
Submitted:
05 December 2025
Posted:
09 December 2025
You are already at the latest version
Abstract
Bloodsucking dipterans are major vectors responsible for the transmission of arboviruses. Additionally, they also carry a lot of insect-specific viruses. High-throughput sequencing technologies has facilitated breakthrough in the study of viruses overall and viruses of haematophagus insects in particular. In this study we used high-throughput sequencing to describe the viromes of bloodsucking dipterans collected in Karelia, northwestern Russia. In addition to various species of Aedes mosquitoes (Diptera: Culicidae), several pool of biting midges (Ceratopogonidae: Culicoides), and blackflies (Simuliidae: Simulium), were analyzed. We managed to assemble and annotate 63 different distinct viruses with complete and partial genomes. The most common viral groups were Durnavirales, Picornavirales, Reovirales, and Mononegavirales, however various other groups of RNA viruses and even one DNA virus were detected. The majority of viruses (78%) were novel viruses. Among known viruses, no known human or animal pathogens were found. Some of the novel viruses can be considered by the International Committee on Taxonomy of Viruses for the demarcation into novel genera.
Keywords:
mosquitoes
; Aedes
; metagenomics
; virome
; Karelia
; Rhabdoviridae
; Phenuiviridae
Introduction
Arthropod-borne viruses (arboviruses) pose a significant threat to people worldwide. Mosquitoes (Diptera: Culicidae) are the major arthropod vectors being responsible for the transmission of over a third of all known arboviruses [1]. For instance, species of the genus Aedes are responsible for the transmission of Dengue [2], Zika [3], and Chikungunya viruses [4]. Members of the genus Culex are responsible for the transmission of West Nile and Japanese Encephalitis viruses [5,6]. The importance of mosquitoes has led to thorough virological surveillance around the world. High-throughput sequencing (HTS) technologies has facilitated breakthrough in the study of viruses overall [7], and virome of vector and non-vector mosquitoes is being actively studied [8,9,10]. The data obtained allowed us to significantly expand our knowledge on viruses associated with mosquitoes. Multiple thousands of viruses were discovered overall across several viral orders, resulted in newly created taxa.
Other bloodsucking dipterans are also being studied, albeit on a much smaller scale. A several studies on the biting midges (Ceratopogonidae) has been conducted, with Culicoides species collected in Australia [11], Brazilian Amazonia [12], Senegal [13], Kenya [14], Greece [15], China [16] and Mexico [17]. Up to several thousands of novel viruses were discovered in biting midges, some of which can be putatively be classified as novel genus by authors [16]. A few recent studies has been conducted on the blackflies (Simuliidae) [18,19]. In a largest study dedicated to Simulium species to date, 55 pools of 10 blackflies, collected in Cameroon, were studied. Authors report that viruses in the blackflies often form deep branches in the phylogenetic trees, suggestive for the discovery of new species and genera [19].
The Republic of Karelia (Karelia) is located in the northwestern Russia, and is a part of Eastern Fennoscandia. Several viruses, such as Sindbis, Inkoo, Tahyna, Khatanga, and Batai are endemic to Fennoscandia and are associated with mosquitoes Aedes communis, Ae. cinereus, Ae. excrucians, Ae. punctor, Ae. hexodontus, Coquillettidia richiardii, Culex pipiens, Cx. torrentium, and Culiseta morsitans [20]. It should be noted, that here we use the classification of Culicidae proposed by Wilkerson et al [21] , in which the genus Aedes includes Ochlerotatus as a subgenus. All these mosquito species are widespread in Karelia and bite human [22,23]. No endemic viruses associated with biting midges and blackflies are known in Fennoscandia, but many species of these dipterans actively attack humans. Moreover, in Karelia, Culicoides species (C. grisescens, C. impunctatus, and C. obsoletus) and Simulium species (S. equinum, S. murmanum, S. ornatum, S. pusillum, and S. truncatum) are numerous and widely distributed [24,25].
Karelia is located directly east of Finland and has a long border with this country. Virome of mosquitoes in Finland is fairly well described. During a large-scale HTS study, viromes of 91 pools comprising of Aedes cantans, Ae. caspius, Ae. communis, Ae. diantaeus, Ae. excrucians, Ae. hexodontus, Ae. intrudens, Ae. pullatus and Ae. punctor/punctodes were studied (Ochlerotatus genus name used in the article). In total, RNA dependent RNA polymerase (RdRp) sequences of 159 distinct viruses were assembled. Out of these, 147 were novel viruses [10].
Here we studied virome of several species of mosquitoes, biting midges, and blackflies, collected in Karelia. Overall, we were able to assemble and annotate 63 different viruses with complete and partial genomes. Several dozen of short sequences with homology to the viral proteins were detected. Majority of detected viruses represented novel viruses, while the rest were viruses previously detected in Finland and Sweden.
Materials and methods
1. Sampling Technique for Dipterans and Pool Composition
We collected bloodsucking dipterans in Loukhsky, Muezersky, and Kondopozhsky districts, as well as in Kostomukshsky Urban Okrug of Karelia from June to August 2023–2024 (Table 1). The previously reviewed techniques for sampling and preparation of dipterans were used [26,27].
Briefly, flying females were collected by a Mosquito Magnet trap (Pioneer design, Octenol as attractant); sampling was performed with 2–24 h exposure time, the trap was examined at two-hour intervals. Collected mosquito females were placed in a freezer at a temperature of −20 °C for 20 minutes. Immediately after, specimens were identified using taxonomic key of Becker et al. [28] and first were frozen at −190 °C and then transferred to -86 °C.
Biting midges and blackflies specimens were divided into equal parts. The first part was frozen at −86 °C and used for high-throughput sequencing, as described below. The second part was fixed in ethanol and used to prepare permanent or non-permanent microscope slides, which are required for correct taxonomic identification. Culicoides specimens were identified using keys of Glukhova [29,30] and Simulium specimens using key of Usova [25] or the same of Yankovsky [31]. The examined specimens (Euparal slides) are deposited in the Zoological Institute of the Russian Academy of Sciences (ZIN RAS, St. Petersburg).
Insects were pooled by species and location (Table 1) and homogenized in saline buffer using a Tissue Lyser 2 (Qiagen, Hilden, Germany) for 10 min at frequency 25 s−1.
2. High-Throughput Sequencing, and Virus Discovery Pipeline
RNA extraction, host rRNA depletion, and library preparation were performed as described earlier [32]. Obtained samples were sequenced (paired-end, 250-nt reads) on the HiSeq1500 (Illumina, San Diego, CA, USA). Obtained raw HTS reads were deposited in the SRA database (PRJNA1366109).
Adapter, low-quality bases (<Q30), 3 leading and trailing nucleotides, as well as short reads (≤35 nt) were trimmed with Trimmomatic v.0.39 [33]. Trimmed reads were de-novo assembled and filtered using two-staged approach using seg, blastn and blastp programs as described previously [32]. Additionally, obtained contigs were compared with high-abundance sequences from the same sequence run in order to control for possible contaminations. Final list of the virus-related hits was manually examined to verify proper assembly and the completeness of virus genomes. In some cases, SeqMan v.7.0.0 (DNAstar Inc., Madison, WI, USA) or UGENE v50.0 [34] were used to produce better virus assembly as described previously. Obtained sequences with complete coding and partial genomes were deposited in GenBank (PX636781-PX636886 and PX633639-PX633699)
3. Estimation of Virus Completeness and Virus Naming
Completeness was of the virus genome was determined manually by comparing ORF composition to closest relatives (according to blastp) and related viruses annotated by International Committee on Taxonomy of Viruses (ICTV). All genomes were divided into three categories: complete coding, partial genome, and genome fragment. Categories are defined as specified previously [32].
A novelty assessment was performed for viruses with a complete coding and partial genome. In our work, we considered virus to be novel if the identity of the ORF encoding the polymerase was below 90% according to blastp (compared to its closest relative). Novel viruses were named. The name of the virus consisted of the name of the region where insect was collected, or nearby geographical features; genus of the insect; and the virus systematic unit. For viruses unclassified to a genus level in our analysis, and in cases when the viral family consists of a single genus, the suffix “-like” was used. For non-novel viruses, name of the closest relative was used.
It should be noted, that final species’ names and taxonomic placement will be determined by the ICTV. Where possible, a virus placement according to ICTV criteria is discussed in the results section.
4. Data Analysis and Visualization
Total virus abundance in the sample was estimated using Bowtie 2 v.2.3.5.1 [35] by aligning reads on an index containing all the virus sequences found in the pool. The reported overall alignment rate was considered the percentage of viral reads in the sample. Individual virus abundance was estimated using coverage values provided by SPAdes v3.13.0 [36] after assembly. For viruses with reported coverage higher than 100, accurate abundance was estimated using Bowtie 2 v.2.3.5.1.
For the phylogenetic analysis, sequence of the ORF encoding the RdRp (VP1 for chaq-like viruses) was extracted from viral contig and translated. Dataset of related sequences was constructed by performing online blastp searches using translated sequence as a query with the nr database. If necessary, sequences from species recognized by the International Committee on Taxonomy of Viruses (ICTV) were added to the analysis.
Phylogenetic trees were constructed from obtained dataset using MAFFT v7.310 [37], TrimAL v1.4. rev 15 [38], IQ-TREE v.2.3.2 [39] and custom python scripts as described earlier [32]. To asses branch support, 1000 bootstrap replicates were performed.
To analyze invertebrate species, where viruses were previously detected, GenBank data base was mined using virus name as organism name. Information containing GenBank ID, virus name, isolation location and host species were extracted from GenBank entry. Genus “Ochlerotatus” was changed to “Aedes” for consistency in all host species. Obtained data was manually checked, and adjusted: “Aedes cf. punctor/punctodes” and “Aedes aff. punctor/punctodes” host species were merged into “Aedes punctor/punctodes”; “mosquito” was replaced with “Culicidae spp.”. Heatmap was visualized using matplotlib library.
Phylogenetic trees were visualized with FigTree v1.4.4. Virus contigs were annotated manually and visualized using the GenomeDrawing tool. Pie charts, heatmaps, and histograms were plotted in python3 using matplotlib library. All figure post-processing was done using GIMP program.
Results
1. High-Throughput Sequencing
The aim of the work was to describe the virome of bloodsucking dipterans collected in Karelia. Overall, 8 pools of dipterans were studies, including 5 pools of mosquitoes, two pools of blackflies and one pool of biting midges. Mosquitoes were pooled by species and sampling location. Overall, four mosquito species were studied: Aedes punctor, Ae. cinereus, Ae. communis, and Ae. dianteus. Blackflies and biting midges were pooled by location, with three species represented in each pool.
We obtained 8–14 million reads per pool (after filtering out low-quality reads), with total percentage of viral reads varied from 0.46% to 11.74%. We found evidence of the presence of at least 138 different viruses in the study (Figure 1, Supplementary Table 1). Number of viruses in pool varied from 12 to 26, with Simulium collected in the Leksozero region containing both the most number of viruses, and the highest number of viral reads (Figure 1).
Since virus richness in our samples was very high, we focused followed analysis on viruses with complete and partial genomes. 63 unique virus ‘species’ with complete and partial genome (according to the criteria stated in the Materials and Methods) were assembled. The majority of them were related to the order of Durnavirales. Other common virus groups were Picornavirales, Reovirales, and Mononegavirales (Figure 1B). For 13 virus ‘species’, several different genomes were assembled, indicating the presence of different variants in our dataset (Figure 2A). Ten virus ‘species’ were found in two pools simultaneously. In several cases, the presence of viruses in different species of insects has been observed.
