
Submitted:
06 December 2025
Posted:
09 December 2025
You are already at the latest version
Abstract
Short-chain fatty acids (SCFAs), acetate, propionate, and butyrate, are microbial me-tabolites with recognised roles in gut and immune homeostasis, but their therapeutic relevance in gastric cancer, particularly in combination with chemotherapeutics, re-mains unclear. This study investigated the antiproliferative synergy between a com-bined SCFA mixture (APB) and doxorubicin (Dox) in AGS gastric adenocarcinoma cells using integrated cellular, molecular, and proteomic approaches. APB and Dox each inhibited cell proliferation, with IC₅₀ values of 568.33 ± 82.56 μg/mL and 0.22 ± 0.04 μg/mL, respectively, and their combination (3000 + 0.27 μg/mL) enhanced cyto-toxicity, achieving 103.46% inhibition and reducing the APB IC₅₀ to 512.80 ± 18.37 μg/mL. Combination Index values confirmed synergistic interactions (CI₅₀ = 0.61; CI₉₅ = 0.13). APB+Dox significantly increased apoptosis (94.83%) with minimal necrosis (4.64%) and induced strong ROS generation comparable to APB alone, while Dox showed limited oxidative effects. Proteomic profiling revealed downregulation of ri-bosomal proteins and cell cycle regulators in Dox and APB+Dox groups, with the com-bination further enhancing apoptosis-related pathways and stress responses. Overall, these findings indicate that SCFA-based interventions, exemplified by APB+Dox, may offer a low-toxicity strategy to potentiate chemotherapy efficacy in gastric cancer through apoptosis induction, redox disruption, and attenuation of drug resistance.
Keywords:
postbiotics
; SCFAs
; gastric cancer
; doxorubicin
; apoptosis
; proteomics
1. Introduction
Gastric cancer remains a major global health concern, ranked as the fifth most commonly diagnosed cancer and the fourth leading cause of cancer-related mortality as of 2020 [1,2]. The high mortality rate is largely due to late diagnosis, with many patients presenting with advanced disease [2]. Despite progress in surgical techniques, recurrence following curative resection remains common, necessitating effective adjuvant treatment strategies [3]. Chemotherapy plays a central role in the management of advanced or metastatic gastric cancer, either as neoadjuvant, adjuvant, or primary treatment [4]. Doxorubicin (Dox), an anthracycline antibiotic, has long been used in combination chemotherapy regimens for gastric cancer [5,6,7]. However, its clinical use is hampered by issues such as drug resistance and severe side effects, including cardiotoxicity, myelosuppression, and mucositis [8,9,10]. Resistance to conventional chemotherapeutics like cisplatin and 5-fluorouracil (5-FU) has further complicated treatment, often leading to suboptimal clinical outcomes [11]. Therefore, there is an urgent need to identify low-toxicity compounds with strong anticancer potential that could be integrated into new combination regimens to circumvent or mitigate drug resistance.
Diet plays a fundamental role in shaping the gut microbiota and, consequently, in maintaining host health and metabolic homeostasis [12]. Diets rich in whole grains, legumes, fruits, and vegetables provide non-digestible carbohydrates (prebiotics) that reach the colon, where they are fermented by beneficial gut bacteria into health-promoting metabolites, including short-chain fatty acids (SCFAs) [13]. Well-known fibre-fermenting bacteria such as Faecalibacterium prausnitzii and Eubacterium rectale produce SCFAs like acetate (A), propionate (P), and butyrate (B), which help maintain gut barrier integrity, modulate immune responses, and exhibit systemic anti-inflammatory effects [13,14,15]. In contrast, Westernised diets low in fibre and high in saturated fats can reduce SCFA production by disrupting the balance of these beneficial microbial species, diminishing their protective effects and contributing to chronic disease risk [12].
The gut microbiota contributes significantly to nutrient metabolism, immune modulation, and protection against pathogens [16]. Beyond maintaining gut health, increasing evidence highlights its critical role as a modulator in cancer prevention and therapy [17]. A key mechanism underlying these protective effects is the production of postbiotics, including SCFAs such as acetate, propionate, and butyrate [18,19]. These SCFAs exert both local and systemic actions relevant to cancer prevention and treatment by influencing cellular proliferation, apoptosis, oxidative stress, and immune pathways [16,17]. SCFAs are primarily generated in the large intestine through microbial fermentation of dietary fibres and are absorbed into systemic circulation, where they can exert effects beyond the gut [20]. Their intestinal concentrations vary along the colon, generally ranging from 70–140 mM in the proximal region to 20–70 mM distally, with total SCFA levels in the colon typically falling between 20 and 140 mM [20]. Butyrate, in particular, is well-documented for its anti-proliferative and pro-apoptotic properties in various cancers through mechanisms such as histone deacetylase (HDAC) inhibition, reactive oxygen species (ROS) modulation, and regulation of apoptotic signalling pathways [21,22]. Propionate and acetate have also demonstrated distinct anticancer activities, including modulation of the NF-κB and Wnt/β-catenin pathways and interference with cancer cell metabolism [23,24].
Sodium butyrate has demonstrated the ability to enhance the effectiveness of various chemotherapeutic agents [25,26,27,28,29,30]. It has been reported to sensitize tumour cells to docetaxel and, when used alongside cisplatin, significantly increases apoptosis in gastric cancer cells through activation of the mitochondrial apoptotic pathway [25,26]. B also improves the impact of 5-FU in colon cancer cells by further disrupting DNA synthesis [27]. In lung cancer, B administered prior to paclitaxel treatment helped restore gut microbiota balance, food intake, and intestinal barrier function, thereby reducing treatment-related side effects [28]. In bladder cancer, co-treatment with B and cisplatin showed synergistic anticancer effects by promoting G1-phase cell cycle arrest and apoptosis through the regulation of proteins such as p21, p27, TRADD, and procaspase-2 [29]. Additionally, propionate has been shown to enhance cisplatin’s cytotoxicity in liver cancer cells by modulating GPR41 signalling pathways [30].
In a study conducted in our lab, B synergised with dexamethasone to enhance antiproliferative effects against AGS gastric cancer cells [31]. A follow-up study evaluated combinations of SCFAs (AP, AB, PB, APB) and their co-treatment with Dex (APB+Dex) [32]. The APB+Dex combination exhibited strong synergistic interactions, targeting multiple tumour-promoting mechanisms, including the disruption of redox balance and the induction of apoptosis. Encouraged by these promising synergistic interactions between SCFAs and chemotherapeutic or immunotherapeutic agents, the current study further investigated the effects of APB, Dox, and their combination (APB+Dox) on AGS gastric adenocarcinoma cells. This novel combination aims to evaluate enhanced safety profile, induction of apoptosis, modulation of oxidative stress, and proteomic reprogramming, offering potential insights into more effective and safer therapeutic strategies for gastric cancer.
2. Results and Discussion
2.1. Antiproliferative Activity of SCFA Combinations, Dox and Their Combination Against AGS Adenocarcinoma Cells:
Magnesium acetate, sodium propionate, and sodium butyrate were used to prepare the APB mixture, reflecting the physiologically relevant salt forms of SCFAs present in the colonic lumen [20]. The concentration range (93.75–3000 μg/mL) captures both physiologically relevant exposures (0.8–10 mM) and supra-physiological conditions (up to ~26.5 mM total SCFAs) frequently used in mechanistic studies to model pharmacological exposure [20]. At the same time, our dosing strategy reflects established in vitro practice. Numerous cancer studies have applied SCFAs across 0.5–10 mM and reported dose-dependent antiproliferative and pro-apoptotic effects. For instance, butyrate and propionate have demonstrated growth inhibition in gastric and breast cancer cells, with IC₅₀ values of ~1.3 mM (B) and ~4.5 mM (P), respectively, and apoptosis induction at higher millimolar concentrations [33,34]
In the current study, APB alone achieved 95.65 ± 7.90% inhibition at 3000 μg/mL (p < 0.05) with an IC₅₀ of 568.33 ± 82.56 μg/mL. Dox alone (p < 0.05) produced 73.51 ± 5.16% inhibition at 0.54 μg/mL with an IC₅₀ of 0.22 ± 0.04 μg/mL (Table 1). The combination of APB and Dox (p < 0.05) resulted in 103.46 ± 2.24% inhibition at 3000 μg/mL APB + 0.27 μg/mL Dox and an IC₅₀ of 512.80 ± 18.37 μg/mL (Table 1). In normal Hs 738.St/Int human intestinal cells, Dox alone reduced viability to 38.37 ± 7.01% at 0.54 μg/mL. APB alone maintained 76.59 ± 8.56% viability, and the APB+Dox combination at 3000 μg/mL + 0.27 μg/mL achieved 64.12 ± 8.76% viability. At 1500 μg/mL APB + 0.136 μg/mL Dox, viability was 92.42 ± 10.66%, compared with 79.67 ± 8.16% for APB alone and 68.52 ± 7.51% for Dox alone. At concentrations ≤ 750 μg/mL APB, combination treatments produced viability values exceeding 100%, with a maximum of 112.90 ± 11.32% at 187.5 μg/mL APB + 0.017 μg/mL Dox (Table 1).
2.2. Synergistic Potential of APB with Dox Against the AGS Cells
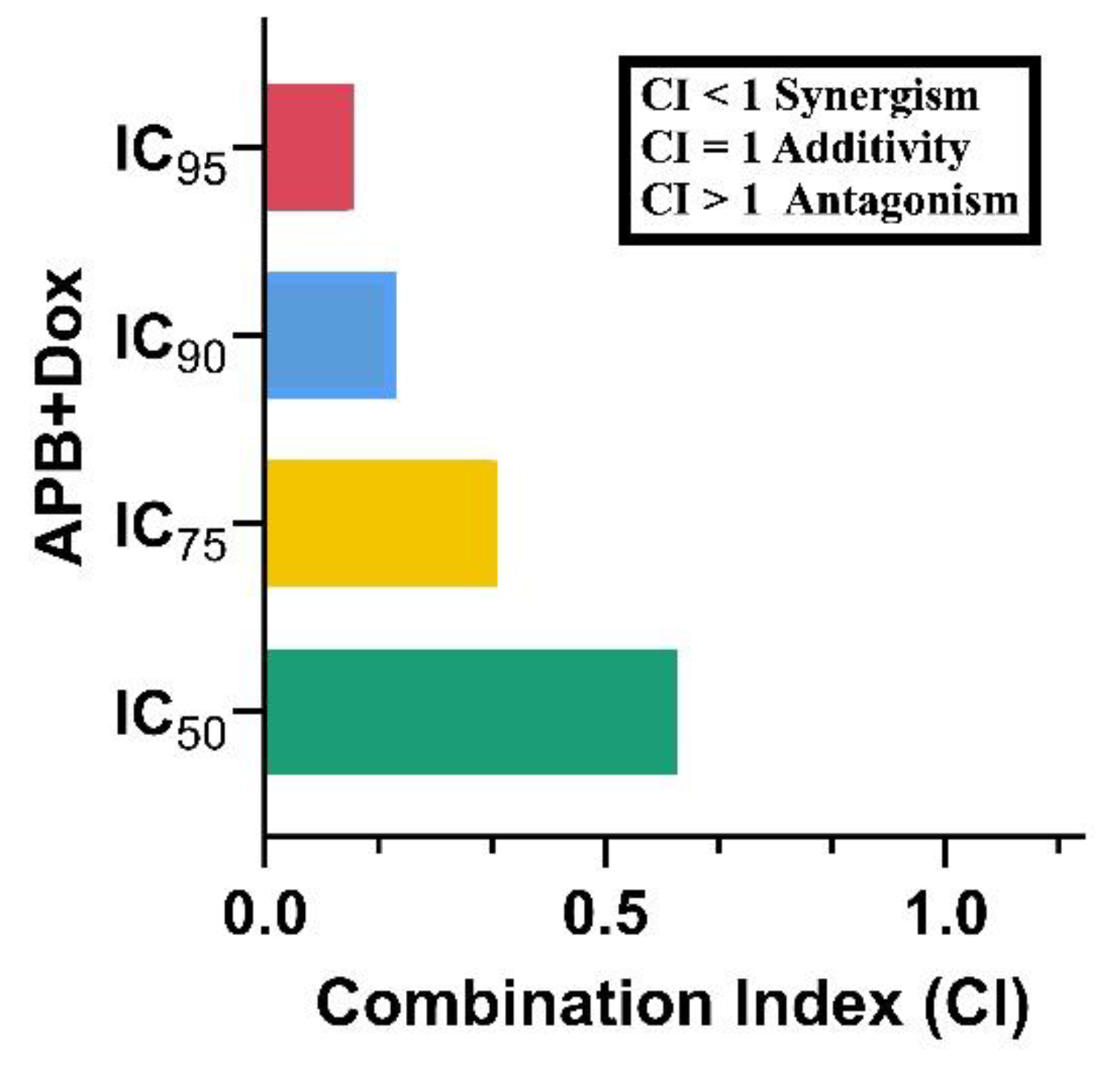
The potential synergistic effects of APB and Dox combination on AGS gastric adenocarcinoma cells was analysed using the CI model [35]. The APB+Dox combination exhibited a pronounced synergistic effect, reflected in a CI value of 0.61 at the IC50 level, further confirming the potential of this combination to enhance therapeutic efficacy against gastric cancer. The IC75 (0.34), IC90 (0.19), and IC95 (0.13) values also showed reduced CI values, indicating that the synergy persists across a range of concentrations. These findings suggested that the inclusion of APB combinations with established chemotherapeutic agents like Dox significantly enhance the inhibitory effects on AGS gastric adenocarcinoma cells.
Figure 1.
CI values of different inhibitory concentrations of APB+Dox on AGS gastric cancer cells.
2.3. Proteomics Study of the AGS Cells Treated with the Synergistic Combination vs Mono Treatments:
Following the promising cell studies and synergy studies, proteomic analysis was conducted in AGS cells after treatment with Dox (compared to the untreated control), and with the APB+Dox combination compared to Dox and APB monotherapies.
2.3.1. Enrichment Analyses of Differentially Expressed Proteins DEPs in Dox-Treated AGS Cells Compared to the Control Untreated Cells:
Dox is a widely used anthracycline chemotherapeutic that exerts its anticancer activity primarily through DNA intercalation, inhibition of topoisomerase II, and generation of ROS, which collectively lead to DNA damage and activation of apoptotic pathways [36]. The current study represents one of the first comprehensive analyses of dysregulated proteins in AGS gastric adenocarcinoma cells following treatment with Dox or its combination with APB.
