Submitted:
31 August 2025
Posted:
01 September 2025
You are already at the latest version
Abstract
CDKL5 deficiency disorder (CDD) is a rare developmental epileptic encephalopathy (DEE) caused by mutations in cyclin-dependent kinase-like 5 (CDKL5). The clinical manifestations include early and severe epilepsy, intellectual disability, motor abnormalities, and cortical visual impairments. The pathophysiological mechanisms underlying CDD are not fully understood and current treatments are limited to symptomatic management and do not target the underlying cause. Understanding the downstream molecular pathways that are disrupted by CDKL5 deficiency may yield additional targets and therapeutic strategies. Previous studies have focused on mapping the differential expression of protein-coding genes and post-translational modifications of CDKL5 targets but the role of noncoding RNAs in CDD is unknown. Here we performed RNA sequencing to define the short non-coding RNA landscape in the hippocampus of the Cdkl5 exon 6 deletion mouse model (12 weeks old heterozygous mice). Our findings catalogue the alterations in the expression level of multiple ncRNA species including microRNAs, tRNAs, piwi-RNAs, snoRNAs, and snRNAs. The findings reveal loss of this single gene has an extensive impact on the noncoding RNA landscape which might contribute to the pathogenesis of CDD and may guide understanding of disease mechanisms and new therapeutic strategies.
Keywords:
CDKL5 deficiency disorder
; hippocampus
; short non-coding RNAs
; microRNAs
; tRNAs
1. Introduction
CDKL5 (cyclin-dependent kinase-like 5) Deficiency Disorder (CDD) is a severe X-linked neurodevelopmental disease characterized by early-onset infantile developmental and epileptic encephalopathy (DEE) syndrome, intellectual disability, motor abnormalities, and cortical visual impairments [1,2,3].CDKL5 pathogenic mutations affect about 1 out of 40 000 to 60 000 births, with females more frequently affected than males [4].These mutations are among the most commonly identified pathogenic findings in epilepsy gene panels [5]. Seizure burden is early in onset and severe in nature, typically beginning within the first weeks to months of postnatal life and being medically intractable [6]. Life expectancy in CDD patients varies depending on disease severity and gender [7], with a high risk of sudden unexpected death in epilepsy (SUDEP) due to higher frequency and severity of seizures [8].
The CDKL5 gene encodes CDKL5 protein, a member of the serine/threonine protein kinase family [9], which is thought to mediate most of its function by phosphorylation of protein targets. CDKL5 expression is highest during early development and localized primarily to the dendrites and nucleus [10]. CDKL5 regulates signal transduction pathways, guides the establishment of neural networks, and influences neuronal morphogenesis and excitatory synaptic input. Pathogenic mutations in CDKL5 cause enzymatic loss of function and impaired kinase catalytic activity. However, little is known of its downstream substrates or the regulatory mechanisms of CDKL5 function [11,12,13]. Current treatments do not target the underlying cause but are limited to symptom management. Currently, therapeutic strategies are focusing on restoring CDKL5 levels or modulating the downstream molecular pathways that are disrupted due to CDKL5 deficiency. Identifying additional targets that could effectively restore the critical functions of CDKL5 remains a crucial step in advancing therapeutic strategies. Gene expression profiling and proteomics have identified some of the disrupted protein-coding genes in CDD but the full extent of altered gene expression in CDD remains uncertain.
Non-coding RNAs (ncRNAs) constitute a diverse family of endogenous RNA molecules that do not encode proteins. It is now understood that only approximately 1.5% of the human genome comprises protein-coding sequence whereas nearly 80% of the remaining genomic regions are transcribed into various forms of ncRNAs, highlighting their potential regulatory and functional significance [14]. Among these, short ncRNAs encompass a wide variety of subtypes, including microRNA (miRNA), small nuclear RNA (snRNA), piwi-interacting RNA (piRNA) and small nucleolar RNA (snoRNA), as well as transfer RNA (tRNA), [15,16,17]. MiRNAs are small non-coding RNAs that refine the transcriptomic landscape through repression and/or degradation of specific mRNA targets [18,19,20]. The principal mechanism of miRNA mediated modulation of protein expression is via sequence specific mRNA degradation [21]. MiRNAs are single stranded RNA molecules ranging in length from 19-25 nucleotides. Complementary nucleotide sequence recognition between miRNA molecules and miRNA binding sites in the 3’UTR (untranslated region) of mRNA targets is imperfect and flexible which enables one miRNA to downregulate expression of hundreds of mRNA targets [22]. It is also possible that miRNA molecules may control CDKL5 or downstream expression of CDKL5 targets which may be contributing to disease burden. On the other hand, transfer RNAs (tRNAs), a major class of ncRNAs comprising 73–90 nucleotides and accounting for 4–10% of total cellular RNA, play an essential role in protein synthesis [23]. tRNAs undergo a wide array of chemical modifications that are essential for numerous cellular processes. These modifications ensure accurate and efficient protein translation and also contribute significantly to the regulation of gene expression and the cellular response to stress [24]. tRNA cleavage is an evolutionarily conserved process that generates tRNA-derived fragments (tRFs) from either precursor or mature tRNAs. These fragments are classified into several subtypes, including tRF-1, tRF-2, tRF-3, tRF-5, and internal tRFs (i-tRFs) [25,26]. tRFs have been associated with several cellular roles and are crucial in normal brain development [27] ensuring several roles such as suppressing protein synthesis, triggering the formation of stress granules, and modulating gene expression [28,29,30,31]. They are also known to orchestrate gene silencing like miRNAs (microRNAs) by binding to Argonaute proteins [32,33,34], and play a vital role in transcription as well as translation [34,35]. Other short ncRNA include small nucleolar RNAs (snoRNAs) which are predominantly localized in the nucleoli of eukaryotic cells and they are primarily transcribed from intronic regions of both protein-coding and non-protein-coding genes [36,37]. Recent studies have highlighted the expanding roles of snoRNAs in cellular regulation, including guiding N4-acetylcytidine modifications, influencing alternative splicing, and exhibiting miRNA-like functions [38]. Also, PIWI-interacting RNAs (piRNAs) consist another important class on short ncRNA, which are single-stranded RNA molecules that play a crucial role in gene expression regulation and RNA silencing [39] [40,41,42]. [39,43,44].
Given that short ncRNAs play a crucial role in regulating a wide range of essential cellular processes and ensure epigenetic modifications across nearly all eukaryotic organisms [45] [46] [47,48]. A number of studies have shown that ncRNAs participate in pathological and physiological processes including the development of the nervous system and synaptic plasticity, as well as learning and memory [49,50]. Notably, ncRNAs are actively involved in regulating the pathophysiological mechanisms underlying epilepsy and exhibit dysregulation during epileptogenesis. This includes prominent roles for miRNA, and tRNA fragments. Moreover, dysregulated levels of various small ncRNAs have been reported in brain tissue from other pediatric epileptic disorders with similarities to CDD, such as Rett syndrome [51,52]. The dysregulation of small ncRNAs in CDD remains unexplored. Therefore, the aim of the current study is to evaluate hippocampal ncRNAs expression including miRNAs, tRNAs, piRNA, snoRNA, and snRNA in the exon 6 excision mouse model of CDD [53]. This model recapitulates several key behavioral and metabolic features of CDD, including impaired motor coordination, spontaneous recurrent seizures, altered social behavior, abnormal visual tracking, and metabolic dysfunction.
2. Results
2.1. Distribution of Small RNAs in Our Samples
To analyse the small ncRNA landscape of CDD, we extracted hippocampus from heterozygous (Het) and wild-type (WT) mice (12 weeks aged female mice). The read distribution across all RNA species revealed the expression levels of various ncRNAs (Figure 2A). Among them, microRNAs (miRNAs) were the most abundantly expressed ncRNAs in both Het and WT hippocampal tissue (Figure 2B), while other ncRNA classes exhibited relatively low expression levels.
