
Submitted:
05 February 2026
Posted:
05 February 2026
You are already at the latest version
Abstract
Mitochondria are a key organelle in maintaining metabolic homeostasis. It not only generates most of the cell's energy through oxidative phosphorylation but also acts as a complex sensor of the redox state and oxygen in the cell. This review thoroughly ana-lyzes the interactions among mitochondrial iron metabolism, mitochondrial reactive oxygen species (mtROS), and lipid peroxidation (LPO), the triggering factors of fer-roptosis, an iron-dependent form of programmed cell death. We point out research showing that intrinsic mitochondrial machinery, such as iron-sulfur (Fe-S) cluster as-sembly and heme metabolism, is both an important cofactor and a master regulator. If these processes are disrupted, they can lead to ferroptosis. Unlike views that focus on the cytosol, we explain that the stability of Fe-S clusters in complexes such as aconitase and respiratory Complex I is crucial for preventing electron leakage and excessive mtROS formation. The Fenton reaction and its direct effect on cardiolipin (CL) oxidation in the inner membrane of mitochondria is a central event in cardiometabolic diseases. Its peroxidation and breakdown make the organelle very unstable and lead to cell death though Ca2+ overload and a significantly decreased reduced/oxidized glutathione ratio. Additionally, the functions of essential iron transporters and glutathione homeostasis are examined, and their dysregulation is correlated with ferroptosis-associated pro-gression of cardiometabolic and neurodegenerative disorders, such as obesity and Alzheimer’s disease. This review focused on the need to revisit the classic bioenergetic core of the mitochondria as a key player in the pathophysiology of metabolic and neu-rodegenerative diseases.
Keywords:
mitochondrial dysfunction
; ferroptosis
; cardiolipin
; complex I
1. Introduction
In the last decade, mitochondria have been repositioned from a secondary target of oxidative damage to a protagonist, integrating iron metabolism, redox signaling, and bioenergetic control to maintain cell health. Disruption of mitochondrial iron handling has been shown to favor the generation of reactive oxygen species (ROS) and membrane lipid oxidation, thereby creating a permissive environment for LPO process, and then, ferroptosis[1]. A key hallmark of this type of cellular death is its high regulation, though a machinery that interconnects antioxidant mechanisms, metabolism, and lipid structures integrity, as evidenced by inhibition by antioxidants and iron chelators[2,3]. Indeed, intracellular iron accumulation and LPO metabolites, such as oxidized lipids, are proposed as biomarkers of ferroptosis in disorders such as hepatic ferroptosis and Alzheimer’s disease (AD) [4,5].
In this context, a mechanistic reassessment of mitochondrial iron homeostasis is required to better understand disease pathogenesis and therapeutic responses. In this review, the interactions among iron homeostasis, the antioxidant machinery, and mitochondrial metabolic function will be examined. The relationship of mtROS generation and ferroptosis is growing in relevance across metabolic, cardiovascular, and neurodegenerative disorders.
2. Healthy Mitochondrial Function Is Crucial for a Metabolically Healthy Cell
Cellular metabolism depends on a healthy mitochondrial function. Mitochondria are dynamic metabolic centers that primarily produce energy for the cell via oxidative phosphorylation (OXPHOS). The generation of ATP via the electron transport chain (ETC) and the maintenance of its membrane potential are hallmarks of healthy mitochondrial function. The ETC is fed by molecules derived from the Krebs cycle, such as NADH and FADH2, which donate electrons to complex I and II, respectively, located in the inner mitochondrial membrane. Structurally, the ETC is a macromolecular supercomplex (respirasome) that organizes its components within the inner mitochondrial membrane to enhance electron flow and reduce the ROS production [6].
The Krebs cycle is not only an essential core of metabolism but also contributes to the cell’s oxygen sensor. Prolyl-hydroxylases (PHDs) are oxygen sensor-regulated by 2-oxoglutarate, succinate, fumarate, and isocitrate, all of which are Krebs ’ cycle intermediates, in order to modulate gene expression and metabolism[7,8]. One of the most characteristic examples is hypoxic-induced factor alpha (HIFα), in which a proline residue can be hydroxylated by oxygen or α-ketoglutarate [9,10]. This signal induces its degradation, which is why HIFα cannot bind to HIFβ to form the active transcription factor dimer[11]. This is a clear connection between metabolically healthy mitochondria and the oxygen sensor capacity.
Mitochondria are also an active part of the cell’s redox homeostasis. ROS generation is part of physiological signal transduction, acting as second messengers, and mtROS are generated during respiration [12]. Both superoxide anion (O₂•⁻) and hydrogen peroxide (H₂O₂) originate from complexes I, II, and III of the respiratory chain, at specific sites. Mitochondria are oriented in specific subcellular locations to conserve energy and minimize mtROS production, thereby reducing potential damage[13]. When an energetic substrate is offered to the cell, electron flux through the ETC increases, raising mitochondrial membrane potential and producing mtROS from the complex I. This overproduction of mtROS inhibits key enzymes and proteins, such as pyruvate dehydrogenase kinase[14]. In this way, pyruvate would not be converted to acetyl-CoA, or mtROS would modify the redox status of uncoupling proteins (UCPs), providing protection against dysregulated hyperpolarization.
Healthy mitochondria have a powerful antioxidant machinery; the glutathione system is present in the mitochondrial matrix as a first line of defense against excess mtROS, which can reach concentrations higher than in the cytosol. Reduced glutathione (GSH) is formed continuously from the reduction of oxidized glutathione (GSSG) and is incorporated by its mitochondrial transport SLC25A39[15]. Both molecules are tripeptides composed of γ-Glu-Cys-Gly; therefore, the cysteine concentration within the matrix is a crucial factor in maintaining adequate GSH levels. This system is powered by glutathione reductase, a NADPH-dependent enzyme that reduces one GSSG to two GSH. A healthy mitochondrion requires sufficient NADPH in its matrix to maintain a GSH/GSSG ratio>10:1[16]. Another clue enzymes are ϒ-Glutamylcysteine synthetase and Glutathione synthetase, both present in the mitochondrial matrix, and both are involved in the de novo synthesis of glutathione. Glutathione peroxidase 4 (GPX4) is present in both the matrix and the inner membrane of the mitochondria. Its activity depends on selenium to reduce H2O2 and peroxidized lipids[17].
Both the respiratory chain and OXPHOS are iron-dependent processes because complexes I, II, and III require Fe-S clusters as cofactors for their function, and cytochrome c and Complex IV contain heme groups[18]. Iron–sulfur clusters vary from simple [1Fe–0S] rubredoxins to complex [8Fe–7S] nitrogenases. Common forms include [2Fe–2S], [3Fe–4S], and [4Fe–4S], typically cysteine-ligated, with histidine ligation in Rieske proteins; the latter are present in the complex III of the ETC [18] (Table 1).
Mitochondrial metabolism works together with heme synthesis. Careful iron management by mitochondria is essential for their own survival and for the metabolic homeostasis of the individual.
3. Mitochondrial Iron Handling
3.1. Fe-S Clusters
Several cellular functions, such as electron transfer in metabolism, depend on metalloproteinases that require Fe-S clusters for activity. The binding of the Fe-S cluster to proteins is through a cysteine or a histidine. Healthy mitochondria have this protein with a Fe-S cluster, which regulates metabolic pathways and serves as a redox sensor. Table 1 shows the Fe-S cluster relevant for mitochondrial metabolism.
3.2. Heme Group
On the other hand, mitochondria play a pivotal role in heme synthesis and its regulation. The pro-oxidant nature of heme could generate oxidative intracellular damage. Its accumulation can disrupt redox balance, as elevated iron and ROS levels can shift the cell’s state from a protective to a pro-death state[39]. The heme group is part of the structure of a large number of enzymes, in and out of the mitochondria. It has been proposed that the enzymes of heme biosynthesis exist as a metabolon, a protein complex formed by interacting enzymes that provide a scaffold for interactions with other pathways[40]. The heme group is a metalloporphyrin composed of a tetrapyrrolic porphyrin ring that coordinates a central iron atom, an essential cofactor for its biochemical function. The canonical heme biosynthetic pathway comprises eight sequentially acting enzymes and is initiated in the mitochondrial matrix by the condensation of succinyl-CoA and glycine to form 5-aminolevulinic acid (ALA), a reaction catalyzed by the pyridoxal-5′-phosphate–dependent enzyme 5-aminolevulinate synthase (ALA)[41]. Then, ALA is transported to the cytosol, where it is converted to porphobilinogen and subsequently processed into coproporphyrinogen III. This intermediate re-enters the mitochondria for oxidation and final iron insertion by ferrochelatase, which inserts ferrous iron into protoporphyrin IX, completing heme synthesis [42].
