
Submitted:
28 February 2026
Posted:
02 March 2026
You are already at the latest version
Abstract
Drought is a major constraint on crop productivity, and microbial inoculants are in-creasingly explored to mitigate plant water stress. However, most inoculant discovery pipelines rely on trait-based screening performed outside the ecological context in which beneficial plant-microbe interactions naturally arise. In natural soils, drought-exposed plants can reshape the rhizosphere environment by altering carbon allocation and root exudation, thereby selectively recruiting microorganisms compatible with water-limited conditions and effectively performing an ecological pre-selection. Here, we captured this process during early seedling establishment and leveraged drought-driven rhizosphere recruitment as a nature-guided strategy to nominate bac-terial inoculant candidates. Tomato seedlings were grown in natural agricultural soil microcosms under well-watered and drought-stressed regimes, and cultivable bacteria were comparatively isolated from rhizosphere and bulk soil fractions. Recruit-ment-prioritized isolates were subsequently characterized through biochemical assays and genome-informed analyses to provide functional and taxonomic context, and were evaluated in early inoculation assays under water stress. Drought-recruited isolates displayed distinct plant-associated responses, and genome-scale taxonomy indicated that one drought-associated isolate represents a genomically distinct lineage within the genus Paracoccus. Together, these findings highlight drought-driven rhizosphere re-cruitment as an ecologically grounded framework for identifying stress-compatible bacterial candidates and uncovering previously undescribed rhizosphere diversity.
Keywords:
drought-driven recruitment
; rhizosphere microbiome
; nature-guided screening
; microbial inoculant
; plant–microbe interactions
1. Introduction
Drought is among the most consequential climate-linked risks to agricultural productivity and food security, with increasing evidence that climate change is intensifying the frequency and severity of water limitation events in many cropping systems [1,2,3]. Because drought constrains plant growth through both direct hydric limitation and downstream metabolic disruption, there is strong interest in scalable strategies that enhance crop performance under water stress beyond genetic improvement or irrigation inputs alone [1,2,3,4].
Plant-associated microbiomes, particularly rhizosphere bacteria, contribute to nutrient acquisition, stress physiology, and resilience under adverse conditions [5,6,7,8]. In response to drought, plants not only adjust their internal physiology but also modify the chemical environment of the rhizosphere through changes in carbon allocation and the composition of root exudates. Root exudates comprise a complex mixture of organic acids, sugars, amino acids, secondary metabolites, and small signaling molecules released into soil [9,10,11,12]. Drought stress can alter both the quantity and quality of these exudates [11], shaping the availability of microbial substrates and chemical cues that influence rhizosphere community assembly [9,10,11,12,13,14]. Root exudates can recruit beneficial microbes through chemotaxis and by modulating microbial gene expression and activity, thereby affecting community composition and function in the rhizosphere. Although exudation-driven signaling is dynamic throughout plant development, early seedling establishment represents a particularly sensitive window, as young roots rapidly initiate microbial recruitment while rhizosphere communities are still assembling. Microbes that establish during this phase may exert disproportionate effects on subsequent rhizosphere trajectories and plant stress responsiveness [9,10,12].
This interplay between host exudation and microbial recruitment under stress has been conceptualized as an ecological “communication” process, including the plant “cry-for-help” hypothesis, whereby drought-induced exudate changes contribute to assembling microbial partners that support stress tolerance [15,16]. Consistent with this view, drought-associated shifts in plant-microbiome assembly can enrich resilient taxa and functional traits, potentially contributing to improved stress adaptation and performance upon re-exposure to water limitation [17,18].
Despite growing mechanistic insight into drought-associated microbiome dynamics, translating these interactions into effective microbial inoculants remains challenging. Traditional inoculant discovery pipelines often deploy trait-first screening approaches in simplified laboratory settings that may not capture the ecological constraints required for persistence and function in natural soils [19,20,21]. In practice, this contributes to variable inoculant performance across environments, reflecting interactions among introduced strains, indigenous microbial communities, soil physicochemistry, and agronomic context [20,21,22].
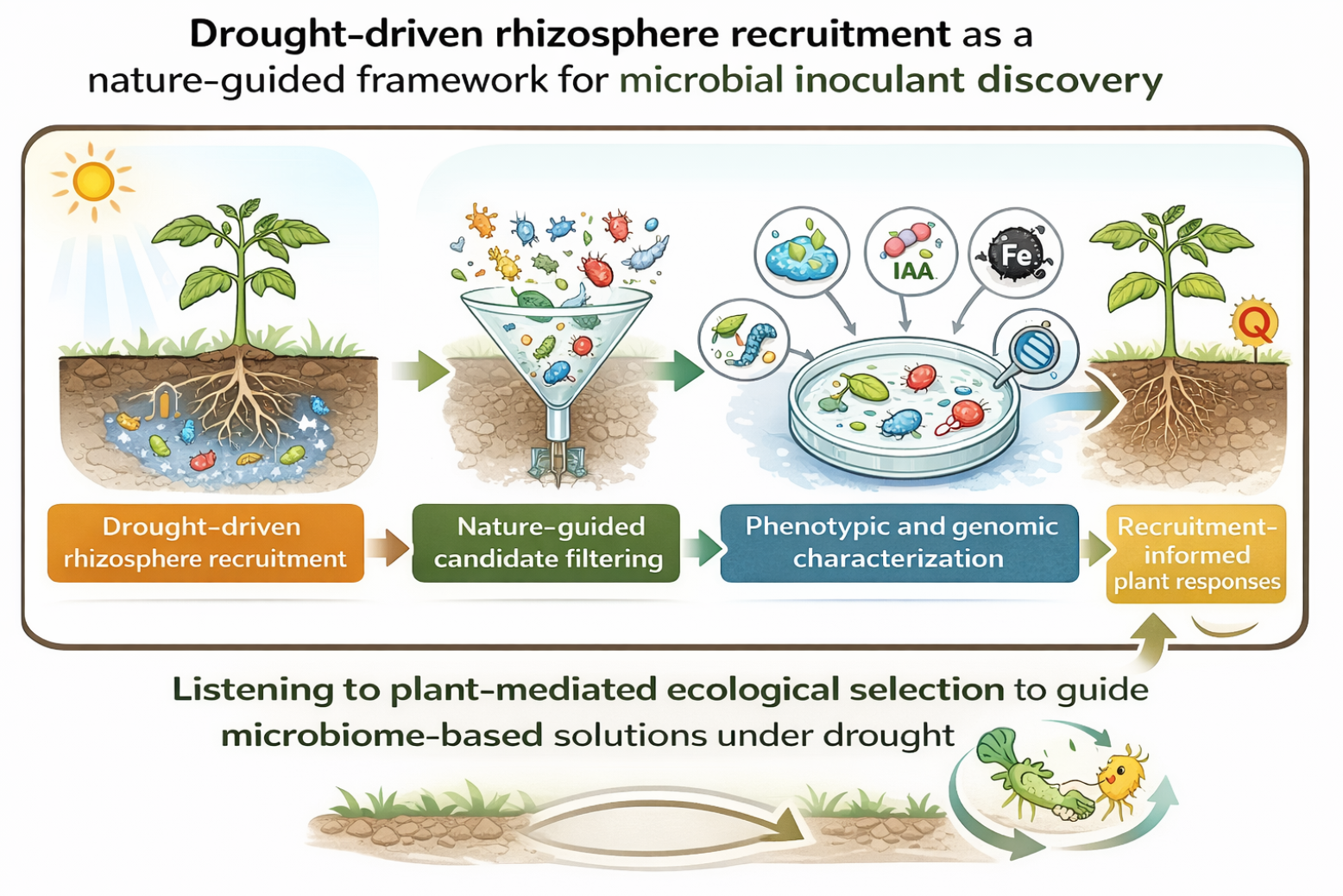
Here, we propose drought-driven rhizosphere recruitment as a nature-guided screening strategy to identify and prioritize bacterial inoculants. The premise is that if plants under drought in natural soil, especially during delicate stages such as seedling emergence, consistently enrich microbial partners suited to water-limited rhizosphere conditions, then those naturally recruited bacteria constitute a rational candidate set for downstream biochemical screening and in planta validation. While drought-driven recruitment can involve multiple microbial kingdoms, we focus here on the cultivable bacterial fraction as a tractable and application-oriented entry point, prioritizing strains that can be readily propagated under standardized laboratory conditions.
Using tomato seedlings grown in natural agricultural soil under controlled water limitation, we (i) isolated cultivable rhizosphere bacteria associated with drought exposure, (ii) prioritized candidates through targeted biochemical assays linked to plant interaction and drought-relevant traits, (iii) leveraged genome-based analyses to support functional potential and taxonomic placement of priority strains, and (iv) validated recruitment-informed candidates via early inoculation assays under water stress. We focused on an early establishment time window, when drought-induced shifts in exudation and recruitment may represent a critical inflection point for plant survival and resilience in natural soil. As an additional outcome of this recruitment-based screening, genome-scale taxonomy supported that one drought-enriched isolate represents a candidate novel species within the genus Paracoccus. Together, this work frames drought-driven recruitment not merely as an observational phenomenon but as a practical and ecologically informed screening pipeline for inoculant discovery.
