
Submitted:
08 May 2025
Posted:
08 May 2025
You are already at the latest version
Abstract
Zebrafish is a well-recognized model for studying human genetic disorders. Recently, we proposed the homozygous cdkl5sa21938 mutant zebrafish as a model of CDKL5 deficiency disorder (CDD), a developmental epileptic encephalopathy with diverse symptoms. This study aimed to explore Cdkl5-associated molecular mechanisms in zebrafish and assess their similarity to those in mammals. We conducted RNA-sequencing on whole cdkl5-/- zebrafish and wild-type siblings at 5 and 35 days post-fertilization (dpf) to compare their gene expression profiles. Most significant differentially expressed genes (DEGs) were related to muscle, neuronal and visual systems which are affected in CDD. Gene Ontology analysis revealed downregulated DEGs enriched in muscle development, extracellular matrix and actin cytoskeleton functions at both stages, while upregulated DEGs were enriched in eye development functions at 35 dpf. KEGG analysis revealed enrichment of downregulated DEGs in focal adhesion and ECM-receptor interaction pathways at both stages. Neuronal development DEGs were mainly downregulated at both stages, while synaptic signaling DEGs were upregulated at 35 dpf. Crossing cdkl5-/- mutants with the Hb9:GFP transgenic line showed fewer motor neurons cells with shorter axons compared to the wild type, which may explain the impaired motor phenotype observed in zebrafish and CDD patients. Moreover, we identified key downregulated DEGs related to cartilage development at both stages and bone development at 35 dpf, potentially explaining the skeletal defects seen in zebrafish and CDD individuals. In conclusion, Cdkl5 loss in zebrafish leads to dysregulation of genes involved in CDKL5-associated functions in mammals, providing new insights into its less studied functions and phenotypes.
Keywords:
Zebrafish model
; Cdkl5
; RNA-seq
; Transcriptomic
; CDKL5 deficiency disorder
1. Introduction
Human cyclin-dependent kinase-like 5 (CDLK5) is encoded by a single-copy gene located on the short arm of the X chromosome. It is widely expressed in many tissues throughout the human body. However, it is most abundant in the brain (specifically in forebrain, cerebral cortex, hippocampus, striatum and olfactory bulb), testicles and thymus[1,2,3]. During the embryonic stages, CDKL5 expression is minimal but it quickly increases in postnatal stages and throughout neuronal development and synaptogenesis, indicating that CDKL5 is crucial for early normal brain development and function[3,4,5]. At subcellular level, it is found both in the nucleus and in the cytoplasm, where it plays specific roles involved in important neuronal processes[6].
CDKL5 is an important protein kinase that is associated with several essential functions through the phosphorylation of its targets and interactions or associations with other proteins[7]. Although the extent of CDKL5 exact functions remains to be elucidated, it is known that in the cytoplasm it regulates axon outgrowth through the interaction with shootin1 (SHTN1)[8]. It is also involved in the regulation of the actin cytoskeleton and dendritic arborization through interaction with Ras-related C3 botulinum toxin substrate 1 (Rac1)[9] and in synapse formation via phosphorylation of amphiphysin 1 (AMPH1)[10,11] and the netrin G1 receptor-protein membrane (NGL-1)[5]. CDKL5 also regulates dendritic microtubule dynamics by direct phosphorylation of microtubule-associated protein 1S (MAP1S)[12,13], EB family member 2 (EB2)[13], Rho guanine nucleotide exchange factor 2 (ARHGEF2)[13] and DLG5 (Discs Large MAGUK Scaffold Protein 5)[12]. Furthermore, by direct phosphorylation of CEP131, CDKL5 also plays a role in ciliogenesis[12]. In the nucleus, CDKL5 phosphorylates the DNA methyltransferase 1 (DNMT1)[14] and the methyl CpG-binding protein 2 (MeCP2), thus regulating gene expression through its effect on DNA methylation [15].
Disruption of CDKL5, resulting in decreased activity, leads to CDKL5 deficiency disorder (CDD), a rare and severe condition classified as a developmental epileptic encephalopathy[16,17]. Mutations in the CDKL5 gene are responsible for the development of this disorder, which can manifest with a broad range of symptoms. It is primarily characterized by early-onset, treatment-resistant seizures (which begin within the first three months of life) and severe neurodevelopmental impairments, including deficits in motor, communication, and cognitive development[16,17,18]. Other common phenotypes include hypotonia, cortical visual impairment, microcephaly, subtle dysmorphic features, scoliosis, sleep disturbances and gastrointestinal problems[16,19]. Currently, there is no cure available for CDD affected individuals and since targeted therapies are scarce, treatment only focus on seizure management and in relieving the symptoms[18,19,20].
Despite extensive efforts to elucidate the molecular factors underlying the onset and progression of CDD, its development is still unclear. Several mouse models have been generated to understand how CDKL5 deficiency leads to neurological defects[21,22,23,24,25]. These models recapitulate several phenotypes of the human disorder such as abnormal eye tracking, impaired learning and memory, motor dysfunction and changes in locomotion[21,24]. However, these studies only focus on the neurological phenotypes of the disorder, thus our current knowledge of CDKL5 function in other organs/tissues besides the brain is still limited. The comprehension of affected gene regulation and signaling pathways is crucial for unraveling the mechanisms underlying both normal physiology and disease states. Few studies on transcriptomic analysis in the context of Cdkl5 deficiency have been reported, and those that exist were performed using specific brain regions from Cdkl5 null mice[26]. Previously, we identified zebrafish as a promising tool to investigate CDD. We then characterized and validated the first cdkl5 mutant zebrafish line (sa21938)[27], suggesting its potential as a valuable model for exploring the underlying mechanisms of CDD and its relevance in high-throughput drug screening[28]. This zebrafish mutant line harbors a nonsense mutation that introduces a premature stop codon, leading to either mRNA degradation or the production of a truncated protein, resulting, among other phenotypes, in altered motor behavior, increased susceptibility to seizures, and skeletal defects[28].
The main objectives of this study were to provide new insights into the molecular mechanisms and pathways linked to Cdkl5 functions with a focus on its unexplored roles and to evaluate whether the cdkl5sa21938 mutant zebrafish line shares molecular similarities with mammalian systems that reproduce the CDD phenotypes. The validation of this model would enable its use to further investigate CDD pathophysiology and contribute to the discovery of new therapeutic molecules. To that end, we investigated the transcriptomic alterations caused by the loss of Cdkl5 function in cdkl5 homozygous zebrafish at five days post-fertilization (dpf) and 35 dpf using high-throughput RNA sequencing approach.
2. Materials and Methods
2.1. Ethics Statement
All animal procedures carry out in the present study were performed in compliance with ARRIVE guidelines (https://arriveguidelines.org) and according to the EU and Portuguese legislation for animal experimentation and welfare (Directives 86/609 CEE and 2010/63/EU; Decreto-Lei 113/2013; Portaria1005/92, 466/95 and 1131/97). This study was approved by the Portuguese Direção-Geral de Alimentação e Veterinária (authorization no. 0421/2021). Qualified operators conducted animal handling and experimentation, prioritizing the minimization of pain, distress, and discomfort.
2.2. Fish Maintenance
Experiments were conducted using the cdkl5sa21938 mutant zebrafish line (obtained through European Zebrafish Resource Center) and their wild-type (WT) siblings. All adult zebrafish and embryos were maintained in the animal facility in a recirculating water system under controlled temperature (28 °C) and lighting (14h light -10h dark cycle). For the experiments, homozygous cdkl5sa21938 (cdkl5-/-) mutant adult fish were incrossed to generate cdkl5-/- embryos, while WT embryos were obtained by crossing adult WT siblings, which were the offspring of heterozygous cdkl5sa21938 mutant incrosses. The cdkl5-/- fish were also crossed with Tg(Hb9:GFP) transgenic reporter fish line[29,30]. The resulting heterozygous zebrafish were incrossed to obtain the homozygous lines, which were confirmed by genotyping.
2.3. RNA Extraction
Total RNA was isolated from embryos and juveniles of homozygous cdkl5sa21938 and their WT siblings at 5 and 35 days post-fertilization, respectively, using the NZYOL reagent (NZYTech, Lisbon, Portugal) and following the manufacturer’s instructions. Each group comprised five biological replicates, prepared from pools of 50 embryos or 7 juveniles. DNase treatment was then performed using the RNase-Free DNase set (QIAGEN, Hilden, Germany) to eliminate any remaining genomic DNA contamination. To increase purity, RNA samples were subjected to the RNeasy kit (QIAGEN) clean-up protocol. The RNA quantity and purity were assessed using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, USA.), and its integrity was verified by electrophoresis on a 1% agarose gel.
2.4. RNA Sequencing (RNA-Seq)
The purified RNA was delivered to Novogene company (Cambridge, UK) for library construction and sequencing. Integrity of RNA samples were analyzed on the Agilent 2100 bioanalyzer and only samples with an RNA integrity number (RIN) > 8 were further used for cDNA library preparation. Briefly, mRNA was separated from total RNA using poly-T oligo-attached magnetic beads and subjected to fragmentation using divalent cations under high temperature. Then, the first strand cDNA was synthesized using random hexamer primers and M-MuLV Reverse Transcriptase, followed by the second strand cDNA synthesis using DNA polymerase I and RNase H. The lasting overhangs were transformed into blunt ends through polymerase and exonuclease reactions. To prepare for hybridization, adaptors containing a hairpin loop structure were ligated to the previously 3’ ends adenylated DNA fragments. The AMPure XP system (Beckman Coulter, Beverly, USA) was used to purify the cDNA fragments with 370~420 bp in length that were then digested with USER enzyme. PCR was carried out using Phusion High-Fidelity DNA polymerase, universal PCR primers and Index (X) primer. The PCR products were purified, and the Agilent Bioanalyzer 2100 system was used to evaluate the library quality. Finally, directional (strand-specific) libraries were sequenced on the Illumina Novaseq platform, generating 150-bp paired-end reads for each sample.
2.5. Bioinformatic Analysis of RNA-Seq Data
For the RNA-Seq data analysis, raw reads in fastq format were first trimmed to obtain clean reads through the elimination of low-quality reads, adapter-containing reads, and ploy-N-containing reads. After filtering, clean reads were aligned to the zebrafish reference genome (Danio rerio GRCz11) using Hisat2 (v2.0.5). For gene expression quantification, featureCounts (v1.5.0-p3) was used to count the number of reads mapped to each gene. Fragments per kilobase per million mapped fragments (FPKM) for each gene were then calculated, considering the gene length and the total number of mapped reads. Genes with FPKM values greater than 1 were considered to be expressed. Analysis of the differential expression between cdkl5 mutant and WT zebrafish was performed using the DESeq2 R package (1.20.0). The resulting p-values were adjusted (padj) according to the method proposed by Benjamini and Hochberg[31] for controlling the false discovery rate. Genes with a p-value < 0.05 and |log2(FoldChange)| > 0 found by DESeq2 were considered as differentially expressed. Gene Ontology (GO) enrichment analysis of differentially expressed genes (DEGs) was performed using the clusterProfiler R package. GO terms with corrected p-value < 0.05 were considered significantly enriched by differential expressed genes. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was performed to identify significantly enriched pathways associated with DEGs. The clusterProfiler package in R was used to test the statistical enrichment of DEGs in KEGG pathways.