Mitochondria regulate heme synthesis through different proteins, such as the complex IV assembly cofactor heme A:farnesyltransferase (COX-10), whose nephron-specific loss leads to mitochondrial dysfunction, severe kidney failure, and premature death, linking impaired mitochondrial respiration to innate immune activation through interferon signaling in renal epithelial cells [43]. In the same context, healthy mitochondria can respond to hypoxia through miR-210, which diminishes cellular heme concentrations and the functionality of mitochondrial and cytosolic heme-dependent proteins by regulating ferrochelatase, the terminal enzyme in heme biosynthesis [44].
Heme is part of the structural conformation of a member of the ETC; complex II (succinate dehydrogenase) requires a heme group only for structural stabilization, because the electrons from FADH₂ flow directly through the [Fe-S] centers to ubiquinone, bypassing this heme[45]. The two b-type hemes (bₗ and bₕ) in the cytochrome b of Complex III are integral to the Q-cycle process[46]. Their unique redox potential establishes a pathway that reverses direction, from the electron pair to ubiquinol. This division allocates one electron to the Rieske cluster and cytochrome c₁, while the other returns through the b-hemes to decrease an additional quinone[47]. This distinctive electron bifurcation is the essential process that allows the complex to translocate protons and save energy. Therefore, without the heme group in complex III, electron transfer could not occur.
The Complex IV or cytochrome c oxidase has a specialized structure with metallic cores, has two heme groups: cytochrome α and cytochrome α3. Cytochrome α carries a formyl group (-CHO), which serves as a transient electron-transfer site in redox homeostasis. A binuclear core with copper (Cu-A and Cu-B) is part of the structure. The last ones are essential for oxygen reduction; Cu-A is an initial electron donor, and cytochrome α3 is associated with the Cu-B center, where molecular oxygen is bound and reduced to water [48]. Both Fe-S clusters and heme groups are essential chemical structures for redox homeostasis, balanced mtROS generation, and mitochondrial metabolic function.
4. Lipoperoxidation Process in Mitochondrial Dysfunction
Several metabolic disturbances characterized by mitochondrial dysfunction include cardiometabolic disease, such as clinical obesity, type 2 diabetes, and metabolically associated steatotic liver disease (MASLD) [49,50,51]. Mitochondrial dysfunction is a set of structural, functional, and molecular defects in mitochondria. Among the main hallmarks are reductions in respiratory capacity and OXPHOS, an increase in oxidative stress that damages membrane lipids, proteins, and mtDNA, alterations in mitochondrial dynamics (disequilibrium between fusion and fission), disruption of energetic metabolism, mtDNA mutations, and activation of apoptosis[52].
The lipoperoxidation process and mitochondrial dysfunction are a vicious cycle of bioregulatory impairment and oxidative stress. Mitochondrial membranes are damaged, amplifying this damage throughout the organelles. Lipoperoxidation occurs when ROS react with polyunsaturated fatty acids (PUFAs) to generate lipid peroxides, a process associated with degenerative chronic diseases with an inflammatory component [53]. LPO occurs in three phases: initiation, propagation, and termination. Initiation could be triggered by both the hydroxyl radical (OH●) or the hydroperoxyl radical (OOH●), ROS-derived by the Fenton reaction or by peroxynitrite (ONOO-), RNS-derived. This step implies the abstraction of a hydrogen atom from the allylic carbon to form a lipid radical (L●). The second phase consists of the reaction of (L●) with oxygen to form a peroxyl radical (LOO●), which can then abstract another allylic carbon to form a new (L●) and a lipid peroxide (LOOH), thereby propagating the chain reaction. The termination phase is characterized by the accumulation of high amounts of peroxyl radical. When they react and form a new bond, the radical is eliminated. Then, the Fenton reaction is a key step in which iron initiates lipid peroxidation [54]. Among the reactive lipid species generated by lipid peroxidation are 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA), both of which are used as biomarkers of lipid peroxidation[55]. Lipoperoxidation in mitochondrial membranes is initiated for the generation of superoxide anion (O₂•⁻) in the ETC. The inner membrane has a microdomain rich in CL, whose structure has linoleic acid (C18:2), and is negatively charged, generating a local environment with low pH relative to the interfacial membrane-matrix. Low pH induces the conversion of O₂•⁻ to hydroperoxyl radical (HO₂•), highly reactive with PUFA, above all CL and phosphatidyl ethanolamine[56]. This reaction triggers the isoprostane pathway of lipoperoxidation, generating a racemic mixture of highly toxic isoprostanes and isoketals, or linoleic acid hydroperoxides. These oxidative products irreversibly alter the structural and functional integrity of respiratory supercomplexes and ATP synthase, compromising oxidative phosphorylation[57].
5. Lipoperoxidation as a Driver of Ferroptosis
Lipid peroxides themselves can induce mitochondrial damage by impairing membrane integrity, disrupting respiratory complex function, opening the mitochondrial permeability transition pore (mPTP), and inducing the release of cytochrome c and mitochondrial swelling, leading to cell death[58]. LPO in the mitochondrial inner membrane alters the integrity and function of calcium transporters, such as Na+/Ca2+ (mNCE). Consequently, Ca2+ is trapped inside, and depolarization further exacerbates the problem, triggering Ca2+ influx through the mitochondrial calcium uniporter (MCU)[59]. This defective Ca2+ homeostasis, a hallmark of mitochondrial dysfunction, could be prevented by mito-ROS scavenging, as reported in ventricular myocytes, restoring Ca2+ homeostasis [60].
Given the highly regulated mitochondrial iron management, iron can be dangerous only when mitochondria are not healthy. Free Fe2+ could react with H2O2 through the Fenton reaction that generates hydroxyl radical (OH●)[54], and lipids such as CL are susceptible to this type of radical species. When a radical attack occurs in CL, lipid peroxidation is initiated in the inner mitochondrial membrane.
Ferroptosis is an iron-dependent mechanism of cell death. This concept was introduced in 2012 by Hirschhorn and Stockwell as a cellular death phenotype morphologically distinct from autophagic, apoptotic, or necrotic cell death [61]. The concept has been enriched with evidence related to the highly regulated process, dependent on GSH homeostasis, in which key enzymes, such as GPX4 and cysteine supply, are crucial for preventing it [62]. Currently, three mechanisms by which ferroptosis can occur have been proposed. One is the inhibition of the glutamate/cystine antiporter system xc− (Figure 1). This transporter imports cystine into the cell, which is used for GSH synthesis. This importer is the rate-limiting substrate for GSH synthesis. Erastin and sorafenib, are drugs that inhibit the system xc− activity, producing the accumulation of peroxidized lipids, and are an effective tool for inducing in vitro ferroptosis[63]. Given that GSH is considered one of the cell’s essential components in counteracting LPO, acting by donating a hydrogen atom to yield a nonradical product, lipid peroxides can also be reduced through a mechanism involving GPX4, with GSH being utilized as a cofactor [64]. Consequently, the second mechanism underlying increased lipid peroxidation is the loss or depletion of GPX4 activity. GPX4 is a selenoprotein containing the rare amino acid selenocysteine at its active site, which requires post-translational modification, such as palmitoylation, to be stable[65]. Following GPX4 action, lipid peroxides are converted to their corresponding alcohols or aldehydes, thereby halting the propagation phase of lipid peroxidation. Consequently, GPX4 is widely recognized as a critical regulator of ferroptosis. Its primary antioxidant function is the reduction of phospholipid hydroperoxides within cellular membranes. Ferroptosis is induced by genetic or pharmacological inhibition of GPX4, leading to elevated lipid peroxidation products that are considered to favor the progression of MASLD. One example of such pharmacological inhibition is RSL3, a ferroptosis inducer whose mechanism of action involves the specific inhibition of GPX4 and has been proposed to target colon cancer[66].
Conversely, genetic conditions such as deletion of Acsl4 or Lpcat3, which encode acyl-CoA synthetase long-chain family member (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), respectively, both involved in PUFA incorporation into membranes, confer ferroptosis resistance [67,68] (Figure 1). Arachidonoyl (AA) rich phospholipids can be oxidized to produce LPO by ACLS4, producing acyl CoA derivatives. Then, LPCAT3 forms phosphatidylethanolamines, which will be substrates for lipoxygenases. In turn, as we might expect, the third mechanism that induces ferroptosis is the inhibition of lipoxygenases. Baicalein is a selective lipoxygenase inhibitor with iron-binding properties and anti-Fenton reaction bioactivity, acting as a negative regulator of ferroptosis[69].