2. Materials and Methods
2.1. Plant Material, Soil, and Experimental Design
Tomato (Solanum lycopersicum L.) seeds (Tres Cantos cultivar; Semillas Fitó, Spain) were used throughout the study. Seeds were surface-sterilized under aseptic conditions by immersion in 20% (v/v) commercial bleach solution for 7 min with gentle agitation, followed by 70% (v/v) ethanol for 3 min, and then rinsed thoroughly (≥3 times) with sterile double-distilled water (ddH2O) [23]. Sterilization efficacy was verified by plating aliquots of the final rinse water on LB agar and by imprinting sterilized seeds on LB agar plates; plates were inspected for microbial growth after incubation.
Sterilized seeds were pre-germinated on sterile, moistened paper towels in closed sterile containers at room temperature until radicle emergence. Five days post-germination, seedlings were transferred to pots containing natural agricultural soil collected from the Oeiras AGROTECH Campus experimental station (Portugal). Soil was homogenized and sieved to remove large debris while preserving its native physicochemical and microbial characteristics, following common practice for controlled soil-based microbiome experiments [24,25,26]. Microcosms were maintained under greenhouse conditions (May 2025).
Drought stress was imposed during early establishment by applying two watering regimes: a well-watered control (maintaining approximately 70% soil water content) and a drought treatment implemented by applying 30% of the irrigation volume used in the control treatment over a two-week period, followed by maintenance under reduced water availability. The total experimental duration was three weeks. Each treatment consisted of 10 biological replicates, with 3 seedlings per replicate.
2.2. Soil Microcosm Design for Recruitment-Based Culturomics
To assess drought-driven microbial recruitment under controlled yet ecologically realistic conditions, soil microcosms were established using natural agricultural soil as the sole substrate. Microcosms consisted of standard pot systems filled with 0.40 L, homogenized soil and assigned to one of three conditions: (i) soil without plants and well-watered conditions, (ii) soil without plants and drought conditions, (iii) soil with tomato seedlings under well-watered conditions, and (iv) soil with tomato seedlings subjected to drought stress (Figure 1). This design enabled direct comparison of cultivable bacterial populations across soil-only and plant-associated environments, while resolving drought-associated enrichment patterns within the same soil matrix. Microcosms were maintained under identical greenhouse conditions [27].
2.3. Rhizosphere and Bulk Soil Sampling
Bulk soil samples were sampled at the beginning of the test (T=0) and later, from plant-free microcosms to provide a baseline reference for soil-associated microbial communities and the physicochemical background of the native soil matrix. Bulk soil was collected from the central pot volume and homogenized prior to downstream processing. On the other hand, rhizosphere soil was collected in parallel at the end of the experimental period using a root-adhering soil collection approach. Seedlings were gently removed from the soil, and loosely attached soil was removed by shaking. Soil tightly adhering to the root surface was collected by brushing roots into sterile containers, operationally defining the rhizosphere fraction [24]. This paired sampling strategy supported inference of recruitment-driven enrichment by contrasting cultivable bacterial recovery in the rhizosphere against the surrounding soil background. This microcosm-based approach enables disentangling the effects of plant presence and drought stress on cultivable bacterial populations while preserving native soil complexity and physicochemical context [24,27].
2.4. Culturomics and Population-Level Discrimination of Bacterial Isolates
Cultivable bacterial communities were assessed using a culturomics-inspired approach, focusing on comparative recovery of bacterial populations across microcosm conditions rather than exhaustive enumeration. Soil suspensions were prepared from bulk soil and rhizosphere samples (root-adhering soil) using 0.25 g of material per sample, suspended in sterile 0.45% (w/v) NaCl solution, serially diluted, and plated exclusively on Luria-Bertani (LB) agar (per liter: 10 g NaCl, 5 g yeast extract, 10 g tryptone, and 15 g agar). Plates were incubated at 28 °C and monitored at 24 and 48 h. Colony-forming units (CFUs) were enumerated for each sampling point and expressed relative to the dry mass (mg) of the original soil material. This single-medium cultivation strategy was intentionally selected to prioritize robust, readily cultivable strains that can be propagated under standardized conditions, thereby facilitating comparative screening across treatments and supporting downstream scalability considerations relevant to potential bioinoculant development. Accordingly, this study does not aim to provide comprehensive coverage of the total rhizosphere microbiome, including fungal communities or nutritionally fastidious bacteria requiring specialized cultivation conditions. We acknowledge that LB-based recovery may bias community representation toward copiotrophic and fast-growing taxa; however, this selective recovery aligns with the application-oriented goal of prioritizing strains with high culturability and scalability potential.
Distinct colonies were selected based on morphology, pigmentation, and growth dynamics (consistent with standard microbiological criteria), and were purified by repeated streaking to obtain axenic cultures [28]. Purified isolates were preserved at −80 °C in 40% (v/v) glycerol. Isolates were catalogued according to their condition of recovery and classified as drought-enriched (isolated exclusively or predominantly from drought-stressed rhizosphere microcosms), plant-associated but drought-independent (recovered from both well-watered and drought-stressed rhizosphere microcosms), or soil-associated (recovered primarily from plant-free bulk soil microcosms). Population-level discrimination was performed by comparing isolate presence–absence patterns and relative recovery frequency across microcosm conditions, enabling identification of candidate taxa selectively associated with plant presence and drought stress. This comparative cultivation strategy prioritizes ecological association and recruitment signatures over absolute abundance and serves as an effective first-pass framework to nominate early inoculant candidates enriched under water limitation [29,30].
2.4.1. Morphological Grouping and Molecular Identification of Cultivable Isolates
Cultivable bacterial isolates recovered from soil microcosms were initially grouped based on colony morphology [28]. Representative isolates from each morphological group were selected for molecular identification to confirm taxonomic consistency and to reduce redundancy among isolates recovered across microcosm conditions, prioritizing morphotypes repeatedly observed across replicate plates and/or conditions.
Template DNA was obtained from individual colonies using a heat-shock extraction protocol, and DNA concentration/purity was evaluated by spectrophotometry (NanoDrop™ One, Thermo Scientific™, USA). Molecular identification was performed by PCR amplification of the bacterial 16S rRNA gene using either the V5–V8 hypervariable region (~700 bp) or the near full-length 16S rRNA gene (~1500 bp). The V5–V8 region was amplified using the universal primer set 779F (5′-AACMGGATTAGATACCCKG-3′) and 1392R (5′-GGTTACCTTGTTACGACTT-3′), whereas near full-length amplification employed primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-CTACGGCTACCTTGTTACGA-3′). PCR reactions were prepared using NZYTaq II 2× Master Mix (Nzytech, Portugal) and run in a Axygen® MaxyGene II Thermal Cycler (Corning, USA) under the following cycling conditions: initial denaturation at 95 °C for 2 min; 40 cycles of denaturation at 95 °C for 3 s, annealing at 45 °C for 30 s, and elongation at 72 °C for 2 min; followed by a final extension step at 72 °C for 7 min. PCR amplification included no-template controls.
Amplification products were verified by electrophoresis on 1% agarose gels and visualized under UV illumination using a Benchmark myGel InstaView Complete Electrophoresis System with Blue LED Illuminator (VWR, Portugal). PCR products were purified and sequenced by Sanger sequencing (GENEWIZ, Azenta Life Sciences). Resulting sequences were quality-checked, trimmed, and assembled where necessary. Taxonomic affiliation was assigned by comparison against reference sequences in the NCBI nucleotide database using BLASTn, and isolate identity was assigned when sequence similarity exceeded 98%. Closest matches were used to support genus-level identification and, where sequence resolution was sufficient, tentative species-level assignment. Isolates reported as “Unidentified” corresponded to cases in which amplification failed repeatedly or sequence quality was insufficient for reliable assignment. These identifications guided selection of representative isolates for downstream genomic and functional analyses. This molecular identification step was used to validate morphology-based grouping and prioritize representative strains for follow-up characterization, rather than to infer community structure or quantitative abundance patterns across treatments.
The 16S rRNA gene sequences generated in this study have been deposited in GenBank under accession numbers PX985972-PX985973. In selected isolates, repeated Sanger sequencing attempts failed despite successful PCR amplification; therefore, whole-genome sequencing was performed to ensure accurate taxonomic resolution.
2.5. Genome Sequencing, Annotation, and Targeted Analysis
Genomic DNA from selected priority isolates was extracted using standard protocols suitable for whole-genome sequencing. Illumina paired-end sequencing was performed, generating high-quality short reads. Raw sequencing data were subjected to quality assessment and preprocessing, including adapter trimming and removal of low-quality bases. De novo genome assembly was performed using a de Bruijn graph-based assembler optimized for bacterial genomes. Assembled contigs were evaluated for quality using standard assembly metrics, including total genome size, number of contigs, and N50 and L50 values, calculated with QUAST [31]. Sequencing reads were mapped back to the final assembly to assess overall coverage and consistency.
The draft genome assemblies and raw sequencing reads have been deposited in the NCBI under BioProject PRJNA1403472 (BioSample SAMN54674915; locus tag prefix ACYAOT). Whole-genome sequencing was undertaken for selected isolates following repeated failure of Sanger sequencing of 16S rRNA PCR products, despite consistent amplification, in order to ensure accurate taxonomic placement.
