Submitted:
30 December 2025
Posted:
31 December 2025
You are already at the latest version
Abstract
Bacillus amyloliquefaciens is an important agricultural microbial resource. This study focuses on the whole genome analysis and functional characterization of B. amyloliquefaciens SH-53, isolated from the Wuliang Mountain National Nature Reserve in Dali, Yunnan. The genomic feature analysis revealed that the genome of SH-53 contains 27 ribosomal RNA operons, 4,078 protein-coding genes, and 250 prophage-related genes. Additionally, 12 biosynthetic gene clusters (BGCs) for secondary metabolites were predicted, of which 7 are novel gene clusters with unknown functions, showing significant differences compared to the known BGCs of conventional biocontrol strains.Functional potential analysis indicates that SH-53 possesses potential antagonistic activity against plant pathogenic bacteria and can colonize the plant rhizosphere through various mechanisms to exert growth-promoting effects. It is capable of synthesizing multiple antibacterial secondary metabolites, indole-3-acetic acid (IAA), iron carriers, secreting amylase, and efficiently utilizing sulfur sources. The genome also harbors a complete core gene network related to the induced systemic resistance (ISR) and supporting genes that maintain secondary metabolism homeostasis.In conclusion, B. amyloliquefaciens SH-53 exhibits rich biocontrol-related characteristics and unique secondary metabolic potential, indicating promising prospects for its development as an excellent biocontrol agent.
Keywords:
whole genome sequencing
; secondary metabolites
; biocontrol
; plant growth promotion
1. Introduction
In recent years, advances in agricultural microbiology have propelled B. amyloliquefaciens to the forefront of research, primarily due to its exceptional biocontrol potential in supporting sustainable plant disease management [1,2]. However, existing studies have largely focused on a limited number of model strains and those isolated from conventional agricultural environments, resulting in a limited understanding of the genomic features and metabolic potentials of strains derived from unique ecosystems [3]. Under the influence of distinctive environmental selection pressures, specialized ecological niches often serve as important sources for novel metabolic pathways and ecological functions through evolution [4,5]. The Wuliang Mountain National Nature Reserve in Dali, Yunnan, as an ecological hotspot with a typical vertical climate zone and high biodiversity, presents a unique opportunity to systematically explore its microbial resource pool, unveiling functional strains that are both novel and efficient.
This study isolates B. amyloliquefaciens SH-53 from the Wuliangshan National Nature Reserve and aims to reveal its potentially evolved unique genetic and functional characteristics. Early research on B. amyloliquefaciens primarily focused on the isolation, identification, and mechanism analysis of core antifungal lipopeptides, such as iturin, fengycin, and surfactin [6]. With the advancement of genomic technologies, research has progressed from phenotypic studies to genotypic analyses, enabling the precise elucidation of the biosynthetic gene clusters, regulatory networks, and secretion mechanisms of key metabolites [7,8]. Current research indicates that the biocontrol efficacy of B. amyloliquefaciens is driven by a synergistic network composed of various secondary metabolites, including lipopeptides, polyene compounds, and bacteriocins [9,10]. For instance, surfactin can enhance the membrane permeability of iturin, while iron carriers inhibit pathogen growth through nutritional competition. This synergistic multi-component, multi-target model is key to achieving efficient pest and disease control [11,12].
Therefore, this study aims to conduct a comprehensive whole-genome analysis and functional validation of B. amyloliquefaciens SH-53, isolated from the special ecological niche of the Wuliangshan National Nature Reserve in Dali, Yunnan. By integrating bioinformatics analyses with physiological and biochemical experiments, the study systematically elucidates the mechanisms by which SH-53 promotes plant growth. Furthermore, from the perspective of “genomic analysis + functional validation,” this research seeks to explore the genetic basis and application potential of SH-53 as a microbial inoculant in greater depth.
2. Materials and Methods
2.1. Genomic Sequencing and Annotation of Strain SH-53
Using the Whole Genome Shotgun (WGS) strategy, libraries with different insert fragment lengths were constructed, and sequencing was performed using a combination of second-generation and third-generation sequencing technologies. Specifically, short reads were generated using the Illumina NovaSeq platform (2×150 bp paired-end reads), while long reads were obtained using the PacBio Sequel platform.After quality control of the sequencing data, genome assembly was performed with software tools such as HiFiasm, Unicycler, and Flye. Additionally, the Pilon software was utilized to correct the contigs obtained from third-generation sequencing using high-quality short reads from Illumina, ultimately yielding a complete and high-quality genome sequence.Following the assembly of the genome, repeat sequences within the genome were predicted de novo using RepeatModeler (v1.0.8) and RepeatMasker (v4.0.5). For functional annotation of protein-coding genes, systematic comparisons were conducted with public databases, including the NCBI non-redundant protein database (NR), eggNOG, KEGG, Swiss-Prot, Gene Ontology (GO), Pfam, the Carbohydrate Active Enzymes database (CAZy), and the Antibiotic Resistance Database (CARD).
2.2. Prediction of NP-BGCs in Strain SH-53 Genome
The BGCs within the genome of strain SH-53 were identified and analyzed using antiSMASH version 8.0.4. This tool facilitated the prediction of the structural formulas of secondary metabolites encoded by the identified gene clusters [13].
2.3. Evaluation of Strain Bioactivity and Seed Germination Promotion
2.3.1. Detection of IAA Production Capability
(1) Qualitative Analysis of IAA Production
To assess the IAA production capability of strain SH-53 qualitatively, a single colony was picked and inoculated into KB medium (containing 20.0 g/L peptone, 1.5 g/L dipotassium hydrogen phosphate (K₂HPO₄·H₂O), 1.5 g/L magnesium sulfate (MgSO₄·7H₂O), 15.0 mL/L glycerol, and 1 g/L L-Tryptophan). The culture was incubated at 30 °C with shaking at 180 rpm for 24 hours under sterile conditions.After incubation, 1 mL of the fermentation broth was taken into a centrifuge tube and rapidly mixed with 4 mL of Sackowcki’s color reagent. The mixture was allowed to stand at room temperature in the dark for 40 minutes to develop color. The color change was then observed and recorded; a pink coloration indicated a positive result, confirming that the strain was capable of secreting IAA.
(2) Quantitation
Standard Curve Preparation: Weigh 10 mg of IAA standard and dissolve it in a small volume of ethanol. Dilute the solution to 100 mL with distilled water to obtain a stock solution with a concentration of 100 μg/mL. Perform gradient dilution to prepare a series of standard solutions with concentrations of 0, 4, 8, 12, 16, 20, and 24 μg/mL (note: the original “24 g/mL” is corrected to μg/mL to conform to experimental logic). Pipette 1 mL of each standard solution, add 4 mL of color reagent, and incubate the mixture at 30 °C in the dark for 40 min to allow color development. Measure the absorbance value at OD₅₃₅ using a spectrophotometer, and plot the standard curve with IAA concentration as the abscissa and OD₅₃₅ value as the ordinate.
Assay Method: Prepare 100 mL of King’s B (KB) medium. Inoculate the nutrient broth (NB) medium (containing 10 g/L peptone, 3 g/L beef extract, 5 g/L NaCl, pH 7.0) with 1% (v/v) of bacterial suspension with a calibrated OD₆₀₀ value. After incubation for 1 day, centrifuge both the bacterial suspension and the blank control at 12,500 r/min for 10 min. Collect 4 mL of the supernatant, add an equal volume (4 mL) of colorimetric reagent, and incubate the mixture in the dark for 40 min. Zero the spectrophotometer with the blank colorimetric solution, then measure the OD₅₃₅ value of each sample. Each treatment is replicated three times to ensure accuracy. Conduct continuous determination for 7 days, record the color changes at different fermentation time points, and calculate the IAA yield of the strain at each time point by referring to the prepared standard curve.
2.3.2. Detection of Siderophore Production Capability of Strains
200 µL of spore suspension with a concentration of 1 x 10^6/mL to 2 x 10^6/mL was added to 200 mL of iron-free Czapek liquid medium. The flask containing the medium (500 mL) was then placed on a shaking incubator (model: HS-200B) at 30 °C and 150 r/min for 48 hours. After cultivation, 2-5 mL of the culture was removed, filtered through a 0.22 µm sterile filter membrane, and mixed with an equal volume of CAS detection solution. After standing for 1 hour, the OD630 was measured using a full-wavelength microplate reader (model: MultiskaGO), recorded as “As.” The OD630 of the un-inoculated liquid medium was measured using the same method as a reference value, recorded as “Ar.” The concentration of siderophores was expressed in siderophore units (SU), calculated as SU = [(Ar - As) / Ar] x 100%. Measurements were repeated three times, and the average value was used for comparative analysis.
2.3.3. Physiological and Biochemical Experiments of Strain
The physiological and biochemical tests were performed on strain SH-53, including hydrogen sulfide (H₂S) production test and starch hydrolysis test [18], with each physiological and biochemical test conducted in triplicate.
H₂S Production Test: LB solid medium was prepared (10 g/L tryptophan, 5 g/L yeast extract, 10 g/L NaCl), and sterilized under high pressure at 121 °C. After cooling to approximately 50 °C, a mixed solution of 1 mL of 10% (w/v) sodium thiosulfate (Na₂S₂O₃) and 1% (w/v) lead acetate (Pb(CH₃COO)₂), both filtered through a 0.22 µm filter membrane for sterilization, was added per 100 mL of medium. The mixture was quickly stirred and poured into Petri dishes. Pure colonies of strain SH-53 were picked with a sterile toothpick and inserted vertically into the depths of the medium. The plates were incubated at 30 °C for 24 hours, and the color change of the medium was observed.
Starch Hydrolysis Test: Strain SH-53 was inoculated onto starch agar plates and incubated at 30 °C for 48 hours. A suitable amount of Lugol’s iodine solution (components: 5.0 g iodine, 10.0 g potassium iodide, made up to 100 mL with distilled water) was then poured over the surface of the plates with colonies. After standing for 1 minute, the results were observed.
2.3.4. Seed Germination Experiment of Strain SH-53
Pure colonies of strain SH-53 preserved on slants were inoculated into 100 mL of NB liquid medium and shaken at 30 °C and 180 r/min for 24 hours. When the OD600 reached approximately 1, this was considered the seed liquid of SH-53. Fermentation was carried out according to the previously optimized formula and culture conditions (sucrose 15.00 g/L, sodium nitrate 15.00 g/L, disodium hydrogen phosphate 5.25 g/L, yeast extract 5.00 g/L, pH 7.5, temperature 30 °C, shaking speed 200 r/min, with a liquid volume of 200 mL for 24 hours) to obtain the fermentation broth of strain SH-53. A blank control group was set up with 2 mL of sterile water, a negative control group with 2 mL of NB liquid medium, treatment group T1 with 2 mL of undiluted fermentation broth, treatment group T2 with 2 mL of 10-fold diluted fermentation broth, and treatment group T3 with 2 mL of 100-fold diluted fermentation broth. Each treatment was repeated three times, and 10 tomato seeds were placed in each plate. The plates were incubated in the dark at 30 °C for 7 days to observe the seed germination status.
3. Results
3.1. Analysis of Genetic Information and Gene Structure Characteristics of the Strain Based on Whole Genome Sequencing
Based on phylogenetic analysis using the universal 16S rRNA primer, the strain SH-53 was subjected to NCBI Blastn search and compared with related species gene sequences in the GenBank database. The results indicated that strain SH-53 belongs to the genus Bacillus, and it resides within the same independent branch as B. amyloliquefaciens DSM7 (Figure 1).
Based on the analysis of Average Nucleotide Identity (ANI) and digital DNA-DNA Hybridization (dDDH), the ANI value between strain SH-53 and B. amyloliquefaciens DSM7 was as high as 99.95%, while the dDDH value was 100%. These values are significantly above the recognized thresholds for prokaryotic species delineation (ANI ≥ 95-96%, dDDH ≥ 70%). This result clearly indicates that strain SH-53 should be classified as B. amyloliquefaciens (Table S1). Furthermore, multi-locus sequence analysis (MLSA) revealed that strain SH-53 is most closely related to B. amyloliquefaciens DSM7 (Table S2).
