
Submitted:
09 December 2025
Posted:
24 December 2025
You are already at the latest version
Abstract
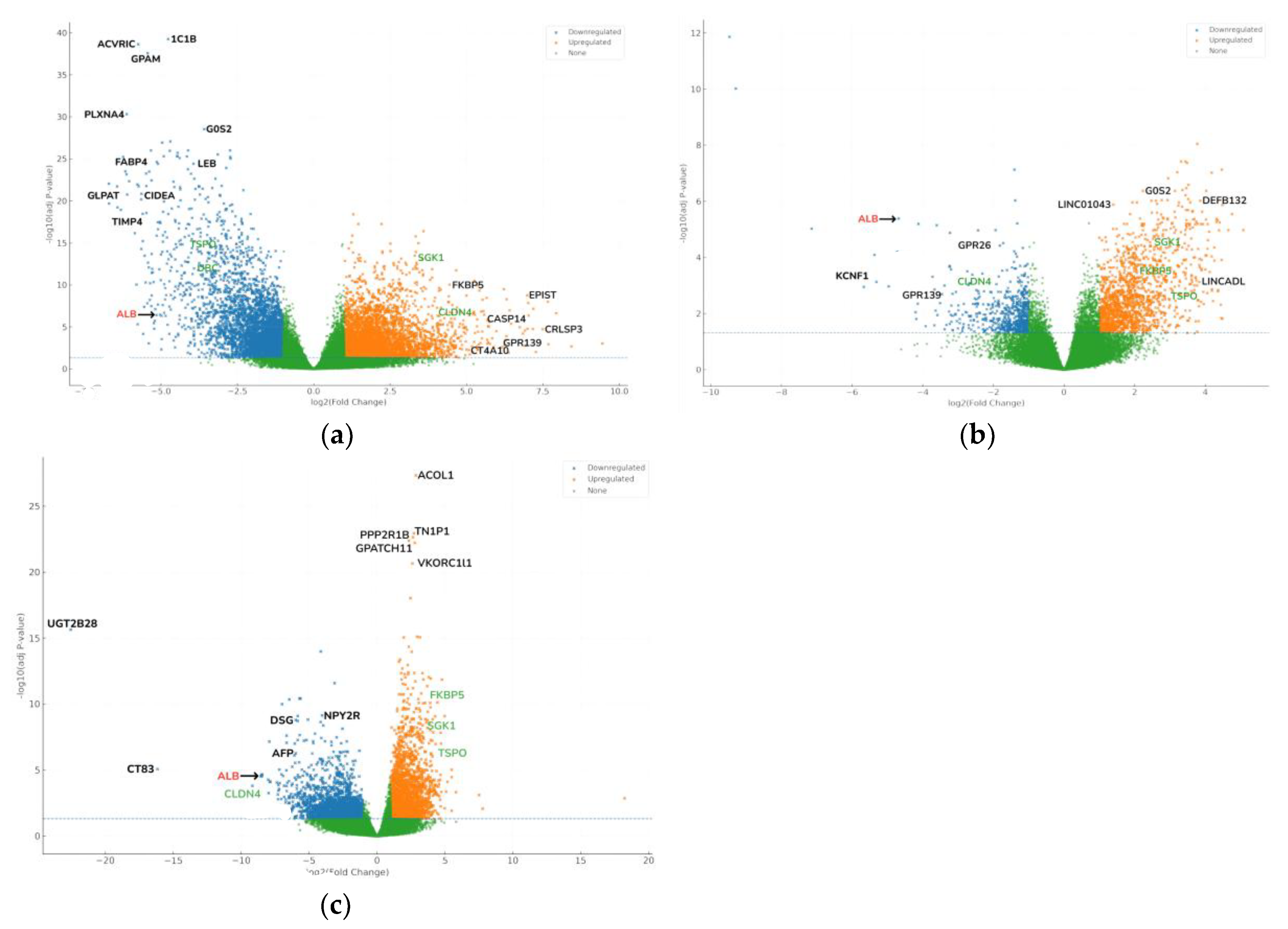
Hydroxyprogesterone (HP) is a synthetic progestogen widely used in obstetric care, and its potential influence on breast cancer biology has become an emerging area of interest. Despite its clinical use, the molecular mechanisms by which HP affects tumor tissue remain insufficiently explored. In this study, transcriptomic profiling was performed to investigate gene expression changes associated with HP in operable breast cancer. Pre-operative 17-OHPC exposure was associated, in normal adjacent tissue (NAT), with activation of steroid-hormone and lipid/xenobiotic-metabolism programs and crosstalk to PI3K–Akt and NF-κB. In NAT, these pathways showed the largest absolute log2 fold-change (|log2FC|); significance is reported as FDR throughout (e.g., FKBP5↑ with HP). In tumor tissue, the dominant signal reflected tight-junction/apical-junction and ECM-receptor remodeling (e.g., CLDN4↑). We prioritized FKBP5 (HP pharmacodynamics) and CLDN4 (tumor baseline) as the main candidates; TSPO and SGK1 are reported as exploratory. These findings provide mechanistic insight into HP’s molecular effects in breast cancer and suggest potential applications in biomarker perioperative management.