2.5.1. Genome-Based Taxonomic Placement and Visualization
Genome-based taxonomic placement of selected isolates was assessed using the Type (Strain) Genome Server (TYGS) [32]. Whole-genome comparisons were performed against available type-strain genomes to identify the closest taxonomic relatives within the genus Paracoccus [24]. Intergenomic distances were calculated using the Genome BLAST Distance Phylogeny (GBDP) approach, and digital DNA-DNA hybridization (dDDH) values were estimated using recommended parameters. Phylogenomic relationships were inferred from genome-scale distance matrices and visualized as balanced minimum-evolution trees with branch support. Species-level relatedness was evaluated using the commonly accepted dDDH threshold of 70%. These analyses were used to assess taxonomic placement and species-level novelty without formal species description.
2.5.2. Identification of Functional Gene Sets
Genes associated with drought adaptation and rhizosphere competence were inferred as a tentative prediction through targeted curation of annotated genome features. Functional categories were defined based on database assignments, with emphasis on pathways previously implicated in rhizosphere colonization, stress tolerance, and plant–microbe interactions. Representative datasets from each functional category were selected for visualization and comparative purposes, including osmoprotection, oxidative stress response, motility and chemotaxis, surface polysaccharide production, and iron acquisition. This curated approach was used for preliminary functional potential and to support figure-based representations rather than comprehensive genome-wide comparisons.
2.6. Biochemical Screening for Plant-Associated and Drought-Relevant Traits
Based on their distribution across microcosm conditions, the rhizosphere isolates were selected for downstream characterization. Priority was given to identified isolates for biosafety reasons, also evaluating their populational changes in enriched/appearing-only/neutral under drought conditions, defining as well stress-independent strains used as internal controls. Selection aimed to balance ecological relevance with feasibility of phenotypic and genomic analyses. Selected isolates were routinely cultured on LB agar or LB broth at 28 °C and maintained as glycerol stocks (20–30% v/v) at −80 °C.
The isolates were then screened for biochemical traits commonly associated with plant, drought-stress mitigation using established qualitative or semi-quantitative assays [33,34]. Indole-related compound production was assessed using Salkowski reagent following standard protocols [35]. Siderophore production was evaluated using chrome azurol S (CAS) agar assays [36]. Biofilm formation was assessed using crystal violet staining in microtiter plates [37]. Moreover, proline production, one of the most relevant protectants against abiotic stresses, was evaluated following previously described approaches [38]. Where appropriate, assays were performed under both standard and drought-mimicking stress conditions to assess stress-dependent trait expression. All assays were conducted using at least 6 independent replicates, and results were interpreted comparatively among isolates to support candidate prioritization rather than as absolute quantitative measurements.
2.7. Quantification of Root Colonization
Root colonization was quantified following the indications in Vilchez et al. 2020 [23], with minor modifications to accommodate tomato seedlings and PEG treatment. Plant material and growth conditions. Tomato seedlings were grown under sterile conditions as explained before, and prepared to test colonization 5 days after germination. Individual strains were grown in LB broth at 30 °C with shaking (220 rpm) for 24 h. Cultures were harvested by centrifugation, washed, and resuspended in 0.45% (w/v) NaCl sterile solution to a standardized density of 106 CFU mL−1. As drought-mimicking stressor, bacterial suspension was supplemented with 15% (w/v) polyethylene glycol (PEG) 6000. Under sterile conditions, seedlings were transferred to sterile tubes containing the bacterial suspension. Tubes were incubated overnight at 26 °C with slow shaking (80 rpm). After overnight incubation, seedlings were rinsed three times with sterile ddH2O to remove loosely attached cells. Then, roots were excised, transferred to sterile tubes, and homogenized using sterile pestles. Homogenates were resuspended in 0.45% NaCl saline solution, and serially diluted (10-fold series). Dilutions were plated on LB agar using the drop-plate method and incubated at 30 °C for 24 h. Colony counts were used to calculate colonization levels, expressed as CFU per root system or normalized to root dry weight (CFU g−1 root DW). Colonization was quantified for each strain under control and PEG conditions using three biological replicates with five seedlings [34].
2.8. Early Inoculation Assays Under Water Stress
To validate the functional relevance of recruitment-informed screening, early inoculation assays were performed using selected priority isolates, and applying protocols previously described [33,34]. Tomato seedlings were inoculated at 5 days post-germination (once transferred to soil mix [turf:vermiculite, 1:1 w:w]) with bacterial suspensions standardized to 106 CFU mL−1 [23]. Inoculation was performed via soil drench to ensure uniform application across treatments. Experimental treatments included non-inoculated controls, plants inoculated with drought-enriched isolates, and plants inoculated with strains not specifically enriched under drought (control). Following inoculation, seedlings were grown under greenhouse conditions, following the same drought regime described above. Ten biological replicates were prepared per condition.
Plant performance under water stress was evaluated by measuring plant height, root length, and dry biomass. Measurements were collected at 14 days-after-treatment corresponding to peak stress exposure.
2.9. Statistical Analysis
All statistical analyses were conducted using GraphPad Prism v9.5.0. Data were tested for normality and homoscedasticity prior to analysis. Treatment effects were assessed using Student t-test or one-way ANOVA, as appropriate. Biological replicates were treated as independent experimental units. Post hoc comparisons were performed using Tukey’s Honest Significant Difference (HSD). Significance thresholds were set at p < 0.05. Graphs were generated using GraphPad Prism v9.5.0 and R v4.5.2.
2.10. Data Availability
The datasets generated and analyzed during the current study are publicly available as follows. The 16S rRNA gene sequences obtained from representative cultivable isolates, corresponding to the V5-V8 hypervariable region or near full-length amplicons, have been deposited in the NCBI GenBank database under accession numbers PX985972–PX985973. The draft annotated genome sequence of Paracoccus sp. strain Q has been deposited in NCBI under BioProject PRJNA1403472 (BioSample SAMN54674915; locus tag prefix ACYAOT; Taxonomy ID: 267). The genome assembly is publicly accessible together with the corresponding raw sequencing reads in the NCBI Sequence Read Archive (SRA) under the same BioProject accession.
Quantitative datasets from biochemical and drought-related phenotypic assays are deposited in Figshare (DOI: 10.6084/m9.figshare.31431346), including raw replicate measurements, experimental metadata, and processed datasets used for statistical analysis. All additional data supporting the findings of this study are available from the corresponding author upon reasonable request.
3. Results
3.1. Population Dynamics of Cultivable Isolates Across Microcosm Conditions
Cultivable bacterial populations differed across soil microcosm conditions in both total abundance and taxonomic composition (Figure 2). Absolute abundance decreased from 90 CFU mg−1 dry soil at T0 to 77 CFU mg−1 in plant-free soil at T1 under regular irrigation (−14%) and to 75 CFU mg−1 under drought (−17%). In seedling-containing microcosms, total recoverable populations reached 86 CFU mg−1 under regular irrigation (−4% vs T0) but declined to 73 CFU mg−1 under drought (−19%), indicating that water limitation reduced the cultivable fraction across both bulk and rhizosphere soils while rhizosphere populations under regular irrigation remained comparatively stable.
Relative abundance analysis revealed clear redistribution of taxa across treatments. At T0, the cultivable fraction was dominated by Pseudomonas fluorescens H (35.6%) and Peribacillus frigoritolerans I (23.3%), together accounting for nearly 60% of recovered isolates, whereas other taxa were present at lower frequencies ranging from 3–13%. Temporal progression in plant-free soil resulted in moderate compositional changes. Under regular irrigation, P. fluorescens H decreased slightly to 30.4% (−15% relative change from T0), whereas drought exposure reduced its contribution further to 22.7% (−36% relative change from T0). In parallel, Pseudomonas putida M increased from 13.3% at T0 to 18.7% under drought (+40% relative increase), and Bacillota representatives collectively expanded their contribution compared with T0.
Rhizosphere samples showed a more even taxonomic distribution compared with bulk soil. Under regular irrigation, P. fluorescens H accounted for 23.3% of isolates (−34% relative to T0), followed by P. putida M (16.3%), Peribacillus simplex J (11.6%), and Priestia megaterium A (7.0%), reflecting reduced dominance of a single taxon relative to bulk soil. Under drought, this redistribution became more pronounced. P. fluorescens H declined to 11.0%, representing a threefold reduction relative to T0, while Bacillota representatives increased markedly, including Peribacillus simplex J and Priestia megaterium A, both reaching 16.4%. Peribacillus frigoritolerans I remained consistently represented across conditions, showing limited variation in relative abundance. The most distinctive shift was observed for Paracoccus sp. Q, which was not recovered from bulk soil or well-watered rhizosphere samples and accounted for 20.5% of isolates exclusively in drought-stressed rhizosphere microcosms.
These contrasting recovery patterns guided selection of representative isolates for downstream biochemical characterization. Priestia megaterium A was prioritized due to its increased representation in seedling-containing microcosms, particularly under drought conditions. Paracoccus sp. Q was selected based on its exclusive recovery from drought-stressed rhizosphere samples, whereas Peribacillus frigoritolerans I was retained as a stress-independent comparator due to its consistent recovery across microcosm conditions.
