54 [27]. In this specific context in mice, prolonged chronic treatment with J54 or (the more relevant) CPT+J54 may result in unexpected levels of pRAD54-T700. Since our working hypothesis is that CPT eventually results in generation of DSBs that require processing via RAD54/HRR, but also that J54 alone can result in accumulation of DNA damage in vivo due to the hypoxic tumor microenvironment, this could be an explanation for the rather unexpected findings of increased pRAD54 signal in two of the J54(+/- )CPT tumor samples
To ensure that J54 was nonetheless inhibiting TLK1 within those tumors, despite the lack of evidence using pRAD54-T700 as reader, we checked the same tissue extracts for pNEK1-T141, which is our most reliable relay for TLK1 activity. Our pNEK1-T141 Ab detects preferentially the phosphorylated form, which runs higher up on SDS/PAGE, but also a bit less efficiently the “lower” un-phosphorylated form (previously demonstrated via overexpression of a T141A mutant [34]). As shown in SI-Figure S2, all representative tumors from the all the mice that included treatment with J54 display a “band shift” change toward the un-phosphorylated NEK1 form. In contrast to the clearly apoptotic hallmark in the tumor tissue from the treated mice, which alone could explain the obvious size regression in the CPT+J54 combination group, as well as in the other groups to a lesser extent, the proliferative capacity (measured by the PCNA expression as an indicator of the % dividing cells) was not affected in any of the 3 tumors from each group (Figure 4). We conclude that heightened apoptotic induction, rather than inhibition of proliferation, is the key explanation for the tumor cytoreduction over the course of treatment.
Discussion
The concept of synthetic lethality has evolved around the central tenet that by affecting two pathways, each one of which is not essential for viability, the result is engendering a non-viable outcome, either via a genetic approach or by pharmacologic targeting. Perhaps its most successful use in cancer therapy is illustrated by the application of PARPi, sometime in combination with other drugs, for the management of gynecological malignancies [35] or for PCa [36-38], particularly in the context of a BRCAness cancer presentation.
In this work we have somewhat expanded on the concept of synthetic lethality by taking advantage of the knowledge of the repair process of CPT-induced lesions (a commonly used chemotherapy) that are known to yield DSBs that are repaired via HRR, a key pathway for their accurate repair, which would otherwise result in lethality from unrepaired damage or engender combination of genomic rearrangements that in most cases would produce non-viable configurations [39]. Specifically, we exploited our knowledge of a regulatory pathway we recently uncovered governing the activity of the key HHR protein, RAD54, by the kinase TLK1 [28]. RAD54 performs critical multiple functions during the HRR process [29], and both its activity and cellular localization were found to be regulated differently by TLK1 during sequential phases of the process. Therefore, the strategy we used was to treat two of the most common CRPC human cell lines with progressively more tolerable doses of CPT in combination with J54, a relatively new TLK1 inhibitor [30, 40]. After a relatively rapid verification, in vitro, of the proof-of-principle of this strategy by determining the viability of these cells, which are normally quite resistant to CPT [41] when challenged in combination with a non-toxic concentration of J54, we then progressed to the more critical xenograft model. For this more critical analysis, we used two methods to follow tumor growth or regression over time: direct measurement of tumors diameters with calipers, and IVIS imaging at midpoint (~1 month) and endpoint (day before sacrifice) of the time course. Direct visual evidence of tumor regression for the most representative group (3 mg/kg CPT+ J54) is displayed in SI-Figure S1, which is best appreciated when viewed in comparison with the endpoint image. Of particular importance is again the radiance scale that at treatment midpoint was 1.89e8 (min) to 3.64e9 (max), showing that the tumors were up to 5 logs more emissive (larger number of cells) than at the endpoint shrinkage stage. But note that even at midpoint, the radiance was already ~1 log lower than that of the control group. The lack of a simple correlation between the phosphorylation of RAD54-T700 (SI-Figure S3) and the tumors regression in mice treated with CPT+J54, made it difficult to conclude mechanistically that the apoptotic response seen in vivo was due to a lapse in HRR consequent to the processing of ICLs. In fact, there could be other possible explanations for the effect observed, as inhibition of TLK1 can suppress tumor growth via other mechanisms (for recent reviews, see [40, 42, 43]).