2.1.1. Dysregulated microRNAs
A total of 1,529 miRNAs were detected in the hippocampal tissue of Het mice compared to WT (Figure 3). The most abundant included miR-9-5p, miR-124-3p, miR-128-3p, miR-132-3p, and miR-134-5p, which are brain-enriched. In CDD mice, 18 miRNAs; miR-150-3p, miR-1249-5p, miR-200c-3p, miR-1969, miR-6537-3p, miR-7000-5p, miR-467a-5p, miR-375-3p, miR-1943-5p, miR-101c, miR-7685-5p, miR-297b-5p, miR-5624-3p, miR-215-3p, miR-144-3p, miR-101a-5p, miR-6929-3p, and miR-671-5p were significantly downregulated, while 12 miRNAs: miR-376c-5p, miR-7046-3p, miR-377-3p, miR-344-5p, miR-505-3p, miR-222-5p, miR-145b, miR-6986-5p, miR-6911-5p, miR-3473e, and miR-384-3p were significantly upregulated.
2.1.1. Transfer RNA
Several tsRNAs were detected in the hippocampi of CDD mice with the most abundant belonged to the tRNA-Gly, tRNA-Glu, and tRNA-Ala families (Figure 4). In CDD mice, several tsRNAs were dysregulated: tRNA-Arg-TCT, tRNA-Glu-CTC, and tRNA-Gly-GCC fragments were upregulated, while tRNA-Ser, tRNA-Gly, and tRNA-Asp fragments were downregulated. This suggests their potential involvement in CDD phenotype regulatory mechanisms.
2.1.3. Other Small ncRNAs
Our results also revealed altered expression of other non-coding RNAs (ncRNAs) in hippocampal samples, including piRNAs (Figure 5A), snoRNAs (Figure 5B), and snRNAs (Figure 5C), which were significantly differentially expressed (p < 0.05) between Het and WT groups, as shown in the volcano plots. These findings suggest that these ncRNAs may also contribute to the molecular alterations associated with CDD.
3. Discussion
CDD is an important albeit rare DEE, characterized by early-onset, intractable seizures and severe neurodevelopmental impairment. In addition to seizures, affected individuals often present with profound intellectual disability, motor dysfunction, and cortical visual impairment. The use of gene and protein profiling techniques have revealed the extensive changes to signaling pathways in CDD [9,53,56]. As expected, various proteins undergo reductions in phosphorylation in models of CDD. What is remarkable is that there are also various changes in other signaling pathways within the brain, likely direct and indirect effects of the loss of CDKL5. Studies are beginning to tease apart how the changes to the protein landscape influence the CDD phenotype [53,56,57]. These studies therefore serve the dual purpose of extending our understanding of how loss of the gene alters the molecular environment in neurons as well as non-neuronal cells through direct and indirect mechanisms, while also providing targets for biomarkers and therapeutics. That is, an approach to improving the treatment of CDD may lie not only with restoring CDKL5 itself but with adjusting the downstream changes or compensating for CDKL5 loss with enhancing the actions of complementary proteins (e.g. CDKL2) [58].
The genomes of humans and mice include extensive and diverse classes of gene that, when active, express RNAs which do not code for proteins but nevertheless perform important regulatory functions in cells. Here we used a well-established mouse model of CDD to profile the small ncRNA landscape [53]. The present study is important because it reveals an additional effect of the loss of CDKL5 is to cause extensive changes to the small ncRNA landscape. Our findings establish the details of how this molecular landscape is adjusted in the hippocampus of mice lacking CDKL5.
Small ncRNAs participate in several biological processes and pathological disease states. They have recently emerged as potential biomarkers, with several studies reported dysregulated expression in brain tissue from animal models of neurologic disorders including epilepsy [37,59,60,61,62]. Notably, altered expression levels of small ncRNAs have been reported in Rett syndrome [51,52,63], a disorder that shares significant overlap in pathophysiology and symptom burden with CDD. To our knowledge, this is the first study to examine ncRNA expression in the hippocampus in the context of CDD. Importantly, our findings reveal dysregulated expression of all the studied classes of ncRNAs, including miRNAs, tRNAs, snoRNAs, pi-RNAs, and snRNAs. The most abundant and well-understood class of ncRNA altered in our samples were miRNAs. Regarding miRNAs expression, our results showed upregulation in several miRNAs including miR-222-5p, miR-376c-5p, miR-377-3p, miR-344-5p, miR-505-3p, miR-145b, miR-3473e, and miR-384-3p while others were downregulated including miR-150-3p, miR-200c-3p, miR-467a-5p, miR-375-3p, miR-101c, miR-297b-5p, miR-215-3p, miR-144-3p, miR-101a-5p, miR-671-5p. Altered expression in specific microRNA levels that modulate injury and repair processes, including changes in neuronal and glial morphology, immune and inflammatory responses, and cellular metabolism has been increasingly reported in brain disorders [64]. MiRNAs shape the transcriptomic landscape through repression and/or degradation of specific mRNA targets [20]. In a mouse model of Rett syndrome with loss of mecp2, which acts upstream of CDKL5 to control its expression [65,66], there is marked dysregulation of miRNA levels including miR-215, miR-375, and miR-101 [67], which were dysregulated in our CDD mice. Moreover, our finding showed upregulated miR-222-5p, while its expression level was significantly reduced in animal models and patients with epilepsy [68,69,70]. Also, miR-344a-5p, miR-671 were upregulated in a rat model of temporal lobe epilepsy [71]. While, the other miRNAs dysregulated in our Het mice were not well documented in epilepsy related disorders.
It is also possible that miRNAs may control CDKL5 or downstream expression of CDKL5 targets which may be contributing to disease burden. While a number of putative miRNA binding sites have been identified in the CDKL5 transcript [72,73], their significance is unclear considering CDKL5 protein levels correlate well with mRNA expression [11]. It is perhaps more feasible, therefore, that downstream targets of CDKL5 are subject to miRNA modulation. Linking the dysregulation of miRNAs to disease, as demonstrated in the current study, may offer a unique therapeutic advantage, through the administration of anti-sense oligonucleotide (ASO) therapy or to normalize the level of downregulated miRNA by introducing synthetic miRNA using adeno-associated virus (AAV)-mediated gene therapy. Therefore, targeting miRNAs that are dysregulated in CDD and target key disease-associated pathways presents a promising opportunity for alternative therapeutic interventions [74].
