Submitted:
13 December 2025
Posted:
17 December 2025
You are already at the latest version
Abstract
The MAPK/ERK signaling pathway is really important and well-conserved in how cells communicate. It controls essential functions like cell growth, differentiation, and survival. When it gets out of whack, it’s often a key player in cancer development, fueling tumor growth, spread, and making treatments less effective across various cancers. Interestingly, this pathway also plays a crucial role in how our brains adapt and process pain, especially in cases of acute and chronic pain, including pain linked to cancer. New findings suggest that these roles aren't just separate; they’re actually connected. Tumor-related factors and stress from treatments can activate MAPK/ERK signaling in both cancer cells and the neurons that sense pain. This connection establishes MAPK/ERK as a fascinating link between cancer progression and the experience of pain. In this review, we’ll look at the current understanding of how the MAPK/ERK axis functions in both cancer and pain, discussing its traditional roles and how they can vary in different contexts. We’ll also dive into how this overlap might impact treatment options, including the existing MAPK/ERK inhibitors used in cancer therapy and their potential use for pain relief. By bringing together insights from cancer biology and pain research, we hope to emphasize the pathway's potential as a dual target for more effective cancer treatment strategies.
Keywords:
analgesia
; cancer progression
; ERK activation
; MAPK pathway
; neuropathic pain
; oncogenic signaling
1. Introduction
The MAPK/ERK pathway is one of the most well-researched signaling pathways in the biological sciences. It plays a key role in how cells respond to signals from outside their environment, which ultimately influences their growth, metabolism, and ability to adapt [1]. This pathway, which has been preserved throughout evolution, takes in signals from various receptors, especially receptor tyrosine kinases (RTKs). It helps manage essential processes like cell growth, differentiation, survival, and programmed cell death under normal conditions [2]. The MAPK/ERK pathway plays a key role in how cells make decisions, which is why it's often prone to going awry and can lead to cancer. In fact, mutations that activate essential parts of this pathway-like RAS, BRAF, and various receptor tyrosine kinases-are some of the most frequently seen genetic changes in many types of human cancers. These mutations can drive unchecked growth, invasion, and resistance to treatment. [3,4]. At the same time, it’s pretty interesting to see that the MAPK/ERK pathway has become a key player in the nervous system, especially when it comes to influencing how neurons behave and how synaptic plasticity works [5]. When it comes to how pain works, the activation of ERK in the peripheral pain receptors and spinal neurons is really important. It acts like a switch that makes us more sensitive to pain. It takes all kinds of harmful signals-like injuries, inflammation, or nerve damage-and turns them into a heightened sensitivity that can lead to ongoing pain [6,7]. It’s interesting how two areas that seem so different-like cancer treatment and pain neuroscience-actually share some important connections. There's a lot of evidence piling up that shows these links aren't just random; they’re often intertwined, particularly when it comes to cancer. The environment around a tumor is full of growth factors, cytokines, and proteases that can boost cancer cell growth while also triggering pain signals in the nearby nerves [8,9]. This sets off a troubling cycle where tumors grow and cause more pain, and the chemicals related to that pain might actually affect the tumor environment. It's interesting and a bit contradictory, but we’ve noticed that some treatments focusing on the MAPK/ERK pathway-though they work well against cancer-can also lead to painful nerve issues. This really shows how complex and context-sensitive that pathway can be [10]. Even though we've made a lot of progress in understanding the MAPK/ERK pathway separately in different fields, we still don't have a combined view that looks at how it connects cancer biology with pain mechanisms. This review is here to fill that void. We'll break down how MAPK/ERK signaling works, look at how it's exploited in cancer, and discuss its crucial role in pain sensitivity. In the end, we’ll talk about the potential of targeting this pathway for treating both cancer and pain, and suggest some future research directions and clinical applications.
2. The Canonical MAPK/ERK Pathway and Its Regulation
The MAPK/ERK pathway acts like a three-step kinase relay that sends signals from the cell membrane to the nucleus and other parts of the cell (see Figure 1). It starts when growth factors, such as EGF and NGF, bind to their specific receptor tyrosine kinases (RTKs). This triggers the receptors to pair up, phosphorylate themselves, and bring in adaptor proteins like GRB2 and SOS. This setup helps activate the small GTPase RAS (including KRAS, NRAS, and HRAS) by loading it with GTP [11]. When RAS is activated, it pulls RAF kinases (like ARAF, BRAF, and CRAF) to the membrane. BRAF stands out as a key player and is the most commonly mutated RAF type seen in cancer. These RAF kinases then go on to phosphorylate and activate MEK1 and MEK2 (which are dual-specificity kinases), and those guys phosphorylate ERK1 and ERK2 (MAPK) at certain threonine and tyrosine sites. The phosphorylation of ERK (often noted as p-ERK) is what really indicates that the pathway is active, making it a crucial marker for both research purposes and clinical diagnostics [13]. When p-ERK gets activated, it pulls away from where it was anchored in the cytoplasm and starts phosphorylating over 100 different substrates in both the cytoplasm and the nucleus, leading to a variety of cellular effects. In the cytoplasm, ERK focuses on proteins that help with quick cellular reactions, like the p90 ribosomal S6 kinase (RSK), which plays a role in protein synthesis and keeping cells alive, as well as cytoskeletal components that manage how cells move around [14]. When p-ERK moves into the nucleus, it gets the transcription factors such as ELK1, c-FOS, and c-JUN phosphorylated and activated. These factors then kickstart the expression of important genes for the cell cycle, like cyclin D1, as well as genes that help with survival and differentiation, which in turn play a role in long-term cell functions [15].