6. Disequilibrium of Mitochondrial Iron Handling in Diseases
6.1. Obesity
A nascent body of evidence suggests mitochondrial iron imbalance as part of the mechanism underlying metabolic disease. Mitoferrin 1 (MFRN1), coded by the Slc25a37 gene, is an importer of iron in the mitochondria. In obesity-resistant mice (mice that do not develop obesity despite being fed a high-fat diet), this gene was overexpressed 4.9-fold compared to control mice. This phenotype had a better fat-to-muscle ratio, increased type 2a oxidative skeletal fibers (fast-twitch), and likely increased the capacity to oxidize fat and enhance ATP production [70]. Considering that skeletal muscle is an active tissue responsible for regulating metabolism, iron import is relevant to maintain the synthesis of mitochondrial Fe-S clusters and heme. Unlikely, overexpression of the Slc25A28 gene, which encodes mitoferrin-2 (MFRN2), enhances diet-induced obesity in mice by increasing fat storage through increased lipogenesis and inhibition of lipolysis. Also, UCP-1 and PGC-1α are downregulated, key proteins for thermogenesis and mitochondrial biogenesis, respectively [71]. MFRN2 is part of the iron transporter SLC25 family and is located in the inner membrane of the mitochondria to import Fe2+ from the cytosol to the mitochondrial matrix. An alteration in Slc25A28 expression could disrupt mitochondrial iron homeostasis, increasing mtROS production and thereby affecting the ETC and energy production in the cell[72].
6.2. MASLD
Some reports suggest that iron stores are implicated in the pathogenesis of MASLD, which is consistent with the role of iron in Fenton reaction-dependent LPO. Hepatic biopsies from patients with MASLD have high hepatic iron deposition as a predictor of advanced fibrosis and histologic damage[73]. Since insulin resistance is a key pathogenic feature of MASLD, significant improvements in insulin levels and the Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) index, and a higher rate of improvement in histological liver damage were reported in iron-depleted patients treated with phlebotomy [74]. Evidence from cell cultures, animal models, and humans shows that iron overload in the organism is a driving force in the MASLD progression. In the presence of hepatic inflammatory infiltrates, the addition of damage-associated molecular patterns (DAMPs) to the increase in LPO and ROS induces ferroptosis. This local environment and iron-mediated signaling can activate hepatic stellate cells (HSCs), which can differentiate into fibroblasts and produce collagen, contributing to the development of fibrosis (Figure 2) [75].
Several proteins regulate iron homeostasis; ferritin is an iron-storing protein, and elevated levels are found in many patients with NAFLD. Also, hepcidin, an intracellular iron sensor, is upregulated in obese mice[76]. In addition, transferrin receptor-1 (TfR1) is upregulated in HFD-fed mice, despite this protein being expected to be downregulated in intracellular iron overload [77]. Upregulation of TfR1 could lead to hepatocellular iron uptake in NAFLD despite increased hepatocellular iron, promoting the Fenton reaction and consequently lipoperoxidation and iron-dependent liver damage.
6.3. Cardiac Dysfunction
A classic model of ischemia/reperfusion (I/R) applied to cardiac tissue; mitochondrial iron imbalance is a protagonist. I/R induces an increase in intracellular iron, which triggers the vicious cycle: mtROS increase- LPO-Calcium intake, MCU activation, opening of mPTP, and cellular death. Various strategies to reduce mitochondrial iron have shown promise in reversing I/R injury. Inhibitors of ferroptosis, iron chelators, and MCU inhibitors are among the pharmacological strategies to induce cardioprotection [78].
This cascade highlights key interrelated mechanisms that are implicated in cellular damage and associated pathologies.
6.4. Neurodegenerative Diseases
The relationship between ferroptosis and the development of neurodegenerative diseases (ND) is a topic of enormous research nowadays. Iron dysregulation and LPO are also present in both the generation and progression of ND, such as Alzheimer’s (AD) and Parkinson’s diseases (PD). The last one is characterized by a loss of dopaminergic neurons in the substantia nigra and the accumulation of α-synuclein (α-syn) misfolding. Then, mtROS increases α-syn aggregation, a hallmark of the disease: Lewy bodies [79]. Increased iron, decreased total GHS levels, and mitochondrial complex I dysfunction are some of the findings reported in the substantia nigra [80,81]. It has been proposed that upregulation of heme oxygenase-1 (HO-1) in Lewy bodies, induced by neuronal stress, is responsible for the accumulation of iron inside mitochondria[82]. Aggregates of α-syn interact with mitochondria, resulting in mitochondrial membrane depolarization, decreased ATP production, mitochondrial fragmentation, and subsequent degradation via cardiolipin–dependent mitophagy [83]. Moreover, mitochondrial dysfunction increases free iron levels and LPO. Another finding is the SLC25A39 degradation, which reduces the GSH uptake, Krebs ’cycle dysregulation increases PUFA synthesis, increasing the substrate offer to ferroptosis, all of these findings promote the dopaminergic neurons’ death[81].
The pathophysiology of the disease comprises the formation of neurofibrillary tangles in neurons, caused by the abnormal aggregation of β-amyloid protein (Aβ) and the phosphorylation of tau protein, which constitute one of the main pathological features of AD[84]. Lactylation is a post-translational modification of proteins, and tau is susceptible to it. Lactylation of tau at K677 was associated with erastin-induced ferroptosis in cells treated with Aβ, opening a new field in which this tau modification could decrease iron levels and inhibit ferroptosis in microglial cells [85].
Mitochondrial dysfunction is part of the pathophysiology of AD. In a mouse model of AD, a decrease of complex I activity was found. The ATP deficit activated AMPK, triggering metabolic reprogramming and altering the membrane phospholipid composition; diacyl- and lyso-phosphatidylcholine levels increase, while ethanolamine plasmalogens and cardiolipin content decrease [86].
The role of iron in the pathophysiology of Alzheimer’s disease has become progressively less controversial, and therapeutic approaches targeting the reduction of iron levels are already being implemented. Iron dysregulation in the brains of patients with AD leads to an excess of free iron, which promotes oxidative stress and lipid peroxidation via the Fenton reaction. Ferritinophagy, a selective autophagy process mediated by nuclear receptor coactivator 4 (NCOA4) that releases iron from ferritin, is considered to act as an upstream trigger of ferroptosis in AD. Therefore, modulation of ferritinophagy and ferroptosis, particularly by targeting nuclear receptor coactivator 4, is a promising therapeutic strategy for intervening in the pathogenesis and progression of AD [87]. Conversely, maneuvers to decrease brain iron content yield contradictory results: using an iron chelator, such as deferiprone, reduced hippocampal iron accumulation in Alzheimer’s patients, but this intervention was detrimental to cognitive function [69]. These observations strongly support the hypothesis that ferroptosis is not merely an epiphenomenon but an active driver of neuronal degeneration in various neurodegenerative diseases. Consequently, therapeutic strategies aimed at modulating iron homeostasis, enhancing antioxidant defenses, or directly inhibiting lipid peroxidation are regarded as promising approaches. However, their clinical translation is considered to require careful evaluation of potential adverse effects.
7. Discussion
There has been a focus on mitochondria not just as passive recipients of oxidative damage but also as active modulators of iron-dependent redox signaling. Classically, the mitochondrion is the motor of the cell; a complex and exquisite regulation of redox, iron, and energy homeostasis is required for healthy function. Indeed, impairment of any of these processes could disrupt equilibrium, triggering a vicious cycle of damage (Figure 2). Then, the highly mitochondrial iron-handling implicated several proteins that protect cells against iron-dependent damage by the Fenton reaction. This review presents a comprehensive paradigm that identifies mitochondrial iron homeostasis and lipid peroxidation as pivotal factors in ferroptosis across diverse disease contexts. Although persuasive data have been compiled associating iron–sulfur cluster disruption, cardiolipin peroxidation, and the overproduction of mitochondrial reactive oxygen species with ferroptosis, certain limitations must be recognized. A significant portion of the mechanistic insights presented have been obtained from in vitro systems or animal models, which may inadequately represent disease complexity and cell-type specificity. There have been many discussions about the therapeutic implications, especially those focusing on mitochondrial iron transport, glutathione metabolism, or lipid peroxidation. Nonetheless, altering these pathways has yielded context-specific outcomes, as iron chelation or antioxidant approaches may interfere with critical physiological processes. Consequently, the translational significance of ferroptosis-targeted therapies must be regarded with circumspection. Future research is necessary to thoroughly investigate the geographical, temporal, and tissue-specific regulation of mitochondrial iron metabolism, preferably utilizing integrated omics and sophisticated in vivo models.