3.2. Biochemical Profiling of Recruitment-Prioritized Isolates Highlights Complementary Plant-Associated and Drought-Relevant Traits
To functionally characterize isolates displaying contrasting recovery patterns across microcosm conditions, representative strains Priestia megaterium A, Peribacillus frigoritolerans I, and Paracoccus sp. Q were screened for biochemical traits commonly associated with plant interaction and drought-relevant responses, including auxin-related compound production, biofilm formation, proline accumulation, and siderophore production (Figure 3). These isolates were selected to represent increased rhizosphere abundance under drought (P. megaterium A), drought-specific recovery (Paracoccus sp. Q), and stress-independent persistence across treatments (P. frigoritolerans I).
Trait expression differed among isolates and across assays. Auxin-related compound production showed a graded distribution, with P. megaterium A displaying the highest IAA-equivalent levels. Relative to P. megaterium A, auxin production was reduced by approximately 20–25% in P. frigoritolerans I and by ~35% in Paracoccus sp. Q. A similar pattern was observed for biofilm formation, where crystal violet retention was strongest in P. megaterium A, while P. frigoritolerans I and Paracoccus sp. Q exhibited comparable but lower levels (~25–30% lower).
In contrast, proline accumulation displayed an inverse distribution. Here, Paracoccus sp. Q showed the highest levels of proline equivalents, exceeding those detected in P. megaterium A and P. frigoritolerans I by more than twofold. Siderophore production further distinguished the isolates, with P. frigoritolerans I exhibiting the strongest CAS reactivity; siderophore units (psu) were roughly threefold higher than those observed in P. megaterium A, whereas activity was not detected in Paracoccus sp. Q under the tested conditions. Together, these results indicate a differential distribution of biochemical traits across the selected isolates, revealing distinct phenotypic profiles within the candidate set.
3.3. Root Colonization of Tomato Seedlings
Root colonization differed among isolates and was affected by PEG-induced osmotic stress (Figure 4). Under control conditions, plants exposed to P. megaterium A displayed the highest colonization levels, exceeding those observed for P. frigoritolerans I and Paracoccus sp. Q by approximately 7.6-fold and 6.8-fold, respectively, whereas P. frigoritolerans I and Paracoccus sp. Q showed comparable colonization.
PEG exposure reduced colonization across all isolates, although the magnitude of reduction differed among treatments. Colonization by P. megaterium A decreased markedly under PEG, reaching approximately 0.17-fold of control levels (~83% reduction). In contrast, colonization by P. frigoritolerans I declined more moderately to 0.63-fold of control levels, while colonization by Paracoccus sp. Q decreased to 0.47-fold of control levels.
As a result, while P. megaterium A displayed substantially higher colonization under control conditions, it also exhibited the strongest proportional decline under PEG stress. Strains P. frigoritolerans I and Paracoccus sp. Q maintained a larger fraction of their control colonization levels, resulting in reduced differences among isolates under osmotic stress. These patterns describe differential colonization dynamics across isolates under osmotic conditions and provided contextual information for subsequent plant growth assays.
3.4. Early Inoculation Assays Support Recruitment-Informed Candidate Prioritization Under Drought
Plant size and biomass differed across watering regimes and inoculation treatments. Plant biomass and length measurements revealed strong treatment-dependent differences under drought stress (Figure 5a,b). In non-inoculated plants, drought reduced shoot dry weight from 45.4 mg under regular watering to 17.6 mg, corresponding to an approximate 61% decrease. Root dry weight showed a similar decline, decreasing from 5.4 mg to 2.0 mg.
Inoculation altered these responses across treatments. Under drought, plants inoculated with P. megaterium A reached 17.1 mg shoot biomass, remaining within the range observed for drought-stressed controls and lower than plants grown under regular watering with the same inoculation (53.5 mg). Plants inoculated with P. frigoritolerans I displayed intermediate shoot biomass under drought (25.4 mg under regular watering), while the highest shoot biomass under drought was recorded in plants inoculated with Paracoccus sp. Q (50.4 mg), exceeding values observed in drought-stressed controls and approaching those observed under regular watering within the same inoculation group (40.2 mg). Root biomass followed comparable patterns. Under drought, plants inoculated with P. megaterium A reached 3.6 mg root biomass, whereas drought-stressed controls remained at 2.0 mg. The highest root biomass under drought was observed in plants inoculated with Paracoccus sp. Q (9.1 mg), exceeding values observed in non-inoculated drought treatments and regular watering controls (5.4 mg).
Shoot length measurements mirrored biomass responses. In non-inoculated plants, drought reduced shoot length from 18.4 cm to 8.6 cm. Under drought, plants inoculated with P. megaterium A reached 12.2 cm shoot length, whereas plants inoculated with P. frigoritolerans I reached 13.8 cm. The greatest shoot length under drought was observed in plants inoculated with Paracoccus sp. Q (18.8 cm), approaching values measured under regular watering within the same inoculation group (15.4 cm). Root length responses were more variable and showed smaller treatment effects. Drought reduced root length from 7.9 cm to 3.6 cm in non-inoculated plants. Under drought, plants inoculated with P. megaterium A reached 5.6 cm root length, while plants inoculated with strain I reached 5.1 cm. Plants inoculated with Paracoccus sp. Q reached 7.1 cm root length under drought, remaining within the range observed under regular watering (7.9 cm).
Representative seedlings illustrated these quantitative trends, with drought-stressed plants inoculated with Paracoccus sp. Q displaying visibly greater shoot development compared with other drought treatments (Figure 6). Together, these measurements demonstrate distinct quantitative growth responses among inoculation treatments under drought.
3.5. Targeted Genomic Analysis Supports Functional Potential and Taxonomic Novelty of a Drought-Recruited Paracoccus Isolate
Given its drought-associated recovery pattern and performance in early inoculation assays, strain Q was selected for genome-informed characterization. The draft genome comprised 4.0 Mb distributed across 78 contigs, with a GC content of 66.55% and 3,798 predicted protein-coding sequences, together with 47 tRNA genes and three rRNA genes. These features support classification of strain Q as a high-quality draft genome suitable for functional interpretation.
Genome-based taxonomic placement assigned strain Q to the genus Paracoccus (Figure 7). Digital DNA–DNA hybridization (dDDH) values relative to available Paracoccus type strains were well below the accepted species delineation threshold, with the highest value observed relative to Paracoccus hibiscisoli (31.1%), suggesting that strain Q represents a genomically distinct lineage within the genus and may correspond to a candidate novel species-level taxon.
Functional annotation revealed gene repertoires consistent with persistence in water-limited rhizosphere environments (Figure 8). Multiple osmoprotection pathways were detected, including trehalose biosynthesis via both the OtsA/OtsB and TreY/TreZ routes (otsA, otsB, treY, treZ), as well as an ectoine biosynthesis cluster (ectABC) and compatible-solute transport systems. Genes linked to glycine betaine and choline uptake further expanded the repertoire of osmolyte-associated functions.
A comprehensive oxidative stress response network was also identified, including catalases (katE, katG), superoxide dismutases (sodA, sodB), and peroxiredoxin-associated functions, supporting resilience under drought-associated oxidative stress. Genes related to motility and chemotaxis were prominent, including structural and regulatory components of the flagellar apparatus and canonical chemotaxis signaling proteins (cheA, cheY, cheW, cheR, cheB), together with multiple methyl-accepting chemotaxis proteins.
Surface-associated persistence functions were represented by polysaccharide-related pathways, including cellulose synthesis (acsAB) and alginate-associated functions, together with additional biofilm-associated genes. The genome also encoded nutrient mobilization and acquisition traits, including a PQQ-dependent glucose dehydrogenase module (pqqABCDE and gcd) linked to mineral phosphate solubilization, phosphate uptake and regulation systems, and transporters for essential ions and micronutrients.
Canonical PGPR markers associated with ethylene modulation and hormone biosynthesis were not supported at the genomic level, as ACC deaminase genes were not detected in the draft annotation. These observations suggest that the drought-associated performance of strain Paracoccus sp. Q may be linked primarily to stress tolerance, rhizosphere persistence, and nutrient-related functions rather than classical hormone modulation pathways.
4. Discussion
Drought-driven changes in the rhizosphere are increasingly recognized as a critical interface through which plants interact with their microbial environment [13,15,39]. Rather than representing passive consequences of stress, these shifts reflect host-mediated processes that reshape the chemical and physical soil environment and, in doing so, influence microbial recruitment and persistence [13,39,40]. Alterations in root exudation patterns under water limitation modify substrate availability and signaling landscapes, thereby restructuring microbial assembly and activity in the rhizosphere [11,39]. Although such recruitment dynamics occur throughout plant development, our results demonstrate that recruitment signatures can be captured and operationalized already at the seedling stage, when microbiome assembly remains highly responsive to environmental cues and host-derived signals [41,42].