Several feedback systems in place carefully control how strong, how long, and where ERK signaling happens, which helps the cells respond just right. Some of the main negative regulators are:
- Dual-specificity phosphatases (DUSPs): There's a group of enzymes that help turn off ERK by removing its phosphate groups, acting like an important off-switch [16].
- Sprouty (SPRY) proteins: These proteins inhibit RAS activation and RAF membrane recruitment, often acting as tumor suppressors [17].
- Feedback phosphorylation: ERK can add phosphate groups to earlier components such as SOS and RAF, which in turn creates feedback loops that decrease the signaling [18].
Furthermore, the MAPK/ERK pathway doesn't work alone. It interacts a lot with other important pathways, especially the PI3K-AKT-mTOR axis. This back-and-forth communication helps integrate signals and can actually cause some paths to compensate, which is a key reason why resistance can happen when you try to block one of these pathways [19,20]. So, the specific outcome of MAPK/ERK signaling really comes from this complex regulatory system. When this system gets messed up-either through mutations or changes in how regulatory parts are expressed-it’s often a key factor in various diseases.
3. Oncogenic Hijacking: The MAPK/ERK Pathway in Cancer Development
The activation of the MAPK/ERK pathway plays a crucial role in cancer development, helping tumors gain important traits. This faulty signaling usually starts from mutations that enhance the function of key parts of the pathway or their regulators, bypassing the usual controls on growth [3,21].
3.1. Key Mutations and Driver Events
The MAPK/ERK pathway is where we see a lot of somatic mutations in cancer. About 25% of all human cancers have activating mutations in KRAS-specifically at codons 12, 13, and 61. This is especially common in pancreatic cancer, where it’s over 90%, as well as in colorectal cancer at around 40% and lung adenocarcinomas at about 30% [22]. Mutations in BRAF, especially the V600E change, show up in around 50% of melanomas and about 10% of colorectal cancers. They can also appear in some thyroid and ovarian cancers. There are also less common but important mutations in NRAS, HRAS, and the MEK1/2 genes, typically depending on the type of tissue involved. Plus, sometimes genes like EGFR or HER2 get amplified or overexpressed, which can activate the pathway without needing mutations in the genes further down the line [24]. These genetic alterations converge to generate a persistent, growth-promoting signal that is largely independent of external stimuli.
3.2. Role in Tumor Hallmarks: Proliferation, Survival, EMT, and Metastasis
When the MAPK/ERK pathway gets turned on in the wrong way, it sets off a bunch of processes that can help tumors grow. It pushes the expression of cyclins, like Cyclin D1, and holds back proteins that usually keep the cell cycle in check, which leads to unchecked cell growth. At the same time, it helps cells survive longer by boosting protective proteins like BCL-2 and MCL-1 while lowering the levels of proteins that promote cell death [26]. One key part of how cancer develops is something called epithelial-mesenchymal transition (EMT). This process gives cancer cells the ability to move and invade other tissues. ERK signaling plays a big role in triggering EMT by activating transcription factors like SNAIL, SLUG, and ZEB1. As a result, the cells lose markers typical of epithelial cells, like E-cadherin, while gaining markers found in mesenchymal cells, such as vimentin and N-cadherin [27]. This reprogramming helps with local invasion and getting into the bloodstream. Also, when ERK gets activated, it boosts metastasis by increasing the release of matrix metalloproteinases (MMPs) that break down the extracellular matrix. Plus, it adjusts integrin signaling, which supports survival away from the original site and helps in spreading to other areas [28].
3.3. Shaping the Tumor Microenvironment
The way MAPK/ERK signaling contributes to cancer isn’t just limited to the cancer cells themselves; it also changes the tumor microenvironment around them. This ERK-driven activity boosts the production of pro-angiogenic factors like VEGF, which leads to forming new blood vessels that often don’t work properly, helping the tumor to grow [29]. Pathway activation in cancer-associated fibroblasts (CAFs) can perpetuate a tumor-promoting stroma through reciprocal cytokine signaling [30]. It's worth noting that recent findings point to a role in how tumors avoid the immune system. Take melanoma, for example-BRAF signaling that drives cancer not only suppresses the presentation of tumor antigens but also encourages the release of cytokines that dampen immune response, creating a sort of 'cold' tumor microenvironment. On a positive note, inhibiting BRAF or MEK can actually counteract some of these effects, which gives us a good reason to think about using targeted therapy alongside immunotherapy [31].

Table 1.