The volcano plot of Dox treatment versus untreated control in AGS cells highlights a range of significantly dysregulated proteins, with several key players implicated in anticancer mechanisms (Figure 2A). Among the most upregulated genes is TP53, a pivotal tumour suppressor involved in cell cycle arrest, DNA repair, and apoptosis, whose activation reinforces Dox's classical cytotoxic role in cancer therapy [37]. HSD11B2, also upregulated, modulates glucocorticoid metabolism and may influence stress hormone signalling within the tumour microenvironment [38]. IKBIP and CEBPB were both found to be elevated in expression, supporting their known roles in regulating apoptosis and inflammatory signalling pathways that suppress tumour survival [39]. ARG2 showed increased expression and has been associated with immune modulation and tumour progression, while ENDOG, another upregulated protein, is a mitochondrial nuclease involved in caspase-independent cell death [40,41].
On the downregulated end, several ribosome biogenesis and RNA processing genes are markedly suppressed. For example, RIOK2, RPL7L1, RRP7A, NOP14, NSA2, and UTP6 are associated with ribosomal RNA synthesis and maturation [42]. Their repression may impair protein translation, contributing to reduced tumour cell proliferation. DDX21 and DDX56, both DEAD-box RNA helicases, are involved in transcriptional regulation and ribosome assembly, and their downregulation may further dampen the biosynthetic capacity of gastric cancer cells [43]. Suppression of MRFAP1, which is linked to chromatin organisation and DNA replication, may enhance DNA damage sensitivity in the presence of Dox [44].
The graphical summary (Figure 2B) shows that Dox triggered tumour suppressive signalling, metabolic and immune reprogramming, and inhibition of stemness-related gene expression. Key regulatory hubs such as TP53, MYC, and TFDP1 orchestrate these changes across core biological processes, including enhanced cell death, lipid oxidation and transcriptional regulation [37,45]. These insights suggested that Dox did more than induce apoptosis, it also reconfigured the cellular state of AGS cells, potentially weakening their proliferative and adaptive capacities and offering opportunities to overcome therapeutic resistance. A key feature of Dox’s action in AGS cells is the activation of TP53-mediated tumour suppression and the repression of oncogenes such as MYC, contributing to reduced cell viability and heightened apoptotic signalling. In parallel, modulation of lipid metabolism genes like KLF15 and ESRRA points to a Dox-induced oxidative stress phenotype, known to provoke mitochondrial dysfunction and DNA damage in gastric cancer cells [46].
Dox also stimulated immune-related genes, notably IFNB1, reflecting activation of antiviral and inflammatory pathways that could enhance tumour immunogenicity [46]. Furthermore, the repression of stemness-associated genes such as POU5F1 and EFNA5 suggested an anticancer stem cell effect, with implications for limiting recurrence and resistance [47].
Among the most significantly inhibited pathways is the “Major pathway of rRNA processing in the nucleolus and cytosol,” alongside suppression of ribosomal protein synthesis and translation-associated machinery (Figure S1). These changes suggested that Dox markedly interfered with ribosome biogenesis and function, a process often hijacked in cancer to support uncontrolled protein synthesis and proliferation. In addition to ribosomal dysregulation, Dox significantly inhibited DNA-related pathways, including DNA replication, repair, and chromatin remodelling (Figure S1). These disruptions reflected Dox’s known mechanism of action as a DNA intercalator and topoisomerase II inhibitor, inducing double-strand breaks and genomic instability in cancer cells. Moreover, suppression of the cell cycle, particularly the G2/M checkpoint and mitotic entry, reinforced Dox’s antiproliferative impact.
Ribosomal Protein Regulation:
Dox treatment resulted in the downregulation of multiple ribosomal proteins (RP), such as RPSA (Log2FC = -0.70), RPL6 (Log2FC = -0.73), RPL10 (Log2FC = -0.64), RPL23 (Log2FC = -0.62), RPS3A (Log2FC = -0.59), RPS6 (Log2FC = -0.59), RPS27 (Log2FC = -0.58), RPL7A (Log2FC = -0.60), RPL13A (Log2FC = -1.04), RPL7L1 (Log2FC = -3.12), and others (Log2FC ranging from -0.58 to -3.12) indicated that Dox suppressed ribosomal function in AGS cells [48,49,50,51,52,53] (Figure 3). These proteins are essential for various aspects of ribosome function, including protein synthesis, ribosome assembly, and translational regulation. Many of the ribosomal proteins affected by Dox, such as RPL6 (Log2FC = -0.73), RPL13A (Log2FC = -1.04), and RPL39 (Log2FC = -0.68), have been linked to progression in gastric cancer [54,55,56]. For example, RPL6 and RPL10 (Log2FC = -0.64) are associated with gastric and colorectal cancers, and RPL23 (Log2FC = -0.62) is known to promote tumorigenesis [54,57,58]. The downregulation of these ribosomal proteins could be interpreted as a therapeutic effect of Dox, potentially slowing down cancer cell proliferation and invasion by interfering with the synthesis of key proteins involved in tumor progression (Figure 3). Other ribosomal proteins, such as RPS3A (Log2FC = -0.59), RPS6 (Log2FC = -0.59), and RPL13A (Log2FC = -1.04), are not only involved in ribosome function but also have roles in cancer cell proliferation and metastasis [49,52]. Specifically, RPS6 (Log2FC = -0.59), which is involved in mTOR signaling, is often activated in gastric cancer, and its downregulation by Dox might impact this critical pathway, potentially reducing cancer cell growth and metastasis [59,60].
Some of the genes affected by Dox, such as RPS27A (Log2FC = -0.63), RPL38 (Log2FC = -0.65), and RPL14 (Log2FC = -0.77), are involved in ribosome stability and function, and their dysregulation is associated with cancer progression and metastasis [50,61,62]. Additionally, serine/threonine-protein kinase SMG enzymes were reported to decrease by a DNA damage inducer, such as Dox [63]. In the current study, genes such as SMG1 (Log2FC = -0.61), SMG6 (Log2FC = -0.80), and SMG7 (Log2FC = -0.92), which are involved in nonsense-mediated mRNA decay (a component of the DNA repair response), show downregulation in response to Dox. This suggested that the drug may influence the cellular DNA damage response in a way that could further reduce the ability of cancer cells to proliferate and survive under stressful conditions.
The downregulation of key ribosomal proteins and associated biogenesis factors—such as RIOK2, UTP6, NSA2, and WDR46—indicated that Dox significantly impairs ribosome maturation and assembly (Figure 3) [64]. RIOK2, for example, is critical for the late-stage maturation of the 40S ribosomal subunit. Its marked downregulation (Log2FC = -4.73) by Dox potentially inhibited the formation of functional ribosomes, reducing the translational capacity of cancer cells [64]. Similarly, UTP6 (Log2FC = -3.99) and UTP14A (Log2FC = -3.24), integral components of the small subunit processome, are involved in pre-rRNA processing [65]. Their suppression disrupts ribosomal RNA synthesis, thereby limiting ribosome production. Downregulation of UTP6 and UTP14A is associated with drug resistance and poor prognosis in gastric cancer [66,67]. Combination therapies (such as APB+Dox) targeting complementary pathways may help overcome resistance.
The downregulation of ribosomal proteins such as DDX56 (Log2FC = -3.12), and RRP7A (Log2FC = -2.83) underscored Dox’s broad impact on ribosome biogenesis [42,68]. These proteins are crucial for various stages of ribosomal assembly, and their inhibition by Dox suggests a comprehensive disruption of this process. The suppression of proteins like WDR46 (Log2FC = -3.27) and NSA2 (Log2FC = -3.61) highlighted Dox's potential ability to induce ribosomal stress, a state that sensitizes cancer cells to apoptosis [69,70]. WDR46, a scaffold protein essential for nucleolar structure and ribosomal RNA processing, is crucial for maintaining the nucleolus, a hub of ribosome biogenesis [69]. Its downregulation disrupts nucleolar integrity, a phenomenon that has been linked to the activation of p53-mediated apoptotic pathways [71]. Similarly, the inhibition of NSA2, which facilitates 60S ribosomal subunit assembly, further compromises ribosomal function, halting protein synthesis and cancer cell growth [72].
DNA-related processes
Dox also impacted DNA-related processes in AGS gastric cancer cells, targeting key proteins involved in DNA repair, replication, and stability. Its effects were evident in multiple pathways that regulate DNA damage response (DDR), replication fork dynamics, and chromatin stability, collectively contributing to its anticancer efficacy (Figure 3). Among the DDR (DNA damage response) proteins, Poly(ADP-ribose) polymerases (PARPs) such as PARP2 (Log2FC = -1.43), PARP4 (Log2FC = -1.04), and PARP12 (Log2FC = -0.94), all of which are central to DNA repair mechanisms, showed significant downregulation. Dox is known to impair DDR by targeting key genes, such as PARPs, a crucial player in DNA repair that interacts with DNA strand breaks [73]. BRCA-encoded protein is crucial for the homologous recombination repair mechanism, which is vital for repairing double-strand breaks caused by Dox [74]. The loss of BRCA1 expression has been shown to enhance resistance to Dox in other cancer models, where BRCA1 deficiency triggers resistance mechanisms [74]. The downregulation of BRCA1 (Log2FC = -0.80) in AGS cells treated with Dox suggested that the DNA damage response (DDR) pathway may be compromised, impairing the cells’ ability to repair DNA effectively. The suppression of these genes potentially compromised the recombination repair, a vital pathway for repairing DNA double-strand breaks. In addition, the reduction in RAD51 (Log2FC = -0.67) further hindered this repair process, leading to increased genomic instability and heightened susceptibility to apoptosis in cancer cells [75]. Interestingly, XPC (Log2FC = 1.13), a gene involved in nucleotide excision repair, was upregulated, suggesting a cellular attempt to counteract the widespread DNA damage. This is in line with a previous study demonstrating the overexpression of XPC gene in Dox-treated cells. [76]
Dox also impacted proteins critical for maintaining DNA metabolism and chromosomal stability. Telomeric repeat-binding factor 1 (TERF1) is a key protein within the telomere complex essential in mediating the interactions between telomeres in mammalian cells [77]. It helps regulate telomere length and stability by binding to the telomeric DNA repeats, thereby facilitating the structural integrity of the telomeres [78]. The observed downregulation of TERF1-encoded protein (Log2FC = -1.22) suggested potential telomere destabilization, which may contribute to genomic instability. In addition, the reduced expression of replication fork-associated genes, such as SMARCAL1 (Log2FC = -0.68) and SMARCAD1 (Log2FC = -0.71), aligned with disruptions in DNA replication fidelity, exacerbating genomic instability [79]. The significant downregulation of SETD2 (Log2FC = -2.02), responsible for histone H3K36 methylation, may further impaired chromatin remodelling and repair, weakening the cell's ability to maintain genomic integrity. [80]. SETD2 mutations contribute to resistance to Dox by impairing the DDR, weakening the apoptotic response to chemotherapy [81]. In gastric cancer, the downregulation of proteins like MUC1 (Log2FC = -0.68), AXIN2 (Log2FC = -1.50), and MGMT (Log2FC = -0.77) potentially indicated the disruption of DNA damage response and signalling pathways, with Dox having a broad impact on these processes [82,83,84,85]. While MGMT is more closely associated with alkylating agents, its downregulation in this context could reflect an indirect effect [84]. Additionally, the suppression of FANCG (Log2FC = -1.99), a key player in the Fanconi anaemia pathway, indicated a probable disruption of DNA repair mechanisms in the AGS gastric cancer cells, contributing to increased DNA damage [86].
SOX9, a transcription factor crucial for cartilage formation and stem cell regulation, also plays roles in cellular responses to DNA damage [87]. STAT6, another transcription factor, is involved in the regulation of immune responses and inflammation, which may be critical in response to Dox-induced cellular stress [88]. The downregulation of SOX9 (Log2FC = -1.33) and STAT6 (Log2FC = -0.84) suggested disruptions in transcriptional regulation related to DNA repair and inflammation [87,88]. Their downregulation could impact cellular responses to DNA damage and inflammatory signals, which aligns with the observed effects of Dox treatment. Additionally, the upregulation of SYVN1 (Log2FC = 1.30), a gene involved in the cellular stress response and protein degradation, indicated an active, though insufficient, compensatory mechanism to mitigate the damage [89]. SYVN1’s role in responding to DNA damage-induced stress further reflected the cell’s attempt to cope with Dox-induced damage, but the response may not be enough to overcome the widespread effects of the drug [89]. Finally, Dox modulated proteins linked to DNA replication complexes and chromatin remodelling. While GINS2 (Log2FC = 1.46) and GINS4 (Log2FC = 1.41) were upregulated, potentially reflecting compensatory mechanisms to sustain replication, the suppression of histone methyltransferases such as ASH1L (Log2FC = -1.26) potentially limited the capacity for chromatin repair and transcription regulation [90,91].
Cell Cycle
Dox significantly impacted the cell cycle in AGS gastric cancer cells by targeting key regulatory pathways and proteins involved in cell cycle progression. From the data provided, Dox induced disruptions at multiple checkpoints, particularly within the G1/S and G2/M phases, thereby promoting cell cycle arrest.
G1/S Phase Arrest
Dox's action began at the G1/S transition, disrupting the progression into the S phase by downregulating critical cyclins and CDKs that control the checkpoint. Key proteins affected include CCND1 (Log2FC = -1.90), CDK4 (Log2FC = -1.34), and CDK6 (Log2FC = -0.77), leading to reduced phosphorylation of Rb protein and failure to activate E2F transcription factors [92,93]. The suppression of E2F4 (Log2FC = -1.71) further inhibited the transcription of S-phase related proteins, compounding the block at this checkpoint (Figure 3). [94]. Moreover, the upregulation of TP53 (Log2FC = 2.94) reinforced this arrest through the activation of p21 (CDKN1A), a cyclin-dependent kinase inhibitor (CDKI), which binds to and inhibits CDK4/6 activity [95]. This halted cell cycle progression and allowed cells to initiate DNA damage repair or apoptosis [95]. The overall inhibition at this stage potentially prevented AGS cells from replicating damaged DNA, a hallmark of effective anticancer action. Interestingly, despite the suppression of key G1/S drivers, the upregulation of CCND3 (Log2FC = 1.03) and CCNE1 (Log2FC = 1.58) suggested that some compensatory mechanisms might be activated in response to Dox-induced stress [96]. However, these attempts were insufficient to overcome the dominant inhibitory effects mediated by Rb and p53.
S Phase Disruption
While the G1/S arrest was prominent, Dox also impacted cells that entered the S phase. The downregulation of replication factors like MCM10 (Log2FC = -0.80) and proteins associated with replication origin licensing disrupted DNA replication initiation [97]. The reduced expression of PCNA (Log2FC = 0.68), a critical replication clamp protein, further hampered replication fork progression [98]. These disruptions created replication stress, which is exacerbated by the suppression of checkpoint kinases CHEK1 (Log2FC = -1.11) and CHEK2 (Log2FC = -1.07) [99]. These kinases are vital for detecting and repairing stalled replication forks [99]. Their downregulation allows replication stress to persist, accumulating DNA damage and driving cells toward apoptosis instead of completing the S phase.