The second most abundant class of small ncRNA detected in the present study was tsRNAs, a key class of noncoding RNAs involved in regulating gene expression and responding to cellular stress [24], that have been linked to neurodevelopmental disorders through genetic mutations that affect tRNA modifications, underscoring their critical role in normal brain development [27]. Under certain circumstances, tRNA can be cleaved into tRNA fragments (tRFs; derived from precursor or mature tRNA). tRFs play crucial roles in suppressing protein translation, initiating stress granule assembly, modulating gene expression [75], regulating protein translation and apoptosis, cell metabolism and epigenetic inheritance [23]. tRFs have also been implicated in various neurological disorders, including stroke, Alzheimer's disease, epilepsy, Parkinson's disease, autism, ALS, and others [23] [76]. Human neurogenetic research has increasingly demonstrated that mutations in genes encoding tRNA biogenesis proteins are associated with brain abnormalities and a variety of neurological disorders [77]. Our findings showed that tRFs are differentially expressed in CDD mice. These include upregulated tRNA-Arg-TCT, tRNA-Glu-CTC, and tRNA-Gly-GCC fragments, while tRNA-Ser, tRNA-Gly, and tRNA-Asp fragments were downregulated. Importantly, elevated levels of tRFs have been detected in the plasma of epileptic patients prior to seizure onset. The 5′GlyGCC and 5′GluCTC tRFs were detected as biomarkers in plasma samples from epilepsy patients [78]. Moreover, Fagan et al. (2021) interestingly reported elevated plasma tRNA fragments which precedes seizures in patients with epilepsy, which can be used as a biomarker to detect the seizure risk in patients with epilepsy [79]. Notably, the levels of tRFs derived from 5′Gly-GCC and 5′Glu-CTC were found to decrease following epileptiform-like activity, suggesting a potential protective or compensatory response within the brain [23]. Moreover, two tRF-5 fragments 5′AlaTGC and 5′GluCTC demonstrated consistent fold changes between pre-seizure and post-seizure samples, highlighting their potential as biomarkers for seizure prediction [78]. Additionally, neuronal tRNAs uncovered that loss of a neuronally enriched arginine tRNA (n-Tr20), a tRNA-Arg with anticodon TCT, alters seizure thresholds and impacts synaptic transmission [80]. The dysregulated tRFs levels in our mice could be linked to changes in tRNA genes or in genes involved in their biosynthesis and modification contributing to CDD pathogenesis. tRFs are also known to mediate gene silencing in a manner similar to miRNAs, primarily through their interaction with Argonaute proteins [32,33]. Indeed, tRNA modifications ensure accurate protein synthesis by stabilizing codon–anticodon interactions, a process essential for neuronal differentiation, synaptic plasticity, and brain function [24], which are relevant to CDD. Moreover, mutations in genes encoding enzymes responsible for tRF biogenesis have been linked to the development of several neurological disorders [79]. Altogether, these findings position tRNA as a promising target in CDD. However, further research is needed to elucidate the precise mechanisms through which tRNA dysregulation may contribute to specific phenotypes in CDD, ultimately guiding the development of effective therapeutic strategies.
Other small ncRNAs altered in our study include snoRNAs, snRNAs and piRNAs. snRNAs are a component of the RNA spliceosome involved in post-transcriptional processing in eukaryotic cells, contributing to the modification of mRNA precursors [81]; [82]. Recent evidence has demonstrated that snoRNAs can perform miRNA-like functions [38], expanding their traditionally recognized roles beyond guiding chemical modifications of rRNA and snRNA. SnoRNAs primarily guide the modification and processing of ribosomal RNA (rRNA) within the nucleolus [83]. Some snoRNAs also directly involved in the nucleolytic processing of rRNA precursors [84]. snoRNAs can also influence cellular processes indirectly by disrupting ribosome and snRNA biogenesis, thereby modulating the expression of protein-coding genes through effects on splicing and translation efficiency [81,85]. Additionally, some snoRNAs are processed into shorter regulatory RNAs, such as miRNAs or piRNAs, which function in gene silencing pathways [86,87]. Thus, the finding that CDD mice display changes in these classes of ncRNA indicates that loss of CDKL5 leads to changes in RNA processing machinery. Since snRNAs are essential for correct spliceosome assembly and activity, their dysregulation raises the possibility that aberrant splicing events may occur in CDD. While widespread spliceosomal dysfunction has not yet been directly reported in CDKL5 deficiency, transcriptomic analyses in CDD models have revealed altered expression of genes involved in RNA metabolism and post-transcriptional regulation [53,88], supporting the hypothesis that splicing impairments may contribute to the molecular pathology. This suggests that defects in ncRNA regulation and spliceosome integrity could represent an underappreciated mechanism underlying neuronal dysfunction in CDD.
A growing body of evidence suggests that nervous system piRNAs play a functional role in processes such as neuronal development, learning, and memory [89,90,91,92], which are altered in both patients and CDD animal models. piRNAs play a crucial role in gene silencing and regulating gene expression [39], [40,41,42], [39,43,44]. Indeed, PIWI-piRNAs recognize and cleave complementary RNA targets, effectively silencing gene expression at the post-transcriptional level and contributing to genomic stability [93,94,95]. The involvement of piRNAs and PIWI proteins has been reported in various CNS pathologies [89,92,96,97,98,99,100,101,102] including Rett syndrome [101], autism spectrum disorders [89] [103] and Alzheimer disease [98,102,104]. Given these roles, it is not surprising that dysregulation of piRNAs may also be involved in the molecular mechanisms underlying CDD, potentially contributing to its characteristic neurological impairments.
In summary, the loss of CDKL5 causes select changes to a spectrum of small noncoding RNA species. The findings may have implications for how loss of the gene disturbs cellular function and may lead to biomarkers for the disease or potential therapies. Small ncRNAs participate in essential biological processes and are increasingly recognized as contributors to neurological disease. Our data extend these insights to CDD, showing that multiple classes of ncRNAs including miRNAs, tsRNAs, snoRNAs, snRNAs, and piRNAs are dysregulated in the hippocampus. The extent of these changes highlights the potential for ncRNA imbalance to disrupt diverse processes ranging from transcriptional regulation and spliceosome activity to ribosome biogenesis, stress responses, and synaptic function. Importantly, because small ncRNAs have been reported as biomarkers and therapeutic targets in other neurological disorders such as epilepsy and Rett syndrome, the dysregulated ncRNA signatures identified here may provide a foundation for biomarker discovery and the development of targeted ncRNA-based therapies in CDD.
Limitations
The present study has some limitations. First, we analyzed only a single time point, sex, and a brain region. It will be important in future work to extend this to determine when these changes first emerge, whether they remain stable over time, and whether other brain regions lacking CDKL5 display similar or distinct alterations in the ncRNA landscape. Second, the study does not provide information on the cellular origin of these ncRNA changes. Although we observed changes in known neuronal miRNAs in the model, it remains unclear which of the various ncRNA changes are occurring specifically in neurons versus other cell types.
4. Materials and Methods
4.1. Animal Care and Ethical Approval
All procedures involving animals were performed in compliance with EU Directive 2010/63/EU on the protection of animals used for scientific purposes. Mice were housed at controlled conditions including a room temperature (20°C–25°C), and humidity (40%–60%), on a 12 h dark–light cycle with ad libitum access to water and food. All procedures were approved by RCSI University of Medicine and Health Sciences’ Research Ethics Committee (REC 1587), and under license from the Ireland Health Products Regulatory Authority (AE19127/P064).
4.2. Animal Model Used in the Study
Mice were bred from two distinct colonies, a wildtype female mouse (Cdkl5+/+) and a CDKL5 exon 6 deletion mice (Cdkl5 -/y) (Jackson Laboratory, USA) (Figure 1A). The off-spring produced resulted in 50% heterozygous female mice (Cdkl5 +/-) and 50% wild-type male mice (Cdkl5 +/y). For this study wildtype males (Cdkl5 +/y) were bred with heterozygous females (Cdkl5 +/-), these mice have a 25% chance of producing a WT female, 25% chance of producing a WT male, 25% chance of producing a Het (female) and a 25% chance of producing a KO male (Figure 1B).
4.3. Small RNA Sequencing of Hippocampal Tissue
Following brain dissection, the hippocampus was collected from 10 wild-type (WT) and 10 heterozygous (Het) mice under sterile conditions. The quality and quantity of RNA were evaluated using the Bioanalyzer RNA 6000 Nano Kit (Agilent). A small RNA library was then constructed from 1 µg of total RNA using the NEBNext® Small RNA Library Prep Set for Illumina (NEB), following the manufacturer's protocol. The size distribution of the library fragments was determined using the Bioanalyzer High Sensitivity DNA Analysis Kit (Agilent), and the library concentration was measured with the KAPA Library Quantification kit (Roche). The libraries were pooled in equal amounts and sequenced on a HiSeq platform with paired-end 150-cycle sequencing (Illumina, MA, USA).
FASTX-Toolkit was used to quality-filter reads and cutadapt was used to remove adaptor sequences. Fastqc and MultiQC was used to ensure high quality sequencing data. Filtered reads were mapped using Bowtie to a list of datasets. First, reads were mapped to tRNA sequences from Genomic tRNA Database (GtRNAdb). Unmapped reads were then mapped to miRNAs from miRBase v22 allowing zero mismatches, but allowing for non-templated 3’ A and T bases. Unmapped reads were then mapped against other relevant small RNA datasets: piRNA, tRNA, snRNA, snoRNA and Y RNA allowing one mismatch. The remaining unmapped reads were mapped to mRNA and rRNA datasets, followed by the mouse genome (mm10). This was done to discover which RNA species were present in the sequencing data.
