
Submitted:
13 February 2026
Posted:
27 February 2026
You are already at the latest version
Abstract
Antimicrobial resistance (AMR) is a growing global concern, exacerbated by the overuse of antibiotics in livestock farming. Honey bees (Apis mellifera), widely used as bioindicators of environmental contamination, may also serve as sentinels for monitoring the environmental spread of antibiotic resistance genes (ARGs). This study investigated the presence of ARGs and the gut microbiota composition of honey bees sampled from 11 apiaries located in a region of Northwestern Italy characterized by intensive livestock farming. PCR and sequencing analyses revealed a widespread presence of tetracycline resistance genes—particularly tet(B) and tet(C)—as well as occasional detection of blaTEM, qnrB, and int1 genes. ARGs were also identified in bacterial colonies isolated from bee guts, notably in Hafnia spp. 16S rRNA gene sequencing of the gut microbiota revealed dominance of genera such as Bartonella, Snodgrassella, Gilliamella, Bombilactobacillus and Lactobacillus. Some samples showed reduced microbial diversity or shifts associated with possible dysbiosis. The findings confirm the potential of honey bees as bioindicators for environmental AMR surveillance and underscore the need for further research to elucidate correlations between ARGs presence and microbial community structure in honey bees from various ecological contexts.
Keywords:
antimicrobial resistance (AMR)
; antibiotic resistance genes (ARGs)
; honey bees
; gut microbiota
; intensive livestock farming
; environmental bioindicators
1. Introduction
Antimicrobial resistance (AMR) is a natural evolutionary phenomenon, but its global increase is strongly driven by the widespread and excessive use of antibiotics [1]. AMR is particularly challenging to control due to the ability of bacteria to exchange genetic material both intra- and interspecifically [2]. The selective pressure induced by antibiotic use, and their subsequent release into the environment, has contributed to the mobilization and horizontal transfer of numerous antibiotic resistance genes (ARGs) among bacterial species [3].
In recent years, several studies have detected ARGs in wildlife and natural environments [4,5,6], highlighting the importance of monitoring organisms that may be exposed to antibiotics indirectly through environmental contamination. Honey bees have been used for over 50 years as bioindicators of environmental pollution, including heavy metals in urban areas, pesticides in agricultural zones, and radionuclides [7]. Bees forage within a range of up to 3 km from their hives, making them highly exposed to environmental contaminants. Although the use of antibiotics is prohibited in beekeeping within the European Union, bees may still come into contact with these compounds in the environment and could also act as potential vectors of ARGs. Recent studies have demonstrated that honey bees can serve as bioindicators of AMR in the environment [8,9,10,11,12]. Moreover, the honey bee gut microbiota—characterized by high bacterial density like that of other animals—has been identified as a hotspot for ARGs.
The honey bee gut microbiota is highly specialized and conserved, comprising nine core bacterial species [13]. In adult worker bees, these bacteria are distributed across specific gut compartments, playing essential roles in nutrient digestion and defense against pathogens [14]. However, the relative abundance of these bacteria can fluctuate seasonally, depending on the diversity and availability of food sources [15].
Disruption of the gut microbial community—often marked by the emergence of opportunistic bacteria such as Enterobacteriaceae—can occur due to external stressors, including antibiotic exposure [16]. Several studies have linked antibiotic exposure to dysbiosis in bees, leading to increased vulnerability to pathogens [17,18,19,20,21,22].
At the global scale, the intensive use of antibiotics in livestock has been one of the most important factors driving AMR [23,24]. Recently, the European Union has opted for a more parsimonious use of antibiotics in livestock, aiming to prevent the occurrence and spread of ARGs [25]. In Italy, the strategy to reduce antibiotic use in livestock is supported by an information system [26], which collects data on drug use at the farm level. Surveillance of antibiotic use will be enhanced by the data collected through ClassyFarm; however, at present, these data are still limited. Therefore, environmental surveillance methods—such as those provided by honey bees—remain a key tool to monitor the spread and occurrence of antibiotic resistance genes.
In this study, we conducted a surveillance across apiaries located in an area characterized by intensive livestock farming. We evaluated the presence of ARGs and analyzed the composition of the gut microbial community in honey bees. Our aim was to investigate the potential role of honey bees as indicators of environmental ARGs contamination and to analyze the gut microbial community structure in apiaries in this sampling area.
2. Materials and Methods
2.1. Study Area and Sampling
Sampling was carried out during the spring and summer seasons of 2021 and 2022, across 11 apiaries located in the Province of Cuneo, Piedmont (Northwestern Italy), an area characterized by a high density of intensive livestock farms (Figure 1). As of 31 December 2021, the density of farm animals in the area was 895 animals/km2 for poultry, 131 for pigs, 62 for cattle, and 8 for sheep or goats. This area is recognized as a hotspot for dairy, beef, and pig farming at the national level in Italy, and it has an intermediate livestock density compared with other European regions [27].
As illustrated in Figure 1, numerous farms housing various food-producing animals are situated in close proximity to the sampled apiaries.
As detailed in Table 1, three hives were sampled at each apiary and four aliquots with about 30 bees were collected for each hive. The beekeepers were interviewed about the technical and sanitary management of the apiaries. Overall, the apiaries were in good condition, with all colonies having an active queen and more than 60% of colony frames populated by bees. However, colonies in Apiaries 5 and 6 were noted to be in poor health due to past depopulation, mortality, robbing, and severe losses caused by Varroa infestation.
2.2. ARGs Determination in Honey Bees Gut
For the determination of antimicrobial resistance genes (ARGs), two aliquots from each sample were used. The honey bee gut was carefully separated from the rest of the body using sterile tweezers. DNA extraction was performed with the Qiamp DNA Mini Kit (Qiagen) following the manual instructions. DNA extracted was quantified setting a spectrophotometer at 260 nm (BioSpectrometer® Invitrogen).