G2/M Phase Arrest
Dox may also profoundly affect the G2/M checkpoint, preventing cells with damaged DNA from entering mitosis. The downregulation of PLK1 (Log2FC = -0.73), a key regulator of mitotic entry and spindle assembly, disrupted mitotic initiation. [100]. Similarly, the suppression of MAD2L2 (Log2FC = -1.10) and BUB1 (Log2FC = -0.88) interfered with spindle checkpoint signalling, leading to improper chromosomal alignment and segregation [101,102]. Separase regulators like ANAPC7 (Log2FC = -0.77), along with the upregulation of PTTG1 (Securin) (Log2FC = 0.90), reflected a broader disruption of chromosome segregation machinery [103,104]. Overexpression of PTTG1 in normal human fibroblasts activates the DNA damage response pathway, leading to p53-dependent cell cycle arrest [104].
Mitotic phase
Mitotic disruption caused by Dox culminates in mitotic phase arrest, a process characterized by cell death following defective mitosis. The TP53 gene, which encodes the p53 protein, plays a central role in cell cycle arrest by stopping the cell cycle and preventing the spread of DNA-damaged cells [105]. Upregulation of TP53 (Log2FC = 2.94) plays a central role in this outcome by enforcing checkpoints and initiating apoptosis. Furthermore, the observed downregulation of NDC80 (Log2FC= -1.08), a kinetochore complex component, potentially disrupted microtubule attachment and chromosome alignment, further impairing mitotic fidelity [106]. Proteins like ESPL1 (Separin) (Log2FC = 0.81) and other regulators of chromosome segregation were affected in a way that enhanced chromosomal instability, creating a lethal environment for cancer cells [107].
2.3.2. Enriched Pathways Using DEPs of APB+Dox Combination Treated AGS Cells vs Mono Treatments:
Figure S2 illustrates the machine learning-based prediction of molecular effects on the malignant neoplasm of the aerodigestive tract signalling pathway, highlighting the impact of APB+Dox treatment compared to the control (Figure S2). This systems-level prediction corresponds closely with proteomic changes identified in the volcano plot (Figure 4A), which compares APB+Dox treatment to individual APB and Dox monotherapies and highlights several differentially expressed genes potentially relevant to the anticancer activity of the combination in AGS gastric adenocarcinoma cells. Notably, several downregulated genes, such as HLA-F, TRIAP1, ENTPD8, TMBIM6, and TMSB10, are known to play roles in cell survival, mitochondrial integrity, and anti-apoptotic processes. HLA-F, a non-classical class I molecule, is implicated in cancer, often upregulated in various types of tumours [108,109,110]. It can be found in cancer cells, potentially allowing them to evade immune surveillance and suppress anti-tumour immune responses [108]. Additionally, HLA-F is associated with poor survival in some cancers, such as glioma and non-small cell lung cancer [108]. TRIAP1 is involved in mitochondrial membrane homeostasis and cell protection from apoptosis, while TMBIM6 is associated with resistance to stress-induced cell death [109,110]. Their suppression suggested enhanced apoptotic susceptibility when APB is used in conjunction with Dox. Additionally, ENTPD8, which has been linked to purinergic signalling and immune evasion in tumours, is also significantly downregulated, potentially improving immunogenicity [111]. On the other hand, genes such as SERPINE1, USP17L4, and TSPAN3 were significantly upregulated. SERPINE1 (plasminogen activator inhibitor-1) is known to modulate extracellular matrix remodelling and may either promote or suppress tumour progression depending on context; however, in this combination, its induction may reflect a stress or reparative response to extensive damage (Figure 4A) [112]. The upregulation of deubiquitinases like USP17L4 and USP17L15 could be compensatory but also point to altered proteostasis under APB+Dox treatment [113].
The graphical summary of APB+Dox vs APB and Dox monotreatment revealed a distinct suppression of tumour-promoting mechanisms in AGS gastric cancer cells, with a particular focus on CD44-driven signalling (Figure 4B). CD44, a well-established marker of cancer stem cells and a facilitator of tumour progression in gastric cancer, is central to this network [114]. It influences key processes such as tumour cell interaction, migration, invasion, and fibrogenesis [114]. Its downregulation, along with that of CD38, suggested a disruption in tumour–stromal interactions and extracellular matrix remodelling—hallmarks of aggressive gastric cancer phenotypes (Figure 4B) [115]. The associated suppression of fibrogenesis-related genes like EPAS1, SMARCD3, and SYVN1 supported the potential of combination therapy to interfere with tumour microenvironment remodelling, which is critical for tumour invasion and metastasis (Figure 4B) [116]. Further, the network indicated transcriptional reprogramming as a major theme, with reduced activity in transcriptional regulators such as KLF4, and EPAS1, which have been implicated in gastric tumour growth and epithelial-to-mesenchymal transition (EMT) [117,118]. The suppression of genes like FOSL1 and SYVN1, which support actin stress fibre formation and cellular motility, suggested impaired cytoskeletal dynamics and reduced metastatic capacity [89,119].
The canonical pathway analysis comparing APB+Dox to the monotreatments (APB and Dox individually) highlighted significant alterations in pathways related to cell cycle regulation, apoptosis, rRNA metabolic processes, and elements associated with invasion of tumour pathways (Figure S2). There was marked upregulation of cell cycle checkpoints, DNA double-strand break response, and mitotic progression pathways, suggesting a strong activation of mechanisms that halted the cell cycle in response to genotoxic stress. This aligns with the mode of action of Dox, which induces DNA damage, and indicates that APB+Dox may enhance this effect to inhibit cancer cell proliferation [120]. Pathways associated with apoptosis and cellular stress, including oxidative stress-induced senescence and the senescence-associated secretory phenotype, were also upregulated, pointing to an increased likelihood of cancer cells undergoing programmed cell death or permanent arrest. Interestingly, rRNA-related processes such as RNA polymerase I transcription and regulation of rRNA expression were activated, possibly reflecting a cellular response to stress or compensatory mechanisms to maintain protein synthesis under damage conditions.
Invasion of tumour pathways:
The APB+Dox combination exerted potent anticancer effects in AGS gastric cancer cells by disrupting multiple pathways involved in tumour invasion, angiogenesis, and epithelial-to-mesenchymal transition (EMT). As illustrated by the network analysis, this treatment resulted in the coordinated downregulation of key genes associated with cell adhesion (e.g., CD44 (–0.96), CD151 (–1.03), ALCAM (–1.43), CLDN4 (0.99), integrins (ITGA2 (–0.60), ITGA6 (–0.65), ITGB1(–0.59)), and extracellular matrix (ECM) remodelling proteins such as MMP14 (–1.07), CTSV (–1.18), and MDK (–2.24) (Figure 5). These changes suggest a marked impairment of tumour–stromal interactions and metastatic capacity. A particularly notable target, MDK (Midkine), a growth factor critical for angiogenesis and vascular remodelling, was significantly downregulated (Log2FC = –2.24), indicating suppressed tumour-supporting vasculature. Similarly, TACSTD2, a gene linked to EMT and cancer progression, was strongly suppressed (Log2FC = –2.49), likely contributing to reduced metastatic potential. Other downregulated factors such as ATP1B1 (Log2FC = –0.74), EPCAM (Log2FC = –1.72), and CD44 (Log2FC = –0.96) further suggest disrupted cellular homeostasis, loss of cancer stemness, and inhibition of tumour recurrence (Figure 5).
Conversely, the upregulation of tumour suppressors like TP53 (1.16) and RBM38 (1.02), and stress regulators such as CLU (1.28), supports a shift towards apoptosis and reduced invasiveness. TGFBR1 (–0.59) although typically involved in growth factor signalling, may under this treatment context exert anti-invasive effects. The network predominantly features predicted inhibitory interactions (blue arrows), reinforcing the hypothesis that APB+Dox disrupts invasion-related pathways through suppression of adhesion, ECM degradation, and cell signalling mechanisms. SNAI1 (Log2FC = 1.71), a key EMT transcription factor, and SERPINE1 (Log2FC = 3.14), a regulator of extracellular matrix remodelling and cell migration, were upregulated. This might suggest a partial or compensatory EMT response despite the broader anti-invasive effects of the treatment (Figure 5). Similarly, GDF15 (Log2FC = 0.91), typically linked to growth factor signalling, and CLDN4 (Log2FC = –0.99), involved in tight junction integrity, displayed expression patterns inconsistent with the predicted inhibition of tumour invasion (Figure 5). These signals may reflect compensatory effect or stress adaptation mechanisms and highlight the complexity of interpreting network-level responses, where isolated pro-invasive signals may persist within an overall suppressive theme.
Apoptosis
The APB+Dox combination exerted a potent anticancer effect on AGS adenocarcinoma cells primarily through apoptosis modulation. Apoptosis is tightly regulated through both intrinsic (mitochondrial-mediated) and extrinsic (death receptor-mediated) pathways [121]. In AGS cells treated with the APB+Dox combination, the expression of pro-apoptotic and anti-apoptotic factors was significantly altered, contributing to the observed therapeutic effects (Figure S4).
Intrinsic Pathway of Apoptosis
The intrinsic apoptotic pathway involves mitochondrial outer membrane permeabilization (MOMP), leading to cytochrome c release and caspase activation [122]. BID (Log2FC = -0.75), a BH3-only protein that activates BAX and BAK, showed a slight downregulation [122]. Despite this, the pro-apoptotic BCL2L11 (BIM, Log2FC = 1.10) was upregulated, tipping the balance in favour of apoptosis by antagonizing anti-apoptotic proteins like BCL-2 and BCL-XL [123]. Concurrently, CASP9 (Log2FC = -0.69), essential for apoptosome formation, was modestly downregulated, potentially modulating the extent of apoptosis [124]. Several genes influencing redox balance and mitochondrial health, such as ERO1B (Log2FC = -2.76) and ENDOG (Log2FC = -0.91), were downregulated. ERO1B's decrease may amplify ER stress-mediated apoptosis, while ENDOG's reduction might partially offset DNA fragmentation effects [125,126]. Additionally, ERN1 (Log2FC = 1.65), associated with the unfolded protein response (UPR), was upregulated, indicating enhanced ER stress that can synergize with mitochondrial apoptosis [127].
Extrinsic Pathway of Apoptosis: The extrinsic pathway is impacted through the modulation of death receptor signalling [122]. TNFRSF10B (TRAIL receptor 2, Log2FC = -1.19) and TRADD (Log2FC = -0.67) were downregulated upon APB+Dox treatment which could dampen direct TRAIL-mediated apoptosis [128,129]. However, TRAIL expression and its downstream signalling may still induce apoptosis in susceptible cells [128]. CASP10 (Log2FC = -0.68), a caspase involved in extrinsic pathway signalling, showed slight downregulation, but this effect may not fully abrogate extrinsic apoptosis.
Crosstalk Between Pathways: The APB+Dox combination facilitated crosstalk between the intrinsic and extrinsic pathways. JUN (Log2FC = 0.60), part of the AP-1 transcription complex, was upregulated, potentially driving pro-apoptotic gene expression [130]. Similarly, TP53 (Log2FC = 1.16), the "guardian of the genome," was upregulated, enhancing both intrinsic and extrinsic apoptosis by potentially inducing genes like BAX, PUMA, and NOXA [131]. TP53 upregulation was also observed in the Dox-only treatment (Log2FC = 2.94), where it contributed to cell cycle arrest via activation of the CDK inhibitor p21 (CDKN1A), which inhibits CDK4/6 and halts progression through the G1/S checkpoint. In APB+Dox group, TP53 activation may also counteract the survival-promoting influence of anti-apoptotic genes such as RAF1 (Log2FC = 0.89) [132]. TP53 acts as a central regulator of cellular fate, integrating multiple stress responses to maintain genomic integrity. As shown in Figure S4, TP53 promotes apoptosis through activation of pro-apoptotic genes such as BAX, PMAIP1 (PUMA), BBC3, and CASP6, while simultaneously inhibiting cell survival by repressing anti-apoptotic factors including BCL2, BCL2L1, and BIRC5 [133,134]. TP53 also induces autophagy via DRAM1 and promotes senescence through SERPINE2, reinforcing its role in halting the proliferation of damaged cells [135,136]. Moreover, TP53 enhanced mitochondrial respiration by activating SCO2 (Log2FC = 0.74), which supports oxidative phosphorylation, while concurrently suppressing glycolysis through upregulation of TIGAR, thereby shifting metabolism away from anaerobic energy production [137,138].
rRNA metabolic process
rRNA Biogenesis and Ribosomal Genes: Ribosome biogenesis is a hallmark of rapidly dividing cancer cells. Key genes involved in rRNA synthesis and ribosome function, including RPL14 (Log2FC = 0.59), UBA52 (Log2FC = 0.60), and RRN3 (Log2FC = 0.76), were upregulated in response to APB+Dox treatment. [139,140,141]. UBA52 has been reported to simultaneously deliver both RPL40 and ubiquitin to the ribosome [140]. These genes play a critical role in driving ribosome assembly and protein synthesis, processes essential for tumour progression. Elevated expression of RPL14 and UBA52 has been linked to increased protein synthesis, metabolic adaptation, and enhanced resistance to cellular stress. Notably, higher levels of RPL14 have also been shown to suppress cell migration and invasion in nasopharyngeal carcinoma [139]. Conversely, RIOK2 (Log2FC = -0.95), a kinase promoting ribosome assembly, was downregulated, potentially disrupting ribosome biogenesis [64]. This downregulation may inhibit tumour growth by reducing ribosomal output and impairing the protein synthesis machinery that supports cancer cell proliferation [64].
RNA Processing and Splicing: The APB+Dox combination influences genes involved in RNA processing and splicing, key processes in post-transcriptional gene regulation. Genes such as SMN2 (Log2FC = 0.73) and ISY1 (Log2FC = 0.69), involved in RNA splicing, were upregulated, suggesting an adaptive response to therapy [142,143]. Increased RNA splicing activity could represent an effort by cancer cells to maintain proper gene expression under therapeutic stress. In contrast, DCAF13 (Log2FC = -1.77), which regulates ubiquitination and degradation of proteins, was significantly downregulated [144]. DCAF13, a substrate receptor in the CUL4-DDB1 E3 ligase, is known for promoting cancer cell migration, invasion, and epithelial–mesenchymal transition [145]. Interestingly, while Dox treatment has been reported to upregulate DCAF13, potentially enhancing metastatic risk, SCFAs may counteract this effect by downregulating DCAF13 (-1.77 for APB+Dox treatment), offering a therapeutic advantage in reducing metastasis during chemotherapy [145]. The APB+Dox combination exerted its anticancer effects by targeting vulnerabilities in the rRNA metabolic process. While some ribosomal and RNA processing genes are upregulated, potentially as a compensatory response, the downregulation of critical genes like RIOK2 and DCAF13 potentially disrupted ribosome biogenesis and protein degradation pathways. This dual modulation leads to metabolic and nucleolar stress, impairing the cancer cell's ability to sustain rapid growth and survival.