4.4. Statistical Analysis
Data analysis was conducted in R using standard plotting packages. Principal component analysis (PCA) was used to evaluate sample clustering and overall variation in ncRNA expression profiles. Heatmaps were generated to highlight differentially expressed miRNAs in Het and WT mice, and volcano plots were used to illustrate the distribution of log2 fold changes versus statistical significance. p < 0,05 was considered statistically significant.
5. Conclusions
The present study revealed significant alterations in the expression of several short non-coding RNAs including microRNAs, tRNAs, snoRNAs, piwi-RNAs, and snRNAs in CDKL5 deficiency disorder. These dysregulations could play a critical role in the pathogenesis of CDD. A deeper understanding of the downstream pathways and mechanisms regulated by these ncRNAs is crucial for the development of targeted and effective treatments for CDD patients.
Author Contributions
Conceptualization, O.M.; methodology, O.M. and E.L, and M.H; software, S.E. M.T.V.; and A.H; validation, OM., and D.C.H.; formal analysis, J. H.; O.M.; B.E.-M.; investigation, J.H, S.E, B.E.-M, and O.M.; resources, O.M. and D.C.H; data curation, A. X, J.H and B.E.-M.; writing—original draft preparation, B.E.-M, A.H and S.B.K; writing—review and editing, O.M. and D.C.H; visualization, O.M.; supervision, O.M. and D.C.H; project administration, O.M.; funding acquisition, O.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Research Ireland (grant number 22/PATH-S/10668) and The Article processing Charges (APC) was funded by Research Ireland, and a Junior Fellowship Programme award from the University of Pennsylvania Orphan Disease Center on behalf of the Loulou Foundation, and CDKL5-UK Alliance- Italy and Ireland.
Institutional Review Board Statement
The animal study protocol was approved by RCSI University of Medicine and Health Sciences’ Research Ethics Committee (REC 1587), and under license from the Ireland Health Products Regulatory Authority (AE19127/P057).
Data Availability Statement
Data are included in the manuscript and supplementary data are available under request.
Acknowledgments
The authors would like to thank Dr. Amaya Sanz Rodriguez, and the FutureNeuro operations team for their valuable support throughout this work. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Leonard H, Downs J, Benke TA, Swanson L, Olson H, Demarest S. CDKL5 deficiency disorder: clinical features, diagnosis, and management. Lancet Neurol. 2022;21(6):563–76. [CrossRef]
- Demarest ST, Olson HE, Moss A, Pestana-Knight E, Zhang X, Parikh S, et al. CDKL5 deficiency disorder: Relationship between genotype, epilepsy, cortical visual impairment, and development. Epilepsia. 2019;60(8):1733–42.
- Fehr S, Wilson M, Downs J, Williams S, Murgia A, Sartori S, et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur J Hum Genet. 2013;21(3):266–73. [CrossRef]
- Daniels C, Greene C, Smith L, Pestana-Knight E, Demarest S, Zhang B, et al. CDKL5 deficiency disorder and other infantile-onset genetic epilepsies. Dev Med Child Neurol. 2024;66(4):456–68.
- Lindy AS, Stosser MB, Butler E, Downtain-Pickersgill C, Shanmugham A, Retterer K, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia. 2018;59(5):1062–71.
- Demarest S, Pestana-Knight EM, Olson HE, Downs J, Marsh ED, Kaufmann WE, et al. Severity assessment in CDKL5 deficiency disorder. Pediatr Neurol. 2019;97:38–42. [CrossRef]
- Benke TA, Demarest S, Angione K, Downs J, Leonard H, Saldaris J, et al. CDKL5 deficiency disorder. GeneReviews®[Internet]. 2024;
- Amin S, Monaghan M, Aledo-Serrano A, Bahi-Buisson N, Chin RF, Clarke AJ, et al. International consensus recommendations for the assessment and management of individuals with CDKL5 deficiency disorder. Front Neurol. 2022;13:874695.
- Van Bergen NJ, Massey S, Quigley A, Rollo B, Harris AR, Kapsa RMI, et al. CDKL5 deficiency disorder: molecular insights and mechanisms of pathogenicity to fast-track therapeutic development. Biochem Soc Trans. 2022;50(4):1207–24.
- Katayama S, Sueyoshi N, Inazu T, Kameshita I. Cyclin-Dependent Kinase-Like 5 (CDKL5): Possible Cellular Signalling Targets and Involvement in CDKL5 Deficiency Disorder. Neural Plast. 2020;2020(1):6970190.
- Kilstrup-Nielsen C, Rusconi L, La Montanara P, Ciceri D, Bergo A, Bedogni F, et al. What we know and would like to know about CDKL5 and its involvement in epileptic encephalopathy. Neural Plast. 2012;2012(1):728267.
- Szafranski P, Golla S, Jin W, Fang P, Hixson P, Matalon R, et al. Neurodevelopmental and neurobehavioral characteristics in males and females with CDKL5 duplications. Eur J Hum Genet. 2015;23(7):915–21.
- Hao S, Wang Q, Tang B, Wu Z, Yang T, Tang J. CDKL5 deficiency augments inhibitory input into the dentate gyrus that can be reversed by deep brain stimulation. J Neurosci. 2021;41(43):9031–46. [CrossRef]
- Huang J, Eilbeck K, Smith B, Blake JA, Dou D, Huang W, et al. The development of non-coding RNA ontology. Int J Data Min Bioinform. 2016;15(3):214–32.
- Ulitsky I. Interactions between short and long noncoding RNAs. FEBS Lett. 2018;592(17):2874–83.
- Dupuis-Sandoval F, Poirier M, Scott MS. The emerging landscape of small nucleolar RNAs in cell biology. Wiley Interdiscip Rev RNA. 2015;6(4):381–97.
- Luteijn MJ, Ketting RF. PIWI-interacting RNAs: from generation to transgenerational epigenetics. Nat Rev Genet. 2013;14(8):523–34.
- Brennan GP, Henshall DC. MicroRNAs as regulators of brain function and targets for treatment of epilepsy. Nat Rev Neurol. 2020;16(9):506–19.
- Henshall DC, Hamer HM, Pasterkamp RJ, Goldstein DB, Kjems J, Prehn JHM, et al. MicroRNAs in epilepsy: pathophysiology and clinical utility. Lancet Neurol. 2016;15(13):1368–76. [CrossRef]
- Mohr AM, Mott JL. Overview of microRNA biology. In: Seminars in liver disease. Thieme Medical Publishers; 2015. p. 3–11. [CrossRef]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466(7308):835–40.
- Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153(3):654–65.
- Mathew BA, Katta M, Ludhiadch A, Singh P, Munshi A. Role of tRNA-derived fragments in neurological disorders: a review. Mol Neurobiol. 2023;60(2):655–71.
- Lv X, Zhang R, Li S, Jin X. tRNA Modifications and Dysregulation: Implications for Brain Diseases. Brain Sci. 2024;14(7):633.
- Zhu L, Liu X, Pu W, Peng Y. tRNA-derived small non-coding RNAs in human disease. Cancer Lett. 2018;419:1–7.
- Jia Y, Tan W, Zhou Y. Transfer RNA-derived small RNAs: potential applications as novel biomarkers for disease diagnosis and prognosis. Ann Transl Med. 2020;8(17):1092.
- Blaze J, Akbarian S. The tRNA regulome in neurodevelopmental and neuropsychiatric disease. Mol Psychiatry. 2022;27(8):3204–13.
- Ivanov P, Emara MM, Villen J, Gygi SP, Anderson P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell. 2011;43(4):613–23.