The selected ARGs are listed in Table 2. The target genes were chosen based on their association with resistance to the following antibiotics classes: tetracyclines, cephalosporins, and fluoroquinolones. Additionally, integrase genes were detected to investigate the phenomenon of multidrug resistance. Simplex PCR end point was performed in a final volume of 25 µL, using 0.5 µM of each primer, 2.5 U of Taq HOT Start type, 2 mM of MgCl2, 0.3 mM of DNTPs mix and 5 µL of DNA sample.
Primers and thermal profile for detecting the genes responsible for cephalosporin resistance were taken from Nagano et al., 2003 [28]. However, primers and thermal profile for amplifying the genes responsible for tetracycline resistance were taken from Ng et al., 2001 [29]. Finally, primers and thermal profile used to amplify the genes responsible for fluoroquinolone resistance were taken from Gay et al., 2006 [30].
The PCR reaction was carried out using a Gene Amp PCR System 9700 (AB System).
Thermal cycling conditions are shown in Table 2.
The PCR products were analysed by electrophoresis on a 2% Agarose gel and amplicons corresponding to the expected sizes of the ARGs were sequenced.
For ARGs identified in the gut, PCR products were purified using the Extractme DNA Clean-Up Kit (Blirt SA, Poland). Sequencing was carried out using the BigDye Terminator v3.1 kit (Thermo Fisher Scientific, US), with the same primers used for PCR. The sequencing products were purified with DyeEX 2.0 Spin Kit (Qiagen, Germany) and capillary electrophoresis was performed using the 3500 Series Genetic Analyzer (Thermo Fisher Scientific, US).
2.3. ARGs Determination in Isolated Colonies
One of the four aliquots for each sample was used for the phenotypical analysis, starting with the separation of the gut. The separated gut were homogenate and to enrich the sample, 500 µL of each homogenate was mixed with 4,5 mL of Nutrient Broth and incubated over night at room temperature. The colture was then inoculated onto two Blood agar plates and incubated at room temperature under aerobic conditions. After 24 hours plates, the plates were examined, and if no growth was observed, the incubation period was extended for up to 72 hours.
To identify isolated colonies, Sanger sequencing was performed. Each colony was diluted in 500 µL of ultrapure water and boiled. The resulting DNA was used for bacterial species identification through the amplification of a 530 bp region of the 16s rDNA using the MicroSEQ™ 500 16S rDNA Sequencing kit (Thermo Fisher Scientific, US).
2.4. Evaluation of Gut Microbial Community Composition
DNA extracted for ARGs determination was quantified with Qubit dsDNA HS Kit (ThermoFisher) and normalized to 5 ng/µL. The protocol suggested by Illumina (16S Metagenomic Sequencing Library Preparation) was followed for 16S ribosomal metabarcoding. The V3-V4 region was amplified with the primers 341F and 806R (Klindworth et al., 2013; Appril et al., 2015), with an adapter added for the attachment of Illumina indexes.
An amplification negative control was included, processed alongside the samples to detect potential contamination. The Amplicon PCR reaction was prepared using 12.5 µl of NEBNext® Q5® Hot Start HiFi 2X Master Mix (BioLabs), 1.25 µl of 10 µM Forward Primer, 1.25 µl of 10 µM Reverse Primer, 7.5 µl of H2O, and 2.5 µl of DNA (5 ng/µL). The thermal cycling conditions were: 98 °C for 30s; 35 cycles of 98 °C for 10s; 55 °C for 30s; 72 °C for 30s; final extension at 72 °C for 2 minutes. PCR products were analysed by electrophoretic run on 2% Agarose gel. The amplicons were purified using magnetic beads (AMPure XP, Beckman) directly on a 96-well plate.
The Index PCR was performed using 25 µl of NEBNext® Q5® Hot Start HiFi 2X Master Mix (BioLabs), 5 µl of Nextera XT Index Primer 1, 5 µl of Nextera XT Index Primer 2 (Illumina), 10 µl of H2O, and 5 µl of purified DNA with the following thermal conditions: 98 °C for 30s; 12 cycles of 98 °C for 10s; 55 °C for 30s; 72 °C for 30s; final extension at 72 °C for 2 minutes.
After purification with magnetic beads (AMPure XP, Beckman), the libraries were quantified using the Qubit dsDNA HS (ThermoFisher) and the average size of the amplicons was evaluated using the BioAnalyzer 2100 with the Agilent High Sensitivity DNA Kit (Agilent Technologies). The libraries were then normalized and pooled together. The pool was quantified using the NEBNext Library Quant Kit for Illumina (BioLabs), normalized to 4 nM, and subjected to paired-end 2x300 sequencing on an Illumina MiSeq platform using the MiSeq Reagent Kit v3 (600-cycle).
2.5. Data Processing and Analysis
The Fastq data obtained from 16S metabarcoding were analysed using the Microbial Genomics Module in CLC Genomics Workbench v. 23.0.2 (Qiagen). The raw data were processed using the “Data QC and OTUs Clustering” and “Estimate Alpha and Beta Diversity” workflow tools. Specifically, trimming was performed for low-quality scores (Qscore <0.05), nucleotide ambiguity (up to 2 nucleotides allowed), adapter sequences, and length. Chimeric reads were removed, duplicate sequences were merged, and the resulting reads were aligned against the SILVA database (v. 138) at a 97% identity threshold. An Operational Taxonomic Units (OTU) table was then created, showing the assigned taxonomy. The profiles of the positive and negative controls were verified to exclude cross-contamination and subsequently removed.
3. Results
3.1. Prevalence of ARGs in Honey Bee Gut
The results of PCR and Sanger sequencing revealed a widespread presence of antibiotic resistance in the honey bee gut, particularly to tetracycline.
The tet (B) target gene was detected in all sample, but its presence was not confirmed by sequencing in three samples due to inconsistent fragment intensities. On the other hand, the tet(C) target gene was found in 54% of the samples. The blaTEM gene was detected in 42% of the samples, and resistance to fluoroquinolones was also observed in 42% of the samples, specifically with the qnrB target gene, although this finding was not confirmed by sequencing.