Keywords:
1. Introduction
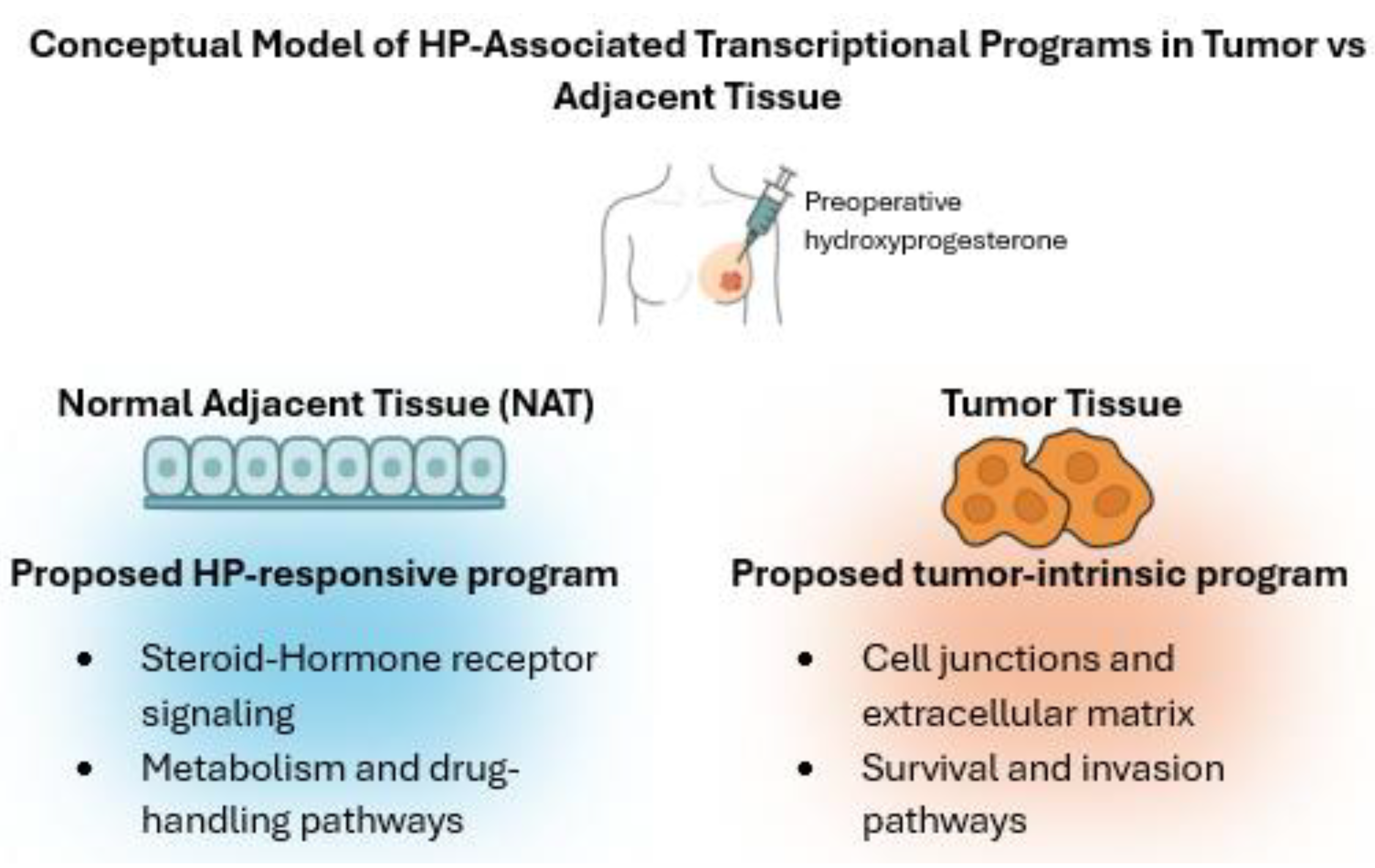
2. Materials and Methods
2.1. Study Design and Overview
2.2. Dataset Description and Sample Grouping
2.3. Data Preprocessing
2.4. Alignment and Quantification
2.5. Differential Expression Analysis
2.6. Functional Enrichment Analysis
2.7. Co-Expression Network Construction (WGCNA)
2.8. Transcriptome-Based Biomarker Prioritization
3. Results
3.1. Quality Control and Read Statistics
3.2. Mapping Efficiency and Gene Coverage
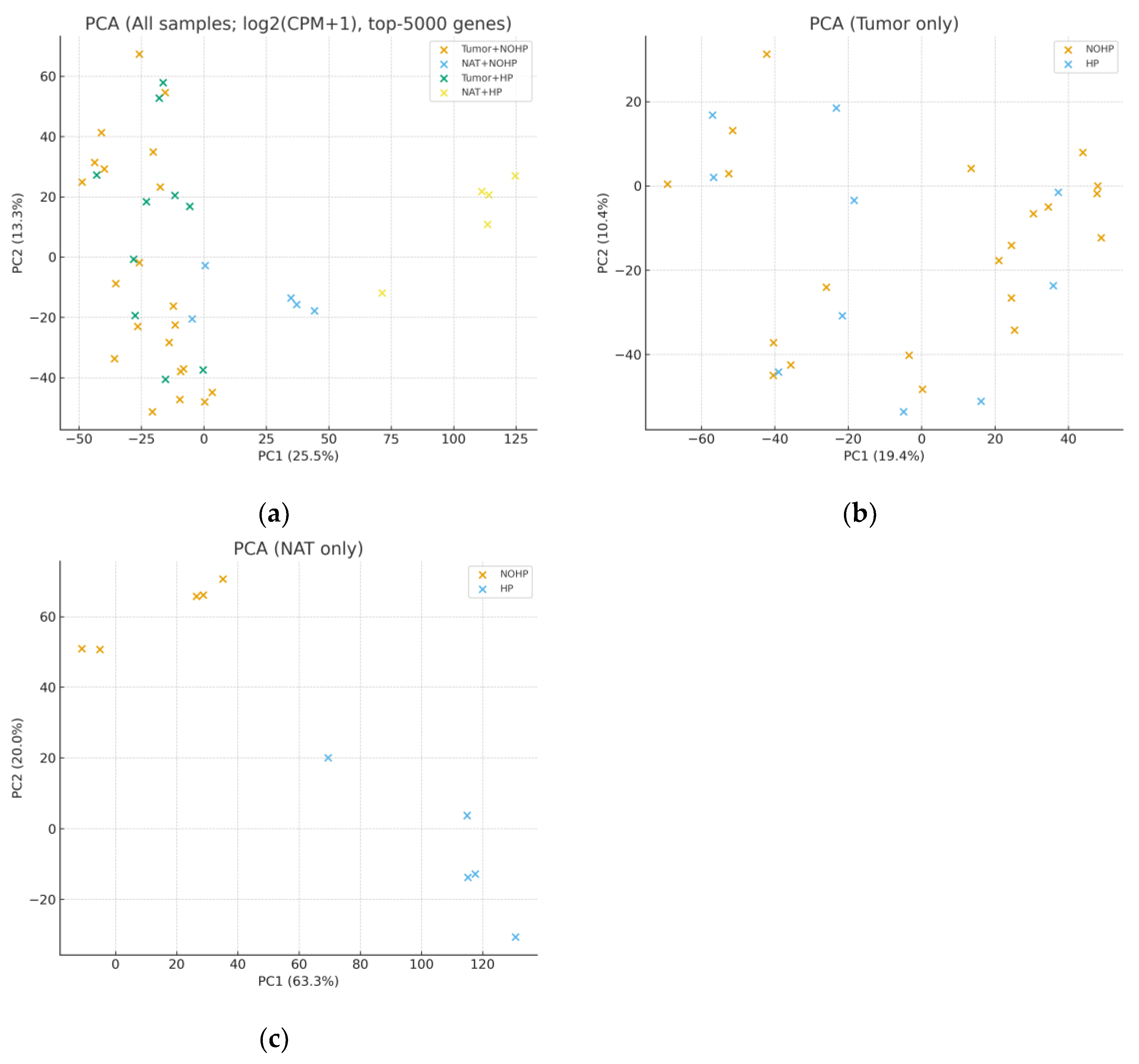
3.3. Transcriptomic Distributions and Principal Component Analysis (PCA)
3.4. Differential Expression and Functional Insights
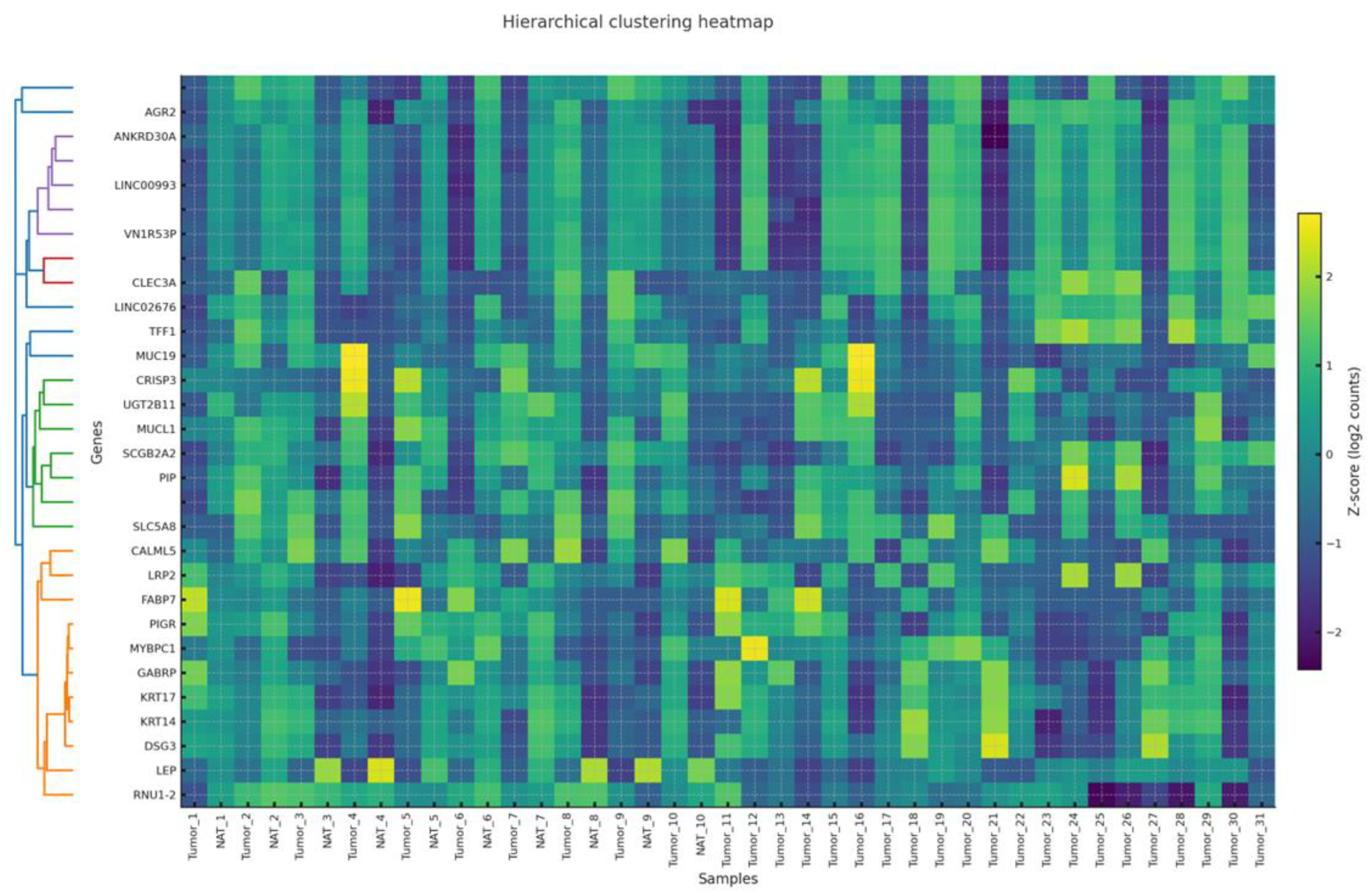
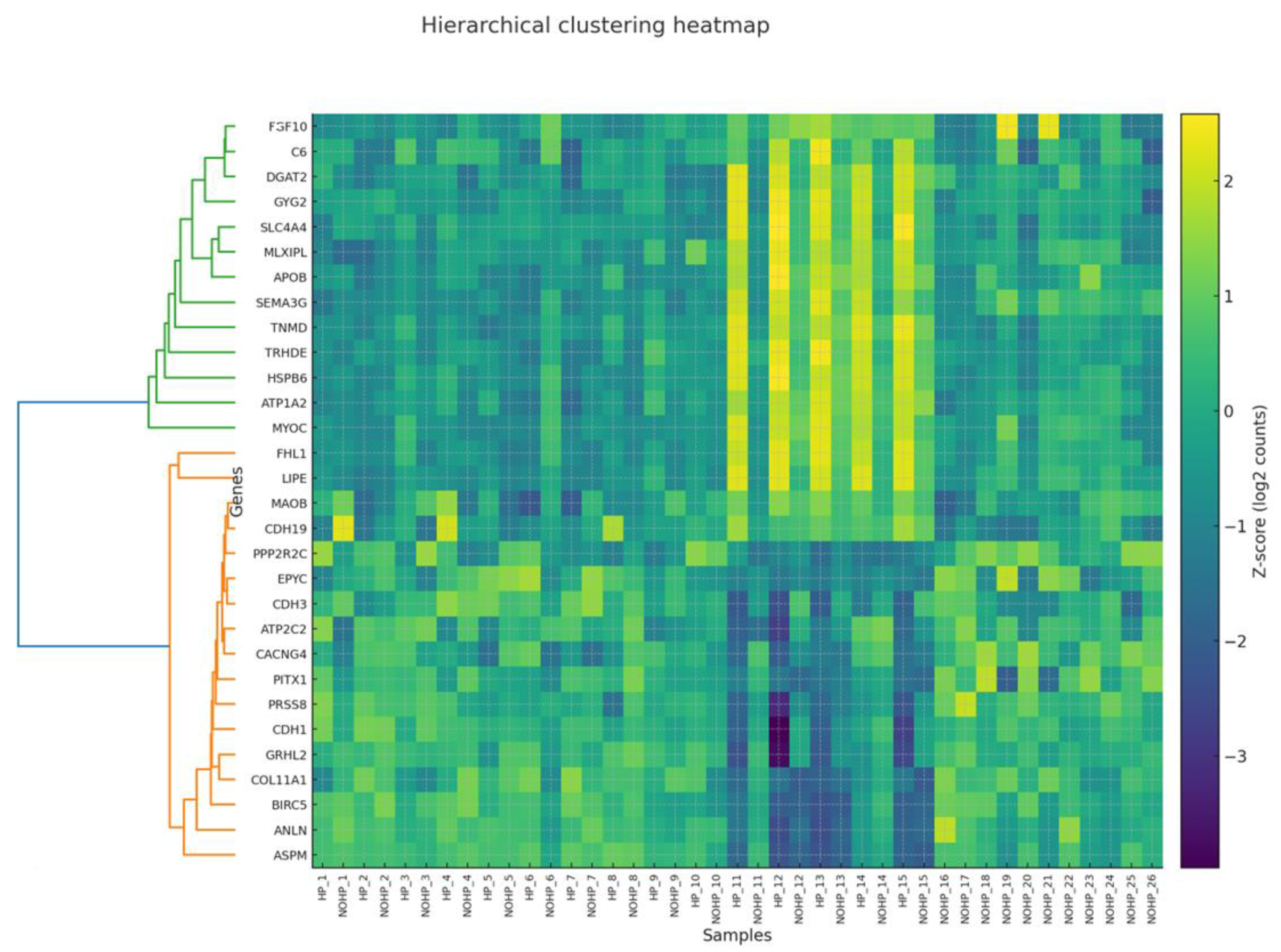
3.5. Co-Expression Modules and Hub Genes (WGCNA)
3.8. Clinical Implications and Biomarker Potential
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Author Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BP | Biological Process (Gene Ontology) |
| CC | Cellular Component (Gene Ontology) |
| DEG | Differentially Expressed Gene |
| ER | Estrogen Receptor |
