1. Introduction
Shrimp aquaculture has expanded rapidly over the last two decades, yet recurrent disease outbreaks driven by opportunistic bacteria continue to constrain productivity, profitability, and sustainability. Among the most consequential threats, acute hepatopancreatic necrosis disease (AHPND) has been tightly linked to
Vibrio parahaemolyticus lineages that became virulent after acquiring a plasmid encoding PirAB-like toxins, which can trigger acute mortality and severe hepatopancreatic lesions in penaeid shrimp. Foundational work demonstrated that pathogenicity can emerge when
V. parahaemolyticus acquires a virulence plasmid expressing a lethal toxin [
1], and subsequent studies established molecular targets for detection and quantification of Pir-like toxin genes and their association with AHPND outbreaks [
2,
3,
4,
5]. Beyond PirAB-mediated pathology, AHPND-causing strains may also maintain antibacterial type VI secretion systems with diverse effector repertoires, enhancing ecological competitiveness within dense aquaculture microbiomes [
6]. Whole-genome sequencing has further clarified geographic origins and dissemination of outbreak-associated lineages, reinforcing the dynamic evolution and mobility of virulence determinants in shrimp production systems [
7]. Importantly, experimental evidence indicates that PirABVP toxins can aggravate broader vibriosis contexts, emphasizing that management must address both pathogen abundance and virulence potential across the culture cycle [
8,
9].
Vibrios are naturally abundant in coastal and marine systems and exhibit substantial genetic and ecological diversity, which makes hatchery water and larval reservoirs interfaces where background
Vibrio populations can expand under intensive husbandry, feed inputs, and fluctuating physicochemical conditions. Comprehensive syntheses describe the biodiversity of vibrios across ecological niches and the drivers of their emergence in aquatic food webs [
10]. For
V. parahaemolyticus specifically, pathogenesis is multifactorial and depends on host susceptibility, environmental conditions, and the accessory genome, which carries a range of fitness determinants and virulence factors beyond Pir toxins [
11]. Consequently, disease mitigation strategies must target not only the presence of
Vibrio spp. but also the ecological context that enables pathogenic dominance, persistence, and expression of virulence determinants.
Sustainable disease control in aquaculture increasingly prioritizes preventive approaches that reinforce microbial and environmental barriers against opportunistic pathogens. Probiotic bacteria are among the most widely investigated tools, acting through competitive exclusion, production of antimicrobials, nutrient competition, and system-level stabilization of rearing conditions. Early frameworks positioned probiotics as biological control agents in aquaculture and highlighted their potential to reduce disease pressure while supporting host performance [
12]. At a conceptual level, the effectiveness of probiotics is rarely attributable to a single mechanism; rather, it emerges from shifts in community interactions and resource partitioning that favor beneficial guilds and suppress pathogen success. Comparative sequence-based approaches have long been used to differentiate microbial pathogens across hosts and settings [
13], and analogous principles underpin culture-independent profiling in aquaculture: probiotic establishment, pathogen suppression, and functional transitions are best evaluated at the level of whole communities rather than individual isolates alone.
Among candidate probiotics,
Bacillus spp. are attractive because spores tolerate stressful rearing conditions and industrial handling, while vegetative cells can produce a spectrum of antimicrobial metabolites and enzymes that influence both pathogens and water quality. Reviews emphasize that reliable probiotics require systematic screening that includes robust identification, safety assessment, and mechanistic evaluation under relevant environmental conditions [
14,
15,
16]. In shrimp systems,
Bacillus subtilis supplementation has been associated with improved growth performance, enhanced digestive enzyme activity, modulation of immune gene expression, and increased resistance under disease challenge [
17]. Administering
Bacillus strains directly in rearing water has also been reported to enhance water quality and increase resistance against
Vibrio infection, supporting the idea that probiotics can reshape the rearing environment as well as the host–microbe interface [
18]. Broader syntheses further highlight the growing evidence base for
Bacillus as probiotics in aquaculture and the mechanistic versatility of this genus [
19]. At the molecular level,
Bacillus lipopeptides represent a key class of bioactive compounds that can inhibit competitors and shape interbacterial interactions, offering a plausible functional bridge between
Bacillus dominance and anti-
Vibrio effects in mixed communities [
20]. Complementary management strategies, such as biofloc technology, likewise rely on microbial community engineering to improve water quality and reduce pathogen pressure, reinforcing the central role of microbiome structure in disease-resilient production [
21].
Despite strong rationale and extensive research, translating probiotics into consistent field performance remains challenging because efficacy depends on strain selection, dosing, formulation, and environmental context. In practice, native consortia derived from the local production environment may offer advantages in ecological compatibility and persistence, but their deployment requires rigorous validation that connects culture-based antagonism phenotypes with community-wide outcomes. Standardized in vitro approaches for antimicrobial activity assessment provide essential first-line evidence of antagonism and help triage candidates prior to in vivo challenge trials or farm-scale implementation [
22]. However, culture-based assays alone cannot resolve the non-culturable fraction of hatchery microbiomes, nor can they quantify how probiotics restructure broader bacterial networks that influence pathogen success and functional potential.
High-throughput sequencing and reproducible bioinformatic pipelines now enable community-level evaluation at a resolution suitable for mechanism-oriented inference in aquaculture systems. QIIME 2 provides an extensible framework for microbiome data science with provenance tracking, supporting transparent processing and analysis [
23]. DADA2 enables high-resolution inference of amplicon sequence variants (ASVs) without reliance on OTU clustering, improving sensitivity to ecological change across treatments and time [
24]. Complementary preprocessing steps, including adapter and quality trimming with Cutadapt and Trimmomatic, support accurate recovery of biological signal from raw reads [
25,
26], while VSEARCH provides a versatile open-source toolkit for key operations such as dereplication and chimera-related procedures when needed [
27]. Taxonomic assignment commonly relies on curated rRNA databases such as SILVA [
28], and primer choice is a critical determinant of coverage and bias in bacterial diversity studies [
29]. In R, phyloseq supports reproducible microbiome analysis and visualization by integrating feature tables, taxonomy, metadata, and derived diversity metrics in a unified structure [
30]. When the analytical aim extends beyond description toward identifying taxa that differ across experimental conditions, DESeq2 offers moderated dispersion estimation and fold-change inference that is widely applied to sequencing count data [
31].