Additionally, the int1 gene cassette, associated with multidrug resistance, was detected in one out of 33 samples. No positivity was observed for the other tested markers. A summary of the identified markers is provided in Table 3.
3.2. Colonies Identification and ARGs
A total of 20 pure colonies were isolated, 17 of which were identified through 16S rDNA region sequencing as belonging to 9 genera: Enterococcus (5 colonies), Hafnia (3 colonies), Bacillus (2 colonies), Pantoea (2 colonies), Metabacillus (1 colony), Paenibacillus (1 colony), Fructobacillus (1 colony), Enterobacter (1 colony), and Staphylococcus (1 colony). Antimicrobial resistance screening performed on individual colonies revealed the presence of tetracycline resistance genes in 3 colonies, all identified as Hafnia and originating from the same apiary and colony (sample 3A). In two of these Hafnia colonies, both tet (B) and tet (C) resistance genes were detected, while the third colony only exhibited resistance to tet (C).
3.3. Gut Microbial Community Composition
The FASTQ files generated by NGS sequencing on the Illumina MiSeq platform were imported into CLC Genomics Workbench v. 23 (Qiagen) for subsequent bioinformatic analysis. After filtering and trimming, a total of 1,012.024 reads were obtained. All samples were deemed suitable for analysis, except for sample 7A, which was excluded from further processing due to insufficient quality (i.e., low read number).
The sequences were clustered into 67 Operational Taxonomic Units (OTUs). The positive control yielded results consistent with expectations, while the negative control and extraction blank showed no signs of contamination.
Analysis of the sequences obtained from wild and farmed samples revealed that the most abundant phyla were Proteobacteria, Firmicutes, and Actinobacteria.
At the class level, Alphaproteobacteria, Gammaproteobacteria, Bacilli, Actinobacteria, and Clostridia were the most represented, accounting for over 98% of the microbiota.
Figure 2 illustrates the taxonomic composition of the microbiota at the genus level for each sample. The five most abundant genera were Bartonella, Gilliamella, Lactobacillus, Snodgrassella, and Bombilactobacillus.
Notably, the presence of Bartonella exhibited highly variable among samples, with proportions ranging from 0% to over 85% of the microbiota.
4. Discussion
The presence of antibiotic resistance genes (ARGs) in the natural environment is now widely recognized, emphasising the urgency of addressing the antimicrobial resistance (AMR) emergency through a One Health approach. All environmental matrices — including water [31,32] soil [33,34], and air [35,36,37] can serve as sources of ARGs. Although ARGs have been detected even in remote areas [38,39], the presence of urban centers and livestock or agricultural farms can create hotspots that intensify the AMR phenomenon [40–42].
Honey bees have emerged as valuable bioindicators for monitoring the environmental presence of ARGs in both natural and anthropogenic contexts, particularly due to the strict regulations on antibiotic use in EU beekeeping that forbid a direct exposure to these drugs for clinical purposes [43]. Their foraging behaviour makes them suitable for localized environmental monitoring, as they typically forage within a 3 km radius from their hive, during which they collect a variety of environmental materials, including potential contaminants.
Various environmental variables are known to promote the spread of ARGs. Recent studies [8,11] have identified correlations between the AMR phenomenon in honey bees and specific landscape features, such as the proximity to livestock farms, agricultural areas, wastewater treatment plants, and hospitals.
Furthermore, analyzing the microbial community composition of honey bees may play an important role in detecting dysbiosis associated with bees exposed to various stressors, compared to healthy individuals [17,19,21,44–46]. This is particularly relevant for the diagnosis and research of disease symptoms in honey bees, which may originate from diverse causes, ranging from pathogens to xenobiotics. Shifts in the microbial community structure have also been linked to territorial and environmental characteristics [47,48].
In this study, the sampling area was characterized by a high density of intensive livestock farms. Approximately 10% of the national livestock population, corresponding to about 1 million Livestock Units (LUs), is reared in the Piedmont region. Within the region, the Province of Cuneo accounts for the highest concentration of livestock, representing the largest zootechnical area in Piedmont.
All sampled apiaries were in close proximity to different livestock farms and, except for apiary 4, 7 and 8, there were more than 15 farms within 1 km radius from the apiaries. These farms rear various species, including cattle, poultry, pigs, sheep, and goats.
The results revealed a widespread presence of tetracycline resistance genes, according to previous studies [9,49,50]. These ARGs are detected both in honey bees gut and in isolated colonies of Hafnia. The global prevalence of tetracycline resistance genes could be attributed to the historic use of tetracyclines in beekeeping prior to the implementation of EU regulations [51]. Moreover, tetracycline is one of the most commonly used antimicrobials in veterinary clinical practice [52].
Gut microbiota analysis showed that the genus Bartonella was the most abundant across samples, followed by Snodgrassella, Gilliamella, Bombilactobacillus, and Lactobacillus. Bartonella spp. were detected in all apiaries except one. Similarly, Gilliamella, Bombilactobacillus, and Snodgrassella were also present in all but one apiary. In contrast, Lactobacillus was found in all colonies with varying abundance. This genus plays a crucial role in carbohydrate metabolism and contributes to host defence through the production of antimicrobial metabolites and sugars [53].
Notably, samples 2A and 2B exhibited reduced abundance of Bartonella, accompanied by an increased presence of Bombilactobacillus. According to previous research [17], a decline in Bartonella spp. abundance may be linked to compromised health status. That study found Bartonella apis to be relatively more abundant in healthy bees compared to bees from collapsing colonies, suggesting a potential role in disease resistance. Bartonella spp. are known to play a role in recycling nitrogenous waste into amino acids and in the degradation of secondary plant metabolites, therefore, a reduction of over 80% in this genus, as observed in some samples, could have implications for digestion and amino acid recovery [17].