Out of 63 unique virus ‘species’, 14 (22%) were not novel viruses (Figure 2C). All of non-novel viruses (except Hebron partiti-like virus) were discovered during large-scale study on the virome of the Aedes species conducted in Finland [10]. Hebron partiti-like virus was discovered in a study conducted on the virome of the mosquitoes in Sweden [40].
In our work, several viruses were detected in the mosquito species, where they had not previously been discovered. For example, Enontekio quenyavirus, Mekrijarvi iflavirus and Hameenlinna phasivirus were detected in Aedes diantaeus in our work, while Ilomantsi partiti-like virus 1 and Hanko iflavirus 1 were detected in Ae. cinereus. For Pedesore iflavirus, Utjoki negevirus 1, Hattula totivirus 3, Hattula partiti-like virus, Hebron partiti-like virus, Joensuu sobemovirus and Ilomantsi deltapartitivirus we discovered them only in mosquito species where they were previously detected.
Some of not novel viruses were very abundant. The percentage of Pedesore iflavirus, Enontekio quenyavirus, Mekrijarvi iflavirus, and Hameenlinna phasivirus reads were 1.72%, 1.21%, 0.93%, 0.66% (Figure 2B), respectively. This may indicate that those viruses are actively replicating; however, future research is needed to prove this.
2. Durnavirales
Viruses related to the order Durnaviruales were the most common in our study, with 17 unique viruses assembled. The majority of those viruses were partiti-like viruses. Classical members of the family Partitiviridae are bisegmented dsRNA viruses with genomes 3–4.8 kbp in length. First segment encodes RdRp, while the second one encodes the coat protein. Family contains five established genera: Alphapartitivirus, Betapartitivirus, Gammapartitivirus, Deltapartitivirus, and Cryspovirus [41]. Recently, a lot of novel partiti-like viruses were discovered using HTS [7].
While a lot of separate first and second segments were assembled, in many cases it was impossible to properly associate them in as a single bisegmented genome. Moreover, in multiple pools, amount of assembled second segment exceeded the amount of first. Thus, the majority of partiti-like viruses we discuss below will be presented only by first RdRp-encoding segment.
According to the phylogenetic analysis done using amino acid sequences of RdRp, novel partiti-like viruses belonged to five distinct clades (Figure 3). Gomselga Aedes partiti-like virus 2 formed a monophyletic clade with several viruses found in different species of mosquitoes, including Ilomantsi partiti-like virus 2.
Panayarvi Aedes partiti-like virus 2 was distantly related to several partiti-like viruses found in various insects. Gomselga Aedes partiti-like virus (GAPV) and Panayarvi Aedes partiti-like virus 1 (PAPV1) were found in both pools of Aedes cinereus. GAPV formed a monophyletic group with Hameenlinna partiti-like virus and Lestijarvi partiti-like virus, discovered in mosquitoes in Finland. Gomselga Culicoides partiti-like virus 1 and Leksozero Simulium partiti-like virus 2 were also related to this clade. PAPV formed monophyletic clade with Verdadero virus and XiangYun partiti-picobirna-like virus 5. Additionally, in the pool of Aedes cinereus we discovered a Gomselga Aedes alphapartiti-like virus. It was related to various viruses related to Alphapartitivirus genus (Supplementary Figure 1)
Additionally, we discovered three chaq-like viruses. Chaq virus was originally described as a small sequence whose presence was correlated with Galbut virus [42,43]. It was hypothesized that it may be a satellite virus. In both Simulium pools, we discovered Gomselga Simulium chaq-like virus. Phylogenetically, it was related to various chaq-like viruses discovered in different species of mosquitoes.
Panayarvi Aedes chaq-like virus was detected in Aedes cinereus pool (pool 14), and Zadnee Aedes chaq-like virus (ZACV) was detected in Aedes communis (pool 12). According phylogenetic analysis based on the VP1 protein, both of those viruses were highly divergent from currently known chaq-like viruses (Figure 4).
3. Picornavirales
All picornavirales-related viruses, discovered in this study, were ifla-like viruses. Family Iflaviridae includes non-enveloped, single-stranded, non-segmented positive-sense RNA viruses. They have genomes that are 9-11 kb in length and contain a single open reading frame [44]. Recently, hundreds of novel ifla-like viruses were discovered using HTS [7].
Apart from viruses that were discovered in other studies, we were able to assemble 6 novel ifla-like viruses. All of them followed typical ifla-like genome layout. Four of them were discovered in Simulium pool (pool 10), one in Culicoides pool, and one in Aedes diantaeus. Viruses were named Gomselga Simulium ifla-like virus 1 – 4 (GSIV1 – 4), Gomselga Culicoides ifla-like virus (GCIV), and Leksozero Aedes ifla-like virus (LAIV). Four of discovered viruses had a complete coding genome, while for the other two partial genome with various completions (Supplementary Figure 2).
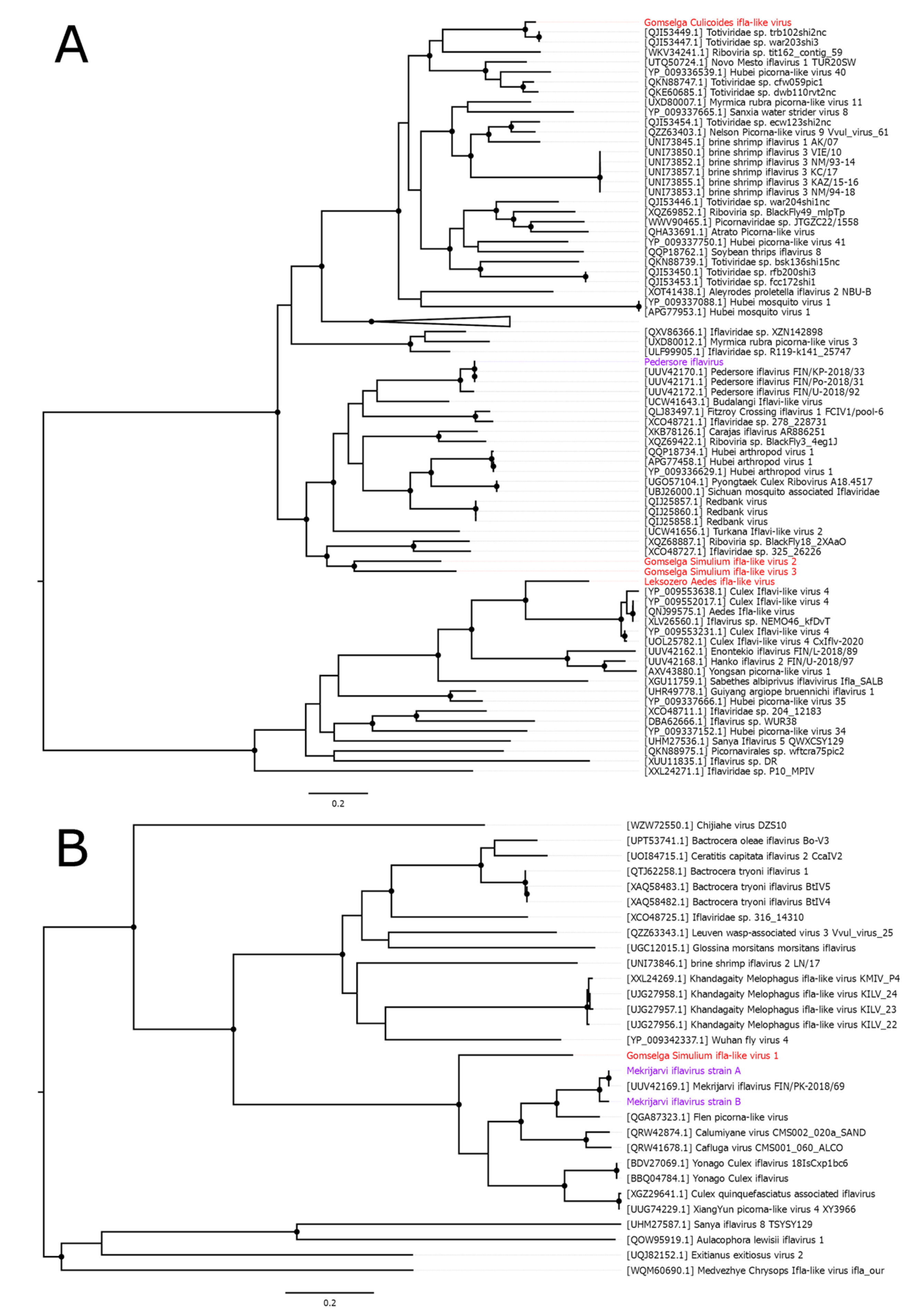
Discovered viruses belong to several distant groups of ifla-like viruses (Figure 4). GCIV formed a monophyletic clade with two viruses discovered in bird metagenome and mistakenly annotated as toti-like viruses. GSIV2 and GSIV3 grouped together and formed monophyletic groups with two viruses discovered in Myotis chinensis bats and Cameroonian blackflies [19]. LAIV grouped together with several variants of Culex Iflavi-like virus 4. GSIV1 was distantly related and formed a monophyletic group with several viruses found in other Diptera members, including Mekrijarvi iflavirus, for which we described two new variants.
Figure 5.
Phylogenetic relationships of novel ifla-like viruses discovered in the study. (A, B) Midpoint-rooted phylogenetic tree constructed using the amino acid sequences of the polyprotein. Nodes with ≥90% bootstrap support are marked, 1000 bootstrap replicates were performed. Some large branches of ifla-like viruses are collapsed for clarity. The scale bar represents the number of amino acid substitutions per site. Discovered viruses are shown in red. Novel variants of already known viruses are in purple.
Figure 5.
Phylogenetic relationships of novel ifla-like viruses discovered in the study. (A, B) Midpoint-rooted phylogenetic tree constructed using the amino acid sequences of the polyprotein. Nodes with ≥90% bootstrap support are marked, 1000 bootstrap replicates were performed. Some large branches of ifla-like viruses are collapsed for clarity. The scale bar represents the number of amino acid substitutions per site. Discovered viruses are shown in red. Novel variants of already known viruses are in purple.
4. Reovirales
Order Reovirales unifies dsRNA viruses with segmented genomes and currently include two large families: Sedoreoviridae and Spinareoviridae. [45,46] Here we found contigs related to Reovirales in every sample we processed. Samples from mosquitoes had contigs distantly related to the genus Dinovernavirus. This genus belongs to the Spinareoviridae family and currently includes a single species - Dinovernavirus aedis. Proposition to classify Aedes albopictus reovirus into the genus is currently under consideration. Member species contain only 9 dsRNA segments [45,46].
Overall, we were able to assemble 7 viruses that belong, according to our criteria, to 3 different ‘species’: Gomselga Aedes dinoverna-like virus 1 (GADV1), Gomselga Aedes dinoverna-like virus 2 (GADV2), and Panayarvi Aedes dinoverna-like virus (PADV). In the 4 cases, we were able to assemble all nine segments for each virus (Figure 6B). Among them, segment 8 was the most distant; even in cases where it was successfully identified it is possible that it is a misidentification.
Both variants of GADV2 were found in different Aedes cinereus pools (pools 9 and 14). However, two other dinoverna-like viruses were detected in multiple species of mosquitoes (Figure 2A). For GADV1, three distinct genetic variants were detected – two in the pool of Ae. punctor (pool 8), and another one in the pool of Ae. communis (pool 12). PADV was found in pools of Ae. cinereus and Ae. diantaeus (Figure 2A).