In addition to taxonomic restructuring, functional interpretation is increasingly central to evaluating whether probiotic-associated community shifts plausibly translate into pathogen suppression. Tools such as PICRUSt2 can infer functional potential from amplicon data [
32], and predicted pathways can be interpreted using reference resources such as KEGG [
33], eggNOG [
34], and the COG database [
35]. Although functional inference from 16S data has limitations, integrating predicted functions with phenotypic assays (e.g., inhibition halos) provides a pragmatic framework to test whether community restructuring aligns with bioactive potential relevant to antimicrobial activity, stress response, or competitive fitness.
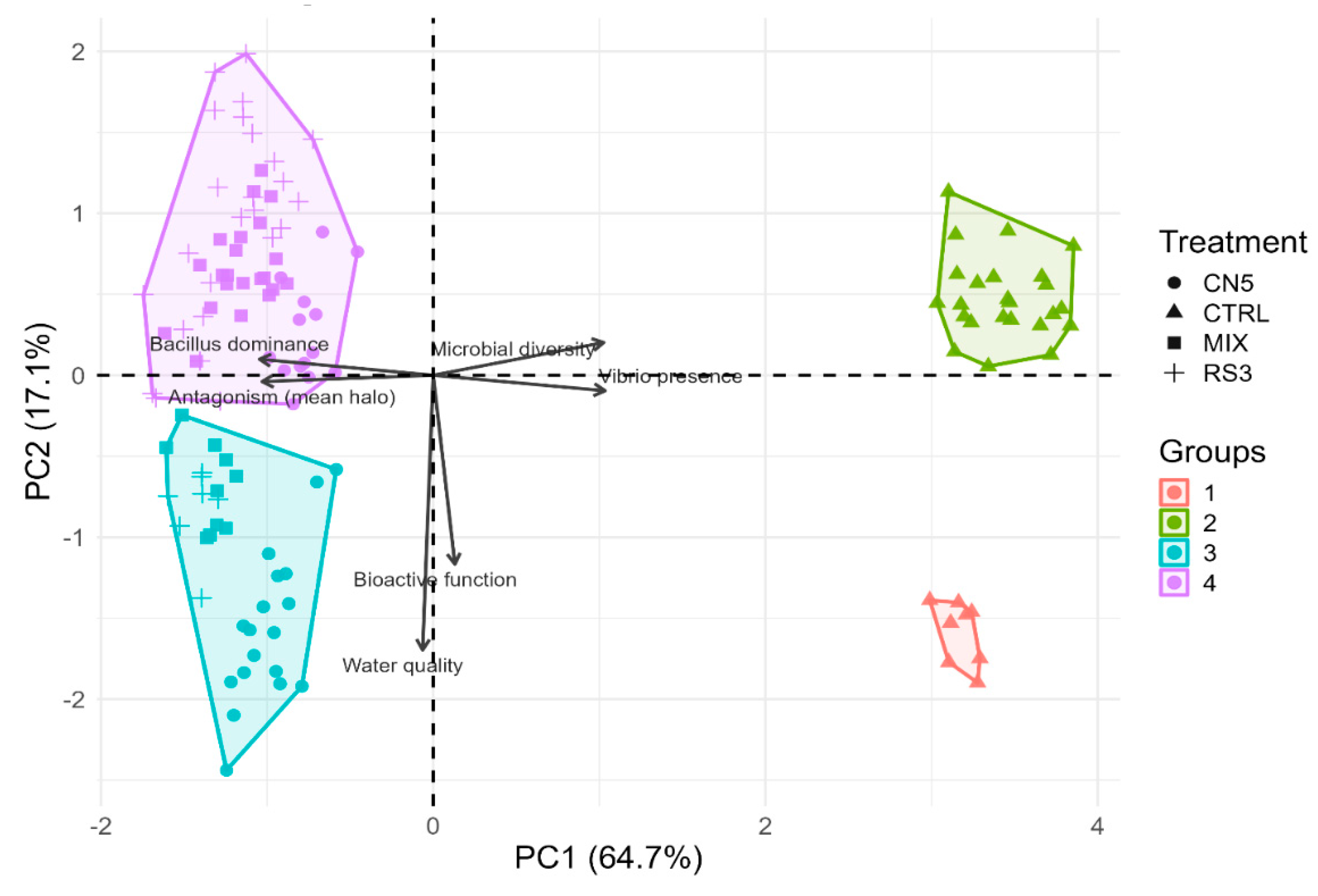
To move from descriptive microbiomics to mechanism-oriented hypotheses about disease control, integrating multivariate and structural modeling is particularly valuable. Phylogeny-aware beta-diversity metrics such as UniFrac support robust comparison of communities across experimental conditions [
36], while ordination approaches summarize complex ecological gradients and highlight treatment-specific regimes. Principal component analysis (PCA) remains a cornerstone method for dimension reduction and visualization of multivariate patterns [
38,
39], with standardized workflows available through tools such as FactoMineR [
40]. Beyond ordination, structural equation modeling (SEM) provides a formal framework to quantify directed relationships among latent constructs and observed indicators when community structure, environmental variation, and functional outcomes interact. Classical reliability metrics such as Cronbach’s alpha support internal consistency assessment of multi-indicator constructs [
41], while convergent and discriminant validity can be evaluated using established criteria including Fornell–Larcker and the heterotrait–monotrait ratio (HTMT) [
42,
43]. For prediction-oriented, multi-construct models in complex biological systems, variance-based SEM using partial least squares (PLS-SEM) is widely used because it can accommodate complex models and does not require strict distributional assumptions [
44,
45,
46]. In parallel, predictive machine-learning approaches can provide complementary validation by testing whether multivariate indices learned from community and environmental data generalize to held-out observations, strengthening confidence that identified drivers are not merely descriptive but also predictive of antagonistic outcomes.