2.6. cDNA Synthesis
Total RNA was reverse-transcribed into cDNA using the Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen, Carlsbad, USA). Briefly, 1µg of RNA was combined with 0.4 µl of oligo(dT) primer (50 mM), 1 µl of dNTPs (10 mM) and DNase/RNase-free water to a final volume of 12 µl, then incubated at 65°C for 5 min. Next, 4 µl of 5× first strand buffer, 2 µl of DTT and 1 µl of Ribolock RNase inhibitor were added, followed by incubation at 37°C for 2 min. Subsequently, 1 µl of M-MLV was added, and the reverse transcription continued for 50 min at 37°C. Finally, the mixture was incubated at 70°C for 15 min to inactivate the enzyme.
2.7. Quantitative Real-Time PCR (qPCR)
Gene-specific amplifications by qPCR were performed in 20 µl reactions containing 10 µl of SensiFAST SYBR Hi-ROX (Meridian Bioscience, Cincinnati, USA), 0.8 µl of each forward and reverse set of primers (10 µM) and 2 μl of cDNA (diluted 1:10). Gene-specific primers are listed in Supplementary Table S1. The qPCR reactions were carried out on a 96-well plate, using a CFX Connect Real-Time PCR Thermocycler (Biorad, Hercules, USA) and under the following conditions: an initial denaturation step at 95°C for 2 min, followed by 40 cycles of amplification (each cycle is 5 s at 95°C, 20 s at 60°C). After the amplification was completed, a melting curve was generated by gradually heating the sample to 95 °C at a rate of 0.1 °C per second, while continuously capturing the fluorescence. For each sample, at least two technical replicates were performed. Relative gene expression levels were determined applying the ΔΔCt comparative method and were normalized to the β-actin and rps18 reference genes for 5 dpf and 35 dpf samples, respectively (Supplementary Table S1).
2.8. Imaging and Analysis of the Tg(hb9: GFP) Embryos
Embryos with 3 dpf were anesthetized in MS-222, oriented on the lateral side on top of 0.8% agarose and fluorescent images were acquired using the ZEISS AXIO Zoom V16 microscope. Images were captured and processed using ZEN 3.7 software. The deconvolution function (deblurring) was used to improve resolution, using the same parameters in all images. The number of GFP+ spinal motor neurons present in three hemisegments after the distal end of the yolk extension were quantified. The length of five motor axons (located immediately after the yolk sac, over the yolk extension) per embryo was measured using the Neuroanatomy/SNT plug-in[32] for ImageJ/Fiji. The average of the five values was calculated for subsequent statistical analysis.
2.9. Statistical Analysis
Statistical analysis was performed using Prism 8 (GraphPad Software). The Kolmogorov-Smirnov test was used to determine the normality of the data. Data with a normal distribution are presented as mean ± SD. One-way analysis of variance (ANOVA) followed by Tukey’s post-test was used to identify significant differences between three groups, while a t-test with Welch’s correction was applied for comparisons between two groups. Data that did not pass the normality test are presented as median with the interquartile region (boxplot). The Kruskal-Wallis test, followed by Dunn’s multiple comparisons test, was used to identify significant differences between three groups, while the Mann-Whitney test was used for comparisons between two groups. Differences were considered statistically significant when p < 0.05.
3. Results
3.1. RNA-seq Data Description and Validation
To identify direct and indirect genes and molecular pathways affected by the potential loss of Cdkl5 function, a high-throughput gene expression analysis through RNA sequencing (RNA-seq) was performed. This analysis compared the transcriptomes of cdkl5-/- mutant zebrafish and their wild-type (WT) siblings at 5 and 35 days post-fertilization with five biological replicates in each group. The RNA-seq data was deposited in the Gene Expression Omnibus (GEO) repository (accession number: GSE294284).
A summary of the RNA-Seq descriptive data is presented in Table 1.
After sequencing, a total of 466,025,230 and 458,390,836 raw reads were generated from 5 dpf embryos and 35 dpf juveniles’ samples, respectively. The obtained sequencing error rate, the quality score of Q30 and the average GC content for all samples validate the good quality of the sequencing. Subsequent data filtering by removal of low-quality reads, adapter-containing reads, and ploy-N-containing reads from 5 dpf embryos and 35 dpf juvenile, identified the high-quality clean reads, which were aligned against the zebrafish reference genome and mapping rates ranging from 87.40% to 92.41% were obtained (Table 1). The percentage of mapped reads should be higher than 70% when an adequate genome is chosen, and no contamination occurs. Therefore, these outcomes indicate that the obtained sequencing data are accurate and reliable.
3.2. Gene Expression Analysis of cdkl5-/- Mutant and WT Siblings’ Zebrafish
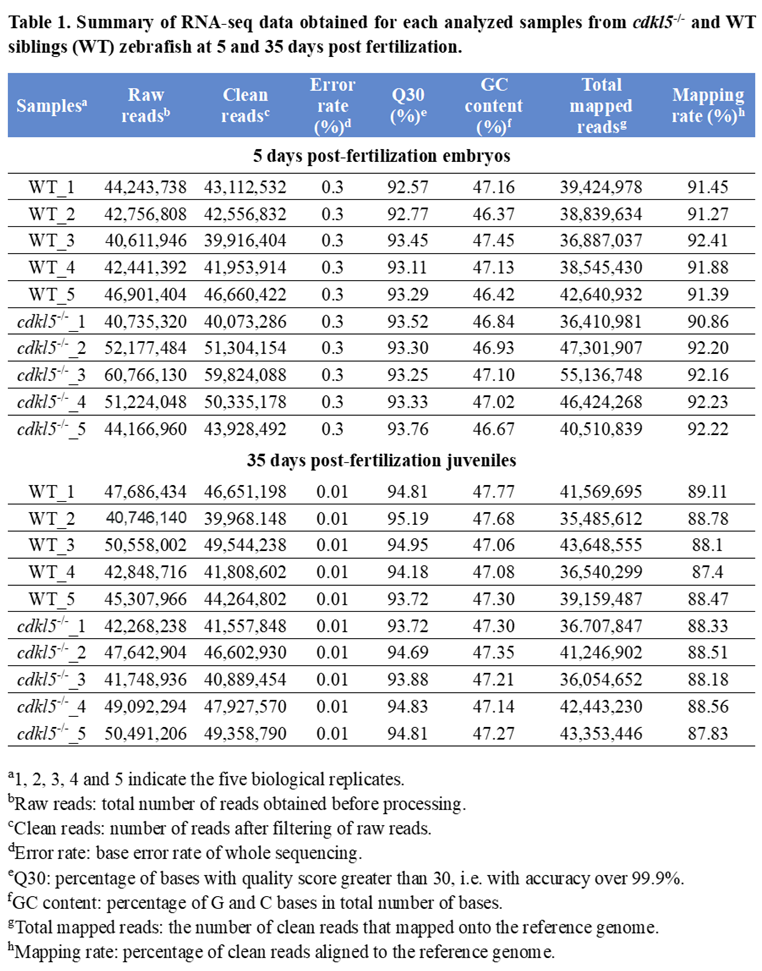
Expression levels for all identified genes in the cdkl5-/- and WT samples from 5 and 35 dpf zebrafish were quantified by calculating the FPKM. The distribution of gene expression levels in the analyzed samples is indicated in the boxplot graphs of Figure 1A,B. As shown, the overall range and distribution of the FPKM values were similar and uniform across the five biological replicates of each group at both developmental ages indicating good standardization and reliability of the data.
The correlation of gene expression levels across samples is crucial for ensuring reliability and appropriate sample selection. If this is the case, the square of the Pearson correlation coefficient (R2) should be greater than 0.8. We performed this analysis, and the correlation coefficients (R2) are displayed as heatmaps in Figure 1C and 1D. As shown, the correlation between biological replicates within the cdkl5-/- and WT groups was high, with R2 values greater than 0.911 and 0.930 for 5 and 35 dpf samples, respectively. This indicates that all data within each group were well correlated and that the biological replicates had good reproducibility.
Additionally, principal component analysis (PCA) was performed on the gene expression values (FPKM) of all samples at 5 and 35 dpf, as shown in Figure 1E,F, respectively. The similarity between samples is indicated by the distance between them, i.e. shorter distances correspond to higher similarity. Overall, in both PCA maps for 5 and 35 dpf zebrafish, the gene expression levels of samples within each group were clustered together; that is, the distance between the five biological replicates from each group was relatively small, indicating similar characteristics. In contrast, the samples from the different cdkl5-/- and WT sibling groups were well segregated, with relatively large separation distances, indicating distinct gene expression profiles between them.
A total of 17,170 and 17,173 genes were found to be expressed in the cdkl5-/- and WT siblings’ samples of 5 dpf zebrafish, respectively (Figure 1G). Among these genes, 16,641 were co-expressed in the two groups, 532 genes were uniquely expressed in WT siblings and 529 genes were specifically expressed in cdkl5-/-. At 35 dpf, 17,681 and 17,503 genes were detected to be expressed in the cdkl5-/- and WT zebrafish, respectively, with 17,045 genes being commonly expressed (Figure 1H), while 458 and 636 genes were exclusively expressed in WT siblings and in cdkl5-/-, respectively.
Altogether, these results demonstrate that the data obtained from RNA-seq in this study is both reliable and reproducible, thus suitable to be used for further bioinformatics analysis. It also indicates that the transcriptomic profile of zebrafish at different developmental stages is altered when Cdkl5 is defecient.
3.3. Identification of Differentially Expressed Genes Between cdkl5-/- and WT Siblings’ Zebrafish
To discover candidate genes affected by the lack of a functional Cdkl5, DESeq2 software was used to identify differentially expressed genes (DEGs) between cdkl5-/- mutant and WT siblings’ zebrafish at both 5 and 35 dpf. A hierarchical clustering analysis based on FPKM data of all DEGs was carried out and heatmaps were generated to observe expression patterns of different genes. An evident separation between the WT siblings and cdkl5-/- groups were observed, indicating that the cdkl5-/- mutant fish displayed a unique gene expression pattern, showing many significant differences from the WT fish at both stages of development analyzed (Figure 2A,B).
At 5 dpf, a total of 2,130 genes were found differently expressed between cdkl5-/- mutant and WT embryos. Among them, 1,178 genes were downregulated, and 952 genes were upregulated in the cdkl5-/- when compared to their WT siblings (Figure 2C). While at 35 dpf, a total of 4,210 genes were identified as significantly dysregulated, out of which 2,141 genes were downregulated and 2,069 were upregulated in the cdkl5-/- mutant juveniles in comparison to the WT (Figure 2D). The detailed information of all DEGs at 5 and 35 dpf is described in Supplementary Tables S2 and S3, respectively. Furthermore, to visualize the abundance and distribution of the differentially expressed genes between the two groups, we have generated a volcano plot for each of the developmental stages (Figure 2E,F).
Since the cdkl5 mutant zebrafish line (sa21938) used in this work harbors a nonsense mutation that may lead to activation of the nonsense-mediated mRNA decay (NMD) mechanism, we specifically examined the expression of cdkl5 between WT and cdkl5-/- groups. Indeed, cdkl5 was one of the genes found to be differentially expressed between the two groups at both stages of development. It was significantly downregulated in both 5 and 35 dpf cdkl5-/- mutant zebrafish (Supplementary Tables S2 and S3).