Of interest are the observations we made about the effect of J54 on the weight and overall health appearance of the mice over the course of the experiment, with the exclusion of the 6 mg/kg CPT two groups for which the toxicity was excessive after 6 doses. Starting from the obvious observation that the mice treated 3 mg/kg CPT progressively lost weight, whereas those that also received J54 did not and appeared healthier and more active, we noted some peculiar differences in the adipose tissue. Whereas the mice treated with 3 mk/kg CPT alone showed either cytostatic or modest cytoreduction of the tumors, but without an obvious infiltration of adipose tissue, the mice that also received J54 displayed a noticeable replacement of the shrunk tumors space with an overgrowth of adipocytes. We suggest that this may be due to the combination of two factors: 1) There is already well described literature of the interplay between cancer cells and locally adjacent adipocytes, which secrete cross-stimulatory adipokines and cytokines [44]. 2) It is well known that several phenothiazine (PTH) antipsychotics (as well as newer atypical ones) cause accumulation of white fat tissue and some weight gain as an unfortunate side-effect [45, 46]. J54 is structurally a PTH, although it was specifically designed to have minimal binding activity to the D2R, explaining its low antidopaminergic properties [30], and such activity is held to be the main mechanism responsible for adipogenesis stimulation. Regardless, other mechanisms have been proposed, such as increase expression of sterol regulatory element-binding protein (SREBP) and very low density lipoprotein (VLDL) genes (rev. in [46]) which could be alternative explanations for the effect of J54 on the localized adipogenesis and overall maintenance of body fat/weight in the face of mice treated with CPT alone. These progressively lost some weight and were visibly thin during the course of treatment likely from the combination of tumor burden and toxicity from CPT, for which it is well know that cachexia is a major issue during the terminal phases of cancer progression.
Overall, we would like to conclude that the use of J54 to achieve pharmacologic synthetic lethality with CPT (one of the most common chemotherapeutic agents) has shown promising results in one model of PCa xenograft, whereby the synthetic targeting of HRR after the generation of DNA adducts and ISLs was shown to greatly improve the efficacy of the therapy in tumor regression. An unexpected benefit was actually the better general health and maintenance of body weight in the most effective dose combination group. While we do not yet know if there are other more subtle side-effects for J54 that in the long run may make its potential clinical use unattainable, so far and at least in mice, we never lost an animal nor have we seen altered behaviors in those treated with a dose of up to 20 mg/kg.
Materials and Methods
- 1.
- Cell Viability Assay
Cell viability was evaluated using the MTT assay, which measures the reduction of 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) by mitochondrial enzymes in live cells. We seeded 20,000 PC-3 and C4-2B cells into 100 μL of medium in 96-well plates and allowed them to adhere for 24 hours. Then, we replaced the media with a fresh medium containing various cisplatin and/or J54 concentrations and incubated for an additional 24 hours. Finally, we added MTT reagent to each well and incubated it for 35 minutes before measuring the absorbance intensity at 490 nm using a spectrophotometer.
- 2.
- Animal Studies
All animals used in this study received humane care based on the recommendations set by the American Veterinary Medical Association, and the Institutional Animal Care and Use Committee of LSU Health Sciences Center at Shreveport approved all the test protocols. Immune-deficient NOD SCID mice (Charles River) were used in this research to host human PCa PC-3 tumors. 0.5*106 human PCa PC-3-Luc cells suspended in matrigel were grafted subcutaneously into the two lower back flanks of the NOD SCID mice. After the tumor sizes of ~150 mm3 were established, the tumor-bearing mice were randomized into eight treatment groups. Mice were treated with vehicle control (PBS), TLK1-RAD54 axis inhibitor J54 (5 mg/kg), and different dosages of CPT (1, 3, and 6 mg/kg) with or without J54. Intraperitoneal (IP) injections of J54 (dissolved in 200 sterile saline with 10% Polysorbate-80 – PS-80) were given bi-weekly. The dose for J54 was based on our previous work [30]. CPT dissolved in 0.9% Sterile-filtered NaCl Solution (Saline) was administered individually at different dosages- (1, 3, and 6 mg/kg) in 100 ul volume by IP injection twice a week. Tumor sizes were measured every other day by using a caliper. The inhibitor treatment lasted 28 days for about nine bi-weekly drug cycles. The tumor-bearing mice were monitored every other day and euthanized if there was an apparent loss in body weight (≥ 20%), abdominal palpitation due to the development of prostate tumors or cancer metastasis, poor body condition, or mice were too sick and unable to reach food and water. The mice were sacrificed at the end of the experiment by CO2 asphyxiation, and the tumors were excised for the tissue western blots.
- 3.