| ER stress | Endoplasmic Reticulum Stress |
| FC | Fold Change |
| FDR | False Discovery Rate |
| GO | Gene Ontology |
| GRCh38 | Genome Reference Consortium Human Build 38 |
| GSEA | Gene Set Enrichment Analysis |
| HP | Hydroxyprogesterone |
| log2FC | Log2 Fold Change |
| MF | Molecular Function (Gene Ontology) |
| NAT | Normal Adjacent Tissue |
| ORA | Overrepresentation Analysis |
| PR | Progesterone Receptor |
| QC | Quality Control |
| RNA-seq | RNA Sequencing |
| rRNA | Ribosomal RNA |
| TMM | Trimmed Mean of M-values |
| WGCNA | Weighted Gene Co-expression Network Analysis |
References
- Sha, R.; Kong, X.; Li, X.; Wang, Y. Global Burden of Breast Cancer and Attributable Risk Factors in 204 Countries and Territories, from 1990 to 2021: Results from the Global Burden of Disease Study 2021. Biomark Res 2024, 12, 87. [Google Scholar] [CrossRef]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and Future Burden of Breast Cancer: Global Statistics for 2020 and 2040. The Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Harper, A.; McCormack, V.; Sung, H.; Houssami, N.; Morgan, E.; Mutebi, M.; Garvey, G.; Soerjomataram, I.; Fidler-Benaoudia, M.M. Global