The top 40 known DEGs, including the 20 most upregulated and the 20 most downregulated genes in the cdkl5-/- mutant compared to the WT based on their significance, are listed in Table 2. Our results showed that the most significantly upregulated gene in cdkl5-/- embryos at 5 dpf was sort1a (sortilin 1a), which encodes a sorting receptor or co-receptor required for the transport of several intracellular proteins from the Golgi apparatus to the cell surface of lysosomes and endosomes[33]. Ligands of sortilin include the neurotensin (NTS), a neuropeptide functioning as neurotransmitter in the brain which is encoded by nts gene[34], that was also one of the most significantly upregulated genes in cdkl5-/- mutant embryos, suggesting that Cdkl5 may be involved in sortilin/neurotensin pathway. Additionally, guca1c (guanylate cyclase activator 1C) and arr3b (arrestin 3b) genes that have important function in visual phototransduction pathway[35,36] were also upregulated. The most significant downregulated gene in mutant embryos was gamt (guanidinoacetate N-methyltransferase), encoding for an enzyme involved in creatine synthesis and whose deficiency in humans causes a disorder characterized by developmental delay, seizures and hypotonia[37]. Furthermore, other downregulated genes with greater significance include: ctsl.1 and col5a1, involved in collagen catabolic processes and extracellular matrix organization[38,39]; myh7ba, involved in actin filament binding activity and microfilament motor activity being important for muscle contraction[40]; foxj3, involved in the regulation of muscle fiber identity and regeneration of skeletal muscle through the activation of Mef2c transcription[41]; dynlt3 that encodes a member of dynein motor protein complex, involved in the transport of organelles and vesicles toward microtubule[42]; klhdc8a, involved in the induction of primary ciliogenesis[43]; mbd1b, encoding a protein involved in epigenetic regulation and its deficiency is associated with reduced neurogenesis[44]; rs1a, involved in the cellular organization of the retina; lrp2b, encoding a multifunctional receptor involved in the development of many organs such as brain and eye[45]. At 35 dpf, 9 out of the 20 most significantly upregulated genes in the cdkl5-/- mutant juveniles were related to visual functions. These included crygm2d18, crygm2d15, crygm2d16, crygm2d11, cryba2a, crygm2d13, and crygm2d14 which belong to the crystallin gene family and are involved in lens development and visual perception[46]; rlbp1b which encode a retinoid-binding protein crucial for the correct function of rod and cone photoreceptors, playing several roles in visual cycle[47]; and lim2.4, which encodes an eye lens protein involved in cell junction organization[48]. The two most significantly upregulated genes were olfml3b and kdm5ba. In the brain, olfml3b encodes a microglia-specific protein whose expression increases during neuroinflammation[49]. The gene KDM5B encodes a histone demethylase involved in the regulation of neuronal, bone and muscle development[50]. Regarding the most significantly downregulated genes in 35 dpf cdkl5-/- juveniles, several were also found to be significantly downregulated in 5 dpf cdkl5-/- mutant embryos, including gnl3l, nucks1a, pir, slc41a1, ccdc120, ugt1b5, dynlt3, myh7ba and cishb. The top two were opn1lw1 and opn1lw2, which encode two red-sensitive cone opsins critical for phototransduction[51]. Other genes among the top 20 significantly downregulated included myhc4, myom2a, and vgll2b, three genes involved in muscle development[52,53,54]. Additionally, vwa1, which encodes an extracellular matrix protein essential for cartilage structure and development was downregulated; knockout of this gene in zebrafish causes cartilage dysmorphologies[55]. The gene nfasca, which encodes a cell adhesion protein, was also downregulated. This gene plays a key role in neurite outgrowth and fasciculation, as well as in the organization of the axon initial segment and nodes of Ranvier[56]. Mutations in this gene in humans cause a neurodevelopmental disorder with motor dysfunction[57].
3.4. Gene Ontology (GO) Enrichment Analysis of the Differentially Expressed Genes Between cdkl5-/- and WT Zebrafish
To investigate the potential biological functions associated with the differentially expressed genes found upon cdkl5 mutation in zebrafish, we performed Gene Ontology (GO) enrichment analysis on DEGs between cdkl5-/- and WT at the two different stages of development analyzed. Gene Ontology is a collection of terms used to describe the functions of genes and classify gene sets. The GO terms are organized into three main categories: biological process (BP), cellular component (CC) and molecular function (MF).
By using the combined list of all 2,130 DEGs found in 5 dpf mutant embryos, GO analysis revealed that the DEGs could be assigned into the three main categories and further classification resulted in the identification of several GO terms. After using a corrected p-value < 0.05, 65 GO terms were considered significantly enriched. Most of them were associated with biological processes (36 terms), followed by cellular component (16 terms) and molecular function (13 terms) (Supplementary Table S4). The ten most enriched GO terms of each category are shown in Figure 3A. Among these categories, the “neuron projection development” GO term belonging to biological process contained the highest number of DEGs (60 genes). In the biological processes, the most significantly enriched GO terms included “myosin filament organization”, “myosin filament assembly” and “striated muscle myosin thick filament assembly”. The most enriched GO terms within the cellular component comprised “actin cytoskeleton”, “myosin complex” and “extracellular matrix”. Regarding the molecular function category, the most significantly enriched GO terms included “extracellular matrix structural constituent”, “actin binding” and “actin filament binding”. GO enrichment analysis of all 4,210 DEGs identified in 35 dpf mutant juveniles showed that 141 GO terms were significantly enriched (padj < 0.05). As in 5 dpf mutant embryos, most of these terms were associated with biological processes (87 terms), followed by cellular components (38 terms) and molecular functions (16 terms) (Supplementary Table S5). The most significantly enriched GO terms in the BP category included “visual perception”, “sensory perception of light stimulus”, “eye development” and “muscle structure development”. Among the CC category, the top significantly enriched GO terms were “contractile fiber”, “sarcomere” and “myofibril”. In the MF category, the most significantly enriched GO terms included “structural constituent of eye lens”, “actin binding” and “protein-containing complex binding” which was also the term with the highest number of DEGs (122 genes) (Figure 3B). Furthermore, several GO terms were similarly significantly enriched in both 5 and 35 dpf cdkl5-/- zebrafish, mainly related to muscle development, extracellular matrix organization and actin binding. In contrast, the main alterations between 5 and 35 dpf cdkl5-/- zebrafish were observed in categories related to the eye, which became particularly enriched at the later stage of development.
To have a better comprehension of which biological functions are being activated and repressed, we have also performed GO enrichment analysis of up- and downregulated DEGs separately. Our results showed that at 5 dpf, significantly enriched GO terms (padj < 0.05) were identified for the downregulated DEGs (Supplementary Table S6 and Figure 4A) which were similar to those identified in the combined analysis. For the upregulated DEGs, only the “neuropeptide hormone activity” GO term was found to be significantly enriched using a padj < 0.05 (Supplementary Table S7 and Figure 4B). At 35 dpf, the top significantly enriched GO terms for the downregulated DEGs were again mostly related to muscle development and contraction (skeletal and cardiac), actin cytoskeleton and extracellular matrix (Supplementary Table S8 and Figure 4C). Additionally, GO terms such as “eye development”, “eye morphogenesis”, “central nervous system development” and “skeletal system development” were also found significantly enriched in downregulated DEGs (Supplementary Table S8). For the upregulated DEGs, the significantly enriched GO terms with higher significance were related to visual and sensory perception, lens and eye development and nervous system processes (Supplementary Table S9 and Figure 4D). Other significantly enriched GO terms in upregulated DEGs were associated with transport of diverse ions, activity of ion gated channel, neurotransmitter transport and secretion, and synaptic signaling (Supplementary Table S9).
Overall, these results indicate that Cdkl5 in zebrafish is involved in important functions associated with the regulation of actin cytoskeleton, skeletal and cardiac muscle development, extracellular matrix organization, neuronal development and eye development.
3.5. KEGG Pathway Enrichment Analysis of the Differentially Expressed Genes Between cdkl5-/- and WT Zebrafish
The Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis is used to identify biological pathways that are significantly overrepresented in a set of genes. To further elucidate the pathways affected by the cdkl5 mutation in zebrafish, we conducted a KEGG pathway analysis on DEGs between cdkl5-/- and WT. Pathways were considered significantly enriched when corrected p-value was less than 0.05.
Separate enrichment analysis of DEGs at 5 dpf revealed three significantly enriched KEGG pathways for the downregulated DEGs, while no significantly enriched pathways were associated with upregulated DEGs. Detailed information on enriched KEGG pathways and involved genes is indicated in Supplementary Table S10. As shown in Figure 5A, downregulated DEGs were associated with “ECM-receptor interaction” (26 DEGs), “focal adhesion” (38 DEGs) and “cardiac muscle contraction” (21 DEGs) pathways, in accordance with what was observed in the GO terms. By enrichment analysis of downregulated DEGs at 35 dpf, eight KEEG pathways were found to be significantly enriched (Figure 5B and Supplementary Table S11). As at 5dpf, the “focal adhesion” (76 DEGs), “ECM-receptor interaction” (37 DEGs) and “cardiac muscle contraction” (28 DEGs) pathways were also enriched in the downregulated DEGs at 35 dpf. Additionally, “regulation of actin cytoskeleton” (55 DEGs), “Wnt signaling pathway” (41 DEGs), “insulin signaling pathway” (36 DEGs), “adherens junction” (34 DEGs) and “ribosome” (31 DEGs) were also identified. For the upregulated DEGs, 16 KEEG pathways were identified as significantly enriched (Figure 5C and Supplementary Table S12). Most of them were related to metabolism, including “drug metabolism-other enzymes”, “retinol metabolism”, “ascorbate and aldarate metabolism”, and “fatty acid metabolism”.
These results further demonstrate that Cdkl5 dysfunction in zebrafish at different stages of development cause downregulation of pathways essential for the interaction between cells and the extracellular matrix, thus affecting tissue organization and development.
3.6. Validation of the RNA-Seq Differential Gene Expression by RT-qPCR
To verify the reliability and accuracy of the differentially expressed genes identified from the RNA-seq results, we randomly selected several DEGs and performed RT-qPCR to assess their expression levels in cdkl5-/- and WT zebrafish (Figure 6). In 5 dpf embryos, the expression levels of the genes cdkl5, ahsa1a, col1a1b, ndr2, rac3a and nts were analyzed. RT-qPCR results indicated that cdkl5, ahsa1a and col1a1b were downregulated, while ndr2, rac3a and nts were upregulated in cdkl5-/- embryos, consistent with the RNA-seq results. In 35 dpf juveniles, the expression levels of cdkl5, mmp9, neb, kcna4, mmp13a and olfml3b were analyzed. Our RT-qPCR results showed a significant decrease in the expression of cdkl5, mmp9, neb and mmp13a, while expression of kcna4 and olfml3b increased in cdkl5-/- compared to their WT siblings, consistent with the RNA-seq results. Altogether, these findings indicate similar expression trends between RNA-seq and RT-qPCR results, therefore confirming the reliability of our RNA-seq data.