Cell Cycle
The APB+Dox combination substantially impacted the cell cycle in AGS gastric cancer cells, disrupting essential processes related to mitosis, chromatin organization, and DNA repair. These effects collectively impair normal cell division, contributing to the anticancer efficacy of the treatment. In the current study, APB+Dox treatment led to marked upregulation of TP53 (log2FC = 1.16) and several key checkpoint regulators, including CHEK2 (log2FC = 0.86), CDC20 (log2FC = 1.24), and CDCA8 (log2FC = 0.74) [133]. It is noteworthy that the Dox treatment resulted in upregulation of TP53 (Log2FC = 2.94) Interestingly, this was accompanied by the downregulation of negative mitotic regulators such as PKMYT1 (log2FC = –0.65) and UBE2N (log2FC = –1.02), suggesting a coordinated shift toward checkpoint activation rather than evasion. Notably, overexpression of PKMYT1 and UBE2N has been associated with poor prognosis in breast and prostate cancers, respectively, supporting their role in tumour progression [146,147]. Additionally, the suppression of UBE2N, UBE2C, and PSMB10 (log2FC = –1.02 to –0.72)—key components of the ubiquitin–proteasome system—indicated inhibition of proteasomal degradation, which may stabilized pro-apoptotic proteins and enhance cell death [146,148,149]. Other mitotic regulators such as SGO2 and PKMYT1, which are crucial for chromosome cohesion and CDK1 inhibition, were also downregulated (log2FC = –0.89 and –0.65, respectively), suggesting further interference with proper mitotic progression [150]. A notable target is KLHL21 (Kelch-like protein 21), which was significantly upregulated (log2FC = 2.06) [151]. KLHL21 is essential for mitotic progression and cytokinesis, ensuring accurate chromosome segregation during cell division. Its upregulation under APB+Dox treatment may counteract KLHL21 loss observed in gastric cancer, which is known to enhance STAT3 activation and promote tumour progression [152]. Restoring KLHL21 expression could improve chemotherapy response and inhibit the transition from metaplasia to dysplasia, offering a novel therapeutic strategy for gastric cancer management [152].
Furthermore, histone cluster genes, including H2BC9, showed significant upregulation (log2FC = 2.51), indicating changes in nucleosome assembly and chromatin structure [153]. These histones play critical roles in DNA replication, repair, and epigenetic regulation [154]. Their altered expression suggested chromatin remodelling in response to APB+Dox, which may contribute to replication stress, cell cycle arrest, or apoptosis during the S phase. In contrast, SLX9, an RNA processing factor involved in ribosome biogenesis, was strongly downregulated (log2FC = –2.51) [155,156]. As ribosome synthesis is tightly linked to cell growth and the G1/S transition, SLX9 suppression likely impaired protein synthesis, leading to growth arrest and enhanced cytotoxicity [155,156]. Finally, several genes involved in DNA repair and genome stability, including WRN (log2FC = 1.84), RRM2 (log2FC = 1.00), and XRCC3 (log2FC = 0.72), were upregulated, reflecting a cellular attempt to mitigate DNA damage induced by treatment [157,158,159]. However, despite these adaptive responses, the extent of DNA damage and stress caused by APB+Dox may exceed the repair capacity, tipping the balance toward apoptosis and tumour regression.
2.4. Flow Cytometric Analyses of Apoptotic Profiles of Mono and Combination Therapies
Recognized as a fundamental barrier to cancer progression, apoptosis, a form of programmed cell death, eliminates potentially malignant cells from the body, making it a central target in the development of many modern anticancer therapies [122]. In this study, flow cytometry was used to distinguish between apoptotic and necrotic cell populations through the application of Annexin V and 7-AAD. Annexin V specifically detects phosphatidylserine, a phospholipid that becomes exposed on the outer leaflet of the plasma membrane during early apoptosis. In contrast, 7-AAD binds strongly to guanine–cytosine-rich regions of double-stranded DNA, allowing for the identification of necrotic cells. The results are classified into four groups: live cells, early-stage apoptotic cells, late-stage apoptotic cells, and necrotic cells. The effects of the mono treatments APB, and Dox along with the combination APB+Dox were investigated using flow cytometric analysis, compared to the negative control (Figure 6).
APB (3000 μg/mL) treatment produced 71.91% late apoptotic cells and 11.81% early apoptotic cells (P < 0.0001), with low necrosis (3.32%) and 12.96% live cells (P < 0.01). Dox (0.54 μg/mL) treatment resulted in 57.40% late apoptotic cells, 3.95% early apoptotic cells (P < 0.0001), and a higher necrotic fraction (38.49%), with only 0.16% live cells. The APB+Dox combination (3000 μg/mL + 0.27 μg/mL) yielded 64.56% late apoptotic cells and 30.27% early apoptotic cells (P < 0.0001), with 4.64% necrotic cells and 0.53% live cells. The necrotic proportion in the combination group was notably lower than in the Dox-only group (38.49%) (Figure 6)
2.5. ROS Production in the AGS Cells After Treatment with Different Concentrations of APB, Dox and APB+Dox:
ROS play a dual role in cancer biology, where their excessive accumulation, commonly referred to as oxidative stress, can contribute to both tumour initiation and progression [160]. Interestingly, manipulating ROS levels within cancer cells, either by promoting their accumulation to toxic levels or by disrupting the redox balance, has emerged as a promising approach in the development of anticancer therapies [160]. TBHP, used as a positive control for ROS induction, produced a significant increase in ROS levels (11.94-fold) compared to the untreated control (Figure 7). At 3000 μg/mL, APB alone induced a 6.14-fold increase in ROS relative to control. The combination of APB+Dox at the same concentration (3000 μg/mL + 0.27 μg/mL) resulted in a similar ROS increase (6.17-fold), indicating that Dox did not markedly alter APB’s pro-oxidative effect at this dose. APB combined with Dox at lower concentrations produced ROS levels of 2.99-fold. In contrast, Dox alone at comparable concentrations yielded lower ROS increases (0.99–1.04-fold).
3. Materials and Methods
3.1. Chemicals and Drug Preparation:
All solvents used in this study were of analytical grade and procured from Sigma Aldrich (Castle Hill, NSW, Australia). Magnesium acetate (A), sodium propionate (P), sodium butyrate (B), and Dox were also obtained from Sigma Aldrich (Castle Hill, NSW, Australia). Additionally, all reagents were prepared following the standard procedures and protocols specified in the assay kits.
3.2. Cell Culture:
The AGS gastric adenocarcinoma (CRL-1739, ATCC) and HS738.St/Int stomach intestinal (CRL-7869, ATCC) cell lines were sourced from the American Type Culture Collection (ATCC, Virgina, USA). AGS cells were maintained in ATCC-formulated F-12K medium (Kaighn's Modification of Ham's F-12), supplemented with 2 mM L-glutamine, 1500 mg/L sodium bicarbonate, 10% foetal bovine serum (Bio-Strategy PTY, Campbellfield, VIC, Australia), and 1% penicillin-streptomycin (Sigma-Aldrich, Castle Hill, NSW, Australia). HS738.St/Int cells were cultured in ATCC-formulated DMEM (Dulbecco's Modified Eagle Medium) containing 4.5 g/L glucose, L-glutamine, sodium pyruvate, 10% foetal bovine serum, and 1% penicillin-streptomycin. Both cell lines were incubated at 37 °C in a humidified atmosphere of 5% CO2, with maintenance performed every 48–72 h to sustain confluent monolayers.
3.3. Cell Viability Assays:
The cell viability of AGS cells after exposure to varying concentrations of SCFAs combinations (APB), Dox and the combined treatment (APB+Dox) was evaluated using the Alamar Blue assay, following previously established protocols [161]. Briefly, 100 µL of AGS cells were seeded at a density of 10⁵ cells/mL into 96-well plates and allowed to adhere for 24 h. The cells were then treated with the test samples and incubated for 72 h. Dox at a concentration of 1 µM was also used as a positive control, while 0.1% DMSO served as a negative control across all plates. After the incubation period, the culture medium was removed, and 100 µL of a freshly prepared 0.1 mg/mL Alamar Blue solution was added to each well. The Alamar Blue solution was made by diluting a 1 mg/mL resazurin stock in PBS at a 1:10 ratio with serum-free media. Fluorescence measurements were obtained using a microplate spectrophotometer (BMG CLARIOstar, Mornington, VIC, Australia) with excitation and emission wavelengths of 555 nm and 595 nm, respectively. Each sample was tested in triplicate, and cell viability in the negative control group was normalized to 100%.
3.4. Synergy:
Dox was combined with the SCFAs mixture APB at a 1:1 ratio to perform combination index (CI) analyses. The potential interactions between Dox and APB were evaluated using the CI model, with calculations conducted via CompuSyn version 2.0 software (Biosoft, CA, USA). The CI calculations were based on the median-effect equation, derived from the mass action law [162]. In this study, the APB+Dox combination was analysed using a six-point dose-response curve within the CI framework.
3.5. Liquid Chromatography-Mass Spectrometry, Label Free Quantification Bottom-Up Proteomics
All reagents were purchased from Thermo Fisher Scientific (Waltham, Massachusetts, USA) unless stated otherwise. Proteomics studies were done following a recently established protocol. AGS gastric adenocarcinoma cells were cultured and treated in T75 flasks with APB, Dox, or their combinations APB+Dox for 24 h. Post-treatment cells were harvested, washed, and lysed in a sodium deoxycholate-based buffer. Protein extraction involved sonication, centrifugation, and acetone precipitation. Protein pellets were reduced, alkylated, quantified, and digested overnight with trypsin. Peptides were desalted, dried, and resuspended in loading buffer before LC-MS analysis. Peptide separation was performed using nano-liquid chromatography (Ultimate 3000 HPLC) with a 75 μm × 45 cm C18 column and a gradient elution protocol. Mass spectrometry was conducted on a Q Exactive HF-X Orbitrap in data-independent acquisition (DIA) mode, capturing full MS scans and sequential fragmentation across m/z windows. Raw data were processed using Spectronaut (v19) with the directDIA+ workflow against the 2023 UniProt human database. Parameters included trypsin digestion, dynamic modifications (oxidation, N-terminal acetylation), and fixed carbamidomethylation. False discovery rates for peptide-spectrum matches, peptides, and protein groups were set at 1%. Quantification was based on MS1 peak area, and differential abundance was assessed using unpaired t-tests. Data were deposited in PRIDE (PXD061824).
3.6. Flow Cytometry
The effects of APB, Dox, and APB+Dox on the apoptosis profiles of AGS adenocarcinoma cells were evaluated using an annexin V and 7-AAD-based apoptosis detection kit (#ab214663, Abcam, Melbourne, VIC, Australia) following established protocols [33,162]. Briefly, AGS gastric cancer cells (1 × 10⁶ cells/10 mL) were seeded in T75 flasks and incubated for 24 hat 37 °C with 5% CO₂. Cells were then treated with APB (3000 µg/mL), doxorubicin (Dox, 0.54 µg/mL), their combination (APB+Dox), and just growth media (untreated control). After 24 h of treatment, apoptosis was assessed using the Annexin V-CF Blue/7-AAD staining kit (Abcam, #ab214663), following the manufacturer’s instructions. Cells were harvested, stained, and analysed via flow cytometry (Novocyte 3000, ACEA Biosciences). Data were gated to exclude debris and identify live, early apoptotic, late apoptotic, and necrotic cells based on Annexin V and 7-AAD fluorescence. Results from three replicates per group were analysed using NovoExpress (v1.5.0).
3.7. ROS Production Analysis:
The impact of Dox, APB and their combination (APB+Dox) on oxidative stress in AGS gastric adenocarcinoma cells was assessed using the H2DCFDA (2′,7′-dichlorofluorescein diacetate) Cellular Reactive Oxygen Species (ROS) Detection Assay Kit (#ab113851; Abcam, Melbourne, VIC, Australia) following a recently established protocol [163]. Briefly, AGS cells were seeded at a density of 2.5 × 10⁵ cells/mL in a 96-well plate and allowed to adhere overnight. To measure ROS levels, the cells were treated with 20 μM H2DCFDA for 45 min. After removing the dye solution, the cells were washed with 1x buffer and then exposed to 750, 1500, and 3000 µg/mL of SCFAs combination APB; 0.54 µg/mL (1 μM) of Dox, APB+Dox (1:1) and 250 μM of tert-butyl hydroperoxide (TBHP; positive control). The cells were incubated at 37 °C for 4 h and fluorescence was measured using a microplate spectrophotometer (BMG CLARIOstar, Mornington, VIC, Australia) at an excitation/emission wavelength of 485/535 nm. The fold change in ROS production was calculated relative to the untreated control, where cells were treated with buffer according to the manufacturer’s instructions.
3.8. Statistical Analysis:
Data collection and analysis were carried out using Microsoft Excel (MS Office) for data management and GraphPad Prism (version 9.0, San Diego, CA, USA) for statistical analysis and visualization. All experiments were conducted in triplicate, with results expressed as the mean ± standard deviation (SD). Statistical significance between mean values was evaluated using a two-way ANOVA, with a significance threshold set at p < 0.05. The IC50 values, representing the concentration of a drug required to inhibit cell growth by 50%, were calculated using GraphPad Prism 9.0. Nonlinear regression analysis was also performed in the same software for IC50 determination. The designation n = 3 corresponds to the number of independent biological replicates included in the analyses.
4. Conclusions
The current study shows that dietary SCFAs, specifically acetate, propionate, and butyrate (APB), can work synergistically with Dox to enhance anticancer activity in AGS gastric cancer cells. Our findings demonstrate that the APB+Dox combination significantly enhances anti-proliferative effects compared to either agent alone, as evidenced by a substantial reduction in IC50 values and a strong synergistic interaction confirmed by Combination Index analysis. Mechanistically, this synergy appears to be mediated through multiple pathways. The APB+Dox combination exerted a synergistic anticancer effect on AGS gastric cancer cells by modulating key pathways involved in cell survival, apoptosis, cell cycle regulation, immune evasion, and tumour microenvironment remodelling.
