
Submitted:
22 February 2024
Posted:
29 February 2024
You are already at the latest version
Abstract
Ferroptosis, a regulated form of cell death characterized by iron-dependent lipid peroxide accumulation, plays a pivotal role in various pathological conditions, including neurodegenerative diseases. While reasonable evidence for ferroptosis exist e.g. in Parkinson’s disease or Alzheimer’s disease, there are only few reports on amyotrophic lateral sclerosis (ALS), a fast progressive and incurable neurodegenerative disorder characterized by progressive motor neuron degeneration. Initial studies, however, highlight that ferroptosis might be significantly involved in ALS. Key features of ferroptosis include oxidative stress, glutathione depletion, mitochondrial and dysfunction alterations in mitochondrial morphology, mediated by proteins such as GPX4, xCT, ACSL4 FSP1, Nrf2 and TfR1. Induction of ferroptosis involves small molecule compounds like erastin and RSL3, which disrupt system Xc- and GPX4 activity, respectively, yielding in lipid peroxidation and cellular demise. Mutations in Fused in Sarcoma (FUS) are associated with familial ALS. Pathophysiological hallmarks of FUS-ALS involve mitochondrial dysfunction and oxidative damage, implicating ferroptosis as putative cell death pathway in motor neuron demise. However, mechanistic understanding of ferroptosis in ALS, particularly FUS-ALS, remains limited. Here, we investigated ferroptosis vulnerability in a FUS-ALS cell model, revealing mitochondrial disturbance and increased susceptibility for ferroptosis in cells harboring the mutant FUS variant. This was accompanied by altered expression of ferroptosis-associated proteins, particularly by dramatic reduction of xCT expression, leading to cellular imbalance of the redox system and increased lipid peroxidation. Iron chelation with deferoxamine as well as inhibition of the mitochondrial calcium uniporter (MCU) significantly alleviated ferroptotic cell death and lipid peroxidation. These findings suggest a link between ferroptosis and FUS-ALS, offering potential therapeutic targets.
Keywords:
amyotrophic lateral sclerosis
; oxidative damage
; mitochondria
; cell death
1. Introduction
Ferroptosis is a form of regulated cell death that is characterized by the iron-dependent accumulation of lipid peroxides (Dixon, Lemberg et al. 2012). This process involves the overwhelming buildup of reactive oxygen species (ROS), particularly lipid peroxides, leading to cellular damage and death (Dixon, Lemberg et al. 2012, Friedmann Angeli, Schneider et al. 2014, Stockwell, Friedmann Angeli et al. 2017). Key features of ferroptosis include the iron-dependent nature of the cell death mechanism, oxidative stress, the depletion of the endogenous antioxidant glutathione (GSH), and alterations in mitochondrial morphology and function (Dixon, Lemberg et al. 2012, Stockwell, Friedmann Angeli et al. 2017).
Numerous proteins, such as glutathione peroxidase 4 (GPX4), cystine-glutamate transporter (system Xc -), ferroptosis suppressor protein-1 (FSP1), LOX, nuclear factor erythroid 2-related factor 2 (Nrf2), and Transferrin receptor protein 1 (TfR1), govern ferroptosis by modulating both iron metabolism and lipid peroxidation (Dixon, Lemberg et al. 2012, Angeli, Shah et al. 2017, Doll, Proneth et al. 2017, Forcina and Dixon 2019, Shi, Zhang et al. 2021). Induction of the ferroptotic cell death can be initiated by small molecule compounds such as erastin or RSL3 (Carracedo, Prieto et al. 1987, Tang and Kroemer 2020). Erastin is an extracellular inhibitor of the glutamate/cystine antiporter system Xc- (xCT), the foremost regulator in the ferroptosis molecular pathway (Carracedo, Prieto et al. 1987, Dixon, Patel et al. 2014).
Inhibition of the system Xc- results in the depletion of cysteine and subsequent depletion of GSH synthesis as cysteine is the rate-limiting factor in GSH synthesis (Carracedo, Prieto et al. 1987, Dixon, Lemberg et al. 2012, Dixon, Patel et al. 2014). Consequently, the reduction in GSH levels diminishes the cellular antioxidant response, thereby leading to an elevation in ROS levels and the induction of caspase-independent cell death (Fleury, Mignotte et al. 2002, He, He et al. 2017). GSH depletion impairs the activity of antioxidant enzymes, such as GPX4. In addition, ferroptosis can be induced by the direct inhibition of GPX4 activity (target of RSL-3), allowing the accumulation of lipid peroxides, which ultimately leads to ferroptotic cell death (Dixon, Lemberg et al. 2012, Angeli, Shah et al. 2017, Stockwell, Friedmann Angeli et al. 2017).
Consequently, various compounds have been formulated, including liproxstatin-1 (Lip-1) and ferrostatin-1 (fer-1), which inhibit ferroptosis by interfering with lipid peroxidation and the associated oxidative stress (Yang, SriRamaratnam et al. 2014, Gleitze, Paula-Lima et al. 2021). Additionally, as ferroptosis is an iron-dependent form of cell death, iron chelators like deferoxamine (DFO) were shown to reduce ferroptosis, (Dixon, Lemberg et al. 2012, Li, Cao et al. 2020, Gleitze, Paula-Lima et al. 2021). Interestingly, it has long been known that abnormalities of the iron metabolism are associated with neurodegenerative diseases and iron chelators such as DFO are subject of clinical trials in different neurodegenerative diseases.
Hallmarks of ferroptosis involve changes in mitochondrial structure and function, encompassing mitochondrial fragmentation, elevated levels of mitochondrial calcium ([Ca2+]m), heightened production of mitochondrial ROS, lipid peroxidation, and perturbation of the mitochondrial membrane potential (ΔΨm). (Munoz, Humeres et al. 2011, Sanmartin, Paula-Lima et al. 2014, Maher, van Leyen et al. 2018, Gleitze, Paula-Lima et al. 2021). Calcium ions play important roles in various cellular processes, including signaling and cell death pathways (Grossmann, Malburg et al. 2023). Lipid peroxidation and dysregulation in cytosolic Ca2+ levels are associated with ferroptosis induction. (Maher, van Leyen et al. 2018, Pedrera, Espiritu et al. 2021). Being the main entry point of Ca2+ into the mitochondrial matrix, the mitochondrial calcium uniporter (MCU) is crucial for mitochondrial calcium level control (Grossmann, Malburg et al. 2023, Marmolejo-Garza, Krabbendam et al. 2023). Inhibition of the MCU with e.g. Ru265, a modified ruthenium compound, is known to exhibit potential neuroprotective effects against hypoxic/ischemic (HI) brain injury. A recent study reported that different inhibitors of the MCU, including Ru265, were able to alleviate erastin-mediated ferroptotic cell death in diverse cell types. However, the potential anti-ferroptotic effects of MCU inhibition in neurodegeneration and particularly in (FUS-) ALS have not been evaluated so far.
Amyotrophic lateral sclerosis (ALS) is a progressive and ultimately fatal neurodegenerative disease marked by the deterioration of upper and lower motor neurons in regions like the motor cortex, brainstem, and spinal cord, leading to mortality typically within 2–5 years of diagnosis (Brown and Al-Chalabi 2017, Wang, Tomas et al. 2022). While the majority of ALS cases are sporadic, about 10-15% are caused by monogenic mutations in several genes, with the most common ones including Superoxide dismutase 1 (SOD1), Tar DNA binding protein (TARDBP), Chromosome 9 open frame 72 (C9ORF72), and Fused in Sarcoma (FUS) (Chen, Wardill et al. 2013, Wang, Tomas et al. 2022). FUS mutations account for the third most common genetic variant, contributing to about 5% of familial ALS cases and up to 1% of sporadic ALS (sALS) cases, but even more in juvenile onset cases (Naumann, Peikert et al. 2019).While the pathophysiology of ALS is not yet fully understood, various cellular and molecular processes were reported to be disturbed, including glutamatergic excitotoxicity, mitochondrial dysfunction, oxidative stress (Turner, Hardiman et al. 2013, Greco, Longone et al. 2019, Valko and Ciesla 2019), all of which are hallmarks of ferroptosis. While ferroptosis has been investigated in many disease conditions including Parkinson’s disease, surprisingly few studies exist on ferroptosis in ALS. Genetic depletion of GPX4, a central repressor of ferroptosis, induced motor phenotypes suggestive for motor neuron diseases accompanied by a more or less selective motor neuron degeneration (Chen, Hambright et al. 2015). GPX4 was reported to be reduced in spinal cord lysates of sporadic and familial ALS patients (Wang, Tomas et al. 2022). GPX4 overexpression in hSOD1G93A mice resulted in delayed disease onset and prolonged survival (Wang, Tomas et al. 2022). In hSOD1-G93A overexpressing cells and mice lipid peroxidation was reported to be induced by TFR1-imported excess free iron, accompanying decreased GSH and mitochondrial membrane dysfunction (Wang, Liang et al. 2023). No data exist so far for FUS-ALS, despite FUS-ALS pathophysiology is particularly characterized by oxidative and mitochondrial damage (Kreiter, Pal et al. 2018, Naumann, Pal et al. 2018, Pal, Glaß et al. 2018, Pal, Kretner et al. 2021, Gunther, Pal et al. 2022).
We thus aimed to investigate the vulnerability to ferroptosis in FUS-ALS using Hela cells, which had been edited to carry BACs with either WT-FUS-eGFP or FUS-P525L-eGFP as previously reported (Patel, Lee et al. 2015).. We systematically investigated cell vulnerability against ferroptosis inducers and inhibitors, cellular redox system and lipid peroxidation in wildtype and FUS-ALS mutant cells. Overall, our study not only highlights the heightened vulnerability of FUS-ALS cells to induced ferroptosis but also provides insights into the potential molecular mechanisms underlying this susceptibility through comprehensive analysis of lipid peroxide levels, expression profiles of ferroptosis-associated proteins, and glutathione levels.