8. Conclusions
Iron in mitochondria is exquisitely regulated by several proteins to prevent its toxic effects when free, as it catalyzes the production of damaging free radicals. Iron overload and oxidative stress can exacerbate its toxicity through lipid peroxidation. Ferroptosis is an iron-dependent form of cell death that, in some diseases, is triggered by lipid peroxidation (in the context of increased PUFA), increased mtROS, and iron dysregulation. The Fenton reaction occurs in this dysregulation, characterized by Fe-S cluster dysfunction and problems in heme metabolism. Locally, mtROS causes peroxidation of cardiolipin, membrane instability, Ca2+ overload, and mitochondrial dysfunction. Disrupted mitochondrial iron homeostasis is therefore a pivotal pathogenic pathway linking metabolic and neurodegenerative diseases.
Author Contributions
Conceptualization and writing, A.E. and J.L.B.; writing—review and editing, M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 4-HNE | 4-hydroxy-2-nonenal |
| AA | Arachidonoyl |
| ACSL4 | Acyl-CoA Synthetase Long-Chain Family Member 4 |
| ALA | 5-aminolevulinic acid |
| ALAS1 | 5-aminolevulinate synthase |
| ATP | Adenosine triphosphate |
| CL | Cardiolipin |
| COX-10 | Heme A:farnesyltransferase (Complex IV assembly cofactor) |
| ETC | Electron transport chain |
| Fe-S | Iron-sulfur |
| GPX4 | Glutathione Peroxidase 4 |
| GSH | Reduced glutathione |
| GSSG | Oxidized glutathione |
| HIFα | Hypoxia-inducible factor alpha |
| HOSS | Homeostatic Oxygen-Sensing System |
| LPCAT3 | Lysophosphatidylcholine Acyltransferase 3 |
| LPO | Lipoperoxidation |
| MASLD | Metabolically Associated Steatotic Liver Disease |
| MCU | Mitochondrial Calcium Uniporter |
| MDA | Malondialdehyde |
| MFRN1 | Mitoferrin 1 |
| MFRN2 | Mitoferrin 2 |
| mNCE | Mitochondrial Na+/Ca2+ Exchanger |
| mPTP | Mitochondrial permeability transition pore |
| mtROS | Mitochondrial reactive oxygen species |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| OXPHOS | Oxidative phosphorylation |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PHDs | Prolyl-hydroxylases |
| PUFAs | Polyunsaturated fatty acids |
| Q-cycle | Quinone cycle (electron transfer mechanism in Complex III) |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive oxygen species |
| SLC25A39 | Solute carrier family 25 member 39 (mitochondrial glutathione transporter) |
| UCP-1 | Uncoupling protein 1 |
| UCPs | Uncoupling proteins |
References
- Zheng, Y.; Sun, J.; Luo, Z.; Li, Y.; Huang, Y. Emerging Mechanisms of Lipid Peroxidation in Regulated Cell Death and Its Physiological Implications. Cell Death Dis 2024, 15, 859. [Google Scholar] [CrossRef]
- Rainey, N.E.; Moustapha, A.; Saric, A.; Nicolas, G.; Sureau, F.; Petit, P.X. Iron Chelation by Curcumin Suppresses Both Curcumin-Induced Autophagy and Cell Death Together with Iron Overload Neoplastic Transformation. Cell Death Discov. 2019, 5, 150. [Google Scholar] [CrossRef]
- Abdullah, H.A.; Moawed, F.S.; Ahmed, E.S.; Abdel Hamid, F.F.; Haroun, R.A.-H. Iron Chelating, Antioxidant and Anti-Apoptotic Activities of Hesperidin and/or Rutin against Induced-Ferroptosis in Heart Tissue of Rats. Int J Immunopathol Pharmacol 2025, 39, 03946320251331873. [Google Scholar] [CrossRef]
- Mayr, E.; Rotter, J.; Kuhrt, H.; Winter, K.; Stassart, R.M.; Streit, W.J.; Bechmann, I. Detection of Molecular Markers of Ferroptosis in Human Alzheimer’s Disease Brains. J Alzheimers Dis 2024, 102, 1133–1154. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Katsumata, Y.; Chu, P.; Morikawa, R.; Nakamoto, N.; Iguchi, K.; Takahashi, K.; Kou, T.; Ito, R.; Taura, K.; et al. Monitoring Ferroptosis in Vivo: Iron-Driven Volatile Oxidized Lipids as Breath Biomarkers. Redox Biology 2025, 86, 103858. [Google Scholar] [CrossRef]
- Guo, R.; Gu, J.; Zong, S.; Wu, M.; Yang, M. Structure and Mechanism of Mitochondrial Electron Transport Chain. Biomedical Journal 2018, 41, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. 2-Oxoglutarate-Dependent Dioxygenases Are Sensors of Energy Metabolism, Oxygen Availability, and Iron Homeostasis: Potential Role in the Regulation of Aging Process. Cell. Mol. Life Sci. 2015, 72, 3897–3914. [Google Scholar] [CrossRef] [PubMed]
- Siddiq, A.; Aminova, L.R.; Ratan, R.R. Hypoxia Inducible Factor Prolyl 4-Hydroxylase Enzymes: Center Stage in the Battle against Hypoxia, Metabolic Compromise and Oxidative Stress. Neurochem Res 2007, 32, 931–946. [Google Scholar] [CrossRef]
- Chan, D.A.; Sutphin, P.D.; Yen, S.-E.; Giaccia, A.J. Coordinate Regulation of the Oxygen-Dependent Degradation Domains of Hypoxia-Inducible Factor 1 Alpha. Mol Cell Biol 2005, 25, 6415–6426. [Google Scholar] [CrossRef] [PubMed]
- Schlisio, S. Neuronal Apoptosis by Prolyl Hydroxylation: Implication in Nervous System Tumours and the Warburg Conundrum. J Cellular Molecular Medi 2009, 13, 4104–4112. [Google Scholar] [CrossRef]
- Schödel, J.; Ratcliffe, P.J. Mechanisms of Hypoxia Signalling: New Implications for Nephrology. Nat Rev Nephrol 2019, 15, 641–659. [Google Scholar] [CrossRef]
- Okoye, C.N.; Koren, S.A.; Wojtovich, A.P. Mitochondrial Complex I ROS Production and Redox Signaling in Hypoxia. Redox Biol 2023, 67, 102926. [Google Scholar] [CrossRef]
- Espinosa, A.; Casas, M.; Jaimovich, E. Energy (and Reactive Oxygen Species Generation) Saving Distribution of Mitochondria for the Activation of ATP Production in Skeletal Muscle. Antioxidants (Basel) 2023, 12, 1624. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.R.; Collins, Y.; Abakumova, I.; Chouchani, E.T.; Baranowski, B.; Fearnley, I.M.; Prime, T.A.; Murphy, M.P.; James, A.M. Inactivation of Pyruvate Dehydrogenase Kinase 2 by Mitochondrial Reactive Oxygen Species. Journal of Biological Chemistry 2012, 287, 35153–35160. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, S.; Tomar, A.; Yen, F.S.; Unlu, G.; Ropek, N.; Weber, R.A.; Wang, Y.; Khan, A.; Gad, M.; et al. Autoregulatory Control of Mitochondrial Glutathione Homeostasis. Science 2023, 382, 820–828. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Marín-Hernández, Á.; Gallardo-Pérez, J.C.; Vázquez, C.; Rodríguez-Enríquez, S.; Saavedra, E. Control of the NADPH Supply and GSH Recycling for Oxidative Stress Management in Hepatoma and Liver Mitochondria. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2018, 1859, 1138–1150. [Google Scholar] [CrossRef]