The proteomic analyses revealed that the combination treatment significantly downregulated tumour-promoting genes such as HLA-F, TRIAP1, TMBIM6, and ENTPD8, suggesting reduced mitochondrial integrity and immune escape, thereby enhancing apoptotic susceptibility. Simultaneously, upregulation of SERPINE1, USP17L4, and components of the TP53 network reflected increased genotoxic stress responses and proteostasis alterations. Network and pathway enrichment analyses highlighted suppression of CD44-centred signalling, epithelial–mesenchymal transition (EMT), and fibrogenesis-related genes, indicating impaired tumour plasticity and reduced metastatic potential. Canonical pathway analysis revealed pronounced activation of DNA damage checkpoints, oxidative stress responses, and cell cycle arrest, supporting the enhanced cytostatic and pro-apoptotic effects of APB+Dox. Apoptosis induction occurred via both intrinsic and extrinsic pathways, with increased expression of pro-apoptotic regulators (TP53, BCL2L11) and downregulation of survival-promoting genes (MDK, TNFRSF10B, DCAF13), tipping the balance toward programmed cell death. The combination also disrupted ribosome biogenesis and RNA processing, impairing protein synthesis and contributing to metabolic stress. Finally, modulation of haemostasis-related factors such as MDK and TACSTD2 further inhibited angiogenesis and tumour-associated signalling. Together, the study demonstrated that APB enhances Dox efficacy by disrupting AGS cell homeostasis at multiple levels, supporting its potential role as a promising adjuvant strategy in gastric cancer therapy.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org.
Author Contributions
Conceptualisation, R.A.E. and D.J.B.; methodology, R.A.E.; investigation, R.A.E.; formal analysis, R.A.E., M.F. and M.A.; data curation, R.A.E.; writing—original draft preparation, R.A.E. and M.F.; writing—review and editing, R.A.E., M.F., M.A., D.C., C.-G.L. and D.J.B.; supervision, D.J.B., D.C. and C.-G.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. We acknowledge the support of Western Sydney University, Australia through the PhD Research Training Program Scholarship (RE) and the Research Support Program Fellowship (DJB) to conduct this research.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors acknowledge and pay respect to the Traditional Owners—the Darug People (Darug Nation: Western Sydney University Westmead and Penrith Campuses), the Bidigal People (Dharawal Nation: Western Sydney University Campbelltown Campus), and the Gadigal People (Eora Nation: The University of Sydney)—of the land on which we research, teach, and collaborate. The authors would also like to acknowledge the University of Sydney Mass Spectrometry Facility for providing access to its instrumentation and assistance with the MS analyses. The authors also acknowledge the Freedman Foundation Metabolomics Facility at the Innovation Centre, Victor Chang Cardiac Research Institute, which is supported by funding from the NSW Government.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sung, H., et al., Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians, 2021. 71(3): p. 209-249.
- Bray, F., et al., Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians, 2018. 68(6): p. 394-424.
- jp, J.G.C.A.j.k.k.-m.a., Japanese gastric cancer treatment guidelines 2018. Gastric cancer, 2021. 24(1): p. 1-21.
- Bang, Y.-J., et al., Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. The Lancet, 2010. 376(9742): p. 687-697. [CrossRef]
- Xu, J., et al., Resveratrol reverses Doxorubicin resistance by inhibiting epithelial-mesenchymal transition (EMT) through modulating PTEN/Akt signaling pathway in gastric cancer. Journal of Experimental & Clinical Cancer Research, 2017. 36: p. 1-14.
- Canani, R.B., et al., Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World journal of gastroenterology: WJG, 2011. 17(12): p. 1519.
- Tacar, O., P. Sriamornsak, and C.R. Dass, Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. Journal of pharmacy and pharmacology, 2013. 65(2): p. 157-170.
- Yonemura, Y., Mechanisms of drug resistance in gastric cancer. Contemporary Approaches Toward Cure of Gastric Cancer. Kanazawa: Maeda Shoten Co. Ltd, 1996: p. 87-91.
- Alkuraishy, H.M., A.I. Al-Gareeb, and H.A. Al-Hussaniy, Doxorubicin-induced cardiotoxicity: molecular mechanism and protection by conventional drugs and natural products. Int J Clin Oncol Cancer Res, 2017. 2(2): p. 31-44.
- Gianni, L., et al., Anthracycline cardiotoxicity: from bench to bedside. Journal of Clinical Oncology, 2008. 26(22): p. 3777-3784. [CrossRef]
- Wang, X., H. Zhang, and X. Chen, Drug resistance and combating drug resistance in cancer. Cancer drug resistance, 2019. 2(2): p. 141.
- Severino, A., et al., The microbiome-driven impact of western diet in the development of noncommunicable chronic disorders. Best Practice & Research Clinical Gastroenterology, 2024: p. 101923. [CrossRef]
- Singh, R.K., et al., Influence of diet on the gut microbiome and implications for human health. Journal of translational medicine, 2017. 15: p. 1-17.
- Deleu, S., et al., Short chain fatty acids and its producing organisms: An overlooked therapy for IBD? EBioMedicine, 2021. 66.
- Fan, Y. and O. Pedersen, Gut microbiota in human metabolic health and disease. Nat Rev Microbiol, 2021. 19(1): p. 55-71.
- Silva, Y.P., A. Bernardi, and R.L. Frozza, The role of short-chain fatty acids from gut microbiota in gut-brain communication. Frontiers in endocrinology, 2020. 11: p. 25. [CrossRef]
- Jaye, K., et al., The role of key gut microbial metabolites in the development and treatment of cancer. Gut Microbes, 2022. 14(1): p. 2038865.
- O'keefe, S.J., Diet, microorganisms and their metabolites, and colon cancer. Nature reviews Gastroenterology & hepatology, 2016. 13(12): p. 691-706.
- Facchin, S., et al., Short-Chain Fatty Acids and Human Health: From Metabolic Pathways to Current Therapeutic Implications. Life, 2024. 14(5): p. 559. [CrossRef]
- Den Besten, G., et al., The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. Journal of lipid research, 2013. 54(9): p. 2325-2340.
- Sun, J., et al., Butyrate as a promising therapeutic target in cancer: From pathogenesis to clinic. International journal of oncology, 2024. 64(4): p. 44. [CrossRef]
- Donohoe, D.R., et al., A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota-and butyrate-dependent manner. Cancer discovery, 2014. 4(12): p. 1387-1397.
- Filippone, A., et al., Sodium propionate contributes to tumor cell growth inhibition through PPAR-γ signaling. Cancers, 2022. 15(1): p. 217. [CrossRef]
- Mirzaei, R., et al., Role of microbiota-derived short-chain fatty acids in cancer development and prevention. Biomedicine & pharmacotherapy, 2021. 139: p. 111619. [CrossRef]
- Chen, M., et al., Sodium butyrate combined with docetaxel for the treatment of lung adenocarcinoma A549 cells by targeting Gli1. OncoTargets and therapy, 2020: p. 8861-8875.
- Li, Y., et al., Combining sodium butyrate with cisplatin increases the apoptosis of gastric cancer in vivo and in vitro via the mitochondrial apoptosis pathway. Frontiers in Pharmacology, 2021. 12: p. 708093. [CrossRef]
- Geng, H.-W., et al., Butyrate suppresses glucose metabolism of colorectal cancer cells via GPR109a-AKT signaling pathway and enhances chemotherapy. Frontiers in molecular biosciences, 2021. 8: p. 634874.
- Cristiano, C., et al., Oral sodium butyrate supplementation ameliorates paclitaxel-induced behavioral and intestinal dysfunction. Biomedicine & Pharmacotherapy, 2022. 153: p. 113528. [CrossRef]
- Maruyama, T., et al., Apoptosis of bladder cancer by sodium butyrate and cisplatin. Journal of Infection and Chemotherapy, 2012. 18(3): p. 288-295.
- Kobayashi, M., et al., A short-chain fatty acid, propionate, enhances the cytotoxic effect of cisplatin by modulating GPR41 signaling pathways in HepG2 cells. Oncotarget, 2018. 9(59): p. 31342.
- Eladwy, R.A., et al., The postbiotic sodium butyrate synergizes the antiproliferative effects of dexamethasone against the AGS gastric adenocarcinoma cells. Frontiers in Nutrition, 2024. 11: p. 1372982.
- Eladwy, R.A., et al., Fuelling the Fight from the Gut: Short-Chain Fatty Acids and Dexamethasone Synergise to Suppress Gastric Cancer Cells. Cancers, 2025. 17(15): p. 2486. [CrossRef]
- Jaye, K., et al., Mechanistic insights into the anti-proliferative action of gut microbial metabolites against breast adenocarcinoma cells. International Journal of Molecular Sciences, 2023. 24(20): p. 15053.
- Semaan, J., et al., Comparative effect of sodium butyrate and sodium propionate on proliferation, cell cycle and apoptosis in human breast cancer cells MCF-7. Breast Cancer, 2020. 27(4): p. 696-705.
- Alsherbiny, M.A., et al., Trustworthy deep neural network for inferring anticancer synergistic combinations. IEEE Journal of Biomedical and Health Informatics, 2021. 27(4): p. 1691-1700.
- Thorn, C.F., et al., Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenetics and genomics, 2011. 21(7): p. 440-446.
- Ozaki, T. and A. Nakagawara, Role of p53 in cell death and human cancers. Cancers, 2011. 3(1): p. 994-1013.
- Finsterwald, C. and C.M. Alberini, Stress and glucocorticoid receptor-dependent mechanisms in long-term memory: from adaptive responses to psychopathologies. Neurobiology of learning and memory, 2014. 112: p. 17-29. [CrossRef]
- Ren, Q., et al., C/EBPβ: the structure, regulation, and its roles in inflammation-related diseases. Biomedicine & Pharmacotherapy, 2023. 169: p. 115938. [CrossRef]
- Martí i Líndez, A.-A. and W. Reith, Arginine-dependent immune responses. Cellular and Molecular Life Sciences, 2021. 78(13): p. 5303-5324.
- Li, L.Y., X. Luo, and X. Wang, Endonuclease G is an apoptotic DNase when released from mitochondria. Nature, 2001. 412(6842): p. 95-99.
- Farooq, M., et al., RRP7A links primary microcephaly to dysfunction of ribosome biogenesis, resorption of primary cilia, and neurogenesis. Nature communications, 2020. 11(1): p. 5816. [CrossRef]
- Calo, E., et al., RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature, 2015. 518(7538): p. 249-253.
- Ray Chaudhuri, A. and A. Nussenzweig, The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nature reviews Molecular cell biology, 2017. 18(10): p. 610-621.
- Matsumura, I., H. Tanaka, and Y. Kanakura, E2F1 and c-Myc in cell growth and death. Cell cycle, 2003. 2(4): p. 332-335. [CrossRef]
- Li, S., et al., Oxidative Stress and Reprogramming of Lipid Metabolism in Cancers. Antioxidants, 2025. 14(2): p. 201. [CrossRef]
- Lathia, J.D. and H. Liu, Overview of cancer stem cells and stemness for community oncologists. Targeted oncology, 2017. 12: p. 387-399.
- Zhang, J., et al., Downregulated RPL6 inhibits lung cancer cell proliferation and migration and promotes cell apoptosis by regulating the AKT signaling pathway. Journal of thoracic disease, 2022. 14(2): p. 507.
- Zhou, C., et al., High RPS3A expression correlates with low tumor immune cell infiltration and unfavorable prognosis in hepatocellular carcinoma patients. American journal of cancer research, 2020. 10(9): p. 2768.
- Li, D., et al., High-fat diet promotes prostate cancer metastasis via RPS27. Cancer & Metabolism, 2024. 12(1): p. 6. [CrossRef]
- Xing, H., et al., High expression of RPL27A predicts poor prognosis in patients with hepatocellular carcinoma. World Journal of Surgical Oncology, 2023. 21(1): p. 209.
- Molavi, G., et al., Moonlight human ribosomal protein L13a downregulation is associated with p53 and HER2/neu expression in breast cancer. Journal of Applied Biomedicine, 2020. 18.
- He, K.J., et al., Pan-cancer analysis of 60S Ribosomal Protein L7-Like 1 (RPL7L1) and validation in liver hepatocellular carcinoma. Translational Oncology, 2024. 40: p. 101844.
- Wu, Q., et al., Downregulation of RPL6 by siRNA inhibits proliferation and cell cycle progression of human gastric cancer cell lines. PloS one, 2011. 6(10): p. e26401. [CrossRef]
- Guo, X., et al., Human ribosomal protein S13 promotes gastric cancer growth through down-regulating p27Kip1. Journal of cellular and molecular medicine, 2011. 15(2): p. 296-306.
- Dave, B., et al., Role of RPL39 in metaplastic breast cancer. JNCI: Journal of the National Cancer Institute, 2017. 109(6): p. djw292. [CrossRef]
- Mao-De, L. and X. Jing, Ribosomal proteins and colorectal cancer. Current genomics, 2007. 8(1): p. 43-49.
- Shi, Y., et al., Ribosomal proteins S13 and L23 promote multidrug resistance in gastric cancer cells by suppressing drug-induced apoptosis. Experimental cell research, 2004. 296(2): p. 337-346.
- Gambardella, V., et al., NRF2 through RPS6 activation is related to anti-HER2 drug resistance in HER2-amplified gastric cancer. Clinical Cancer Research, 2019. 25(5): p. 1639-1649.
- Gambardella, V., et al., NRF2 activation via PI3K/AKT/mTOR/RPS6 causes resistance to anti-HER2 agents among HER2 amplified gastric cancer. Annals of Oncology, 2018. 29: p. viii224. [CrossRef]
- Qin, Y., et al., Expression and bioinformatics analysis of RPL38 protein and mRNA in gastric cancer. Cellular and Molecular Biology, 2023. 69(13): p. 256-261.
- Huang, X.-P., et al., Alteration of RPL14 in squamous cell carcinomas and preneoplastic lesions of the esophagus. Gene, 2006. 366(1): p. 161-168.
- Sato, H. and R.H. Singer, Cellular variability of nonsense-mediated mRNA decay. Nature communications, 2021. 12(1): p. 7203.
- Matsuzaki, Y., et al., RIOK2 contributes to cell growth and protein synthesis in human oral squamous cell carcinoma. Current Oncology, 2022. 30(1): p. 381-391. [CrossRef]
- Black, J.J., et al., Utp14 interaction with the small subunit processome. RNA, 2018. 24(9): p. 1214-1228.
- Li, K.K., et al., Overexpression of U three protein 14a (UTP14a) is associated with poor prognosis of esophageal squamous cell carcinoma. Thoracic Cancer, 2019. 10(11): p. 2071-2080.
- Zhang, Y., et al., Hypermethylation and downregulation of UTP6 are associated with stemness properties, chemoradiotherapy resistance, and prognosis in rectal cancer: a co-expression network analysis. Frontiers in cell and developmental biology, 2021. 9: p. 607782.
- Wang, J., et al., DEAD-box helicase 56 functions as an oncogene promote cell proliferation and invasion in gastric cancer via the FOXO1/p21 Cip1/c-Myc signaling pathway. Bioengineered, 2022. 13(5): p. 13970-13985.