2. Material and Methods
2.1. Cell Culture
HeLa cell lines with either WT-FUS-eGFP or FUS-P525L-eGFP were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS, 1% Pen/Strep; Thermo Fisher Scientific, Braunschweig, Germany). All the experimental cells tested on a monthly base for Mycoplasma contamination using the Plasmo Test_Detection Kit (InvivoGen).
2.2. Live Cell Imaging
For analysis of the mitochondrial membrane potential (MMP) and lipid peroxidation, FUS-eGFP HeLa cells were seeded into 8 well µ-slides (Ibidi: 80806-90) at a density of 10,000 cells per well. Cells were grown in DMEM + 10% FBS + 1% Pen/Strep for 24 hours. On the day of the MMP measurement, cells were stained with 50 nM Tetramethylrhodamine Ethyl Ester (TMRE; Invitrogen: T669) and 1 nM MitoTracker Deep Red (Invitrogen: M22426) for 30 min at 37°C. Live-cell imaging was performed on a Zeiss inverted AxioObserver.Z1 microscope with LSM 900 module, 63x oil high-resolution objective, at 37°C and 5% CO2. While for analysis of the lipid peroxidation, cells were treated with specific concentrations of different compounds including (1S, 3R)-RSL3 (Merk, SML2234), erastin (Merk, 329600-5M) and Ru265 (Merck/Sigma-Aldrich, SML2991) for 24 h. The level of lipid peroxidation within the cell was assessed using the fluorescent dye boron-dipyrromethene BODIP 665/676 (Thermo Fisher, B3932, Germany). On the next day, the growth medium containing different compounds was removed from the wells and cells were incubated with equal volumes of normal growth medium. Afterwords, cells were stained with 2 µM BODIPY 665/676 dye for 30 min at 37°C. Live-cell imaging was conducted on a Zeiss inverted AxioObserver.Z1 microscope with LSM 900 module, using 10x resolution objective, and full environment control of 37°C and 5% CO2. Image analysis was performed using ImageJ. Data acquired from live-cell imaging were evaluated using macros in Fiji. Masks are depicted in the respective figures. Statistical analysis was performed using GraphPad Prism 8 software. Data shown represent the mean ± s.d. of n=3, (one-way ANOVA) was used for all experiments to determine statistical differences between groups. *P < 0.001, was considered significant. Data values represent mean ± SDTEV unless indicated otherwise.
2.3. PrestoBlue Cell Viability Assay
To analyse cell viability, cells were seeded into 96-well plate (735-0465, VWR) at a density of (2000 cells/well) in 100 μL of medium per well in triplicate per each group treatment and incubated overnight at 37°C with 5 % CO2. The next day, cells were treated with increasing concentrations of different compounds including (1S, 3R)-RSL3, and erastin with and without liproxstatin-1 (Lip-1) and Deferoxamine (DFO) for 24 h. The cell viability was analyzed using PrestoBlue Viability Assay (Thermo Fisher Scientific) according to the manufacturer’s protocol. Resazurin, the active ingredient of PrestoBlue reagent, is converted to resorufin in the intracellular reducing environment of live cells. Conversion of resazurin to resorufin changes its color and absorbance. PrestoBlue diluted 1:10 with culture medium was incubated with the cells at 37°C with 5% CO2 for 15 min. Absorbance at (560 nm excitation and 590 nm emission) was read on Microplate Reader (CM Sunrise™, Tecan) after 15 min incubation at 37°C with 5 % CO2. Measurements within each line were normalized to the average values of untreated treated wells of each cell line.
2.4. Measurement of GSH Levels
A luciferase-based assay used to analyze total glutathione (GSH) and oxidized glutathione (GSSG) levels in the experimental cell lines. The assay was performed following the protocol of Promega® GSH/GSSG Assay. Cells were seeded into 96-well plate (735-0465, VWR) at a density of (2000 cells/well) in 100 μL of medium per well. The no-cells control was used to normalise the background signal from the assay chemistry, which were then subtracted from vehicle and test signals to give net values. Absorbance at (555 nm excitation and 565 nm emission) was read on Microplate Reader (CM Sunrise™, Tecan) with in the incubation time.
2.5. Immunoblotting
Cells were collected via trypsinization and centrifuged at 4 °C for 15 min at max speed. Supernatant was transferred to a new tube and stored at −20 °C until further use. Cells were washed with 1x PBS and lysed in RIPA buffer, containing 125mM NaCL (1.06404.1000; EMSURE), 25mM Tris-Cl (pH 7.4) 1% Triton X-100 (X100-500M; Sigma-Aldrich), 0.5% sodium deoxycholate (D6750- 500G; Sigma-Aldrich), 0.1% SDS (L5750-1KG; Sigma-Aldrich) and 1x complete EDTA-free protease inhibitors. Protein amount was measured by BCA assay (Thermo Fisher Scientific). Immunoblot analysis was performed using the 12 % SDS gel was transferred to a ready to use Trans-Blot® Turbo™ PVDF membranes (Bio-Rad). Afterwords, protein transfer was conducted using the standard program (30 min, 25 V, 1 A) in the semi-dry Trans-Blot® Turbo™ Transfer System (Bio-Rad). After that, the blotting cassette were disassembled, and the membranes were placed in chamber and washed three time with TBS-T before blocking was started. The membranes were then incubated under constant shaking at room temperature for 1 h in blocking buffer (5% non-fat milk) dissolved in Tris-Buffered Saline supplemented with 0.1% Tween-20 (TBS-T). Next, the membranes were then incubated with primary antibody diluted according to manufacturer`s protocol in the corresponding blocking solution for an overnight incubation at 4°C on a shaker. Next day three washes were carried out with TBS-T for 5 minutes at room temperature. The membrane was then incubated with secondary antibody diluted according to manufacturer`s protocol at room temperature for 1 hour on a shaker. Finally, the membranes were washed again 3 times with TBS-T for 5 minutes. To visualise, the membrane was placed in a transparent plastic wrap and Clarity™ Western ECL Blotting Substrate (Bio-Rad) was applied to the membrane for 1 min. The excess reagent was removed, and the blot was scanned using an imaging system from LI-COR Biosciences. To redevelop with different antibody, antibodies were stripped from the PVDF membrane with 0.4 M NaOH for 7 min. After all, protein bands were quantified using the Image Lab software (Bio-Rad).Data acquired from live-cell imaging were evaluated using ImageJ. Statistical analysis was performed using GraphPad Prism 8 software. Data shown represent the mean ±s.d. of n=3, (two-way ANOVA) was used for all experiments to determine statistical differences between groups and *P < 0.001, was considered significant.
2.6. Statistics
All data displayed are mean ± s.d. unless otherwise indicated in the figure legend. Statistical analysis was conducted using GraphPad Prism 8.0 software. All experiments were independently repeated at least three times (n represents the number of independent biological replicates).
3. Results
3.1. Mitochondrial Depolarization in FUS -Mutated Cells
Mitochondrial dysfunction is a characteristic of ferroptosis. FUS-ALS pathophysiology was associated with significant mitochondrial dysfunction. Specifically, axonal mitochondrial transport was shown to be impaired as well as mitochondrial depolarization was obvious together with impacted metabolic state of FUS-mutated neurons (Naumann, Pal et al. 2018, Zimyanin, Pielka et al. 2023). We first investigated whether this mitochondrial phenotype is also seen in HeLa cells engineered to carry either wildtype FUS-eGFP or P525L FUS-eGFP. Thus, we investigated whether or not the inner membrane potential – known to be an important indicator of proper mitochondrial function – is disturbed in FUS mutant HeLa lines.
For this, we used Tetramethylrhodamine ethyl ester (TMRE), a cell-permeable fluorescent dye that accumulates in active mitochondria, as a marker for mitochondrial inner membrane potential relevant for mitochondrial integrity, relative to Mitotracker deep red. Hela-FUS-P525L cells showed an almost halved membrane potential compared to Hela-FUS-WT cells (Figure 1A), thereby confirming the mitochondrial dysfunction being specifically associated with FUS-ALS across different cell types.
3.2. Increased Sensitivity to Ferroptosis in FUS Mutated Cells
Ferroptosis leads to the reduction in cell viability via inducing oxidative cell death (Dixon, Lemberg et al. 2012). Having shown mitochondrial depolarization in FUS-ALS mutants, we hypothesised that ferroptosis might be significantly involved in FUS-ALS pathophysiology. Glutathione peroxidase 4 (GPX4) is a key enzyme of ferroptosis, mediating lipid peroxidation (Conrad and Friedmann Angeli 2015), whereas system Xc- cystine/glutamate antiporter (xCT), plays a key role in controlling the intracellular redox state. Inhibition of xCT leads to upregulation of ROS production upstream of GPX4 (Lewerenz, Hewett et al. 2013).
To investigate the vulnerability of FUS-mutated cells to ferroptosis, we conducted a comprehensive investigation into the dose-dependent response of cell death induced by RSL3 and erastin, two well-known ferroptosis inducers either interfering with GPX4 or xCT, respectively. Following treatment with varying concentrations of RSL3 and erastin for a duration of 24 hours, both HeLa-FUS-P525L and HeLa-FUS-WT cells exhibited a notable change in morphology suggestive for cell death (Figure 2A, C) and cell viability loss in a dose-dependent manner (Figure 2B, D). Cell morphology changes and cell proliferation was significantly reduced in the treated cells, particularly in HeLa-FUS-P525L (Figure 2A, C). Of note, HeLa-FUS-P525L cells were significantly more vulnerable against ferroptosis induction compared to HeLa-FUS-WT cells (Figure 2B, D). These findings suggests that FUS mutant cells exhibit heightened vulnerability to key ferroptosis inducers.