-
Western Blots
- Tissue Western Blot: Western blots were performed in three biological replicates for the tumors excised from the different treatment groups, including control (PBS), cisplatin (3mg/kg), J54 (5mg/kg) and the combination of the PC-3 grafted NOD SCID mice. The frozen tumor tissues were disrupted with the Bioruptor® Plus sonication device (Diagenode; Cat. No. B01020001), homogenized, and lyzed in the ice-cold RIPA lysis buffer system (Santa Cruz Biotechnology; Cat. No. SC-24948). The samples were clarified by centrifugation at 13,000 rpm for 20 mins in the refrigerated setting. The supernatant was collected, transferred into fresh 1.5 ml microfuge tubes, flash-frozen, and stored at -800C until further use. The total protein concentration was measured using a Pierce™ BCA protein assay kit (Thermo Scientific; Cat. No. 23225) with bovine serum albumin (BSA) as a standard control. An equal loading amount of 15 µg was calculated for each protein sample. The sample supernatant was denatured with 1X Laemmli Buffer for 10 mins at 950C and separated using 12% Mini PROTEAN TGX protein gel (BioRad; Cat. No. 4568084) at 100 volts for 120 mins. The proteins were transferred to the Immun-Blot PVDF membrane (BioRad; Cat. No. 1620177) using a Mini Trans-Blot Cell (BioRad; Cat. No. 1703930) at 100 volts for 150-180 mins on ice. The membrane was blocked with 5% Non-fat dry milk (Cell Signaling Technology; Cat. No. 9999S) in 1X Tris-buffered saline with Tween-20 (TBST) for 1 hour at room temperature. Following blocking, the membrane was washed once with 1X TBST and incubated with mouse anti-PCNA (PC10) monoclonal antibody (Santa Cruz Biotechnology; Cat. No. SC-56; 1:1000 dilution) and mouse anti-PARP-1 (F-2) monoclonal antibody (Santa Cruz Biotechnology; Cat. No. SC-8007; 1:1000 dilution) in 5% BSA in 1X TBST overnight at 40C with gentle rocking. The next day, after washing four times with 1X TBST, the membrane was incubated with horse anti-mouse antibody (Cell Signaling Technology; Cat. No. 7076S: 1:2000 dilution) labeled with horseradish peroxidase in 5% BSA in 1X TBST for 1-1.5 hours at room temperature. After incubation, the membrane was washed four times with 1X TBST, and the reactive bands were detected using Pierce™ ECL Western Blotting Substrate (Thermo Scientific; Cat. No. 32106) on ChemiDoc MP Imaging System (BioRad; Cat. No. 12003154).
- Cell Western Blot: The western blot for PC-3 cells was performed as described above but with minor modifications. Briefly, 3*106 PC-3 cells (control and drug-treated) were collected, washed twice with ice-cold PBS, and lyzed with RIPA lysis buffer system. The lysate was vortexed and centrifuged at 13,000 rpm for 10 min to remove cell debris. The total protein was estimated, and 30 μg of the cell lysate was loaded onto an SDS-PAGE gel. The separated proteins were transferred to the membrane using a wet transfer apparatus. The complete transfer was ensured by checking the membrane for uniform background staining. The membrane was then incubated in a blocking solution (e.g., 5% non-fat milk in TBST) for 1 h at room temperature to block non-specific binding sites, followed by primary antibody (custom-made anti-pRAD54 rabbit polyclonal; Thermo Scientific; Cat. No. AB1991; 1:1000) incubation in blocking solution overnight at 4°C. The next day, the membrane was washed 3X with TBST for 10 min each to remove excess primary antibody. Further, it was incubated with goat anti-rabbit HRP-conjugated secondary antibody diluted in a blocking solution for 1 h at room temperature. Next, the membrane was washed 3X with TBST for 10 min each to remove excess secondary antibody, and the bands were detected using the ECL chemiluminescent substrate.
Author Contributions
Conceptualization by ADB, SB, and OO, manuscript writing: ADB, SB, and OO; methodology: ADB, SB, XY and OO; experiment performance, analysis and interpretation ADB, SB, XY and OO.
Institutional Review Board Statement
N/A.
IACUC approval
This study “ Combining CPT with targeting TLK/RAD54 axis in prostate Cancer” (P-23-039) was approved on May 12, 2023.
Data Availability Statement
Description of all data and materials can be found in the referenced article. No additional data is withheld from the public.
Acknowledgements
We like to thank Lucia Solitro in the animal care facility of LSU Health Shreveport for the expert and dedicated IVIS analysis of our tumor regression studies. Special thanks to Brian Latimer for assistance in working with the Synergy4 plate reader. We also like to thank the Research Core Facility Genomics Core and animal facility of LSU Health Shreveport. Funding was provided by a Louisiana CCRI grant LSU-2022-CCRI-6.
Conflicts of Interest
The authors declare No conflicts.