It is also noteworthy that sample 6A exhibited the least diverse microbiota, consisting of only three genera: Bartonella, Lactobacillus, and Serratia. The presence of Serratia, which can act as a pathogen, is particularly significant in bees that may already be experiencing compromised health conditions.
5. Conclusions
This study confirms that honey bees could come into contact with ARGs, supporting their potential use as bioindicators for monitoring the presence of these environmental contaminants, in an area characterized by intensive livestock farming.
The analysis of gut microbial community composition may serve as a valuable indicator for honey bee health status in potentially contaminated areas. However, numerous environmental and biological variables influence the composition of the gut microbiota, often complicating the interpretation of results.
Further research, involving a larger number of samples and comparisons between bees from different environmental contexts, is needed to better understand this complex phenomenon and to clarify whether the presence of ARGs can be reliably correlated with shifts in microbial community composition in this territorial context.
This work confirms the importance of monitoring studies to support surveillance of AMR and its environmental dissemination.
Author Contributions
Conceptualization, R.Z. and M.G.; methodology, R.Z., M.G., C.B., S.P.; software, C.B., S.P.; formal analysis, L.C.; investigation, R.Z., C.B., C.G., S.P.; resources, C.G.; data curation, R.Z. and S.P.; writing—original draft preparation, S.O.; writing—review and editing, C.B., L.C., A.T., S.P.; supervision, S.P. and M.G.; funding acquisition, A.D. and M.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Italian Ministry of Health, grant number IZSPLV 03/19 RC.
Institutional Review Board Statement
The authors confirm that the ethical policies of the journal, as noted on the journal’s author guidelines page, have been adhered to. No ethical approval was required referring to DIRECTIVE 2010/63/EU OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 22 September 2010 on the protection of animals used for scientific purposes.
Data Availability Statement
The data supporting the findings of this study are available within the manuscript. Additional information is available from the corresponding author upon reasonable request.
Acknowledgments
The authors express their sincere gratitude to the beekeepers involved in this project for their collaboration and for sharing valuable insights into local beekeeping practices.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| LD | Linear dichroism |
References
- OECD. Antimicrobial resistance – tackling the burden in the EU. Organization for Economic Co-operation and Development, 2022.
- Vittecoq M., Godreuil S., Prugnolle F., Durand P., Brazier L., Renaud N., Arnal A., Aberkane S., Jean-Pierre H., Gauthier-Clerc M., Thomas F. and Renaud F. Antimicrobial resistance in wildlife. Journal of Applied Ecology 2016, 53(2), 519–529. [CrossRef]
- Larsson, D. G. J., & Flach, C. F. Antibiotic resistance in the environment. Nature Reviews Microbiology 2022, 20, 257–269. [CrossRef]
- Allen, H. K., Donato, J., Wang, H. H., Cloud-Hansen, K. A., Davies, J., & Handelsman, J. Call of the wild: Antibiotic resistance genes in natural environments. Nature Reviews Microbiology 2010, 8(4), 251–259. [CrossRef]
- Schwartz, T., Kohnen, W., Jansen, B., & Obst, U. Detection of antibiotic-resistant bacteria and their resistance genes in wastewater, surface water, and drinking water biofilms. FEMS Microbiology Ecology 2003, 43(3), 325–335. [CrossRef]
- Wang W., Weng L., Luo T., Wang Q., Yang G., and Jin Y. Antimicrobial and the Resistances in the Environment: Ecological and Health Risks, Influencing Factors, and Mitigation Strategies. Toxics 2023, 11,185. [CrossRef]
- Celli, G., & Maccagnani, B. Honey bees as bioindicators of environmental pollution. Bulletin of Insectology 2003, 56(1), 137–139. [CrossRef]
- Cenci-Goga B. T., Sechi P., Karama M., Ciavarella R., Pipistrelli M .V., Goretti E., Elia A. C., Gardi T., Pallottini M., Rossi R., Selvaggi R., Grispoldi L.Cross-sectional study to identify risk factors associated with the occurrence of antimicrobial resistance genes in honey bees (Apis mellifera) in Umbria, Central Italy. Environmental Science and Pollution Research 2020, 27, 9637–9645. [CrossRef]
- Cilia G., Resci I., Scarpellini R., Zavatta L., Albertazzi S., Bortolotti L., Nanetti A., Piva S. Antimicrobial-Resistant Environmental Bacteria Isolated Using a Network of Honey Bee Colonies (Apis mellifera L. 1758). Transboundary and Emerging Diseases 2023. [CrossRef]
- Piva S., Giacometti F., Marti E., Massella E, Cabbri R., Galuppi R. and Serraino A. Could honey bees signal the spread of antimicrobial resistance in the environment? Letters in Applied Microbiology 2019, 70, 349—355. [CrossRef]
- Resci I., Zavatta L., Piva S., Mondo E., Albertazzi S., Nanetti A., Bortolotti L., Cilia G. Predictive statistical models for monitoring antimicrobial resistance spread in the environment using Apis mellifera (L. 1758) colonies. Environmental Research 2024, 248. [CrossRef]
- Saccà M.L., Resci I., Cilia G. Phenotypic and genotypic antimicrobial resistance patterns in honey bee (Apis mellifera L.) bacterial symbionts. Environmental Science and Pollution Research 2024. [CrossRef]
- Raymann, K., Bobay, L. M., & Moran, N. A. Antibiotics reduce genetic diversity of core species in the honeybee gut microbiome. Molecular Ecology 2018, 27(8), 2057–2066. [CrossRef]