From an ecological standpoint, pathogen control in shrimp hatcheries is best viewed as management of a dynamic microbial meta-community rather than suppression of a single taxon. Virulence plasmids and toxin repertoires explain why specific
V. parahaemolyticus lineages can trigger AHPND [
1,
2,
3,
4,
5], yet disease expression is shaped by background community competition, resource availability, and physicochemical stressors that modulate host susceptibility and microbial growth rates [
6,
7,
8,
9,
10,
11]. Accordingly, interventions that shift the community toward stable, competitive, and functionally protective states may reduce the probability that toxigenic vibrios reach critical abundances or express virulence determinants. Within this logic,
Bacillus-based probiotics remain among the most practical tools for community-level engineering because spores withstand storage and delivery constraints, and
Bacillus metabolism can contribute both direct antagonism and indirect system stabilization [
14,
15,
16,
17,
18,
19,
20,
21]. Nevertheless, variability across farms persists, often reflecting mismatches between probiotic strains and local conditions—an argument for prioritizing native consortia that are already adapted to the production environment.
Objective of this study. In this context, the present study aimed to evaluate native probiotic consortia (CN5 and RS3), individually and as a mixed consortium (MIX), for their capacity to suppress
V. parahaemolyticus and restructure hatchery water microbiomes across a 30-day time course, relative to a no-probiotic control (CTRL). Specifically, we combined (i) standardized in vitro antagonism screening to quantify anti-
Vibrio activity [
22], (ii) 16S rRNA gene (V3–V4) amplicon profiling with reproducible pipelines to characterize taxonomic and diversity shifts [
23,
24,
25,
26,
27,
28,
29,
30,
31], (iii) functional inference to estimate bioactive potential relevant to pathogen suppression [
32,
33,
34,
35], and (iv) integrative multivariate and structural modeling (PCA and PLS-SEM) to test mechanistic relationships linking water quality, microbial diversity,
Bacillus dominance,
Vibrio presence, inferred bioactive function, and antagonistic outcomes [
36,
38,
39,
40,
41,
42,
43,
44,
45,
46]. By integrating phenotype, community composition, inferred function, and mechanistic modeling, this work seeks to provide a high-resolution, field-relevant evaluation of native probiotic consortia as scalable biocontrol tools for disease-resilient shrimp hatchery management.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, B.P-G., K. R-P., T.T-G., S. S-V. and E. R-N.; methodology, B.P-G., K. R-P., T. T-G, W. B-Q., S. V-A. and E. R-N.; software, R.P-P., S. S-V., S. V-A. and D. O-F.; validation, R.P-P., S. S-V., S.V-A. and M.V-V.; formal analysis, R.P-P., S. S-V., S. V-A. and M. V-V.; investigation, B.P-G., K. R-P, T. T-G. D. O-F. and E. R-N.; resources, B. P-G. and D. O-F.; data curation, R.P-P., S. S-V., S. V-A. and M. V-V.; writing—original draft preparation, B. P-G.; writing—review and editing, B.P-G., K. R-P. S. V-A. and M. V-V.; visualization, B.P-G., K. R-P., T. T-G., W. B-Q., and E. R-N.; supervision, B.P-G., K. R-P., W. B-Q., and D. O-F. and E. R-N.; project administration, E.R-N.; funding acquisition, B. P-G. All authors have read and agreed to the published version of the manuscript
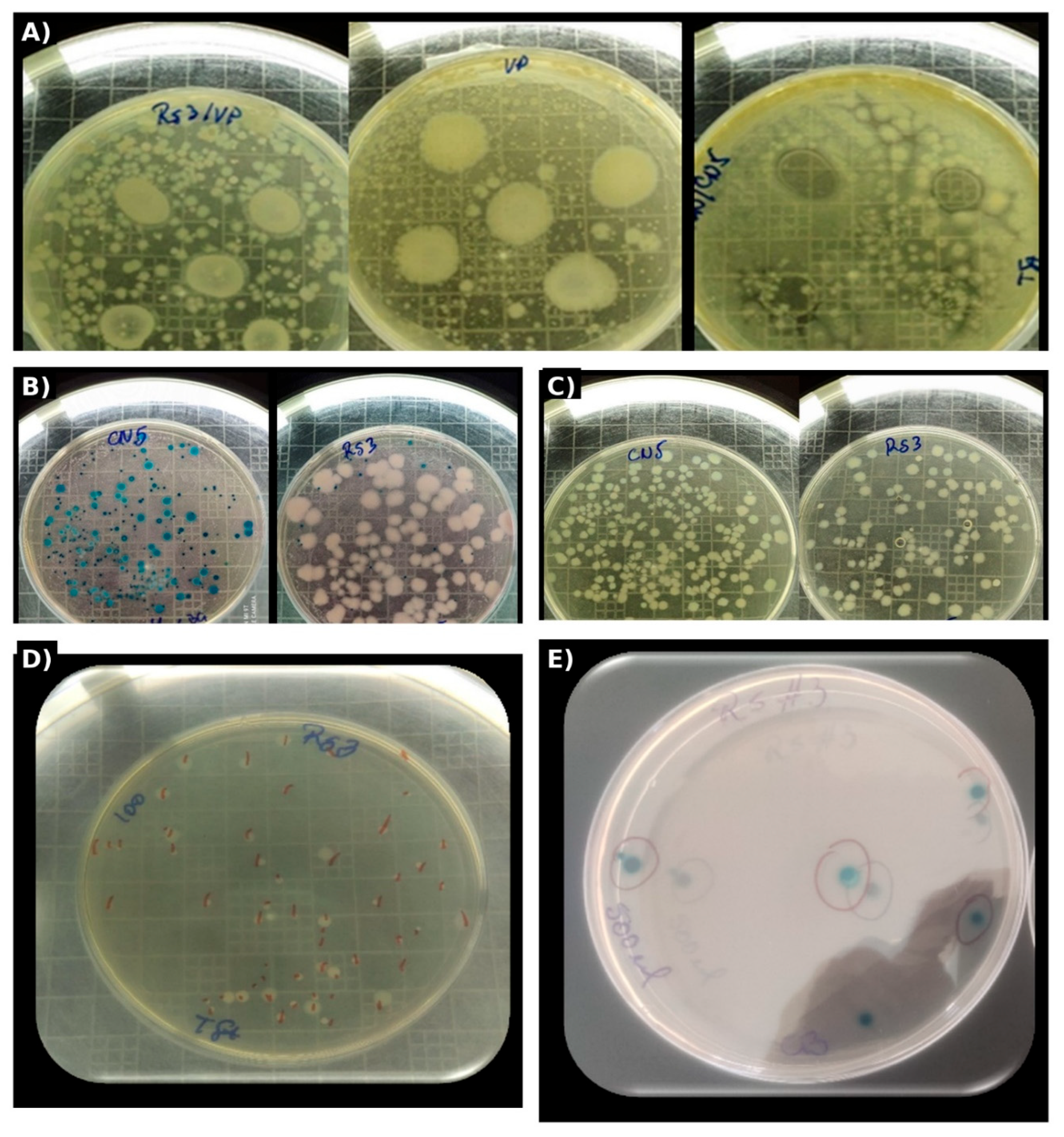
Figure 1.