References
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Nyberg, K.A.; Michelson, R.J.; Putnam, C.W.; Weinert, T.A. Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet 2002, 36, 617–656. [Google Scholar] [CrossRef] [PubMed]
- Setton, J.; Zinda, M.; Riaz, N.; Durocher, D.; Zimmermann, M.; Koehler, M.; Reis-Filho, J.S.; Powell, S.N. Synthetic lethality in cancer therapeutics: the next generation. Cancer Discov 2021, 11, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin Cancer Res 2020, 26, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Tsujino, T.; Takai, T.; Hinohara, K.; Gui, F.; Tsutsumi, T.; Bai, X.; Miao, C.; Feng, C.; Gui, B.; Sztupinszki, Z.; et al. CRISPR screens reveal genetic determinants of PARP inhibitor sensitivity and resistance in prostate cancer. BioRxiv 2022, Preprint. [Google Scholar] [CrossRef]
- Adams, M.N.; Ashton, N.W.; Paquet, N.; O’Byrne, K.; Richard, D.J. Mechanisms of cisplatin resistance: DNA repair and cellular implications. In Advances in Drug Resistance Research, Morais, C., Ed. Nova Science Publishers, Inc.: 2014; pp. 1-37.
- Kiss, R.C.; Xia, F.; Acklin, S. Targeting DNA damage response and repair to enhance therapeutic index in cisplatin-based cancer treatment. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Jones, D.; Lee, S.H.; Williamson, E.A.; Hromas, R. Drugging the Cancers Addicted to DNA Repair. J Natl Cancer Inst 2017, 109. [Google Scholar] [CrossRef]
- Schmid, S.; Omlin, A.; Higano, C. Activity of platinum-based chemotherapy in patients with advanced prostate cancer with and without DNA repair gene aberrations (vol 3, e2021692, 2020). Jama Network Open 2020, 3. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Sarangi, P.; D'Andrea, A.D. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol 2016, 17, 337–349. [Google Scholar] [CrossRef]
- Hashimoto, S.; Anai, H.; Hanada, K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes Environ 2016, 38, 9. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.; Albers, P. Testicular germ cell tumors: pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2011, 7, 43–53. [Google Scholar] [CrossRef]
- Perše, M. Cisplatin Mouse Models: Treatment, Toxicity and Translatability. Biomedicines 2021, 9, 1406. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: an intimate relationship. Clinics (Sao Paulo) 2018, 73, e478s. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Gu, F.X.; Langer, R.; Farokhzad, O.C.; Lippard, S.J. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A 2008, 105, 17356–17361. [Google Scholar] [CrossRef]
- Selemenakis, P.; Sharma, N.; Uhrig, M.E.; Katz, J.; Kwon, Y.; Sung, P.; Wiese, C. RAD51AP1 and RAD54L Can Underpin Two Distinct RAD51-Dependent Routes of DNA Damage Repair via Homologous Recombination. Frontiers in Cell and Developmental Biology 2022, 10. [Google Scholar] [CrossRef]
- Mota, J.M.; Barnett, E.; Nauseef, J.T.; Nguyen, B.; Stopsack, K.H.; Wibmer, A.; Flynn, J.R.; Heller, G.; Danila, D.C.; Rathkopf, D.; et al. Platinum-Based Chemotherapy in Metastatic Prostate Cancer With DNA Repair Gene Alterations. JCO Precis Oncol 2020, 10.1200/po.19.00346, 355–366. [Google Scholar] [CrossRef]
- Schmid, S.; Omlin, A.; Higano, C.; Sweeney, C.; Martinez Chanza, N.; Mehra, N.; Kuppen, M.C.P.; Beltran, H.; Conteduca, V.; Vargas Pivato de Almeida, D.; et al. Activity of Platinum-Based Chemotherapy in Patients With Advanced Prostate Cancer With and Without DNA Repair Gene Aberrations. JAMA network open 2020, 3, e2021692–e2021692. [Google Scholar] [CrossRef]
- Sillje, H.H.; Takahashi, K.; Tanaka, K.; Van Houwe, G.; Nigg, E.A. Mammalian homologues of the plant Tousled gene code for cell-cycle-regulated kinases with maximal activities linked to ongoing DNA replication. Embo J 1999, 18, 5691–5702. [Google Scholar] [CrossRef]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. The TLK1-Nek1 axis promotes prostate cancer progression. Cancer Lett 2019, 453, 131–141. [Google Scholar] [CrossRef]
- Sunavala-Dossabhoy, G.; Fowler, M.; De Benedetti, A. Translation of the radioresistance kinase TLK1B is induced by gamma-irradiation through activation of mTOR and phosphorylation of 4E-BP1. Bmc Molecular Biology 2004, 5. [Google Scholar] [CrossRef]