Patterns and Trends in Breast Cancer Incidence and Mortality across 185 Countries. Nat Med 2025, 31, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, L.; Gathani, T. Understanding Breast Cancer as a Global Health Concern. The British Journal of Radiology 2022, 95, 20211033. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.Q.; Li, J.L.; Fisher, S.G.; Yao, K.A.; David, S.P.; Huo, D. Declination of Treatment, Racial and Ethnic Disparity, and Overall Survival in US Patients With Breast Cancer. JAMA Netw Open 2024, 7, e249449. [Google Scholar] [CrossRef]
- Teklemariam, A.B.; Muche, Z.T.; Agidew, M.M.; Mulu, A.T.; Zewde, E.A.; Baye, N.D.; Adugna, D.G.; Maru, L.; Ayele, T.M. Receptor Tyrosine Kinases and Steroid Hormone Receptors in Breast Cancer: Review of Recent Evidences. Metabolism Open 2024, 24, 100324. [Google Scholar] [CrossRef]
- Chakravorty, G.; Ahmad, S.; Godbole, M.S.; Gupta, S.; Badwe, R.A.; Dutt, A. Deciphering the Mechanisms of Action of Progesterone in Breast Cancer. Oncotarget 2023, 14, 660–667. [Google Scholar] [CrossRef]