- Chen, T.-H.; Wang, H.-C.; Chang, C.-J.; Lee, S.-Y. Mitochondrial Glutathione in Cellular Redox Homeostasis and Disease Manifestation. Int J Mol Sci 2024, 25, 1314. [Google Scholar] [CrossRef] [PubMed]
- Read, A.D.; Bentley, R.E.; Archer, S.L.; Dunham-Snary, K.J. Mitochondrial Iron-Sulfur Clusters: Structure, Function, and an Emerging Role in Vascular Biology. Redox Biol 2021, 47, 102164. [Google Scholar] [CrossRef]
- Meinhardt, S.W.; Kula, T.; Yagi, T.; Lillich, T.; Ohnishi, T. EPR Characterization of the Iron-Sulfur Clusters in the NADH: Ubiquinone Oxidoreductase Segment of the Respiratory Chain in Paracoccus Denitrificans. J Biol Chem 1987, 262, 9147–9153. [Google Scholar] [CrossRef]
- Kang, P.T.; Chen, C.-L.; Lin, P.; Zhang, L.; Zweier, J.L.; Chen, Y.-R. Mitochondrial Complex I in the Post-Ischemic Heart: Reperfusion-Mediated Oxidative Injury and Protein Cysteine Sulfonation. J Mol Cell Cardiol 2018, 121, 190–204. [Google Scholar] [CrossRef]
- Child, S.A.; Reddish, M.J.; Glass, S.M.; Goldfarb, M.H.; Barckhausen, I.R.; Guengerich, F.P. Functional Interactions of Adrenodoxin with Several Human Mitochondrial Cytochrome P450 Enzymes. Arch Biochem Biophys 2020, 694, 108596. [Google Scholar] [CrossRef] [PubMed]
- Schulz, V.; Freibert, S.-A.; Boss, L.; Mühlenhoff, U.; Stehling, O.; Lill, R. Mitochondrial [2Fe-2S] Ferredoxins: New Functions for Old Dogs. FEBS Lett 2023, 597, 102–121. [Google Scholar] [CrossRef]
- Nett, J.H.; Hunte, C.; Trumpower, B.L. Changes to the Length of the Flexible Linker Region of the Rieske Protein Impair the Interaction of Ubiquinol with the Cytochrome Bc1 Complex. Eur J Biochem 2000, 267, 5777–5782. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Liu, J.; Knapp, M.; Donnan, P.H.; Boggs, D.G.; Bridwell-Rabb, J. Custom Tuning of Rieske Oxygenase Reactivity. Nat Commun 2023, 14, 5858. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.K.; Pain, J.; Singh, P.; Dancis, A.; Pain, D. Mitochondrial Glutaredoxin Grx5 Functions as a Central Hub for Cellular Iron-Sulfur Cluster Assembly. Journal of Biological Chemistry 2025, 301, 108391. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, X.; Lai, Y.; Xu, J.; Zhang, Y.; Zhou, S.; Feng, Z.; Yu, L.; Tang, Y.; Wang, W.; et al. Structure of the Human Respiratory Complex II. Proc. Natl. Acad. Sci. U.S.A. 2023, 120, e2216713120. [Google Scholar] [CrossRef]
- Brown, N.M.; Kennedy, M.C.; Antholine, W.E.; Eisenstein, R.S.; Walden, W.E. Detection of a [3Fe-4S] Cluster Intermediate of Cytosolic Aconitase in Yeast Expressing Iron Regulatory Protein 1. Insights into the Mechanism of Fe-S Cluster Cycling. J Biol Chem 2002, 277, 7246–7254. [Google Scholar] [CrossRef]
- Deck, K.M.; Vasanthakumar, A.; Anderson, S.A.; Goforth, J.B.; Kennedy, M.C.; Antholine, W.E.; Eisenstein, R.S. Evidence That Phosphorylation of Iron Regulatory Protein 1 at Serine 138 Destabilizes the [4Fe-4S] Cluster in Cytosolic Aconitase by Enhancing 4Fe-3Fe Cycling. J Biol Chem 2009, 284, 12701–12709. [Google Scholar] [CrossRef]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Kennedy, M.C. Mitochondrial Aconitase Is a Source of Hydroxyl Radical. An Electron Spin Resonance Investigation. J Biol Chem 2000, 275, 14064–14069. [Google Scholar] [CrossRef]
- Le Breton, N.; Wright, J.J.; Jones, A.J.Y.; Salvadori, E.; Bridges, H.R.; Hirst, J.; Roessler, M.M. Using Hyperfine Electron Paramagnetic Resonance Spectroscopy to Define the Proton-Coupled Electron Transfer Reaction at Fe-S Cluster N2 in Respiratory Complex I. J Am Chem Soc 2017, 139, 16319–16326. [Google Scholar] [CrossRef]
- Arias-Mayenco, I.; González-Rodríguez, P.; Torres-Torrelo, H.; Gao, L.; Fernández-Agüera, M.C.; Bonilla-Henao, V.; Ortega-Sáenz, P.; López-Barneo, J. Acute O2 Sensing: Role of Coenzyme QH2/Q Ratio and Mitochondrial ROS Compartmentalization. Cell Metab 2018, 28, 145–158.e4. [Google Scholar] [CrossRef]
- Lushchak, O.V.; Piroddi, M.; Galli, F.; Lushchak, V.I. Aconitase Post-Translational Modification as a Key in Linkage between Krebs Cycle, Iron Homeostasis, Redox Signaling, and Metabolism of Reactive Oxygen Species. Redox Rep 2014, 19, 8–15. [Google Scholar] [CrossRef]
- Brazzolotto, X.; Gaillard, J.; Pantopoulos, K.; Hentze, M.W.; Moulis, J.M. Human Cytoplasmic Aconitase (Iron Regulatory Protein 1) Is Converted into Its [3Fe-4S] Form by Hydrogen Peroxide in Vitro but Is Not Activated for Iron-Responsive Element Binding. J Biol Chem 1999, 274, 21625–21630. [Google Scholar] [CrossRef]
- Xu, Y.; Xue, D.; Kyani, A.; Bankhead, A.; Roy, J.; Ljungman, M.; Neamati, N. First-in-Class NADH/Ubiquinone Oxidoreductase Core Subunit S7 (NDUFS7) Antagonist for the Treatment of Pancreatic Cancer. ACS Pharmacol Transl Sci 2023, 6, 1164–1181. [Google Scholar] [CrossRef] [PubMed]
- Dunham-Snary, K.J.; Hong, Z.G.; Xiong, P.Y.; Del Paggio, J.C.; Herr, J.E.; Johri, A.M.; Archer, S.L. A Mitochondrial Redox Oxygen Sensor in the Pulmonary Vasculature and Ductus Arteriosus. Pflugers Arch - Eur J Physiol 2016, 468, 43–58. [Google Scholar] [CrossRef]
- Hameedi, M.A.; Grba, D.N.; Richardson, K.H.; Jones, A.J.Y.; Song, W.; Roessler, M.M.; Wright, J.J.; Hirst, J. A Conserved Arginine Residue Is Critical for Stabilizing the N2 FeS Cluster in Mitochondrial Complex I. Journal of Biological Chemistry 2021, 296, 100474. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, H. Mechanical Unfolding Pathway of the High-Potential Iron-Sulfur Protein Revealed by Single-Molecule Atomic Force Microscopy: Toward a General Unfolding Mechanism for Iron-Sulfur Proteins. J Phys Chem B 2018, 122, 9340–9349. [Google Scholar] [CrossRef] [PubMed]
- Crofts, A.R. The Modified Q-Cycle: A Look Back at Its Development and Forward to a Functional Model. Biochim Biophys Acta Bioenerg 2021, 1862, 148417. [Google Scholar] [CrossRef] [PubMed]
- Pethő, D.; Hendrik, Z.; Nagy, A.; Beke, L.; Patsalos, A.; Nagy, L.; Póliska, S.; Méhes, G.; Tóth, C.; Potor, L.; et al. Heme Cytotoxicity Is the Consequence of Endoplasmic Reticulum Stress in Atherosclerotic Plaque Progression. Sci Rep 2021, 11, 10435. [Google Scholar] [CrossRef]