- Emara MM, Ivanov P, Hickman T, Dawra N, Tisdale S, Kedersha N, et al. Angiogenin-induced tRNA-derived stress-induced RNAs promote stress-induced stress granule assembly. J Biol Chem. 2010;285(14):10959–68. [CrossRef]
- Chen Q, Yan M, Cao Z, Li X, Zhang Y, Shi J, et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science (80- ). 2016;351(6271):397–400.
- Dhahbi JM, Spindler SR, Atamna H, Yamakawa A, Boffelli D, Mote P, et al. 5′ tRNA halves are present as abundant complexes in serum, concentrated in blood cells, and modulated by aging and calorie restriction. BMC Genomics. 2013;14:1–14.
- Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009;10(2):94–108. [CrossRef]
- Kuscu C, Kumar P, Kiran M, Su Z, Malik A, Dutta A. tRNA fragments (tRFs) guide Ago to regulate gene expression post-transcriptionally in a Dicer-independent manner. Rna. 2018;24(8):1093–105. [CrossRef]
- Keam SP, Hutvagner G. tRNA-derived fragments (tRFs): emerging new roles for an ancient RNA in the regulation of gene expression. Life. 2015;5(4):1638–51.
- Sobala A, Hutvagner G. Transfer RNA-derived fragments: origins, processing, and functions. Wiley Interdiscip Rev RNA. 2011;2(6):853–62.
- Dieci G, Preti M, Montanini B. Eukaryotic snoRNAs: a paradigm for gene expression flexibility. Genomics. 2009;94(2):83–8.
- Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–74. [CrossRef]
- Huang Z hao, Du Y ping, Wen J tao, Lu B feng, Zhao Y. snoRNAs: functions and mechanisms in biological processes, and roles in tumor pathophysiology. Cell Death Discov. 2022;8(1):259.
- Ozata DM, Gainetdinov I, Zoch A, O’Carroll D, Zamore PD. PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet. 2019;20(2):89–108.
- Wang J, Shi Y, Zhou H, Zhang P, Song T, Ying Z, et al. piRBase: integrating piRNA annotation in all aspects. Nucleic Acids Res. 2022;50(D1):D265–72.
- Stoyko D, Genzor P, Haase AD. Hierarchical length and sequence preferences establish a single major piRNA 3′-end. Iscience. 2022;25(6).
- Perera BPU, Tsai ZTY, Colwell ML, Jones TR, Goodrich JM, Wang K, et al. Somatic expression of piRNA and associated machinery in the mouse identifies short, tissue-specific piRNA. Epigenetics. 2019;14(5):504–21.
- Wang X, Ramat A, Simonelig M, Liu MF. Emerging roles and functional mechanisms of PIWI-interacting RNAs. Nat Rev Mol Cell Biol. 2023;24(2):123–41.
- Czech B, Munafò M, Ciabrelli F, Eastwood EL, Fabry MH, Kneuss E, et al. piRNA-guided genome defense: from biogenesis to silencing. Annu Rev Genet. 2018;52(1):131–57.
- Mirzaei S, Gholami MH, Hushmandi K, Hashemi F, Zabolian A, Canadas I, et al. The long and short non-coding RNAs modulating EZH2 signaling in cancer. J Hematol Oncol. 2022;15(1):18. [CrossRef]
- Aalto AP, Pasquinelli AE. Small non-coding RNAs mount a silent revolution in gene expression. Curr Opin Cell Biol. 2012;24(3):333–40.
- Redis RS, Calin GA. SnapShot: non-coding RNAs and metabolism. Cell Metab. 2017;25(1):220.
- E Nicolas F. Role of ncRNAs in development, diagnosis and treatment of human cancer. Recent Pat Anticancer Drug Discov. 2017;12(2):128–35.
- Karnati HK, Panigrahi MK, Gutti RK, Greig NH, Tamargo IA. miRNAs: key players in neurodegenerative disorders and epilepsy. J Alzheimer’s Dis. 2015;48(3):563–80.
- Nwaobi SE, Lin E, Peramsetty SR, Olsen ML. DNA methylation functions as a critical regulator of Kir4. 1 expression during CNS development. Glia. 2014;62(3):411–27.
- Petazzi P, Sandoval J, Szczesna K, Jorge OC, Roa L, Sayols S, et al. Dysregulation of the long non-coding RNA transcriptome in a Rett syndrome mouse model. RNA Biol. 2013;10(7):1197–203. [CrossRef]
- Obiols-Guardia A, Guil S. The role of noncoding RNAs in neurodevelopmental disorders: the case of Rett syndrome. Neuroepigenomics aging Dis. 2017;23–37.
- Wang ITJ, Allen M, Goffin D, Zhu X, Fairless AH, Brodkin ES, et al. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc Natl Acad Sci. 2012;109(52):21516–21.
- Rishik S, Hirsch P, Grandke F, Fehlmann T, Keller A. miRNATissueAtlas 2025: an update to the uniformly processed and annotated human and mouse non-coding RNA tissue atlas. Nucleic Acids Res. 2025;53(D1):D129–37. [CrossRef]
- Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48(D1):D127–31.
- Schroeder E, Yuan L, Seong E, Ligon C, DeKorver N, Gurumurthy CB, et al. Neuron-type specific loss of CDKL5 leads to alterations in mTOR signaling and synaptic markers. Mol Neurobiol. 2019;56(6):4151–62.
- Zhu ZA, Li YY, Xu J, Xue H, Feng X, Zhu YC, et al. CDKL5 deficiency in adult glutamatergic neurons alters synaptic activity and causes spontaneous seizures via TrkB signaling. Cell Rep. 2023;42(10). [CrossRef]
- Silvestre M, Dempster K, Mihaylov SR, Claxton S, Ultanir SK. Cell type-specific expression, regulation and compensation of CDKL5 activity in mouse brain. Mol Psychiatry. 2024;1–13. [CrossRef]
- Eddy SR. Non–coding RNA genes and the modern RNA world. Nat Rev Genet. 2001;2(12):919–29.
- Chen Y, Mateski J, Gerace L, Wheeler J, Burl J, Prakash B, et al. Non-coding RNAs and neuroinflammation: implications for neurological disorders. Exp Biol Med. 2024;249:10120.
- Wang SW, Liu Z, Shi ZS. Non-coding RNA in acute ischemic stroke: mechanisms, biomarkers and therapeutic targets. Cell Transplant. 2018;27(12):1763–77.
- Salvatori B, Biscarini S, Morlando M. Non-coding RNAs in nervous system development and disease. Front cell Dev Biol. 2020;8:273.
- Siqueira E, Velasco CD, Tarrasón A, Soler M, Srinivas T, Setién F, et al. NEAT1-mediated regulation of proteostasis and mRNA localization impacts autophagy dysregulation in Rett syndrome. Nucleic Acids Res. 2025;53(4):gkaf074.
- Henshall DC. MicroRNAs Fine-Tune Brain and Body Communication in Health and Disease. Brain-Body Connect. 2025;311–37.
- Mari F, Azimonti S, Bertani I, Bolognese F, Colombo E, Caselli R, et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet. 2005;14(14):1935–46.
- Carouge D, Host L, Aunis D, Zwiller J, Anglard P. CDKL5 is a brain MeCP2 target gene regulated by DNA methylation. Neurobiol Dis. 2010;38(3):414–24.
- Urdinguio RG, Fernandez AF, Lopez-Nieva P, Rossi S, Huertas D, Kulis M, et al. Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics. 2010;5(7):656–63. [CrossRef]
- Ashhab MU, Omran A, Gan N, Kong H, Peng J, Yin F. microRNA s (9, 138, 181A, 221, and 222) and mesial temporal lobe epilepsy in developing brains. Transl Neurosci. 2013;4(3):357–62.