3.3. Liproxstatin-1 Suppressed Ferroptosis in HeLa-FUS Cells
Liproxstatin-1 (Lip-1) is a pharmacological agent known for its role in mitigating cell death induced by ferroptosis inducers such as RSL3 and erastin (Friedmann Angeli, Schneider et al. 2014).
To further substantiate that ferroptosis significantly contributes to cell death, we aimed at elucidating whether ferroptosis-selective inhibitors such as Lip-1 can counteract cell death, we conducted co-treatment experiments using both HeLa-FUS-P525L and HeLa-FUS-WT cells (Figure 3).
These experiments involved subjecting the cells to varying concentrations of both erastin and RSL3, with and without a single concentration of Lip-1 (Figure 3). Intriguingly, our results unveiled that ferroptotic cell death in both cell lines induced by different concentrations of the xCT inhibitor erastin was completely inhibited at concentrations of (500 nM) Lip-1 (Figure 3A, B). Similar inhibitory effect was observed at concentrations of (500 nM) Lip-1 in both cell lines treated with variable concentration of GXP4 inhibitor RSL3 (Figure 3C, D). These results further suggest a significant role of ferroptosis pathway in cellular demise in FUS-ALS.
3.4. FUS Mutated Cells Exhibit Misregulation of Key Factors of Ferroptosis
To further elucidate possible explanations for the increased vulnerability against ferroptosis in FUS-ALS, we next conducted an analysis of the expression of main ferroptosis proteins GPX4, xCT, ACSL4, and FSP1 (Figure 4A, B). GPX4 knockout studies revealed motor neuron disease phenotypes as consequence of (selective) motor neuron loss (Chen, Hambright et al. 2015). Furthermore, GPX4 was reported to be reduced in spinal cord lysates of sporadic and familial ALS patients (Wang, Tomas et al. 2022).
Interestingly, the immunoblot analysis indicated only a non-significant decrease in the protein expression levels of GPX4, but a significant reduction of xCT, and FSP1 expression, while there was an increase in ACSL4 protein expression in HeLa-FUS-P525L cells compare to HeLa-FUS-WT cells (Figure 4A, B).
3.5. FUS Mutations Lead to Disturbance of Cellular Redox Defense System Downstream of xCT
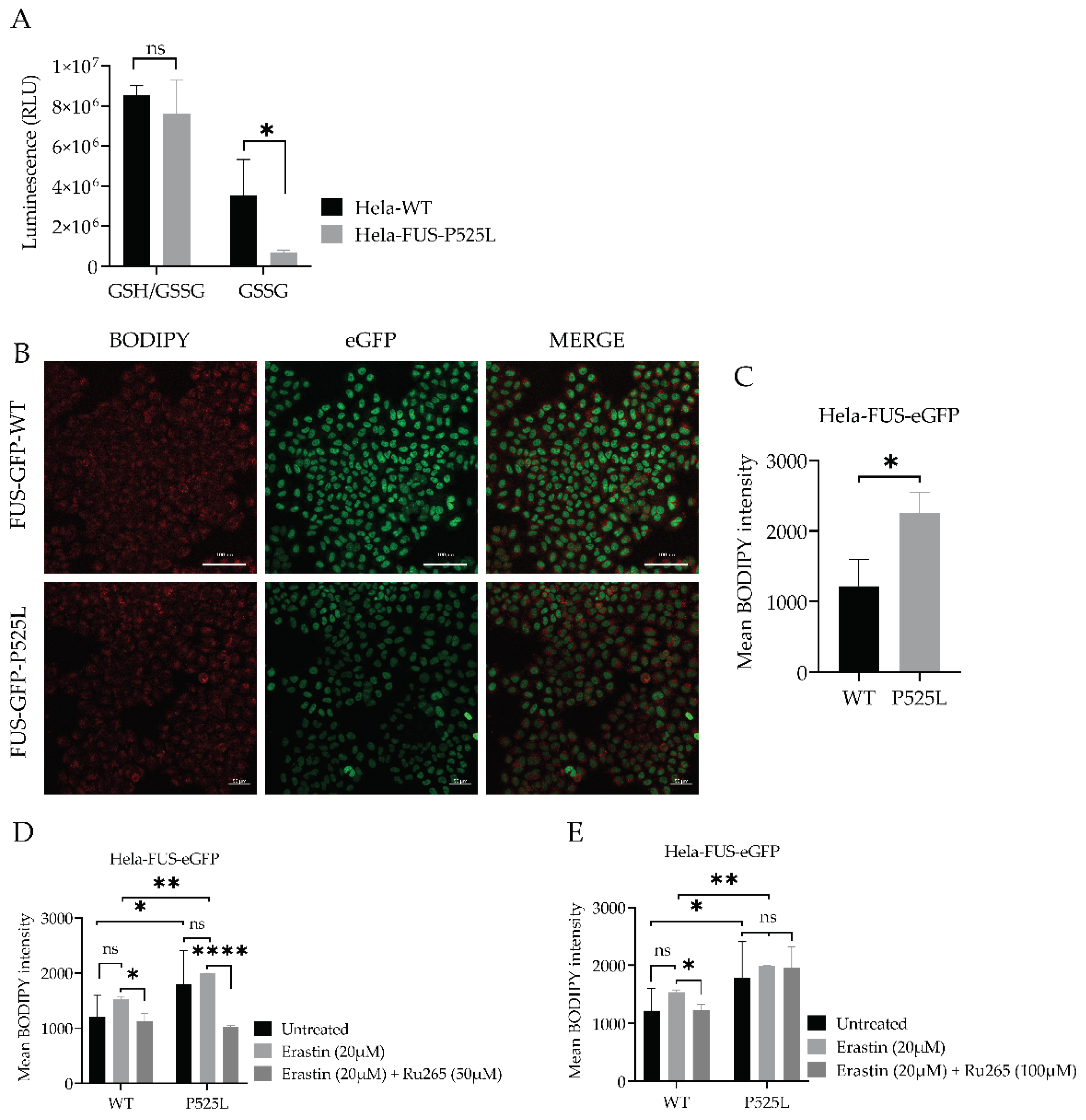
Having shown that xCT is dramatically reduced in FUS ALS cells, we analysed this pathway further. xCT plays a crucial role in the cellular uptake of cystine, which is necessary for the synthesis of GSH, an important antioxidant in cells (Dixon, Lemberg et al. 2012). Ferroptosis is an iron- and ROS-dependent form of regulated cell death. Free iron is a strong oxidant, able to generate highly reactive hydroxyl radicals, especially through the Fenton reaction. DFO categorized as iron chelator, has gained attention for its ability to reduce the availability of free iron within the intracellular environment. By curtailing the Fenton reaction, a pivotal mechanism driving lipid peroxidation and consequent cell demise by ferroptosis, DFO has shown promise to reduce ferroptotic cell death in various studies (Chen, Yu et al. 2020). We thus asked whether DFO is able to restore ferroptosis in FUS-ALS cells as well. We conducted co-treatment experiments using both HeLa-FUS-P525L and HeLa-FUS-WT cells (Figure 4). Our results, revealed that the induction of ferroptotic cell death in both cell lines by variable concentrations of erastin was prevented when treated with (100 uM) DFO (Figure 4C, D). These outcomes underline the significant contribution of the ferroptosis induced cell death observed in FUS-ASL. Downregulation of xCT results in decreased cellular uptake of cystine, leading to reduced synthesis of GSH. Glutathione is a key antioxidant that helps neutralizing ROS and protects cells from oxidative damage (Kwon, Cha et al. 2019). However, when cellular levels of reduced glutathione is depleted or compromised, the balance between oxidants and antioxidants shifts towards oxidative stress, leading to the accumulation of lipid peroxides (Cao and Dixon 2016). GSSG accumulation due to imbalanced redox conditions can contribute to increased susceptibility to lipid peroxidation (Pizzino, Irrera et al. 2017). We thus evaluated the level of GSH by comparatively analysing the cellular GSH level in both HeLa-FUS-P525L and HeLa-FUS-WT cells (Figure 5). The obtained data revealed significant reduction in the level of oxidized glutathione (GSSG) in HeLa-FUS-P525L cells. These results suggest that decreased GSH level due to xCT downregulation render cells more vulnerable to oxidative stress and lipid peroxidation, ultimately leading to the induction of ferroptosis. Although GSSG reduction itself does not directly induce lipid peroxidation, its accumulation due to imbalanced redox conditions can contribute to increased susceptibility to lipid peroxidation. The accumulation of lipid peroxidation products in the cellular membrane can lead to membrane disruption and cell death mediated by ferroptosis. Lipid hydroperoxide, a marker of oxidative stress, indicates an imbalance between the production of ROS and the cell’s antioxidant defense mechanisms (Nita and Grzybowski 2016). We thus investigated whether there is increased level of lipid peroxidation in case of FUS mutants. Here we utilized the lipid peroxidation sensor, BODIPY 665/676, a commercially available fluorescent dye, to assess lipid peroxide accumulation in living cells (Naguib 2000). BODIPY 665/676 demonstrates altered fluorescence upon interaction with peroxyl radicals. Laser scanning microscopy (LSM) revealed higher levels of lipid peroxides that accumulate primarily in the plasma membrane of Hela-FUS-P525L cells compared to Hela-FUS-WT (Figure 5). Consistent with this observation, a significant increased level of lipid peroxides was detected in the HeLa-FUS-P525L cells compared to Hela-FUS-WT cells (Figure 5B, C). Next, we analysed the intensity of lipid peroxides in both cell lines following the treatment with xCT inhibitor erastin (Dixon et al. in 2012) (Figure 5D, E). Surprisingly, the inhibition of xCT did not result in a significant further increase in lipid peroxidation compare to untreated cells (Figure 5D, E).