A–E. Differential growth of CN5 and RS3 on TSA and Chromagar™ Bacillus and qualitative evidence of competitive exclusion against V. parahaemolyticus in simultaneous inhibition assays.
Figure 1.
A–E. Differential growth of CN5 and RS3 on TSA and Chromagar™ Bacillus and qualitative evidence of competitive exclusion against V. parahaemolyticus in simultaneous inhibition assays.
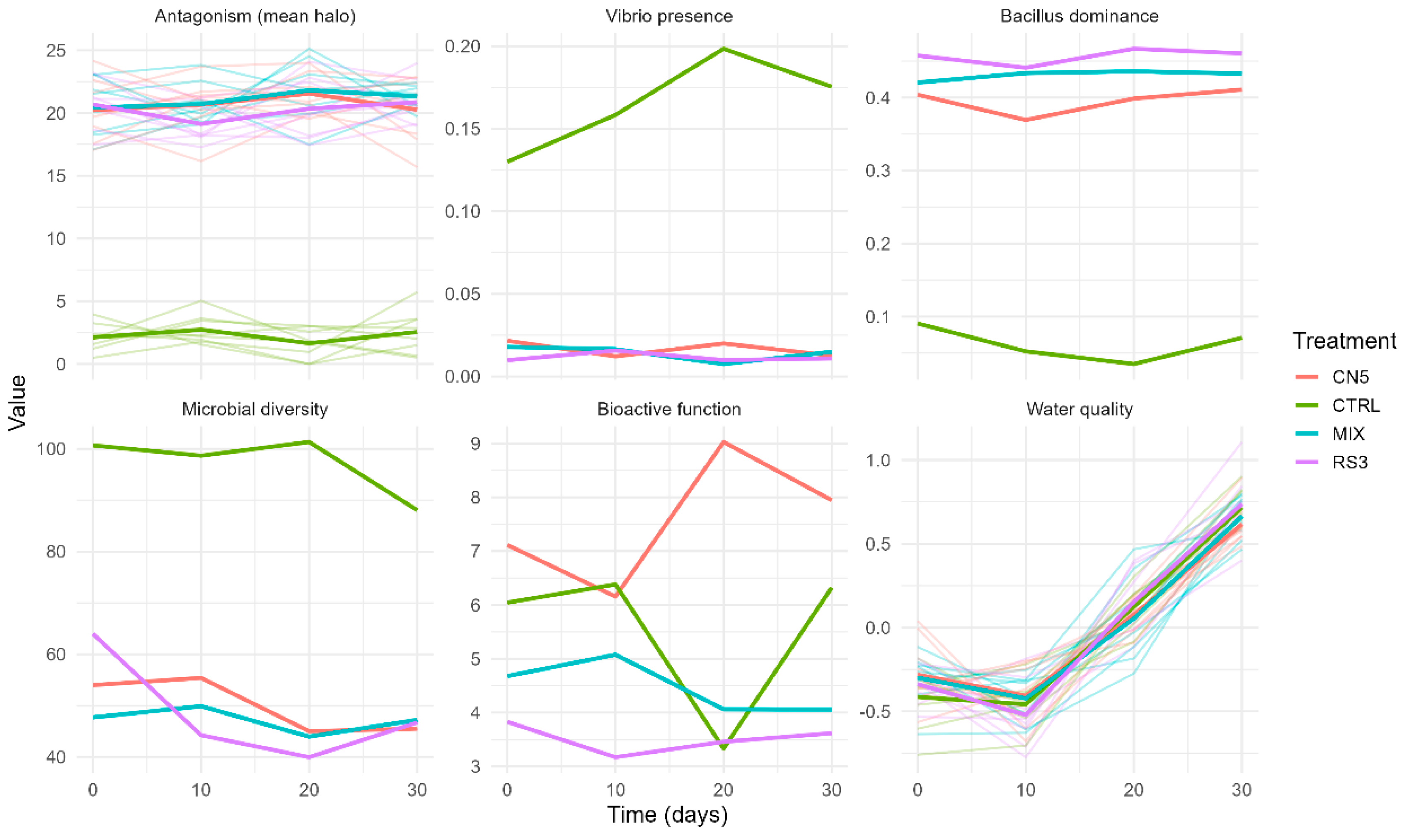
Figure 2.
Temporal dynamics (replicates + treatment mean) of antagonism and SEM-aligned indices across time (0–30 days). Thin lines represent individual replicates and thick lines represent treatment means. Panels show: antagonism (mean inhibition halo), Vibrio presence, Bacillus dominance, microbial diversity, bioactive function, and water quality (z-index).
Figure 2.
Temporal dynamics (replicates + treatment mean) of antagonism and SEM-aligned indices across time (0–30 days). Thin lines represent individual replicates and thick lines represent treatment means. Panels show: antagonism (mean inhibition halo), Vibrio presence, Bacillus dominance, microbial diversity, bioactive function, and water quality (z-index).