Phylogenic analysis based on RdRp-encoding ORF shows that all viruses discovered here are grouped together with genus Dinovernavirus and related viruses (Figure 6A). At the same time, they are quite distant from previously discovered viruses. GADV1 and GADV2 were very abundant in some pools. The percentage of GADV1 reads reached 1.38% in pool 8, while the percentage of GADV2 reads reached 1.23% in pool 14. This may indicate that those viruses are actively replicating; however, future research is needed to prove this. PADV was also one of the most abundant viruses in the study, reaching 0.52% of total reads in pool 14.
In Simulium blackflies we also were able to detect several contigs with ORFs related to Reovirales. Further analysis allowed us to recover two segments (RdRp-encoding segment 1 and partial segment 5) in pool 10, and three segments (partial segment 1, segments 3 and 5) in pool 14 (Figure 7B). We concluded that each set of contigs belong to a single virus and named this viruses Gomselga Simulium reo-like virus and Leksozero Simulium reo-like virus. Those viruses grouped together and formed a monophyletic clade with Shenzhen reo-like virus 2 and Nephila clavipes virus 6. This group was relatively distantly related to both genus Phytoreovirus and genus Orbivirus (Figure 7A).
In Culicoides we discovered multiple contigs, all related to the Cimodo virus which is an unclassified virus isolated from mosquitoes collected in a rainforest in Côte d’Ivoire in 2014. The genome of the Cimodo virus is divided into 12 segments. It is proposed to form a novel genus within the subfamily Spinareovirinae [47].
We were able to assemble and detect 7 contigs (see Figure 7D) with some level of homology to Cimodo virus segments 1-6 and 8. We concluded that all this contigs belong to a single virus and named it Gomselga Culicoides reo-like virus (GCRV). Phylogenetic analysis based on the RdRp-encoding ORF shown, that GCRV is quite distantly related to the Cimodo and cimodo-like viruses (Figure 7C). Such phylogenetic divergence is likely to be a reason we were able to identify only 7 segments. GCRV is likely have more segments, but they are far too divergent from known viruses to detect with blastp analysis alone.
5. Mononegavirales
The order Mononegavirales unifies viruses with negative-sense, non-segmented, linear RNA genomes. At the time of writing, 11 families are officially recognized by ICTV within Mononegavirales [48]. In the current work, all assembled complete coding genomes belonged to the family Rhabdoviridae, as well as 1 partial genome that was related to the family Xinmoviridae. All of the discovered viruses can be considered novel according to our criteria.
The family Xinmoviridae mostly includes viruses discovered in various species of insects using HTS, and contains multiple genera that include viruses found in different species of mosquitoes [49]. In the pool of Aedes communis, we discovered a contig containing several proteins with homology to the xinmovirid protein sequences. The contig was 13091 nt in length and encoded 5 ORFs (Figure 8B), including typical for xinmovirid L (RdRp), N, and G proteins.
According to the blastp analysis of L protein, it was closest to the Hanko anphevirus with 77% identity. The phylogenetic analysis, which was performed using the amino acid sequences of the L protein, shows that our contig forms monophyletic group with the genus Culivirus (Figure 8A).
We judged this contig to represent a genome of the novel virus, and we named it the Zadnee Aedes Culivirus (ZAC). For the ORF encoding N protein, we were unable to locate the start codon, and thus we consider this genome only partial. However, alignment with other related N proteins shows, that missing starting codon may be the result of the error during sequencing or assembly.
All other mononegaviruses in our study are firmly grouped with the family Rhabdoviridae. Contigs related to rhabdoviruses were discovered in both Simulium pools, as well as pools of Aedes communis and Ae. diantaeus. They all showed typical genome organization for rhabdoviruses (Figure 8D, F). According to the phylogenetic analysis, the contig discovered in the Ae. communis pool grouped with members of the genus Merhavirus, while the contig from Ae. diantaeus grouped with members of the genus Stangrhavirus. Those contigs were named Zadnee Aedes Merhavirus (ZAM) and Leksozero Aedes Stangrhavirus (LAS), respectively. After classification, small U1 protein (located between G and L ORFs) typical for genus Stangrhavirus was annotated in the LAS genome.
In two Simulium pools, 5 contigs with homology to proteins of the family Rhabdoviridae were found. Two of them, both discovered in the second Simulium pool (collected from Leksozero), showed high homology with viruses from genus Almendravirus. However one contig (Figure 9B), had shortened L ORF and Simulium rRNA sequence. Thus, this contig is either inserted in the host genome, or a product of an assembly error. The second contig contained complete ORFs typical for the genus Almendravirus, including small U1 ORF between L and G genes. We judged this contig to represent a genome of the novel virus, and we named it the Leksozero Simulium Almendravirus (LSA). Phylogenetically, it grouped within genus Almendravirus, and formed a monophyletic group with Balsa virus, Xiangshan rhabdo-like virus 1, and unspecified almendravirus detected in the blackflies (Figure 9A).
Other 3 contigs had also had typical genome organization for rhabdoviruses, and had about 38-39% identity to the Orius laevigatus rhabdovirus 3. However, L proteins from contigs had 94% identity, and therefore, according to our criteria, they represented a single novel virus. We named both contigs Gomselga Simulium Rhabdo-like virus (GSRV). The third contig was named Leksozero Simulium Rhabdo-like virus 1 (LSRV). Both variants of GSRV, and LSRV, grouped together on the phylogenetic tree.
All discovered here rhabdo-like viruses has coding-complete genome and can be considered by the ICTV for classification. Genera Merhavirus, Stangrhavirus,and Almendravirus have six very similar species demarcation criteria: 10% amino acid sequence divergence in the protein N (5% for Almendravirus), 10% amino acid sequence divergence in the protein L, 15% amino acid sequence divergence in the protein G, significant differences in numbers and locations of ORFs, the possibility to distinguish viruses in the neutralization tests and differences in hosts [50].
We evaluated if our viruses are satisfying ICTV species demarcation criteria, except the neutralization tests (Supplementary Table 2). According to blastp analysis, ZAM, LAS, and LSA are sufficiently divergent from the closest blastp hit in L, G, and N to be considered a novel species. Moreover, LAS and LSA were found in novel hosts, compared to established viral species of the genera. However, there are some yet unclassified viruses within genus Almendravirus that were discovered in Simuliidae species [19]. ZAM is discovered in the Aedes communis, the same species where Hattula rhabdovirus (Merhavirus hattula) were discovered. Overall, we believe that the demarcation of a new species for ZAM, LAS, and LSA can be considered.
Our phylogenetic analysis shows, that GSRV and LSRV form a well-supported monophyletic clade outside of established Rhabdoviridae genera. The closest genus to it is Betathriprhavirus, which contains viruses that infect trips. According to genus demarcation criteria within family Rhabdoviridae, novel genus must form a monophyletic clade in well-supported trees using full-length L sequences, as well as significant differences in genome sequence and architecture, antigenicity and ecological properties [50]. GSRV and LSRV differs in genome structure from Betathriprhavirus, because they don’t encode additional ORF in between L and G genes. They are also found in Simulium blackflies, not in trips, making them ecologically distinct. Thus, after additional analysis, the demarcation of the genus for the GSRV/LSRV clade can be considered.
6. Other Virus Groups
In addition to the above-mentioned virus groups, we have discovered multiple RNA viruses belonging to Negeviruses, Bunyaviricetes, Sobelivirales, Ghabrivirales, Wolframvirales, Nodamuvirales. We have also assembled a single viral genome belonging to quenyaviruses, from the family Flaviviridae, and the orders Nidovirales and Martellivirales. Among the discovered viruses several are of particular interest.
Several negev-like viruses were assembled, with four of them being novel viruses. Three of them were found in a single Simulium sp. pool (pool 10), and followed general ORF layout of the negev-like viruses. However, the ORF layout in contig found in Aedes cinereus pool was not typical. First, it was a partial genome, with gaps estimated to be on both ends of the sequence (Supplementary Figure S3). Second, its RdRp-encoding ORF was divided in two by UGA codon. It is not common for negeviruses to express ORFs via stop codon read-through, thus it is possible that this contig represents not a virus, but a host genetic element.
We assembled a complete coding genome of a noda-like virus in the Simulium sp. pool. This virus, named Leksozero Simulium noda-like virus (LSNV), was distantly related (58% identity in RdRp-encoding ORF according to the blastp) to the Sanxia water strider virus 17 and had a similar ORF layout (Supplementary Figure 3) with it. Curiously, it is the most abundant virus in our study. The percentage of LSNV reads was 9.34% in the pool. Such high abundance indicates that this virus is likely to be actively replicating in Simulium blackflies.
In the Simulium sp. pool (pool 15) we assembled genomes of two novel bunya-like viruses. The first virus, named Leksozero Simulium phenui-like virus (LSPV), was distantly related (26% identity according to the blastp of the L protein) to the Catch-me-cave virus (Uukuvirus uukuniemiense). We were able to annotate all three segments of the LSPV. The percentage of LSPV reads was 0.82% in the pool – one of the highest in the study. This may indicate that LSPV is actively replicating in Simulium blackflies; however, future research is needed to prove this.
It should be noted, that the assembly quality for this particular virus was lower than average across this study. First, the second segment was originally assembled in a circular form, and manual assembly and re-annotation were performed to obtain the final sequence. Second, contig that was annotated as the third segment of LSPV was very distant from closest relative (33.6% identity with 10-4 E-value in N protein). However, it exhibited comparable average coverage compared to the segments 1 and 2. Based on this additional evidence, we concluded that this contig is segment 3 of the LSPV. If our annotation of the segment 3 is correct, then both putative NSs and putative N proteins there are coded in the same direction, with putative NSs ORF preceding N ORF (Figure 10).
The second novel bunya-like virus, discovered in the Simulium sp. pool (pool 15), was distantly related to the Buffalo Bayou virus (35% identity according to the blastp of the L protein). This virus was named Leksozero Simulium bunya-like virus. Interestingly, while only a single conting RdRp-encoding contig was discovered, we assembled two close contigs, both encoding G protein, as well as two variants of the segment 3 (Figure 10). The first one was 746 nt in length and contained a single N protein ORF. The second variant encoded an additional ORF in the reverse orientation. Unfortunately, without additional data, it is impossible to determine which segment 3 variant belong to the genomic RNA.
While most discovered here viruses were RNA viruses, we assembled a single complete coding genome of a DNA virus – Gomselga Aedes draupnir-like virus. It is likely belong to the family Draupnirviridae with closest relative (according to blastp) being Mosquito circovirus found in Aedes vexans in Germany.
Discussion
In this work we studied viromes of bloodsucking dipterans collected in Karelia. Several widespread species of Aedes mosquitoes were studied, due to their importance as vectors for human and animal pathogens. Additionally, we analyzed viromes of blackflies (Simulium) and biting midges (Culicoides). Number of viruses in pool varied from 12 to 26, with 63 unique viruses with complete or partial genome assembled. Among them, no known human or animal pathogens were found.
Out of 63 unique viruses, 22% were not novel and were mostly related to viruses discovered in mosquitoes in Finland [10]. The percentage is even higher (41%), if we consider only viruses detected in mosquitoes, because all partial and complete coding viruses found in blackflies and biting midges were novel viruses. It was reported, that there is no overlap in viruses between large-scale HTS virome studies done in Finland and in Sweden [10,51]. However, there is a big overlap in mosquito viruses found in Finland and Karelia. This can be explained by the fact that insect collection points in Finland are much closer geographically to the mosquito collection points in our work than to the mosquito collection points in Sweden. Nevertheless, we were able to discover multiple novel viruses even in mosquito species already studied in Finland (Aedes communis and Ae. diantaeus).