Key Genomic Alterations in the MAPK/ERK Pathway Across Major Cancers.
| Cancer Type | RAS Mutation Frequency | BRAF Mutation Frequency | MEK (MAP2K1/2) Mutation Frequency | Primary Alterations & Notes |
| Melanoma | 15-30% (NRAS) [32,33] | 40-50% (V600E/K) [12] | 5-8% [34] | BRAF V600 is the classic driver; it defines a major therapeutic subtype. |
| Colorectal Cancer (CRC) | ~40% (KRAS) [22] | 8-12% (V600E) [23] | <2% [35] | KRAS mutations predict resistance to anti-EGFR therapy. |
| Non-Small Cell Lung Cancer (NSCLC) | 25-30% (KRAS) [22] | 2-4% (V600E & non-V600) [36] | Rare | KRAS G12C is a recent therapeutic target. BRAF V600E defines a small subset. |
| Pancreatic Ductal Adenocarcinoma (PDAC) | >90% (KRAS) [22] | Rare | Rare | KRAS mutation is near-universal and a critical early event. |
| Thyroid Cancer (Papillary) | 10-20% (NRAS, HRAS) [37] | 45-60% (V600E) [37] | Rare | BRAF V600E correlates with aggressive features. |
| Ovarian Cancer (Low-Grade Serous) | 20-35% (KRAS) [38] | 30-50% (BRAF) [38] | Reported [39] | Distinct from high-grade serous, mutations are common in this subtype. |
| Hairy Cell Leukemia | Very Rare | ~100% (V600E) [40] | N/A | BRAF V600E is a disease-defining genetic lesion. |
4. The Signaling Nexus of Pain: MAPK/ERK in Nociception and Sensitization
Unlike its function in helping cancer cells multiply, the MAPK/ERK pathway plays a key role in the nervous system by regulating how neurons adapt. This is crucial in understanding the changes that can result in chronic pain [5,41]. Its activation in response to noxious stimuli occurs in a temporally and spatially coordinated manner across different cell types within the pain pathway.
4.1. Peripheral Sensitization: From Inflammation to Injury
Peripheral sensitization happens when nociceptive neurons, or nociceptors, become more sensitive and easier to trigger at the injury or inflammation site. When there’s damage, a variety of pain-causing substances, like nerve growth factor (NGF), bradykinin, prostaglandins, and ATP, are released. These substances then activate specific receptors (like TrkA, B2, EP, and P2X) on the ends of the nociceptors. It's important to note that these receptors are linked to the MAPK/ERK signaling pathway [42]. When ERK gets activated in the endings of peripheral nerves, it triggers the phosphorylation and regulation of important ion channels that play a role in signaling pain. For instance, when ERK phosphorylates the TRPV1 channel, it becomes more sensitive to heat and capsaicin. At the same time, the phosphorylation of certain sodium channels (like Naᵥ1.7 and Naᵥ1.8) ramps up the excitability of neurons and boosts action potential firing [43,44]. This results in primary hyperalgesia (increased pain from noxious stimuli) and allodynia (pain from normally non-painful stimuli).
4.2. Central Sensitization: Spinal Cord and Supraspinal Plasticity
Central sensitization refers to an increase in how sensitive neurons in the central nervous system, especially in the spinal dorsal horn, can become depending on activity. It's a key factor in persistent pain. When harmful signals come from the body, they cause the release of glutamate and neuropeptides like substance P from the nerve endings, which then activate NMDA receptors and G-protein coupled receptors on spinal neurons. This leads to a flow of calcium that turns on various kinases inside the cells, including ERK [45].
Phospho-ERK in dorsal horn neurons then contributes to central sensitization by:
- Enhancing Synaptic Efficacy: Adding phosphate groups to postsynaptic receptors, like NMDA and AMPA receptors, helps control their activity and encourages them to be inserted into the membrane, which in turn boosts the strength of synaptic transmission [46].
- Regulating Transcription: It's about getting to the nucleus to trigger the expression of pain-related genes like c-Fos, Cox-2, and prodynorphin, which can result in lasting changes in function [47].
It's worth noting that ERK activation isn't just limited to neurons. Microglia and astrocytes in the spinal cord, which get activated when there's nerve damage or inflammation, also show a strong level of ERK phosphorylation. In these glial cells, ERK signaling plays a key role in producing and releasing pro-inflammatory cytokines like TNF-α, IL-1β, and IL-6, as well as chemokines. This process ends up increasing neuronal excitability and neuroinflammation, leading to this ongoing cycle of pain [48,49]. This glial ERK activation is a critical contributor to the maintenance of neuropathic pain states.
5. Convergence: The MAPK/ERK Axis as a Shared Hub in Cancer and Pain
The way the MAPK/ERK pathway gets out of whack in both cancer and pain seems to point to more than just a coincidence; it actually highlights a common molecular basis that allows these issues to affect one another directly, especially when we're talking about cancer pain and neuropathy caused by treatment.
5.1. Mechanistic Overlap in Cellular Processes
Basically, the MAPK/ERK pathway regulates similar processes downstream in both cancer cells and neurons/glia, but it leads to different results in terms of how they appear (see Figure 2). In both contexts, ERK activation:
- Amplifies Inflammatory Signaling: In the tumor microenvironment and nervous system, when ERK gets activated in immune cells like macrophages and in glial cells such as microglia and astrocytes, it triggers the release of pro-inflammatory cytokines like TNF-α, IL-1β, and IL-6. This sets off a feedback loop that not only encourages tumor growth but also increases pain sensitivity at the same time [48,49].