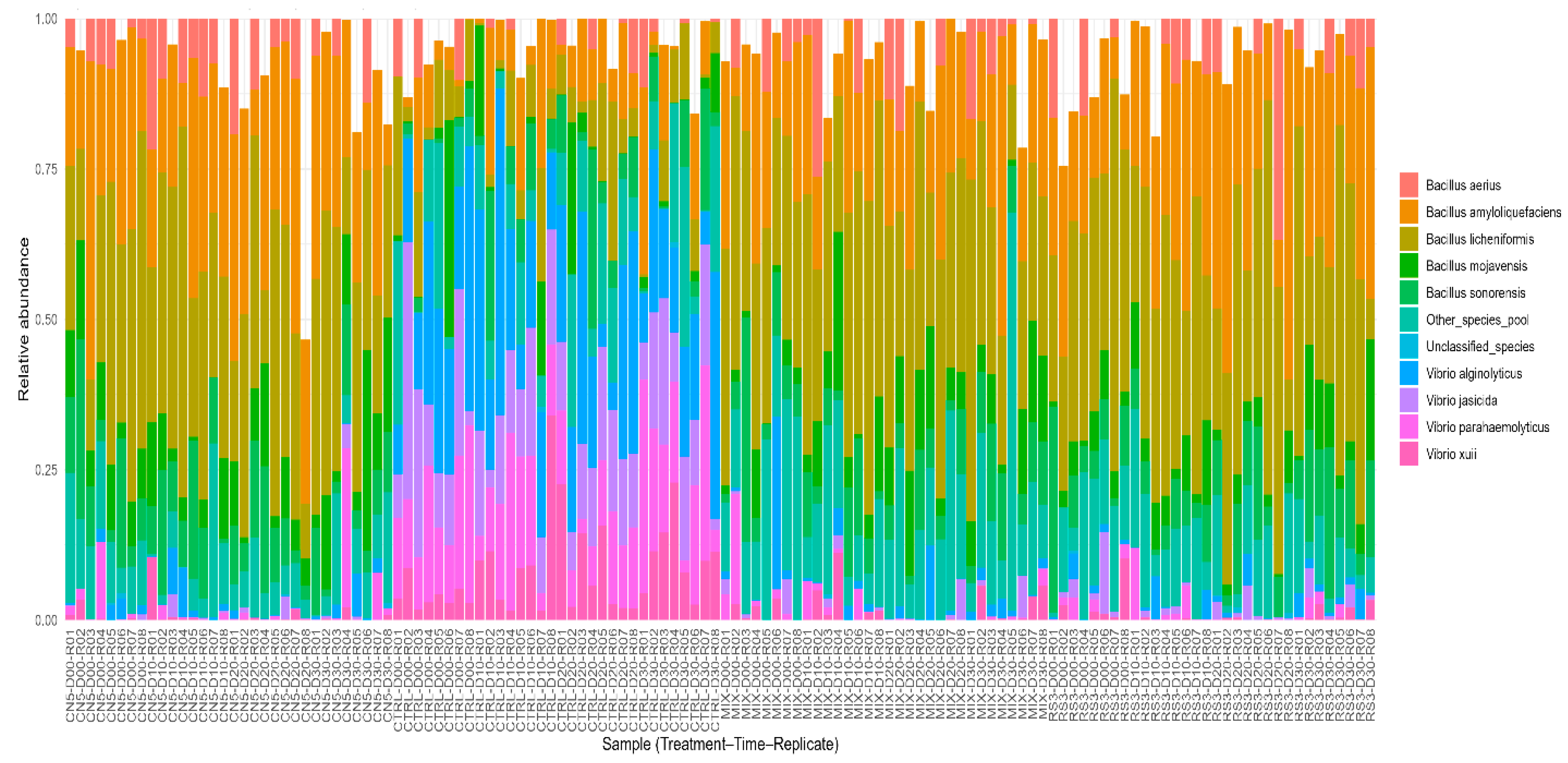
Figure 3.
Species-level community composition across treatments and time (n = 128). Each bar represents one observation (Treatment–Time–Replicate). Colors denote relative abundances of detected species and aggregated categories (“Other_species_pool” and “Unclassified_species”).
Figure 3.
Species-level community composition across treatments and time (n = 128). Each bar represents one observation (Treatment–Time–Replicate). Colors denote relative abundances of detected species and aggregated categories (“Other_species_pool” and “Unclassified_species”).
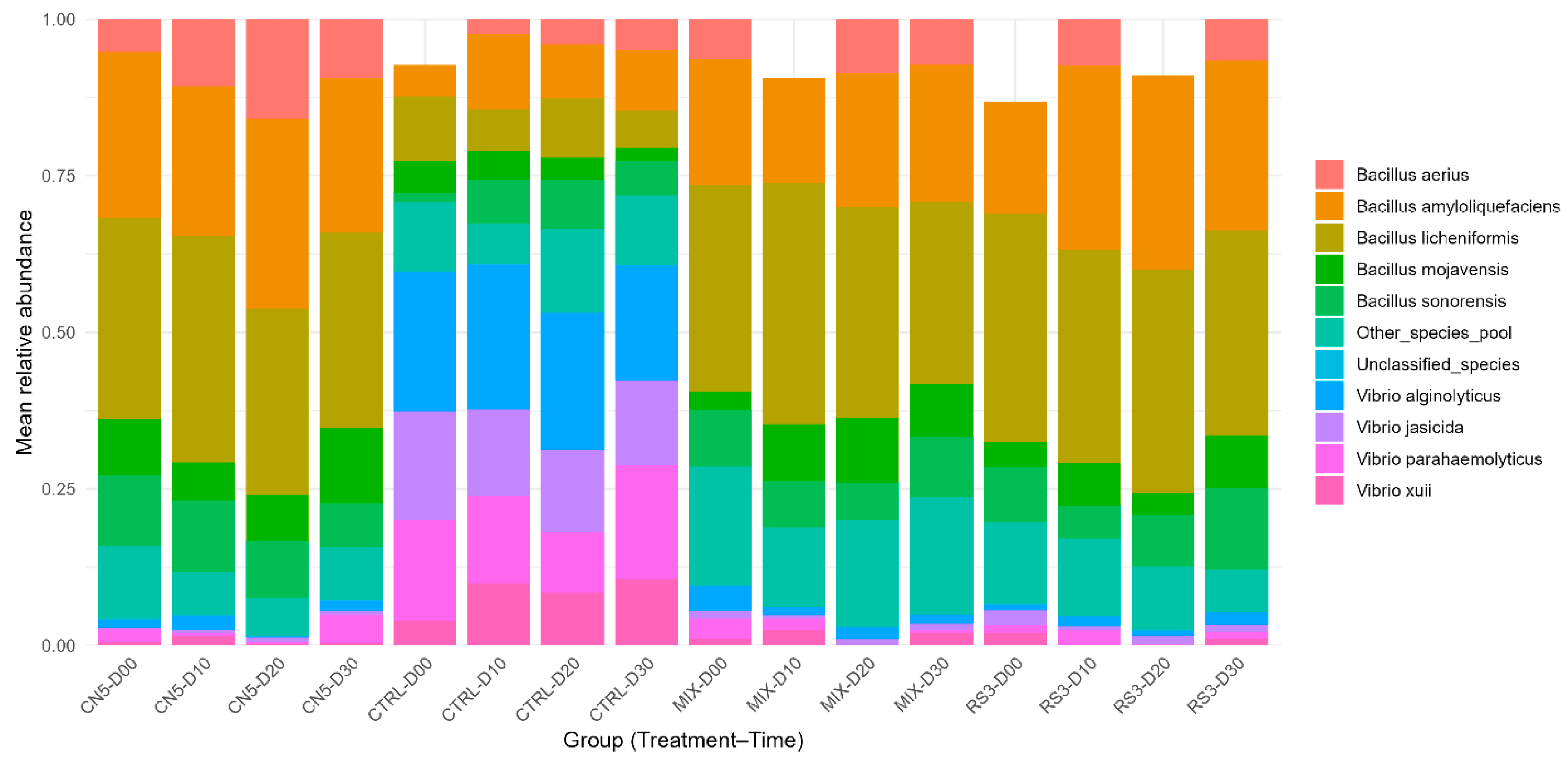
Figure 4.