3.7. Impact of Cdkl5 Loss on Zebrafish Nervous System
It is widely known that CDKL5 has important roles in the brain and its deficiency in humans causes neurological defects, a main characteristic of CDKL5 deficiency disorder. To further assess the impact of cdkl5 disruption on the nervous system of zebrafish, we specifically investigated the differentially expressed genes associated with neuronal functions between cdkl5-/- and WT zebrafish. A subset of these DEGs is listed in Table 3.
Several DEGs related to neuronal development were found and many contributed to the enrichment of GO terms at both stages of development. At 5 dpf, GO terms such as “regulation of nervous system development”, “neuron projection development” and “axon development” were significantly enriched in a higher number of downregulated genes compared to upregulated genes. Accordingly, DEGs related to neuronal development were also mainly downregulated at 35 dpf and contributed to the enrichment of GO terms such as “central nervous system development”. Most of these genes play important roles in neuronal morphogenesis, neurite outgrowth, dendritic arborization and axon guidance. For instance, the genes nfasca, ntn1a, nav2b and plxnb2a were downregulated, while stmn1b and pacsin1a were upregulated at both developmental stages. Additionally, at 5 dpf, slit1a, cntf, casp3a, pcdh18b, mycbp2 and zc4h2 were downregulated, whereas draxina, nefmb, nefla, stmn3, stmn4, srcin1b, amigo1, ndr2 and fez1 were upregulated. At 35 dpf, genes that were downregulated included slit2, pax6b, srgap2, prickle1a and prickle2b. Interestingly, neuronal-related CDKL5 known targets or interactors were also identified as differentially expressed. This included the downregulated genes iqgap1 at both developmental stages, arhgef2, dlg5a and rac1b at 5 dpf and smad3a at 35 dpf, as well as the upregulated shtn1 at 35 dpf (Table 3).
Moreover, genes involved in oligodendrocyte development and myelination were found mainly downregulated at both development stages but only contributed to the enrichment of “glial cell development” and “oligodendrocyte development” GO terms at 35 dpf. For instance, the expression of tfeb, notch3, prx, daam2 and plp1b was found to be altered, in both 5 and 35 dpf cdkl5-/- zebrafish. They were all downregulated, except for plp1b which was upregulated at 5 dpf and downregulated at 35 dpf. In addition, lama2 and actr10 were also downregulated at 35 dpf (Table 3).
Several DEGs associated with synaptic transmission were identified, with these effects being particularly pronounced at 35 dpf, where the upregulated genes significantly contributed to enrichment of many related GO terms including “synaptic signaling”, “neurotransmitter transport”, “neurotransmitter secretion” and “synaptic vesicle exocytosis”. Genes involved in synaptic vesicle exocytosis and neurotransmission release, which is crucial process of neurotransmission, were found mainly upregulated. Among them were syt12, snap25a and stx12l at both developmental stages and syt2a, syt6b, syt11b, rab3ab, rph3ab, rimbp2, unc13a, sv2ca, napbb at 35 dpf (Table 3). Additionally, several DEGs encoding ion channels playing essential role in neuronal transmission[58,59] were dysregulated in cdkl5-/- at both developmental stages but with a greater number identified at 35 dpf, where upregulated genes also contributed to the enrichment of GO terms related to ion channel activity. These included calcium, sodium, and potassium voltage-gated channels, which are activated by alterations in electrical membrane potential, as well as ligand-gated ion channels like GABAA, glutamate, glycine and nicotinic acetylcholine receptors, activated by the neurotransmitter binding (Table 3).
Overall, these findings suggest that the cdkl5 disruption impairs neuronal development and synaptic neurotransmission in zebrafish.
3.8. Effects of Cdkl5 Deficiency on Zebrafish Motor Neurons
In our previous work, we showed that cdkl5-/- mutant zebrafish exhibit an altered swimming performance compared to WT siblings, characterized by shorter distances travelled. Potential causes for the observed phenotype might be related to muscle weakness caused by defects in the neurons innervating the muscle. Therefore, we have investigated the development of motoneurons in the spinal cord by crossing the zebrafish mutants with hb9:GFP transgenic line. The green fluorescence protein (GFP) expression is controlled by hb9 gene promoter, which encodes a transcription factor crucial for differentiation of postmitotic motor neurons, thereby specifically marking primary and secondary motoneurons. Here, we analyzed the number of hb9+ motoneuron cells and the axonal projection length of the caudal primary (CaP) motoneurons at 3 dpf by fluorescence microscopy (Figure 7). Our results showed that the average number of motor neurons (hb9+ cells) per hemisegment is significantly lower in cdkl5-/- embryos when compared to WT siblings (Figure 7B). Additionally, the axon length of CaP motoneurons in cdkl5-/- mutants was significantly smaller than that of WT siblings (Figure 7C).
3.9. Impact of cdkl5 Loss on Zebrafish Skeletal System
In our previous phenotypic analyses of the cdkl5-/- mutant zebrafish, we identified skeletal abnormalities, including craniofacial cartilage defects and impaired bone development[28]. Therefore, we investigated dysregulated genes associated with the skeletal system that might contribute to these phenotypes.
Several skeletal-related genes were found mostly downregulated in cdkl5-/- and many contributed to the significant enrichment of GO terms such as “skeletal system development”, “cartilage development” and “extracellular matrix” both at 5 and 35 dpf. Furthermore, GO terms such as “bone development”, “ossification” and “regulation of bone mineralization” were only significantly enriched in downregulated genes at 35 dpf. A subset of identified DEGs are listed in Table 4.
Downregulated genes involved in chondrogenesis and cartilage development in cdkl5-/- zebrafish included its major regulators sox6 at both developmental stages and sox9a at 5 dpf and sox9b at 35dpf, as well as mbtps1, creb3l2 and skia at 5 dpf and chsy1 at 35 dpf. Expression of the well-known osteogenic markers sp7 and runx2a/b was decreased in cdkl5-/- juveniles, as well as genes involved in mineralization such as tmem119b at 5dpf and tmem119a, ano6 and ptk2bb at 35 dpf. Interestingly, genes with both roles in cartilage and bone development were also identified, including mef2cb at both developmental stages, yap1 and foxl1 at 5 dpf, and runx1, mef2ca, prdm1a/b, panx3, ccn2b, pthlha, nr3c1 and rflna/b at 35 dpf (Table 4).
Dysregulated genes belonging to signaling pathways that are implicated in cartilage development, osteoblast differentiation and bone formation such as Wntless (Wnt), bone morphogenetic protein (BMP) and transforming growth factor beta (TGFβ) signaling pathways were identified (Table 4). In fact, Wnt-related downregulated genes in cdkl5-/- juveniles contributed to the significant enrichment of the Wnt signaling KEGG pathway at 35 dpf. These genes included wnt7bb, wnt7aa, wnt16, lrp5, lrp6, dkk2, dvl1a, dvl2, fzd1, fzd4, frzb, ctnnb1, ctnnb2, bcl9, tcf7, tle2b/c, nkd1, nkd2a and kremen1 (Table 4).
A wide diversity of genes encoding components of the extracellular matrix (ECM) crucial for the development and maintenance of the cartilage and bone integrity have been identified. Among them, several collagen genes were downregulated in cdkl5-/- at both developmental stages, including col1a1b and col1a2, which are the main constituents of bone ECM, col5a1, col6a2, col9a1b, col10a1a, col11a1b and col12a1a. The main collagen type in cartilage, col2a1a/b, was also downregulated at 5 dpf. Other key downregulated ECM constituents included the proteoglycans encoded genes dcn at both stages of development, aspn at 5 dpf and hspg2 at 35 dpf; the glycoproteins encoded genes fn1a, fbln2, and emilin1b at 5 dpf and fbln1 and comp at 35 dpf; the thrombospondins encoded genes thbs1, thbs2a, thbs3b and thbs4 at both developmental stages. The expression of spp1 and bglapl, two major bone ECM non-collagenous genes, was reduced at 5 and 35 dpf, respectively, in cdkl5-/- mutant zebrafish. Furthermore, genes encoding proteins crucial for ECM organization and stabilization through interactions with ECM components were also found to be downregulated, including hapln1 and tgfbi at both developmental stages, matn1 at 5 dpf, and vwa1 and postna/b at 35 dpf (Table 4).
The expression of genes encoding enzymes of the metalloproteinase family responsible for the degradation of ECM proteins, such as matrix metalloproteinases (MPMs) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS), was reduced in cdkl5-/-. These included mmp14a and adamts16 at both developmental stages, mmp15a/b at 5 dpf and mmp2, mmp9, mmp11b, mmp13a, adamts5, adamts7, adamts10 and adamts15b at 35dpf. Moreover, at 5 dpf, genes encoding proteases of the cathepsin family were also downregulated in cdkl5-/- embryos, including ctsk, ctsl.1 and ctsbb. The expression of the sh3pxd2b gene, which encodes an adaptor protein involved in ECM remodeling, also decreased at 5 dpf (Table 4).
Altogether, these results demonstrate that cdkl5 dysfunction leads to the reduction of genes essential for normal cartilage and bone formation, consistent with the phenotypes observed in cdkl5-/- mutant zebrafish.
4. Discussion
In humans, pathogenic variants of CDKL5 cause a severe developmental and epileptic encephalopathy, accompanied by multisystemic comorbidities as a consequence of CDKL5 deficiency[17]. However, its molecular mechanisms are still poorly understood, particularly those associated with the less studied phenotypes. To decipher potential cdkl5 molecular mechanisms, RNA-seq was performed in the present study to obtain the whole transcriptomic profile of a cdkl5 mutant zebrafish previously identified as a suitable model for CDD[28,60], both at a more initial and at a later stage of development (5 and 35 dpf). Our findings revealed the dysregulation of several genes in cdkl5-/- mutant zebrafish along with enriched GO functions and pathways implicated in diverse functions and tissues at both stages of development. The most significant DEGs in both conditions were linked to muscle, neuronal and visual functions, and GO analysis further highlighted the involvement of the DEGs in muscle and neuronal development at both stages and in eye development at 35dpf. These results indicate a Cdkl5 role within these systems in zebrafish, consistent to the associated phenotypes observed in humans. Although the mechanisms underlying the visual dysfunctions in CDD, such as poor eye contact and lack of visual tracking, are not well understood, they are believed to be linked with cerebral visual impairment[61]. Here, we identified the upregulation of genes crucial for visual perception, including crystalline genes encoding structural constituent of eye lens and genes involved in the phototransduction cascade within the retina. Whether this upregulation is due to Cdlkl5 loss or a compensatory mechanism in attempt to counteract potential cerebral visual dysfunctions should be further investigated.
Additionally, GO terms related to ECM organization and actin cytoskeleton regulation were also predominant at both stages. Consistently, KEGG enrichment analysis identified the ECM-receptor interaction and focal adhesion as the most significant downregulated pathways affected by Cdkl5 loss, at both stages of development, and the regulation of actin cytoskeleton pathway was also enriched at 35 dpf. The ECM, focal adhesions and actin cytoskeleton form an interconnected system fundamental for tissue development through the regulation of cell adhesion, migration, proliferation and differentiation, suggesting that their disruption might account for CDD underlying mechanism.