3.6. Inhibition of Mitochondrial Calcium Uniporter Alleviates Lipid Peroxidation
Inhibition of xCT was reported to lead to mitochondrial fragmentation, mitochondrial calcium overload, increased mitochondrial ROS production, and disruption of the mitochondrial membrane potential (Marmolejo-Garza, Krabbendam et al. 2023). The observation that mitochondrial dysfunction is characteristic of ferroptosis (Figure 1A,B) makes preservation of mitochondrial function a potential therapeutic option against ferroptotic cell death. Calcium overload has been shown to play a significant role in cell death caused by e.g. ferroptosis (Maher, van Leyen et al. 2018, Marmolejo-Garza, Krabbendam et al. 2023).
Luciferase-based assay used to analyze total glutathione (GSH) and oxidized glutathione (GSSG) levels in both cell lines. Data shown represent the mean ± s.d. p<0.001 (two-way ANOVA), normalized with vehicle (untreated control) of n = 3 wells of a 96-well plate, from three independent experiments. (B) Live cell fluorescence imaging of lipid peroxides in HeLa-FUS-eGFP-WT and HeLa-FUS-eGFP-P525L. Scale bars 100uM. (C) Lipid peroxides florescence intensity relative to baseline without any treatment in HeLa-FUS-eGFP-WT compare to HeLa-FUS-eGFP-P525L. (D&E) Lipid peroxides fluorescence intensity after erastin 20uM treatment for 24 h with and without Ru265 (50 µM) and Ru265 (100 µM) in both HeLa-FUS-eGFP-WT and HeLa-FUS-eGFP-P525L. Control untreated (no erastin and no Ru265). Data are from a minimum of 3 stage positions. For statistical analysis, each stage position counted as one data entry. Data are mean +- s.d., p<0.001 (two-way ANOVA).
Mitochondrial calcium levels are controlled via the MCU, which represents the main entry point of Ca2+ into the mitochondrial matrix (Grossmann, Malburg et al. 2023, Marmolejo-Garza, Krabbendam et al. 2023). A recent report suggested that inhibition of the MCU might be protective against lipid peroxidation and ferroptosis in wild type cells (Marmolejo-Garza, Krabbendam et al. 2023). Having shown both, mitochondrial dysfunction (Figure 1) and increased vulnerability against ferroptosis (Figure 2 & Figure 3), we hypothesized that inhibition of the MCU might also be protective in FUS-ALS cell lines. The ruthenium compound Ru265 is a commonly studied pharmacological inhibitor of the MCU and inhibitor of lipid peroxide generation (Huang, MacMillan et al. 2023, Marmolejo-Garza, Krabbendam et al. 2023). To this end, we treated cells with erastin with or without Ru265. A significantly reduced intensity of lipid peroxides was observed in the cells co-treated with 20 µM erastin and 50 µM Ru265 (Figure 5D), while 100 µM Ru265 increased cell death in FUS mutants (Figure 5E). Co-treatment of Ru265 with erastin appeared to decrease cellular lipid peroxides in both, wild type and FUS mutant cell lines. These results demonstrated that FUS mutations lead to increased vulnerability against ferroptosis most likely by impairing cellular redox signaling downstream of xCT. Both, iron chelation as well as interfering with the MCU might be interesting targets to alleviate ferroptosis in FUS-ALS.
4. Discussion
The results of our study reveal compelling evidence indicating an association between FUS mutations and heightened susceptibility to ferroptosis, a regulated form of necrotic cell death characterized by the accumulation of lipid peroxides (Dixon, Lemberg et al. 2012). Our results further suggest that downregulation of xCT due to FUS mutation led to disturbed GSH metabolism and increased lipid peroxidation, which could be alleviated by either iron chelation or MCU inhibition.
Ferroptosis characterized by the reduction in cell viability due to oxidative cell death, is governed by key enzymes such as glutathione peroxidase 4 (GPX4) and the cystine/glutamate antiporter system Xc- (Dixon, Patel et al. 2014). Understanding the cellular mechanisms underlying ferroptosis susceptibility in the context of FUS mutations is crucial for advancing therapeutic interventions in FUS-related neurodegenerative diseases. To investigate the vulnerability of FUS-mutated cells to ferroptosis, we utilized a model system of HeLa cells expressing either FUS-P525L mutant or WT-FUS. Our comprehensive investigation into the dose-dependent response of cell death induced by ferroptosis inducers RSL3 and erastin revealed a notable induction of cell death in both FUS-mutated and wild-type cells in a dose-dependent manner (Figure 2). Previous research by Do Van et al. (2016) and Ayala and Muñoz (2019) has implicated ferroptosis in various neurodegenerative disorders, including ALS (Ayala, Munoz et al. 2014, Do Van, Gouel et al. 2016). They have shown that oxidative stress and lipid peroxidation, hallmarks of ferroptosis, contribute to neuronal death and disease pathology. Interestingly, FUS-P525L HeLa cells exhibited significantly compromised cellular viability compared to FUS-WT HeLa cells to both erastin and RSL3, particularly at lower concentrations of RSL3 supports the notion that ferroptosis plays a role in FUS-related neurodegeneration. (Figure 2B, D). Liproxstatin-1 (Lip-1) is a well-known inhibitor of ferroptosis (Friedmann Angeli, Schneider et al. 2014). Co-treatment experiments revealed that cell death induced by varying concentrations of erastin and RSL3 was completely inhibited in the presence of Lip-1, further supporting ferroptosis as underlying cell death mechanism (Figure 3A-D). This differential susceptibility to ferroptosis suggests that FUS mutations may predispose cells to heightened oxidative stress and lipid peroxidation, leading to increased susceptibility to ferroptosis-mediated cell death.
As mentioned above, the regulation of key proteins involved in ferroptosis, such as xCT and GPX4, plays a crucial role in determining the susceptibility of cells to this form of cell death (Dixon, Lemberg et al. 2012, Conrad and Friedmann Angeli 2015). Interestingly, we noted a reduction of xCT as the most misregulated finding in case of FUS-ALS (Figure 4A, B). FSP1 was also significantly reduced in FUS-ALS, whereas GPX4 was not significantly reduced (Figure 4A, B). This notable decrease in xCT protein expression raises our attention, as compromised xCT can result in decreased cellular uptake of cystine, leading to reduced synthesis of GSH. We therefore evaluated the cellular levels of GSH, a key antioxidant involved in neutralizing ROS and protecting cells from oxidative damage (Kwon, Cha et al. 2019) (Figure 5A). Here we found a significant reduction in the level of GSSG in HeLa-FUS-P525L cells compared to HeLa-FUS-WT cells (Figure 5A). Although, GSSG depletion does not have a direct effect on inducing lipid peroxidation, however, the observed reduction in GSSG levels suggests a depletion or compromise in cellular levels of reduced glutathione leading to imbalanced redox conditions. However, when cellular levels of reduced glutathione are depleted or compromised, the balance between oxidants and antioxidants shifts towards oxidative stress, leading to the accumulation of lipid peroxides (Cao and Dixon 2016). Here we assume that xCT downregulation can possibly aggregate imbalanced redox conditions and subsequently induce ferroptotic cell death. Similar to our findings, Sato et al. (2018) and Lewerenz et al. (2013) have demonstrated the critical role of xCT in maintaining cellular redox homeostasis by regulating the import of cystine for glutathione synthesis (Lewerenz, Ates et al. 2018, Sato, Kusumi et al. 2018). This is in line with the finding that the FUS-mutation itself was already sufficient to show increased lipid peroxidation, which was not further increased by xCT inhibition (Figure 5D, E). Polyunsaturated fatty acids (PUFAs) have been implicated in various neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) (Fitzgerald, O’Reilly et al. 2014). ACSL4 is known to promote the incorporation of polyunsaturated fatty acids (PUFAs) into phospholipids, rendering cells more susceptible to lipid peroxidation and ferroptosis (Doll, Proneth et al. 2017). Thus, the elevated ACSL4 expression in HeLa-FUS-P525L cells may have possibly contributed to the increased levels of lipid peroxides observed in these cells.
The pathophysiology of FUS-ALS significantly involves oxidative and mitochondrial damage. Interestingly, inhibition of xCT was reported to lead to diverse mitochondrial phenotypes, such as mitochondrial fragmentation, mitochondrial calcium overload, increased mitochondrial ROS production and disruption of the mitochondrial membrane potential (Marmolejo-Garza, Krabbendam et al. 2023). A recent report suggested that inhibition of the MCU might be protective against lipid peroxidation and ferroptosis in wild type cells (Marmolejo-Garza, Krabbendam et al. 2023). Excitingly, MCU inhibition was able to alleviate lipid peroxidation in both, wildtype and FUS-ALS cells (Figure 5D, E). The observed protective effects of MCU inhibition against lipid peroxidation provide further support for targeting mitochondrial dysfunction in ALS therapy.
In conclusion, our study sheds light on the role of lipid peroxidation and ferroptosis in the pathogenesis of FUS-associated ALS. The observed accumulation of lipid peroxides and dysregulation of ferroptosis-associated proteins, coupled with heightened vulnerability to ferroptotic cell death in cells expressing the FUS-P525L mutant, highlight the potential involvement of ferroptosis in FUS-related neurodegenerative diseases. In addition, the protective effects of Ru265, Lip-1 and DFO against ferroptotic cell death proposed that modulating lipid peroxidation and iron availability could mitigate cell death in FUS-associated ALS. However, further research is required to elucidate the intricate molecular mechanisms underlying ferroptosis and validate the efficacy of targeted interventions in FUS-ALS. These efforts might pave the way for the development of novel therapeutic approaches aimed at modulating ferroptosis pathways and improving outcomes for patients with FUS-related neurodegenerative diseases.