In our work, several not novel viruses we discovered were detected in mosquitoes where they had not previously been found. Additionally, novel dinoverna-like GADV1 and PADV were discovered in two different species of mosquitoes (Figure 2A). Mosquito specific RNA viruses are often considered vertically transmitted [52]. Discovery of the virus in several mosquito species may occur via intermediate vertebrate host, making them arboviruses. However, other routes of horizontal transmission (not involving vertebrate hosts), were also described [52], in particular for cypoviruses (Reoviridae) [53,54]. Overall, viruses discovered in multiple species of mosquitoes simultaneously can be a suitable object for future research on possible routes of horizontal transmission.
The most prevalent viruses in our research were those belonging to the order Durnavirales. In addition to multiple partiti-like viruses, 4 chaq-like viruses were identified. Chaq-like viruses are hypothesized to be satellites of partitiviruses [42,43,55]. Another hypothesis is that they are optional partitivirus segments not strictly required for replication [55]. In our study, all chaq-like viruses were detected in pools with other partiti-like viruses, confirming previous studies.
Some novel viruses we found here are of particular interest. GSRV and LSRV form a well-supported monophyletic clade outside of established Rhabdoviridae genera, have a different ORF layout compared to closest genus, and were discovered in Simulium blackflies, making them ecologically distinct. Combined with a determined complete coding sequence for both viruses, it makes those viruses potential candidates for demarcation of new genus in the family Rhabdoviridae.
LSPV has novel ORF layout in the 3rd segment, compared to the other established viruses in the family Phenuiviridae. According to the blastp analysis, it is very distantly related to known phenuiviruses. While we believe that we assembled a complete coding genome, additional confirmation, involving sequencing of the genome termini, is probably necessary before classification. In both cases, final decision on virus on nomenclature and classification of the abovementioned cases can only be made by the ICTV.
Overall, our work significantly expanded known biodiversity of viruses. Among other, we identified several variants of three dinoverna-like viruses, several new potential species of family Rhabdoviridae, as well as viruses that may even represent new genera in families Rhabdoviridae and Phenuiviridae. Given the relatively small number of pools we studied, it shows that despite extensive research, haemotophagous insects remain a suitable target for the search of novel viruses.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
The Russian Science Foundation and Karelian Venture Capital Fund, grant no. 23–14-20020, https://rscf.ru/project/23-14-20020 (fieldwork in Karelia and HTS); Chumakov FSC R&D IBP RAS (Institute of Poliomyelitis) fundamental research assignment № FNZG-2024-0008 (data analysis).
References
- De Almeida, P.P.; Aguiar, E.R.G.R.; Armache, J.N.; Olmo, R.P.; Marques, T. The Virome of Vector Mosquitoes. Curr Opin Virol 2021, 49, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Myers, M.F.; et al. The Global Distribution and Burden of Dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Wikan, N.; Smith, D.R. Zika Virus: History of a Newly Emerging Arbovirus. Lancet Infect Dis 2016, 16, e119–e126. [Google Scholar] [CrossRef] [PubMed]
- Martelossi-Cebinelli, G.; Carneiro, J.A.; Yaekashi, K.M.; Bertozzi, M.M.; Bianchini, B.H.S.; Rasquel-Oliveira, F.S.; Zanluca, C.; Duarte dos Santos, C.N.; Arredondo, R.; Blackburn, T.A.; et al. A Review of the Biology of Chikungunya Virus Highlighting the Development of Current Novel Therapeutic and Prevention Approaches. Pathogens 2025, 14, 1–55. [Google Scholar] [CrossRef]
- Brault, A.C. Changing Patterns of West Nile Virus Transmission: Altered Vector Competence and Host Susceptibility. Vet Res 2009, 40, 43. [Google Scholar] [CrossRef]
- Gubler, D.J. Emerging Vector-Borne Flavivirus Diseases: Are Vaccines the Solution? Expert Rev Vaccines 2011, 10, 563–565. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.; Tian, J.; Chen, L.; Chen, X.; Li, C.; Qin, X.; Li, J.; Cao, J.; Eden, J.; et al. Redefining the Invertebrate RNA Virosphere. Nature 2016, 1–12. [Google Scholar] [CrossRef]
- Marseille, R.; Nebbak, A.; Monteil-bouchard, S.; Berenger, J.; Almeras, L.; Parola, P.; Desnues, C. Virome Diversity among Mosquito Populations in a Sub-Urban Region of Marseille, France. Viruses 2021, 13, 1–20. [Google Scholar] [CrossRef]
- Atoni, E.; Wang, Y.; Karungu, S.; Waruhiu, C.; Zohaib, A.; Obanda, V.; Agwanda, B.; Mutua, M.; Xia, H.; Yuan, Z. Metagenomic Virome Analysis of Culex Mosquitoes from Kenya and China. Viruses 2018, 10, 1–15. [Google Scholar] [CrossRef]
- Nguyen, P.T.T.; Culverwell, C.L.; Suvanto, M.T.; Korhonen, E.M.; Uusitalo, R.; Vapalahti, O.; Smura, T.; Huhtamo, E. Characterisation of the RNA Virome of Nine Ochlerotatus Species in Finland. Viruses 2022, 14, 1–64. [Google Scholar] [CrossRef]
- Sharpe, S.R.; Madhav, M.; Klein, M.J.; Blasdell, K.R.; Paradkar, P.N.; Lynch, S.E.; Eagles, D.; López-Denman, A.J.; Ahmed, K.A. Characterisation of the Virome of Culicoides Brevitarsis Kieffer (Diptera : Ceratopogonidae), a Vector of Bluetongue Virus in Australia. 2025, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Sivla, S.; Aragao, C.; Gorayeb, I.; Cruz, A.; Dias, D.; Nascimento, B.; Chiang, J.; Casseb, L.; Neto, J.; et al. Investigation of RNA Viruses in Culicoides Latreille, 1809 ( Diptera : Ceratopogonidae ) in a Mining Complex in the Southeastern Region of the Brazilian Amazon. Viruses 2024, 16. [Google Scholar] [CrossRef] [PubMed]
- Temmam, S.; Monteil-Bouchard, S.; Robert, C.; Baudoin, J.-P.; Sambou, M.; Aubadie-ladrix, M.; Labas, N.; Raoult, D.; Mediannikov, O.; Desnues, C. Characterization of Viral Communities of Biting Midges and Identification of Novel Thogotovirus Species and Rhabdovirus Genus. Viruses 2016, 8, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Langat, S.K.; Eyase, F.; Bulimo, W.; Lutomiah, J.; Oyola, S.O.; Imbuga, M.; Sang, R. Profiling of RNA Viruses in Biting Midges (Ceratopogonidae) and Related Diptera from Kenya Using Metagenomics and Metabarcoding Analysis. mSphere 2021, 6, e00551-21. [Google Scholar] [CrossRef]
- Konstantinidis, K.; Bampali, M.; Williams, M.D.C.; Dovrolis, N.; Gatzidou, E.; Papazilakis, P.; Nearchou, A.; Veletza, S.; Karakasiliotis, I. Dissecting the Species-Specific Virome in Culicoides of Thrace. Front Microbiol 2022, 13, 1–16. [Google Scholar] [CrossRef]
- Yang, L.; Wu, W.; Cai, S.; Wang, J.; Kuang, G.; Yang, W.; Wang, J.; Han, X.; Pan, H.; Shi, M.; et al. Transcriptomic Investigation of the Virus Spectrum Carried by Midges in Border Areas of Yunnan Province. Viruses 2024, 16, 1–14. [Google Scholar] [CrossRef]
- Laredo-Tiscareño, S.V.; Garza-Hernandez, J.A.; Tangudu, C.S.; Dankaona, W.; Rodríguez-Alarcón, C.A.; Adame-Gallegos, J.R.; De Luna Santillana, E.J.; Huerta, H.; Gonzalez- Peña, R.; Rivera-Martínez, A.; et al. Discovery of Novel Viruses in Culicoides Biting Midges in Chihuahua, Mexico. Viruses 2024, 16, 1–18. [Google Scholar] [CrossRef]
- Kobayash, D.; Murota, K.; Faizah, A.N.; Amoa-Bosompem, M.; Higa, Y.; Hayashi, T.; Tsuda, Y.; Sawabe, K.; Isawa, H. RNA Virome Analysis of Hematophagous Chironomoidea Ies (Diptera : Ceratopogonidae and Simuliidae) Collected in Tokyo, Japan. Med. Entomol. Zool Vol. 2020(71), 225–243. [CrossRef]
- De Coninck, L.; Hadermann, A.; Colebunders, R.; Njamnshi, K.G.; Njamnshi, A.K.; Mokili, J.L.; Fodjo, J.; Matthijnssens, J. Cameroonian Blackflies (Diptera : Simuliidae) Harbour a Plethora of RNA Viruses. Virus Evol 2025, 11, 1–10. [Google Scholar] [CrossRef]
- Wilkman, L.; Ahlm, C.; Evander, M.; Lwande, O.W. Mosquito-Borne Viruses Causing Human Disease in Fennoscandia—Past, Current, and Future Perspectives. Front Med (Lausanne) 2023, 10. [Google Scholar] [CrossRef]
- Wilkerson, RC; Linton, YM; Strickman, DA. Mosquitoes of the World.; Hopkins University Press: Baltimore, 2021; Vol. 1,2. [Google Scholar]
- Khalin, A. V.; Aibulatov, S. V.; Filonenko, I. V. Mosquito Distribution in Northwestern Russia: Species of the Genera Anopheles Meigen, Coquillettidia Dyar, Culex L., and Culiseta Felt (Diptera, Culicidae). Entomol Rev 2021, 101, 308–330. [Google Scholar] [CrossRef]
- Khalin, A. V.; Aibulatov, S. V.; Filonenko, I. V. Mosquito Distribution in Northwestern Russia: Species of the Genus Aedes Meigen (Diptera, Culicidae). Entomol Rev 2021, 101, 1060–1095. [Google Scholar] [CrossRef]
- Glukhova, V. M. The Biting Midges of Karelia. In Fauna of Karelian lakes. Invertebrates; Academy of Sciences USSR Publishing House: Moscow, Leningrad., 1965; pp. 278–283. [Google Scholar]
- Usova, Z.V. Black Flies of Karelia and the Murmansk Region; USSR Academy of Sciences Publishing House: Moscow, Leningrad, 1961. [Google Scholar]
- Khalin, A.V.; Aibulatov, S.V. Preparation Techniques for the Mosquitoes and the Blackflies (Diptera: Culicidae: Simuliidae). Proceedings of the Zoological Institute RAS 2024, 328, 139–164. [Google Scholar] [CrossRef]
- Khalin, A. V.; Aibulatov, S. V.; Przhiboro, A.A. Sampling Techniques for Bloodsucking Dipterans (Diptera: Culicidae, Simuliidae, Ceratopogonidae, Tabanidae). Entomol Rev 2021, 101, 1219–1243. [Google Scholar] [CrossRef]
- Becker, N.; Petrić, D.; Zgomba, M.; Boase, C.; Madon, M.B.; Dahl, C.; Kaiser, A. Mosquitoes: Identification, Ecology and Control; Springer International Publishing: Cham, 2020; ISBN 978-3-030-11622-4. [Google Scholar]
- Glukhova, V. M. Bloodsucking Midges of the Genera Culicoides and Forcipomyia (Ceratopogonidae). ; 5a ed.; 1989; Vol. 3.