Mean species-level composition by treatment and time. Stacked bars represent mean relative abundances (n = 8 replicates per treatment–time group) for CTRL, CN5, MIX, and RS3 at 0, 10, 20, and 30 days.
Figure 4.
Mean species-level composition by treatment and time. Stacked bars represent mean relative abundances (n = 8 replicates per treatment–time group) for CTRL, CN5, MIX, and RS3 at 0, 10, 20, and 30 days.
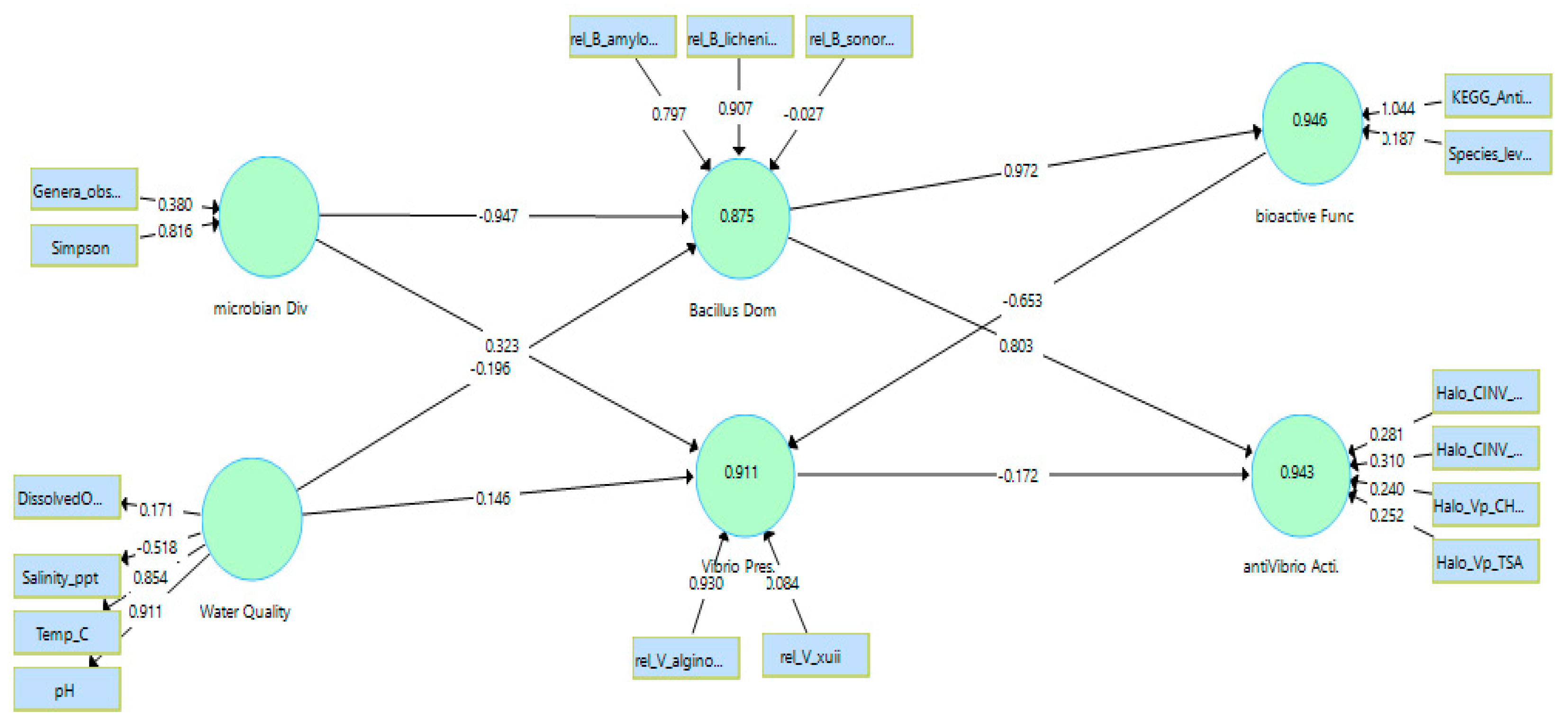
Figure 6.
PLS-SEM structural model linking water quality, microbial diversity, Bacillus dominance, bioactive function, Vibrio presence, and anti-Vibrio activity. Values on endogenous constructs represent R²; values on arrows represent standardized path coefficients (β).