- Kwong, W. K., & Moran, N. A. Gut microbial communities of social bees. Nature Reviews Microbiology 2016, 14(6), 374–384. [CrossRef]
- Kešnerová, L., Emery O., Troilo M., Liberti J., Erkosar B., Engel E. Gut microbiota structure differs between honeybees in winter and summer. The ISME Journal 2020, 14 (3), 801–814. [CrossRef]
- Baffoni, L., Alberoni D., Gaggia F., Braglia C., Stanton C., Ross P. R., Di Gioia D. Honeybee Exposure to Veterinary Drugs: How Is the Gut Microbiota Affected? Microbiology Spectrum 2021, 9 (1). [CrossRef]
- Li J. H., Evans J.D., Li W. F., Zhao Y. Z., DeGrandi-Hoffman G, Huang S. K., Li Z. G., Hamilton M., Chen Y.P. New evidence showing that the destruction of gut bacteria by antibiotic treatment could increase the honey bee’s vulnerability to Nosema infection. PLOS One 2017. [CrossRef]
- Liu Y., Jia S., Wu Y., Zhou N., Xie Y., Wei R., Huang Z., Chen Y., Hu F., Zheng H. Tetracycline-induced gut community dysbiosis and Israeli Acute Paralysis Virus infection synergistically negatively affect honeybees. Ecotoxicology and Environmental Safety 2024. [CrossRef]
- Mallory E., Freeze G., Daisley B.A. and Allen-Vercoe E. Revisiting the role of pathogen diversity and microbial interactions in honeybee susceptibility and treatment of Melissococcus plutonius infection. Front. Vet. Sci. 2024, 11:1495010. [CrossRef]
- Raymann, K., Shaffer, Z., & Moran, N. A. Antibiotic exposure perturbs the gut microbiota and elevates mortality in honeybees. PLoS Biology 2017, 15(3). [CrossRef]
- Tian, B., Fadhil, N. H., Powell, J. E., Kwong, W. K., & Moran, N. A. Long-term exposure to antibiotics has caused accumulation of resistance genes in the honeybee gut microbiota. mBio 2012, 3(6). [CrossRef]
- Mulchandani R., Wang Y., Gilbert M., Van Boeckel T. P. Global trends in antimicrobial use in food-producing animals: 2020 to 2030. PLOS Global Public Health, 2023. [CrossRef]
- Tomassone L., Scali F., Formenti N., Alborali G. L., Aragrande M., Canali M., Romanelli C., Suprani V and De Meneghi D. Evaluation of ‘ClassyFarm’, the Italian integrated surveillance system of livestock farms, in the context of antimicrobial use and antimicrobial resistance. Italian Journal of Animal Science 2024, 23(1), 1426–1438. [CrossRef]
- DG Health and Food Safety. Report on the implementation of the Farm to Fork strategy. European Commission 2022.
- Neumann, K., Elbersen, B.S., Verburg, P.H. Modelling the spatial distribution of livestock in Europe. Landscape Ecol 2009, 24, 1207–1222. [CrossRef]
- Nagano N., Shibata N., Saitou Y., Nagano Y. and Arakawa Y. Nosocomial Outbreak of Infections by Proteus mirabilis That Produces Extended-Spectrum CTX-M-2 Type –Lactamase. Journal of clinical microbiology 2003, 41 (12), 5530–5536. [CrossRef]
- Ng, L. K., Martin, I., Alfa, M., & Mulvey, M. Multiplex PCR for the detection of tetracycline resistant genes. Molecular and Cellular Probes 2001, 15(4), 209–215. [CrossRef]
- Gay K., Robicsek A., Strahilevitz J., Park C. H., Jacoby G., Barrett T.J., Medalla F., Chiller T.M. and Hooper D. C. Plasmid-mediated quinolone resistance in non-Typhi serotypes of Salmonella enterica. Clinical Infectious Diseases 2006, 43(3), 297–304. [CrossRef]
- Kelbrick M., Hesse E. and O’ Brien S. Cultivating antimicrobial resistance: how intensive agriculture ploughs the way for antibiotic resistance. Microbiology 2023, 169 (8). [CrossRef]
- Zhang Y., Su J. Q., Liao H.,Breed M. F., Yao H.,Shangguan H., Li H. Z., Sun X., Zhu Y.G. Increasing Antimicrobial Resistance and Potential Human Bacterial Pathogens in an Invasive Land Snail Driven by Urbanization. Environmental Science and Technology 2023, 57, 18, 7273–7284. [CrossRef]
- Waseem H., Williams M.R., Stedtfeld R.D., Hashsham S.A. Antimicrobial Resistance in the Environment. Water Environ Res 2017, 89(10):921-941. [CrossRef]
- Rogowska J., Gałęzowska G., Zimmermann A. Challenges and Current Trends in Preventing Antimicrobial Resistance in EU Water Law Context. Antibiotics (Basel) 2024, 14(1):18. [CrossRef]
- Agarwal V., Yue Y., Zhang X., Feng X., Tao Y., Wang J. Spatial and temporal distribution of endotoxins, antibiotic resistance genes and mobile genetic elements in the air of a dairy farm in Germany. Environ Pollut. 2023, 1;336:122404. [CrossRef]
- Azaglo G.S.K., Khogali M., Hann K., Pwamang J.A., Appoh E., Appah-Sampong E., Agyarkwa M.A., Fiati C., Kudjawu J., Hedidor G.K., Akumwena A., Timire C., Tweya H., Opintan J.A., Harries A.D. Bacteria and Their Antibiotic Resistance Profiles in Ambient Air in Accra, Ghana, February 2020: A Cross-Sectional Study. Trop Med Infect Dis 2021, 6(3):110. [CrossRef]
- de Rooij M.M.T., Hoek G., Schmitt H., Janse I., Swart A., Maassen C.B.M., Schalk M., Heederik D.J.J., Wouters I.M. Insights into Livestock-Related Microbial Concentrations in Air at Residential Level in a Livestock Dense Area. Environ Sci Technol 2019, 53(13):7746-7758 . [CrossRef]
- Martinez, J. L. Environmental pollution by antibiotics and by antibiotic resistance determinants. Environmental Pollution 2009, 157(11), 2893–2902. [CrossRef]
- Van Goethem, M. W., et al. A reservoir of ‘historical’ antibiotic resistance genes in remote pristine Antarctic soils. Microbiome 2018, 6, 40. [CrossRef]
- Wellington, E. M., et al. The role of the natural environment in the emergence of antibiotic resistance in Gram-negative bacteria. The Lancet Infectious Diseases 2013, 13(2), 155–165. [CrossRef]
Figure 1.