- Glukhova, V. M. Culicoides (Diptera, Ceratopogonidae) of Russia and Adjacent Lands. Journal of Dipterological Research 2005, 16, 3–75. [Google Scholar]
- Yankovsky, A.V. A Key for the Identification of Blackflies (Diptera: Simuliidae) of Russia and Adjacent Countries (Former USSR). Handbooks for the Identification of the Fauna of Russia Published by Zoological Institute, Russian Academy of Sciences, 170; ZIN: Sankt-Petersburg, 2002. [Google Scholar]
- Litov, A.G.; Semenyuk, I.I.; Belova, O.A.; Polienko, A.E.; Van Thinh, N.; Karganova, G.G.; Tiunov, A. V Extensive Diversity of Viruses in Millipedes Collected in the Dong Nai Biosphere Reserve (Vietnam). Viruses 2024, 16, 1–25. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Team., the U. Unipro UGENE: A Unified Bioinformatics Toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S. Fast Gapped-Read Alignment with Bowtie 2. Nat Methods 2013, 9, 357–359. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. Journal of Computational Biology 2012, 19, 455–477. [Google Scholar] [CrossRef]
- 3Kazutaka Katoh; Daron M. Standley MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol Biol Evol 2013, 30, 772–780. [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. TrimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol Biol Evol 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-baez, A.S.; Holmes, E.C.; Charon, J.; Pettersson, J.H.; Hesson, J.C. Meta-Transcriptomics Reveals Potential Virus Transfer between Aedes Communis Mosquitoes and Their Parasitic Water Mites. Virus Evol 2022, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vainio, E.J.; Chiba, S.; Ghabrial, S.A.; Maiss, E.; Roossinck, M.; Sabanadzovic, S.; Suzuki, N.; Xie, J.; Nibert, M.; Consortium, I.R. ICTV Virus Taxonomy Profile: Partitiviridae. Journal of General Virology 2017, 7, 17–18. [Google Scholar] [CrossRef]
- Webster, C.L.; Waldron, F.M.; Robertson, S.; Crowson, D.; Ferrari, G.; Quintana, J.F.; Brouqui, J.M.; Bayne, E.H.; Longdon, B.; Buck, A.H.; et al. The Discovery, Distribution, and Evolution of Viruses Associated with Drosophila Melanogaster. PLoS Biol 2015, 13, 1–33. [Google Scholar] [CrossRef]
- Shi, M.; White, V.L.; Schlub, T.; Eden, J.; Hoffmann, A.A.; Holmes, E.C. No Detectable Effect of Wolbachia w Mel on the Prevalence and Abundance of the RNA Virome of Drosophila Melanogaster. Proc. R. Soc. B 2018, 285. [Google Scholar] [CrossRef]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Gu, D.M.A.; Hashimoto, Y.; Herrero, S.; De Miranda, J.R.; Ryabov, E. ICTV Virus Taxonomy Profile : Iflaviridae. Journal of General Virology 2017, 527–528. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Attoui, H.; Bányai, K.; Brussaard, C.P.D.; Danthi, P.; del Vas, M.; Dermody, T.S.; Duncan, R.; Fāng, Q.; Johne, R.; et al. ICTV Virus Taxonomy Profile: Spinareoviridae 2022. Journal of General Virology 2022, 103. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Attoui, H.; Bányai, K.; Brussaard, C.P.D.; Danthi, P.; Del Vas, M.; Dermody, T.S.; Duncan, R.; Fāng, Q.; Johne, R.; et al. ICTV Virus Taxonomy Profile: Sedoreoviridae 2022. Journal of General Virology 2022, 103. [Google Scholar] [CrossRef]
- Hermanns, K.; Zirkel, F.; Kurth, A.; Drosten, C.; Junglen, S. Cimodo Virus Belongs to a Novel Lineage of Reoviruses Isolated from African Mosquitoes. Journal of General Virology 2014, 95. [Google Scholar] [CrossRef]
- Amarasinghe, G.K.; Ayllón, M.A.; Bào, Y.; Basler, C.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the Order Mononegavirales: Update 2019. Arch Virol 2019, 164, 1967–1980. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, S.R.; Paraskevopoulou, S. ICTV Virus Taxonomy Profile: Xinmoviridae 2023. Journal of General Virology 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.; Freitas-astúa, J.; Walker, P.J.; Astúa, J.F.-; Bejerman, N.; Blasdell, K.R.; Breyta, R.; Dietzgen, R.G. ICTV Virus Taxonomy Profile : Rhabdoviridae 2022. Journal of General Virology 2022, 103, 0–2. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, P.; Hayer, J.; Lundén, H.; Hesson, J.C.; Blomström, A.-L. Viromics Reveal a Number of Novel RNA Viruses in Swedish Mosquitoes. Viruses 2019, 11, 1–18. [Google Scholar] [CrossRef]
- Agboli, E.; Leggewie, M.; Altinli, M.; Schnettler, E. Mosquito-Specific Viruses—Transmission and Interaction. Viruses 2019, 11, 1–26. [Google Scholar] [CrossRef]
- Green, T.B.; Shapiro, A.; White, S.; Rao, S.; Mertens, P.P.C.; Carner, G.; Becnel, J.J. Molecular and Biological Characterization of a Cypovirus from the Mosquito Culex Restuans. J Invertebr Pathol 2011, 91, 27–34. [Google Scholar] [CrossRef]
- Shapiro, A.; Green, T.; Rao, S.; White, S.; Carner, G.; Mertens, P.P.C.; Becnel, J.J. Morphological and Molecular Characterization of a Cypovirus (Reoviridae) from the Mosquito Uranotaenia Sapphirina (Diptera : Culicidae). J Virol 2005, 79, 9430–9438. [Google Scholar] [CrossRef]
- Cross, S.T.; Maertens, B.L.; Dunham, T.J.; Rodgers, C.P.; Brehm, A.L.; Miller, M.R.; Williams, A.M.; Foy, B.D.; Stenglein, D. Partitiviruses Infecting Drosophila Melanogaster and Aedes Aegypti Exhibit Efficient Biparental Vertical Transmission. J Virol 2020, 94, 1–17. [Google Scholar] [CrossRef]
Figure 1.
(A) Total number of viruses (top) and abundance of virus-containing reads (bottom) in each studied pool. Distinct virus groups are colored. Viral genome fragments are marked by crosses. (B) Pie-chart of unique virus ‘species’ with complete and partial genomes assembled in the study. Distinct virus groups are colored. ‘Species’ are defined according to criteria stated in the Materials and Methods.
Figure 1.
(A) Total number of viruses (top) and abundance of virus-containing reads (bottom) in each studied pool. Distinct virus groups are colored. Viral genome fragments are marked by crosses. (B) Pie-chart of unique virus ‘species’ with complete and partial genomes assembled in the study. Distinct virus groups are colored. ‘Species’ are defined according to criteria stated in the Materials and Methods.
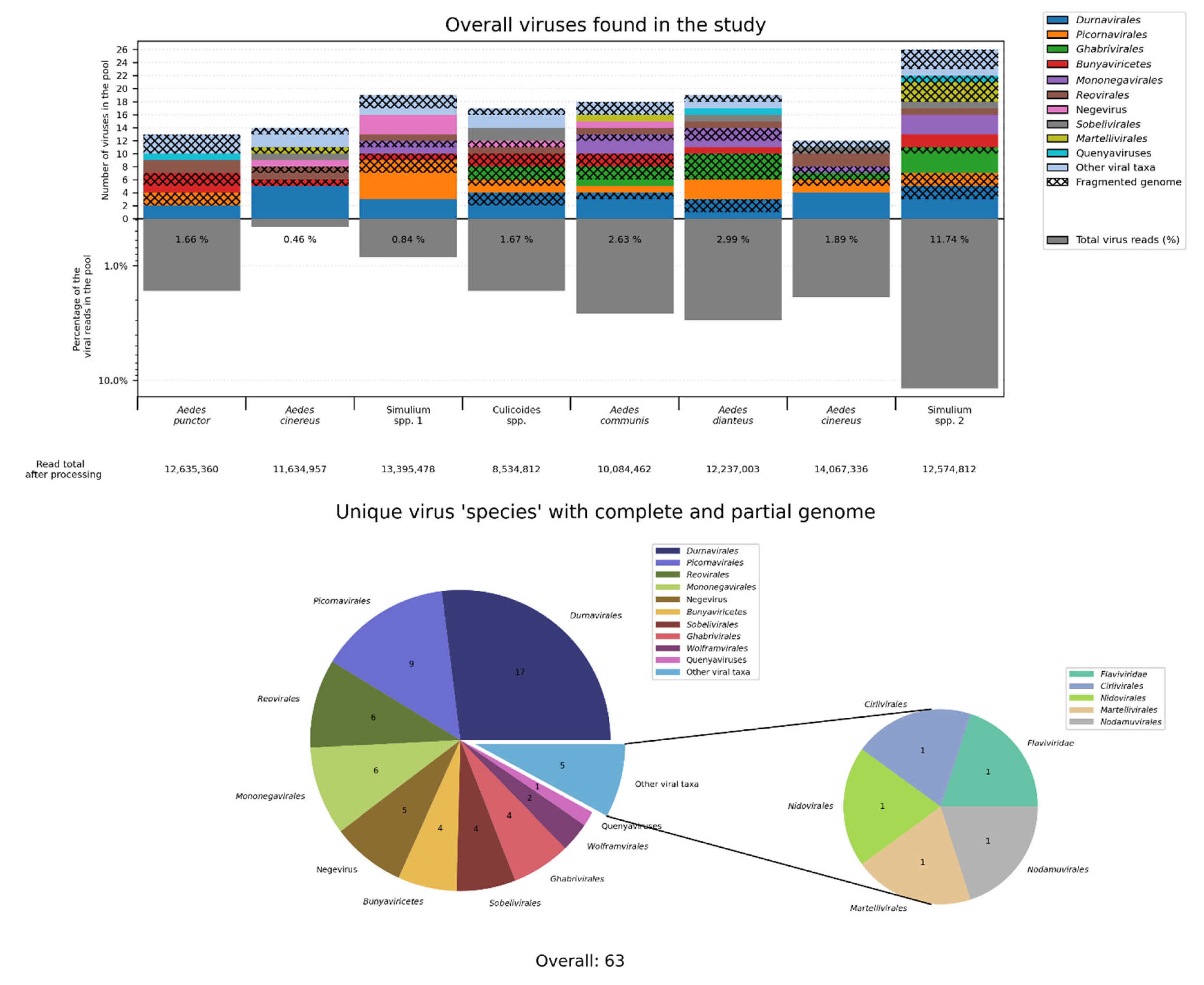
Figure 2.
Virus occurrence and abundance. (A) A heatmap representation of viruses for which multiple genome variants were found in our research. (B) The most abundant viruses discovered in this work. (C) A heatmap showing insect species where virus has been detected, both in literature and in this research.
Figure 2.
Virus occurrence and abundance. (A) A heatmap representation of viruses for which multiple genome variants were found in our research. (B) The most abundant viruses discovered in this work. (C) A heatmap showing insect species where virus has been detected, both in literature and in this research.

Figure 3.
Phylogenetic relationships of novel partiti-like viruses discovered in the study. Midpoint-rooted phylogenetic tree constructed using the amino acid sequences of the RdRp, with 1000 bootstrap replicates performed. Novel viruses are shown in red. Novel variants of already known viruses are in purple. Magnified subtrees containing novel viruses are shown on the right. Nodes with ≥90% bootstrap support are marked. The scale bar represents the number of amino acid substitutions per site.
Figure 3.
Phylogenetic relationships of novel partiti-like viruses discovered in the study. Midpoint-rooted phylogenetic tree constructed using the amino acid sequences of the RdRp, with 1000 bootstrap replicates performed. Novel viruses are shown in red. Novel variants of already known viruses are in purple. Magnified subtrees containing novel viruses are shown on the right. Nodes with ≥90% bootstrap support are marked. The scale bar represents the number of amino acid substitutions per site.