4.1. Deficiency of Cdkl5 in Zebrafish Causes Alterations in Genes Associated with Muscle System
Individuals with CDD exhibit severe impaired gross motor function with abnormal muscle tone (hypotonia)[17]. Previously, we have also demonstrated that the locomotor behavior of this cdkl5 mutant zebrafish was impaired, suggesting muscle weakness caused by disturbances in the neurons controlling muscle system functions and/or in the muscle itself. Accordingly, RNA-seq data showed that loss of Cdkl5 led to the downregulation of many key genes involved in muscle development and differentiation that was consistent throughout both developmental stages. Indeed, functional enrichment analysis highlighted the enrichment of muscle development-related GO terms such as muscle cell differentiation and skeletal muscle tissue development. Therefore, our findings suggest a key role of Cdkl5 for normal skeletal muscle development. Nevertheless, Serrano et al. found no obvious defects in the muscle organization or patterning of this cdkl5 mutant zebrafish at 2 and 6 dpf[60]. Possible reasons for this inconsistency could be the sensitivity of the method used to detect subtle changes or the appearance of defects later in development.
Interestingly, GO and KEGG pathway analysis revealed the enrichment of downregulation genes involved in cardiac muscle development and cardiac muscle contraction, respectively. Although CDKL5 function in the cardiovascular system is poorly investigated, a few studies reported cardiac abnormalities in CDD patients and Cdkl5+/− female mice, such as prolonged QT interval and arrythmia, [62,63]. Further studies are needed to investigate Cdkl5 function in muscle.
4.2. Cdkl5 Deficiency in Zebrafish Leads to Dysregulation of Genes Involved in Neuronal Functions
Normal brain development requires precise control of neuronal morphogenesis and signaling. Defects in dendrite formation and arborization, axon outgrowth and guidance, and synapse formation result in the dysfunction of neural circuits that are the cause of several neurodevelopmental disorders[64,65,66,67]. A major clinical presentation of CDD is severe neurodevelopmental impairment that affects both cognitive and motor functions. This study revealed that the loss of Cdkl5 in zebrafish led to the dysregulation of several key genes involved in nervous system development at both developmental stages, as well as the contribution of several related-GO enrichment functions. Specifically, we observed both the downregulation and upregulation of genes with key roles in neurite outgrowth and axon/dendritic arborization and in axon guidance. We reported the downregulation of genes such as nfasca, zc4h2, srgap2, prickle1a and prickle2b which are described as positive regulators of neurite outgrowth and branching, as well as mycbp2, which regulates axon extension/guidance and synapse formation[68,69,70,71,72]. Disruption or pathogenic variants in their human orthologs, leading to loss-of-function, have been associated with neurodevelopmental disorders whose phenotypes resemble CDD, including developmental delay, seizures, motor impairment, hypotonia, intellectual disability and facial dysmorphism[70,73,74,75,76,77]. In addition, Cdkl5 loss in zebrafish caused an alteration in the expression of genes encoding proteins known to interact or be phosphorylated by CDKL5 in mammals and playing a role in these processes. These included IQGAP1 and RAC1 which are crucial regulators of actin cytoskeleton that together with CDKL5 form a complex involved in dendritic arborization; DLG5 which plays a role in cell adhesion, cell polarity and transmission of extracellular signals to the cytoskeleton, thereby regulating dendritic spine morphogenesis and synaptogenesis. ARHGEF2, which regulates microtubule and actin cytoskeleton dynamics, as well as focal adhesion, influencing dendritic spine morphology; and SHTN1, which is involved in axon outgrowth[7]. Altogether, our results are consistent with previous studies using other systems such as knockout mice, cultured neurons and human iPSC-derived neurons, where neuronal CDKL5 loss caused defects in axon outgrowth, dendrite arborization and synaptogenesis. Therefore, indicating a conservation in CDKL5 molecular mechanisms between mammals and zebrafish.
Since the reduced swimming behavior of cdkl5-/- mutant zebrafish might be caused by defects in motoneurons, we have investigated the effect of cdkl5 dysfunction on the motoneurons development using hb9:GFP transgenic line. cdkl5-/- embryos displayed a reduced number of motoneuron cells and shorter caudal primary axons at 3 dpf, suggesting that loss of Cdkl5 impairs motor neuron development and reduces axonal outgrowth. Accordingly, previous studies showed that CDKL5 knockdown in primary hippocampal neurons and cultured cortical neurons or CDKL5 knockout in hippocampal neurons from mice resulted in the reduction of axons total length[8,9,78]. Moreover, Serrano et al. showed that through the crossing with islet1:EGFP line, this cdkl5 mutant zebrafish presented reduced branching in the middle primary motor neuron at 6 dpf. However, in contrast to our results, no differences in the number of motoneuron cells were observed. This could be due to the distinct markers used to identify motoneurons or the different stages of development that were analyzed. Altogether, defects in motor neurons, along with the defective expression of genes crucial for muscle development, could explain the impaired locomotor behavior in cdkl5-/- mutant zebrafish.
Genes involved in the oligodendrocyte development and myelination were downregulated in cdkl5-/- mutant zebrafish from early stages. This glial-cell type is responsible for the formation of myelin sheath wrapping the axons, that is essential for effective neuronal transmission in the central nervous system[79]. Defects in oligodendrocytes development and maintenance impairing myelination have been implicated with several neurological disorders, including leukodystrophies and autism spectrum disorder (ASD) [80,81,82], that is a feature of CDD. These findings suggest that Cdkl5 might play a role in differentiation and maturation of oligodendrocytes and myelination in zebrafish. Interestingly, white matter alterations and abnormal myelination were observed in CDD individuals[16]. Nevertheless, the function of CDKL5 in oligodendrocyte development and myelination process has not yet been investigated.
Precise synaptic transmission is essential for proper brain function. This process begins in the presynaptic neuron, where an action potential opens Ca²⁺ channels. The Ca2+ influx triggers neurotransmitters release from the synaptic vesicles (SV) into the synaptic cleft, then interacting with their receptors on the postsynaptic neuron. For this release to happen, the SVs need to be mobilized to the active zone, docked, and primed for membrane fusion[83,84,85]. Abnormalities affecting any of these key modulators of synaptic function have been implicated in the pathogenesis of many neurodevelopmental diseases including epilepsy, intellectual disability and ASD[86]. Previous studies indicated that Cdkl5 loss impaired synaptic transmission in mice[87,88,89,90,91,92]. Consistent with this, our transcriptomic analysis revealed that cdkl5 disruption in zebrafish led to the dysregulation of several genes involved in various steps of the synaptic transmission process, predominantly showing an upregulation pattern. While some of these genes were affected at 5 dpf, most changes occurred later in development, becoming more evident at 35 dpf as the nervous system matures. Genes encoding diverse types of proteins playing crucial roles in the regulation of SV exocytosis and neurotransmission release were overexpressed in cdkl5-/- mutant zebrafish. Among these were synaptotagmin genes encoding SV membrane proteins, as well as the vesicle docking-regulators rab3ab and rph3ab, the vesicle priming-promoters rimbp2 and unc13a, and the vesicle fusion-contributors snap25a, sv2ca, stx11b and napb. Interestingly, previous study reported a SV2C overexpression in tissue from human epileptic individuals[93]. Moreover, gain-of-function variants in RPH3A and UNC13A have been detected in individuals with phenotypes ranging from epilepsy and developmental delay to ASD[94,95]. These phenotypes are part of the main clinical presentation of individuals with CDKL5 deficiency disorder and an increased susceptibility to seizures was observed in this cdkl5-/- zebrafish mutant. Therefore, our results suggest that Cdkl5 plays a role in presynaptic function and its deficiency in zebrafish might enhance SV exocytosis, resulting in aberrant neurotransmitter release thus impairing neuronal transmission. So far, few studies have investigated the presynaptic role of CDKL5, and its only known presynaptic target is amphiphysin 1 that is involved in SV endocytosis and recycling[10]. Additionally, it was reported that loss of CDKL5 in primary hippocampal neurons of rats led to slower SV endocytosis, while SV exocytosis remained unaffected[96]. This inconsistency may be due to differences in the specific cell type analyzed, the developmental stage studied, or even species differences.
Ion channels are also known to play a crucial role in synaptic neurotransmission. Their abnormal activity, whether caused by gain- or loss-of-function (GOF; LOF), is associated with numerous diseases classified as channelopathies[97,98]. Here, we identified several DEGs encoding both voltage- and ligand-gated ion channels, most of which were overexpressed in cdkl5-/- mutant zebrafish. Voltage-gated calcium channels control the Ca2+ influx into cells triggered by membrane depolarization. We observed the upregulation of genes such as cacna1aa, cacna1c, cacna1da encoding the calcium channels subunits Cav2.1, Cav1.2 and Cav1.3, whose GOF mutations in humans leads to excessive Ca2+ entry and increased neuron excitability causing developmental disorders such as epilepsy or ASD[99,100,101]. Recently, Cav2.3 subunit encoded by CACNA1E was identified as a target of CDKL5 in humans and mice and loss of its phosphorylation leads to channel GOF causing increased calcium influx and increased neuronal excitability[102]. Additionally, we identified the upregulation of voltage-gated sodium channel genes with key roles in the generation and propagation of action potentials, including scn8aa/b, scn1ba/b genes whose mutations in humans are associated with developmental and epileptic encephalopathies (DEE)[103,104,105,106]. Typically, mutations in SCN8A result in gain of function, causing higher sodium currents and channel hyperactivity[104,105,107]. Interestingly, a recent study showed the effectiveness of sodium channel blockers to control seizures in CDD individuals[108]. Furthermore, we have also observed the upregulation of voltage-gated sodium channel genes such as kcna1a, kcnq3, kcnb1, kcnc1b and kcnd1 whose orthologs in humans have been implicated in epilepsy syndromes like DEE by either LOF or GOF mutations[109,110,111,112,113,114]. Accordingly, organoids derived from CDD individuals displayed neuronal hyperexcitability due to voltage-gated channels dysfunction, marked by increased Na+ and K+ current densities and premature Na+ channels opening[115]. This might suggest common molecular mechanisms for the seizure pathophysiology in CDD between fish and mammals. This study also revealed the overexpression of ligand-gated ion channel in cdkl5-/- zebrafish comprising both glutamate and GABA neurotransmitter receptors, that mediate excitatory (E) and inhibitory (I) synaptic neurotransmission, respectively. The impaired function of these types of receptors leading to disrupted E/I balance is linked with neurodevelopmental disorders[116]. Among identified upregulated genes were the glutamate receptor grin1a and gria3a/b and the GABA receptor gabra1, gabrd and gabrg2 whose both LOF and GOF mutations in human orthologs are known to cause neurodevelopmental phenotypes including epilepsy, cognitive impairment and/or ASD[117,118,119,120,121,122,123]. This study suggests that loss of Cdkl5 in zebrafish enhances glutamatergic and GABAergic synaptic neurotransmission. In accordance, research using CDKL5 mouse models has revealed disturbances in the E/I balance across multiple brain regions. Consistently, overexpression of glutamate receptors has been reported, leading to enhanced glutamatergic neurotransmission[90,91,124]. Selective loss of CDKL5 in forebrain glutamatergic neurons triggered spontaneous seizures in conditional Nex-cKO mice, due to increased excitability in dentate gyrus granule cells (DGCs), accompanied by increased inhibitory activity, which may be a compensatory response to counteract the E/I imbalance[88]. Similar observations were made in CA1 pyramidal neurons, following CDKL5 ablation in forebrain glutamatergic neurons of Cdkl5 Emx1-cKO mice, leading to hippocampal associated learning and memory deficits[92]. In contrast, CDKL5 deficiency in forebrain GABAergic neurons resulted in enhanced glutamatergic transmission and hypercitability in CA1 pyramidal neurons, leading to autistic features[91]. In addition, CDKL5 loss reduced and augmented excitatory and inhibitory transmission, respectively, decreasing the E/I ration in DGCs of Cdkl5−/y mice thus impairing hippocampal learning and memory[89]. Increased inhibitory GABAergic transmission was also reported in primary visual cortex and perirhinal cortex of Cdkl5−/y knockout mice, which might underline the CDD visual impairments and novel object recognition memory deficits, respectively[87,125]. Altogether, our findings indicate a CDKL5 role in the regulation of ion channels’ activity, supporting prior studies stating that CDD is in part a channelopathy.