- Rodrigues, I.; Fernandes, R.; Ferreira, A.; Pereira, D.; Fernandes, R.; Soares, R.; Luís, C. Is Progesterone Receptor a Neglected Feature in Breast Cancer? A Retrospective Study Analysing the Clinicopathological Characteristics of Breast Cancer Based on Progesterone Receptor Status. Clinical Breast Cancer 2025, 25, e331–e340. [Google Scholar] [CrossRef]
- Health Centre (Cube 34B), University of Calabria 87030 Arcavacata di Rende (CS) Italy.; Aquila, S. 17 Hydroxyprogesterone Progesterone Receptor B Signalling Disrupts the Metabolic Reprogramming in Breast Cancer Cell Lines. J Oncology 2022, 2.
- Fedotcheva, T.A.; Fedotcheva, N.I.; Shimanovsky, N.L. Progesterone as an Anti-Inflammatory Drug and Immunomodulator: New Aspects in Hormonal Regulation of the Inflammation. Biomolecules 2022, 12, 1299. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Fountzilas, E.; Bleris, L.; Kurzrock, R. Transcriptomics and Solid Tumors: The next Frontier in Precision Cancer Medicine. Seminars in Cancer Biology 2022, 84, 50–59. [Google Scholar] [CrossRef]
- Van De Stolpe, A.; Verhaegh, W.; Blay, J.-Y.; Ma, C.X.; Pauwels, P.; Pegram, M.; Prenen, H.; De Ruysscher, D.; Saba, N.F.; Slovin, S.F.; et al. RNA Based Approaches to Profile Oncogenic Pathways From Low Quantity Samples to Drive Precision Oncology Strategies. Front. Genet. 2021, 11, 598118. [Google Scholar] [CrossRef]
- Ji, J.-H.; Ahn, S.G.; Yoo, Y.; Park, S.-Y.; Kim, J.-H.; Jeong, J.-Y.; Park, S.; Lee, I. Prediction of a Multi-Gene Assay (Oncotype DX and Mammaprint) Recurrence Risk Group Using Machine Learning in Estrogen Receptor-Positive, HER2-Negative Breast Cancer—The BRAIN Study. Cancers 2024, 16, 774. [Google Scholar] [CrossRef]
- Hristova, V.A.; Chan, D.W. Cancer Biomarker Discovery and Translation: Proteomics and Beyond. Expert Review of Proteomics 2019, 16, 93–103. [Google Scholar] [CrossRef]
- ERP135222 - SRA - NCBI. Available online: https://www.ncbi.nlm.nih.gov/sra/?term=ERP135222.
- PureLink RNA Mini Kit - US. Available online: https://www.thermofisher.com/us/en/home/life-science/dna-rna-purification-analysis/rna-extraction/rna-types/total-rna-extraction/purelink-rna-mini-kit.html.