- Hong, H., et al., RNA-seq and integrated network analysis reveals the hub genes and key pathway of paclitaxel inhibition on Adriamycin resistant diffuse large B cell lymphoma cells. Bioengineered, 2022. 13(3): p. 7607-7621. [CrossRef]
- Zhang, H., et al., NSA2, a novel nucleolus protein regulates cell proliferation and cell cycle. Biochemical and biophysical research communications, 2010. 391(1): p. 651-658.
- Hirai, Y., et al., Nucleolar scaffold protein, WDR 46, determines the granular compartmental localization of nucleolin and DDX 21. Genes to Cells, 2013. 18(9): p. 780-797.
- Paternoga, H., et al., Mutational analysis of the Nsa2 N-terminus reveals its essential role in ribosomal 60S subunit assembly. International Journal of Molecular Sciences, 2020. 21(23): p. 9108. [CrossRef]
- Zaremba, T., et al., Doxorubicin-induced suppression of poly (ADP-ribose) polymerase-1 (PARP-1) activity and expression and its implication for PARP inhibitors in clinical trials. Cancer chemotherapy and pharmacology, 2010. 66: p. 807-812.
- De Luca, P., et al., BRCA1 loss induces GADD153-mediated doxorubicin resistance in prostate cancer. Molecular Cancer Research, 2011. 9(8): p. 1078-1090.
- Zhang, X., et al., RAD51 is a potential marker for prognosis and regulates cell proliferation in pancreatic cancer. Cancer cell international, 2019. 19: p. 1-11.
- Busatto, F.F., et al., Cell growth analysis and nucleotide excision repair modulation in breast cancer cells submitted to a protocol using doxorubicin and paclitaxel. Life Sciences, 2021. 268: p. 118990. [CrossRef]
- Beier, F., et al., Conditional TRF1 knockout in the hematopoietic compartment leads to bone marrow failure and recapitulates clinical features of dyskeratosis congenita. Blood, The Journal of the American Society of Hematology, 2012. 120(15): p. 2990-3000.
- Schneider, R.P., et al., TRF1 is a stem cell marker and is essential for the generation of induced pluripotent stem cells. Nature communications, 2013. 4(1): p. 1946.
- Lugli, N., S.K. Sotiriou, and T.D. Halazonetis, The role of SMARCAL1 in replication fork stability and telomere maintenance. DNA repair, 2017. 56: p. 129-134. [CrossRef]
- Bhattacharya, S., et al., Elevated levels of the methyltransferase SETD2 causes transcription and alternative splicing changes resulting in oncogenic phenotypes. Frontiers in Cell and Developmental Biology, 2022. 10: p. 945668.
- Mar, B.G., et al., SETD2 alterations impair DNA damage recognition and lead to resistance to chemotherapy in leukemia. Blood, The Journal of the American Society of Hematology, 2017. 130(24): p. 2631-2641.
- Lan, Y., W. Ni, and G. Tai, Expression of MUC1 in different tumours and its clinical significance. Molecular and Clinical Oncology, 2022. 17(6): p. 1-10.
- Lee, D.H., I.H. Jeong, and B. Jang, Elevated expression of Axin2 in intestinal metaplasia and gastric cancers. Journal of Pathology and Translational Medicine, 2023. 57(6): p. 315. [CrossRef]
- Lei, Y., et al., Inhibition of MGMT-mediated autophagy suppression decreases cisplatin chemosensitivity in gastric cancer. Biomedicine & Pharmacotherapy, 2020. 125: p. 109896.
- Kim, Y.-I., et al., MUC1 expressions and its prognostic values in US gastric cancer patients. Cancers, 2023. 15(4): p. 998.
- Nepal, M., et al., Fanconi anemia signaling and cancer. Trends in cancer, 2017. 3(12): p. 840-856.
- Panda, M., S.K. Tripathi, and B.K. Biswal, SOX9: An emerging driving factor from cancer progression to drug resistance. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer, 2021. 1875(2): p. 188517. [CrossRef]
- Gooch, J.L., B. Christy, and D. Yee, STAT6 mediates interleukin-4 growth inhibition in human breast cancer cells. Neoplasia, 2002. 4(4): p. 324-331.
- Shi, Y., et al., E3 ubiquitin ligase SYVN1 is a key positive regulator for GSDMD-mediated pyroptosis. Cell death & disease, 2022. 13(2): p. 106.
- Hu, H., L. Ye, and Z. Liu, GINS2 regulates the proliferation and apoptosis of colon cancer cells through PTP4A1. Molecular Medicine Reports, 2022. 25(4): p. 1-9.
- Xu, B., et al., Novel role of ASH1L histone methyltransferase in anaplastic thyroid carcinoma. Journal of Biological Chemistry, 2020. 295(26): p. 8834-8845.
- Nardone, V., et al., CDK4, CDK6/cyclin-D1 complex inhibition and radiotherapy for cancer control: a role for autophagy. International Journal of Molecular Sciences, 2021. 22(16): p. 8391. [CrossRef]
- Fassl, A., Y. Geng, and P. Sicinski, CDK4 and CDK6 kinases: From basic science to cancer therapy. Science, 2022. 375(6577): p. eabc1495.
- Chimploy, K., et al., E2F4 and ribonucleotide reductase mediate S-phase arrest in colon cancer cells treated with chlorophyllin. International journal of cancer, 2009. 125(9): p. 2086-2094. [CrossRef]
- Engeland, K., Cell cycle regulation: p53-p21-RB signaling. Cell Death & Differentiation, 2022. 29(5): p. 946-960.
- Jardim, D.L., et al., Cyclin pathway genomic alterations across 190,247 solid tumors: leveraging large-scale data to inform therapeutic directions. The Oncologist, 2021. 26(1): p. e78-e89.
- Quan, Y., et al., Cell-cycle-regulated interaction between Mcm10 and double hexameric Mcm2-7 is required for helicase splitting and activation during S phase. Cell reports, 2015. 13(11): p. 2576-2586. [CrossRef]
- Malkas, L.H., et al., A cancer-associated PCNA expressed in breast cancer has implications as a potential biomarker. Proceedings of the National Academy of Sciences, 2006. 103(51): p. 19472-19477.
- Bartek, J. and J. Lukas, Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer cell, 2003. 3(5): p. 421-429.
- Mouery, R.D., et al., Proteomic analysis reveals a PLK1-dependent G2/M degradation program and a role for AKAP2 in coordinating the mitotic cytoskeleton. Cell reports, 2024. 43(8).
- Iwai, H., et al., A bacterial effector targets Mad2L2, an APC inhibitor, to modulate host cell cycling. Cell, 2007. 130(4): p. 611-623.
- Malureanu, L.A., et al., BubR1 N terminus acts as a soluble inhibitor of cyclin B degradation by APC/CCdc20 in interphase. Developmental cell, 2009. 16(1): p. 118-131.
- Kang, Y., et al., Expression of anaphase-promoting complex7 in fibroadenomas and phyllodes tumors of breast. Human pathology, 2009. 40(1): p. 98-107.
- Hsu, Y.-H., et al., Overexpression of the pituitary tumor transforming gene induces p53-dependent senescence through activating DNA damage response pathway in normal human fibroblasts. Journal of Biological Chemistry, 2010. 285(29): p. 22630-22638. [CrossRef]
- Marei, H.E., et al., p53 signaling in cancer progression and therapy. Cancer cell international, 2021. 21(1): p. 703.
- Wang, J.-X., et al., Implications of the NDC80 complex on the tumor immune microenvironment and cell growth in pan-cancer. Journal of Cancer, 2024. 15(19): p. 6364. [CrossRef]
- Liu, Z., et al., ESPL1 is a novel prognostic biomarker associated with the malignant features of glioma. Frontiers in Genetics, 2021. 12: p. 666106.
- Lin, A., et al., HLA-F expression is a prognostic factor in patients with non-small-cell lung cancer. Lung Cancer, 2011. 74(3): p. 504-509.
- Yan, J., et al., miR-107 inhibits the proliferation of gastric cancer cells in vivo and in vitro by targeting TRIAP1. Frontiers in Genetics, 2022. 13: p. 855355.
- Robinson, K.S., et al., TMBIM6/BI-1 is an intracellular environmental regulator that induces paraptosis in cancer via ROS and Calcium-activated ERAD II pathways. Oncogene, 2025. 44(8): p. 494-512. [CrossRef]
- Chen, L., Bleomycin alters intratumoral immune response of EBV-associated gastric cancer by ENTPD8 and PCOLCE2. 2023.
- Su, Y.-H., et al., Obesity promotes radioresistance through SERPINE1-mediated aggressiveness and DNA repair of triple-negative breast cancer. Cell Death & Disease, 2023. 14(1): p. 53. [CrossRef]
- de La Vega, M., et al., The deubiquitinating enzyme USP17 is essential for GTPase subcellular localization and cell motility. Nature communications, 2011. 2(1): p. 259.
- Hou, W., et al., CD44 is a prognostic biomarker and correlated with immune infiltrates in gastric cancer. BMC Medical Genomics, 2022. 15(1): p. 225.
- Nishina, H., et al., Cell surface antigen CD38 identified as ecto-enzyme of NAD glycohydrolase has hyaluronate-binding activity. Biochemical and biophysical research communications, 1994. 203(2): p. 1318-1323.
- Wang, K., et al., Fibrogenesis-driven tumor progression in clear cell renal cell carcinoma: prognostic, therapeutic implications and the dual role of neuropilin-1. Cancer Cell International, 2025. 25: p. 179.
- Subbalakshmi, A.R., et al., KLF4 induces mesenchymal–epithelial transition (MET) by suppressing multiple EMT-inducing transcription factors. Cancers, 2021. 13(20): p. 5135.
- Baj, J., et al., Mechanisms of the epithelial–mesenchymal transition and tumor microenvironment in Helicobacter pylori-induced gastric cancer. Cells, 2020. 9(4): p. 1055. [CrossRef]
- Wang, W., et al., CYTOR facilitates formation of FOSL1 phase separation and super enhancers to drive metastasis of tumor budding cells in head and neck squamous cell carcinoma. Advanced Science, 2024. 11(4): p. 2305002.
- Kciuk, M., et al., Doxorubicin—an agent with multiple mechanisms of anticancer activity. Cells, 2023. 12(4): p. 659.
- Jan, R., Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Advanced pharmaceutical bulletin, 2019. 9(2): p. 205. [CrossRef]
- Elmore, S., Apoptosis: a review of programmed cell death. Toxicologic pathology, 2007. 35(4): p. 495-516.
- Dai, Y. and S. Grant, BCL2L11/Bim as a dual-agent regulating autophagy and apoptosis in drug resistance. Autophagy, 2015. 11(2): p. 416-418.
- An, H.-K., et al., CASP9 (caspase 9) is essential for autophagosome maturation through regulation of mitochondrial homeostasis. Autophagy, 2020. 16(9): p. 1598-1617.
- Chen, P., et al., Biological mechanisms and clinical significance of endoplasmic reticulum oxidoreductase 1 alpha (ERO1α) in human cancer. Journal of Experimental & Clinical Cancer Research, 2024. 43(1): p. 71. [CrossRef]
- Choi, Y.N., et al., Nuclear endonuclease G controls cell proliferation in ovarian cancer. FEBS Open bio, 2023. 13(4): p. 655-669.
- Zhang, W., et al., Endoplasmic reticulum stress—a key guardian in cancer. Cell Death Discovery, 2024. 10(1): p. 343.
- Tian, X., et al., Targeting apoptotic pathways for cancer therapy. The Journal of clinical investigation, 2024. 134(14).
- Pobezinskaya, Y.L. and Z. Liu, The role of TRADD in death receptor signaling. Cell Cycle, 2012. 11(5): p. 871-876.
- Bossy-Wetzel, E., L. Bakiri, and M. Yaniv, Induction of apoptosis by the transcription factor c-Jun. The EMBO journal, 1997.
- Wang, H., et al., Targeting p53 pathways: mechanisms, structures, and advances in therapy. Signal transduction and targeted therapy, 2023. 8(1): p. 92.
- Dorard, C., et al., RAF1 contributes to cell proliferation and STAT3 activation in colorectal cancer independently of microsatellite and KRAS status. Oncogene, 2023. 42(20): p. 1649-1660.
- Wang, H., et al., Targeting p53 pathways: Mechanisms, structures and advances in therapy. Signal transduction and targeted therapy, 2023. 8(1): p. 92.
- Nechiporuk, T., et al., The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer discovery, 2019. 9(7): p. 910-925.
- Xie, X., et al., Autophagy is induced through the ROS-TP53-DRAM1 pathway in response to mitochondrial protein synthesis inhibition. Autophagy, 2012. 8(7): p. 1071-1084. [CrossRef]
- Liu, Y., et al., p53 regulates hematopoietic stem cell quiescence. Cell stem cell, 2009. 4(1): p. 37-48.
- Kruse, J.-P. and W. Gu, p53 aerobics: the major tumor suppressor fuels your workout. Cell metabolism, 2006. 4(1): p. 1-3.
- Bensaad, K., et al., TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell, 2006. 126(1): p. 107-120.
- Zhang, Z., et al., Human/eukaryotic ribosomal protein L14 (RPL14/eL14) overexpression represses proliferation, migration, invasion and EMT process in nasopharyngeal carcinoma. Bioengineered, 2021. 12(1): p. 2175-2186.
- Martín-Villanueva, S., et al., Ubiquitin and ubiquitin-like proteins and domains in ribosome production and function: chance or necessity? International Journal of Molecular Sciences, 2021. 22(9): p. 4359.
- Stepanchick, A., et al., DNA binding by the ribosomal DNA transcription factor rrn3 is essential for ribosomal DNA transcription. Journal of Biological Chemistry, 2013. 288(13): p. 9135-9144.
- Lauria, F., et al., SMN-primed ribosomes modulate the translation of transcripts related to spinal muscular atrophy. Nature cell biology, 2020. 22(10): p. 1239-1251.
- Jaiswal, A.S., et al., The splicing component ISY1 regulates APE1 in base excision repair. DNA repair, 2020. 86: p. 102769. [CrossRef]
- Wei, S., et al., DCAF13 inhibits the p53 signaling pathway by promoting p53 ubiquitination modification in lung adenocarcinoma. Journal of Experimental & Clinical Cancer Research, 2024. 43(1): p. 3.
- Sun, Z., et al., Doxorubicin promotes breast cancer cell migration and invasion via DCAF13. FEBS Open Bio, 2022. 12(1): p. 221-230. [CrossRef]
- Yang, B., et al., UBE2N promotes cell viability and glycolysis by promoting Axin1 ubiquitination in prostate cancer cells. Biology Direct, 2024. 19(1): p. 35. [CrossRef]
- Li, H., et al., Overexpression of PKMYT1 associated with poor prognosis and immune infiltration may serve as a target in triple-negative breast cancer. Frontiers in Oncology, 2023. 12: p. 1002186.