5.2. Clinical Intersection: Cancer-Induced and Therapy-Induced Pain
This biological process has clear effects on clinical outcomes. Take cancer-induced bone pain (CIBP), for instance. Tumor cells in the bone release substances like endothelin-1, nerve growth factor (NGF), and protons, which activate receptor tyrosine kinases (RTKs) and acid-sensing ion channels on nerve endings. This activation sets off MAPK/ERK signaling pathways in both the tumor cells-accelerating the breakdown of bone-and in the sensory nerves, leading to increased sensitivity and spontaneous pain [50,51]. Likewise, when a tumor presses on or invades nerves, it can lead to neuropathic pain through the activation of ERK in both the damaged neurons and the nearby glial cells [52]. Interestingly, therapy-induced peripheral neuropathy (TIPN) is one of the main side effects that limits the doses of several cancer treatments, including certain MAPK/ERK pathway inhibitors. Even though BRAF/MEK inhibitors are good at stopping cancer signals, they can also interfere with the same pathway in healthy sensory neurons and Schwann cells that support them. This interference in normal ERK signaling, which is essential for keeping neurons healthy, ensuring mitochondrial function, and helping with axonal transport, might end up causing axonal degeneration and neuropathic pain [10,53]. This highlights the delicate balance required in therapeutic targeting: inhibiting pathological signaling in cancer without compromising essential functions in normal tissues.
6. Therapeutic Targeting of the MAPK/ERK Axis
MAPK/ERK signaling is really important in both cancer and pain management, which opens up some interesting treatment possibilities. What’s developed for one area could actually work well in the other. Plus, the field of pathway inhibitors is changing quickly.
6.1. MAPK/ERK Inhibitors in Clinical Oncology: A Comprehensive List
Focusing on blocking this particular pathway has really changed the game for treating various cancers, especially those with BRAF V600 mutations. The inhibitors are categorized based on which part of the cascade they aim for (see Table 2).
6.2. Evidence for MAPK/ERK Modulation in Pain Management
Research before clinical trials strongly suggests that blocking the MAPK/ERK pathway could help with pain relief (see Table 3). Using MEK inhibitors like U0126, PD0325901, or SL327, either directly in the spinal fluid or systemically, seems to reduce pain responses in various models of inflammation, nerve damage, and pain caused by cancer [54,55]. We've noticed that these effects are linked to lower levels of p-ERK in the DRG neurons and spinal cord, along with a drop in pro-pain mediators. What's interesting is that some clinical cases suggest there could be a real-world application here. For example, patients with BRAF-mutant melanoma who are on BRAF/MEK inhibitors sometimes mention that their existing cancer pain gets better, though this is more of an anecdote and can be complicated by the shrinkage of their tumors [56]. More systematically, the MEK inhibitor selumetinib is under investigation for pain associated with plexiform neurofibromas in NF1 [57].
6.3. Challenges: Resistance, Toxicity, and the Therapeutic Window
Turning MAPK/ERK inhibition into a workable pain relief approach has a lot of challenges. In cancer treatment, dealing with acquired resistance is a big issue. This often happens when the pathway gets reactivated-like with MEK1/2 mutations or BRAF amplification-or when other pathways, such as PI3K-AKT, get activated [58]. These mechanisms might also reduce how effective pain relief is over time. Toxicity is definitely a big worry, too. Pathway inhibitors can lead to various side effects, like skin issues (rashes, squamous cell carcinoma), eye problems (retinopathy), heart issues (QT prolongation, cardiomyopathy), and digestive troubles (diarrhea, colitis) [59]. Most relevantly, as noted, they can cause therapy-induced peripheral neuropathy (TIPN) [10,53]. This sets up a tricky situation where a medication meant to help with cancer pain could actually cause neuropathic pain instead, limiting its effectiveness. So, if we're thinking about repurposing it, we really need to think through things like how much to give, how to give it (like whether it should be localized or systemic), and which patients to choose. The goal is to get the most benefit while keeping nerve damage to a minimum.
Future Perspectives and Conclusions
The MAPK/ERK pathway plays a crucial role in both cancer development and pain sensitivity. This presents a rare chance for treatment: by focusing on a single pathway, we could tackle both tumor growth and pain relief at the same time. But taking advantage of this opportunity isn't easy; there are major hurdles to get over, like resistance to the pathway, possible nerve damage, and a limited therapeutic window.
Moving forward, we need to focus on a few important strategies. First up, precision clinical trials should consider pain metrics as key indicators when testing MAPK/ERK inhibitors. Then, developing predictive biomarkers, like measuring phospho-ERK levels, could help us pinpoint which patients are most likely to gain dual benefits. Also, we should look into innovative drug delivery systems—think tumor-targeted nanoparticles or localized neural formulations—to boost effectiveness while reducing side effects throughout the body. Lastly, considering smart combination therapies that pair pathway inhibitors with immunomodulators or specific pain relievers might help broaden the therapeutic window and lead to better patient outcomes.
In the end, shifting our approach to more integrated, pathway-focused strategies could pave the way for more tailored and comprehensive cancer treatment. The goal is not just to prolong life but also to significantly improve the quality of life for patients.