3.2. Exploration of Bioactivity and Mechanism Analysis Based on Whole Genome Sequencing
The whole genome sequencing results of B. amyloliquefaciens SH-53 (Figure 2) show that its chromosome length is 4,007,020 bp, with a GC content of 45.99% and encoding 4,078 genes. The average length of the coding genes is 864.68 bp, and the chromosome contains 27 rRNA, 87 tRNA, 83 sRNA, and 11 genomic islands encoding a total of 446 genes. These data provide a critical foundation for further investigation of the bioactivity of this strain and its corresponding mechanisms of action.
3.3. Gene Annotation Results
Using the NCBI nr, KEGG, Swiss Prot, GO, COG, TCDB, Pfam, CAZy, and CARD databases, the predicted gene protein sequences were compared with various functional databases using Diamond (E-value ≤ 1e−5). The highest scoring results were selected for annotation (with default identity ≥ 40% and coverage ≥ 40%). The final annotation statistics are shown in Figure 3. A significant number of genes had functional annotations in the NCBI nr, Swiss Prot, Pfam, COG, GO, and KEGG databases, totaling 4,076, 3,655, 3,504, 3,155, 2,410, and 2,875, respectively, which accounts for 99.95%, 89.63%, 85.92%, 77.37%, 59.10%, and 53.21% of the total genes. In the CAZy database, a total of 133 genes were annotated, representing 3.26%. In the TCDB database, 775 genes were annotated, accounting for 19.00%, and in the CARD database, 163 genes were annotated, making up 4.00% of the total gene count.
3.3.1. Analysis Results of COG Database Genes in Strain SH-53
The proteins of B. amyloliquefaciens SH-53 were annotated using the COG database. Figure 4 displays the classification results of the 3,155 genes annotated in strain SH-53 according to COG. The most abundant category was amino acid transport and metabolism, with a total of 320 genes, accounting for 10.14% of the annotated genes. This was followed by transcription, which included 302 genes, representing 9.57% of the annotated genes. Other categories that received annotation include carbohydrate transport and metabolism (263 genes, 8.34%), translation, ribosomal structure and biogenesis (232 genes, 7.35%), cell wall/membrane/envelope biogenesis (213 genes, 6.75%), and signal transduction mechanisms (198 genes, 6.28%).
Additionally, 153 genes (accounting for 4.85%) were identified to have unknown functions, warranting further investigation. Among the 23 subcategories, aside from the functionally unknown genes (S, 4.31%), those belonging to amino acid transport and metabolism (E, 9.02%), transcription (K, 8.52%), carbohydrate transport and metabolism (G, 7.42%), general function prediction (R, 7.16%), and translation, ribosomal structure and biogenesis (J, 6.54%) also exhibited significant abundance. This reveals potential advantages of the strain in metabolic processes, environmental adaptation, and competitive survival.
3.3.2. Gene Analysis Results of Strain SH-53 in the GO Database
Using the GO database, a total of 2,410 genes from strain SH-53 were annotated, accounting for 59.10% of the total annotated genes. This database classifies all annotated genes into three categories (Figure 5), with some genes appearing in multiple classifications. The genes relate to cellular components (1,227 genes, 28.00%), molecular functions (1,890 genes, 43.13%), and biological processes (1,265 genes, 28.87%), allowing for overlap among the categories.In the cellular component functional annotation, 1,227 genes were successfully annotated, with membrane-related genes being the most abundant, exceeding 400 in number. Additionally, genes closely associated with the plasma membrane and cytoplasm were effectively annotated. In the biological process functional annotation, a total of 1,265 genes were annotated, with phosphorylation processes representing the largest number of annotated biological processes within this category. For molecular function annotations, a total of 1,890 genes received annotations, with ATP binding activity identified as a core annotated entry within this category.
3.3.3. Results of KEGG Database Gene Analysis of Strain SH-53
The KEGG annotation database is organized into six primary categories: cellular processes, environmental information processing, genetic information processing, human diseases, metabolism, and biological systems. Among these categories, the metabolism section contained the most extensive information, encompassing a total of 2,875 annotated genes (as illustrated in Figure 6). Notably, when genes were simultaneously annotated across multiple categories, the metabolism category represented the highest proportion, accounting for 2,170 genes (69.84%). This finding aligns closely with the annotation results derived from the COG database.
Excluding the 842 genes assigned to the global and overview maps category, the top three subcategories within the six major categories were carbohydrate metabolism (269 genes, 8.66%), amino acid metabolism (233 genes, 7.50%), and the metabolism of cofactors and vitamins (214 genes, 6.89%). These results largely correspond with the findings from the GO and COG databases, indicating that the strain demonstrates a robust capacity for environmental adaptability and competitive nutrient acquisition. This insight underscores the strain’s significant biological value, particularly in industrial applications and host-microbe interactions.
3.3.4. Annotation of Carbohydrate-Active Enzymes in Strain SH-53
The carbohydrate-active enzymes in strain SH-53 were classified into six main categories: accessory activities, carbohydrate-binding modules, carbohydrate esterases, glycoside hydrolases, glycosyltransferases, and polysaccharide lyases. Among these, the genes encoding glycosyltransferases were notably represented, with a total of 44 genes annotated in this category. The gene counts for the other five categories were as follows: accessory activities (10 genes), carbohydrate esterases (4 genes), glycoside hydrolases (31 genes), glycosyltransferases (44 genes), and polysaccharide lyases (4 genes), as depicted in Figure 7. This distribution indicates a diverse enzymatic toolkit related to carbohydrate metabolism, which may play a crucial role in the strain’s functionality and potential applications.
Further analysis revealed that the genome of strain SH-53 contains multiple key genes involved in the starch degradation pathway, aligning closely with the high proportion of glycoside hydrolases found in its CAZyme annotations. For instance, the amyA gene encodes a precursor form of α-amylase (1,4-α-D-glucan glucanohydrolase), an enzyme that directly hydrolyzes the α-1,4-glycosidic bonds in starch, making it a core enzyme in the initial stages of starch degradation. Additionally, the malL and malZ genes encode highly conserved α-glucosidases that further hydrolyze the oligosaccharides (such as maltose) produced from starch degradation into glucose, providing the strain with a readily available carbon source. This enzymatic capability enhances strain SH-53’s potential for efficient starch utilization and suggests its application in processes involving carbohydrate metabolism.
It is noteworthy that this strain also contains genes such as sacA and sacC, which encode proteins from glycoside hydrolase family 32 and sucrose-6-phosphate hydrolase, respectively. These enzymes are capable of catalyzing the hydrolysis of sucrose and its derivatives, effectively breaking down sucrose-6-phosphate into glucose-6-phosphate and fructose. This functionality not only broadens the carbon source utilization range of SH-53, allowing it to utilize disaccharides like sucrose, but also corroborates the starch degradation capabilities observed in experiments (Figure 7).Considering both the CAZyme annotations and the functional analysis of key genes, it is evident that SH-53 possesses a diverse carbohydrate metabolism capacity. Its genetic basis for enzyme production highlights its significant potential, particularly in the degradation and utilization of starch and sucrose. This enzymatic versatility enhances the strain’s applicability in various industrial and biotechnological processes involving carbohydrate substrates.
3.3.5. Prediction of NP BGCs in the Genome of Strain SH-53
Using the antiSMASH v8.0.4 bioinformatics analysis platform, we predicted secondary metabolite BGCs in the whole genome sequence of B. amyloliquefaciens SH-53. A total of 12 potential secondary metabolite biosynthetic gene clusters were identified (Table 2), and their genomic localization is illustrated in Figure 8. These clusters include 3 types of terpenes, 3 types of transAT-PKS, 1 type of lanthipeptide III, 2 types of NRPS, and 2 types of PKS. Among these gene clusters, 5 showed high similarity to known compound gene clusters, namely surfactin, bacillaene, fengycin, bacillibactin, and bacilysin, as depicted in the biosynthetic gene cluster map shown in Figure S1. The remaining 7 gene clusters did not find homologous gene clusters, which may provide potential metabolic reserves for the strain to cope with various stress pressures in complex environments, thereby allowing it to better adapt to different ecological environments and exert biocontrol functions. Preliminary antagonistic experiments indicated that strain SH-53 exhibited good inhibitory effects against various plant pathogenic bacteria [14]. The secondary metabolite prediction results based on the genome in this study well explain this phenotype. We speculate that its broad-spectrum antagonistic ability against pathogens is not reliant on a single mechanism but is rather the result of the synergistic action of various metabolites, including these lipopeptides (NRPS/PKS), bacteriocins (lanthipeptides), and siderophores. These abundant BGCs provide a significant metabolic potential for competing for nutrients, occupying ecological niches, and efficiently performing biocontrol functions in complex rhizosphere environments.

Table 1.
Secondary metabolite biosynthetic gene clusters in strain SH-53 predicted by antiSMASH.
| Cluster ID | Nucleotide Length (bp) | Gene Cluster Type | Most similar known cluster | Similarity Confidence |
| 1 | 65,411 | NRPS | surfactin | High |
| 2 | 22,598 | lanthipeptide-class-iii | ||
| 3 | 41,245 | PKS-like | butirosin A/butirosin B | |
| 4 | 20,744 | terpene | ||
| 5 | 110,124 | NRPS,T3PKS,transAT-PKS | bacillaene | High |
| 6 | 110,422 | NRPS,betalactone,transAT-PKS | fengycin | High |
| 7 | 21,884 | terpene | ||
| 8 | 41,101 | T3PKS | ||
| 9 | 20,891 | terpene-precursor | ||
| 10 | 45,529 | NRPS | ||
| 11 | 65,250 | NRP-metallophore,NRPS,RiPP-like,terpene-precursor | bacillibactin | High |
| 12 | 41,419 | other | bacilysin | High |
Figure 8.
Genomic localization of secondary metabolite biosynthetic gene clusters in B. amyloliquefaciens SH-53.
Figure 8.
Genomic localization of secondary metabolite biosynthetic gene clusters in B. amyloliquefaciens SH-53.
3.3.6. Gene Analysis of ISR in the Strain
Based on the annotation results from the KEGG database, we further annotated the ISR genes (Table S3). The results showed that the genome of B. amyloliquefaciens SH-53 contains a series of genes associated with plant root colonization and biofilm formation. The genome includes a variety of core genes related to ISR, such as lipopeptide antibiotic synthesis genes (ppsD, ppsE, ituA, ituB, ituC, licA, licB, licC), antimicrobial peptide (bacteriocin) synthesis and transport genes (bacG, bacF, bacE, bacD), as well as quorum sensing and regulation genes (luxS, ahlD, comA, slrR). Additionally, we identified many genes associated with iron nutrition competition, including siderophore synthesis genes (entC, entE) and siderophore transport genes (fecE, fhuA, efeO). These genes can inhibit pathogen growth by chelating soil iron ions, thereby indirectly enhancing plant disease resistance. The genome also contains genes responsible for synthesizing volatile ISR signal molecules (butB, acoA, acoB) as well as metabolic support genes that maintain secondary metabolite homeostasis (murA, atpA, talB, amyA). Therefore, B. amyloliquefaciens SH-53 can enhance plant immunity and induce systemic resistance through the synergistic action of multiple types of signaling molecules, while exhibiting excellent biocontrol potential.
3.3.7. Antibiotic Resistance Analysis of Strain SH-53 (CARD)
The genome of strain SH-53 was compared with the CARD database, resulting in the identification of 268 resistance genes (Table S4). The specific classification of these resistance genes is illustrated in Figure 9. Among these, the proportion of resistance genes was relatively high for peptide antibiotics (11.51%), tetracycline antibiotics (9.59%), macrolide antibiotics (8.63%), fluoroquinolone antibiotics (6.95%), penicillins (6.71%), and disinfectants and preservatives (6.24%).
3.4. Evaluation of Biological Activity and Seed Germination Promotion by Strain SH-53
3.4.1. Evaluation of Biological Activity of Strain SH-53
The KEGG database annotation results indicate that strain SH-53 possesses multiple potentials for plant growth promotion, including the secretion of IAA, production of hydrogen sulfide (H2S), phosphate transport and assimilation, and synthesis of siderophores. Previous studies have also demonstrated that the strain exhibits biological activities such as amylase production and nitrogen fixation [14].