- Piel, R.B.; Dailey, H.A.; Medlock, A.E. The Mitochondrial Heme Metabolon: Insights into the Complex(Ity) of Heme Synthesis and Distribution. Molecular Genetics and Metabolism 2019, 128, 198–203. [Google Scholar] [CrossRef]
- Belot, A.; Puy, H.; Hamza, I.; Bonkovsky, H.L. Update on Heme Biosynthesis, Tissue-Specific Regulation, Heme Transport, Relation to Iron Metabolism and Cellular Energy. Liver Int 2024, 44, 2235–2250. [Google Scholar] [CrossRef] [PubMed]
- Obi, C.D.; Bhuiyan, T.; Dailey, H.A.; Medlock, A.E. Ferrochelatase: Mapping the Intersection of Iron and Porphyrin Metabolism in the Mitochondria. Front Cell Dev Biol 2022, 10, 894591. [Google Scholar] [CrossRef]
- Baek, J.-H.; Gomez, I.G.; Wada, Y.; Roach, A.; Mahad, D.; Duffield, J.S. Deletion of the Mitochondrial Complex-IV Cofactor Heme A:Farnesyltransferase Causes Focal Segmental Glomerulosclerosis and Interferon Response. The American Journal of Pathology 2018, 188, 2745–2762. [Google Scholar] [CrossRef] [PubMed]
- Qiao, A.; Khechaduri, A.; Kannan Mutharasan, R.; Wu, R.; Nagpal, V.; Ardehali, H. MicroRNA-210 Decreases Heme Levels by Targeting Ferrochelatase in Cardiomyocytes. J Am Heart Assoc 2013, 2, e000121. [Google Scholar] [CrossRef] [PubMed]
- Oyedotun, K.S.; Sit, C.S.; Lemire, B.D. The Saccharomyces Cerevisiae Succinate Dehydrogenase Does Not Require Heme for Ubiquinone Reduction. Biochim Biophys Acta 2007, 1767, 1436–1445. [Google Scholar] [CrossRef]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, Function, and Assembly of Heme Centers in Mitochondrial Respiratory Complexes. Biochim Biophys Acta 2012, 1823, 1604–1616. [Google Scholar] [CrossRef]
- Crofts, A.R. The Cytochrome Bc1 Complex: Function in the Context of Structure. Annu. Rev. Physiol. 2004, 66, 689–733. [Google Scholar] [CrossRef]
- Llases, M.-E.; Morgada, M.N.; Vila, A.J. Biochemistry of Copper Site Assembly in Heme-Copper Oxidases: A Theme with Variations. Int J Mol Sci 2019, 20, 3830. [Google Scholar] [CrossRef]
- LeFort, K.R.; Rungratanawanich, W.; Song, B.-J. Contributing Roles of Mitochondrial Dysfunction and Hepatocyte Apoptosis in Liver Diseases through Oxidative Stress, Post-Translational Modifications, Inflammation, and Intestinal Barrier Dysfunction. Cell Mol Life Sci 2024, 81, 34. [Google Scholar] [CrossRef]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial Dysfunction in Type 2 Diabetes Mellitus: An Organ-Based Analysis. Am J Physiol Endocrinol Metab 2019, 316, E268–E285. [Google Scholar] [CrossRef] [PubMed]
- Pliouta, L.; Lampsas, S.; Kountouri, A.; Korakas, E.; Thymis, J.; Kassi, E.; Oikonomou, E.; Ikonomidis, I.; Lambadiari, V. Mitochondrial Dysfunction in the Development and Progression of Cardiometabolic Diseases: A Narrative Review. JCM 2025, 14, 3706. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Sauceda, C.; Webster, N.J.G. Mitochondrial Dysfunction in Obesity and Reproduction. Endocrinology 2021, 162, bqaa158. [Google Scholar] [CrossRef]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox Lipid Reprogramming Commands Susceptibility of Macrophages and Microglia to Ferroptotic Death. Nat Chem Biol 2020, 16, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Aust, S.D. The Requirement for Iron (III) in the Initiation of Lipid Peroxidation by Iron (II) and Hydrogen Peroxide. J Biol Chem 1987, 262, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Mas-Bargues, C.; Escrivá, C.; Dromant, M.; Borrás, C.; Viña, J. Lipid Peroxidation as Measured by Chromatographic Determination of Malondialdehyde. Human Plasma Reference Values in Health and Disease. Arch Biochem Biophys 2021, 709, 108941. [Google Scholar] [CrossRef]
- Panov, A.V.; Dikalov, S.I. Cardiolipin, Perhydroxyl Radicals, and Lipid Peroxidation in Mitochondrial Dysfunctions and Aging. Oxid Med Cell Longev 2020, 2020, 1323028. [Google Scholar] [CrossRef]
- Stavrovskaya, I.G.; Baranov, S.V.; Guo, X.; Davies, S.S.; Roberts, L.J.; Kristal, B.S. Reactive Gamma-Ketoaldehydes Formed via the Isoprostane Pathway Disrupt Mitochondrial Respiration and Calcium Homeostasis. Free Radic Biol Med 2010, 49, 567–579. [Google Scholar] [CrossRef]
- Mendoza, A.; Patel, P.; Robichaux, D.; Ramirez, D.; Karch, J. Inhibition of the mPTP and Lipid Peroxidation Is Additively Protective Against I/R Injury. Circ Res 2024, 134, 1292–1305. [Google Scholar] [CrossRef]
- Fefelova, N.; Wongjaikam, S.; Pamarthi, S.H.; Siri-Angkul, N.; Comollo, T.; Kumari, A.; Garg, V.; Ivessa, A.; Chattipakorn, S.C.; Chattipakorn, N.; et al. Deficiency of Mitochondrial Calcium Uniporter Abrogates Iron Overload-Induced Cardiac Dysfunction by Reducing Ferroptosis. Basic Res Cardiol 2023, 118, 21. [Google Scholar] [CrossRef]
- Hamilton, S.; Terentyeva, R.; Perger, F.; Hernández Orengo, B.; Martin, B.; Gorr, M.W.; Belevych, A.E.; Clements, R.T.; Györke, S.; Terentyev, D. MCU Overexpression Evokes Disparate Dose-Dependent Effects on Mito-ROS and Spontaneous Ca2+ Release in Hypertrophic Rat Cardiomyocytes. Am J Physiol Heart Circ Physiol 2021, 321, H615–H632. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, T.; Stockwell, B.R. The Development of the Concept of Ferroptosis. Free Radic Biol Med 2019, 133, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological Inhibition of Cystine-Glutamate Exchange Induces Endoplasmic Reticulum Stress and Ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Chen, B.; Fan, P.; Song, X.; Duan, M. The Role and Possible Mechanism of the Ferroptosis-Related SLC7A11/GSH/GPX4 Pathway in Myocardial Ischemia-Reperfusion Injury. BMC Cardiovasc Disord 2024, 24, 531. [Google Scholar] [CrossRef]
- Huang, B.; Wang, H.; Liu, S.; Hao, M.; Luo, D.; Zhou, Y.; Huang, Y.; Nian, Y.; Zhang, L.; Chu, B.; et al. Palmitoylation-Dependent Regulation of GPX4 Suppresses Ferroptosis. Nat Commun 2025, 16, 867. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front Pharmacol 2018, 9, 1371. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat Chem Biol 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Tang, D. Signaling Pathways and Defense Mechanisms of Ferroptosis. FEBS J 2022, 289, 7038–7050. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.-Q.; Zhao, L.; Hong, C.; Zou, Q.-M.; Su, J.-X.; Li, S.-J.; Zhou, X.-F.; Li, Z.-S.; Deng, B.; Cao, J.; et al. Baicalein Triggers Ferroptosis in Colorectal Cancer Cells via Blocking the JAK2/STAT3/GPX4 Axis. Acta Pharmacol Sin 2024, 45, 1715–1726. [Google Scholar] [CrossRef]