References
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- McKay, M.M.; Morrison, D.K. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113–3121. [Google Scholar] [CrossRef]
- Prior, Ian A.; Hood, Fiona E.; Hartley, James L. The frequency of Ras mutations in cancer. Cancer research 2020, 80.14, 2969–2974. [Google Scholar] [CrossRef]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef]
- Ji, R.-R.; Gereau, R.W.; Malcangio, M.; Strichartz, G.R. MAP kinase and pain. Brain Res. Rev. 2009, 60, 135–148. [Google Scholar] [CrossRef]
- Obata, K.; Noguchi, K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. 2004, 74, 2643–2653. [Google Scholar] [CrossRef]
- Zhuang, Z.-Y.; Gerner, P.; Woolf, C.J.; Ji, R.-R. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain 2005, 114, 149–159. [Google Scholar] [CrossRef]
- Mantyh, Patrick. Bone cancer pain: causes, consequences, and therapeutic opportunities. PAIN® 2013, 154, S54–S62. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.L.; Hamamoto, D.T.; Simone, D.A.; Wilcox, G.L. Mechanism of Cancer Pain. Mol. Interv. 2010, 10, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Adamsky, K.; Weizer, O.; Amariglio, N. Bibliography Current World Literature Vol 12 No 2 March 2005. Clinical Genetics 2004, 65, 378–383. [Google Scholar]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Roskoski, Robert, Jr. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacological research 2012, 66.2, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Anjum, R.; Blenis, J. The RSK family of kinases: emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008, 9, 747–758. [Google Scholar] [CrossRef]
- Murphy, L.O.; Blenis, J. MAPK signal specificity: the right place at the right time. Trends Biochem. Sci. 2006, 31, 268–275. [Google Scholar] [CrossRef]
- Caunt, Christopher J.; Keyse, Stephen M. Dual-specificity MAP kinase phosphatases (MKPs) Shaping the outcome of MAP kinase signalling. The FEBS journal 2013, 280.2, 489–504. [Google Scholar] [CrossRef]
- Mason, J.M.; Morrison, D.J.; Basson, M.A.; Licht, J.D. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006, 16, 45–54. [Google Scholar] [CrossRef]
- Lake, D.; Corrêa, S.A.L.; Müller, J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef]
- Yarden, Y.; Pines, G. The ERBB network: at last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Meloche, S.; Pouysségur, J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007, 26, 3227–3239. [Google Scholar] [CrossRef]
- Balmanno, K.; Cook, S.J. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2008, 16, 368–377. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Reddy, K.B.; Nabha, S.M.; Atanaskova, N. Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev. 2003, 22, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Meadows, K.N.; Bryant, P.; Pumiglia, K. Vascular Endothelial Growth Factor Induction of the Angiogenic Phenotype Requires Ras Activation. J. Biol. Chem. 2001, 276, 49289–49298. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.; Nguyen, T.; Gundre, E.; Ogunlusi, O.; El-Sobky, M.; Giri, B.; Sarkar, T.R. Cancer-associated fibroblasts: The chief architect in the tumor microenvironment. Front. Cell Dev. Biol. 2023, 11, 1089068. [Google Scholar] [CrossRef] [PubMed]
- Hu-Lieskovan, Siwen; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF V600E melanoma. Science translational medicine 2015, 7.279, 279ra41–279ra41. [Google Scholar] [CrossRef] [PubMed]
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; De Giorgi, V.; Massi, D.; Fonsatti, E.; Staibano, S.; Nappi, O.; et al. BRAF/NRAS Mutation Frequencies Among Primary Tumors and Metastases in Patients With Melanoma. J. Clin. Oncol. 2012, 30, 2522–2529. [Google Scholar] [CrossRef]
- Edlundh-Rose, E.; Egyházi, S.; Omholt, K.; Månsson-Brahme, E.; Platz, A.; Hansson, J.; Lundeberg, J. NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma Res. 2006, 16, 471–478. [Google Scholar] [CrossRef] [PubMed]
- I Nikolaev, S.; Rimoldi, D.; Iseli, C.; Valsesia, A.; Robyr, D.; Gehrig, C.; Harshman, K.; Guipponi, M.; Bukach, O.; Zoete, V.; et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat. Genet. 2011, 44, 133–139. [Google Scholar] [CrossRef]
- Murugan, A.K.; Dong, J.; Xie, J.; Xing, M. MEK1 mutations, but not ERK2 mutations, occur in melanomas and colon carcinomas, but none in thyroid carcinomas. Cell Cycle 2009, 8, 2122–2124. [Google Scholar] [CrossRef] [PubMed]
- Cardarella, S.; Ogino, A.; Nishino, M.; Butaney, M.; Shen, J.; Lydon, C.; Yeap, B.Y.; Sholl, L.M.; Johnson, B.E.; Jänne, P.A. Clinical, Pathologic, and Biologic Features Associated with BRAF Mutations in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2013, 19, 4532–4540. [Google Scholar] [CrossRef]