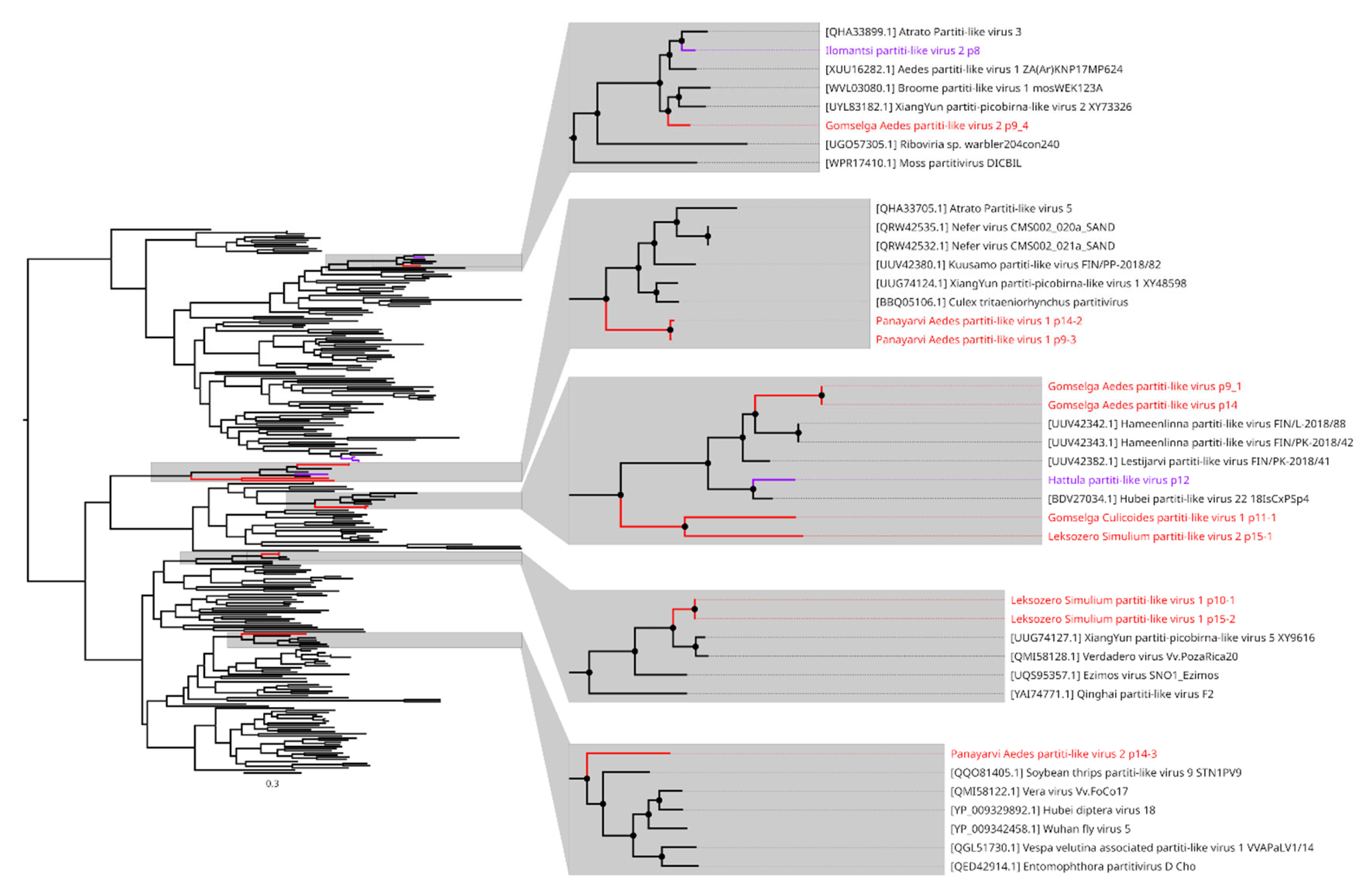
Figure 4.
Phylogenetic relationships and genome structure of chaq-like viruses. (A) Midpoint-rooted phylogenetic tree constructed using the amino acid sequences of the VP1. Nodes with ≥90% bootstrap support are marked, 1000 bootstrap replicates were performed. The scale bar represents the number of amino acid substitutions per site. Discovered viruses are shown in red. (B) Genome scheme of discovered chaq-like viruses. ORFs are shown in purple.
Figure 4.
Phylogenetic relationships and genome structure of chaq-like viruses. (A) Midpoint-rooted phylogenetic tree constructed using the amino acid sequences of the VP1. Nodes with ≥90% bootstrap support are marked, 1000 bootstrap replicates were performed. The scale bar represents the number of amino acid substitutions per site. Discovered viruses are shown in red. (B) Genome scheme of discovered chaq-like viruses. ORFs are shown in purple.
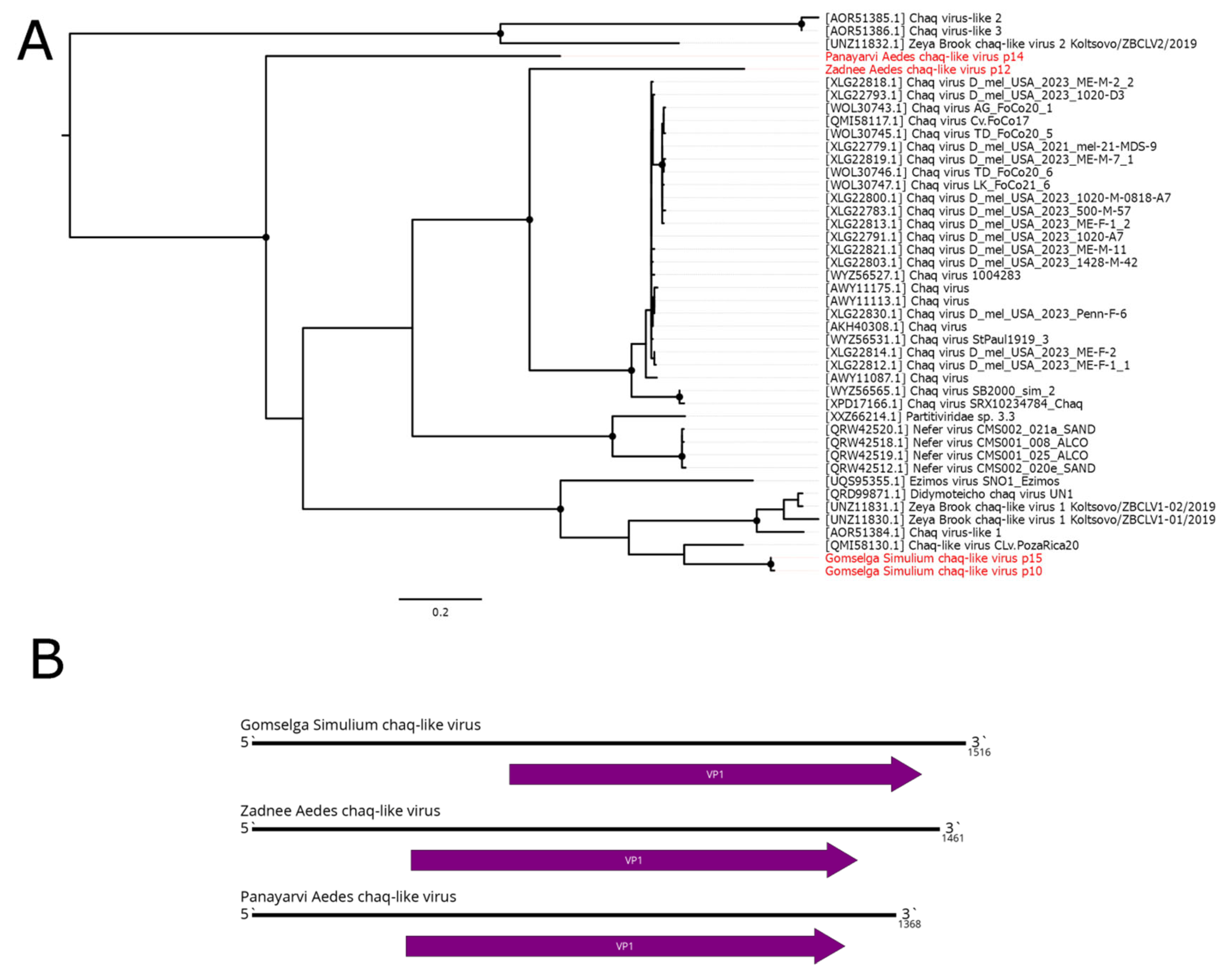
Figure 6.
Phylogenetic relationships and genome structure of dimoverna-like viruses discovered in mosqitoes. (A) Midpoint-rooted phylogenetic tree constructed using the amino acid sequences of the RdRp. Nodes with ≥90% bootstrap support are marked, 1000 bootstrap replicates were performed. The scale bar represents the number of amino acid substitutions per site. Discovered viruses are shown in red. (B) Genome scheme of the Gomselga Aedes dinoverna-like virus 1. ORFs are shown in purple. RdRp-encoding ORF is marked in green. Segment 8, which were not identified in some viruses is marked by red frame.
Figure 6.
Phylogenetic relationships and genome structure of dimoverna-like viruses discovered in mosqitoes. (A) Midpoint-rooted phylogenetic tree constructed using the amino acid sequences of the RdRp. Nodes with ≥90% bootstrap support are marked, 1000 bootstrap replicates were performed. The scale bar represents the number of amino acid substitutions per site. Discovered viruses are shown in red. (B) Genome scheme of the Gomselga Aedes dinoverna-like virus 1. ORFs are shown in purple. RdRp-encoding ORF is marked in green. Segment 8, which were not identified in some viruses is marked by red frame.