- Yousefi MJ, Rezvanimehr A, Saleki K, Mehrani A, Barootchi E, Ramezankhah M, et al. Inflammation-related microRNA alterations in epilepsy: a systematic review of human and animal studies. Rev Neurosci. 2025; [CrossRef]
- Rajabi M, Kalantar SM, Mojodi E, Salehi M, Firouzabadi RD, Etemadifar SM, et al. Assessment of circulating miRNA-218, miRNA-222, and miRNA-146 as biomarkers of polycystic ovary syndrome in epileptic patients receiving valproic acid. Biomed Res Ther. 2023;10(9):5884–95.
- Szydlowska K, Bot A, Nizinska K, Olszewski M, Lukasiuk K. Circulating microRNAs from plasma as preclinical biomarkers of epileptogenesis and epilepsy. Sci Rep. 2024;14(1):708.
- Rehmsmeier M, Steffen P, Höchsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. Rna. 2004;10(10):1507–17.
- Vorozheykin PS, Titov II. Web server for prediction of miRNAs and their precursors and binding sites. Mol Biol. 2015;49:755–61.
- Christopher AF, Kaur RP, Kaur G, Kaur A, Gupta V, Bansal P. MicroRNA therapeutics: discovering novel targets and developing specific therapy. Perspect Clin Res. 2016;7(2):68–74.
- Hogg MC, Raoof R, El Naggar H, Monsefi N, Delanty N, O’Brien DF, et al. Elevation of plasma tRNA fragments precedes seizures in human epilepsy. J Clin Invest. 2019;129(7):2946–51.
- Jirström E, Matveeva A, Baindoor S, Donovan P, Ma Q, Morrissey EP, et al. Effects of ALS-associated 5’tiRNAGly-GCC on the transcriptomic and proteomic profile of primary neurons in vitro. Exp Neurol. 2025;385:115128.
- Schaffer AE, Pinkard O, Coller JM. tRNA metabolism and neurodevelopmental disorders. Annu Rev Genomics Hum Genet. 2019;20(1):359–87.
- McArdle H, Hogg MC, Bauer S, Rosenow F, Prehn JHM, Adamson K, et al. Quantification of tRNA fragments by electrochemical direct detection in small volume biofluid samples. Sci Rep. 2020;10(1):7516.
- Fagan SG, Helm M, Prehn JHM. tRNA-derived fragments: A new class of non-coding RNA with key roles in nervous system function and dysfunction. Prog Neurobiol. 2021;205:102118.
- Kapur M, Ganguly A, Nagy G, Adamson SI, Chuang JH, Frankel WN, et al. Expression of the Neuronal tRNA n-Tr20 Regulates Synaptic Transmission and Seizure Susceptibility. Neuron. 2020 Oct;108(1):193-208.e9. [CrossRef]
- Karijolich J, Yu YT. Spliceosomal snRNA modifications and their function. RNA Biol. 2010;7(2):192–204. [CrossRef]
- Zhang Z, Huang R, Lai Y. Expression signature of ten small nuclear RNAs serves as novel biomarker for prognosis prediction of acute myeloid leukemia. Sci Rep. 2023;13(1):18489.
- Matera AG, Terns RM, Terns MP. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat Rev Mol cell Biol. 2007;8(3):209–20.
- Watkins NJ, Bohnsack MT. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA. 2012;3(3):397–414.
- Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3(3):179–92. [CrossRef]
- Falaleeva M, Stamm S. Processing of snoRNAs as a new source of regulatory non-coding RNAs: snoRNA fragments form a new class of functional RNAs. Bioessays. 2013;35(1):46–54.
- Zhong F, Zhou N, Wu K, Guo Y, Tan W, Zhang H, et al. A SnoRNA-derived piRNA interacts with human interleukin-4 pre-mRNA and induces its decay in nuclear exosomes. Nucleic Acids Res. 2015;43(21):10474–91.
- Ricciardi S, Ungaro F, Hambrock M, Rademacher N, Stefanelli G, Brambilla D, et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1–PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat Cell Biol. 2012;14(9):911–23. [CrossRef]
- Zhao P ping, Yao M jin, Chang S yuan, Gou L tao, Liu M fang, Qiu Z long, et al. Novel function of PIWIL1 in neuronal polarization and migration via regulation of microtubule-associated proteins. Mol Brain. 2015;8:1–12.
- Ghosheh Y, Seridi L, Ryu T, Takahashi H, Orlando V, Carninci P, et al. Characterization of piRNAs across postnatal development in mouse brain. Sci Rep. 2016;6(1):25039.
- Nandi S, Chandramohan D, Fioriti L, Melnick AM, Hébert JM, Mason CE, et al. Roles for small noncoding RNAs in silencing of retrotransposons in the mammalian brain. Proc Natl Acad Sci. 2016;113(45):12697–702. [CrossRef]
- Leighton LJ, Wei W, Marshall PR, Ratnu VS, Li X, Zajaczkowski EL, et al. Disrupting the hippocampal Piwi pathway enhances contextual fear memory in mice. Neurobiol Learn Mem. 2019;161:202–9.
- Yang Z, Chen KM, Pandey RR, Homolka D, Reuter M, Janeiro BKR, et al. PIWI slicing and EXD1 drive biogenesis of nuclear piRNAs from cytosolic targets of the mouse piRNA pathway. Mol Cell. 2016;61(1):138–52.
- De Fazio S, Bartonicek N, Di Giacomo M, Abreu-Goodger C, Sankar A, Funaya C, et al. The endonuclease activity of Mili fuels piRNA amplification that silences LINE1 elements. Nature. 2011;480(7376):259–63. [CrossRef]
- Reuter M, Berninger P, Chuma S, Shah H, Hosokawa M, Funaya C, et al. Miwi catalysis is required for piRNA amplification-independent LINE1 transposon silencing. Nature. 2011;480(7376):264–7.
- Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature. 2011;479(7374):534–7.
- Blaudin de Thé F, Rekaik H, Peze-Heidsieck E, Massiani-Beaudoin O, Joshi RL, Fuchs J, et al. Engrailed homeoprotein blocks degeneration in adult dopaminergic neurons through LINE-1 repression. EMBO J. 2018;37(15):e97374. [CrossRef]
- Sun W, Samimi H, Gamez M, Zare H, Frost B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat Neurosci. 2018;21(8):1038–48.
- Schulze M, Sommer A, Plötz S, Farrell M, Winner B, Grosch J, et al. Sporadic Parkinson’s disease derived neuronal cells show disease-specific mRNA and small RNA signatures with abundant deregulation of piRNAs. Acta Neuropathol Commun. 2018;6:1–18.
- Zhan L, Chen M, Pang T, Li X, Long L, Liang D, et al. Attenuation of Piwil2 induced by hypoxic postconditioning prevents cerebral ischemic injury by inhibiting CREB2 promoter methylation. Brain Pathol. 2023;33(1):e13109. [CrossRef]
- Saxena A, Tang D, Carninci P. piRNAs warrant investigation in Rett Syndrome: an omics perspective. Dis Markers. 2012;33(5):261–75.
- Qiu W, Guo X, Lin X, Yang Q, Zhang W, Zhang Y, et al. Transcriptome-wide piRNA profiling in human brains of Alzheimer’s disease. Neurobiol Aging. 2017;57:170–7. [CrossRef]
- Iossifov I, O’roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–21.
- Roy J, Sarkar A, Parida S, Ghosh Z, Mallick B. Small RNA sequencing revealed dysregulated piRNAs in Alzheimer’s disease and their probable role in pathogenesis. Mol Biosyst. 2017;13(3):565–76. [CrossRef]
Figure 2.
Small RNA landscape of hippocampal tissue extracted from 10 heterozygous (Het) and 10 wildtype (WT) mice (A). Images showing the percentage of different small RNA species detected in tissue samples obtained from Het and WT mice. Note, majority of reads mapped to miRNAs (B).
Figure 2.