- Xing, M. B. R. A. F. BRAF mutation in thyroid cancer. Endocrine-related cancer 2005, 12.2, 245–262. [Google Scholar] [CrossRef]
- Singer, G.; Oldt, R., III; Cohen, Y.; Wang, B.G.; Sidransky, D.; Kurman, R.J.; Shih, I.-M. Mutations in BRAF and KRAS Characterize the Development of Low-Grade Ovarian Serous Carcinoma. J. Natl. Cancer Inst. 2003, 95, 484–486. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.-L.; Shih, I.-M.; Mao, T.-L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent Mutations of Chromatin Remodeling Gene ARID1A in Ovarian Clear Cell Carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef]
- Tiacci, Enrico; et al. BRAF mutations in hairy-cell leukemia. New England Journal of Medicine 2011, 364.24, 2305–2315. [Google Scholar] [CrossRef]
- Ji, R.-R.; Xu, Z.-Z.; Gao, Y.-J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov. 2014, 13, 533–548. [Google Scholar] [CrossRef]
- Pezet, S.; McMahon, S.B. NEUROTROPHINS: Mediators and Modulators of Pain. Annu. Rev. Neurosci. 2006, 29, 507–538. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Oxford, G.S. Phosphoinositide-3-kinase and mitogen activated protein kinase signaling pathways mediate acute NGF sensitization of TRPV1. Mol. Cell. Neurosci. 2007, 34, 689–700. [Google Scholar] [CrossRef]
- Hudmon, A.; Choi, J.-S.; Tyrrell, L.; Black, J.A.; Rush, A.M.; Waxman, S.G.; Dib-Hajj, S.D. Phosphorylation of Sodium Channel Nav1.8 by p38 Mitogen-Activated Protein Kinase Increases Current Density in Dorsal Root Ganglion Neurons. J. Neurosci. 2008, 28, 3190–3201. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Kohno, T.; Zhuang, Z.-Y.; Brenner, G.J.; Wang, H.; Van Der Meer, C.; Befort, K.; Woolf, C.J.; Ji, R.-R. Ionotropic and Metabotropic Receptors, Protein Kinase A, Protein Kinase C, and Src Contribute to C-Fiber-Induced ERK Activation and cAMP Response Element-Binding Protein Phosphorylation in Dorsal Horn Neurons, Leading to Central Sensitization. J. Neurosci. 2004, 24, 8310–8321. [Google Scholar] [CrossRef]
- Wei, F.; Qiu, C.-S.; Kim, S.J.; Muglia, L.; Maas, J.W.; Pineda, V.V.; Xu, H.-M.; Chen, Z.-F.; Storm, D.R.; Muglia, L.J.; et al. Genetic Elimination of Behavioral Sensitization in Mice Lacking Calmodulin-Stimulated Adenylyl Cyclases. Neuron 2002, 36, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Befort, K.; Brenner, G.J.; Woolf, C.J. ERK MAP Kinase Activation in Superficial Spinal Cord Neurons Induces Prodynorphin and NK-1 Upregulation and Contributes to Persistent Inflammatory Pain Hypersensitivity. J. Neurosci. 2002, 22, 478–485. [Google Scholar] [CrossRef]
- Zhuang, Z.-Y.; Wen, Y.-R.; Zhang, D.-R.; Borsello, T.; Bonny, C.; Strichartz, G.R.; Decosterd, I.; Ji, R.-R. A Peptide c-Jun N-Terminal Kinase (JNK) Inhibitor Blocks Mechanical Allodynia after Spinal Nerve Ligation: Respective Roles of JNK Activation in Primary Sensory Neurons and Spinal Astrocytes for Neuropathic Pain Development and Maintenance. J. Neurosci. 2006, 26, 3551–3560. [Google Scholar] [CrossRef]
- Gao, Y.-J.; Ji, R.-R. Targeting Astrocyte Signaling for Chronic Pain. Neurotherapeutics 2010, 7, 482–493. [Google Scholar] [CrossRef]
- Mantyh, P.W. The neurobiology of skeletal pain. Eur. J. Neurosci. 2014, 39, 508–519. [Google Scholar] [CrossRef]
- Zheng, X.-Q.; Wu, Y.-H.; Huang, J.-F.; Wu, A.-M. Neurophysiological mechanisms of cancer-induced bone pain. J. Adv. Res. 2021, 35, 117–127. [Google Scholar] [CrossRef]
- Ji, R.R.; Woolf, C.J. Neuronal Plasticity and Signal Transduction in Nociceptive Neurons: Implications for the Initiation and Maintenance of Pathological Pain. Neurobiol. Dis. 2001, 8, 1–10. [Google Scholar] [CrossRef]
- Staff, N.P.; Grisold, A.; Grisold, W.; Windebank, A.J. Chemotherapy-induced peripheral neuropathy: A current review. Ann. Neurol. 2017, 81, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.-Y.; Xu, H.; Clapham, D.E.; Ji, R.-R. Phosphatidylinositol 3-Kinase Activates ERK in Primary Sensory Neurons and Mediates Inflammatory Heat Hyperalgesia through TRPV1 Sensitization. J. Neurosci. 2004, 24, 8300–8309. [Google Scholar] [CrossRef]
- Duan, C.; Zhu, Y.; Zhang, Z.; Wu, T.; Shen, M.; Xu, J.; Gao, W.; Pan, J.; Wei, L.; Su, H.; et al. Esketamine inhibits the c-Jun N-terminal kinase pathway in the spinal dorsal horn to relieve bone cancer pain in rats. Mol. Pain 2024, 20. [Google Scholar] [CrossRef]
- Falchook, G.S.; Lewis, K.D.; Infante, J.R.; Gordon, M.S.; Vogelzang, N.J.; DeMarini, D.J.; Sun, P.; Moy, C.; A Szabo, S.; Roadcap, L.T.; et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 782–789. [Google Scholar] [CrossRef]
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of Selumetinib in Neurofibromatosis Type 1–Related Plexiform Neurofibromas. New Engl. J. Med. 2016, 375, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
Figure 1.
The Canonical MAPK/ERK Signaling Pathway and Key Regulatory Nodes.