Apiary locations and number of livestock farms in the surrounding area. The map shows the study area in the Piedmont region (northwestern Italy), including the local health authority units (ASL) CN1 and CN2. In the map, the red scale indicates the number of livestock farms within each municipality. The bar plot shows the number of farms located within a 1 km radius of the sampled apiaries.
Figure 1.
Apiary locations and number of livestock farms in the surrounding area. The map shows the study area in the Piedmont region (northwestern Italy), including the local health authority units (ASL) CN1 and CN2. In the map, the red scale indicates the number of livestock farms within each municipality. The bar plot shows the number of farms located within a 1 km radius of the sampled apiaries.
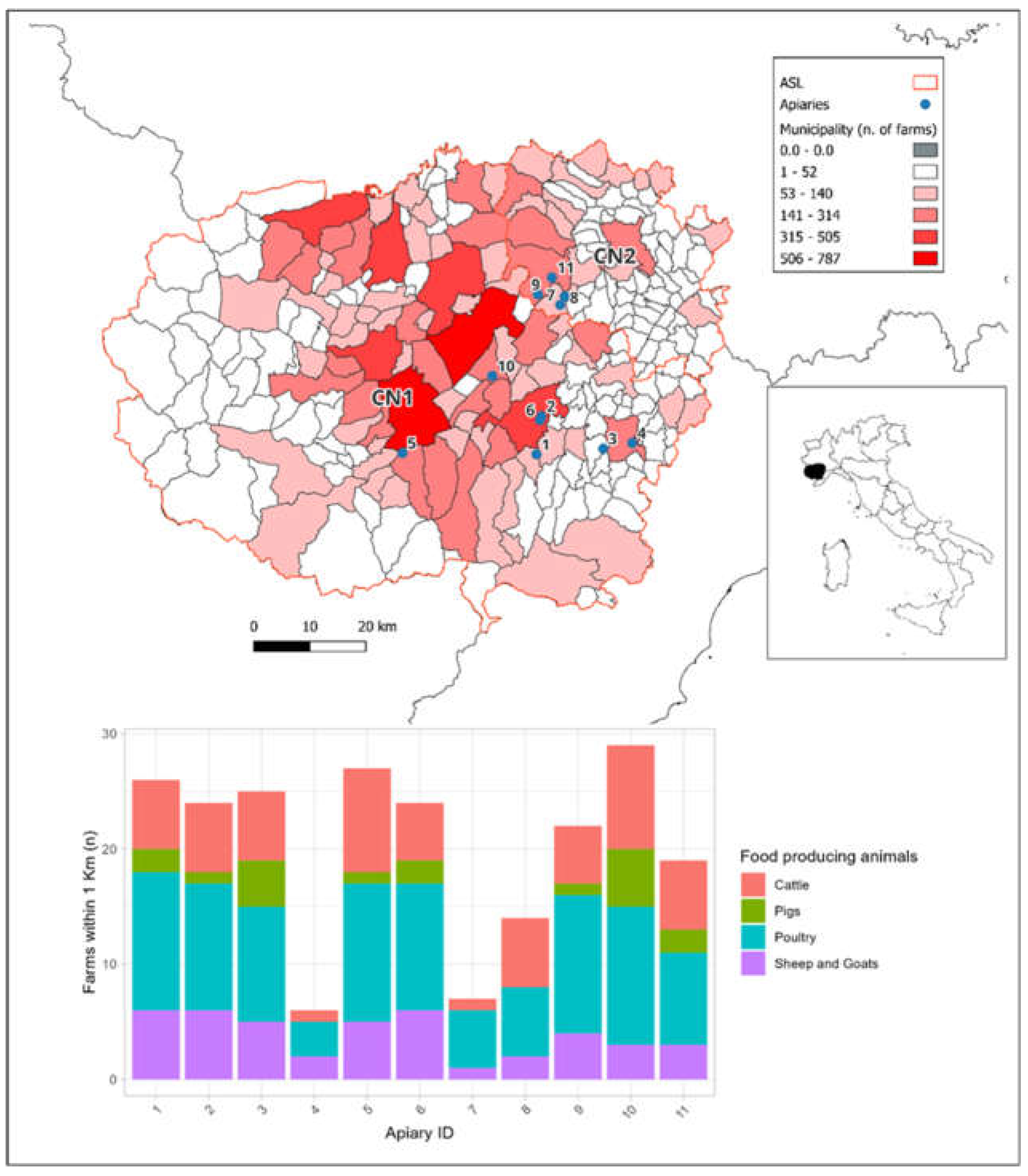
Figure 2.
Relative abundance of bacterial communities at the genus level for each individual sample. The legend shows the 15 most abundant genera.
Figure 2.
Relative abundance of bacterial communities at the genus level for each individual sample. The legend shows the 15 most abundant genera.