4.3. Loss of Cdkl5 in Zebrafish Disrupts the Expression of Genes Associated with the Skeletal System
Discrete skeletal phenotypes have been observed in CDD individuals, such as dysmorphic facial features and scoliosis[17,19]. Nevertheless, mechanisms underlying their occurrence are still not known, as no investigations have been conducted to specifically address this issue. We previously reported the skeletal abnormalities in the cdkl5-/- mutant zebrafish[28], and in the present study, we gained insights into the molecular mechanisms underlying these defects.
During development, cartilage is the first skeletal tissue to form, a process that occurs when mesenchymal stem cells (MSCs) differentiate into chondrocytes, which produce an ECM[126,127,128,129]. Bone formation initiates with MSCs and can occur through different forms involving osteoblast's deposition of an ECM and its later mineralization[127,128,129]. In line with impaired cartilage and bone formation, GO enrichment analysis revealed enrichment of downregulated DEGs involved in cartilage development from the earliest developmental stage analyzed, as well as in bone development, ossification and mineralization at the later stage. Loss of Cdkl5 reduced the expression of master regulators of early chondrogenesis like sox9 and sox6, which are crucial for chondrocyte proliferation and differentiation by inducing the expression of cartilaginous ECM components[126]. Consistently, the expression of main cartilage ECM components was impaired including the collagen genes col2a1, col9a1b and col11a1 whose expression is known to be regulated by SOX9[130,131,132], as well as proteoglycans and glycoproteins encoding genes. Abnormal ECM synthesis and composition have been linked to several diseases associated with craniofacial malformation and skeletal defects[133,134]. Overall, the altered craniofacial cartilage structures in the cdkl5-/- mutant zebrafish might be the result of a decreased expression of crucial regulators of chondrogenesis and cartilage ECM constituents supporting our previous hypothesis that Cdkl5 plays an important role in cartilage formation and development. Moreover, we observed reduced expression of col10a1 and genes encoding ECM- degrading proteases such as mmp13, mmp9 and adamts5 which are well-established markers of terminally differentiated hypertrophic chondrocytes[135,136,137]. This suggests that Cdkl5 loss impairs endochondral ossification, as the expression of these genes in hypertrophic chondrocytes is required for cartilage replacement by bone.
Intramembranous ossification, which occurs through the direct differentiation of MSCs into mature osteoblasts capable of producing bone ECM[129], also appears to be regulated by Cdkl5 in zebrafish. Loss of Cdkl5 resulted in decreased expression of key osteogenic markers, including runx2 and sp7, which encode transcription factors crucial for early differentiation from MSCs to pre-osteoblast, as well as major bone ECM-encoding genes such as the early osteoblast marker col1a1b, the middle to late marker spp1 and the late marker bglapl[138]. Signaling pathways involved in skeletal development were affected in cdkl5-/- mutant zebrafish, particularly the Wnt signaling pathway, which was significantly downregulated at 35 dpf, as determined by KEGG analysis. Studies in human and animal models have demonstrated that Wnt signaling induces bone formation, and its dysregulation is associated with bone disorders such as osteoporosis[139,140,141,142]. These findings indicate that the impaired or delayed mineralization phenotype of cdkl5-/- mutant zebrafish results from defective osteogenic differentiation, suggesting that Cdkl5 plays an important role in bone development.
5. Conclusion
In summary, this study contributes to gain insights into the Cdkl5 molecular mechanisms through the identification of genes, functions and pathways affected by its disruption. Overall, our findings highlight the key role of Cdkl5 in neuronal development and synaptic neurotransmission, suggesting a common Cdkl5 function between zebrafish and mammalian models. Moreover, we demonstrate that cdkl5 disruption causes motor neuron defects that in combination with abnormal muscle development and structure may contribute to the observed gross motor impairments. Finally, this study provides novel evidence of Cdkl5 role in cartilage and bone development, potentially underlying the skeletal defects observed in mutant zebrafish and individuals with CDD. Altogether, these results emphasize the cdkl5-/- mutant zebrafish as a valuable model for CDD and highlight its potential for screening novel drugs to treat the disorder’s symptoms.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Natércia Conceição and M. Leonor Cancela; Formal analysis, Tatiana Varela; Funding acquisition, M. Leonor Cancela; Investigation, Tatiana Varela; Methodology, Tatiana Varela, Débora Varela; Project administration, M. Leonor Cancela; Resources, Natércia Conceição and M. Leonor Cancela; Supervision, Natércia Conceição and M. Leonor Cancela; Writing – original draft, Tatiana Varela; Writing – review & editing, Débora Varela, Natércia Conceição and M. Leonor Cancela.
Data Availability Statement
The raw and processed RNA-seq data was deposited in the Gene Expression Omnibus (GEO) database under the accession number GSE294284.
Acknowledgments
This work was supported by a research grant from the University of Pennsylvania Orphan Disease Center (CDKL5-22-103-02) on behalf of the Loulou Foundation and received national funds from the Portuguese Foundation for Science and Technology (FCT) through the projects UIDB/04326/2020, UIDP/04326/2020, LA/P/0101/2020 (CCMAR) and 2022.06526.PTDC. Tatiana Varela (https://doi.org/10.54499/SFRH/BD/144230/2019) and Débora Varela (SFRH/BD/141918/2018) are recipients of Ph.D. fellowships from FCT.
Conflicts of Interest
The authors declare no competing interests.
References
- Hector, R.D.; Dando, O.; Landsberger, N.; Kilstrup-Nielsen, C.; Kind, P.C.; Bailey, M.E.S.; Cobb, S.R. Characterisation of CDKL5 Transcript Isoforms in Human and Mouse. PLOS ONE 2016, 11, e0157758. [CrossRef]
- Kilstrup-Nielsen, C.; Rusconi, L.; La Montanara, P.; Ciceri, D.; Bergo, A.; Bedogni, F.; Landsberger, N. What We Know and Would Like to Know about CDKL5 and Its Involvement in Epileptic Encephalopathy. Neural Plast. 2012, 2012, 1–11. [CrossRef]
- Rusconi, L.; Salvatoni, L.; Giudici, L.; Bertani, I.; Kilstrup-Nielsen, C.; Broccoli, V.; Landsberger, N. CDKL5 Expression Is Modulated during Neuronal Development and Its Subcellular Distribution Is Tightly Regulated by the C-terminal Tail. J. Biol. Chem. 2008, 283, 30101–30111. [CrossRef]
- Chen, Q.; Zhu, Y.-C.; Yu, J.; Miao, S.; Zheng, J.; Xu, L.; Zhou, Y.; Li, D.; Zhang, C.; Tao, J.; et al. CDKL5, a Protein Associated with Rett Syndrome, Regulates Neuronal Morphogenesis via Rac1 Signaling. J. Neurosci. 2010, 30, 12777–12786. [CrossRef]
- Ricciardi, S.; Ungaro, F.; Hambrock, M.; Rademacher, N.; Stefanelli, G.; Brambilla, D.; Sessa, A.; Magagnotti, C.; Bachi, A.; Giarda, E.; et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1–PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat. Cell Biol. 2012, 14, 911–923. [CrossRef]
- Rusconi, L.; Kilstrup-Nielsen, C.; Landsberger, N. Extrasynaptic N-Methyl-d-aspartate (NMDA) Receptor Stimulation Induces Cytoplasmic Translocation of the CDKL5 Kinase and Its Proteasomal Degradation. 2011, 286, 36550–36558. [CrossRef]
- Van Bergen, N. J. et al. CDKL5 deficiency disorder: molecular insights and mechanisms of pathogenicity to fast-track therapeutic development. Biochem Soc Trans 50, 1207–1224 (2022).
- Nawaz, M.S.; Giarda, E.; Bedogni, F.; La Montanara, P.; Ricciardi, S.; Ciceri, D.; Alberio, T.; Landsberger, N.; Rusconi, L.; Kilstrup-Nielsen, C. CDKL5 and Shootin1 Interact and Concur in Regulating Neuronal Polarization. PLOS ONE 2016, 11, e0148634. [CrossRef]
- Chen, Q.; Zhu, Y.-C.; Yu, J.; Miao, S.; Zheng, J.; Xu, L.; Zhou, Y.; Li, D.; Zhang, C.; Tao, J.; et al. CDKL5, a Protein Associated with Rett Syndrome, Regulates Neuronal Morphogenesis via Rac1 Signaling. J. Neurosci. 2010, 30, 12777–12786. [CrossRef]
- Sekiguchi, M.; Katayama, S.; Hatano, N.; Shigeri, Y.; Sueyoshi, N.; Kameshita, I. Identification of amphiphysin 1 as an endogenous substrate for CDKL5, a protein kinase associated with X-linked neurodevelopmental disorder. Arch. Biochem. Biophys. 2013, 535, 257–267. [CrossRef]
- Katayama, S.; Sueyoshi, N.; Kameshita, I. Critical Determinants of Substrate Recognition by Cyclin-Dependent Kinase-like 5 (CDKL5). Biochemistry 2015, 54, 2975–2987. [CrossRef]
- Muñoz, I.M.; E Morgan, M.; Peltier, J.; Weiland, F.; Gregorczyk, M.; Brown, F.C.; Macartney, T.; Toth, R.; Trost, M.; Rouse, J. Phosphoproteomic screening identifies physiological substrates of the CDKL 5 kinase. EMBO J. 2018, 37. [CrossRef]
- Baltussen, L.L.; Negraes, P.D.; Silvestre, M.; Claxton, S.; Moeskops, M.; Christodoulou, E.; Flynn, H.R.; Snijders, A.P.; Muotri, A.R.; Ultanir, S.K. Chemical genetic identification of CDKL 5 substrates reveals its role in neuronal microtubule dynamics. EMBO J. 2018, 37, e99763. [CrossRef]
- Kameshita, I.; Sekiguchi, M.; Hamasaki, D.; Sugiyama, Y.; Hatano, N.; Suetake, I.; Tajima, S.; Sueyoshi, N. Cyclin-dependent kinase-like 5 binds and phosphorylates DNA methyltransferase 1. Biochem. Biophys. Res. Commun. 2008, 377, 1162–1167. [CrossRef]
- Mari, F.; Azimonti, S.; Bertani, I.; Bolognese, F.; Colombo, E.; Caselli, R.; Scala, E.; Longo, I.; Grosso, S.; Pescucci, C.; et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum. Mol. Genet. 2005, 14, 1935–1946. [CrossRef]
- Bahi-Buisson, N.; Nectoux, J.; Rosas-Vargas, H.; Milh, M.; Boddaert, N.; Girard, B.; Cances, C.; Ville, D.; Afenjar, A.; Rio, M.; et al. Key clinical features to identify girls with CDKL5 mutations. Brain 2008, 131, 2647–2661. [CrossRef]
- Fehr, S.; Wilson, M.; Downs, J.; Williams, S.; Murgia, A.; Sartori, S.; Vecchi, M.; Ho, G.; Polli, R.; Psoni, S.; et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet. 2012, 21, 266–273. [CrossRef]
- Jakimiec, M.; Paprocka, J.; Śmigiel, R. CDKL5 Deficiency Disorder—A Complex Epileptic Encephalopathy. Brain Sci. 2020, 10, 107. [CrossRef]
- Leonard, H.; Downs, J.; A Benke, T.; Swanson, L.; Olson, H.; Demarest, S. CDKL5 deficiency disorder: clinical features, diagnosis, and management. Lancet Neurol. 2022, 21, 563–576. [CrossRef]
- Olson, H.E.; Daniels, C.I.; Haviland, I.; Swanson, L.C.; Greene, C.A.; Denny, A.M.M.; Demarest, S.T.; Pestana-Knight, E.; Zhang, X.; Moosa, A.N.; et al. Current neurologic treatment and emerging therapies in CDKL5 deficiency disorder. J. Neurodev. Disord. 2021, 13, 1–11. [CrossRef]
- Amendola, E. et al. Mapping Pathological Phenotypes in a Mouse Model of CDKL5 Disorder. PLoS One 9, e91613 (2014).