Small RNA landscape of hippocampal tissue extracted from 10 heterozygous (Het) and 10 wildtype (WT) mice (A). Images showing the percentage of different small RNA species detected in tissue samples obtained from Het and WT mice. Note, majority of reads mapped to miRNAs (B).
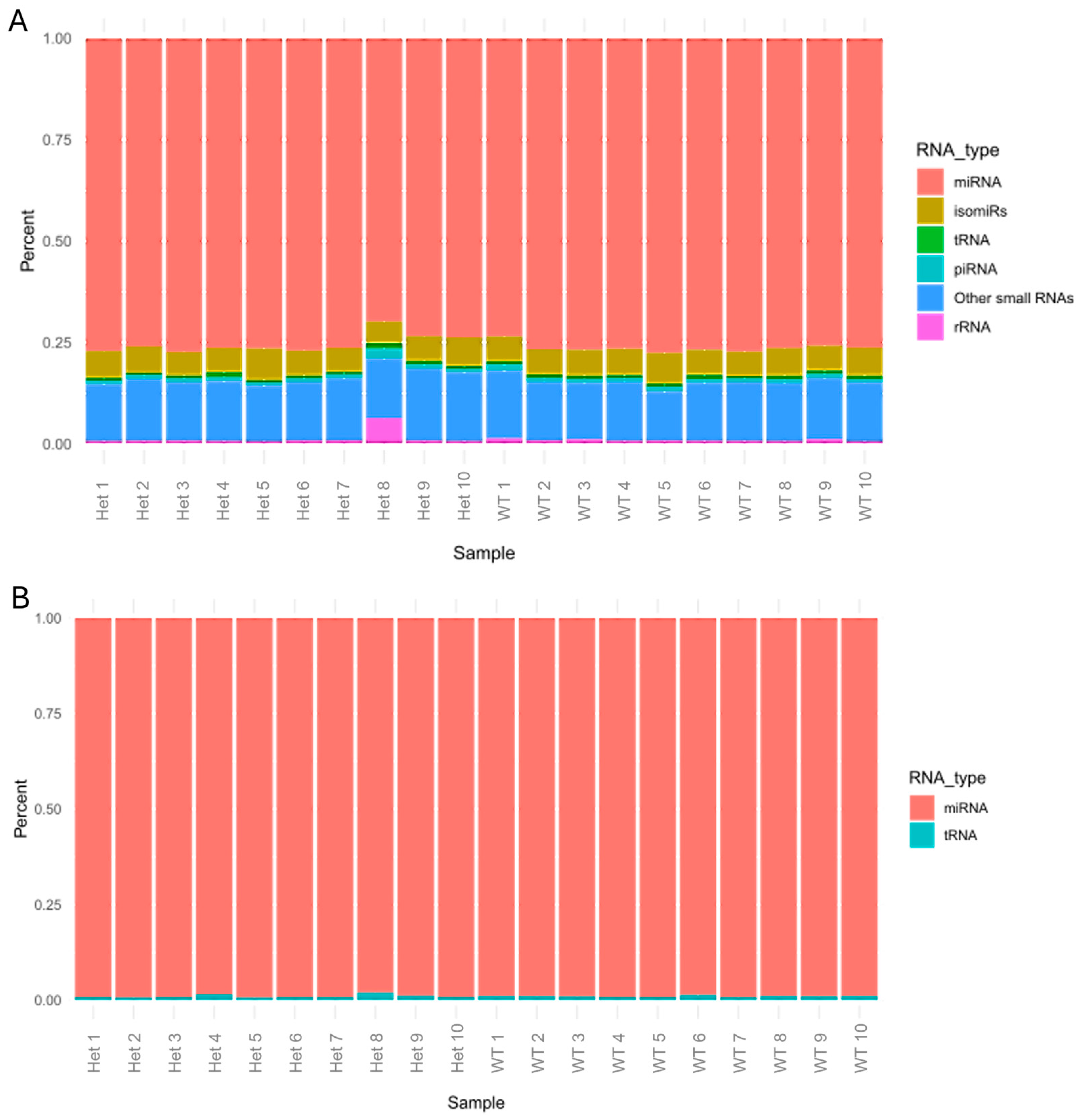
Figure 3.
(A) Volcano plots depicting miRNAs expression 10 wildtype female (WT) mice compared to 10 heterozygous (Het; CDD) female mice using Next Generation Sequencing (NGS). (B) heatmap showing the top 50 microRNAs expression. (C) the upregulated (blue bars) and downregulated (red bars) microRNAs in CDD (Het) mice. (D) Scatter plot illustrating the log fold change vs average hippocampal expression taken from miRNA tissue atlas [54]. Each point represents a single miRNA with its colour/size according to its predicted targets from miRDB (mouse mirdb data base, [55]).
Figure 3.
(A) Volcano plots depicting miRNAs expression 10 wildtype female (WT) mice compared to 10 heterozygous (Het; CDD) female mice using Next Generation Sequencing (NGS). (B) heatmap showing the top 50 microRNAs expression. (C) the upregulated (blue bars) and downregulated (red bars) microRNAs in CDD (Het) mice. (D) Scatter plot illustrating the log fold change vs average hippocampal expression taken from miRNA tissue atlas [54]. Each point represents a single miRNA with its colour/size according to its predicted targets from miRDB (mouse mirdb data base, [55]).
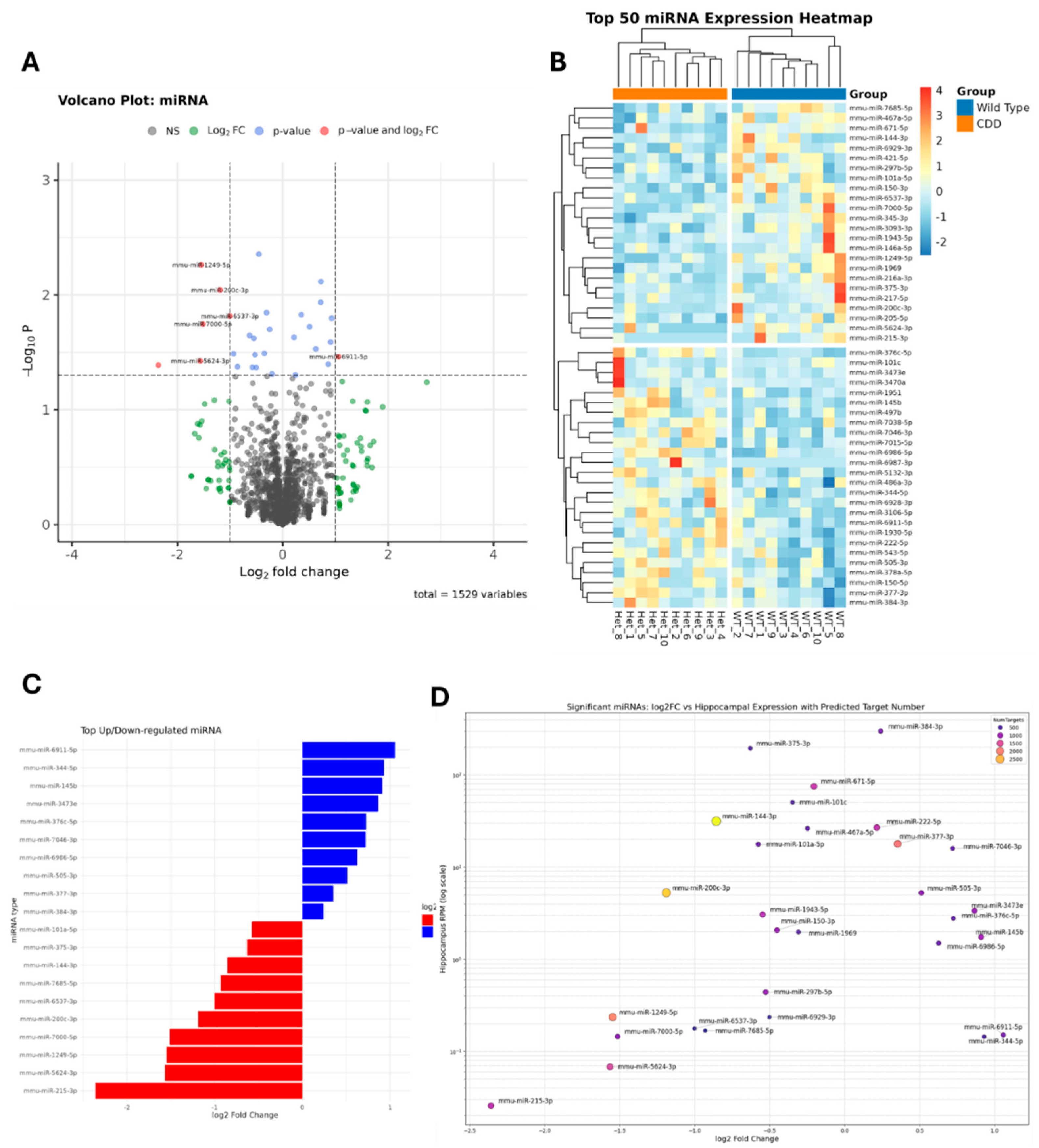
Figure 4.