Author Contributions
M.I. and A.H. conceived the project and outlined the study. M.I. and D.G. performed the methodology, carried out the formal analysis and data curation. M.I. and A.H. wrote the original draft. All authors reviewed, and edited the manuscript. A.H administrated the project and carried out the funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
A.H. is supported by the Hermann and Lilly Schilling-Stiftung für medizinische Forschung im Stifterverband.
Acknowledgments
HeLa FUS-eGFP cell line was kindly provided by Dr. Ina Poser/Prof. Anthony A. Hyman, Max Planck Institute for Molecular Cell Biology and Genetics, Dresden, Germany. The cell line uses a BAC system to overexpress FUS c-terminally tagged with eGFP. We acknowledge the great help of Jette Abel with all kind of cell culture work.
Conflicts of Interest
The authors declare that they have no competing interests related to the direct applications of this research.
References
- Angeli, J. P. F., R. Shah, D. A. Pratt and M. Conrad (2017). "Ferroptosis Inhibition: Mechanisms and Opportunities." Trends Pharmacol Sci 38(5): 489-498. [CrossRef]
- Ayala, A., M. F. Munoz and S. Arguelles (2014). "Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal." Oxid Med Cell Longev 2014: 360438. [CrossRef]
- Brown, R. H. and A. Al-Chalabi (2017). "Amyotrophic Lateral Sclerosis." N Engl J Med 377(2): 162-172. [CrossRef]
- Cao, J. Y. and S. J. Dixon (2016). "Mechanisms of ferroptosis." Cell Mol Life Sci 73(11-12): 2195-2209. [CrossRef]
- Carracedo, A., J. M. Prieto, L. Concheiro and J. Estefania (1987). "Isoelectric focusing patterns of some mammalian keratins." J Forensic Sci 32(1): 93-99. [CrossRef]
- Chen, L., W. S. Hambright, R. Na and Q. Ran (2015). "Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis." J Biol Chem 290(47): 28097-28106. [CrossRef]
- Chen, T. W., T. J. Wardill, Y. Sun, S. R. Pulver, S. L. Renninger, A. Baohan, E. R. Schreiter, R. A. Kerr, M. B. Orger, V. Jayaraman, L. L. Looger, K. Svoboda and D. S. Kim (2013). "Ultrasensitive fluorescent proteins for imaging neuronal activity." Nature 499(7458): 295-300. [CrossRef]
- Chen, X., C. Yu, R. Kang and D. Tang (2020). "Iron Metabolism in Ferroptosis." Front Cell Dev Biol 8: 590226. [CrossRef]
- Conrad, M. and J. P. Friedmann Angeli (2015). "Glutathione peroxidase 4 (Gpx4) and ferroptosis: what’s so special about it?" Mol Cell Oncol 2(3): e995047. [CrossRef]
- Dixon, S. J., K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison, 3rd and B. R. Stockwell (2012). "Ferroptosis: an iron-dependent form of nonapoptotic cell death." Cell 149(5): 1060-1072. [CrossRef]
- Dixon, S. J., D. N. Patel, M. Welsch, R. Skouta, E. D. Lee, M. Hayano, A. G. Thomas, C. E. Gleason, N. P. Tatonetti, B. S. Slusher and B. R. Stockwell (2014). "Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis." Elife 3: e02523. [CrossRef]
- Do Van, B., F. Gouel, A. Jonneaux, K. Timmerman, P. Gele, M. Petrault, M. Bastide, C. Laloux, C. Moreau, R. Bordet, D. Devos and J. C. Devedjian (2016). "Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC." Neurobiol Dis 94: 169-178. [CrossRef]
- Doll, S., B. Proneth, Y. Y. Tyurina, E. Panzilius, S. Kobayashi, I. Ingold, M. Irmler, J. Beckers, M. Aichler, A. Walch, H. Prokisch, D. Trumbach, G. Mao, F. Qu, H. Bayir, J. Fullekrug, C. H. Scheel, W. Wurst, J. A. Schick, V. E. Kagan, J. P. Angeli and M. Conrad (2017). "ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition." Nat Chem Biol 13(1): 91-98. [CrossRef]
- Fitzgerald, K. C., E. J. O’Reilly, G. J. Falcone, M. L. McCullough, Y. Park, L. N. Kolonel and A. Ascherio (2014). "Dietary omega-3 polyunsaturated fatty acid intake and risk for amyotrophic lateral sclerosis." JAMA Neurol 71(9): 1102-1110. [CrossRef]
- Fleury, C., B. Mignotte and J. L. Vayssiere (2002). "Mitochondrial reactive oxygen species in cell death signaling." Biochimie 84(2-3): 131-141. [CrossRef]
- Forcina, G. C. and S. J. Dixon (2019). "GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis." Proteomics 19(18): e1800311. [CrossRef]
- Friedmann Angeli, J. P., M. Schneider, B. Proneth, Y. Y. Tyurina, V. A. Tyurin, V. J. Hammond, N. Herbach, M. Aichler, A. Walch, E. Eggenhofer, D. Basavarajappa, O. Radmark, S. Kobayashi, T. Seibt, H. Beck, F. Neff, I. Esposito, R. Wanke, H. Forster, O. Yefremova, M. Heinrichmeyer, G. W. Bornkamm, E. K. Geissler, S. B. Thomas, B. R. Stockwell, V. B. O’Donnell, V. E. Kagan, J. A. Schick and M. Conrad (2014). "Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice." Nat Cell Biol 16(12): 1180-1191. [CrossRef]
- Gleitze, S., A. Paula-Lima, M. T. Nunez and C. Hidalgo (2021). "The calcium-iron connection in ferroptosis-mediated neuronal death." Free Radic Biol Med 175: 28-41. [CrossRef]
- Greco, V., P. Longone, A. Spalloni, L. Pieroni and A. Urbani (2019). "Crosstalk Between Oxidative Stress and Mitochondrial Damage: Focus on Amyotrophic Lateral Sclerosis." Adv Exp Med Biol 1158: 71-82. [CrossRef]
- Grossmann, D., N. Malburg, H. Glass, V. Weeren, V. Sondermann, J. F. Pfeiffer, J. Petters, J. Lukas, P. Seibler, C. Klein, A. Grunewald and A. Hermann (2023). "Mitochondria-Endoplasmic Reticulum Contact Sites Dynamics and Calcium Homeostasis Are Differentially Disrupted in PINK1-PD or PRKN-PD Neurons." Mov Disord 38(10): 1822-1836. [CrossRef]
- Gunther, R., A. Pal, C. Williams, V. L. Zimyanin, M. Liehr, C. von Neubeck, M. Krause, M. G. Parab, S. Petri, N. Kalmbach, S. L. Marklund, J. Sterneckert, P. Munch Andersen, F. Wegner, J. D. Gilthorpe and A. Hermann (2022). "Alteration of Mitochondrial Integrity as Upstream Event in the Pathophysiology of SOD1-ALS." Cells 11(7). [CrossRef]
- He, L., T. He, S. Farrar, L. Ji, T. Liu and X. Ma (2017). "Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species." Cell Physiol Biochem 44(2): 532-553. [CrossRef]
- Huang, Z., S. N. MacMillan and J. J. Wilson (2023). "A Fluorogenic Inhibitor of the Mitochondrial Calcium Uniporter." Angew Chem Int Ed Engl 62(6): e202214920. [CrossRef]
- Kreiter, N., A. Pal, X. Lojewski, P. Corcia, M. Naujock, P. Reinhardt, J. Sterneckert, S. Petri, F. Wegner, A. Storch and A. Hermann (2018). "Age-dependent neurodegeneration and organelle transport deficiencies in mutant TDP43 patient-derived neurons are independent of TDP43 aggregation." Neurobiol Dis 115: 167-181. [CrossRef]
- Kwon, D. H., H. J. Cha, H. Lee, S. H. Hong, C. Park, S. H. Park, G. Y. Kim, S. Kim, H. S. Kim, H. J. Hwang and Y. H. Choi (2019). "Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway." Antioxidants (Basel) 8(4). [CrossRef]
- Lewerenz, J., G. Ates, A. Methner, M. Conrad and P. Maher (2018). "Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System." Front Neurosci 12: 214. [CrossRef]
- Lewerenz, J., S. J. Hewett, Y. Huang, M. Lambros, P. W. Gout, P. W. Kalivas, A. Massie, I. Smolders, A. Methner, M. Pergande, S. B. Smith, V. Ganapathy and P. Maher (2013). "The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities." Antioxid Redox Signal 18(5): 522-555. [CrossRef]
- Li, J., F. Cao, H. L. Yin, Z. J. Huang, Z. T. Lin, N. Mao, B. Sun and G. Wang (2020). "Ferroptosis: past, present and future." Cell Death Dis 11(2): 88. [CrossRef]