- Liu, P.-F., et al., UBE2C is a potential biomarker for tumorigenesis and prognosis in tongue squamous cell carcinoma. Diagnostics, 2020. 10(9): p. 674. [CrossRef]
- Rouette, A., et al., Expression of immunoproteasome genes is regulated by cell-intrinsic and–extrinsic factors in human cancers. Scientific reports, 2016. 6(1): p. 34019.
- Deng, M., et al., High SGO2 expression predicts poor overall survival: a potential therapeutic target for hepatocellular carcinoma. Genes, 2021. 12(6): p. 876. [CrossRef]
- Maerki, S., et al., The Cul3–KLHL21 E3 ubiquitin ligase targets Aurora B to midzone microtubules in anaphase and is required for cytokinesis. Journal of Cell Biology, 2009. 187(6): p. 791-800.
- Huang, X.-B., et al., KLHL21 suppresses gastric tumourigenesis via maintaining STAT3 signalling equilibrium in stomach homoeostasis. Gut, 2024. 73(11): p. 1785-1798.
- Golebiowski, F. and K.S. Kasprzak, Inhibition of core histones acetylation by carcinogenic nickel (II). Molecular and cellular biochemistry, 2005. 279: p. 133-139. [CrossRef]
- Loscalzo, J. and D.E. Handy, Epigenetic modifications: basic mechanisms and role in cardiovascular disease (2013 Grover Conference series). Pulmonary circulation, 2014. 4(2): p. 169-174.
- Chaillou, T., T.J. Kirby, and J.J. McCarthy, Ribosome biogenesis: emerging evidence for a central role in the regulation of skeletal muscle mass. Journal of cellular physiology, 2014. 229(11): p. 1584-1594.
- Hwang, S.-P. and C. Denicourt, The impact of ribosome biogenesis in cancer: from proliferation to metastasis. NAR cancer, 2024. 6(2): p. zcae017. [CrossRef]
- Cheng, J., et al., XRCC 3 is a promising target to improve the radiotherapy effect of esophageal squamous cell carcinoma. Cancer science, 2015. 106(12): p. 1678-1686.
- Chen, L., et al., WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging cell, 2003. 2(4): p. 191-199.
- Zuo, Z., et al., Ribonucleotide reductase M2 (RRM2): Regulation, function and targeting strategy in human cancer. Genes & diseases, 2024. 11(1): p. 218-233.
- Hayes, J.D., A.T. Dinkova-Kostova, and K.D. Tew, Oxidative stress in cancer. Cancer cell, 2020. 38(2): p. 167-197.
- Ahmed, S.A., R.M. Gogal Jr, and J.E. Walsh, A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: an alternative to [3H] thymidine incorporation assay. Journal of immunological methods, 1994. 170(2): p. 211-224. [CrossRef]
- Alsherbiny, M.A., et al., Synergistic interactions of cannabidiol with chemotherapeutic drugs in mcf7 cells: Mode of interaction and proteomics analysis of mechanisms. International journal of molecular sciences, 2021. 22(18): p. 10103.
- Dissanayake, I.H., et al., Antiproliferative effects of Australian native plums against the MCF7 breast adenocarcinoma cells and UPLC-qTOF-IM-MS-driven identification of key metabolites. Food Bioscience, 2023. 54: p. 102864. [CrossRef]
Figure 2.
Pathway enrichment analysis using differentially expressed proteins in Dox-treated AGS cells compared to control cells. (A) Volcano plot showing significantly regulated proteins (Absolute Log2FC ≥ 0.58 and Q ≤ 0.05) (B) Graphical summary of top predictions in Ingenuity Pathway Analysis (IPA) interpret report as derived from IPA core analysis.
Figure 2.
Pathway enrichment analysis using differentially expressed proteins in Dox-treated AGS cells compared to control cells. (A) Volcano plot showing significantly regulated proteins (Absolute Log2FC ≥ 0.58 and Q ≤ 0.05) (B) Graphical summary of top predictions in Ingenuity Pathway Analysis (IPA) interpret report as derived from IPA core analysis.
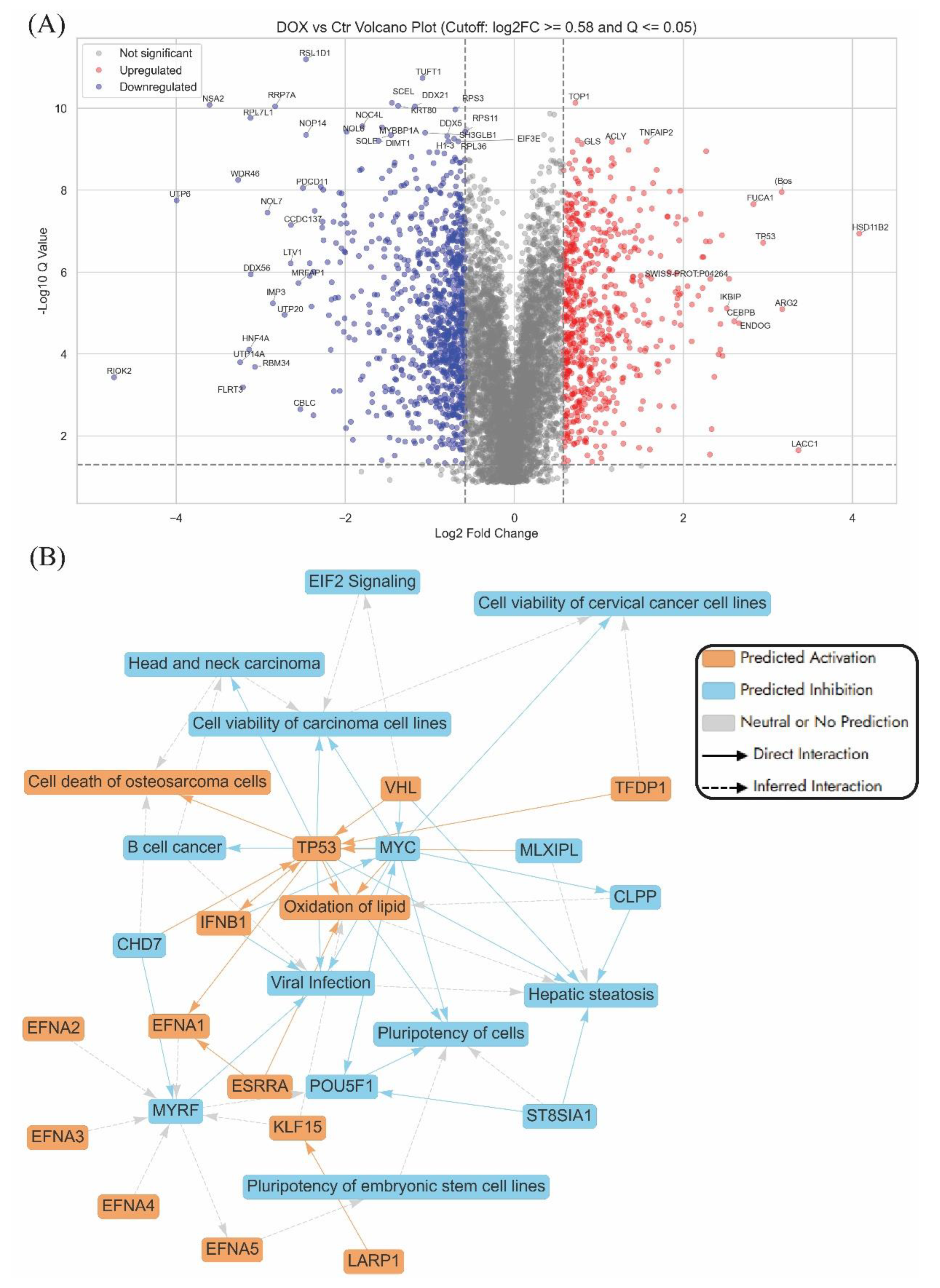
Figure 3.
A schematic representation of the major pathway of rRNA processing in the nucleolus and cytosol. Proteins indicated in red were significantly upregulated, while proteins indicated in blue is downregulated in Dox treated AGS cells compared to control untreated cells. Figure was generated by Qiagen Ingenuity Pathway Analysis (IPA).
Figure 3.
A schematic representation of the major pathway of rRNA processing in the nucleolus and cytosol. Proteins indicated in red were significantly upregulated, while proteins indicated in blue is downregulated in Dox treated AGS cells compared to control untreated cells. Figure was generated by Qiagen Ingenuity Pathway Analysis (IPA).
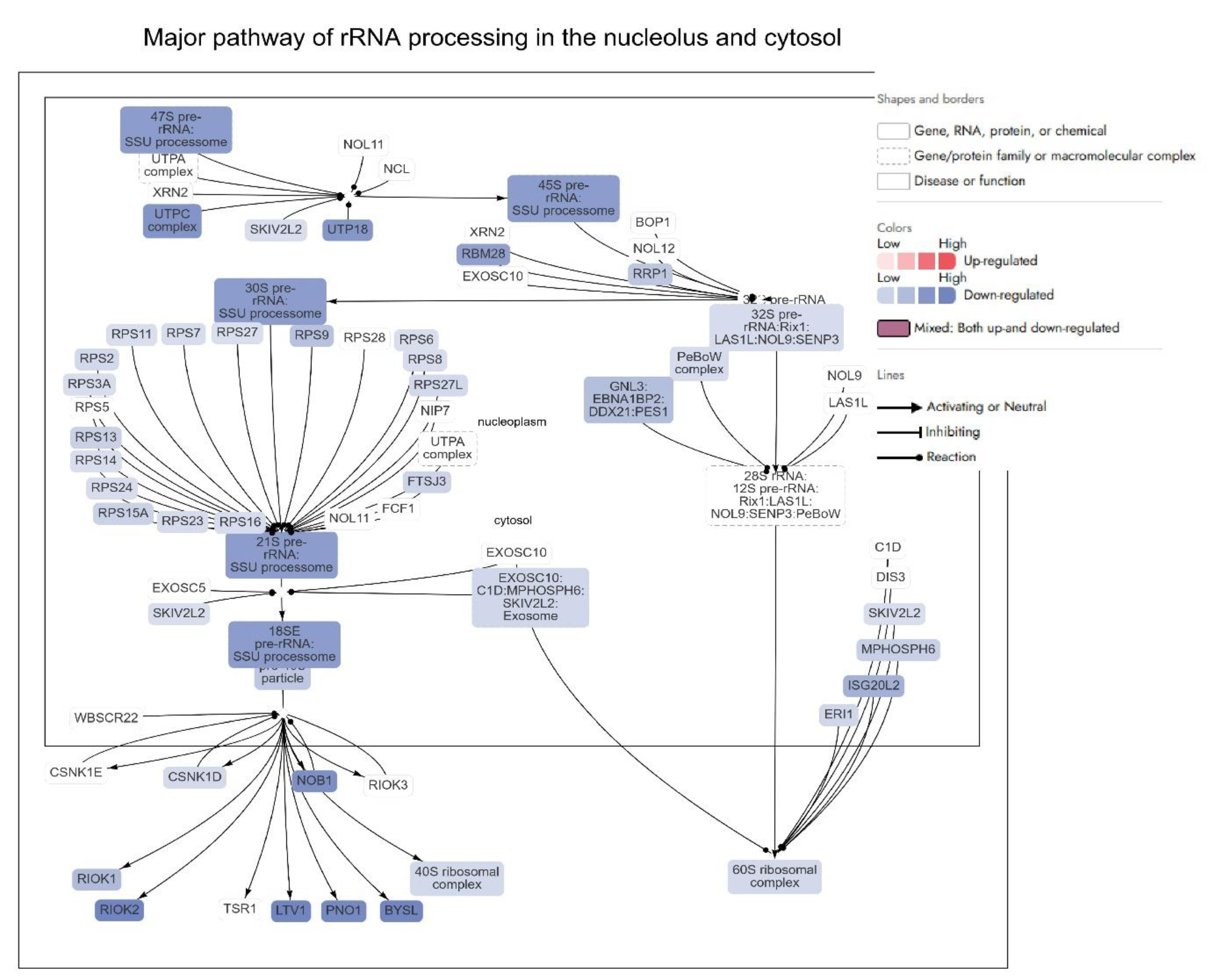
Figure 4.
Pathway enrichment analysis using differentially expressed proteins in APB+Dox -treated AGS cells compared to APB and Dox (Q ≤0.05) (A) Volcano plot showing significantly regulated proteins (Absolute Log2FC ≥ 0.58 and Q ≤ 0.05) (B) Graphical summary of top predictions in an IPA Interpret report presented in the form of a simple network.
Figure 4.
Pathway enrichment analysis using differentially expressed proteins in APB+Dox -treated AGS cells compared to APB and Dox (Q ≤0.05) (A) Volcano plot showing significantly regulated proteins (Absolute Log2FC ≥ 0.58 and Q ≤ 0.05) (B) Graphical summary of top predictions in an IPA Interpret report presented in the form of a simple network.