A complete set of tryptophan (IAA precursor) biosynthesis genes, trpABCDEFG, was identified in the genome of strain SH-53, indicating that it can autonomously synthesize IAA precursors without the need for exogenous tryptophan, possessing a complete biosynthetic pathway (Figure S3). Additionally, key genes related to the indole-3-pyruvic acid (IPA) pathway, including ipdC (which encodes indole-3-pyruvic acid decarboxylase, EC:4.1.1.74) and aldehyde dehydrogenase genes (EC:1.2.1.3), as well as genes in the indole-3-acetamide (IAM) pathway, such as amiE (amidase, EC:3.5.1.4) and iaaH (indole-3-acetamide hydrolase, EC:3.5.1.-) were also identified in the genome (Table S5). This suggests that strain SH-53 may efficiently synthesize IAA through multiple pathways. Functional screening experiments further confirmed the ability of strain SH-53 to produce IAA (Figure S5A).
Strain SH-53 exhibits a significant ability to secrete siderophores. Qualitative detection using the CAS plate method demonstrated that the color of the reaction system changed from blue to orange-red upon inoculation with this strain (Figure S5B), confirming its capability to secrete siderophores. Quantitative analysis revealed that the siderophore activity unit (SU) reached as high as 94.95% ± 0.07%, indicating a strong ability for siderophore secretion.
The genome of strain SH-53 contains two complete gene clusters involved in siderophore biosynthesis: the entABCEF and dhbBEF clusters (Table S6). These clusters are responsible for encoding non-ribosomal peptide synthetases (NRPS) and modifying enzymes required for the synthesis of two efficient iron chelators, enterobactin and bacillibactin. Moreover, a complete catechol-type siderophore synthesis pathway was annotated (Figure S4). These catechol-type siderophores exhibit a high affinity for trivalent iron ions (Fe3+), enabling them to effectively capture trace amounts of iron in the environment.
Analysis of the genome of B. amyloliquefaciens SH-53 indicates that it has a complete genetic basis for sulfur metabolism. This strain contains multiple sets of key genes involved in the utilization of different sulfur sources. Notably, the sat-cysC-cysH-cysJ gene cluster constitutes the sulfate assimilation pathway, responsible for the stepwise reduction of inorganic sulfate (SO42-) to hydrogen sulfide (H2S), which is subsequently used for the synthesis of sulfur-containing organic compounds such as cysteine. The cysE-cysK genes encode key enzymes in the cysteine synthesis pathway, catalyzing the reaction between hydrogen sulfide and O-acetylserine to form cysteine. Additionally, the ssuABCD gene cluster encodes a complete sulfonate transport and oxidation system, allowing the strain to utilize organic sulfonates as alternative sulfur sources. Genes such as metA and mccB are involved in methionine synthesis and sulfur cycle regulation. The presence of soxC suggests that the strain may participate in sulfur oxidation processes (Table S7).Collectively, these sulfur metabolism-related genes enhance the adaptability and competitive capacity of SH-53 in fluctuating sulfur environments in the rhizosphere. Functional screening experiments also confirmed the ability of strain SH-53 to produce hydrogen sulfide (Figure S5C).
Analysis of the genome of B. amyloliquefaciens SH-53 demonstrates that it possesses a genetic basis for the efficient utilization of various inorganic nitrogen sources. This strain contains a complete nitrate reduction system, composed of genes such as narGHI, nasADE, and narK (Table S8), which enables the stepwise reduction of nitrate (NO3-) to ammonium (NH4+) in the environment. In addition, SH-53 has a complete ammonium assimilation pathway with genes including glnA, gltBD, and gudB, allowing for the efficient incorporation of ammonium into amino acid metabolism. Together, these nitrogen metabolism-related genes enhance the adaptability and competitive capability of SH-53 in fluctuating nitrogen environments in the rhizosphere. This genetic foundation not only supports the strain’s growth but also contributes to its potential role in promoting plant growth by improving nitrogen availability.
The analysis of the starchase in the genome of strain SH-53 indicates its genetic potential to utilize complex carbohydrates present in the environment. This strain contains multiple sets of key genes involved in the degradation of starch and glycogen. Among them, the amyA gene, which encodes a typical α-amylase, is responsible for the initial hydrolysis of starch, and its products are further degraded into glucose by the α-glucosidases encoded by malL and malZ. Meanwhile, the treC and sacA genes handle products from other carbohydrate metabolic pathways by hydrolyzing trehalose-6-phosphate and sucrose-6-phosphate, respectively, thereby providing accessible carbon sources and energy for the cells (Table S9). Preliminary experiments have also confirmed the amylase production capability of strain SH-53.
The genome of strain SH-53 contains genes involved in phosphate transport and assimilation, such as phoA and phoD, which are capable of hydrolyzing various organic phosphonates in the environment, decomposing them, and releasing accessible inorganic phosphate. Additionally, genes like pstS, pstA, pstC, and pstB have been annotated, representing a complete high-affinity phosphate-specific ABC transport system that efficiently uptake trace amounts of inorganic phosphate from the environment under phosphate-limiting conditions. Furthermore, the two-component regulatory system gene phoR encodes a histidine kinase that initiates the phosphate stress response under phosphorus deficiency, upregulating the expression of related genes such as the aforementioned alkaline phosphatases (phoA/phoD) and the phosphate transport system (pstSCAB). This adaptation allows the strain to respond effectively to phosphate-limited environments (TableS10).
3.4.2. Determination of Seed Germination-Promoting Traits of Strain SH-53
Previous research has confirmed that strain SH-53 possesses biofilm formation and root colonization abilities [15]; however, it remains unclear whether this strain influences seed germination during the early stages. Therefore, this study conducted qualitative tests on plant growth-promoting (PGP) bioactivity. The experimental results indicate that strain SH-53 has a strong capacity to promote seed germination. Under treatment with the undiluted fermentation broth of strain SH-53 (T1), the germination rate of tomato seeds reached a maximum of 60.00%, which is an increase of 10% compared to the blank control group. The 100-fold diluted fermentation broth treatment group (T3) showed the best promoting effect on root length, with an increase of 21.09% compared to the control group (Figure 10). These results demonstrate that strain SH-53 has a good ability to promote plant growth and possesses potential for further development as a plant growth promoter.
Table 2.
Effects of strain SH-53 on seed germination rate and root length.
| Treatment | CK | NB | T1 | T2 | T3 |
| Germinationrate/% | 50.00±17.32a | 33.33±15.28b | 60.00±17.32a | 53.33±15.28a | 40.00±10.00b |
| Rootlength/cm | 7.97±2.66b | 0.46±0.24c | 5.56±1.20b | 7.77±2.57b | 10.10±3.19a |
Note: CK: Sterile water; NB: NB medium; T1: Original bacterial fermentation broth; T2: 10-fold diluted bacterial fermentation broth; T3: 100-fold diluted bacterial fermentation broth. Data in the table represent mean ± standard error; Different letters indicate significant differences (P < 0.05).
4. Discussion
This study reveals the significant biocontrol potential of SH-53 as a plant root-associated growth-promoting bacterium (PGPR) through whole-genome analysis. Notably, compared to other excellent biocontrol strains, SH-53 exhibits two extremely remarkable genomic features: First, it has as many as 27 ribosomal RNA operons (rRNA operons), which is much higher than the reference strains SQR9 (7), FZB42 (10), DSM7 (10), and B. subtilis 168 (10). The copy number of rRNA operons is directly related to the growth rate of bacteria, suggesting that SH-53 may possess the potential for rapid niche colonization [16]. Secondly, SH-53 contains 4,078 protein-coding genes, which is on par with the recognized strong biocontrol strain SQR9 (4,078) and significantly exceeds the number in FZB42 (3,693) and DSM7 (3,921). This indicates that SH-53 has similarly robust metabolic functions and secondary metabolite synthesis potential [17].
Additionally, the genome of SH-53 contains 250 prophage-related genes, a number comparable to the high levels found in B. subtilis 168 (268 genes) and SQR9 (218 genes), and significantly higher than that of FZB42 (44 genes). This suggests that the genome of SH-53 has undergone active horizontal gene transfer events, potentially endowing it with additional functional advantages, such as novel stress resistance or antibacterial genes [18,19].
These results indicate that SH-53 not only possesses genetic coding potential comparable to that of top-performing biocontrol strains but also benefits from an exceptional competitive colonization advantage due to its high output of rRNA operons. These findings reveal, from a genomic perspective, the mechanisms by which this strain exerts its functions in biocontrol and plant growth promotion.
B. amyloliquefaciens, as an important representative of PGPR, is renowned for its exceptional ability to produce antibacterial substances and promote plant growth [20,21]. Numerous studies have demonstrated that this strain is effective in controlling various plant diseases. For instance, the strain FZB42 can effectively inhibit sheath blight in rice caused by Rhizoctonia solani [22] and gray mold in strawberries caused by Botrytis cinerea [23]. The strain SQR9 exhibits strong antagonistic effects against the pathogen Fusarium oxysporum responsible for cucumber wilt [24]. In addition, B. amyloliquefaciens can produce a variety of antibacterial substances, such as surfactin, iturin, and fengycin, which have been widely applied in agricultural production, showcasing its significant potential in biocontrol [25].
The biocontrol efficacy of B. amyloliquefaciens primarily arises from the secretion of various secondary metabolites. In the whole-genome analysis of the SH-53 strain, we identified a total of 12 BGCs for secondary metabolites, of which 5 were highly similar to known BGCs responsible for the synthesis of Surfactin, Bacillaene, Fengycin, Bacillibactin, and Bacilysin. Notably, an important finding is that the remaining 7 BGCs show very low similarity to known clusters, a characteristic that significantly distinguishes SH-53 from conventional biocontrol strains and likely reflects the adaptive evolution of SH-53 to its native environment in the unique ecological niche of the Wuliang Mountain Nature Reserve [26].
Based on the KEGG database annotation results, we systematically mined the ISR related genes in B. amyloliquefaciens SH-53. The results indicated that, in addition to the known antibacterial secondary metabolite synthesis genes, the genome of SH-53 contains a series of core genes associated with systemic resistance induction (Table S8), laying a molecular foundation for its biocontrol functionalities.
We identified multiple key genes that mediate ISR, which collectively form a core network regulating plant immunity and the balance of rhizosphere microecology through various metabolic pathways. Firstly, the gene clusters responsible for the synthesis of lipopeptide antibiotics (ppsD, ppsE, ituA-ituC, licA-licC) can produce critical ISR signaling molecules such as fengycin, iturin, and surfactin. These molecules not only directly activate plant defense pathways but also exhibit antibacterial properties, enhancing plant disease resistance from dual dimensions [27,28,29].Secondly, the synthesis and transport genes for antimicrobial peptides (bacteriocin) (bacG, bacF, bacE, bacD) are involved in the modification of bacteriocin precursors, formation of active groups, and their extracellular secretion. By disrupting the metabolism of pathogenic microorganisms, these genes contribute to both antibacterial activity and the induction of plant defense mechanisms, enriching the ISR-mediated pathways [30,31].Finally, quorum sensing and regulatory genes synergistically optimize biocontrol efficacy through multiple pathways: luxS synthesizes the AI-2 precursor to mediate interspecies communication [32], ahlD degrades pathogenic bacteria, and AHLs maintain microbial community balance [33]. Additionally, comA and slrR promote biofilm formation to ensure rhizosphere colonization [34,35].
Additionally, we discovered genes related to iron nutrient competition (siderophore synthesis genes entC and entE; transport genes fecE, fhuA, and efeO), which chelate iron ions to create a nutritional competitive advantage, inhibiting the growth of pathogenic bacteria and indirectly activating the plant jasmonic acid defense pathway [36,37,38]. Moreover, SH-53 possesses genes for the synthesis of volatile ISR signaling molecules. The gene butB catalyzes the synthesis of substances such as 2,3-butanediol and ethephon, which induce plant defenses at a distance. The genes acoA and acoB regulate the metabolic steady state of ethephon to ensure its immune-inducing functions [39,40].
SH-53 also contains support genes that maintain the stability of secondary metabolism, including murA, atpA, talB, and amyA. Specifically, murA is responsible for the synthesis of the lipid peptidoglycan modification precursor [41]; atpA generates ATP to provide energy for the synthesis and secretion of signaling molecules [42]; talB participates in the pentose phosphate pathway, providing raw materials for nucleic acids and coenzymes [43]; and amyA degrades starch to serve as a carbon source, maintaining the carbon source balance necessary for the growth of the strain and secondary metabolism [44].