- Maher, P., K. van Leyen, P. N. Dey, B. Honrath, A. Dolga and A. Methner (2018). "The role of Ca(2+) in cell death caused by oxidative glutamate toxicity and ferroptosis." Cell Calcium 70: 47-55. [CrossRef]
- Marmolejo-Garza, A., I. E. Krabbendam, M. D. A. Luu, F. Brouwer, M. Trombetta-Lima, O. Unal, S. J. O’Connor, N. Majernikova, C. R. S. Elzinga, C. Mammucari, M. Schmidt, M. Madesh, E. Boddeke and A. M. Dolga (2023). "Negative modulation of mitochondrial calcium uniporter complex protects neurons against ferroptosis." Cell Death Dis 14(11): 772. [CrossRef]
- Munoz, P., A. Humeres, C. Elgueta, A. Kirkwood, C. Hidalgo and M. T. Nunez (2011). "Iron mediates N-methyl-D-aspartate receptor-dependent stimulation of calcium-induced pathways and hippocampal synaptic plasticity." J Biol Chem 286(15): 13382-13392. [CrossRef]
- Naguib, Y. M. (2000). "Antioxidant activities of astaxanthin and related carotenoids." J Agric Food Chem 48(4): 1150-1154. [CrossRef]
- Naumann, M., A. Pal, A. Goswami, X. Lojewski, J. Japtok, A. Vehlow, M. Naujock, R. Gunther, M. Jin, N. Stanslowsky, P. Reinhardt, J. Sterneckert, M. Frickenhaus, F. Pan-Montojo, E. Storkebaum, I. Poser, A. Freischmidt, J. H. Weishaupt, K. Holzmann, D. Troost, A. C. Ludolph, T. M. Boeckers, S. Liebau, S. Petri, N. Cordes, A. A. Hyman, F. Wegner, S. W. Grill, J. Weis, A. Storch and A. Hermann (2018). "Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation." Nat Commun 9(1): 335. [CrossRef]
- Naumann, M., K. Peikert, R. Gunther, A. J. van der Kooi, E. Aronica, A. Hubers, V. Danel, P. Corcia, F. Pan-Montojo, S. Cirak, G. Haliloglu, A. C. Ludolph, A. Goswami, P. M. Andersen, J. Prudlo, F. Wegner, P. Van Damme, J. H. Weishaupt and A. Hermann (2019). "Phenotypes and malignancy risk of different FUS mutations in genetic amyotrophic lateral sclerosis." Ann Clin Transl Neurol 6(12): 2384-2394. [CrossRef]
- Nita, M. and A. Grzybowski (2016). "The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults." Oxid Med Cell Longev 2016: 3164734. [CrossRef]
- Pal, A., H. Glaß, M. Naumann, N. Kreiter, J. Japtok, R. Sczech and A. Hermann (2018). "High content organelle trafficking enables disease state profiling as powerful tool for disease modelling." Sci Data 5: 180241. [CrossRef]
- Pal, A., B. Kretner, M. Abo-Rady, H. Glabeta, B. P. Dash, M. Naumann, J. Japtok, N. Kreiter, A. Dhingra, P. Heutink, T. M. Bockers, R. Gunther, J. Sterneckert and A. Hermann (2021). "Concomitant gain and loss of function pathomechanisms in C9ORF72 amyotrophic lateral sclerosis." Life Sci Alliance 4(4). [CrossRef]
- Patel, A., H. O. Lee, L. Jawerth, S. Maharana, M. Jahnel, M. Y. Hein, S. Stoynov, J. Mahamid, S. Saha, T. M. Franzmann, A. Pozniakovski, I. Poser, N. Maghelli, L. A. Royer, M. Weigert, E. W. Myers, S. Grill, D. Drechsel, A. A. Hyman and S. Alberti (2015). "A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation." Cell 162(5): 1066-1077. [CrossRef]
- Pedrera, L., R. A. Espiritu, U. Ros, J. Weber, A. Schmitt, J. Stroh, S. Hailfinger, S. von Karstedt and A. J. Garcia-Saez (2021). "Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics." Cell Death Differ 28(5): 1644-1657. [CrossRef]
- Pizzino, G., N. Irrera, M. Cucinotta, G. Pallio, F. Mannino, V. Arcoraci, F. Squadrito, D. Altavilla and A. Bitto (2017). "Oxidative Stress: Harms and Benefits for Human Health." Oxid Med Cell Longev 2017: 8416763. [CrossRef]
- Sanmartin, C. D., A. C. Paula-Lima, A. Garcia, P. Barattini, S. Hartel, M. T. Nunez and C. Hidalgo (2014). "Ryanodine receptor-mediated Ca(2+) release underlies iron-induced mitochondrial fission and stimulates mitochondrial Ca(2+) uptake in primary hippocampal neurons." Front Mol Neurosci 7: 13. [CrossRef]
- Sato, M., R. Kusumi, S. Hamashima, S. Kobayashi, S. Sasaki, Y. Komiyama, T. Izumikawa, M. Conrad, S. Bannai and H. Sato (2018). "The ferroptosis inducer erastin irreversibly inhibits system x(c)- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells." Sci Rep 8(1): 968. [CrossRef]
- Shi, Z., L. Zhang, J. Zheng, H. Sun and C. Shao (2021). "Ferroptosis: Biochemistry and Biology in Cancers." Front Oncol 11: 579286. [CrossRef]
- Stockwell, B. R., J. P. Friedmann Angeli, H. Bayir, A. I. Bush, M. Conrad, S. J. Dixon, S. Fulda, S. Gascon, S. K. Hatzios, V. E. Kagan, K. Noel, X. Jiang, A. Linkermann, M. E. Murphy, M. Overholtzer, A. Oyagi, G. C. Pagnussat, J. Park, Q. Ran, C. S. Rosenfeld, K. Salnikow, D. Tang, F. M. Torti, S. V. Torti, S. Toyokuni, K. A. Woerpel and D. D. Zhang (2017). "Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease." Cell 171(2): 273-285. [CrossRef]
- Tang, D. and G. Kroemer (2020). "Ferroptosis." Curr Biol 30(21): R1292-R1297. [CrossRef]
- Turner, M. R., O. Hardiman, M. Benatar, B. R. Brooks, A. Chio, M. de Carvalho, P. G. Ince, C. Lin, R. G. Miller, H. Mitsumoto, G. Nicholson, J. Ravits, P. J. Shaw, M. Swash, K. Talbot, B. J. Traynor, L. H. Van den Berg, J. H. Veldink, S. Vucic and M. C. Kiernan (2013). "Controversies and priorities in amyotrophic lateral sclerosis." Lancet Neurol 12(3): 310-322. [CrossRef]
- Valko, K. and L. Ciesla (2019). "Amyotrophic lateral sclerosis." Prog Med Chem 58: 63-117. [CrossRef]
- Wang, D., W. Liang, D. Huo, H. Wang, Y. Wang, C. Cong, C. Zhang, S. Yan, M. Gao, X. Su, X. Tan, W. Zhang, L. Han, D. Zhang and H. Feng (2023). "SPY1 inhibits neuronal ferroptosis in amyotrophic lateral sclerosis by reducing lipid peroxidation through regulation of GCH1 and TFR1." Cell Death Differ 30(2): 369-382. [CrossRef]
- Wang, T., D. Tomas, N. D. Perera, B. Cuic, S. Luikinga, A. Viden, S. K. Barton, C. A. McLean, A. L. Samson, A. Southon, A. I. Bush, J. M. Murphy and B. J. Turner (2022). "Ferroptosis mediates selective motor neuron death in amyotrophic lateral sclerosis." Cell Death Differ 29(6): 1187-1198. [CrossRef]
- Yang, W. S., R. SriRamaratnam, M. E. Welsch, K. Shimada, R. Skouta, V. S. Viswanathan, J. H. Cheah, P. A. Clemons, A. F. Shamji, C. B. Clish, L. M. Brown, A. W. Girotti, V. W. Cornish, S. L. Schreiber and B. R. Stockwell (2014). "Regulation of ferroptotic cancer cell death by GPX4." Cell 156(1-2): 317-331. [CrossRef]
- Zimyanin, V. L., A. M. Pielka, H. Glass, J. Japtok, D. Grossmann, M. Martin, A. Deussen, B. Szewczyk, C. Deppmann, E. Zunder, P. M. Andersen, T. M. Boeckers, J. Sterneckert, S. Redemann, A. Storch and A. Hermann (2023). "Live Cell Imaging of ATP Levels Reveals Metabolic Compartmentalization within Motoneurons and Early Metabolic Changes in FUS ALS Motoneurons." Cells 12(10). [CrossRef]
Figure 1.
Mitochondrial depolarization in FUS -mutated cells (A) HeLa cells were stained with TMRE and MitoTracker deep red for live cell imaging. (B) The mean TMRE signal intensity was quantified using MitoTracker deep red as counterstain for detection of mitochondria. Data are from 6 independent biological replicates (N=6). Data indicated as mean ± SEM, * p<0.001 (Wilcoxon matched-pairs signed rank test).
Figure 1.
Mitochondrial depolarization in FUS -mutated cells (A) HeLa cells were stained with TMRE and MitoTracker deep red for live cell imaging. (B) The mean TMRE signal intensity was quantified using MitoTracker deep red as counterstain for detection of mitochondria. Data are from 6 independent biological replicates (N=6). Data indicated as mean ± SEM, * p<0.001 (Wilcoxon matched-pairs signed rank test).