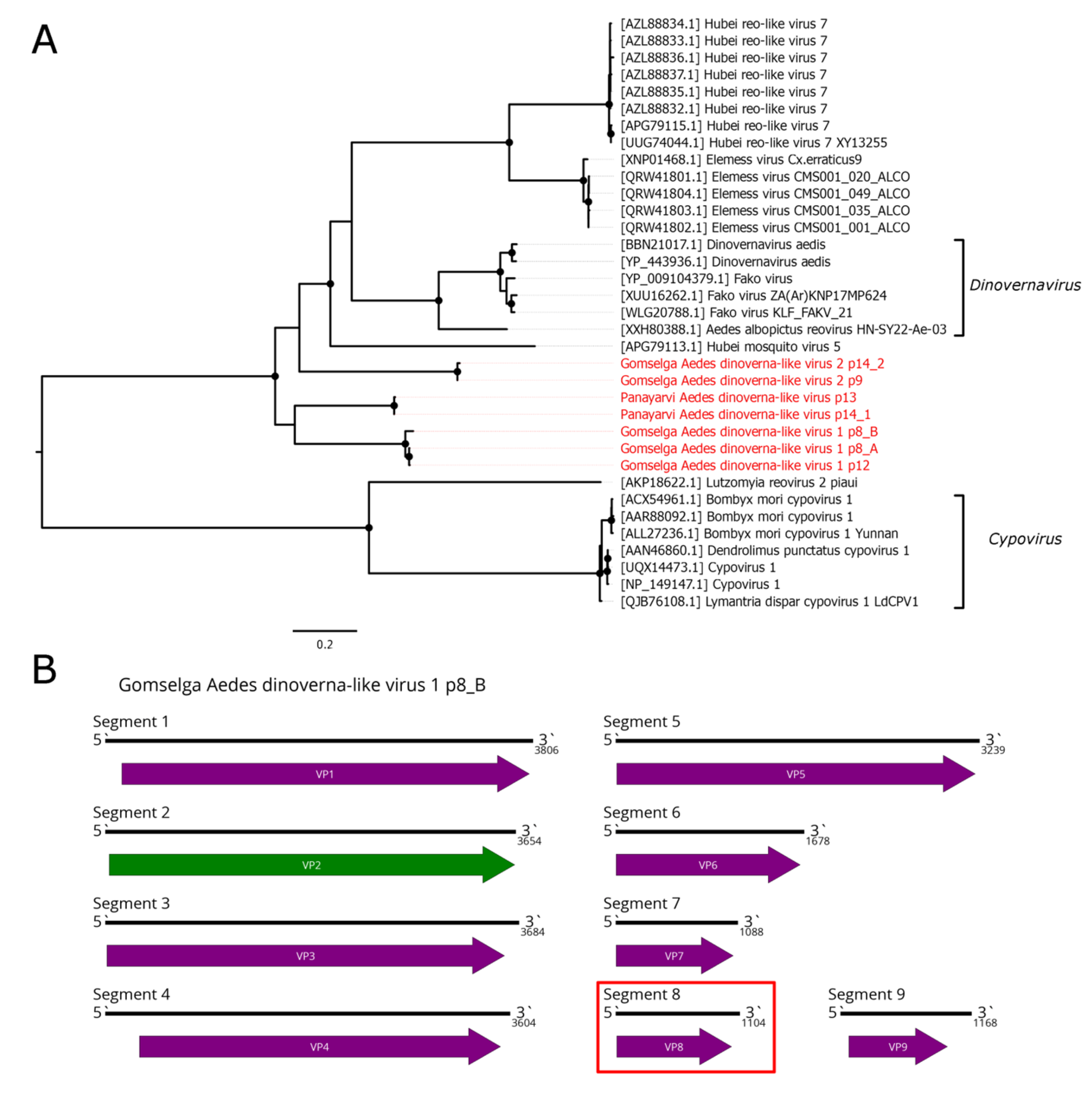
Figure 7.
Phylogenetic relationships and genome structure of reo-like viruses discovered in blackflies and biting midges. (A, C) Midpoint-rooted phylogenetic tree constructed using the amino acid sequences of the RdRp. Nodes with ≥90% bootstrap support are marked, 1000 bootstrap replicates were performed. The scale bar represents the number of amino acid substitutions per site. Discovered viruses are shown in red. (B) Genome scheme of the discovered viruses. ORFs are shown in purple. RdRp-encoding ORF is marked in green. Estimated gaps are in grey.
Figure 7.
Phylogenetic relationships and genome structure of reo-like viruses discovered in blackflies and biting midges. (A, C) Midpoint-rooted phylogenetic tree constructed using the amino acid sequences of the RdRp. Nodes with ≥90% bootstrap support are marked, 1000 bootstrap replicates were performed. The scale bar represents the number of amino acid substitutions per site. Discovered viruses are shown in red. (B) Genome scheme of the discovered viruses. ORFs are shown in purple. RdRp-encoding ORF is marked in green. Estimated gaps are in grey.
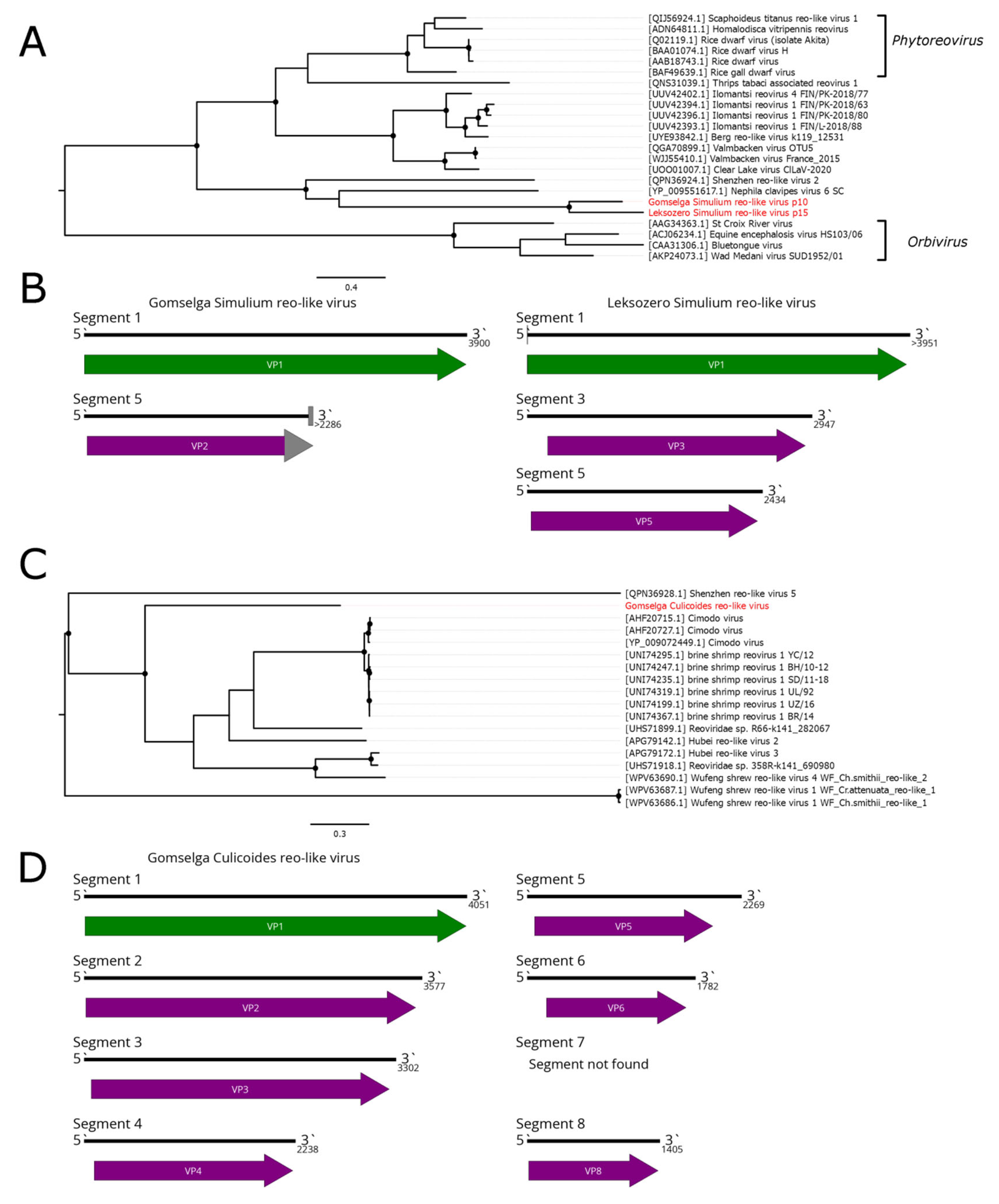
Figure 8.
Phylogenetic relationships and genome structure of mononegaviruses discovered in mosqitoes. (A, C, E) Midpoint-rooted phylogenetic trees constructed using the amino acid sequences of the L protein (Mononegavirales RdRp). Nodes with ≥90% bootstrap support are marked, 1000 bootstrap replicates were performed. The scale bar represents the number of amino acid substitutions per site. Discovered viruses are shown in red. (B,D,F) Genome scheme of discovered viruses. ORFs are shown in purple. RdRp-encoding ORF is marked in green. Gray blocks indicate estimated gaps.
Figure 8.
Phylogenetic relationships and genome structure of mononegaviruses discovered in mosqitoes. (A, C, E) Midpoint-rooted phylogenetic trees constructed using the amino acid sequences of the L protein (Mononegavirales RdRp). Nodes with ≥90% bootstrap support are marked, 1000 bootstrap replicates were performed. The scale bar represents the number of amino acid substitutions per site. Discovered viruses are shown in red. (B,D,F) Genome scheme of discovered viruses. ORFs are shown in purple. RdRp-encoding ORF is marked in green. Gray blocks indicate estimated gaps.
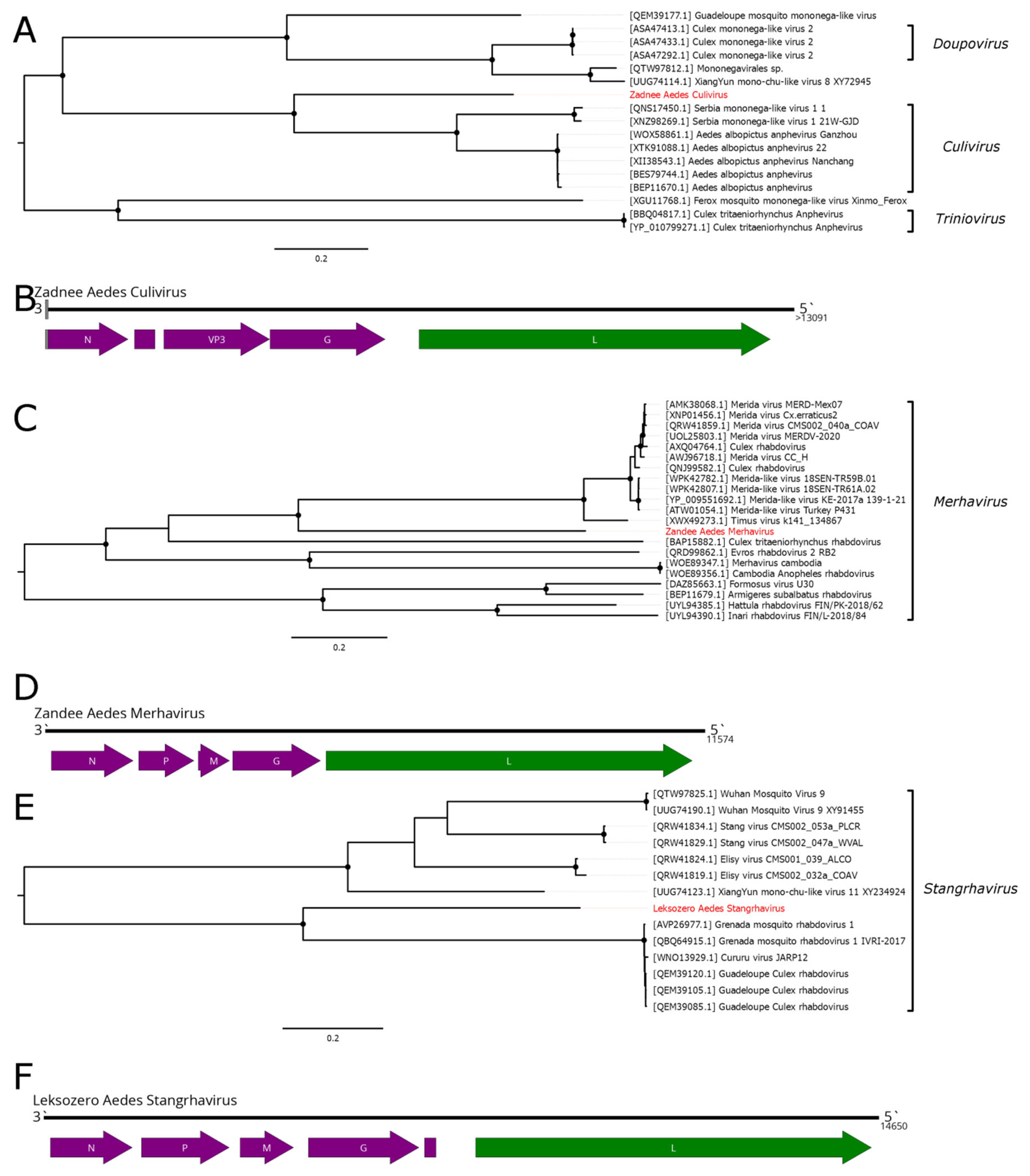
Figure 9.