- Groth, A.; Lukas, J.; Nigg, E.A.; Sillje, H.H.; Wernstedt, C.; Bartek, J.; Hansen, K. Human Tousled like kinases are targeted by an ATM- and Chk1-dependent DNA damage checkpoint. Embo J 2003, 22, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; DeFatta, R.; Anthony, C.; Sunavala, G.; De Benedetti, A. A translationally regulated Tousled kinase phosphorylates histone H3 and confers radioresistance when overexpressed. Oncogene 2001, 20, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.P.; De Benedetti, A. TLK1B promotes repair of UV-damaged DNA through chromatin remodeling by Asf1. BMC Mol Biol 2006, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Takayama, Y.; Kokuryo, T.; Yokoyama, Y.; Ito, S.; Nagino, M.; Hamaguchi, M.; Senga, T. Silencing of Tousled-like kinase 1 sensitizes cholangiocarcinoma cells to cisplatin-induced apoptosis. Cancer Lett 2010, 296, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Kwon, Y.; Shabestari, A.B.; Chikhale, R.; Chen, J.; Wiese, C.; Sung, P.; De Benedetti, A. TLK1-mediated RAD54 phosphorylation spatio-temporally regulates Homologous Recombination Repair. Nucleic Acids Research 2023. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.-D.; Li, X.; Rolfsmeier, M.; Zhang, X.-P. Rad54: the Swiss Army knife of homologous recombination? Nucleic Acids Research 2006, 34, 4115–4125. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Bhoir, S.; Chikhale, R.V.; Hussain, J.; Dwyer, D.; Bryce, R.A.; Kirubakaran, S.; De Benedetti, A. Generation of Phenothiazine with Potent Anti-TLK1 Activity for Prostate Cancer Therapy. iScience 2020, 23, 101474. [Google Scholar] [CrossRef]
- Castri, P.; Lee, Y.J.; Ponzio, T.; Maric, D.; Spatz, M.; Bembry, J.; Hallenbeck, J. Poly(ADP-ribose) polymerase-1 and its cleavage products differentially modulate cellular protection through NF-kappaB-dependent signaling. Biochim Biophys Acta 2014, 1843, 640–651. [Google Scholar] [CrossRef]
- Los, M.; Mozoluk, M.; Ferrari, D.; Stepczynska, A.; Stroh, C.; Renz, A.; Herceg, Z.; Wang, Z.Q.; Schulze-Osthoff, K. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell 2002, 13, 978–988. [Google Scholar] [CrossRef]
- Ronald, S.; Awate, S.; Rath, A.; Carroll, J.; Galiano, F.; Dwyer, D.; Kleiner-Hancock, H.; Mathis, J.M.; Vigod, S.; De Benedetti, A. Phenothiazine Inhibitors of TLKs Affect Double-Strand Break Repair and DNA Damage Response Recovery and Potentiate Tumor Killing with Radiomimetic Therapy. Genes Cancer 2013, 4, 39–53. [Google Scholar] [CrossRef]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of androgen independent prostate tumors. International journal of cancer 2019, 145, 1055–1067. [Google Scholar] [CrossRef]
- Lightfoot, M.; Montemorano, L.; Bixel, K. PARP Inhibitors in Gynecologic Cancers: What Is the Next Big Development? Curr Oncol Rep 2020, 22, 29. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan Geethakumari, P.; Schiewer, M.J.; Knudsen, K.E.; Kelly, W.K. PARP Inhibitors in Prostate Cancer. Curr Treat Options Oncol 2017, 18, 37. [Google Scholar] [CrossRef] [PubMed]
- Stopsack, K.H. Efficacy of PARP Inhibition in Metastatic Castration-resistant Prostate Cancer is Very Different with Non-BRCA DNA Repair Alterations: Reconstructing Prespecified Endpoints for Cohort B from the Phase 3 PROfound Trial of Olaparib. Eur Urol 2021, 79, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Teyssonneau, D.; Margot, H.; Cabart, M.; Anonnay, M.; Sargos, P.; Vuong, N.-S.; Soubeyran, I.; Sevenet, N.; Roubaud, G. Prostate cancer and PARP inhibitors: progress and challenges. J Hematol Oncol 2021, 14, 51–51. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Löbrich, M. Misrepair of radiation-induced DNA double-strand breaks and its relevance for tumorigenesis and cancer treatment (review). Int J Oncol 2002, 21, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Bhoir, S.; De Benedetti, A. Targeting Prostate Cancer, the ‘Tousled Way&rsquo. International Journal of Molecular Sciences 2023, 24, 11100. [Google Scholar]
- Gumulec, J.; Balvan, J.; Sztalmachova, M.; Raudenska, M.; Dvorakova, V.; Knopfova, L.; Polanska, H.; Hudcova, K.; Ruttkay-Nedecky, B.; Babula, P.; et al. Cisplatin-resistant prostate cancer model: Differences in antioxidant system, apoptosis and cell cycle. Int J Oncol 2014, 44, 923–933. [Google Scholar] [CrossRef]
- Khalil, M.I.; De Benedetti, A. Tousled-like kinase 1: a novel factor with multifaceted role in mCRPC progression and development of therapy resistance. Cancer Drug Resistance 2022, 5, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; De benedetti, A. Untousling the Role of Tousled like Kinase 1 in DNA Damage Repair. In Preprints, Preprints: 2023. [CrossRef]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Carli, M.; Kolachalam, S.; Longoni, B.; Pintaudi, A.; Baldini, M.; Aringhieri, S.; Fasciani, I.; Annibale, P.; Maggio, R.; Scarselli, M. Atypical Antipsychotics and Metabolic Syndrome: From Molecular Mechanisms to Clinical Differences. Pharmaceuticals (Basel) 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Dayabandara, M.; Hanwella, R.; Ratnatunga, S.; Seneviratne, S.; Suraweera, C.; de Silva, V.A. Antipsychotic-associated weight gain: management strategies and impact on treatment adherence. Neuropsychiatr Dis Treat 2017, 13, 2231–2241. [Google Scholar] [CrossRef]
Figure 1.