A central insight emerging from this work is that drought itself can function as an ecological filter. In natural soil, drought narrowed the cultivable candidate space while enriching a reproducible subset of bacteria associated with stressed seedlings [13,43,44]. This narrowing effect is conceptually important because it positions the plant-soil system as an initial selection layer that integrates stress tolerance, rhizosphere competence, and host compatibility before any laboratory characterization. Similar stress-driven enrichment patterns have been observed in community-level drought microbiome studies, where taxa exhibiting osmotic resilience, metabolic flexibility, and surface-associated lifestyles become disproportionately represented under water limitation [45,46]. However, such ecological filtering has rarely been translated into isolate-level discovery strategies, and our study demonstrates how recruitment signatures can be operationalized to guide candidate prioritization. Although the study relied on a single agricultural soil, this design intentionally minimized edaphic variability to enable resolution of recruitment signatures attributable to plant presence and water limitation. Future multi-site validation will be necessary to assess the generality of recruitment-informed candidates across soil types and climatic contexts. The use of a single cultivation medium further constrained isolate recovery; however, this selective window was consistent with the application-oriented focus of identifying robust, readily cultivable strains. Expanding cultivation diversity in future work may reveal additional recruitment-associated taxa with complementary functional traits.
The functional coherence observed among drought-recruited isolates further supports the value of recruitment-informed discovery. Phenotypes associated with rhizosphere persistence and stress adaptation, including biofilm formation, osmolyte-related responses, and iron acquisition strategies, were distributed across the selected isolates. Importantly, these traits were not used as screening criteria but emerged as descriptive features of isolates already selected through recruitment. This distinction is particularly relevant given growing recognition that classical PGPR trait screening often fails to predict inoculant establishment or performance in complex soils [47,48]. By integrating ecological compatibility upstream of functional characterization, recruitment-based pipelines may reduce the disconnect between laboratory trait expression and soil-based functionality that has historically limited microbial inoculant translation.
Root colonization assays provided additional insight into strain-specific interaction dynamics under osmotic stress. Although Priestia megaterium A exhibited higher colonization under non-stress conditions, its pronounced proportional decline under PEG contrasted with the comparatively stable colonization of strains P. frigoritolerans I and Paracoccus sp. Q [49,50,51,52,53]. These observations reinforce the notion that colonization magnitude and stress resilience represent distinct dimensions of rhizosphere competence. Previous work has shown that beneficial effects on plant performance do not necessarily correlate with maximal root colonization density but may instead depend on metabolic compatibility, spatial niche occupation, or functional interactions within the rhizosphere community [5,54]. Accordingly, colonization data in this study provide contextual characterization of persistence dynamics rather than predictive screening metrics.
Early inoculation assays offered functional validation of the recruitment-informed framework. Drought reduced plant growth and biomass in non-inoculated seedlings, consistent with established impacts of water limitation on early developmental processes and carbon allocation. Inoculated treatments displayed distinct quantitative responses, with Paracoccus sp. Q consistently associated with the strongest plant performance under drought conditions. Although causality cannot be inferred from these assays alone, the observed responses indicate that recruitment-informed candidate selection can translate into measurable phenotypic differences during early establishment. This developmental window is increasingly recognized as a critical phase for microbiome-mediated modulation of plant stress resilience, as early colonizers may shape subsequent community assembly trajectories and functional interactions [41,55,56]. The inoculation assays were conducted during early seedling establishment, a deliberately short experimental window selected to capture recruitment-informed effects during a highly plastic developmental phase. While longer-term and field-based assessments will be required to evaluate persistence and agronomic relevance, early-stage responses provide a sensitive proxy for candidate prioritization.
Genome-informed characterization provided further ecological context for the drought-recruited Paracoccus isolate. Functional annotations revealed gene repertoires associated with osmoprotection, oxidative stress mitigation, chemotaxis, and nutrient mobilization, pathways widely implicated in microbial adaptation to drying soils and rhizosphere persistence [9,33,38,46,57]. Notably, the absence of canonical hormone-modulation genes highlights that beneficial plant responses may arise from alternative ecological mechanisms such as stress buffering, metabolic cross-feeding, or resource mobilization. This observation aligns with emerging perspectives emphasizing that plant growth promotion in natural soils often reflects complex ecological interactions rather than single-trait mechanisms [8,42,58,59].
The recovery of a Paracoccus strain representing a candidate novel species among drought-recruited isolates illustrates an additional outcome of recruitment-based approaches: the discovery of ecologically relevant microbial diversity. Members of the genus Paracoccus are widely distributed across soil and plant-associated habitats and are recognized for metabolic versatility, including stress tolerance, denitrification capacity, and diverse carbon utilization pathways [51,53]. However, their roles in drought-associated rhizosphere interactions remain comparatively underexplored. The detection of a potentially novel lineage exclusively within drought-stressed rhizosphere samples suggests that stress-driven recruitment can expose functionally important microbial taxa that may be underrepresented in culture collections and trait-based screening pipelines. Culturomics studies have similarly demonstrated that selective environmental contexts can facilitate the recovery of previously undescribed taxa with specialized ecological functions [29,30,60,61].
Collectively, these findings support a conceptual shift in microbial inoculant discovery toward ecology-informed frameworks. Recruitment-based approaches position candidate strains as outcomes of plant-driven selection processes rather than externally optimized inputs, aligning with growing calls to incorporate ecological realism into microbiome applications in agriculture [62,63,64]. By observing how plants restructure their microbial environment under stress and redeploying those interactions experimentally, it becomes possible to identify microbial partners refined through repeated plant-soil interactions. In this sense, recruitment-guided discovery can be interpreted as a strategy that leverages natural selection operating at the rhizosphere interface to inform inoculant design. The identification and functional characterization of Paracoccus sp. Q exemplify this perspective. Its drought-associated recruitment pattern, plant performance outcomes, and genomic features collectively suggest compatibility with water-limited rhizosphere conditions, while its potential taxonomic novelty underscores the exploratory value of recruitment-based pipelines. More broadly, this work demonstrates how early-stage plant–microbe interactions can be captured and translated into candidate discovery strategies that bridge ecological observation and application. Future studies integrating exudate profiling, community-resolved sequencing, and multi-season validation will further clarify recruitment mechanisms and assess the stability of recruitment-informed candidates across environmental contexts.
5. Conclusions
This study demonstrates that drought-driven rhizosphere recruitment can be leveraged as an ecologically informed framework to identify readily cultivable bacterial candidates with functional relevance under water stress. By integrating natural soil microcosms, comparative cultivation, biochemical and colonization characterization, genome-informed analysis, and early seedling-stage validation, we show that drought can act as an ecological filter that narrows the candidate space and enriches bacteria compatible with drought-altered rhizosphere conditions.
Although exploratory, this recruitment-centered approach provides an alternative to conventional trait-first inoculant discovery pipelines by embedding ecological compatibility upstream of functional evaluation. The recovery of a drought-associated Paracoccus isolate representing a genomically distinct lineage further illustrates the capacity of recruitment-guided strategies to reveal previously undescribed, stress-associated microbial diversity while identifying candidates with measurable plant-associated effects.
Together, these findings support a perspective in which microbial inoculant discovery is grounded in plant-driven ecological selection and rhizosphere feedback processes. By aligning candidate identification with naturally occurring plant–microbe interactions, recruitment-informed pipelines may contribute to the development of microbiome-based solutions that are ecologically compatible and functionally relevant under water limitation. This framework provides a foundation for future mechanistic studies and translational evaluation of recruitment-selected candidates across environmental contexts. In this sense, recruitment-guided discovery can be viewed as a strategy that listens to plant-mediated ecological selection, enabling identification of microbial partners refined by natural rhizosphere dynamics rather than solely by laboratory screening.
Author Contributions
Conceptualization, K.N. and J.I.V.; methodology, K.N., A.S. and J.I.V.; software, K.N., A.S., L.A.L.d.C. and J.I.V.; validation, K.N., A.S., D.S., and M.L.C.; formal analysis, K.N., A.S., M.L.C. and J.I.V.; investigation, K.N., L.W., M.L.C., D.S., H.C., A.S., M.M., M.L.C.; resources, J.I.V.; data curation, K.N., H.C., A.S., M.M., M.L.C. and J.I.V.; writing—original draft preparation, K.N. and J.I.V.; writing—review and editing, K.N. and J.I.V.; visualization, J.I.V.; supervision, J.I.V.; project administration, J.I.V.; funding acquisition, J.I.V. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by FCT - Fundação para a Ciência e a Tecnologia, I.P., through Green-it Bioresources for Sustainability R&D Unit (UID/04551/2025, DOI: 10.54499/UID/04551/2025; UID/PRR/04551/2025, DOI: 10.54499/UID/PRR/04551/2025) and LS4FUTURE Associated Laboratory (LA/P/0087/2020, DOI: 10.54499/LA/P/0087/2020). The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Data Availability Statement
Genome sequence data generated in this study have been deposited in NCBI under BioProject accession number PRJNA1403472 (BioSample SAMN54674915; locus tag prefix ACYAOT). The draft genome assembly of Paracoccus sp. strain Q, including structural and functional annotation, is publicly available in GenBank under the same BioProject, together with the corresponding raw sequencing reads in the NCBI Sequence Read Archive (SRA). Partial 16S rRNA gene sequences of representative isolates (V5–V8 region or near full-length amplicons) have been deposited in GenBank under accession numbers PX985972–PX985973. Quantitative datasets from biochemical and drought-related phenotypic assays, including raw replicate measurements, experimental metadata, and processed datasets used for statistical analyses, are publicly available in Figshare (DOI: 10.6084/m9.figshare.31431346). All additional data supporting the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
The authors thank ITQB NOVA (NOVA University of Lisbon, Oeiras, Portugal) and GREEN-IT Research Unit for access to greenhouse facilities and supporting infrastructure. The authors also thank Semillas Fitó for kindly providing the tomato seeds used in this study, with special thanks to Dr. Narváez (Seed Technologist, Semillas Fitó).