- Fuchs, C.; Gennaccaro, L.; Trazzi, S.; Bastianini, S.; Bettini, S.; Martire, V.L.; Ren, E.; Medici, G.; Zoccoli, G.; Rimondini, R.; et al. Heterozygous CDKL5 Knockout Female Mice Are a Valuable Animal Model for CDKL5 Disorder. Neural Plast. 2018, 2018, 1–18. [CrossRef]
- Okuda, K.; Takao, K.; Watanabe, A.; Miyakawa, T.; Mizuguchi, M.; Tanaka, T. Comprehensive behavioral analysis of the Cdkl5 knockout mice revealed significant enhancement in anxiety- and fear-related behaviors and impairment in both acquisition and long-term retention of spatial reference memory. PLOS ONE 2018, 13, e0196587. [CrossRef]
- Judy, W. I.-T. et al. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proceedings of the National Academy of Sciences 109, 21516–21521 (2012).
- Jhang, C.-L.; Huang, T.-N.; Hsueh, Y.-P.; Liao, W. Mice lacking cyclin-dependent kinase-like 5 manifest autistic and ADHD-like behaviors. Hum. Mol. Genet. 2017, 26, 3922–3934. [CrossRef]
- Liao, W.; Lee, K.-Z. CDKL5-mediated developmental tuning of neuronal excitability and concomitant regulation of transcriptome. Hum. Mol. Genet. 2023, 32, 3276–3298. [CrossRef]
- Vitorino, M.; Cunha, N.; Conceição, N.; Cancela, M.L. Expression pattern of cdkl5 during zebrafish early development: implications for use as model for atypical Rett syndrome. Mol. Biol. Rep. 2018, 45, 445–451. [CrossRef]
- Varela, T.; Varela, D.; Martins, G.; Conceição, N.; Cancela, M.L. Cdkl5 mutant zebrafish shows skeletal and neuronal alterations mimicking human CDKL5 deficiency disorder. Sci. Rep. 2022, 12, 1–12. [CrossRef]
- de Sena-Tomás, C. et al. Neutrophil immune profile guides spinal cord regeneration in zebrafish. Brain Behav Immun 120, 514–531 (2024).
- Flanagan-Steet, H.; Fox, M.A.; Meyer, D.; Sanes, J.R. Neuromuscular synapses can form in vivo by incorporation of initially aneural postsynaptic specializations. Development 2005, 132, 4471–4481. [CrossRef]
- Benjaminit, Y. & Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Statist. Soc. B vol. 57 (1995).
- Arshadi, C.; Günther, U.; Eddison, M.; Harrington, K.I.S.; Ferreira, T.A. SNT: a unifying toolbox for quantification of neuronal anatomy. Nat. Methods 2021, 18, 374–377. [CrossRef]
- Petersen, C.M.; Nielsen, M.S.; Nykjær, A.; Jacobsen, L.; Tommerup, N.; Rasmussen, H.H.; Røigaard, H.; Gliemann, J.; Madsen, P.; Moestrup, S.K. Molecular Identification of a Novel Candidate Sorting Receptor Purified from Human Brain by Receptor-associated Protein Affinity Chromatography. J. Biol. Chem. 1997, 272, 3599–3605. [CrossRef]
- Mazella, J. et al. The 100-KDa Neurotensin Receptor Is Gp95/Sortilin, A Non-G-Protein-Coupled Receptor*. http://www.jbc.org (1998).
- Sakuma, H.; Murakami, A.; Fujimaki, T.; Inana, G. Isolation and characterization of the human X-arrestin gene. Gene 1998, 224, 87–95. [CrossRef]
- Morales-Cámara, S.; Alexandre-Moreno, S.; Bonet-Fernández, J.-M.; Atienzar-Aroca, R.; Aroca-Aguilar, J.-D.; Ferre-Fernández, J.-J.; Méndez, C.-D.; Morales, L.; Fernández-Sánchez, L.; Cuenca, N.; et al. Role of GUCA1C in Primary Congenital Glaucoma and in the Retina: Functional Evaluation in Zebrafish. Genes 2020, 11, 550. [CrossRef]
- Libell, J.L.; Lakhani, D.A.; Balar, A.B.; Khan, M.; Carpenter, J.S.; Joseph, J.T. Guanidinoacetate N-methyltransferase deficiency: Case report and brief review of the literature. Radiol. Case Rep. 2023, 18, 4331–4337. [CrossRef]
- Vizovišek, M.; Fonović, M.; Turk, B. Cysteine cathepsins in extracellular matrix remodeling: Extracellular matrix degradation and beyond. Matrix Biol. 2019, 75-76, 141–159. [CrossRef]
- Kahai, S., Vary, C. P. H., Gao, Y. & Seth, A. Collagen, type V, α1 (COL5A1) is regulated by TGF-β in osteoblasts. Matrix Biology 23, 445–455 (2004).
- Peter, A.K.; Rossi, A.C.; Buvoli, M.; Ozeroff, C.D.; Crocini, C.; Perry, A.R.; Buvoli, A.E.; Lee, L.A.; Leinwand, L.A. Expression of Normally Repressed Myosin Heavy Chain 7b in the Mammalian Heart Induces Dilated Cardiomyopathy. J. Am. Hear. Assoc. 2019, 8, e013318. [CrossRef]
- Alexander, M.S.; Shi, X.; Voelker, K.A.; Grange, R.W.; Garcia, J.A.; Hammer, R.E.; Garry, D.J. Foxj3 transcriptionally activates Mef2c and regulates adult skeletal muscle fiber type identity. Dev. Biol. 2010, 337, 396–404. [CrossRef]
- Lo, K.W.-H.; Kogoy, J.M.; Pfister, K.K. The DYNLT3 Light Chain Directly Links Cytoplasmic Dynein to a Spindle Checkpoint Protein, Bub3. J. Biol. Chem. 2007, 282, 11205–11212. [CrossRef]
- Lee, D.; Gimple, R.C.; Wu, X.; Prager, B.C.; Qiu, Z.; Wu, Q.; Daggubati, V.; Mariappan, A.; Gopalakrishnan, J.; Sarkisian, M.R.; et al. Superenhancer activation of KLHDC8A drives glioma ciliation and hedgehog signaling. J. Clin. Investig. 2023, 133. [CrossRef]
- Zhao, X.; Ueba, T.; Christie, B.R.; Barkho, B.; McConnell, M.J.; Nakashima, K.; Lein, E.S.; Eadie, B.D.; Willhoite, A.R.; Muotri, A.R.; et al. Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc. Natl. Acad. Sci. 2003, 100, 6777–6782. [CrossRef]
- Fisher, C.E.; Howie, S.E. The role of megalin (LRP-2/Gp330) during development. Dev. Biol. 2006, 296, 279–297. [CrossRef]
- Greiling, T.M.; Houck, S.A.; Clark, J.I. The zebrafish lens proteome during development and aging. 2009, 15, 2313–2325.
- Lima de Carvalho, J. R. et al. Effects of deficiency in the RLBP1-encoded visual cycle protein CRALBP on visual dysfunction in humans and mice. Journal of Biological Chemistry 295, 6767–6780 (2020).
- Irum, B.; Khan, S.Y.; Ali, M.; Kaul, H.; Kabir, F.; Rauf, B.; Fatima, F.; Nadeem, R.; Khan, A.O.; Al Obaisi, S.; et al. Mutation in LIM2 Is Responsible for Autosomal Recessive Congenital Cataracts. PLOS ONE 2016, 11, e0162620. [CrossRef]
- Yadav, H.; Bakshi, A.; Anamika; Singh, V.; Paul, P.; Murugan, N.A.; Maurya, S.K. Co-localization and co-expression of Olfml3 with Iba1 in brain of mice. J. Neuroimmunol. 2024, 394, 578411. [CrossRef]
- Yoo, J.; Kim, G.W.; Jeon, Y.H.; Kim, J.Y.; Lee, S.W.; Kwon, S.H. Drawing a line between histone demethylase KDM5A and KDM5B: their roles in development and tumorigenesis. Exp. Mol. Med. 2022, 54, 2107–2117. [CrossRef]
- Chinen, A.; Hamaoka, T.; Yamada, Y.; Kawamura, S. Gene Duplication and Spectral Diversification of Cone Visual Pigments of Zebrafish. Genetics 2003, 163, 663–675. [CrossRef]
- Lamber, E.P.; Guicheney, P.; Pinotsis, N. The role of the M-band myomesin proteins in muscle integrity and cardiac disease. J. Biomed. Sci. 2022, 29, 1–15. [CrossRef]
- Liu, G.; Ito, T.; Kijima, Y.; Yoshitake, K.; Asakawa, S.; Watabe, S.; Kinoshita, S. Zebrafish Danio rerio myotomal muscle structure and growth from a spatial transcriptomics perspective. Genomics 2022, 114, 110477. [CrossRef]
- Chen, H.; Maeda, T.; Mullett, S.J.; Stewart, A.F. Transcription cofactor Vgl-2 is required for skeletal muscle differentiation. Genesis 2004, 39, 273–279. [CrossRef]
- Niu, X.; Zhang, F.; Ping, L.; Wang, Y.; Zhang, B.; Wang, J.; Chen, X. vwa1 Knockout in Zebrafish Causes Abnormal Craniofacial Chondrogenesis by Regulating FGF Pathway. Genes 2023, 14, 838. [CrossRef]
- Kriebel, M.; Wuchter, J.; Trinks, S.; Volkmer, H. Neurofascin: A switch between neuronal plasticity and stability. Int. J. Biochem. Cell Biol. 2012, 44, 694–697. [CrossRef]
- Efthymiou, S.; Salpietro, V.; Malintan, N.; Poncelet, M.; Kriouile, Y.; Fortuna, S.; De Zorzi, R.; Payne, K.; Henderson, L.B.; Cortese, A.; et al. Biallelic mutations in neurofascin cause neurodevelopmental impairment and peripheral demyelination. Brain 2019, 142, 2948–2964. [CrossRef]
- Cooper, E.C.; Jan, L.Y. Ion channel genes and human neurological disease: Recent progress, prospects, and challenges. Proc. Natl. Acad. Sci. 1999, 96, 4759–4766. [CrossRef]
- Weir, C. J. Ion channels, receptors, agonists and antagonists. Anaesthesia and Intensive Care Medicine 14, 410–416 (2013).