In the known metabolic products, the SH-53 strain retains a typical framework of antibacterial mechanisms: Bacilysin disrupts the synthesis of the pathogenic bacterial cell wall by inhibiting the GlmS enzyme [45]; Bacillaene effectively prevents the formation of biofilms by pathogenic bacteria [46]; Fengycin possesses a concentration-dependent dual action mechanism, inducing fungal apoptosis while also directly damaging cell membranes [47,48]; Bacillibactin restricts pathogenic bacterial growth through an iron ion competition strategy [49]. However, the most valuable finding of this study is the presence of seven functionally uncharacterized secondary BGCs within the SH-53 genome. These unannotated BGCs not only dominate in number but also suggest that this strain may have evolved novel antibacterial mechanisms that surpass conventional metabolic frameworks, thereby bestowing it with broader biocontrol potential [50].
Particularly noteworthy is that these 7 uncharacterized BGCs not only outnumber the known clusters but also indicate that SH-53 may produce novel antibacterial substances or signaling molecules that have yet to be reported. This rich diversity of BGCs, especially the high proportion of unknown clusters, distinguishes SH-53 from conventional agricultural strains, offering broader mechanistic possibilities and developmental prospects for its application in biocontrol [51].
Bacillus promotes plant growth through various mechanisms, one key role being the direct regulation of plant growth and development by secreting plant hormones such as auxins, cytokinins, and gibberellins [52,53]. For example, rhizosphere bacteria of Bacillus can significantly enhance root growth and expansion by secreting endogenous synthesized IAA and activating the plant’s endogenous auxin synthesis pathway [54]. Genome annotation has revealed that SH-53 possesses a complete tryptophan biosynthesis pathway (trpABCDEFG gene cluster), enabling it to autonomously synthesize IAA precursors, thereby regulating plant growth and development [55]. The ability to secrete amylase is one of the important mechanisms through which many beneficial strains promote plant growth [56]. By secreting amylase, the strain can degrade organic matter in the soil, releasing simple sugars that are available for plant absorption, thus improving soil structure and providing additional nutrient sources for plants [57]. The strain SH-53 has been annotated to contain 7 genes involved in amylase secretion [58].Iron is an essential micronutrient for plant growth, and its bioavailability is often limited by soil pH and iron forms [58,59]. The strain can secrete iron carriers (such as siderophores or pyoverdine) that dissolve insoluble iron compounds in the soil, increasing the available iron content and thereby enhancing the iron absorption capacity of plants [60]. Six iron carrier synthesis genes were identified in SH-53. Additionally, the bioavailability of sulfur typically restricts plant growth; strains with sulfur-oxidizing capabilities can oxidize insoluble sulfides or organic sulfur compounds in the soil, releasing sulfate that is available for plant absorption, thus increasing the available sulfur content in the soil and indirectly promoting plant growth and development. The sulfur metabolism ability of SH-53 also provides strong support for its plant growth-promoting effects [61]. Genomic analysis revealed that SH-53 has been annotated with 14 sulfur metabolism-related genes, which can efficiently utilize sulfur sources in the environment through various sulfur metabolism pathways, such as cysteine synthesis, sulfite metabolism, and sulfate assimilation [62]. In summary, the strain SH-53 has comprehensive potential in promoting plant growth and maintaining plant health through multiple mechanisms, including the tryptophan-IAA synthesis pathway, amylase production, iron nutrient supply mediated by iron carriers, and sulfur nutrient supply mediated by sulfur metabolism, providing a theoretical basis for its application in agricultural production.
The resistance genes of B. amyloliquefaciens SH-53 were analyzed. According to the alignment results from the CARD database, 268 resistance genes were annotated, covering five main functional categories: drug efflux pumps, target modification, antibiotic inactivation, membrane structure alteration, and regulatory systems. The ABC superfamily transport proteins (such as macB, bcrA, patA/patB) and MFS transport proteins (such as blt, mreA) comprise the core efflux system of the strain. The former can enhance the resistance to azole compounds, while the latter mediates the efflux of a variety of compounds, including caprolactam and azoles [63,64].
In the rhizospheric microecosystem, the competition for microbial resources is intense, and the antibacterial metabolic products of closely related Bacillus species and pathogenic bacteria are major stress factors for the survival of strains. Previous studies have shown that B. velezensis FZB42 and SQR9 can inhibit the growth of closely related strains by secreting antimicrobial substances [65]. This study confirms that the abundant resistance genes carried by SH-53 not only enable it to tolerate antimicrobial substances in the rhizosphere but also allow it to avoid self-toxicity effects from its own antimicrobial products, thereby maintaining metabolic homeostasis. Coupled with its significant inhibitory ability against closely related Bacillus strains, this provides critical ecological support for its biocontrol applications [66,67].
In summary, the experimental and analytical results indicate that this study confirms that B. amyloliquefaciens SH-53 is a plant growth-promoting rhizobacterium with significant application potential. Furthermore, the genomic information of strain SH-53 may help to elucidate the molecular mechanisms underlying its antimicrobial activity.
5. Conclusions
Strain SH-53 was isolated from the Yunnan Wuliangshan National Nature Reserve, and based on whole-genome analysis and multilocus sequence analysis (MLSA), it was confirmed to be B. amyloliquefaciens. This strain exhibits antagonistic effects against plant pathogens and can colonize the plant rhizosphere and promote plant growth through various mechanisms. It possesses the ability to produce multiple antibacterial secondary metabolites, synthesize auxin (IAA), secrete amylase, synthesize iron chelators, and efficiently utilize sulfur sources.The strain B. amyloliquefaciens SH-53 contains 27 ribosomal RNA operons and 4,078 protein-coding genes, and 250 prophage-related genes were also identified. Additionally, the genome of SH-53 harbors 12 BGCs for secondary metabolites, of which 7 are novel gene clusters with unknown functions, significantly differing from conventional biocontrol strains. It also includes a complete core gene network related to ISR and supporting genes that ensure the homeostasis of secondary metabolism. The enrichment of novel secondary metabolite biosynthetic gene clusters within the genome of B. amyloliquefaciens SH-53 provides critical genetic resources for the exploration of new antibacterial active substances and lays a molecular foundation for elucidating the evolutionary mechanisms of plant rhizosphere-promoting and biocontrol functions. Furthermore, its complete ISR regulatory network and secondary metabolism homeostasis supporting system further ensure the strain’s ability to maintain stable biocontrol efficacy in complex field environments. In summary, this study clarifies that this strain possesses core potential to be developed as an excellent biocontrol agent.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Supplementary Table; Figure S1: Predicted results of secondary metabolite biosynthesis gene clusters in the B. amyloliquefaciens SH-53 genome; Figure S2: Predicted structures of secondary metabolites by B. amyloliquefaciens SH-53; Figure S3: Tryptophan biosynthesis pathway of strain SH-53; Figure S4: Biosynthetic pathway of siderophores in strain SH-53; Figure S5: Evaluation of additional biological activities of strain SH-53. (A) Indole-3-acetic acid (IAA) production; (B) Siderophore production; (C) Hydrogen sulfide production; Table S1: ANI and dDDH values between the genome of B. amyloliquefaciens SH-53 and reference genomes of the genus Bacillus; Table S2: Genomic comparison among B.amyloliquefaciens SH-53,B.velezensis SQR9,B.velezensis FZB42,B.amyloliquefaciens DSM7,and B.subtilis 168; Table S3: Genes involved in tryptophan (IAA precursor) metabolism in the genome of B. amyloliquefaciens SH-53; Table S4: Genes involved in siderophore metabolism in the B. amyloliquefaciens SH-53 genome; Table S5: Genes involved in sulfur metabolism in the B. amyloliquefaciens SH-53 genome; Table S6: Genes involved in nitrogen metabolism in the B. amyloliquefaciens SH-53 genome; Table S7: Genes involved in amylase secretion in the B. amyloliquefaciens SH-53 genome; Table S8: Genes involved in phosphate metabolism in the B. amyloliquefaciens SH-53 genome.
Author Contributions
Conceptualization, J.J., Y.W., X.L., W.P., T.P., Z.S., F.H and P.Y.; methodology, P.Y.; software, J.J.; formal analysis, J,J.; investigation, Y.W., T.P. and W.P.; resources, Y.W., Z.S. and F.H.; writing—original draft, J.J.; writing—review and editing, J.J. and P.Y.; funding acquisition, P.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Yunnan Province “Xingdian Talent” Support Program (XDYC-CYCX-2002-0071), and the Central Government Guided Local Science and Technology Development Fund (202407AC110006).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Genome sequences were deposited in the GenBank database under accession number CP187258.1 (https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_049677765.1/).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kazerooni, E.; Maharachchikumbura, S.; Al-Sadi, A.; Kang, S.; Yun, B.; Lee, I. Biocontrol potential of Bacillus amyloliquefaciens against Botrytis pelargonii and Alternaria alternata on Capsicum annuum. Journal of fungi 2021, 7, 472. [Google Scholar] [CrossRef]