(A) Volcano plots depicting tRNAs expression using Next Generation Sequencing (NGS). Downstream analysis of 10 wildtype female (WT) mice compared to 10 heterozygous (Het; CDD) female mice. (B) heatmap showing the top 50 tRNAs expression. (C) the unregulated (blue bars) and downregulated (red bars) tRNAs in CDD (Het) mice.
Figure 4.
(A) Volcano plots depicting tRNAs expression using Next Generation Sequencing (NGS). Downstream analysis of 10 wildtype female (WT) mice compared to 10 heterozygous (Het; CDD) female mice. (B) heatmap showing the top 50 tRNAs expression. (C) the unregulated (blue bars) and downregulated (red bars) tRNAs in CDD (Het) mice.
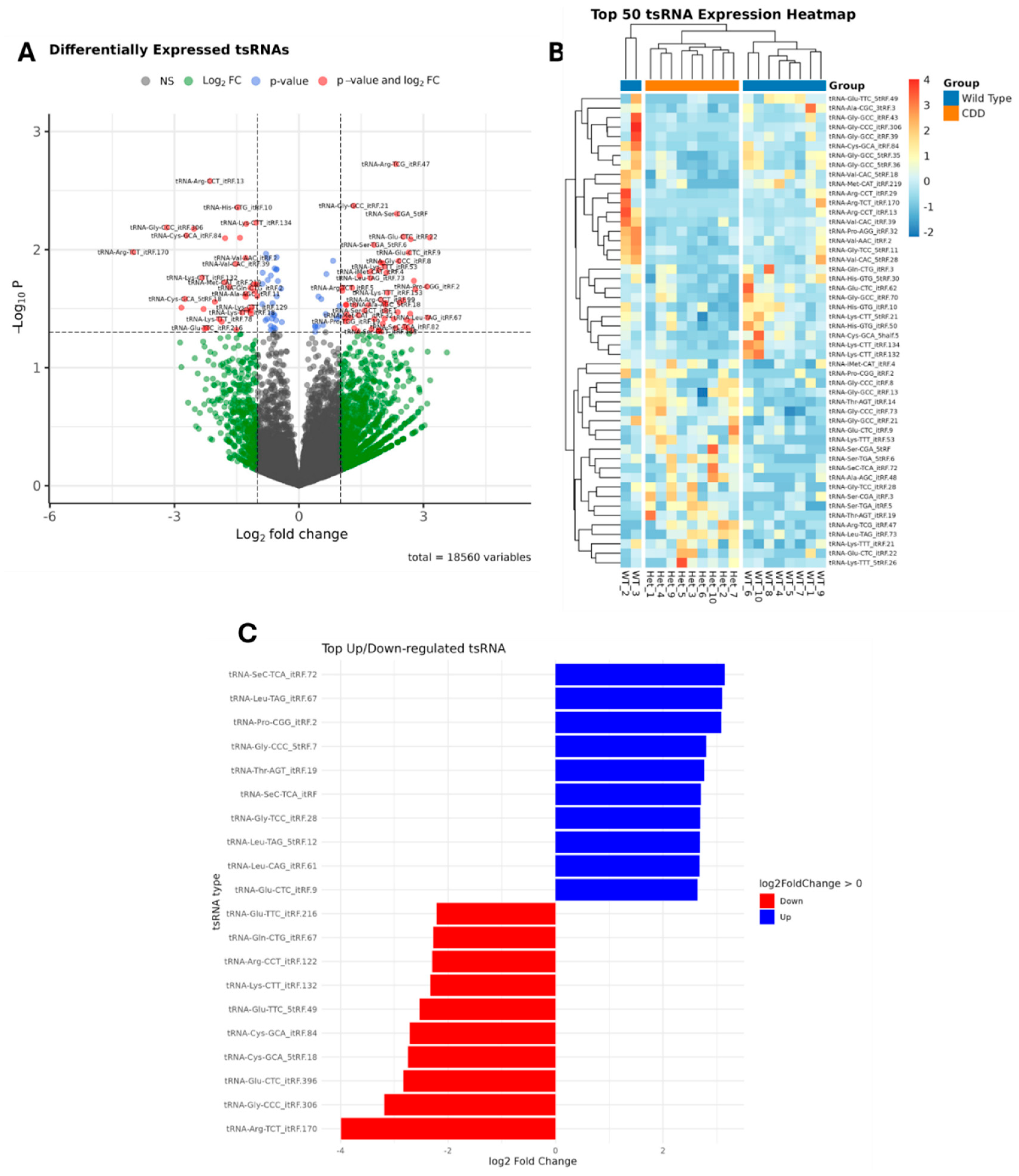
Figure 5.
Volcano plots of other ncRNAs differentially expressed including piRNAs (A), snoRNA (B) and snRNA (C) between heterozygous (Het) and wild-type (WT) mice. Downstream analysis of 10 wildtype female mice compared to 10 heterozygous female mice. All mice used were age matched. Dots colored red show statically significant difference (p< 0.05).
Figure 5.
Volcano plots of other ncRNAs differentially expressed including piRNAs (A), snoRNA (B) and snRNA (C) between heterozygous (Het) and wild-type (WT) mice. Downstream analysis of 10 wildtype female mice compared to 10 heterozygous female mice. All mice used were age matched. Dots colored red show statically significant difference (p< 0.05).
Figure 1.
Breeding Scheme used to develop the initial experimental colony (A) and CDKL5 experimental mice (B). (A) The offspring produced from breading wild-type (WT) female mice (Cdkl5+/+) with a knock-out (KO) male mice (Cdkl5 -/y) result in 50% heterozygous female (Het) mice (Cdkl5 +/-) and 50% WT male mice (Cdkl5 +/y) population. (B) A WT male mouse (CDKL5 +/y) is bread with Het female mice (CDKL5 +/-). The offspring produced by the breading scheme include WT female (CDKL5 +/+), WT male (CDKL5+/y), Het female (CDKL5 +/-) and KO male (CDKL5 -/y), each offspring has equal probability.
Figure 1.
Breeding Scheme used to develop the initial experimental colony (A) and CDKL5 experimental mice (B). (A) The offspring produced from breading wild-type (WT) female mice (Cdkl5+/+) with a knock-out (KO) male mice (Cdkl5 -/y) result in 50% heterozygous female (Het) mice (Cdkl5 +/-) and 50% WT male mice (Cdkl5 +/y) population. (B) A WT male mouse (CDKL5 +/y) is bread with Het female mice (CDKL5 +/-). The offspring produced by the breading scheme include WT female (CDKL5 +/+), WT male (CDKL5+/y), Het female (CDKL5 +/-) and KO male (CDKL5 -/y), each offspring has equal probability.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