- Home - SRA - NCBI. Available online: https://www.ncbi.nlm.nih.gov/sra.
- Krueger, F.; James, F.; Ewels, P.; Afyounian, E.; Schuster-Boeckler, B. FelixKrueger/TrimGalore: V0.6.7 - DOI via Zenodo 2021.
- Babraham Bioinformatics - FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat Biotechnol 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR : A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS: A Journal of Integrative Biology 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Research 2019, gkz1021. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. CP in Bioinformatics 2016, 54. [Google Scholar] [CrossRef]
- Chatterjee, S.; Chaubal, R.; Maitra, A.; Gardi, N.; Dutt, A.; Gupta, S.; Badwe, R.A.; Majumder, P.P.; Pandey, P. Pre-Operative Progesterone Benefits Operable Breast Cancer Patients by Modulating Surgical Stress. Breast Cancer Res Treat 2018, 170, 431–438. [Google Scholar] [CrossRef]
- Kovalchuk, A.; Ilnytskyy, Y.; Rodriguez-Juarez, R.; Katz, A.; Sidransky, D.; Kolb, B.; Kovalchuk, O. Growth of Triple Negative and Progesterone Positive Breast Cancer Causes Oxidative Stress and Down-Regulates Neuroprotective Transcription Factor NPAS4 and NPAS4-Regulated Genes in Hippocampal Tissues of TumorGraft Mice—an Aging Connection. Front. Genet. 2018, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- González-Espinoza, A.; Zamora-Fuentes, J.; Hernández-Lemus, E.; Espinal-Enríquez, J. Gene Co-Expression in Breast Cancer: A Matter of Distance. Front. Oncol. 2021, 11, 726493. [Google Scholar] [CrossRef]
- Yadav, N.; Sunder, R.; Desai, S.; Dharavath, B.; Chandrani, P.; Godbole, M.; Dutt, A. Progesterone Modulates the DSCAM-AS1/miR-130a/ESR1 Axis to Suppress Cell Invasion and Migration in Breast Cancer. Breast Cancer Res 2022, 24, 97. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat Commun 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network Medicine: A Network-Based Approach to Human Disease. Nat Rev Genet 2011, 12, 56–68. [Google Scholar] [CrossRef]
- Wochnik, G.M.; Rüegg, J.; Abel, G.A.; Schmidt, U.; Holsboer, F.; Rein, T. FK506-Binding Proteins 51 and 52 Differentially Regulate Dynein Interaction and Nuclear Translocation of the Glucocorticoid Receptor in Mammalian Cells. Journal of Biological Chemistry 2005, 280, 4609–4616. [Google Scholar] [CrossRef] [PubMed]
- Murakami-Nishimagi, Y.; Sugimoto, K.; Kobayashi, M.; Tachibana, K.; Kojima, M.; Okano, M.; Hashimoto, Y.; Saji, S.; Ohtake, T.; Chiba, H. Claudin-4-Adhesion Signaling Drives Breast Cancer Metabolism and Progression via Liver X Receptor β. Breast Cancer Res 2023, 25, 41. [Google Scholar] [CrossRef]
- Hemler, M.E. Tetraspanin Proteins Promote Multiple Cancer Stages. Nat Rev Cancer 2014, 14, 49–60. [Google Scholar] [CrossRef]
- Selvaraj, V.; Stocco, D.M.; Tu, L.N. Minireview: Translocator Protein (TSPO) and Steroidogenesis: A Reappraisal. Molecular Endocrinology 2015, 29, 490–501. [Google Scholar] [CrossRef]
- Papadopoulos, V.; Fan, J.; Zirkin, B. Translocator Protein (18 kDa): An Update on Its Function in Steroidogenesis. J Neuroendocrinology 2018, 30, e12500. [Google Scholar] [CrossRef]
- Günzel, D.; Yu, A.S.L. Claudins and the Modulation of Tight Junction Permeability. Physiological Reviews 2013, 93, 525–569. [Google Scholar] [CrossRef]
- Kahle, K.T.; MacGregor, G.G.; Wilson, F.H.; Van Hoek, A.N.; Brown, D.; Ardito, T.; Kashgarian, M.; Giebisch, G.; Hebert, S.C.; Boulpaep, E.L.; et al. Paracellular Cl- Permeability Is Regulated by WNK4 Kinase: Insight into Normal Physiology and Hypertension. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 14877–14882. [Google Scholar] [CrossRef] [PubMed]