- Ying, T.; Wu, P.; Gao, L.; Wang, C.; Zhang, T.; Liu, S.; Huang, R. Isolation and characterization of a new strain of Bacillus amyloliquefaciens and its effect on strawberry preservation. LWT 2022, 165, 113712. [Google Scholar] [CrossRef]
- Zhang, N.; Yang, D.; Kendall, J.; Borriss, R.; Druzhinina, I.; Kubicek, C.; Shen, Q.; Zhang, R. Comparative genomic analysis of Bacillus amyloliquefaciens and Bacillus subtilis reveals evolutional traits for adaptation to plant-associated habitats. Frontiers in microbiology. 2016, 7, 2039. [Google Scholar] [CrossRef]
- Hua, Z.; Qu, Y.; Zhu, Q.; Zhou, E.; Qi, Y.; Yin, Y.; Rao, Y.; Tian, Y.; Li, Y.; Liu, L.; Castelle, C.; Hedlund, B.; Shu, W.; Knight, R.; Li, W. Genomic inference of the metabolism and evolution of the archaeal phylum Aigarchaeota. Nat Commun. 2018, 9, 2832. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, J.; Huang, X.; Zhang, J.; Li, Q.; Lyu, L.; Ju, F.; Li, J.; Zhang, S. Exploring the role of organotrophic microbes in geochemical cycling of cold seep sediments. The Innovation Geoscience. 2025, 3, 100123–1. [Google Scholar] [CrossRef]
- Lu, Q.; Xie, Y.; Gu, F.; Tu, C.; He, J.; Guo, Q.; Wu, Y.; Xu, M.; Liu, J. Potential of Bacillus amyloliquefaciens QY-1 as a biocontrol agent of Botrytis cinerea in postharvest blueberry. Physiological and Molecular Plant Pathology 2023, 128, 102117. [Google Scholar] [CrossRef]
- Liang, L.; Fu, Y.; Deng, S.; Wu, Y.; Gao, M. Genomic, antimicrobial, and aphicidal traits of Bacillus velezensis ATR2, and its biocontrol potential against ginger rhizome rot disease caused by Bacillus pumilus. Microorganisms 2021, 10, 63. [Google Scholar] [CrossRef]
- Zhao, X.; Kuipers, O. Identification and classification of known and putative antimicrobial compounds produced by a wide variety of Bacillales species. BMC genomics. 2016, 17.1, 882. [Google Scholar] [CrossRef]
- Luo, L.; Zhao, C.; Wang, E.; Raza, A.; Yin, C. Bacillus amyloliquefaciens as an excellent agent for biofertilizer and biocontrol in agriculture: An overview for its mechanisms. Microbiological research. 2022, 259, 127016. [Google Scholar] [CrossRef]
- Tanaka, K.; Amaki, Y.; Ishihara, A.; Nakajima, H. Synergistic effects of [Ile7] surfactin homologues with bacillomycin D in suppression of gray mold disease by Bacillus amyloliquefaciens biocontrol strain SD-32. Journal of agricultural and food chemistry 2015, 63, 5344–5353. [Google Scholar] [CrossRef]
- Chen, X.; Koumoutsi, A.; Scholz, R.; Schneider, K.; Vater, J.; Süssmuth, R.; Piel, J.; Borriss, R. Genome analysis of Bacillus amyloliquefaciens FZB42 reveals its potential for biocontrol of plant pathogens. Journal of biotechnology 2009, 140(1-2), 27–37. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, H.; You, J.; Yang, J.; Zhang, Q.; Zhao, J.; Aimaier, R.; Zhang, J.; Han, S.; Zhao, H.; Zhao, H. Transcriptome and metabolome analyses reveal that Bacillus subtilis BS-Z15 lipopeptides mycosubtilin homologue mediates plant defense responses. Frontiers in Plant Science 2023, 13, 1088220. [Google Scholar] [CrossRef]
- Zeng, Q.; Xie, J.; Li, Y.; Chen, X.; Gu, X.; Yang, P.; Hu, G.; Wang, Q. Organization, evolution and function of fengycin biosynthesis gene clusters in the Bacillus amyloliquefaciens group. Phytopathology Research 2021, 3, 26. [Google Scholar] [CrossRef]
- Wang, N.; Liao, Y.; Shi, Z.; Shen, Y.; Yang, T.; Feng, L.; Yi, X.; Tang, J.; Chen, Q.; Yang, P. Identification of Three Strains of Bacillus from the Forest Soil of Wuliang Mountain and Mining of Their Bioactivities. Biotechnology Bulletin 2024, 40.2, 277–288. [Google Scholar]
- Wang, N.; Liao, Y.; Shen, Y.; Zhao, J.; Chen, Q.; Yang, P. Optimization of fermentation conditions and biocontrol effect of Bacillus amyloliquefaciens SH-53 from rhizosphere of rumex. Journal of Plant Protection 2024, 51, 817–829. [Google Scholar]
- Roller, B.; Stoddard, S.; Schmidt, T. Exploiting rRNA operon copy number to investigate bacterial reproductive strategies. Nat Microbiol. 2016, 1, 16160. [Google Scholar] [CrossRef]
- Belbahri, L.; Bouket, A.; Rekik, I.; Alenezi, F.; Vallat, A.; Luptakova, L.; Petrovova, E.; Oszako, T.; Cherrad, S.; Vacher, S.; Rateb, M. Comparative genomics of Bacillus amyloliquefaciens strains reveals a core genome with traits for habitat adaptation and a secondary metabolites rich accessory genome. Frontiers in microbiology 2017, 8, 1438. [Google Scholar] [CrossRef]
- She, T.; Tan, D.; Balcazar, J.; Friman, V.; Wang, D.; Zhu, D.; Ye, M.; Sun, M.; Yuan, S.; Hu, F. Phage-mediated horizontal transfer of Salmonella enterica virulence genes with regulatory feedback from the host. iMeta 2025, e70042. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Liu, C.; Zhou, S.; Liu, C.; Eldridge, D.; Ai, C.; Wilhelm, S.; Singh, B.; Liang, X.; Radosevich, M.; Yang, Q.; Tang, X.; Wei, Z.; Friman, V.; Gillings, M.; Delgado-Baquerizo, M.; Zhu, Y. Prophage-encoded antibiotic resistance genes are enriched in human-impacted environments. Nature communications. 2025, 15, 8315. [Google Scholar] [CrossRef]
- Cai, Y.; Tao, H.; Gaballa, A.; Pi, H.; Helmann, J. The extracytoplasmic sigma factor σX supports biofilm formation and increases biocontrol efficacy in Bacillus velezensis 118. Scientific Reports 2025, 15, 5315. [Google Scholar] [CrossRef]
- Glaeser, S.; Kämpfer, P. Multilocus sequence analysis (MLSA) in prokaryotic taxonomy. Systematic and applied microbiology 2015, 38, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Hartmann, A.; Gao, X.; Borriss, R. Biocontrol mechanism by root-associated Bacillus amyloliquefaciens FZB42–a review. Frontiers in microbiology 2015, 6, 780. [Google Scholar] [CrossRef] [PubMed]
- Sylla, J.; Alsanius, B.; Krüger, E.; Reineke, A.; Strohmeier, S.; Wohanka, W. Leaf microbiota of strawberries as affected by biological control agents. Phytopathology. 2013, *103*(10), 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shao, J.; Li, B.; Yan, X.; Shen, Q.; Zhang, R. Contribution of bacillomycin D in Bacillus amyloliquefaciens SQR9 to antifungal activity and biofilm formation. Applied and environmental microbiology. 2013, 79, 808–815. [Google Scholar] [CrossRef]
- Belbahri, L.; Bouket, A.; Rekik, I.; Alenezi, F.; Vallat, A.; Luptakova, L.; Petrovova, E.; Oszako, T.; Cherrad, S.; Vacher, S.; Rateb, M. Comparative genomics of Bacillus amyloliquefaciens strains reveals a core genome with traits for habitat adaptation and a secondary metabolites rich accessory genome. Frontiers in microbiology 2017, 8, 1438. [Google Scholar] [CrossRef]
- Liu, H.; Prajapati, V.; Prajapati, S.; Bais, H.; Lu, J. Comparative genome analysis of Bacillus amyloliquefaciens focusing on phylogenomics, functional traits, and prevalence of antimicrobial and virulence genes. Frontiers in Genetics 2021, 12, 724217. [Google Scholar] [CrossRef]
- Lam, V.; Meyer, T.; Arias, A.; Ongena, M.; Höfte, M. Contribution of Bacillus cyclic lipopeptides iturin and fengycin to control rice blast caused by Pyricularia oryzae in potting and acid sulfate soils by direct antagonism and induced systemic resistance. Microorganisms 2021, 9, 1441. [Google Scholar] [CrossRef]
- Lv, J.; Da, R.; Cheng, Y.; Tuo, X.; Wei, J.; Jiang, K.; Monisayo, A.; Han, B. Mechanism of antibacterial activity of Bacillus amyloliquefaciens C-1 lipopeptide toward anaerobic Clostridium difficile. BioMed Research International 2020, 2020, 3104613. [Google Scholar] [CrossRef]
- Yuan, Q.; Yang, P.; Liu, Y.; Tabl, K.; Guo, M.; Zhang, J.; Wu, A.; Liao, Y.; Huang, T.; He, W. Iturin and fengycin lipopeptides inhibit pathogenic Fusarium by targeting multiple components of the cell membrane and their regulative effects in wheat. Journal of Integrative Plant Biology 2025, 67, 2184–2197. [Google Scholar] [CrossRef]
- Lam, V.; Meyer, T.; Arguelles Arias, A.; Ongena, M.; Oni, F.; Höfte, M. Bacillus cyclic lipopeptides iturin and fengycin control rice blast caused by Pyricularia oryzae in potting and acid sulfate soils by direct antagonism and induced systemic resistance. Microorganisms 2021, 9, 1441. [Google Scholar] [CrossRef]
- Parker, J.; Walsh, C. Action and timing of BacC and BacD in the late stages of biosynthesis of the dipeptide antibiotic bacilysin. Biochemistry 2013, 52, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Feng, H.; Chen, L.; Zhang, H.; Dong, X.; Xiong, Q.; Zhang, R. Root-secreted spermine binds to Bacillus amyloliquefaciens SQR9 histidine kinase KinD and modulates biofilm formation. Molecular Plant-Microbe Interactions 2020, 33, 423–432. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, J.; Guo, Y.; Wang, Y.; Peng, Y.; Wang, Y.; Hu, M.; Lin, W.; Wu, Z. Effects of quorum sensing and quorum quenching mediated by AHLs on plant-rhizosphere microbial interactions. Chinese Journal of Eco-Agriculture. 2024, 32, 1–14. [Google Scholar]
- Jia, R.; Wang, Y.; Wang, H.; Ji, X.; Sadiq, F.; Wang, X.; Zhang, G. The role of quorum sensing effector ComA in regulating biofilm formation and surfactin production in Bacillus subtilis ASAG 010. Food Bioscience 2025, 106842. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, N.; Xia, L.; Li, Q.; Shao, J.; Shen, Q.; Zhang, R. ResDE two-component regulatory system mediates oxygen limitation-induced biofilm formation by Bacillus amyloliquefaciens SQR9. Applied and Environmental Microbiology 2018, 84, e02744-17. [Google Scholar] [CrossRef]