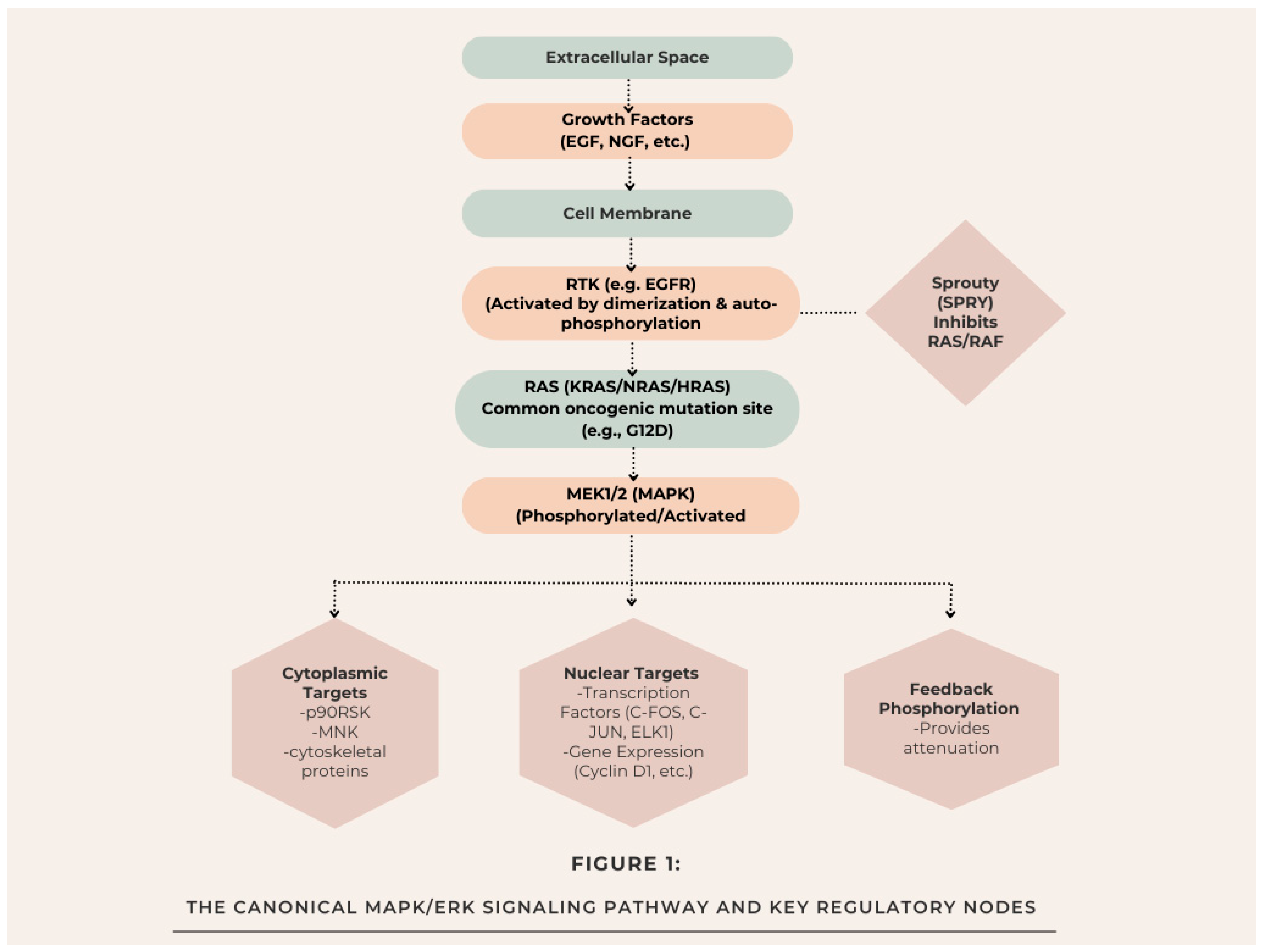
Figure 2.
Cancer and Pain MAPK Pathway Schematic.
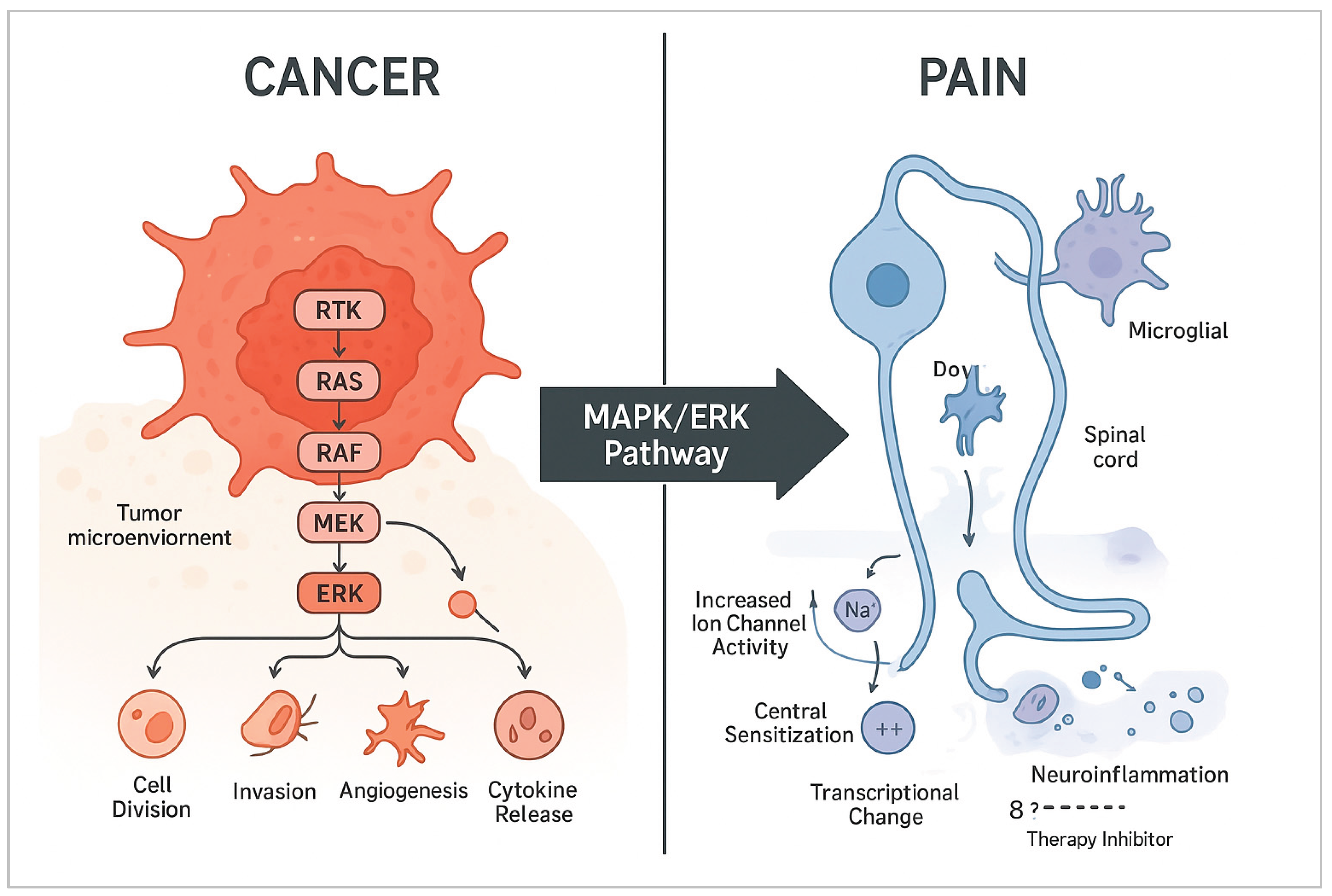
Table 2.
Clinically Approved and Investigational MAPK/ERK Pathway Inhibitors.
| Target | Drug Name (Examples) | Status (Key Indications) | Notable Clinical Context |
| BRAF (Monotherapy) | Vemurafenib, Dabrafenib, Encorafenib | FDA-Approved: *BRAF V600E/K* mutant Melanoma, NSCLC, Thyroid Cancer. | First-generation inhibitors: effective but prone to resistance. Encorafenib is often used in combination for CRC. |
| MEK1/2 (Monotherapy) | Trametinib, Cobimetinib, Binimetinib, Selumetinib | FDA-Approved: Trametinib/Cobi/Bini for BRAF mutant Melanoma (in combo). Selumetinib: Neurofibromatosis Type 1 (NF1). | Selumetinib shows activity in low-grade serous ovarian cancer and pediatric gliomas. |
| BRAF + MEK (Combination) | Dabrafenib + Trametinib, Vemurafenib + Cobimetinib, Encorafenib + Binimetinib | FDA-Approved: Standard of care for BRAF mutant Melanoma, NSCLC. Encorafenib+Binimetinib is also for CRC. | Combination improves efficacy, reduces cutaneous toxicities (e.g., squamous cell carcinoma) vs. BRAF monotherapy. |
| ERK1/2 (Direct) | Ulixertinib (BVD-523), MK-8353, LY3214996 | Phase I/II Trials (Various solid tumors, incl. BRAF/NRAS mutant). | Designed to overcome resistance to upstream BRAF/MEK inhibitors. Emerging safety and efficacy data. |