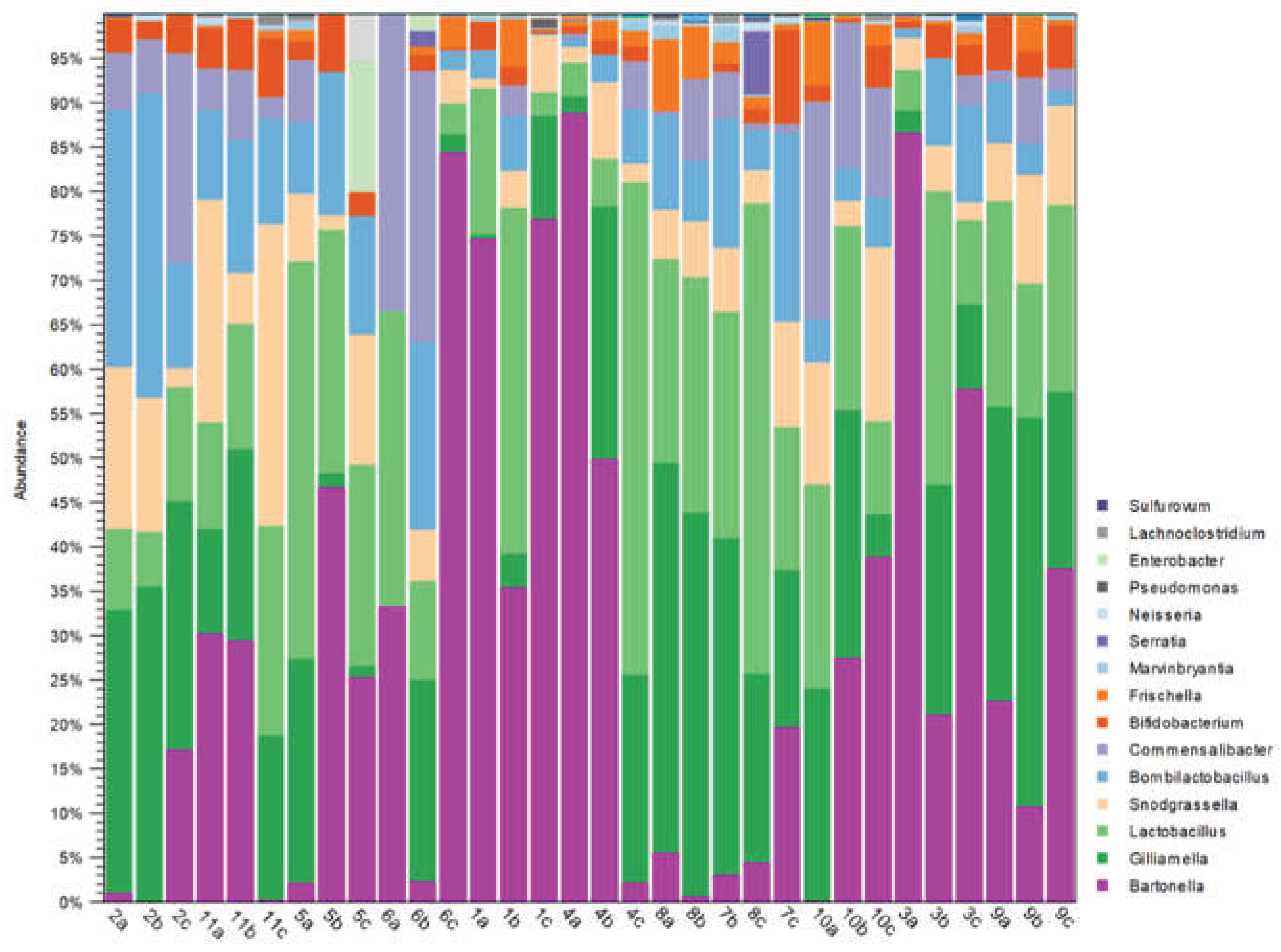

Table 1.
Sampling number and apiary localization.
| Apiary ID | Colony ID | Sampling Date | Municipality | Coordinate WGS 84 |
| 1 | A | 2022-07-27 | Monastero di Vasco (CN) | |
| B | 44.348637 (N) 7.832953 (E) |
|||
| C | ||||
| 2 | A | 2021-09-15 | Mondovì (CN) | |
| B | 44.410084 (N) 7.843101 (E) |
|||
| C | ||||
| 3 | A | 2022-07-27 | Monbasiglio (CN) | |
| B | 44.359357 (N) 7.982282 (E) |
|||
| C | ||||
| 4 | A | 2022-07-27 | Malpotremo (CN) | |
| B | 44.368907 (N) 8.047212 (E) |
|||
| C | ||||
| 5 | A | 2022-07-26 | Boves (CN) | |
| B | 44.347433 (N) 7.532728 (E) |
|||
| C | ||||
| 6 | A | 2022-07-27 | Mondovì (CN) | |
| B | 44.404124 (N) 7.839416 (E) |
|||
| C | ||||
| 7 | A | 2022-09-05 | Narzole (CN) | |
| B | 44.589716 (N) 7.881601 (E) |
|||
| C | ||||
| 8 | A | 2022-09-05 | Narzole (CN) | |
| B | 44.603442 (N) 7.891445 (E) |
|||
| C | ||||
| 9 | A | 2021-05-26 | Cherasco (CN) | |
| B | 44.605938 (N) 7.830899 (E) |
|||
| C | ||||
| 10 | A |
2022-09-04 |
Sant’Albano Stura (CN) | |
| B | 44.473888 (N) 7.731156 (E) |
|||
| C | ||||
| 11 | A | 2021-09-15 | Cherasco (CN) | |
| B | 44.633413 (N) 7.862137 (E) |
|||
| C |
Table 2.