- Shao, Z.; Gu, S.; Zhang, X.; Xue, J.; Yan, T.; Guo, S.; Pommier, T.; Jousset, A.; Yang, T.; Xu, Y.; Shen, Q.; Wei, Z. Siderophore interactions drive the ability of Pseudomonas spp. consortia to protect tomato against Ralstonia solanacearum. Horticulture Research 2024, 11, uhae186. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Wei, Z.; Shao, Z.; Friman, V.; Cao, K.; Yang, T.; Kramer, J.; Wang, X.; Li, M.; Mei, X.; Xu, Y.; Shen, Q.; Kümmerli, R.; Jousset, A. Competition for iron drives phytopathogen control by natural rhizosphere microbiomes. Nature Microbiology. 2020, 5, 1002–1010. [Google Scholar] [CrossRef]
- Montejano-Ramírez, V.; Valencia-Cantero, E. Cross-talk between iron deficiency response and defense establishment in plants. International Journal of Molecular Sciences 2023, 24, 6236. [Google Scholar] [CrossRef] [PubMed]
- Neilands, J.B. Siderophores: structure and function of microbial iron transport compounds. Journal of Biological Chemistry 1995, 270, 26723–26726. [Google Scholar] [CrossRef]
- Xu, X.; Qiao, W.; Dong, Y.; Yang, H.; Xu, H.; Qiao, M. 3-Butanediol dehydrogenase is more efficient than acetoin reductase at metabolizing reserve carbon to improve carbon cycling pathways in Lactococcus lactis N8. International Journal of Biological Macromolecules 2025, 299, 140023. [Google Scholar] [CrossRef]
- Ryu, C.; Farag, M.; Hu, C.; Reddy, M.; Kloepper, J.; Paré, P. Bacterial volatiles induce systemic resistance in Arabidopsis. Plant Physiology. 2004, 134, 1017–1026. [Google Scholar] [CrossRef]
- Mihalovits, L.; Ferenczy, G.; Keserű, G. Catalytic mechanism and covalent inhibition of UDP-N-Acetylglucosamine Enolpyruvyl Transferase (MurA): implications to the design of novel antibacterials. Journal of Chemical Information and Modeling 2019, 59, 5161–5173. [Google Scholar] [CrossRef] [PubMed]
- Nirody, J.; Budin, I.; Rangamani, P. ATP synthase: Evolution, energetics, and membrane interactions. Journal of General Physiology. 2020, 152, e201912475. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Zhao, W.; Liu, D.; Yang, R.; Cui, Z.; Zou, D.; Li, D.; Wei, X.; Xiong, H.; Niu, C. Screening, identification, engineering, and characterization of Bacillus-derived α-amylase for effective tobacco starch degradation. International Journal of Biological Macromolecules 2024, 282, 137364. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, X.; Wu, M.; Ge, S. Molecular insights into the antifungal mechanism of bacilysin. Journal of molecular modeling 2018, 24, 118. [Google Scholar] [CrossRef]
- Erega, A.; Stefanic, P.; Dogsa, I.; Danevčič, T.; Simunovic, K.; Klančnik, A.; Možina, S.; Mulec, M. Bacillaene mediates the inhibitory effect of Bacillus subtilis on Campylobacter jejuni biofilms. Applied and Environmental Microbiology 2021, 87, e02955-20. [Google Scholar] [CrossRef]
- Deng, Y.; Chen, Z.; Chen, Y.; Wang, J.; Xiao, R.; Wang, X.; Liu, B.; Chen, M.; He, J. Lipopeptide C17 fengycin B exhibits a novel antifungal mechanism by triggering metacaspase-dependent apoptosis in Fusarium oxysporum. Journal of Agricultural and Food Chemistry 2024, 72, 7943–7953. [Google Scholar] [CrossRef]
- Tang, Q.; Bie, X.; Lu, Z.; Lv, F.; Tao, Y.; Qu, X. Effects of fengycin from Bacillus subtilis fmbJ on apoptosis and necrosis in Rhizopus stolonifer. Journal of Microbiology 2014, 52, 675–680. [Google Scholar] [CrossRef]
- Lyng, M.; Jørgensen, J.; Schostag, M.; Jarmusch, S.; Aguilar, D.; Lozano-Andrade, C.; Kovács, Á. Competition for iron shapes metabolic antagonism between Bacillus subtilis and Pseudomonas marginalis. The ISME journal. 2024, 18, wrad001. [Google Scholar] [CrossRef]
- Wang, J.; Xie, X.; Li, B.; Yang, L.; Song, F.; Zhou, Y.; Jiang, M. Complete genome analysis and antimicrobial mechanism of Bacillus velezensis GX0002980 reveals its biocontrol potential against mango anthracnose disease. Microbiology Spectrum 2025, 13, e02685-24. [Google Scholar] [CrossRef]
- Moshe, M.; Gupta, C.; Sela, N.; Minz, D.; Banin, E.; Frenkel, O.; Cytryn, E. Comparative genomics of Bacillus cereus sensu lato spp. biocontrol strains in correlation to in-vitro phenotypes and plant pathogen antagonistic capacity. Frontiers in Microbiology 2023, 14, 996287. [Google Scholar] [CrossRef]
- Yang, J.; Li, S.; Zhou, X.; Du, C.; Fang, J.; Li, X.; Zhao, J.; Ding, F.; Wang, Y.; Zhang, Q.; Wang, Z.; Liu, J.; Dong, G.; Zhang, J.; Xu, F.; Xu, W. Bacillus amyloliquefaciens promotes cluster root formation of white lupin under low phosphorus by mediating auxin levels. Plant Physiology 2025, 197, kiae676. [Google Scholar] [CrossRef]
- Sorokan, A.; Veselova, S.; Benkovskaya, G.; Maksimov, I. Endophytic strain Bacillus subtilis 26D increases levels of phytohormones and repairs growth of potato plants after colorado potato beetle damage. Plants 2021, 10, 923. [Google Scholar] [CrossRef] [PubMed]
- Hussein, K.; Kadhum, N.; Yassera, Y. The role of bacteria Bacillus subtilis in improving rooting response of Mung bean (Vigna ratiata) cuttings. Journal of Contemporary Medical Sciences 2016, 2, 88–92. [Google Scholar]
- Li, Y.; Gao, M.; Zhang, W.; Liu, Y.; Wang, S.; Zhang, H.; Li, X.; Yu, S.; Lu, L. Halotolerant Enterobacter asburiae A103 isolated from the halophyte Salix linearistipularis: Genomic analysis and growth-promoting effects on Medicago sativa under alkali stress. Microbiological Research 2024, 289, 127909. [Google Scholar] [CrossRef]
- Li, Y.; Hu, Q.P. Studying of the promotion mechanism of Bacillus subtilis QM3 on wheat seed germination based on β-amylase. Open Life Sciences 2020, 15, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Dong, A.; Xiong, G.; Liu, Y.; Hossain, M.S.; Liu, S.; Qiu, D. Production of highly active extracellular amylase and cellulase from Bacillus subtilis ZIM3 and a recombinant strain with a potential application in tobacco fermentation. Frontiers in Microbiology 2020, 11, 1539. [Google Scholar] [CrossRef]
- Khan, S.; Kaur, K.; Kumar, V.; Tiwari, S. Iron transport and homeostasis in plants: current updates and applications for improving human nutrition values and sustainable agriculture. Plant Growth Regulation 2023, 100, 373–390. [Google Scholar] [CrossRef]
- Saleem, A.; Zulfiqar, A.; Saleem, M.; Ali, B.; Saleem, M.; Ali, S.; Tufekci, E.; Tufekci, A.; Rahimi, M.; Mostafa, R. Alkaline and acidic soil constraints on iron accumulation by Rice cultivars in relation to several physio-biochemical parameters. BMC Plant Biology 2023, 23, 397. [Google Scholar] [CrossRef]
- Wang, N.; Wang, T.; Chen, Y.; Wang, M.; Lu, Q.; Wang, K.; Dou, Z.; Chi, Z.; Qiu, W.; Dai, J.; Niu, L.; Cui, J.; Wei, Z.; Zhang, F.; Kümmerli, R.; Zuo, Y. Microbiome convergence enables siderophore-secreting-rhizobacteria to improve iron nutrition and yield of peanut intercropped with maize. Nature communications 2024, 15, 839. [Google Scholar] [CrossRef]
- Vishnu; Sharma, P.; Kaur, J.; Gosal, S.; Walia, S. Characterization of Sulfur Oxidizing Bacteria and Their Effect on Growth Promotion of Brassica napus L. Journal of Basic Microbiology 2024, 64, e2400239. [Google Scholar] [CrossRef]
- Liu, H.; Hou, Y.; Wang, Y.; Li, Z. Enhancement of sulfur conversion rate in the production of L-cysteine by engineered Escherichia coli. Journal of Agricultural and Food Chemistry 2019, 68, 250–257. [Google Scholar] [CrossRef]
- Kohli, A.; Gupta, V.; Krishnamurthy, S.; Hasnain, S.; Prasad, R. Specificity of drug transport mediated by CaMDR1: a major facilitator of Candida albicans. Journal of Biosciences 2001, 26, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Pata, J.; Sharma, S.; Monk, B.; Falson, P.; Prasad, R. Directed mutational strategies reveal drug binding and transport by the MDR transporters of Candida albicans. Journal of Fungi 2021, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xu, Z.; Zhang, G.; Xia, L.; Dong, X.; Li, Q.; Liles, M.; Shao, J.; Shen, Q.; Zhang, R. A genomic island in a plant beneficial rhizobacterium encodes novel antimicrobial fatty acids and a self-protection shield to enhance its competition. Environ. Microbiol. 2019, 21, 3455–3471. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zhou, C.; Qiu, F.; Peng, D.; Wang, X.; Li, X. Acid-resistant Bacillus velezensis effectively controls pathogenic Colletotrichum capsici and improves plant health through metabolic interactions. Applied and Environmental Microbiology 2025, 91, e00340-25. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Zeng, L.; Xu, T.; Sun, S.; Yan, S.; Yang, L.; Huang, Y.; Jia, J.; Dou, T. Antibiotic resistance genes in bacteria: Occurrence, spread, and control. Journal of Basic Microbiology 2021, 61, 1049–1070. [Google Scholar] [CrossRef]
Figure 1.
phylogene ticanalysis basedon 16S rRNA.
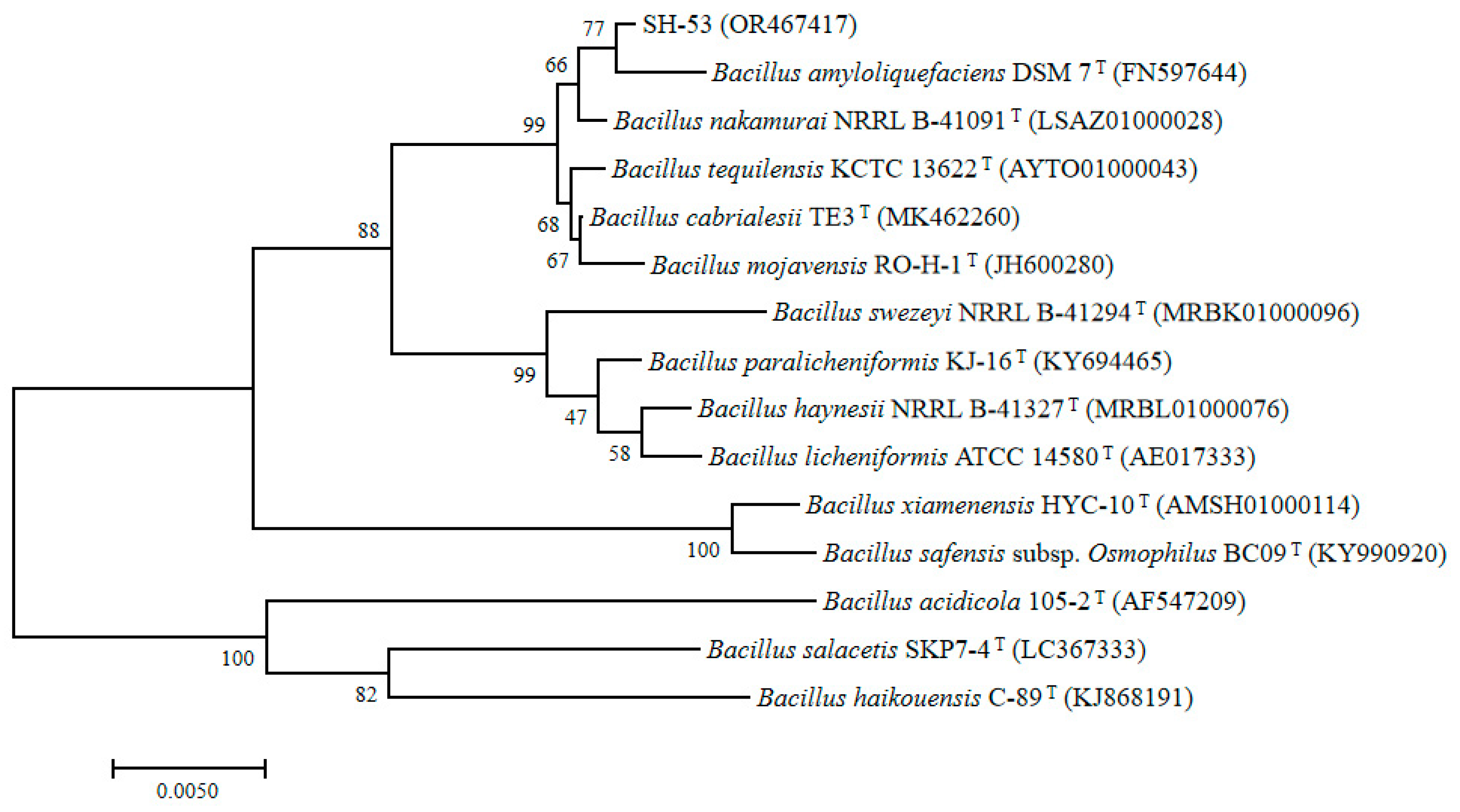
Figure 2.
Genomic circle map of B. amyloliquefaciens SH-53.Note: From the inside out, the first circle represents the scale; the second circle represents the GC skew; the third circle represents the GC content; the fourth and seventh circles represent the COG categories to which each CDS belongs; and the fifth and sixth circles represent the positions of CDS, tRNA, and RNA on the genome.
Figure 2.
Genomic circle map of B. amyloliquefaciens SH-53.Note: From the inside out, the first circle represents the scale; the second circle represents the GC skew; the third circle represents the GC content; the fourth and seventh circles represent the COG categories to which each CDS belongs; and the fifth and sixth circles represent the positions of CDS, tRNA, and RNA on the genome.
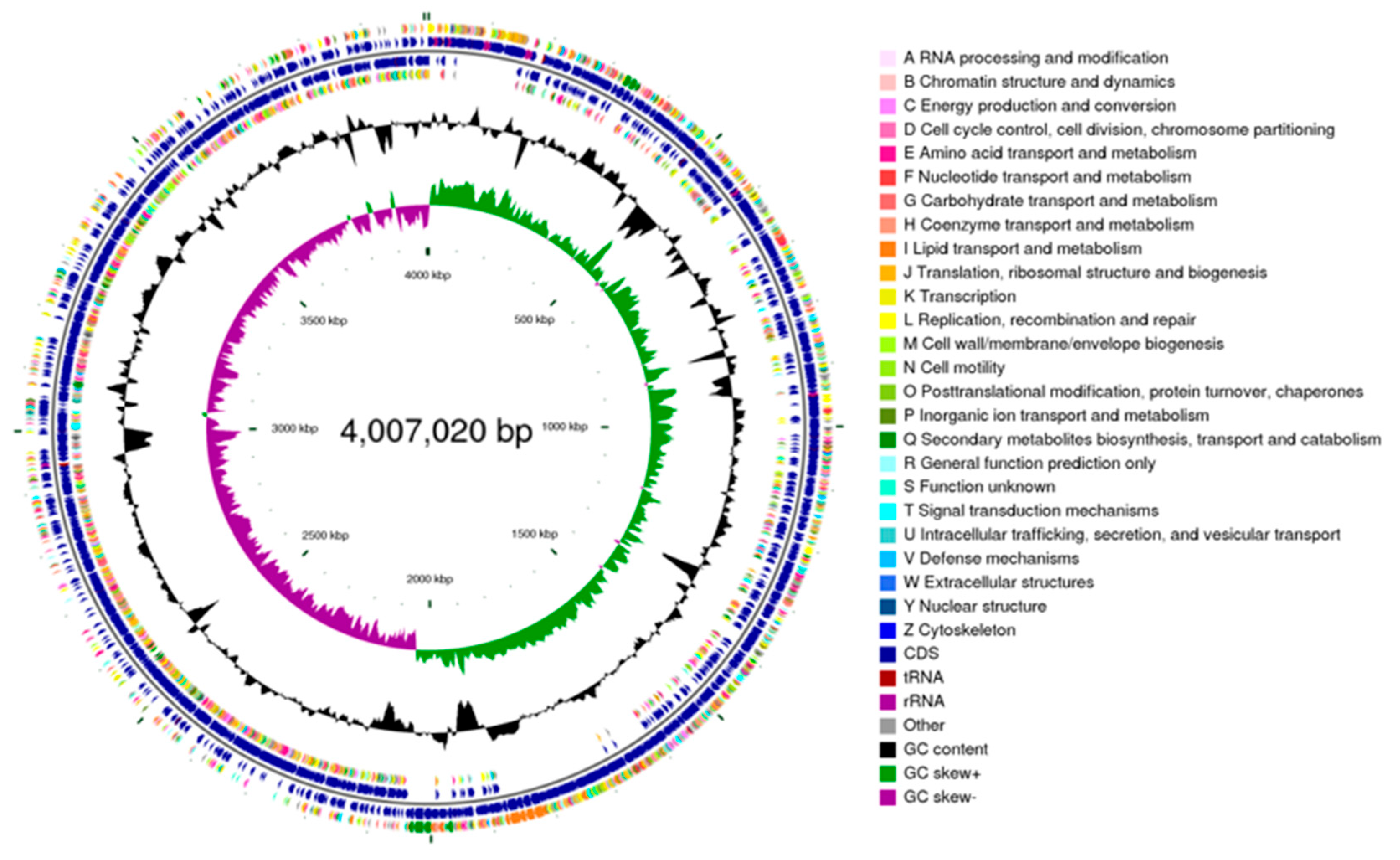
Figure 3.