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Caretta, M.A.; Mukherji, A.; Arfanuzzaman, M.; Betts, R.A.; Gelfan, A.; Hirabayashi, Y.; Lissner, T.K.; Liu, J.L.G.E.; Morgan, R.; Mwanga, S.; et al. IPCC. Climate Change 2022: Impacts, Adaptation and Vulnerability (AR6 WGII), Chapter 4: Water 2022, 197–377.
- Pörtner, H.-O.; Roberts, D.C.; Tignor, M.; Poloczanska, E.S.; Mintenbeck, K.; Alegría, A.; Craig, M.; Langsdorf, S.; Löschke, S.; Möller, V.; et al. IPCC. Climate Change 2022: Impacts, Adaptation and Vulnerability (AR6 WGII) – Fact Sheet: Food and Water; Intergovernmental Panel on Climate Change: Cambridge, UK and New York, NY, USA, 2022. [Google Scholar]
- OECD. Global Drought Outlook: Trends, Impacts and Policies to Adapt to a Drier World 2025.
- Kraklow, V.A.; Paff, K.; Comeau, D.; Solander, K.; Pitts, T.R.; Price, S.F.; Xu, C. Impact of drought on global food security by 2050. Nature Communications 2025. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant-microbiome interactions: From community assembly to plant health. Nature reviews. Microbiology 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; van der Putten, W.H. Going back to the roots: the microbial ecology of the rhizosphere. Nature Reviews Microbiology 2013, 11, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Anand, G.; Gaur, R.; Yadav, D. Plant-microbiome interactions for sustainable agriculture: a review. Physiology and Molecular Biology of Plants 2021, 27, 165–179. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; Ver Loren van Themaat, E.; Schulze-Lefert, P. Structure and functions of the bacterial microbiota of plants. Annual review of plant biology 2013, 64, 807–838. [Google Scholar] [CrossRef]
- Rizaludin, M.S.; Stopnisek, N.; Raaijmakers, J.M.; Garbeva, P. The chemistry of stress: Understanding the ‘cry for help’ of plant roots. Metabolites 2021, 11. [Google Scholar] [CrossRef]
- Rolfe, S.A.; Griffiths, J.; Ton, J. Crying out for help with root exudates: adaptive mechanisms by which stressed plants assemble health-promoting soil microbiomes. Current Opinion in Microbiology 2019, 49, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; de Vries, F.T. Plant root exudation under drought: Implications for ecosystem functioning. New Phytol 2020, 225, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.X.; Chen, S.J.; Hong, X.Y.; Wang, L.Z.; Wu, H.M.; Tang, Y.Y.; Gao, Y.Y.; Hao, G.F. Plant exudates-driven microbiome recruitment and assembly facilitates plant health management. FEMS microbiology reviews 2025, 49. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yao, Z.; Sun, Y.; Wang, E.; Tian, C.; Sun, Y.; Liu, J.; Sun, C.; Tian, L. Current studies of the effects of drought stress on root exudates and rhizosphere microbiomes of crop plant species. International journal of molecular sciences 2022, 23. [Google Scholar] [CrossRef]
- Lin, H.A.; Coker, H.R.; Howe, J.A.; Tfaily, M.M.; Nagy, E.M.; Antony-Babu, S.; Hague, S.; Smith, A.P. Progressive drought alters the root exudate metabolome and differentially activates metabolic pathways in cotton (Gossypium hirsutum). Frontiers in Plant Science 2023, 14, 1244591. [Google Scholar] [CrossRef]
- Kazarina, A.; Sarkar, S.; Adams, B.; Vogt, B.; Rodela, L.; Pogranichny, S.; Powell, S.; Wiechman, H.; Heeren, L.; Reese, N.; et al. Interaction of plant-derived metabolites and rhizobiome functions enhances drought stress tolerance. Genome biology 2025, 26, 310. [Google Scholar] [CrossRef]
- Tong, Y.; Zheng, X.; Hu, Y.; Wu, J.; Liu, H.; Deng, Y.; Lv, W.; Yao, H.; Chen, J.; Ge, T. Root exudate-mediated plant–microbiome interactions determine plant health during disease infection. Agriculture, Ecosystems and Environment 2024, 370, 109056. [Google Scholar] [CrossRef]
- Naylor, D.; DeGraaf, S.; Purdom, E.; Coleman-Derr, D. Drought and host selection influence bacterial community dynamics in the grass root microbiome. ISME J 2017, 11, 2691–2704. [Google Scholar] [CrossRef]
- Xie, J.; Dawwam, G.E.; Sehim, A.E.; Li, X.; Wu, J.; Chen, S.; Zhang, D. Drought stress triggers shifts in the root microbial community and alters functional categories in the microbial gene pool. Frontiers in microbiology 2021, 12, 744897. [Google Scholar] [CrossRef]
- Adeniji, A.; Fadiji, A.E.; Li, S.; Guo, R. From lab bench to farmers’ fields: Co-creating microbial inoculants with farmers input. Rhizosphere 2024, 31, 100920. [Google Scholar] [CrossRef]
- Copeland, C.; Schulze-Lefert, P.; Ma, K.W. Potential and challenges for application of microbiomes in agriculture. Plant Cell 2025, 37. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Rodríguez, A.M.; Parra Cota, F.I.; Cira Chávez, L.A.; García Ortega, L.F.; Estrada Alvarado, M.I.; Santoyo, G.; de Los Santos-Villalobos, S. Microbial inoculants in sustainable agriculture: Advancements, challenges, and future directions. Plants 2025, 14. [Google Scholar] [CrossRef] [PubMed]
- Hartman, K.; van der Heijden, M.G.A.; Wittwer, R.A.; Banerjee, S.; Walser, J.C.; Schlaeppi, K. Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome 2018, 6, 14. [Google Scholar] [CrossRef]
- Vilchez, J.I.; Yang, Y.; Yi, D.; Zhang, H. Measurements of root colonized bacteria species. Bio-protocol 2021, 11, e3976. [Google Scholar] [CrossRef]
- Caracciolo, A.B.; Bottoni, P.; Grenni, P. Microcosm studies to evaluate microbial potential to degrade pollutants in soil and water ecosystems. Microchemical Journal 2013, 107, 126–130. [Google Scholar] [CrossRef]
- Xiong, C.; Lu, Y. Microbiomes in agroecosystem: Diversity, function and assembly mechanisms. Environ Microbiol Rep 2022, 14, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Niza Costa, M.; Gil, T.; Teixeira, R.; Rodrígues dos Santos, A.S.; Rebelo Romão, I.; Sequero López, C.; Vílchez, J.I. Combined use of a bacterial consortium and early-colonizing plants as a treatment for soil recovery after fire: A model based on Los Guájares (Granada, Spain) Wildfire. Biology 2023, 12, 1093. [Google Scholar] [CrossRef] [PubMed]
- Nicolitch, O.; Feucherolles, M.; Churin, J.L.; Fauchery, L.; Turpault, M.P.; Uroz, S. A microcosm approach highlights the response of soil mineral weathering bacterial communities to an increase of K and Mg availability. Scientific reports 2019, 9, 14403. [Google Scholar] [CrossRef]
- Breakwell, D.; MacDonald, B.; Adams, C.; Smith, K.; Robison, R.J.A.S.f.M.U. Colony morphology protocol. In Proceedings of the ASM Conference for Undergraduate Educators, San Diego, CA, USA, 28.