- Milhem, F.; Hamilton, L.M.; Skates, E.; Wilson, M.; Johanningsmeier, S.D.; Komarnytsky, S. Biomarkers of Metabolic Adaptation to High Dietary Fats in a Mouse Model of Obesity Resistance. Metabolites 2024, 14, 69. [Google Scholar] [CrossRef]
- Guan, H.; Xiao, L.; Hao, K.; Zhang, Q.; Wu, D.; Geng, Z.; Duan, B.; Dai, H.; Xu, R.; Feng, X. SLC25A28 Overexpression Promotes Adipogenesis by Reducing ATGL. J Diabetes Res 2024, 2024, 5511454. [Google Scholar] [CrossRef]
- Han, R.; Liu, L.; Wang, Y.; Wu, R.; Yang, Y.; Zhao, Y.; Jian, L.; Yuan, Y.; Zhang, L.; Gu, Y.; et al. Microglial SLC25A28 Deficiency Ameliorates the Brain Injury After Intracerebral Hemorrhage in Mice by Restricting Aerobic Glycolysis. Inflammation 2024, 47, 591–608. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V. Nonalcoholic Steatohepatitis Clinical Research Network Relationship between the Pattern of Hepatic Iron Deposition and Histological Severity in Nonalcoholic Fatty Liver Disease. Hepatology 2011, 53, 448–457. [Google Scholar] [CrossRef]
- Valenti, L.; Fracanzani, A.L.; Dongiovanni, P.; Bugianesi, E.; Marchesini, G.; Manzini, P.; Vanni, E.; Fargion, S. Iron Depletion by Phlebotomy Improves Insulin Resistance in Patients with Nonalcoholic Fatty Liver Disease and Hyperferritinemia: Evidence from a Case-Control Study. Am J Gastroenterol 2007, 102, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Anastasopoulos, N.-A.; Barbouti, A.; Goussia, A.C.; Christodoulou, D.K.; Glantzounis, G.K. Exploring the Role of Metabolic Hyperferritinaemia (MHF) in Steatotic Liver Disease (SLD) and Hepatocellular Carcinoma (HCC). Cancers (Basel) 2025, 17, 842. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Kim, M.S.; Han, S.N. Diet-Induced Obesity Leads to Decreased Hepatic Iron Storage in Mice. Nutr Res 2011, 31, 915–921. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Lanti, C.; Gatti, S.; Rametta, R.; Recalcati, S.; Maggioni, M.; Fracanzani, A.L.; Riso, P.; Cairo, G.; Fargion, S.; et al. High Fat Diet Subverts Hepatocellular Iron Uptake Determining Dysmetabolic Iron Overload. PLoS One 2015, 10, e0116855. [Google Scholar] [CrossRef]
- Ravingerová, T.; Kindernay, L.; Barteková, M.; Ferko, M.; Adameová, A.; Zohdi, V.; Bernátová, I.; Ferenczyová, K.; Lazou, A. The Molecular Mechanisms of Iron Metabolism and Its Role in Cardiac Dysfunction and Cardioprotection. Int J Mol Sci 2020, 21, 7889. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Romano, R.; Coelho-Júnior, H.J.; Bucci, C.; Marzetti, E. Mitochondrial Dysfunction, Protein Misfolding and Neuroinflammation in Parkinson’s Disease: Roads to Biomarker Discovery. Biomolecules 2021, 11, 1508. [Google Scholar] [CrossRef]
- Flønes, I.H.; Toker, L.; Sandnes, D.A.; Castelli, M.; Mostafavi, S.; Lura, N.; Shadad, O.; Fernandez-Vizarra, E.; Painous, C.; Pérez-Soriano, A.; et al. Mitochondrial Complex I Deficiency Stratifies Idiopathic Parkinson’s Disease. Nat Commun 2024, 15, 3631. [Google Scholar] [CrossRef]
- Wang, W.; Thomas, E.R.; Xiao, R.; Chen, T.; Guo, Q.; Liu, K.; Yang, Y.; Li, X. Targeting Mitochondria-Regulated Ferroptosis: A New Frontier in Parkinson’s Disease Therapy. Neuropharmacology 2025, 274, 110439. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Heme Oxygenase-1: Role in Brain Aging and Neurodegeneration. Exp Gerontol 2000, 35, 821–830. [Google Scholar] [CrossRef]
- Lurette, O.; Martín-Jiménez, R.; Khan, M.; Sheta, R.; Jean, S.; Schofield, M.; Teixeira, M.; Rodriguez-Aller, R.; Perron, I.; Oueslati, A.; et al. Aggregation of Alpha-Synuclein Disrupts Mitochondrial Metabolism and Induce Mitophagy via Cardiolipin Externalization. Cell Death Dis 2023, 14, 729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int J Biol Sci 2021, 17, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- An, X.; He, J.; Xie, P.; Li, C.; Xia, M.; Guo, D.; Bi, B.; Wu, G.; Xu, J.; Yu, W.; et al. The Effect of Tau K677 Lactylation on Ferritinophagy and Ferroptosis in Alzheimer’s Disease. Free Radic Biol Med 2024, 224, 685–706. [Google Scholar] [CrossRef]
- Monteiro-Cardoso, V.F.; Oliveira, M.M.; Melo, T.; Domingues, M.R.M.; Moreira, P.I.; Ferreiro, E.; Peixoto, F.; Videira, R.A. Cardiolipin Profile Changes Are Associated to the Early Synaptic Mitochondrial Dysfunction in Alzheimer’s Disease. J Alzheimers Dis 2015, 43, 1375–1392. [Google Scholar] [CrossRef]
- Chen, Z.; Zheng, N.; Wang, F.; Zhou, Q.; Chen, Z.; Xie, L.; Sun, Q.; Li, L.; Li, B. The Role of Ferritinophagy and Ferroptosis in Alzheimer’s Disease. Brain Res 2025, 1850, 149340. [Google Scholar] [CrossRef]
Figure 1.
Mechanisms by which ferroptosis could be produced. The pathways for eliminating LPO products (represented as PUFA-OOH●) include system Xc-, GSH/GPX4 axis, and Fenton reaction. Cystine uptake is produced by system Xc-, is catalyzed to GSH/GSS recycling. GPX4 converts GSH to GSSH, reducing LPO and inhibiting ferroptosis. Main inhibitors are shown. PUFA, polyunsaturated fatty acids; LD, lipid droplet; ACSL4, acyl-CoA synthetase long-chain family member; GR, glutathione reductase; LPCAT3, lysophosphatidylcholine acyltransferase 3; GPX4, glutathione peroxidase 4.
Figure 1.
Mechanisms by which ferroptosis could be produced. The pathways for eliminating LPO products (represented as PUFA-OOH●) include system Xc-, GSH/GPX4 axis, and Fenton reaction. Cystine uptake is produced by system Xc-, is catalyzed to GSH/GSS recycling. GPX4 converts GSH to GSSH, reducing LPO and inhibiting ferroptosis. Main inhibitors are shown. PUFA, polyunsaturated fatty acids; LD, lipid droplet; ACSL4, acyl-CoA synthetase long-chain family member; GR, glutathione reductase; LPCAT3, lysophosphatidylcholine acyltransferase 3; GPX4, glutathione peroxidase 4.
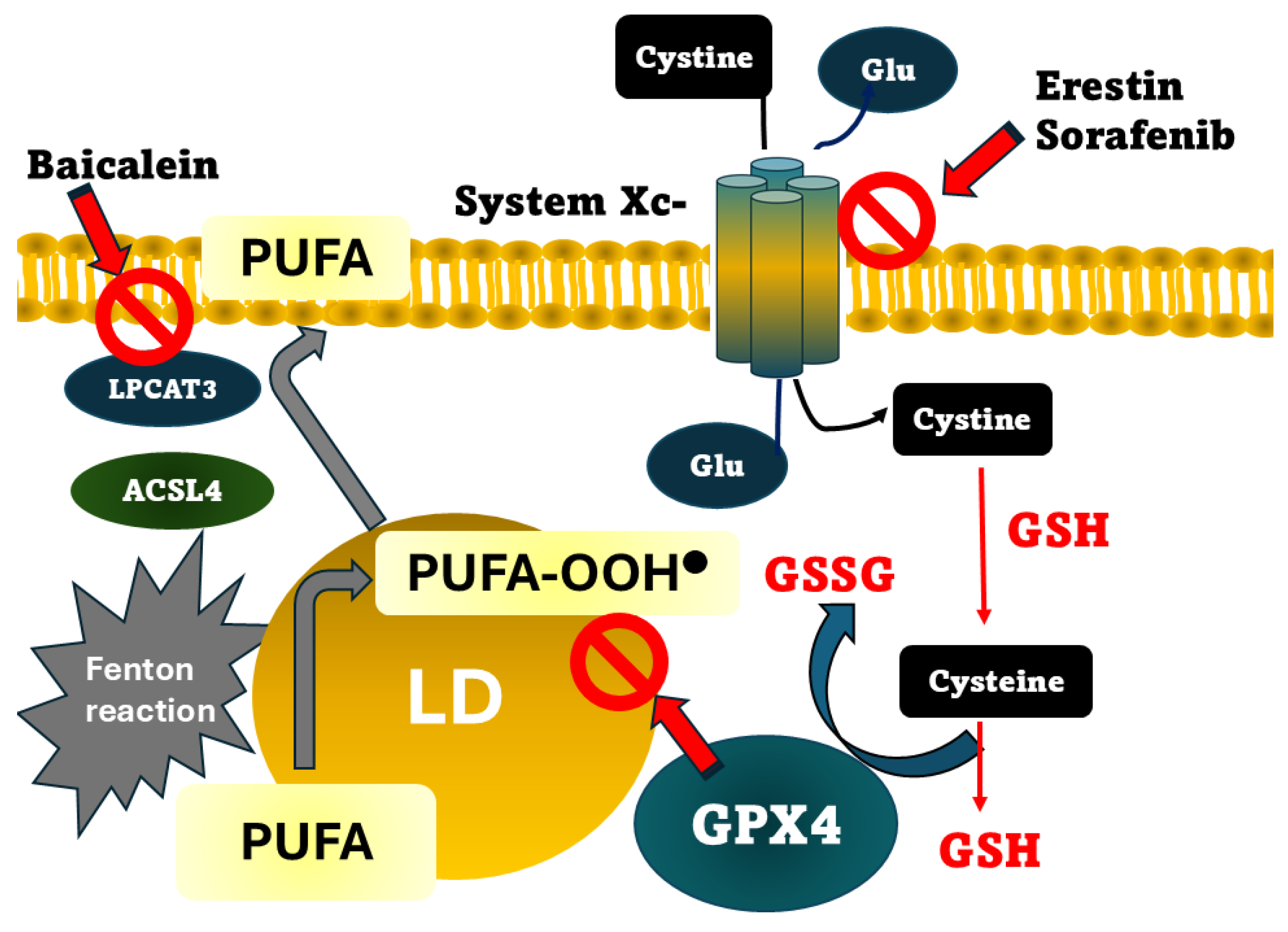
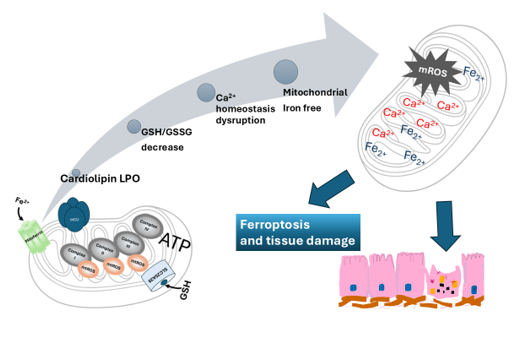
Figure 2.