Viability of PC3 and C4-2B cells treated with CPT or in combination with J54.
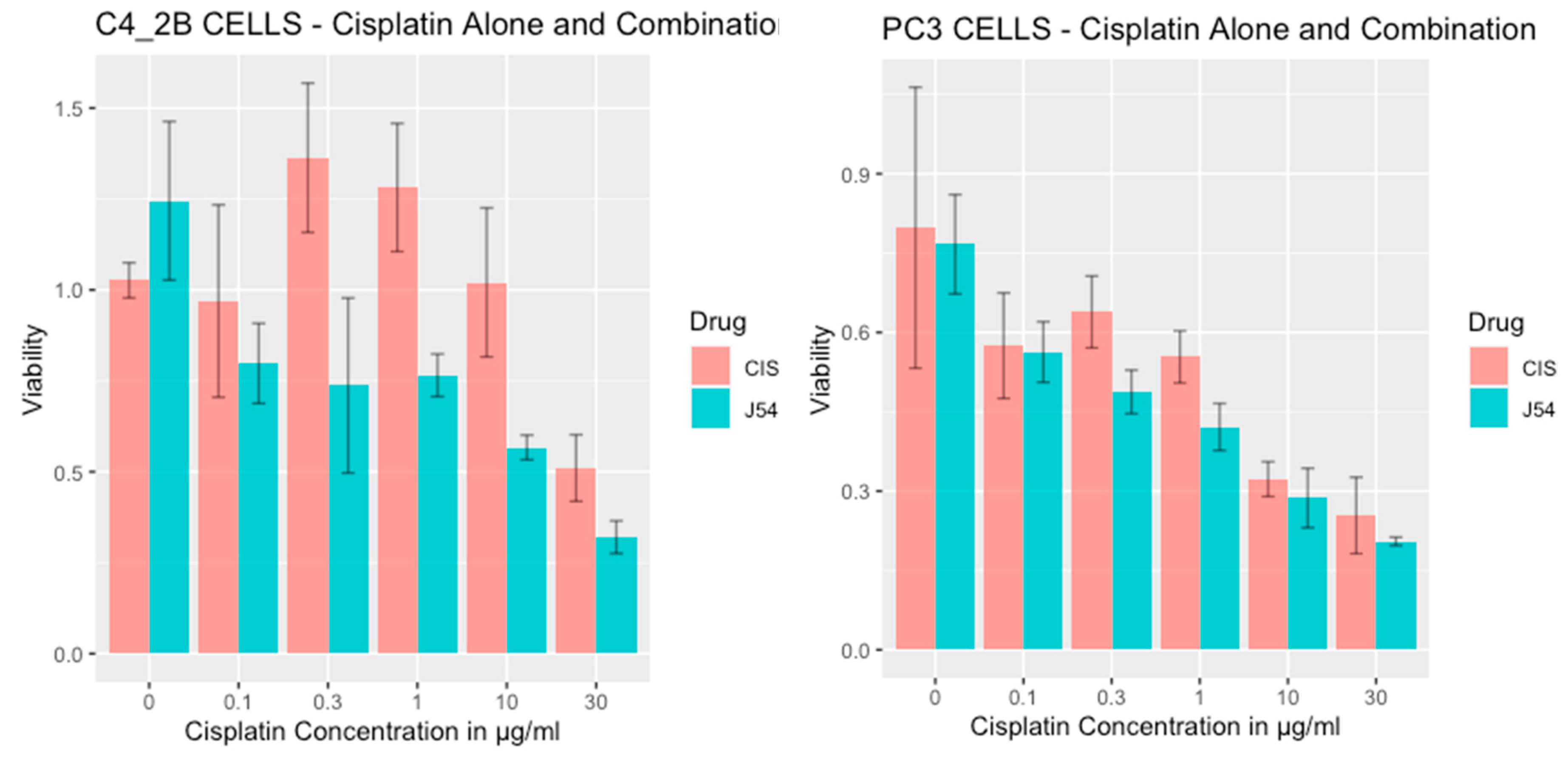
Figure 2.