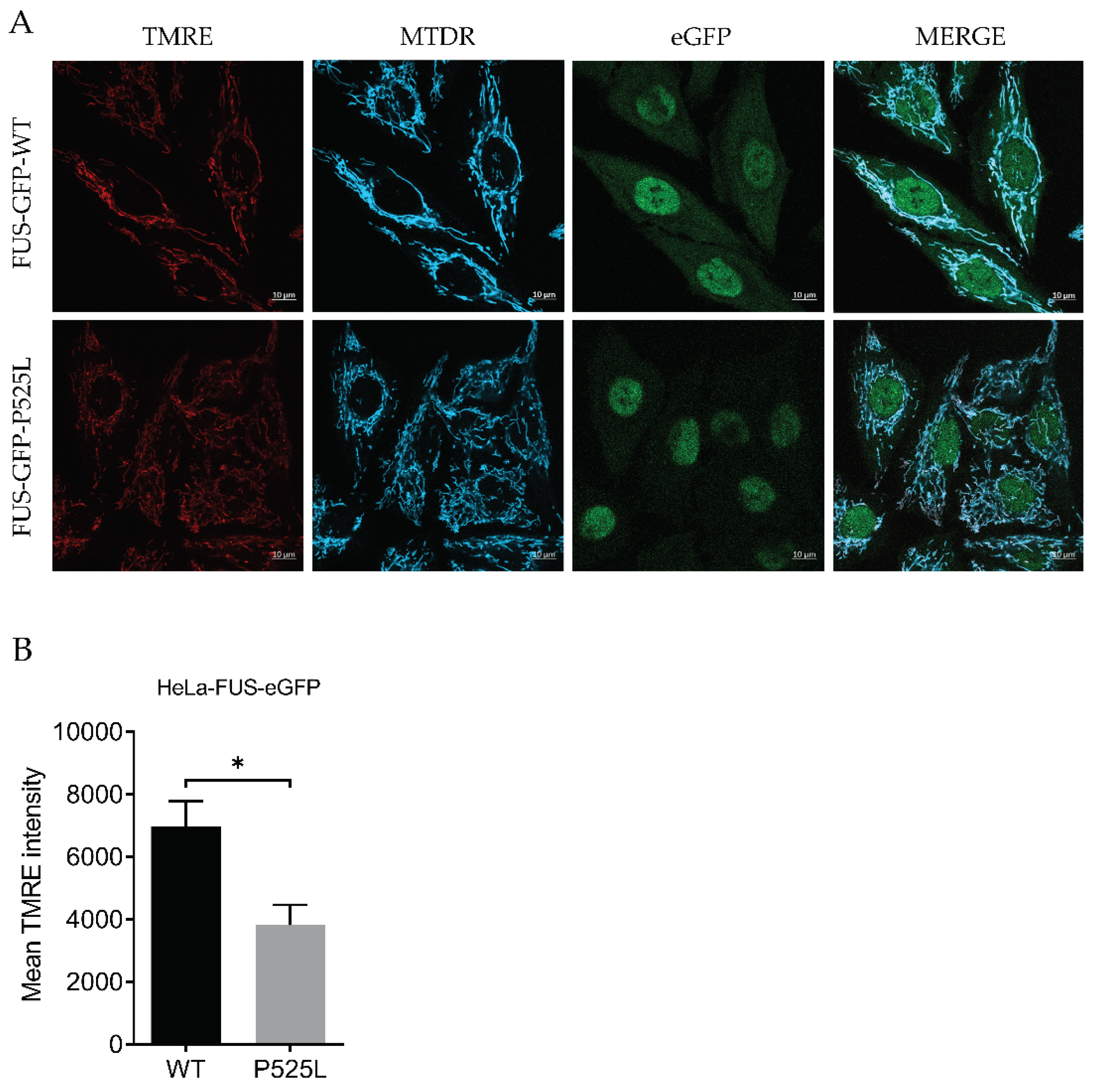
Figure 2.
Inhibition of GPX4 and xCT enhances the vulnerability to ferroptosis in FUS mutated cells. (A & C) Microscope images of both HeLa-FUS-eGFP-WT and HeLa-FUS-eGFP-P525L of untreated and treated with different concentration of RSL3 and erastin. Scale bar = 100 μm. (B &D) Dose dependent toxicity of oxidative cell death inducing agents (RSL3 and Erastin). Both cell lines were treated with increasing concentration of GPX4 inhibitor RSL3 and system xCT inhibitor Erastin. Cell viability was measured 24 h.
Figure 2.
Inhibition of GPX4 and xCT enhances the vulnerability to ferroptosis in FUS mutated cells. (A & C) Microscope images of both HeLa-FUS-eGFP-WT and HeLa-FUS-eGFP-P525L of untreated and treated with different concentration of RSL3 and erastin. Scale bar = 100 μm. (B &D) Dose dependent toxicity of oxidative cell death inducing agents (RSL3 and Erastin). Both cell lines were treated with increasing concentration of GPX4 inhibitor RSL3 and system xCT inhibitor Erastin. Cell viability was measured 24 h.
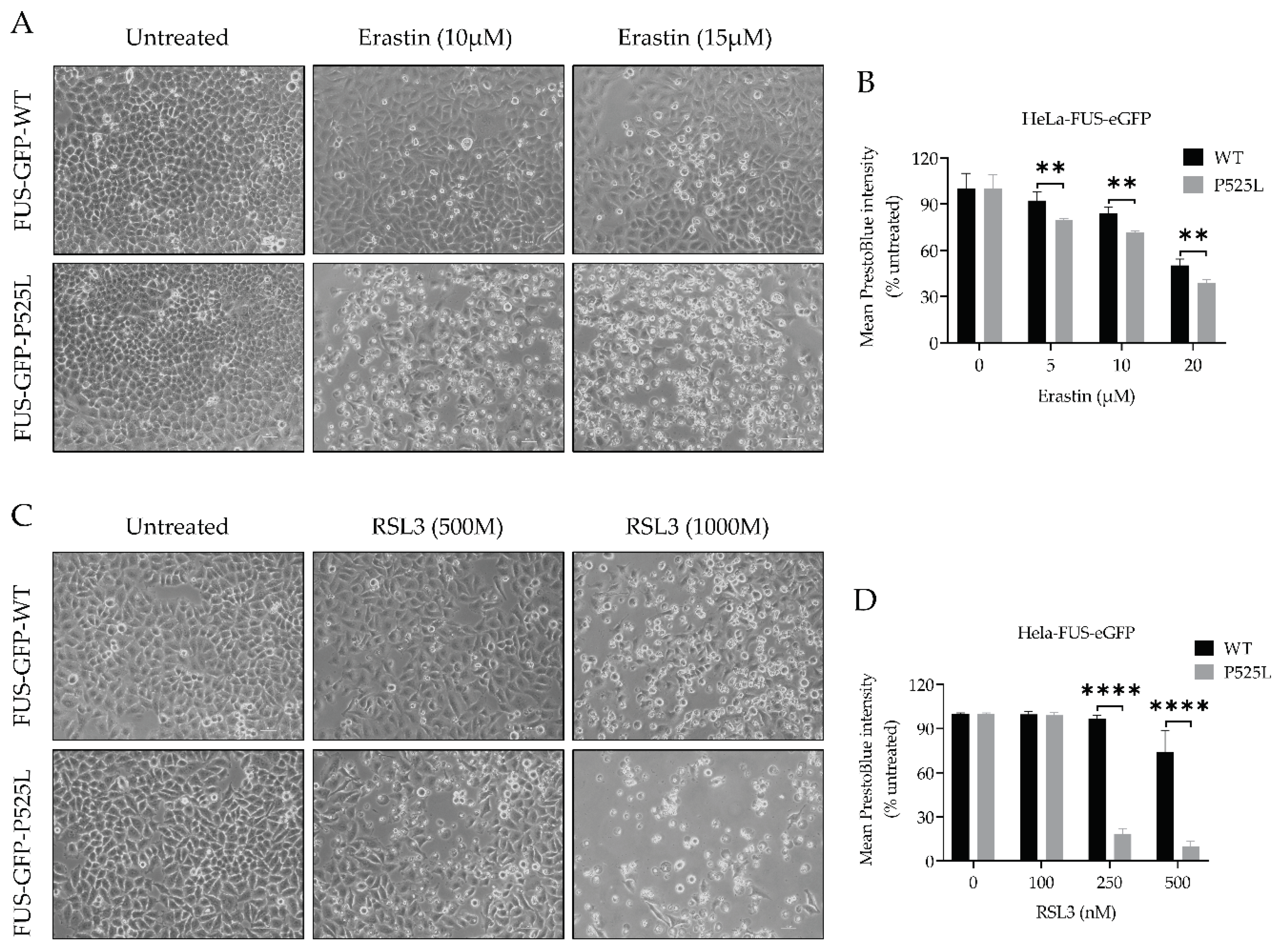
Figure 3.
Cell death induced by inhibiting GPX4 and xCT is inhibited by Liproxstatin-1 and Deferoxamine. Dose-dependent toxicity of ferroptosis inducing compounds (RSL3 and erastin). Both HeLa-FUS-eGFP-WT and HeLa-FUS-eGFP-P525L cell lines were cultured at a density of 2500 cells per well (96 well plate) and grown for 24 h. (A, B) Both cell lines were treated with variable concentration erastin with or without the ferroptosis inhibitor Lip-1 (500 nm). (C, D) Both cell lines were treated with variable concentration RSL3 with or without the ferroptosis inhibitor Lip-1 (500 nm). Data shown represent the mean ± s.d. p<0.001 (two-way ANOVA), of n = 3 wells of a 96-well plate, from three independent experiments. Cell viability was assessed 24 h after the treatment using Presto Blue method.
Figure 3.
Cell death induced by inhibiting GPX4 and xCT is inhibited by Liproxstatin-1 and Deferoxamine. Dose-dependent toxicity of ferroptosis inducing compounds (RSL3 and erastin). Both HeLa-FUS-eGFP-WT and HeLa-FUS-eGFP-P525L cell lines were cultured at a density of 2500 cells per well (96 well plate) and grown for 24 h. (A, B) Both cell lines were treated with variable concentration erastin with or without the ferroptosis inhibitor Lip-1 (500 nm). (C, D) Both cell lines were treated with variable concentration RSL3 with or without the ferroptosis inhibitor Lip-1 (500 nm). Data shown represent the mean ± s.d. p<0.001 (two-way ANOVA), of n = 3 wells of a 96-well plate, from three independent experiments. Cell viability was assessed 24 h after the treatment using Presto Blue method.