| RAF (Pan-RAF or Paradox Breaker) | LXH254, Tovorafenib (DAY101) | Clinical Trials. Tovorafenib in pediatric low-grade glioma. | Aim to inhibit all RAF isoforms or avoid paradoxical activation in RAS-mutant cells. |
| SHP2 (Upstream Node) | RMC-4630, TNO155 | Phase I/II Trials (e.g., with Osimertinib in NSCLC, with MEKi in KRAS mutants). | Targets node connecting RTKs to RAS; potential in KRAS-driven and resistant cancers. |
Table 3.
Pharmacological Modulators of the MAPK/ERK Pathway in Preclinical Pain Research.
| Compound / Drug Class | Target | Pain Model (Example) | Observed Analgesic Effect & Proposed Mechanism |
| U0126, PD0325901 | MEK1/2 | Neuropathic pain (SNL, CCI), Inflammatory pain (CFA), Cancer-induced bone pain. | Reversal of mechanical allodynia & thermal hyperalgesia. ↓ p-ERK in DRG/spinal cord; ↓ cytokine production in glia. |
| SL327 | MEK1/2 | Formalin test, Capsaicin-induced hyperalgesia. | Attenuation of phase 2 formalin response & capsaicin-evoked sensitization. Blocks central sensitization. |
| ASN007 (ERK1/2 inhibitor) | ERK1/2 | Inflammatory pain (CFA). | Dose-dependent reduction in pain hypersensitivity. More direct target than MEK inhibitors. |
| Schwann Cell-derived Exosomes | N/A (Modulate pathway) | Neuropathic pain (SNI). | Alleviate pain by delivering miRNAs that suppress RAS/MAPK signaling in neurons. |
| Peripheral Opioids (e.g., Morphine) | µ-opioid receptor (Gᵢ) | Various | Analgesia is partly via inhibition of cAMP/PKA, leading to reduced downstream ERK activation in neurons. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