List of ARGs with their Nucleotide sequences and thermal cycling details.
| Type of resistance |
Target gene | Nucleotide sequence 5’-3’ |
AMPL (bp) |
Amplification |
|---|---|---|---|---|
| Tetracyclines | tet A | GCTACATCCTGCTTGCCTTC CATAGATCGCCGTGAAGAGG |
210 | 94 °C × 60’’, 55 °C × 60″, 72 °C × 90″, 72 °C × 7’ × 35 cycles |
| Tetracyclines | tet B | TTGGTTAGGGGCAAGTTTTG GTAATGGGCCAATAACACCG |
659 |
94 °C × 60’’, 55 °C × 60″, 72 °C × 90″, 72 °C × 7’ × 35 cycles |
| Tetracyclines | tet C | CTTGAGAGCCTTCAACCCAG ATGGTCGTCATCTACCTGCC |
418 | 94 °C × 60’’, 55 °C × 60″, 72 °C × 90″, 72 °C × 7’× 35 cycles |
| Tetracyclines | tet D |
AAACCATTACGGCATTCTGC GACCGGATACACCATCCATC |
787 |
94 °C × 60’’, 55,5 °C × 60″, 72 °C × 90″, 72 °C × 7’ × 35 cycles |
| Tetracyclines | tet E |
AAACCACATCCTCCATACGC AAATAGGCCACAACCGTCAG |
278 |
94 °C × 60’’, 55 °C × 60″, 72 °C × 90″, 72 °C × 7’ × 30 cycles |
| Tetracyclines | tet G |
GCTCGGTGGTATCTCTGCTC AGCAACAGAATCGGGAACAC |
468 |
94 °C × 60’’, 55 °C × 60″, 72 °C × 90″, 72 °C × 7’ × 35 cycles |
| Fluoroquinolones | qnrA |
ATTTCTCACGCCAGGATTTG GATCGGCAAAGGTTAGGTCA |
516 |
94 °C × 45’’, 53 °C × 45″, 72 °C × 60″, 72 °C × 7’ × 32 cycles |
| Fluoroquinolones | qnrB |
GATCGTGAAAGCCAGAAAGG ACGATGCCTGGTAGTTGTCC |
469 |
94 °C × 45’’, 53 °C × 45″, 72 °C × 60″, 72 °C × 7’ × 32 cycles |
| Fluoroquinolones | qnrS |
ACGACATTCGTCAACTGCAA TAAATTGGCACCCTGTAGGC |
417 |
94 °C × 45’’, 53 °C × 45″, 72 °C × 60″, 72 °C × 7’ × 32 cycles |
| Fluoroquinolones | gyrA |
ATGAGCGACCTTGCGAGAGAAATTACACCG TTCCATCAGCCCTTCAATGCTGATGTCTTC |
630 |
94 °C × 30’’, 55 °C × 30″, 72 °C × 30″, 72 °C × 7’ × 30 cycles |
| Cephalosporines | blaTEM |
CCGTGTCGCCCTTATTCC AGGCACCTATCTCAGCGA |
823 |
94 °C × 60’’, 55 °C × 60″, 72 °C × 90″, 72 °C × 7’ × 30 cycles |
| Cephalosporines | blaSHV |
ATTTGTCGCTTCTTTACTCGC TTATGGCGTTACCTTTGACC |
526 |
94 °C × 60’’, 55 °C × 60″, 72 °C × 90″, 72 °C × 7’ × 30 cycles |
| Cephalosporines | blaCTX-M-1 |
CGGTGCTGAAGAAAAGTG TACCCAGCGTCAGATTAC |
353 |
94 °C × 60’’, 55 °C × 60″, 72 °C × 90″, 72 °C × 7’ × 30 cycles |
| Cephalosporines | blaCTX-M-2 |
ACGCTACCCCTGCTATTT CCTTTCCGCCTTCTGCTC |
764 |
94 °C × 60’’, 55 °C × 60″, 72 °C × 90″, 72 °C × 7’ × 30 cycles |
| Cephalosporines | blaCTX-M-9 |
GCAGATAATACGCAGGTG CGCCGTGGTGGTGTCTCT |
392 |
94 °C × 60’’, 55 °C × 60″, 72 °C × 90″, 72 °C × 7’ × 30 cycles |
| Multiresistance | int 1 |
GGGTCAAGGATCTGGATTTCG ACATGGGTGTAAATCATCGTC |
483 |
94 °C × 30’’, 62 °C × 30″, 72 °C × 30″, 72 °C × 7’× 30 cycles |
| Multiresistance | int 2 |
CACGGATATGCGACAAAAAGGT GTAGCAAACGAGTGACGAAATG |
788 |
94 °C × 30’’, 62 °C × 30″, 72 °C × 30″, 72 °C × 7’ × 30 cycles |
Table 3.
Presence of ARGs in honey bee gut samples analysed with Simplex PCR end point.
| Sample ID | blaTEM | tet B | tet C | qnrB | int1 |
|---|---|---|---|---|---|
| 1A | - | + | - | - | - |
| 1B | - | + | + | + Not confirmed | - |
| 1C | - | + | + Not confirmed | + Not confirmed | - |
| 2A | + | + Not confirmed | + | + Not confirmed | - |
| 2B | + | + | + | + Not confirmed | - |
| 2C | + | + | + | - | + |
| 3A | + | + | + | + Not confirmed | - |
| 3B | - | + | + | - | - |
| 3C | - | + | - | + Not confirmed | - |
| 4A | + | + | + Not confirmed | - | - |
| 4B | + | + | + | + Not confirmed | - |
| 4C | + | + | + | + Not confirmed | - |
| 5A | + | + | + Not confirmed | + Not confirmed | - |
| 5B | + | + | - | + Not confirmed | - |
| 5C | - | + | + | + Not confirmed | - |
| 6A | - | + | + Not confirmed | - | - |
| 6B | - | + | + | - | - |
| 6C | - | + | + Not confirmed | - | - |
| 7A | + | + | - | + Not confirmed | - |
| 7B | - | + | + | - | - |
| 7C | - | + | + | - | - |
| 8A | - | + | + | - | - |
| 8B | - | + | + | - | - |
| 8C | - | + | + | - | - |
| 9A | + | + | + Not confirmed | - | - |
| 9B | - | + Not confirmed | - | - | - |
| 9C | + | + Not confirmed | + | - | - |
| 10A | - | + | + Not confirmed | - | - |
| 10B | - | + | + Not confirmed | - | - |
| 10C | - | + | - | + Not confirmed | - |
| 11A | + | + | + | - | - |
| 11B | + | + | + | + Not confirmed | - |
| 11C | - | + | + Not confirmed | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