Figure 6.
PLS-SEM structural model linking water quality, microbial diversity, Bacillus dominance, bioactive function, Vibrio presence, and anti-Vibrio activity. Values on endogenous constructs represent R²; values on arrows represent standardized path coefficients (β).
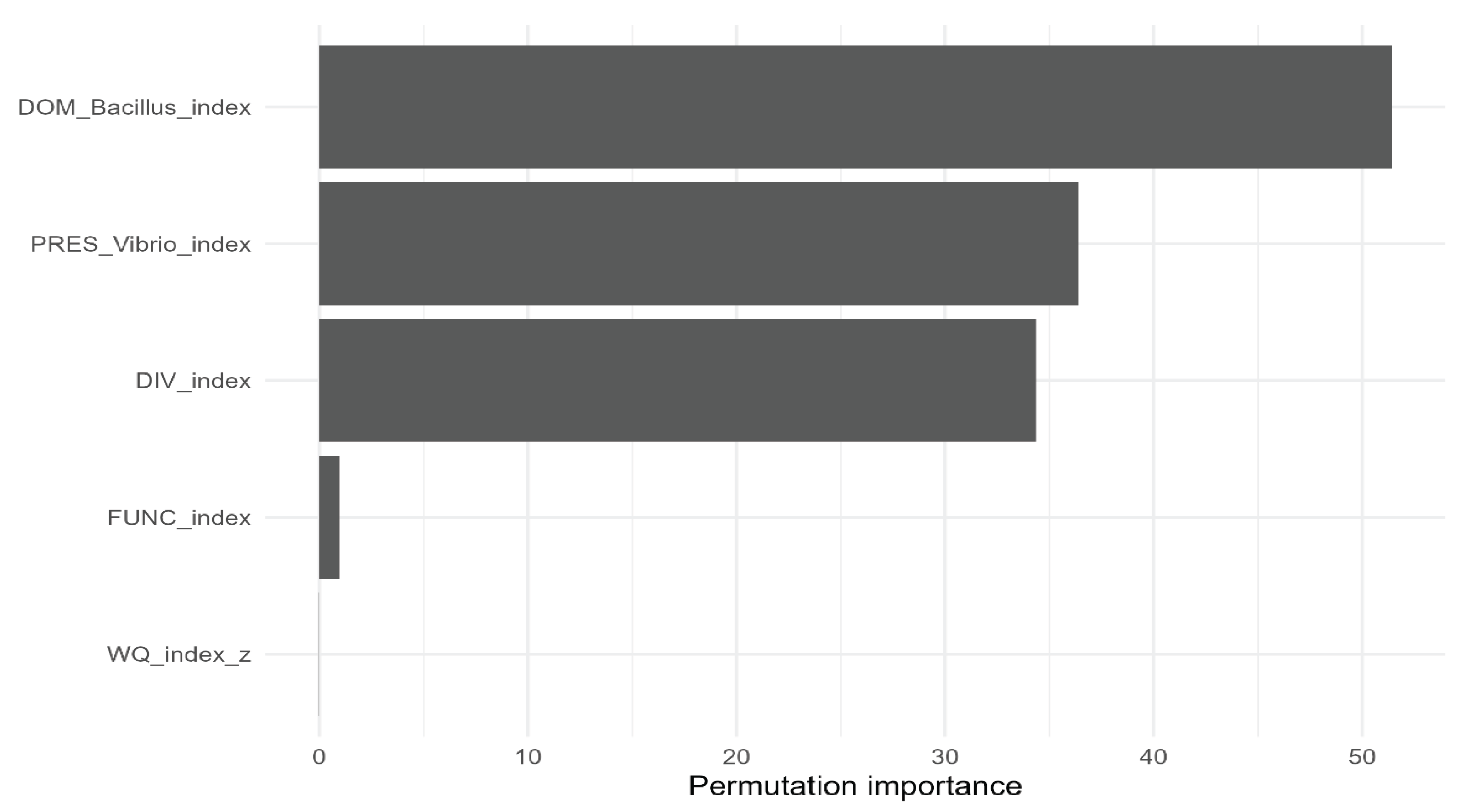
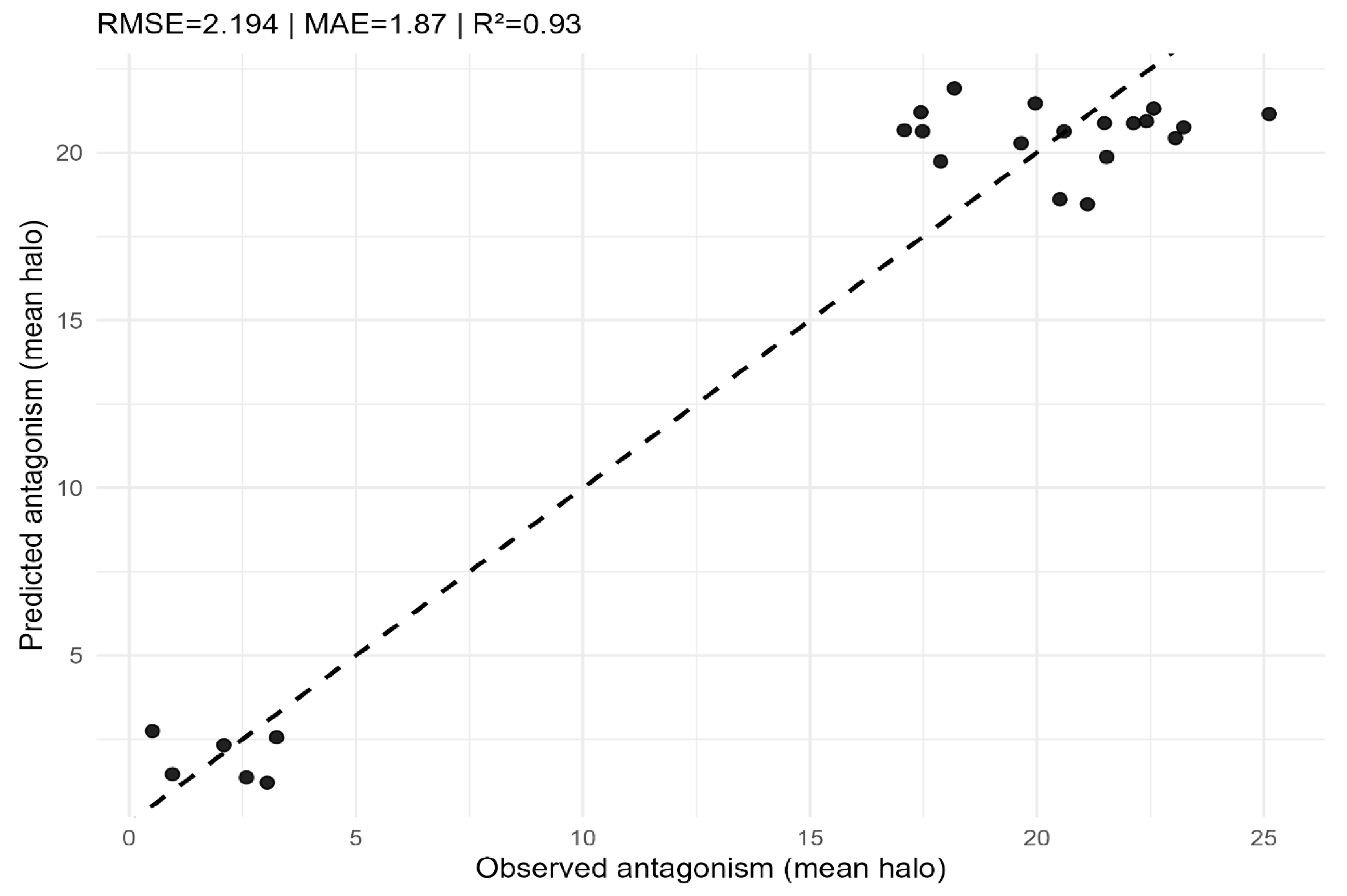
Figure 7.
Random Forest regression: observed vs. predicted antagonism (mean halo) on the test set. The dashed line represents the 1:1 reference. Performance metrics (RMSE, MAE, R²) are reported in the panel.
Figure 7.
Random Forest regression: observed vs. predicted antagonism (mean halo) on the test set. The dashed line represents the 1:1 reference. Performance metrics (RMSE, MAE, R²) are reported in the panel.
Table 1.
Experimental design and sampling structure (n = 128).
Table 1.
Experimental design and sampling structure (n = 128).
| Factor |
Levels |
Details |
| Time |
4 |
0, 10, 20, 30 days (10-day intervals) |
| Treatment |
4 |
CN5, RS3, MIX (CN5+RS3), CTRL |
| Replication |
8 |
Independent biological replicates per treatment–time |
| Total observations |
128 |
4 × 4 × 8 |
Table 2.
Evaluated variables, measurement, and analytical mapping.
Table 2.
Evaluated variables, measurement, and analytical mapping.
| Domain (SEM-aligned) |
Operational definition |
Data source |
Notes / related refs. |
| Anti-Vibrio activity |
Mean inhibition halo (mm) |
Culture assay |
In vitro antimicrobial evaluation [22] |
|
Vibrio presence |
Relative abundance index of Vibrio spp. |
16S taxonomic table |
Vibrio ecology & identification [10,11] |
|
Bacillus dominance |
Relative abundance index of Bacillus spp. |
16S taxonomic table |
Bacillus probiotics in shrimp [17,18,19] |
| Microbial diversity |
Observed genera + Simpson (index) |
ASV table |
Alpha diversity via phyloseq workflow [30] |
| Bioactive function |
Aggregated inferred pathways (e.g., antimicrobial-related) |
PICRUSt2 + KEGG + eggNOG/COG |
Functional inference [32,33,34,35] |
| Water quality |