Distribution of functional annotation of strain SH-53 genes across databases.
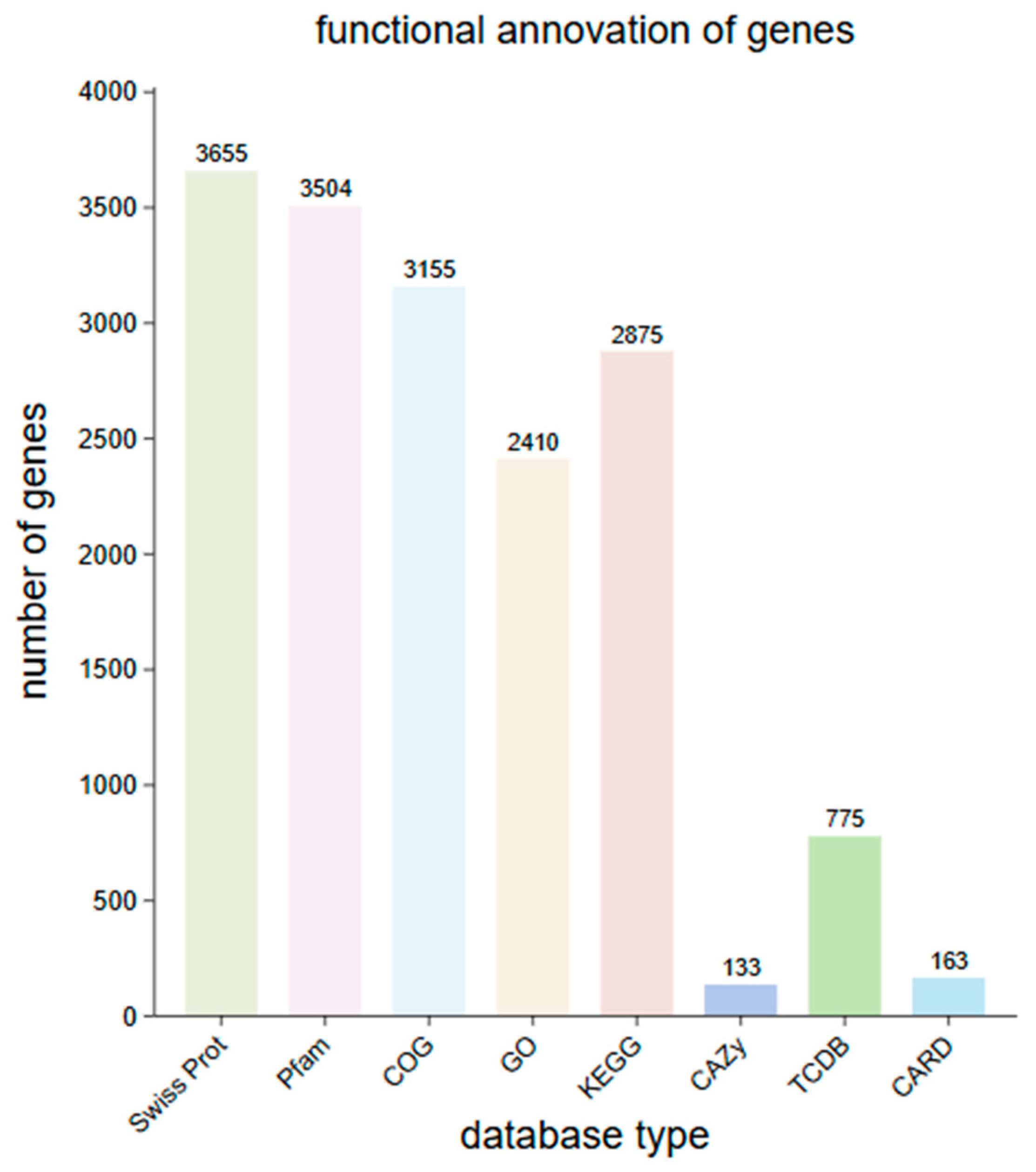
Figure 4.
GOG annotation classification results statistics of B. amyloliquefaciens SH-53 genome,The COG functional annotation scontain 2 3categories.
Figure 4.
GOG annotation classification results statistics of B. amyloliquefaciens SH-53 genome,The COG functional annotation scontain 2 3categories.
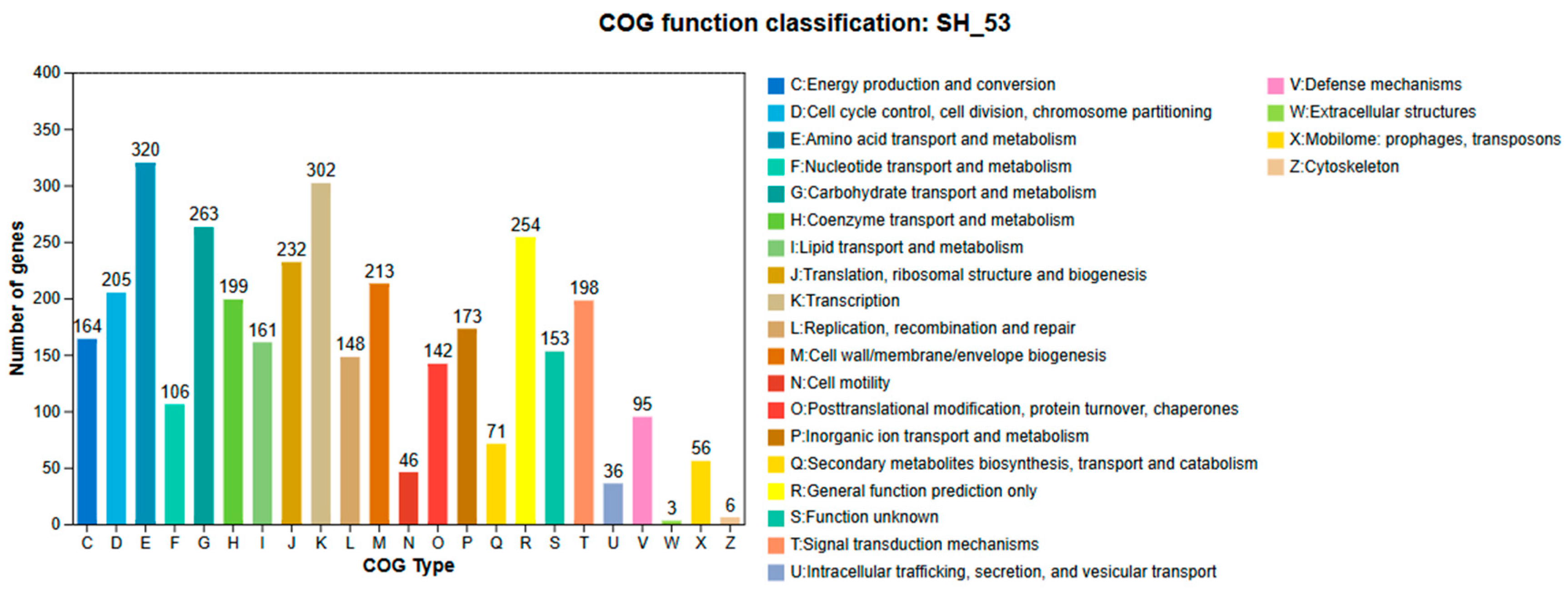
Figure 5.
GO annotation classification results statisticsof B. amyloliquefaciens SH-53 genome.
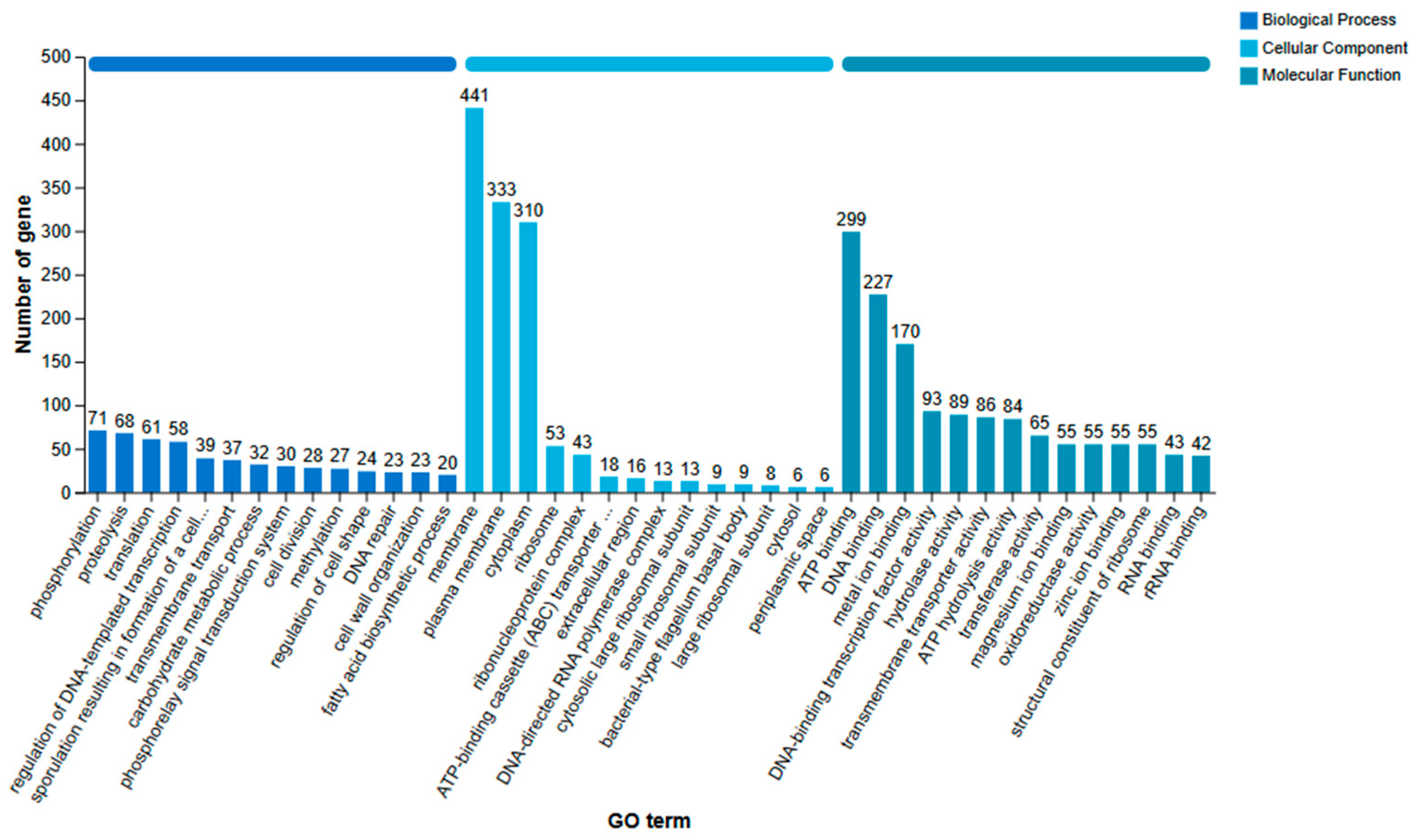
Figure 6.