RAD54-T700 phosphorylation is induced by DNA damage (CIPT) w/o induction of hsp70 expression but is limited by concomitant J54 treatment.
Figure 2.
RAD54-T700 phosphorylation is induced by DNA damage (CIPT) w/o induction of hsp70 expression but is limited by concomitant J54 treatment.
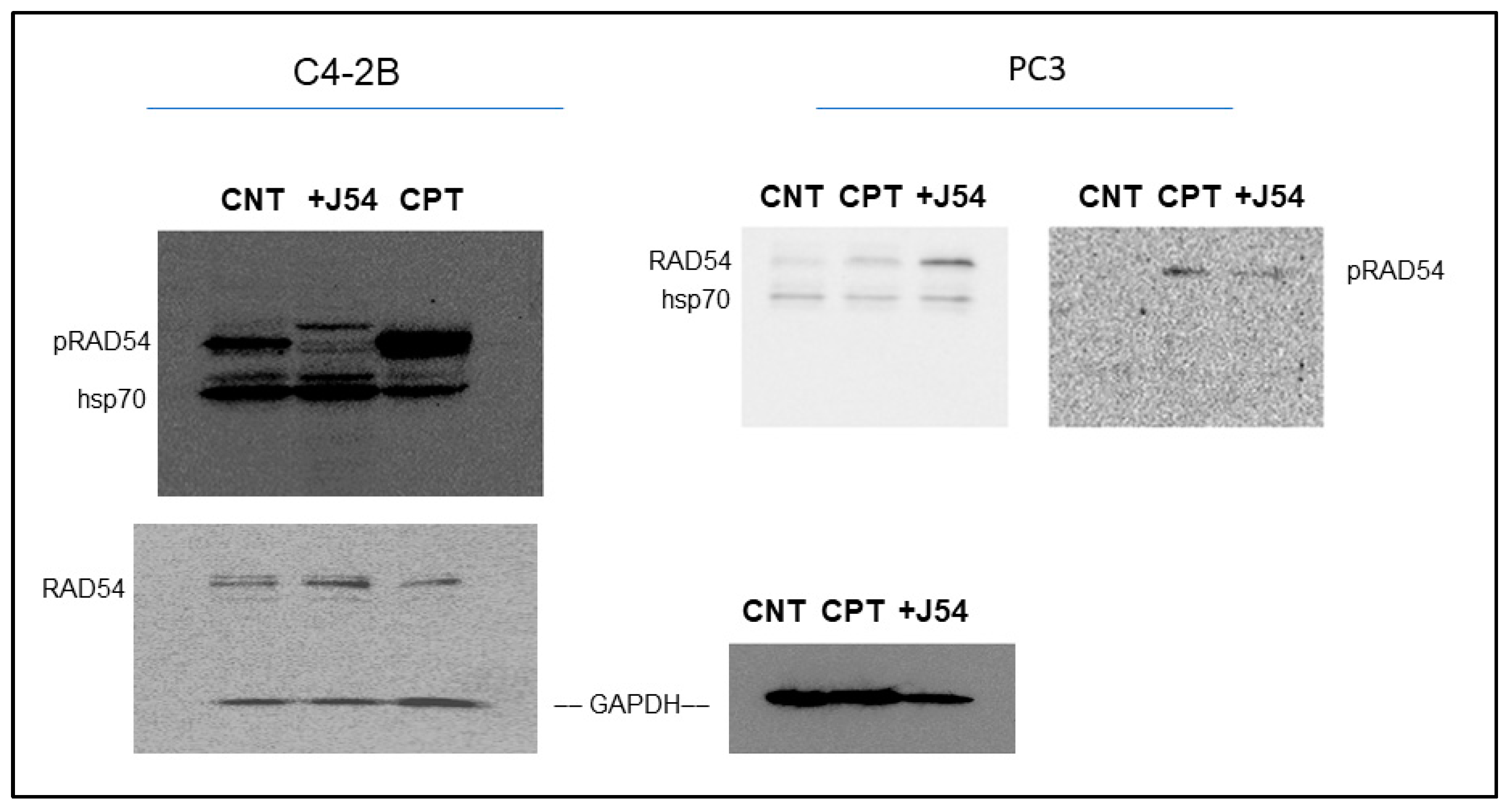
Figure 3.
Time course of tumor progression in PC3-Luc xenografts. The pattern of tumor growth was studied over time with calipers (bottom left) or by one final analysis with the IVIS system one day before sacrifice. Body weights in time-lapse were also recorded (bottom right). NI is not inoculated. Also note the important parameter of radiance divided by the full body area (label on top of rectangles) which in essence represents the total burden of tumor cells for the animal.