- Clagnan, E.; Costanzo, M.; Visca, A.; di Gregorio, L.; Tabacchioni, S.; Colantoni, E.; Sevi, F.; Sbarra, F.; Bindo, A.; Nolfi, L.; et al. Culturomics- and metagenomics-based insights into the soil microbiome preservation and application for sustainable agriculture. Front. Microbiol. 2024, 15–2024. [Google Scholar] [CrossRef]
- Li, S.; Lian, W.-H.; Han, J.-R.; Ali, M.; Lin, Z.-L.; Liu, Y.-H.; Li, L.; Zhang, D.-Y.; Jiang, X.-Z.; Li, W.-J.; et al. Capturing the microbial dark matter in desert soils using culturomics-based metagenomics and high-resolution analysis. npj Biofilms and Microbiomes 2023, 9, 67. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics (Oxford, England) 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun 2019, 10, 2182. [Google Scholar] [CrossRef]
- Gil, T.; Teixeira, R.; Sousa, A.; d’Oliveira Palmeiro, M.A.; Cruz Coimbra de Matos, A.; Niza Costa, M.; Ferrer, M.V.; Rodrígues dos Santos, A.S.; Sequero López, C.; Rebelo Romão, I.; et al. Isolation and characterization of culturable osmotolerant microbiota in hypersaline and hypergypsic soils as new treatment for osmotic stress in plants. Soil Systems. 2023, 7, 86. [Google Scholar] [CrossRef]
- Niza-Costa, M.; Rodríguez-dos Santos, A.S.; Rebelo-Romão, I.; Ferrer, M.V.; Sequero López, C.; Vílchez, J.I. Geographically Disperse, Culturable Seed-Associated Microbiota in forage plants of alfalfa (Medicago sativa L.) and pitch clover (Bituminaria bituminosa L.): Characterization of beneficial inherited strains as plant stress-tolerance enhancers. Biology 2022, 11, 1838. [Google Scholar] [CrossRef]
- Anguiano-Cabello, J.; Flores-Olivas, A.; Ochoa-Fuentes, Y.; Arredondo-Valdés, R.; Portugal, V. Fast detection of auxins by microplate technique. American Journal of Plant Sciences 2017, 08, 171–177. [Google Scholar] [CrossRef]
- Arora, N.K.; Verma, M. Modified microplate method for rapid and efficient estimation of siderophore produced by bacteria. 3 Biotech 2017, 7, 381. [Google Scholar] [CrossRef]
- O’Toole, G.A. Microtiter dish biofilm formation assay. In Journal of visualized experiments: JoVE; 2011. [Google Scholar] [CrossRef]
- Goswami, G.; Hazarika, D.J.; Chowdhury, N.; Bora, S.S.; Sarmah, U.; Naorem, R.S.; Boro, R.C.; Barooah, M. Proline confers acid stress tolerance to Bacillus megaterium G18. Scientific reports 2022, 12, 8875. [Google Scholar] [CrossRef]
- Canarini, A.; Kaiser, C.; Merchant, A.; Richter, A.; Wanek, W. Root exudation of primary metabolites: Mechanisms and their roles in plant responses to environmental stimuli. Frontiers in plant science 2019, 10, 157. [Google Scholar] [CrossRef]
- Feng, H.; Fu, R.; Luo, J.; Hou, X.; Gao, K.; Su, L.; Xu, Y.; Miao, Y.; Liu, Y.; Xu, Z.; et al. Listening to plant’s Esperanto via root exudates: Reprogramming the functional expression of plant growth-promoting rhizobacteria. New Phytol 2023, 239, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Zhalnina, K.; Louie, K.B.; Hao, Z.; Mansoori, N.; da Rocha, U.N.; Shi, S.; Cho, H.; Karaoz, U.; Loqué, D.; Bowen, B.P.; et al. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nature Microbiology 2018, 3, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. PNAS 2015, 112, E911-920. [Google Scholar] [CrossRef]
- Ait-El-Mokhtar, M.; Meddich, A.; Baslam, M. Plant-microbiome interactions under drought—insights from the molecular machinist’s toolbox. Front. Sustain. Food Syst. 2023, 7. [Google Scholar] [CrossRef]
- Gil, T.; Romão, I.R.; Gomes, J.d.C.; Vergara-Diaz, O.; de Carvalho, L.A.L.; Sousa, A.; Kasa, F.; Teixeira, R.; Mateus, S.; Katamadze, A.; et al. Comparing native and non-native seed-isolated strains for drought resilience in maize (Zea mays L.). Plant Stress 2024, 12, 100462. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Chiniquy, D.; Pierroz, G.; Deng, S.; Gao, C.; Diamond, S.; Simmons, T.; Wipf, H.M.-L.; Caddell, D. Genome-resolved metagenomics reveals role of iron metabolism in drought-induced rhizosphere microbiome dynamics. Nature communications 2021, 12, 3209. [Google Scholar] [CrossRef] [PubMed]
- Naylor, D.; Coleman-Derr, D. Drought stress and root-associated bacterial communities. Frontiers in plant science 2018, 8–2017. [Google Scholar] [CrossRef]
- Berg, G.; Kusstatscher, P.; Abdelfattah, A.; Cernava, T.; Smalla, K. Microbiome modulation-toward a better understanding of plant microbiome response to microbial inoculants. Frontiers in microbiology 2021, 12, 650610. [Google Scholar] [CrossRef] [PubMed]
- Compant, S.; Cambon, M.C.; Vacher, C.; Mitter, B.; Samad, A.; Sessitsch, A. The plant endosphere world – bacterial life within plants. Environ Microbiol. 2021, 23, 1812–1829. [Google Scholar] [CrossRef]
- Vilchez, J.I.; Tang, Q.; Kaushal, R.; Wang, W.; Lv, S.; He, D.; Chu, Z.; Zhang, H.; Liu, R.; Zhang, H. Complete genome sequence of Bacillus megaterium strain TG1-E1, a plant drought tolerance-enhancing bacterium. Microbiol Resour Announc. 2018, 7. [Google Scholar] [CrossRef]
- Vílchez, J.I.; Tang, Q.; Kaushal, R.; Wang, W.; Lv, S.; He, D.; Chu, Z.; Zhang, H.; Liu, R.; Zhang, H. Genome sequence of Bacillus megaterium strain YC4-R4, a plant growth-promoting rhizobacterium isolated from a high-salinity environment. Genome Announcements 2018, 6, 10.1128/genomea.00527–00518. [Google Scholar] [CrossRef]
- Sahoo, B.; Ningthoujam, R.; Chaudhuri, S. Isolation and characterization of a lindane degrading bacteria Paracoccus sp. NITDBR1 and evaluation of its plant growth promoting traits. J International Microbiology 2019, 22, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Świątczak, J.; Kalwasińska, A.; Brzezinska, M.S. Plant growth–promoting rhizobacteria: Peribacillus frigoritolerans 2RO30 and Pseudomonas sivasensis 2RO45 for their effect on canola growth under controlled as well as natural conditions. Frontiers in plant science 2024, 14–2023. [Google Scholar] [CrossRef]
- Ayim, B.Y.; Shaffique, S.; Khan, M.S.; Khan, M.A.; Jeon, J.R.; Kang, S.M.; Lee, I.J. Evaluating the combined effect of the Paracoccus acridae BA106 and naringenin to confer drought stress in soybean via activation of nucleic acid machinery to confer protection for future food security. International journal of biological macromolecules 2025, 328, 147172. [Google Scholar] [CrossRef]
- Acuña, J.J.; Rilling, J.I.; Inostroza, N.G.; Zhang, Q.; Wick, L.Y.; Sessitsch, A.; Jorquera, M.A. Variovorax sp. strain P1R9 applied individually or as part of bacterial consortia enhances wheat germination under salt stress conditions. Scientific reports 2024, 14, 2070. [Google Scholar] [CrossRef]
- Hardoim, P.R.; Hardoim, C.C.P.; van Overbeek, L.S.; van Elsas, J.D. Dynamics of seed-borne rice endophytes on early plant growth stages. PloS one 2012, 7, e30438. [Google Scholar] [CrossRef]
- Özkurt, E.; Mariush, Z.; Landermann-Habetha, D.; Stukenbrock, E.H. Ecological assembly dynamics of the seed-borne microbiome in cultivated and wild wheat. bioRxiv 2024, 2024.2012.2027.630520. [Google Scholar] [CrossRef]
- Patil, J.R.; Mhatre, K.J.; Yadav, K.; Yadav, L.S.; Srivastava, S.; Nikalje, G.C. Flavonoids in plant-environment interactions and stress responses. Discover Plants 2024, 1, 68. [Google Scholar] [CrossRef]
- Chesneau, G.; Laroche, B.; Préveaux, A.; Marais, C.; Briand, M.; Marolleau, B.; Simonin, M.; Barret, M. Single seed microbiota: Assembly and transmission from parent plant to seedling. mBio 2022, 13, e0164822. [Google Scholar] [CrossRef] [PubMed]
- Ning, D.; Yuan, M.; Wu, L.; Zhang, Y.; Guo, X.; Zhou, X.; Yang, Y.; Arkin, A.P.; Firestone, M.K.; Zhou, J. A quantitative framework reveals ecological drivers of grassland microbial community assembly in response to warming. Nature Communications 2020, 11, 4717. [Google Scholar] [CrossRef]
- Mitter, E.K.; Tosi, M.; Obregón, D.; Dunfield, K.E.; Germida, J.J. Rethinking crop nutrition in times of modern microbiology: Innovative biofertilizer technologies. Front. Sustain. Food Syst. 2021, 5. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, Q.; Ruan, W.; Guo, Y.; Zhuang, Y.; Li, Y.; Ruan, Z.J.A. Culturomics and amplicon-based metagenomic insights into the bacteria of soils with high yield of Oryza sativa L. subsp. japonica. Agronomy 2023, 13, 2867. [Google Scholar] [CrossRef]
- Liu, H.; Li, J.; Carvalhais, L.C.; Percy, C.D.; Prakash Verma, J.; Schenk, P.M.; Singh, B.K. Evidence for the plant recruitment of beneficial microbes to suppress soil-borne pathogens. New Phytol. 2021, 229, 2873–2885. [Google Scholar] [CrossRef]
- Lyu, D.; Msimbira, L.A.; Nazari, M.; Antar, M.; Pagé, A.; Shah, A.; Monjezi, N.; Zajonc, J.; Tanney, C.A.S.; Backer, R.; et al. The coevolution of plants and microbes underpins sustainable agriculture. Microorganisms 2021, 9. [Google Scholar] [CrossRef]
- Santoyo, G. How plants recruit their microbiome? New insights into beneficial interactions. Journal of Advanced Research 2022, 40, 45–58. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Soil microcosm framework to resolve rhizosphere recruitment during early seedling establishment under drought. (a) Overview of the soil microcosm conditions used to disentangle plant presence and drought effects within a constant soil background, including plant-free bulk soil controls (Regular and Drought) and tomato seedling-containing microcosms under well-watered (Regular + Seedling) and drought-stressed (Drought + Seedling) regimes. (b) Representative seedlings recovered at the end of the experimental period illustrating early developmental responses to contrasting water availability and providing the ecological context for defining cultivable rhizosphere bacterial populations during initial community assembly under stress.