KEGG annotation classification results statistics of B. amyloliquefaciens SH-53 genome.
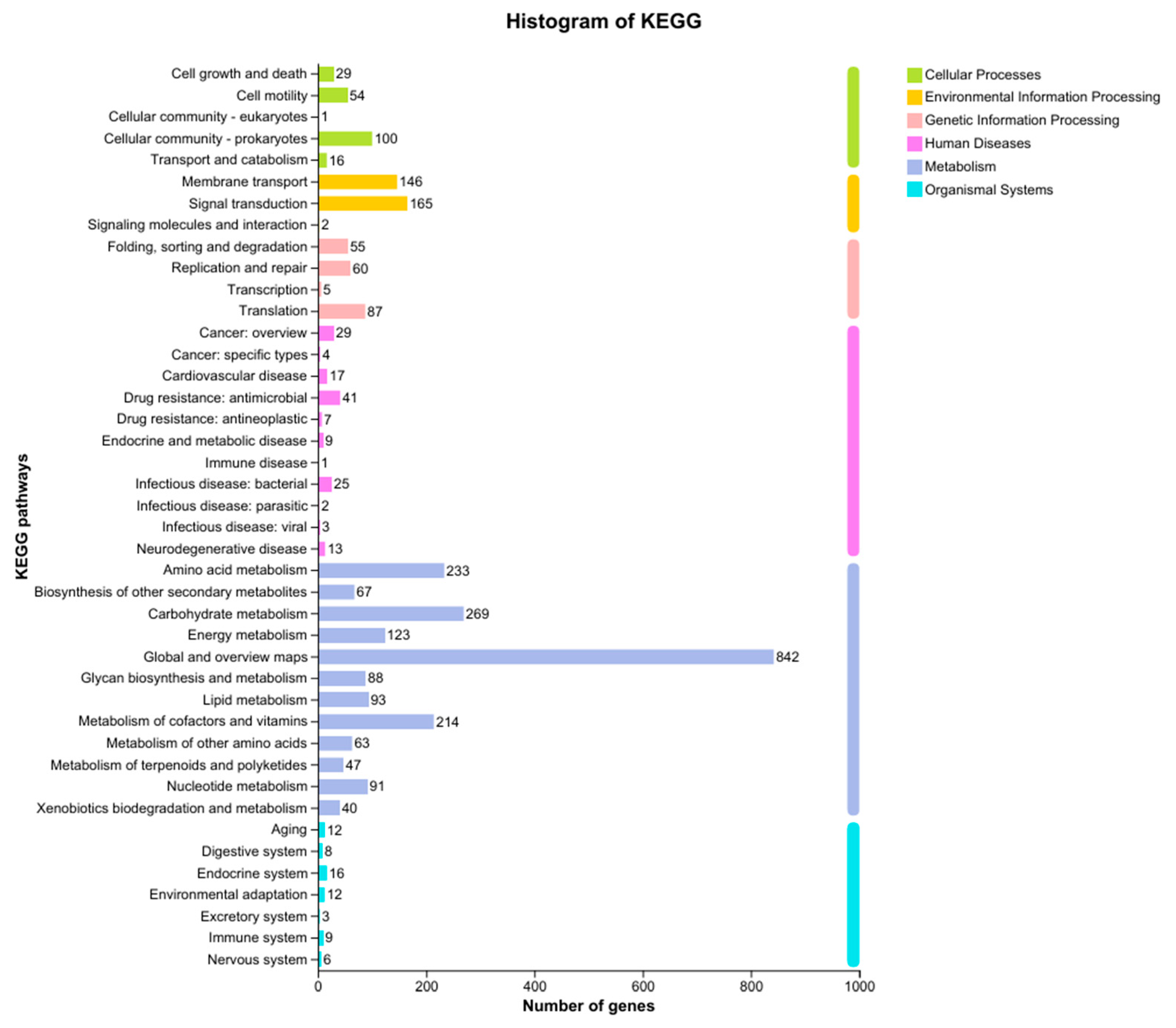
Figure 7.
Annotation of carbohydrate-active enzymes.
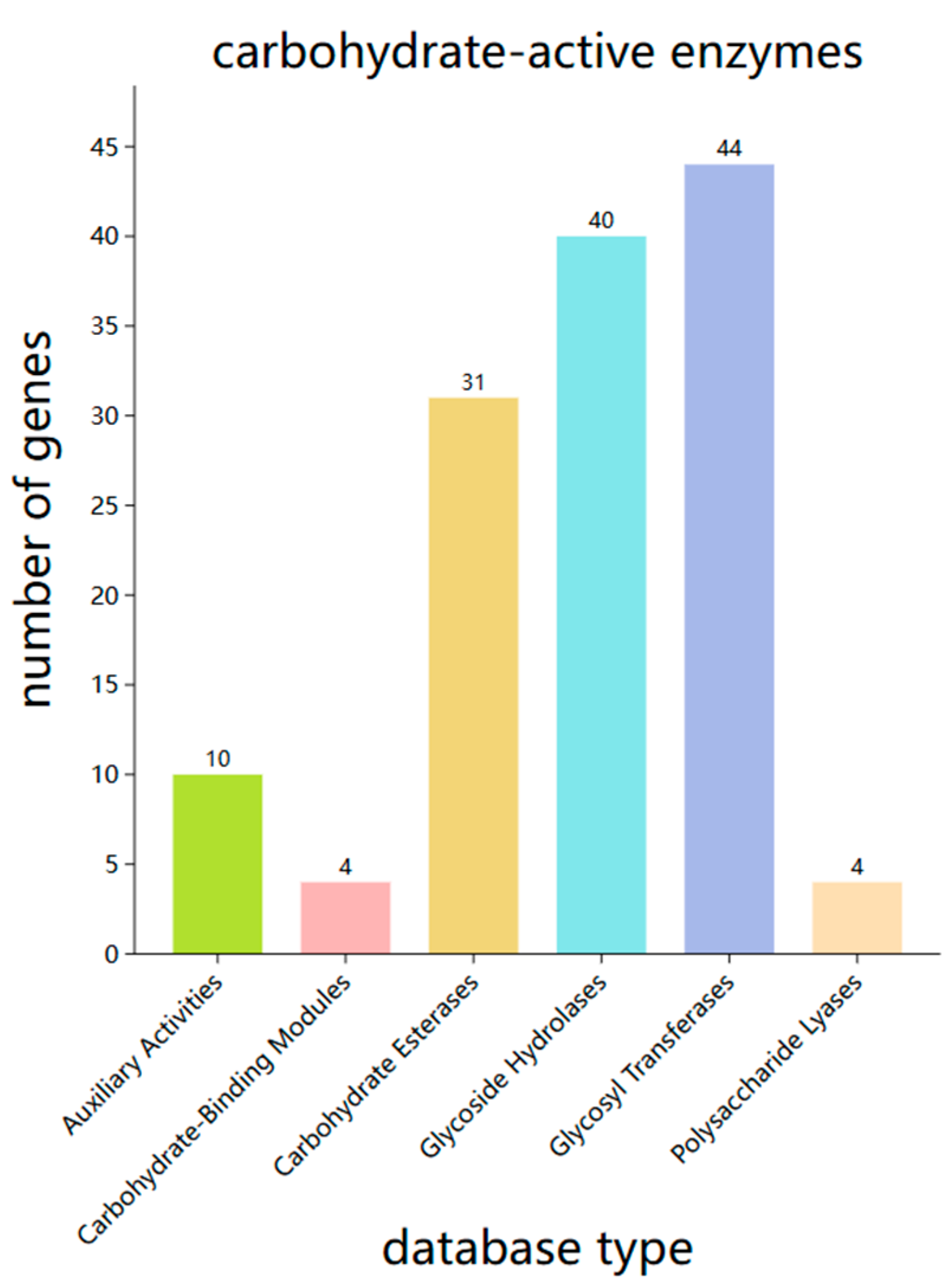
Figure 9.
Functional annotation results of the SH-53 genome based on the CARD database.
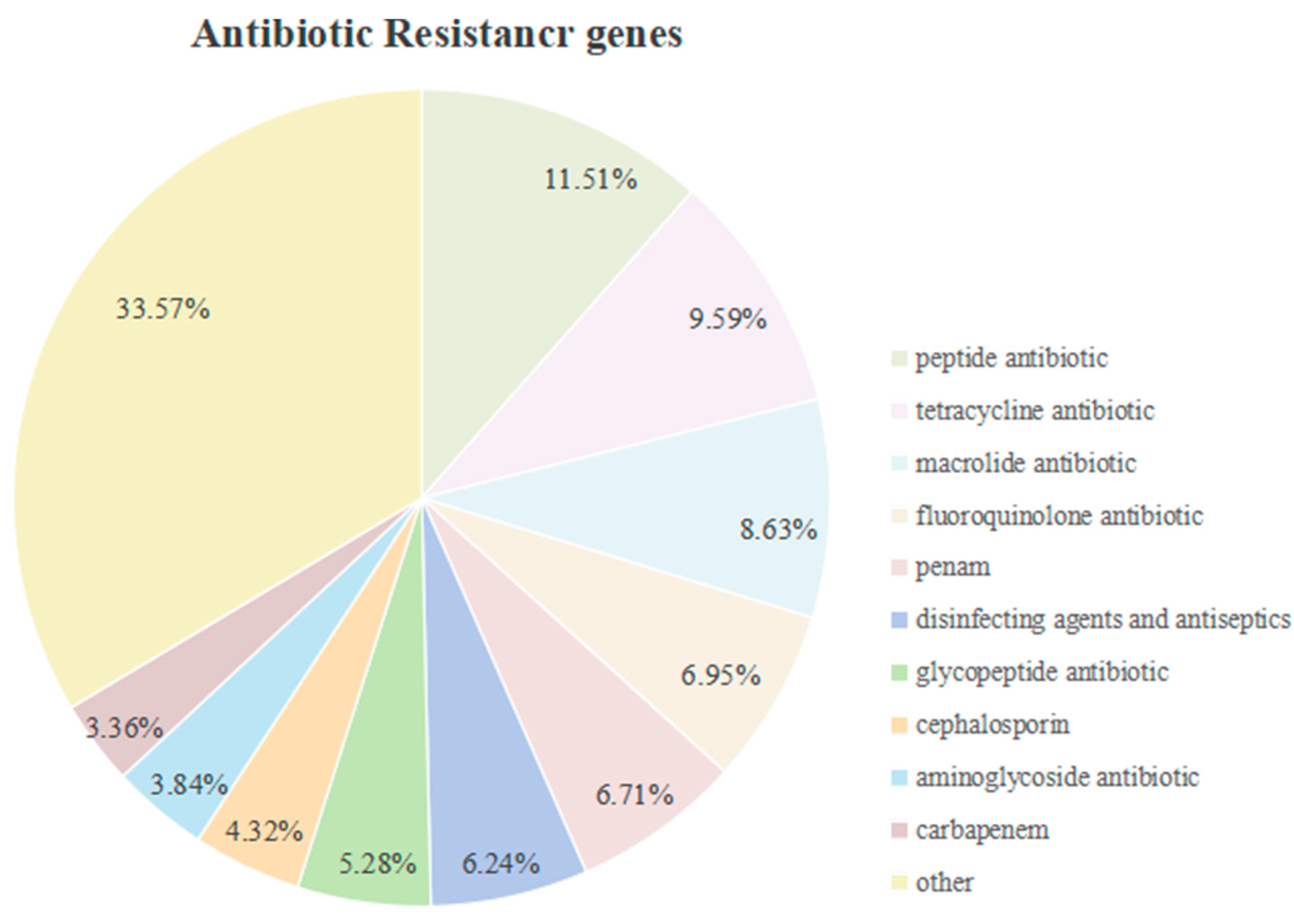
Figure 10.
Effect of strain SH-53 on tomato seed germination.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