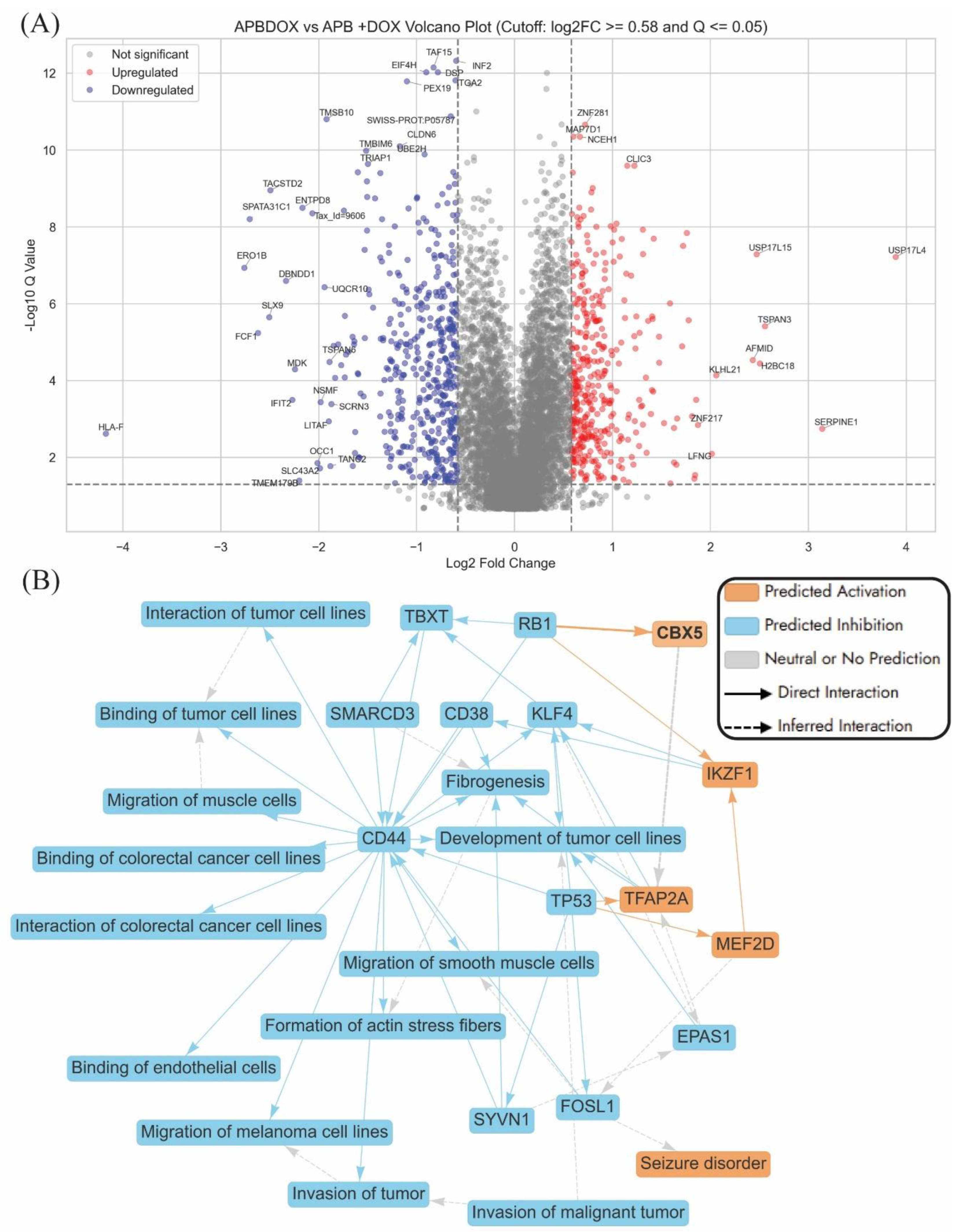
Figure 5.
Network analysis of differentially expressed proteins involved in tumour progression following APB+Dox treatment in AGS cells compared to monotreatments APB and Dox.
Figure 5.
Network analysis of differentially expressed proteins involved in tumour progression following APB+Dox treatment in AGS cells compared to monotreatments APB and Dox.
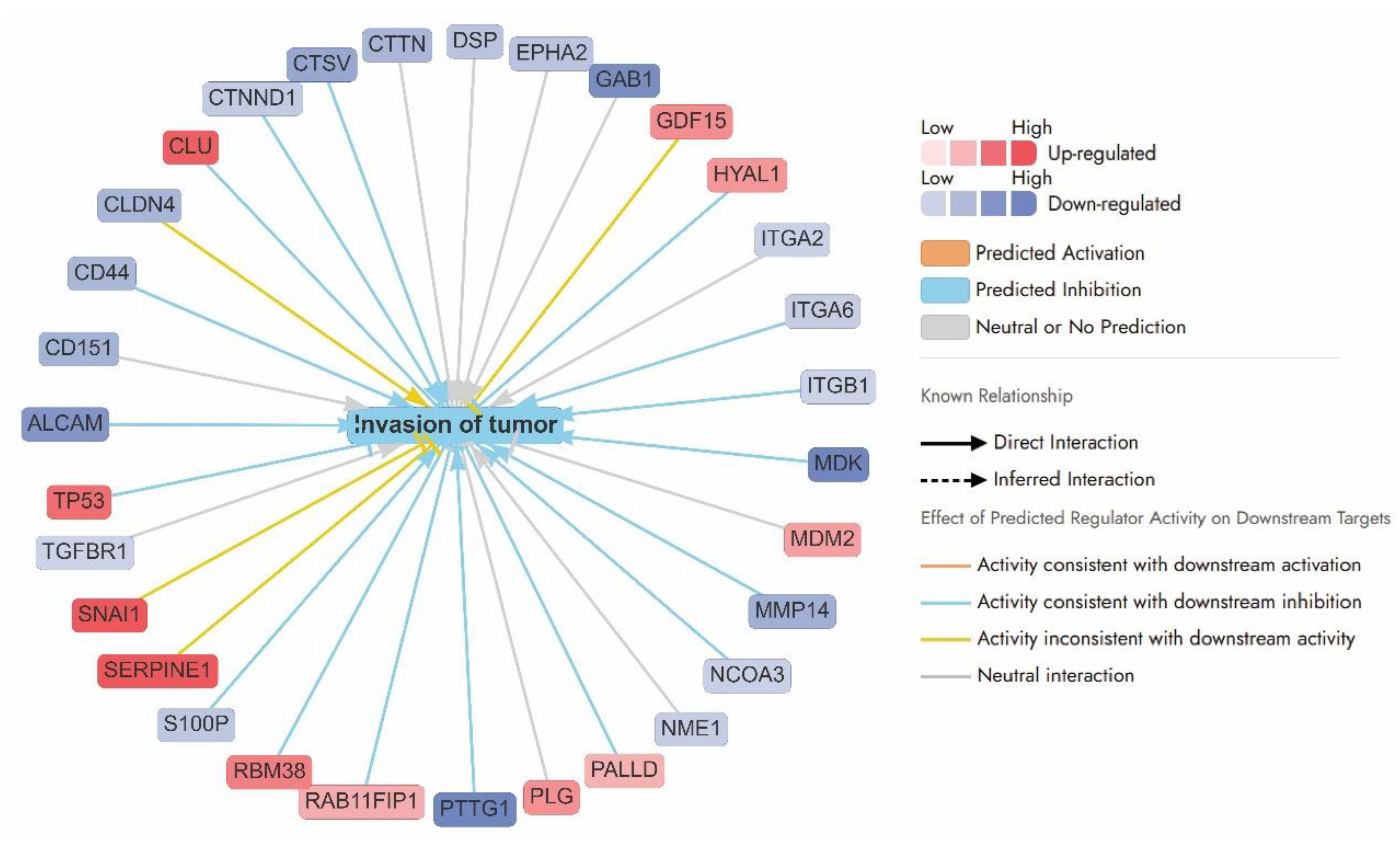
Figure 6.
Flow cytometric assessment of the apoptotic profiles of the AGS gastric cancer cells after 24 h of treatment A) The live, early apoptotic, late apoptotic, and necrotic cell percentages after 24 h treatment with APB (3000 µg/mL), Dox (0.54 µg/mL), APB+Dox (3000.27 µg/mL), and control (n = 4). * Indicates 0.01 < p-value < 0.05; ** indicates p < 0.01; *** indicates p < 0.001; **** indicates p < 0.0001 compared to the negative control. B) Represented are the density plots of each drug treatment that is most representative of the average data from the flow cytometric analyses, with Q3-1 = necrotic cells, Q3-2 = late-stage apoptotic cells, Q3-3 = live cells, and Q3-4 = early-stage apoptotic cell.
Figure 6.
Flow cytometric assessment of the apoptotic profiles of the AGS gastric cancer cells after 24 h of treatment A) The live, early apoptotic, late apoptotic, and necrotic cell percentages after 24 h treatment with APB (3000 µg/mL), Dox (0.54 µg/mL), APB+Dox (3000.27 µg/mL), and control (n = 4). * Indicates 0.01 < p-value < 0.05; ** indicates p < 0.01; *** indicates p < 0.001; **** indicates p < 0.0001 compared to the negative control. B) Represented are the density plots of each drug treatment that is most representative of the average data from the flow cytometric analyses, with Q3-1 = necrotic cells, Q3-2 = late-stage apoptotic cells, Q3-3 = live cells, and Q3-4 = early-stage apoptotic cell.
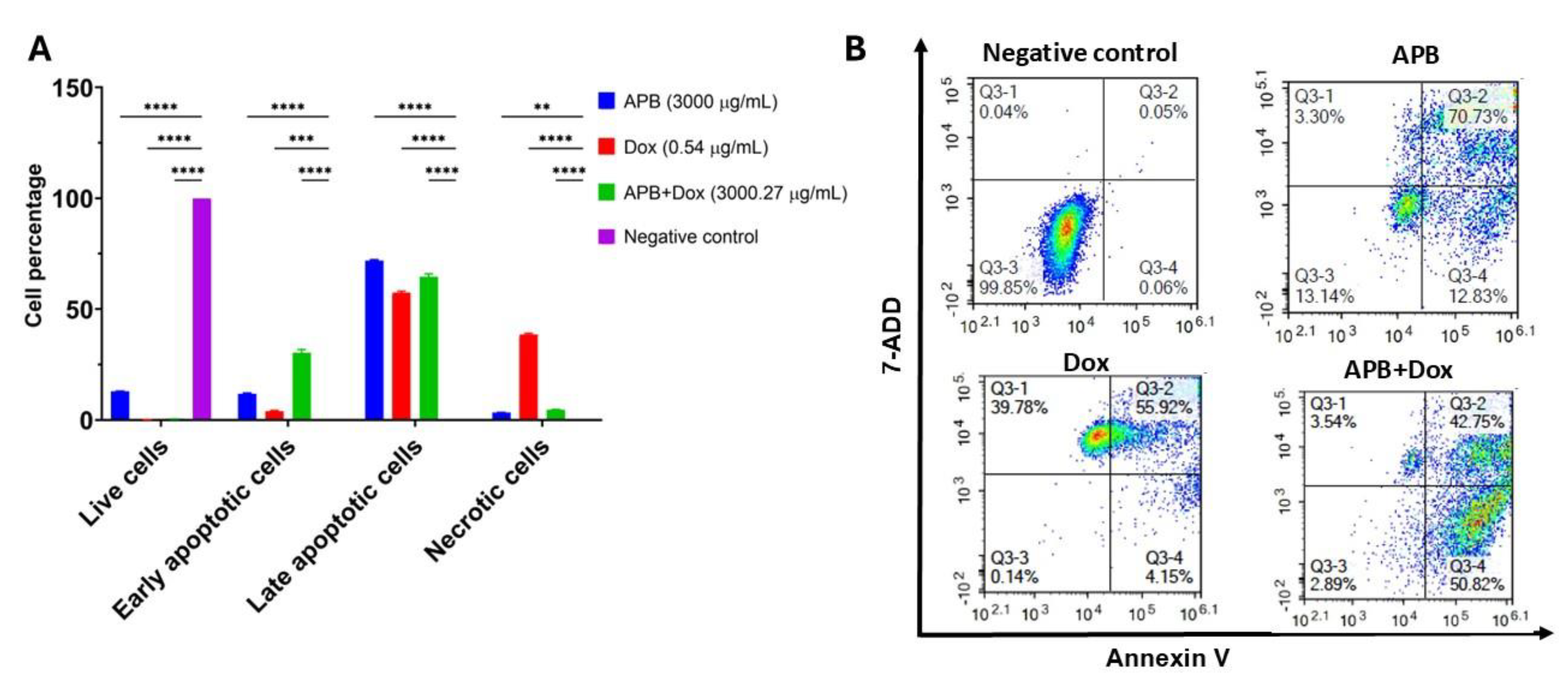
Figure 7.
Fold change in ROS generation following treatment with various concentrations: 3000:0.27 μg/mL, 1500:0.14 μg/mL, and 750:0.07 μg/mL of APB+Dox. Additionally, Doxorubicin (0.54, 0.27, 0.14 μg/mL) and tert-Butyl hydroperoxide (TBHP) (22.5 μg/mL or 250 μM) are included for comparative purposes. The values are expressed as mean ± SD. The values are expressed as mean ± SD. *indicates value of p: 0.01 < value of p <0.05, **indicates value of p ≤0.01, ****indicates p < 0.0001 compared to untreated control.
Figure 7.
Fold change in ROS generation following treatment with various concentrations: 3000:0.27 μg/mL, 1500:0.14 μg/mL, and 750:0.07 μg/mL of APB+Dox. Additionally, Doxorubicin (0.54, 0.27, 0.14 μg/mL) and tert-Butyl hydroperoxide (TBHP) (22.5 μg/mL or 250 μM) are included for comparative purposes. The values are expressed as mean ± SD. The values are expressed as mean ± SD. *indicates value of p: 0.01 < value of p <0.05, **indicates value of p ≤0.01, ****indicates p < 0.0001 compared to untreated control.


Table 1.
Cell growth inhibition (%) against the AGS gastric adenocarcinoma and cell viability (%) of the Hs 738.St/Int normal intestine cell lines at different concentrations of APB combination, doxorubicin (Dox), combination APB+Dox and combination for 72 h using the Alamar Blue assay (n = 3).
Table 1.
Cell growth inhibition (%) against the AGS gastric adenocarcinoma and cell viability (%) of the Hs 738.St/Int normal intestine cell lines at different concentrations of APB combination, doxorubicin (Dox), combination APB+Dox and combination for 72 h using the Alamar Blue assay (n = 3).
| Conc. μg/mL |
Cell viability (%) HS738.St/Int |
Conc. μg/mL |
Cell Growth Inhibition (%) of AGS cels | Cell viability (%) HS738.St/Int |
Conc. μg/mL |
Cell Growth Inhibition (%) of AGS cels | Cell viability (%) HS738.St/Int |
|---|---|---|---|---|---|---|---|
| APB | Dox | APB+Dox | |||||
| 3000 | 76.59 ± 8.56 a | 0.54 | 73.51 ± 5.16 a | 38.37 ± 7.01 a | 3000 + 0.27 | 103.46 ± 2.24 a | 64.12 ± 8.76 a |
| 1500 | 79.67 ± 8.16 a | 0.27 | 34.69 ± 2.96 b | 62.84 ± 11.53 b | 1500 +0.136 | 102.51 ± 9.05 a | 92.42 ± 10.66 b |
| 750 | 80.98 ± 9.19 a | 0.136 | 20.77 ± 7.29 c | 68.52 ± 7.51 b | 750+0.068 | 85.89 ± 8.58 b | 100.54 ± 8.51 b |
| 375 | 85.80 ± 13.04 a | 0.068 | 17.29 ± 7.96 c | 71.51 ± 7.81 b | 375+0.034 | 50.34 ± 8.49 c | 105.92 ± 11.80 b |
| 187.5 | 103.99 ± 10.54 b | 0.034 | 9.77 ± 6.90 c | 87.28 ± 9.25 c | 187.5+0.017 | 28.04 ± 7.40 d | 112.90 ± 11.32 b |
| 93.75 | 117.67 ± 13.09 b | 0.017 | 1.21 ± 3.84 c | 89.33 ± 11.72 c | 93.75+0.0085 | 27.29 ± 11.68 d | 114.72 ± 11.16 b |
| IC50 | >3000 | IC50 | 0.22 ± 0.04 | >0.27 | IC50 | 512.80± 18.37 | >3000.27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