Figure 1.
Soil microcosm framework to resolve rhizosphere recruitment during early seedling establishment under drought. (a) Overview of the soil microcosm conditions used to disentangle plant presence and drought effects within a constant soil background, including plant-free bulk soil controls (Regular and Drought) and tomato seedling-containing microcosms under well-watered (Regular + Seedling) and drought-stressed (Drought + Seedling) regimes. (b) Representative seedlings recovered at the end of the experimental period illustrating early developmental responses to contrasting water availability and providing the ecological context for defining cultivable rhizosphere bacterial populations during initial community assembly under stress.

Figure 2.
Recruitment dynamics of cultivable rhizosphere isolates across soil treatments. (a) Absolute abundance of cultivable bacterial isolates expressed as colony-forming units (CFU) per mg dry soil and (b) corresponding relative abundance (%) across soil treatments, including control soil at time 0 (T0), control soil at T1 under regular watering and drought, and soil with tomato seedlings at T1 under regular watering and drought. Stacked bars represent the contribution of individual isolates to the total cultivable community within each treatment. Isolate identities are indicated by color-coded segments as shown in the legend.
Figure 2.
Recruitment dynamics of cultivable rhizosphere isolates across soil treatments. (a) Absolute abundance of cultivable bacterial isolates expressed as colony-forming units (CFU) per mg dry soil and (b) corresponding relative abundance (%) across soil treatments, including control soil at time 0 (T0), control soil at T1 under regular watering and drought, and soil with tomato seedlings at T1 under regular watering and drought. Stacked bars represent the contribution of individual isolates to the total cultivable community within each treatment. Isolate identities are indicated by color-coded segments as shown in the legend.

Figure 3.
Biochemical characterization of recruitment-prioritized rhizosphere isolates. Biochemical traits associated with plant interaction and drought tolerance were assessed in representative isolates Priestia megaterium A, Peribacillus frigoritolerans I, and Paracoccus sp. Q. Auxin-related compound production is shown in panel (a) and expressed as IAA equivalents (µg mL−1). Biofilm formation capacity is presented in panel (b) following crystal violet staining and reported as optical density at 550 nm (OD550). Osmolyte-associated proline production is depicted in panel (c) and expressed as proline equivalents (µg mL−1). Siderophore production is shown in panel (d), quantified using the chrome azurol S (CAS) assay and expressed as percent siderophore units (psu). Bars represent mean values ± variation across biological replicates, and letters indicate statistically significant differences among isolates (p < 0.05).
Figure 3.
Biochemical characterization of recruitment-prioritized rhizosphere isolates. Biochemical traits associated with plant interaction and drought tolerance were assessed in representative isolates Priestia megaterium A, Peribacillus frigoritolerans I, and Paracoccus sp. Q. Auxin-related compound production is shown in panel (a) and expressed as IAA equivalents (µg mL−1). Biofilm formation capacity is presented in panel (b) following crystal violet staining and reported as optical density at 550 nm (OD550). Osmolyte-associated proline production is depicted in panel (c) and expressed as proline equivalents (µg mL−1). Siderophore production is shown in panel (d), quantified using the chrome azurol S (CAS) assay and expressed as percent siderophore units (psu). Bars represent mean values ± variation across biological replicates, and letters indicate statistically significant differences among isolates (p < 0.05).

Figure 4.
Root colonization of tomato seedlings by selected isolates. Root colonization by isolates represented by red bars (P. megaterium A), orange bars (P. frigoritolerans I), and yellow bars (Paracoccus sp. Q), quantified as colony-forming units (CFU) per mg root dry weight. Solid bars indicate colonization under regular conditions, whereas patterned bars represent colonization in the presence of 15% PEG as a drought-mimicking osmotic treatment. Bars correspond to mean values and error bars indicate standard deviation of biological replicates (n = 5). Different letters denote statistically significant differences among treatments according to one-way ANOVA followed by a post hoc test (p < 0.05).
Figure 4.
Root colonization of tomato seedlings by selected isolates. Root colonization by isolates represented by red bars (P. megaterium A), orange bars (P. frigoritolerans I), and yellow bars (Paracoccus sp. Q), quantified as colony-forming units (CFU) per mg root dry weight. Solid bars indicate colonization under regular conditions, whereas patterned bars represent colonization in the presence of 15% PEG as a drought-mimicking osmotic treatment. Bars correspond to mean values and error bars indicate standard deviation of biological replicates (n = 5). Different letters denote statistically significant differences among treatments according to one-way ANOVA followed by a post hoc test (p < 0.05).

Figure 5.
Plant growth responses to bacterial inoculation under regular watering and drought. (a) Shoot and root length and (b) shoot and root dry weight of tomato seedlings grown under regular watering or drought conditions following inoculation with isolates represented by grey bars (non-inoculated control), red bars (P. megaterium A), orange bars (P. frigoritolerans I), and yellow bars (Paracoccus sp. Q). Solid bars correspond to plants grown under regular watering, whereas patterned bars indicate drought-exposed treatments. Bars represent mean values and error bars indicate standard deviation of biological replicates (n = 5). Different letters denote statistically significant differences among treatments within each panel and organ (one-way ANOVA with post hoc test, p < 0.05).
Figure 5.
Plant growth responses to bacterial inoculation under regular watering and drought. (a) Shoot and root length and (b) shoot and root dry weight of tomato seedlings grown under regular watering or drought conditions following inoculation with isolates represented by grey bars (non-inoculated control), red bars (P. megaterium A), orange bars (P. frigoritolerans I), and yellow bars (Paracoccus sp. Q). Solid bars correspond to plants grown under regular watering, whereas patterned bars indicate drought-exposed treatments. Bars represent mean values and error bars indicate standard deviation of biological replicates (n = 5). Different letters denote statistically significant differences among treatments within each panel and organ (one-way ANOVA with post hoc test, p < 0.05).

Figure 6.
Representative phenotypes of tomato seedlings treated with the selected candidate strains. Representative images of tomato seedlings grown under regular watering (a, b) ans drought conditions (c, d) following inoculation with the indicated treatments (Mock, non-inoculated control; A, P. megaterium A; I, P. frigoritolerans I; Q, Paracoccus sp. Q). Panels (a) and (c) show uprooted seedlings to visualize shoot and root architecture, whereas panels (b) and (d) show seedlings grown in pots. Images correspond to representative individuals from independent biological replicates included in the quantitative analyses. Scale bars represent 5 cm.
Figure 6.
Representative phenotypes of tomato seedlings treated with the selected candidate strains. Representative images of tomato seedlings grown under regular watering (a, b) ans drought conditions (c, d) following inoculation with the indicated treatments (Mock, non-inoculated control; A, P. megaterium A; I, P. frigoritolerans I; Q, Paracoccus sp. Q). Panels (a) and (c) show uprooted seedlings to visualize shoot and root architecture, whereas panels (b) and (d) show seedlings grown in pots. Images correspond to representative individuals from independent biological replicates included in the quantitative analyses. Scale bars represent 5 cm.

Figure 7.
Genome-based phylogenetic placement of drought-recruited Paracoccus sp. Q. Phylogenomic tree inferred from TYGS analysis showing the position of strain Q relative to representative Paracoccus species. Bootstrap values are indicated at nodes. Colored blocks represent genome-associated metadata. The tree supports assignment of strain Q to the genus Paracoccus.
Figure 7.
Genome-based phylogenetic placement of drought-recruited Paracoccus sp. Q. Phylogenomic tree inferred from TYGS analysis showing the position of strain Q relative to representative Paracoccus species. Bootstrap values are indicated at nodes. Colored blocks represent genome-associated metadata. The tree supports assignment of strain Q to the genus Paracoccus.

Figure 8.
Functional repertoire of the drought-recruited Paracoccus sp. Q genome. Distribution of predicted gene functions associated with plant interaction, colonization, stress response, nutrient mobilization, and competitive traits based on PLaBAse annotation. Bars represent the proportion of genes assigned to each functional category within the draft genome.
Figure 8.
Functional repertoire of the drought-recruited Paracoccus sp. Q genome. Distribution of predicted gene functions associated with plant interaction, colonization, stress response, nutrient mobilization, and competitive traits based on PLaBAse annotation. Bars represent the proportion of genes assigned to each functional category within the draft genome.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