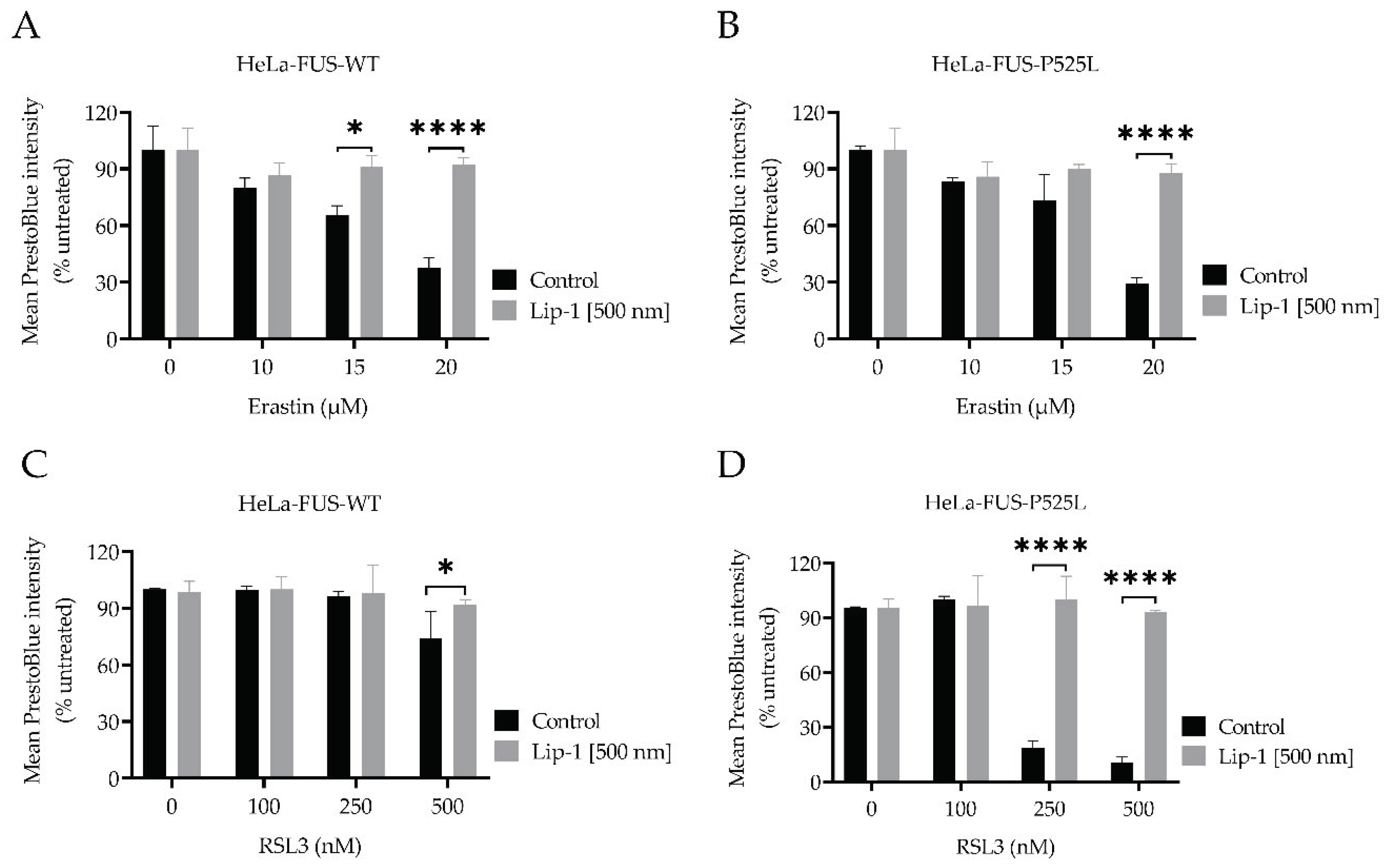
Figure 4.
Mutated FUS in HeLa cells does not influence the protein expression of established ferroptotic factors. (A) Immunoblot analysis of both HeLa-FUS-eGFP-WT and HeLa-FUS-eGFP-P525L cell lines using FUS monoclonal antibody. Western blot analysis revealed FUS expressing both HeLa cell lines. Protein expression analysis of GPX4, XCT, FSP1 and ACSL4 using specific antibodies demonstrated the regulation of the analysed genes in HeLa-FUS-eGFP-P525L cell line compared HeLa-FUS-eGFP-WT. (B) The expression of the key ferroptotic genes are regulated, though the detected expression level is significant. Relative gene expression was normalised to the reference gene B-Actin (N = 3). Data shown from three independent experiments represents the mean ±s.d. of n=3. (C, D) Both cell lines were treated with variable concentration of erastin with or without the ferroptosis inhibitor DFO (100 µM). Data shown represent the mean ± s.d. p<0.001 (two-way ANOVA), of n = 3 wells of a 96-well plate, from three independent experiments. Cell viability was assessed 24 h after the treatment using Presto Blue method.
Figure 4.
Mutated FUS in HeLa cells does not influence the protein expression of established ferroptotic factors. (A) Immunoblot analysis of both HeLa-FUS-eGFP-WT and HeLa-FUS-eGFP-P525L cell lines using FUS monoclonal antibody. Western blot analysis revealed FUS expressing both HeLa cell lines. Protein expression analysis of GPX4, XCT, FSP1 and ACSL4 using specific antibodies demonstrated the regulation of the analysed genes in HeLa-FUS-eGFP-P525L cell line compared HeLa-FUS-eGFP-WT. (B) The expression of the key ferroptotic genes are regulated, though the detected expression level is significant. Relative gene expression was normalised to the reference gene B-Actin (N = 3). Data shown from three independent experiments represents the mean ±s.d. of n=3. (C, D) Both cell lines were treated with variable concentration of erastin with or without the ferroptosis inhibitor DFO (100 µM). Data shown represent the mean ± s.d. p<0.001 (two-way ANOVA), of n = 3 wells of a 96-well plate, from three independent experiments. Cell viability was assessed 24 h after the treatment using Presto Blue method.
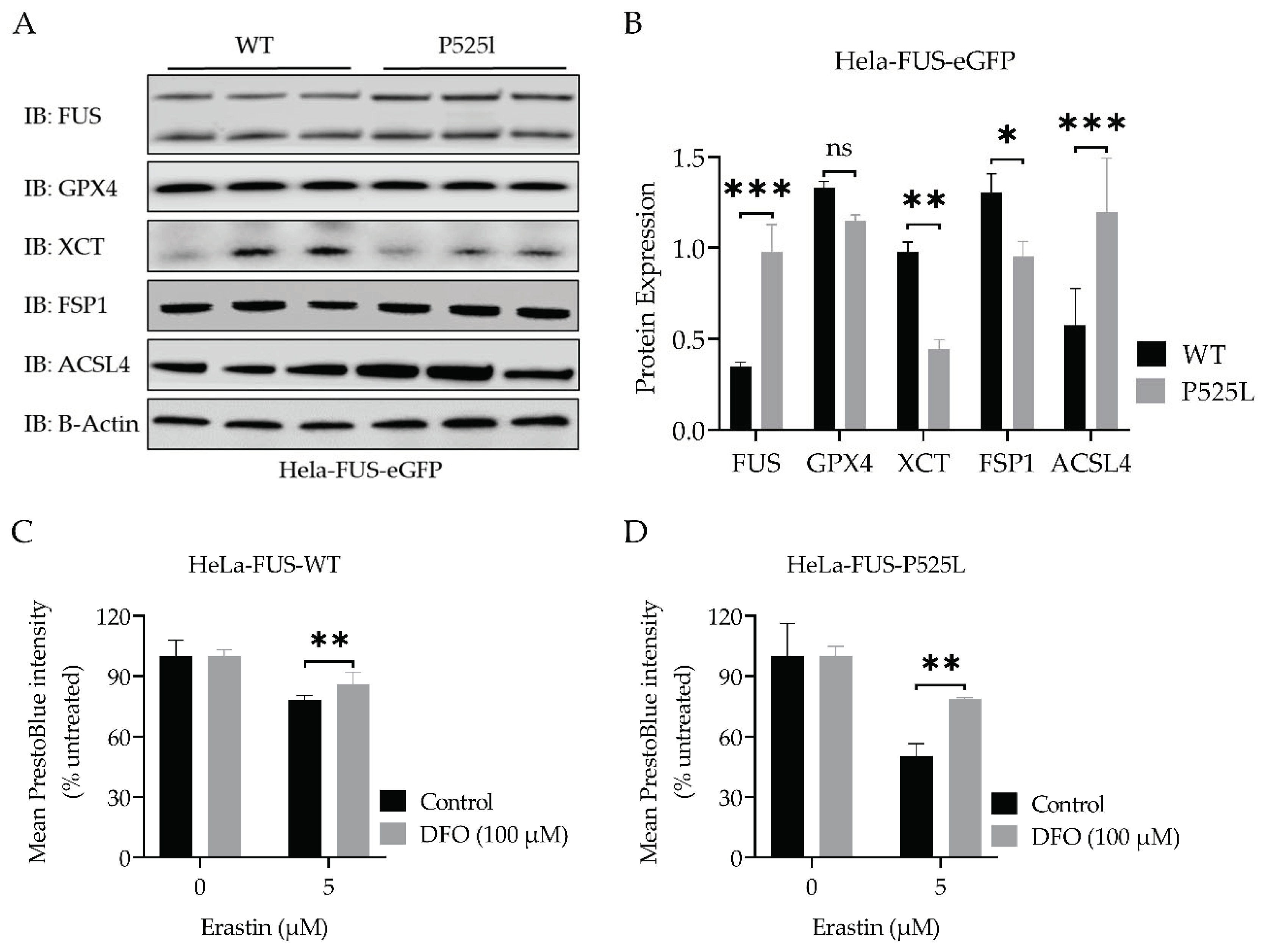
Figure 5.
Mitochondrial calcium uniporter inhibition mitigates FUS mutation-induced redox defense disruption.
Figure 5.
Mitochondrial calcium uniporter inhibition mitigates FUS mutation-induced redox defense disruption.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