Phylogenetic relationships and genome structure of rhabdo-like viruses discovered in Simulium species. (A) Phylogenetic tree constructed using the amino acid sequences of the L protein (Rhabdoviridae RdRp). Nodes with ≥90% bootstrap support are marked, 1000 bootstrap replicates were performed. The scale bar represents the number of amino acid substitutions per site. Phylogenetic tree is rooted on members of genera Vesiculovirus and Alpharicinrhavirus for clarity only. Discovered viruses are shown in red. Novel branch of Simulium specific rhabdo-like are in marked in red (B) Genome scheme of discovered viruses and a rhabdo-like element. ORFs are shown in purple. RdRp-encoding ORF is marked in green. Cellular genes are shown in blue.
Figure 9.
Phylogenetic relationships and genome structure of rhabdo-like viruses discovered in Simulium species. (A) Phylogenetic tree constructed using the amino acid sequences of the L protein (Rhabdoviridae RdRp). Nodes with ≥90% bootstrap support are marked, 1000 bootstrap replicates were performed. The scale bar represents the number of amino acid substitutions per site. Phylogenetic tree is rooted on members of genera Vesiculovirus and Alpharicinrhavirus for clarity only. Discovered viruses are shown in red. Novel branch of Simulium specific rhabdo-like are in marked in red (B) Genome scheme of discovered viruses and a rhabdo-like element. ORFs are shown in purple. RdRp-encoding ORF is marked in green. Cellular genes are shown in blue.
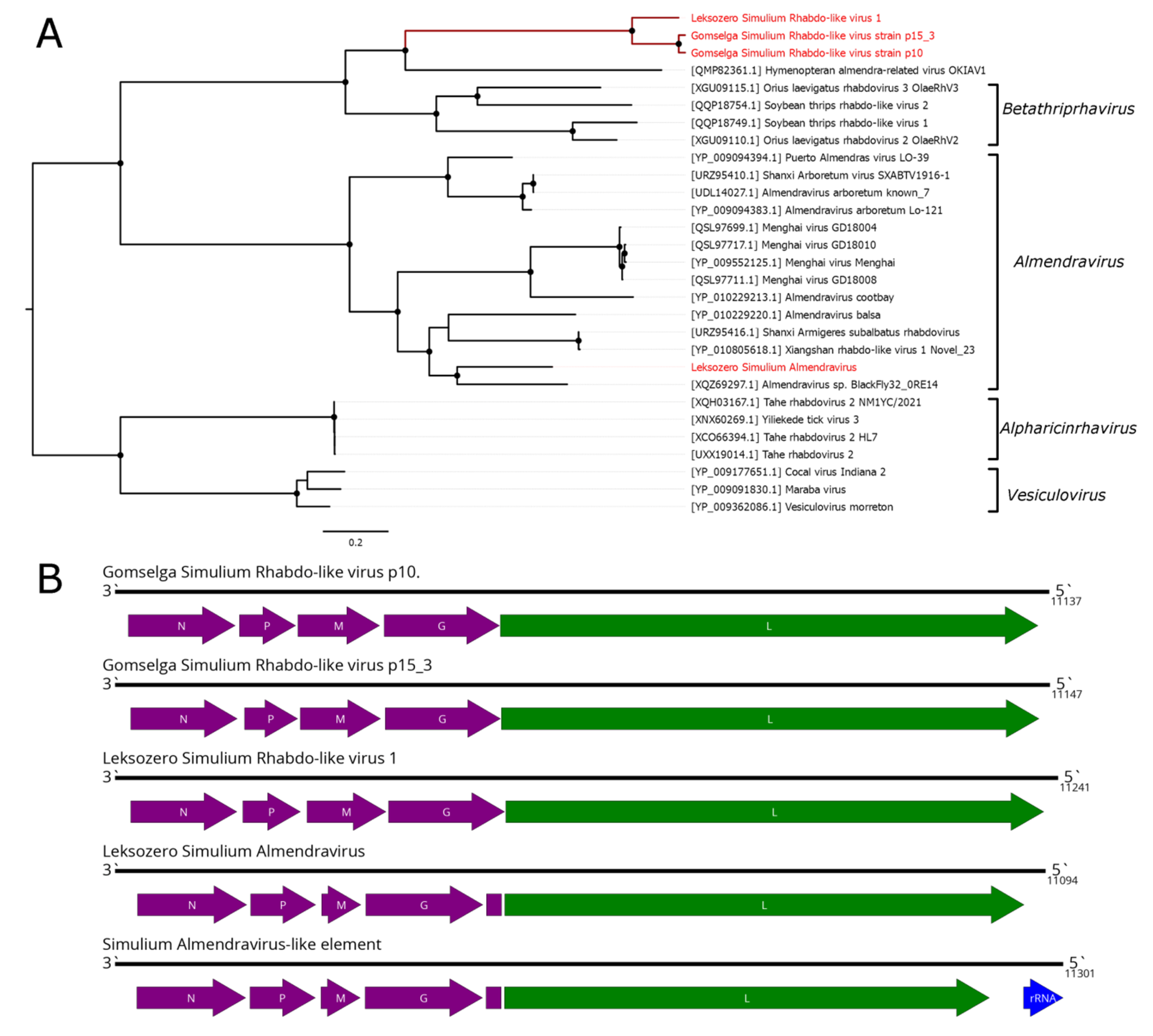
Figure 10.
Genome structure of Leksozero Simulium phenui-like virus and Leksozero Simulium bunya-like virus. ORFs are shown in purple, with RdRp-encoding ORFs marked in green. Estimated gaps are maked in grey.
Figure 10.
Genome structure of Leksozero Simulium phenui-like virus and Leksozero Simulium bunya-like virus. ORFs are shown in purple, with RdRp-encoding ORFs marked in green. Estimated gaps are maked in grey.
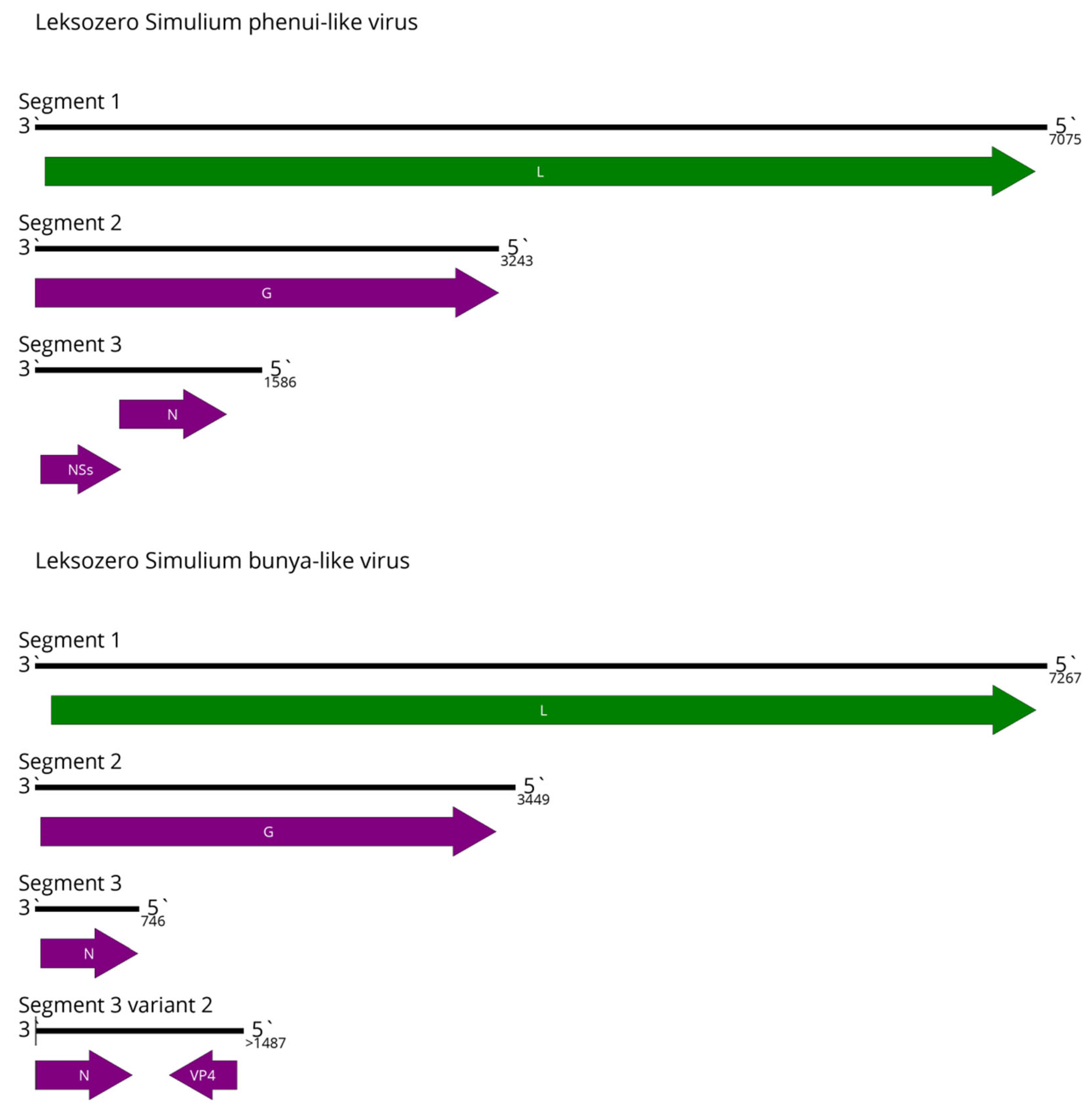

Table 1.
Data on the examined material.
| Pool | Specimens in the Pool | Species | Location | Coordinates | Collection date |
|---|---|---|---|---|---|
| 8 | 60 | Aedes punctor (Kirby, 1837) | Kondopozhsky District, Village of Gomselga | 62.0683°N, 33.9592°E | 21.08.2024 |
| 9 | 34 | Aedes cinereus Meigen, 1818 | 31.07.2024; 20.08.2024 | ||
| 10 | 40 | Simulium equinum (Linnaeus, 1758); S. noelleri Friederichs, 1920; S. reptans (Linnaeus, 1758) | 31.07.2024 | ||
| 11 | 60 | Culicoides punctatus (Meigen, 1804); C. impunctatus Goetghebuer, 1920; C. obsoletus (Meigen, 1818) | 01.08.2024 | ||
| 12 | 60 | Aedes communis (De Geer, 1776) | Kostomukshsky Urban Okrug, Near the Village of Pongaguba | 65.0442°N, 30.3567°E | 18.07.2023 – 19.07.2023 |
| 13 | 60 | Aedes diantaeus Howard, Dyar et Knab, 1913 | Muezersky District, Western shore of Lake Leksozero | 63.7898°N, 30.8640°E | 15.07.2023 - 17.07.2023 |
| 14 | 40 | Aedes cinereus Meigen, 1818 | Loukhsky District, Paanajärvi National Park | 66.2437°N, 30.5639°E | 25.06.2024 |
| 15 | 40 | Simulium reptans (Linnaeus, 1758); S. truncatum (Lundström, 1911); S. subpusillum Rubtsov, 1940 | Muezersky District, Western shore of Lake Leksozero | 63.7898°N, 30.8640°E | 15.07.2023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