Figure 3.
Time course of tumor progression in PC3-Luc xenografts. The pattern of tumor growth was studied over time with calipers (bottom left) or by one final analysis with the IVIS system one day before sacrifice. Body weights in time-lapse were also recorded (bottom right). NI is not inoculated. Also note the important parameter of radiance divided by the full body area (label on top of rectangles) which in essence represents the total burden of tumor cells for the animal.
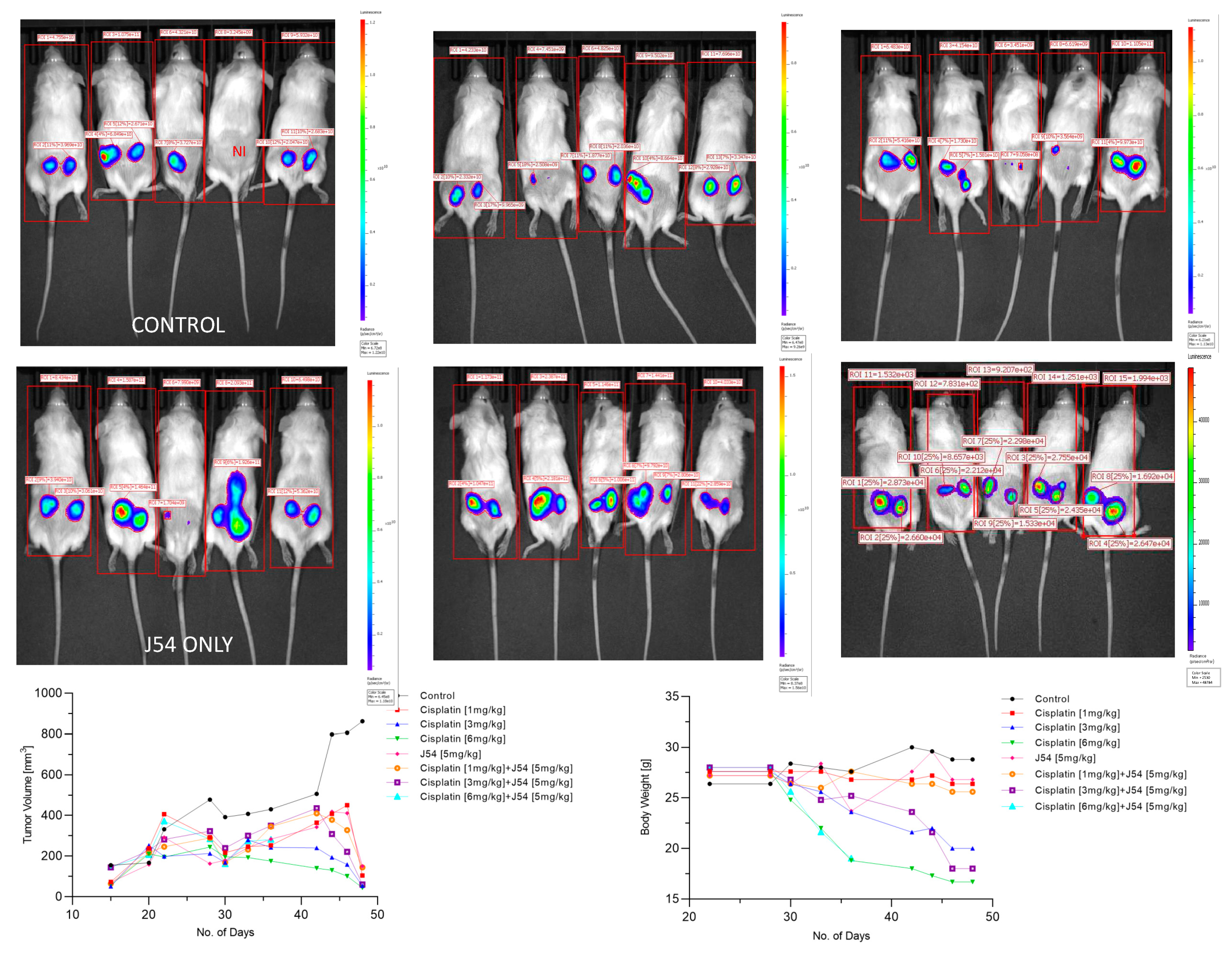
Figure 4.
Western blot analysis of PARP and Cleaved PARP from three sets of independent tumors from all treatment groups, and a separately probed for PCNA and tubulin for loading control.
Figure 4.
Western blot analysis of PARP and Cleaved PARP from three sets of independent tumors from all treatment groups, and a separately probed for PCNA and tubulin for loading control.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