- Lashhab, R.; Essuman, G.; Chavez-Canales, M.; Alexander, R.T.; Cordat, E. Expression of the Kidney Anion Exchanger 1 Affects WNK4 and SPAK Phosphorylation and Results in Claudin-4 Phosphorylation. Heliyon 2023, 9, e22280. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Gu, Y.-J.; Cao, Z.-H.; Bian, X.-J.; Lan, T.; Sang, J.-R.; Jiang, L.; Wang, Y.; Qian, H.; Chen, Y.-C. Endogenous cGMP-Dependent Protein Kinase Reverses EGF-Induced MAPK/ERK Signal Transduction through Phosphorylation of VASP at Ser239. Oncology Letters 2012, 4, 1104–1108. [Google Scholar] [CrossRef]
- Zuzga, D.S.; Pelta-Heller, J.; Li, P.; Bombonati, A.; Waldman, S.A.; Pitari, G.M. Phosphorylation of Vasodilator-stimulated Phosphoprotein Ser239 Suppresses Filopodia and Invadopodia in Colon Cancer. Intl Journal of Cancer 2012, 130, 2539–2548. [Google Scholar] [CrossRef] [PubMed]
- Gai, J.; Ji, M.; Shi, C.; Li, W.; Chen, S.; Wang, Y.; Li, H. FoxO Regulates Expression of ABCA6, an Intracellular ATP-Binding-Cassette Transporter Responsive to Cholesterol. The International Journal of Biochemistry & Cell Biology 2013, 45, 2651–2659. [Google Scholar] [CrossRef]
- Nagy, Z.; Kovács, I.; Török, M.; Tóth, D.; Vereb, G.; Buzás, K.; Juhász, I.; Blumberg, P.M.; Bíró, T.; Czifra, G. Function of RasGRP3 in the Formation and Progression of Human Breast Cancer. Mol Cancer 2014, 13, 96. [Google Scholar] [CrossRef]
- Yang, D.; Tao, J.; Li, L.; Kedei, N.; Tóth, Z.E.; Czap, A.; Velasquez, J.F.; Mihova, D.; Michalowski, A.M.; Yuspa, S.H.; et al. RasGRP3, a Ras Activator, Contributes to Signaling and the Tumorigenic Phenotype in Human Melanoma. Oncogene 2011, 30, 4590–4600. [Google Scholar] [CrossRef] [PubMed]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the Dynamic HSP90 Complex in Cancer. Nat Rev Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Ge, L.; Hoa, N.T.; Wilson, Z.; Arismendi-Morillo, G.; Kong, X.-T.; Tajhya, R.B.; Beeton, C.; Jadus, M.R. Big Potassium (BK) Ion Channels in Biology, Disease and Possible Targets for Cancer Immunotherapy. International Immunopharmacology 2014, 22, 427–443. [Google Scholar] [CrossRef]
- Cormerais, Y.; Massard, P.A.; Vucetic, M.; Giuliano, S.; Tambutté, E.; Durivault, J.; Vial, V.; Endou, H.; Wempe, M.F.; Parks, S.K.; et al. The Glutamine Transporter ASCT2 (SLC1A5) Promotes Tumor Growth Independently of the Amino Acid Transporter LAT1 (SLC7A5). Journal of Biological Chemistry 2018, 293, 2877–2887. [Google Scholar] [CrossRef] [PubMed]

| Group | Tissue type | HP exposure | Sample count |
|---|---|---|---|
| NAT HP− | NAT | Not exposed | 5 |
| NAT HP+ | NAT | Exposed | 5 |
| Tumor HP− | Tumor | Not exposed | 13 |
| Tumor HP+ | Tumor | Exposed | 18 |
| Module color | Gene count | Example hub gene |
|---|---|---|
| Black | 14 | MAGEA11 |
| Blue | 201 | PAX6 |
| Brown | 96 | ABCC8 |
| Cyan | 24 | C1orf106 |
| Green | 43 | ITGA5 |
| Grey | 70 | PATE3 |
| Red | 78 | PROM1 |
| Turquoise | 258 | HSP90AA1 |
| Yellow | 82 | ESR1 |
| Module color | Gene count | Example hub gene |
|---|---|---|
| Black | 57 | BARX2 |
| Blue | 187 | ACTA1 |
| Brown | 101 | RASGRP3 |
| Cyan | 21 | ABCA6 |
| Green | 47 | PRKG1 |
| Grey | 72 | SH2D6 |
| Red | 78 | PROM1 |
| Turquoise | 251 | KIF5B |
| Yellow | 84 | SLC1A5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