z-index of DO, salinity, temperature, pH |
Field/lab measures |
Modeled as latent construct in SEM |
Table 3.
Culture-based isolation and screening workflow.
Table 3.
Culture-based isolation and screening workflow.
| Step |
Medium / method |
Purpose |
Output |
| Serial dilution |
10⁻¹–10⁻² in sterile saline |
Reduce density; isolate colonies |
Dilution series |
| Plating |
TSA; Chromagar™ Bacillus
|
General heterotrophs vs. Bacillus enrichment |
Mixed vs. Bacillus-like morphotypes |
| Morphotyping |
Colony traits |
Select distinct candidates |
Candidate isolates |
| Purification & storage |
Re-streak; −80 °C stocks |
Preserve strains/consortia |
CN5, RS3 isolate pools |
Table 4.
Sequencing and bioinformatics pipeline (reproducibility map).
Table 4.
Sequencing and bioinformatics pipeline (reproducibility map).
| Stage |
Tool / database |
Key operation |
Ref. |
| Denoising & ASVs |
DADA2 (via QIIME2) |
Error-correction, ASV inference |
[23,24] |
| Trimming |
Cutadapt; Trimmomatic |
Adapter removal; quality trimming |
[25,26] |
| Auxiliary ops |
VSEARCH |
Dereplication / support operations |
[27] |
| Taxonomy |
SILVA v138 |
Classifier training & assignment |
[28] |
| R integration |
phyloseq |
Alpha diversity; composition |
[30] |
| Differential abundance |
DESeq2 |
Count-model testing |
[31] |
Table 5.
Statistical and modeling plan (what was used vs. not used).
Table 5.
Statistical and modeling plan (what was used vs. not used).
| Analysis objective |
Method |
Output |
| Assay comparisons |
Anderson–Darling; Levene; one-way ANOVA; Tukey (α=0.05) |
Group differences |
| Community composition |
Relative abundance summaries |
Taxa profiles |
| Differential abundance |
DESeq2 |
log2FC + adjusted p |
| Beta-diversity |
UniFrac |
Distance matrix |
| Ordination |
PCA (FactoMineR) |
PC scores/loadings |
| Unsupervised regimes |
k-means in PC space |
Cluster membership |
| Causal/latent modeling |
PLS-SEM |
β paths, R², validity |
| Predictive validation |
Random Forest regression |
RMSE/MAE/R² + importance |