Hallmarks of mitochondrial dysfunction associated with mitochondrial iron homeostasis disturbance. The current picture shows that mitochondrial iron dysregulation is a primary trigger of cardiolipin peroxidation and increased mtROS production. Additionally, Ca2+ overload and a significantly decreased GSH/GSSG ratio are depicted as critical hallmarks of this pathological cycle. These interconnected dysfunctions are associated with the progression of neurodegenerative diseases, obesity, and MASLD.
Figure 2.
Hallmarks of mitochondrial dysfunction associated with mitochondrial iron homeostasis disturbance. The current picture shows that mitochondrial iron dysregulation is a primary trigger of cardiolipin peroxidation and increased mtROS production. Additionally, Ca2+ overload and a significantly decreased GSH/GSSG ratio are depicted as critical hallmarks of this pathological cycle. These interconnected dysfunctions are associated with the progression of neurodegenerative diseases, obesity, and MASLD.
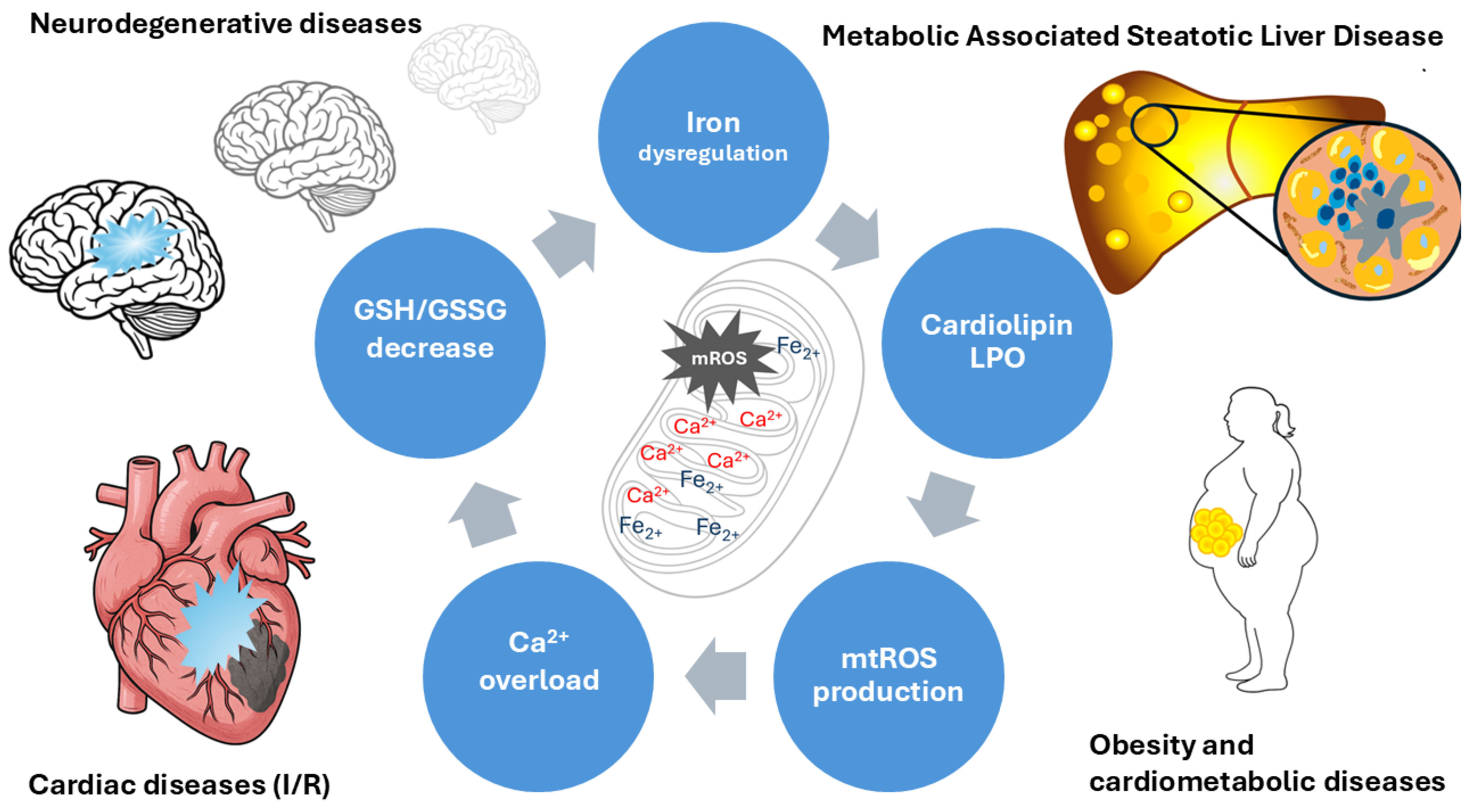

Table 1.
Fe-S clusters relevant for mitochondrial metabolism.
| Cluster | Mitochondrial location | Metabolic function | References |
| [2Fe-2S] | Complex I (N1a, N1b clusters) | Electron Transfer: Mediates a single electron jumps within the ETC and key redox-active components. | [19,20] |
| Complex II | Involved in the biosynthesis of steroids, heme and lipoyl cofactors. | [21,22] | |
| Complex III (Rieske protein) | The Rieske cluster moves physically to facilitate electron transfer from ubiquinol to cytochrome c. Tune the activity of monooxygenase TsaM. | [23,24] | |
| Mitochondrial matrix | Regulator: Molecular sensors (Cysteine Desulfurase 1, NFS1; Iron-Sulfur Cluster Scaffold Protein, ISCU and Glutaredoxin-related protein 5, GLRX5) incorporated into SLC25A39 (GSH transport) | [15,25] | |
| [3Fe-4S] | Complex II (terminal cluster) |
Electron Transfer: Aligned near the quinone binding site in Complex II. | [26] |
| Mitochondrial Aconitase (inactive form) | Redox Sensing: The inactive aconitase contains this cluster; it transitions to the [4Fe-4S] upon acquiring a labile iron atom. | [27,28] | |
| [4Fe-4S] | Complex I (N2, N3, N4, N5, N6a, N6b clusters) | Enzyme Catalysis: catalyzes the conversion of citrate to isocitrate via aconitase in the Krebs cycle | [29,30] |
| Complex II (middle cluster Mitochondrial) |
Oxygen Sensing: The N2 cluster in subunit Ndufs2 acts as a redox-sensitive oxygen sensor. | [31] | |
| Aconitase (active form) | Electron Tunneling: Forms a tunneling chain over 95 Å in the Complex I to drive proton pumping | [32,33] | |
| Cluster N2 | Complex I (Subunits NDUFS7/NDUFS2) | Terminal Sink: Acts as the high-potential electron sink that reduces ubiquinone to ubiquinol | [34] |
| Homeostatic oxygen sensing system (HOSS) Regulation: Vital for homeostatic oxygen-sensing systems in pulmonary arteries and the carotid body | [35,36] | ||
| Rieske | Complex III (Iron Sulfur Protein) | Bifurcated Electron Flow: Participates in the “high potential pathway” of the Q-cycle, transferring electrons to cytochrome c1 | [37,38] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