- Serrano, R.J.; Lee, C.; Douek, A.M.; Kaslin, J.; Bryson-Richardson, R.J.; Sztal, T.E. Novel preclinical model for CDKL5 deficiency disorder. Dis. Model. Mech. 2022, 15. [CrossRef]
- Quintiliani, M.; Ricci, D.; Petrianni, M.; Leone, S.; Orazi, L.; Amore, F.; Gambardella, M.L.; Contaldo, I.; Veredice, C.; Perulli, M.; et al. Cortical Visual Impairment in CDKL5 Deficiency Disorder. Front. Neurol. 2022, 12, 805745. [CrossRef]
- Loi, M.; Bastianini, S.; Candini, G.; Rizzardi, N.; Medici, G.; Papa, V.; Gennaccaro, L.; Mottolese, N.; Tassinari, M.; Uguagliati, B.; et al. Cardiac Functional and Structural Abnormalities in a Mouse Model of CDKL5 Deficiency Disorder. Int. J. Mol. Sci. 2023, 24, 5552. [CrossRef]
- Stansauk, J.; Fidell, A.; Benke, T.; Schaffer, M.; Demarest, S.T. Analysis of electrocardiograms in individuals with CDKL5 deficiency disorder. Am. J. Med Genet. Part A 2022, 191, 108–111. [CrossRef]
- Lin, Y.-C.; Koleske, A.J. Mechanisms of Synapse and Dendrite Maintenance and Their Disruption in Psychiatric and Neurodegenerative Disorders. Annu. Rev. Neurosci. 2010, 33, 349–378. [CrossRef]
- Melom, J.E.; Littleton, J.T. Synapse development in health and disease. Curr. Opin. Genet. Dev. 2011, 21, 256–261. [CrossRef]
- Xiong, G.-J.; Sheng, Z.-H. Presynaptic perspective: Axonal transport defects in neurodevelopmental disorders. J. Cell Biol. 2024, 223. [CrossRef]
- Engle, E.C. Human Genetic Disorders of Axon Guidance. Cold Spring Harb. Perspect. Biol. 2010, 2, a001784–a001784. [CrossRef]
- Rathjen, F.G.; Wolff, J.; Chang, S.; Bonhoeffer, F.; Raper, J.A. Neurofascin: A novel chick cell-surface glycoprotein involved in neurite-neurite interactions. Cell 1987, 51, 841–849. [CrossRef]
- Koticha, D.; Babiarz, J.; Kane-Goldsmith, N.; Jacob, J.; Raju, K.; Grumet, M. Cell adhesion and neurite outgrowth are promoted by neurofascin NF155 and inhibited by NF186. Mol. Cell. Neurosci. 2005, 30, 137–148. [CrossRef]
- Hirata, H.; Nanda, I.; van Riesen, A.; McMichael, G.; Hu, H.; Hambrock, M.; Papon, M.-A.; Fischer, U.; Marouillat, S.; Ding, C.; et al. ZC4H2 Mutations Are Associated with Arthrogryposis Multiplex Congenita and Intellectual Disability through Impairment of Central and Peripheral Synaptic Plasticity. Am. J. Hum. Genet. 2013, 92, 681–695. [CrossRef]
- Guerrier, S.; Coutinho-Budd, J.; Sassa, T.; Gresset, A.; Jordan, N.V.; Chen, K.; Jin, W.-L.; Frost, A.; Polleux, F. The F-BAR Domain of srGAP2 Induces Membrane Protrusions Required for Neuronal Migration and Morphogenesis. Cell 2009, 138, 990–1004. [CrossRef]
- Okuda, H.; Miyata, S.; Mori, Y.; Tohyama, M. Mouse Prickle1 and Prickle2 are expressed in postmitotic neurons and promote neurite outgrowth. FEBS Lett. 2007, 581, 4754–4760. [CrossRef]
- Kvarnung, M.; Shahsavani, M.; Taylan, F.; Moslem, M.; Breeuwsma, N.; Laan, L.; Schuster, J.; Jin, Z.; Nilsson, D.; Lieden, A.; et al. Ataxia in Patients With Bi-Allelic NFASC Mutations and Absence of Full-Length NF186. Front. Genet. 2019, 10, 896. [CrossRef]
- Smigiel, R.; Sherman, D.L.; Rydzanicz, M.; Walczak, A.; Mikolajkow, D.; Krolak-Olejnik, B.; Kosińska, J.; Gasperowicz, P.; Biernacka, A.; Stawinski, P.; et al. Homozygous mutation in the Neurofascin gene affecting the glial isoform of Neurofascin causes severe neurodevelopment disorder with hypotonia, amimia and areflexia. Hum. Mol. Genet. 2018, 27, 3669–3674. [CrossRef]
- Ahn, J.Y.; Kim, S.Y.; Lim, B.C.; Kim, K.J.; Chae, J.-H. Variable Phenotypes of ZC4H2-Associated Rare Disease in Six Patients. Ann. Child Neurol. 2022, 30, 120–126. [CrossRef]
- Bassuk, A.G.; Wallace, R.H.; Buhr, A.; Buller, A.R.; Afawi, Z.; Shimojo, M.; Miyata, S.; Chen, S.; Gonzalez-Alegre, P.; Griesbach, H.L.; et al. A Homozygous Mutation in Human PRICKLE1 Causes an Autosomal-Recessive Progressive Myoclonus Epilepsy-Ataxia Syndrome. Am. J. Hum. Genet. 2008, 83, 572–581. [CrossRef]
- Bayat, A.; Iqbal, S.; Borredy, K.; Amiel, J.; Zweier, C.; Barcia, G.; Kraus, C.; Weyhreter, H.; Bassuk, A.G.; Chopra, M.; et al. PRICKLE2 revisited—further evidence implicating PRICKLE2 in neurodevelopmental disorders. Eur. J. Hum. Genet. 2021, 29, 1235–1244. [CrossRef]
- Fuchs, C.; Medici, G.; Trazzi, S.; Gennaccaro, L.; Galvani, G.; Berteotti, C.; Ren, E.; Loi, M.; Ciani, E. CDKL5 deficiency predisposes neurons to cell death through the deregulation of SMAD3 signaling. Brain Pathol. 2019, 29, 658–674. [CrossRef]
- Masson, M.; Nait-Oumesmar, B. Emerging concepts in oligodendrocyte and myelin formation, inputs from the zebrafish model. Glia 2023, 71, 1147–1163. [CrossRef]
- Elitt, M.S.; Barbar, L.; Shick, H.E.; Powers, B.E.; Maeno-Hikichi, Y.; Madhavan, M.; Allan, K.C.; Nawash, B.S.; Gevorgyan, A.S.; Hung, S.; et al. Suppression of proteolipid protein rescues Pelizaeus–Merzbacher disease. Nature 2020, 585, 397–403. [CrossRef]
- Galvez-Contreras, A.Y.; Zarate-Lopez, D.; Torres-Chavez, A.L.; Gonzalez-Perez, O. Role of Oligodendrocytes and Myelin in the Pathophysiology of Autism Spectrum Disorder. Brain Sci. 2020, 10, 951. [CrossRef]
- Khelfaoui, H.; Ibaceta-Gonzalez, C.; Angulo, M.C. Functional myelin in cognition and neurodevelopmental disorders. Cell. Mol. Life Sci. 2024, 81, 1–14. [CrossRef]
- Ashery, U. et al. Chapter Two - The molecular mechanisms underlying synaptic transmission: A view of the presynaptic terminal. in The Synapse (eds. Pickel, V. & Segal, M.) 21–109 (Academic Press, Boston, 2014). [CrossRef]
- Südhof, T.C. THE SYNAPTIC VESICLE CYCLE. Annu. Rev. Neurosci. 2004, 27, 509–547. [CrossRef]
- Murthy, V. N. & De Camilli, P. Cell biology of the presynaptic terminal. Annu Rev Neurosci 26, 701–728 (2003).
- Casillas-Espinosa, P.M.; Powell, K.L.; O’brien, T.J. Regulators of synaptic transmission: Roles in the pathogenesis and treatment of epilepsy. Epilepsia 2012, 53, 41–58. [CrossRef]
- Pizzo, R.; Gurgone, A.; Castroflorio, E.; Amendola, E.; Gross, C.; Sassoè-Pognetto, M.; Giustetto, M. Lack of Cdkl5 Disrupts the Organization of Excitatory and Inhibitory Synapses and Parvalbumin Interneurons in the Primary Visual Cortex. Front. Cell. Neurosci. 2016, 10, 261. [CrossRef]
- Wang, H.; Zhu, Z.; Li, Y.; Lou, S.; Yang, G.; Feng, X.; Xu, W.; Huang, Z.; Cheng, X.; Xiong, Z. CDKL5 deficiency in forebrain glutamatergic neurons results in recurrent spontaneous seizures. Epilepsia 2021, 62, 517–528. [CrossRef]
- Hao, S.; Wang, Q.; Tang, B.; Wu, Z.; Yang, T.; Tang, J. CDKL5 Deficiency Augments Inhibitory Input into the Dentate Gyrus That Can Be Reversed by Deep Brain Stimulation. J. Neurosci. 2021, 41, 9031–9046. [CrossRef]
- Okuda, K.; Kobayashi, S.; Fukaya, M.; Watanabe, A.; Murakami, T.; Hagiwara, M.; Sato, T.; Ueno, H.; Ogonuki, N.; Komano-Inoue, S.; et al. CDKL5 controls postsynaptic localization of GluN2B-containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol. Dis. 2017, 106, 158–170. [CrossRef]
- Tang, S.; Terzic, B.; Wang, I.-T.J.; Sarmiento, N.; Sizov, K.; Cui, Y.; Takano, H.; Marsh, E.D.; Zhou, Z.; Coulter, D.A. Altered NMDAR signaling underlies autistic-like features in mouse models of CDKL5 deficiency disorder. Nat. Commun. 2019, 10, 2655. [CrossRef]
- Tang, S.; Wang, I.-T.J.; Yue, C.; Takano, H.; Terzic, B.; Pance, K.; Lee, J.Y.; Cui, Y.; Coulter, D.A.; Zhou, Z. Loss of CDKL5 in Glutamatergic Neurons Disrupts Hippocampal Microcircuitry and Leads to Memory Impairment in Mice. J. Neurosci. 2017, 37, 7420–7437. [CrossRef]
- Crèvecœur, J.; Kaminski, R.M.; Rogister, B.; Foerch, P.; Vandenplas, C.; Neveux, M.; Mazzuferi, M.; Kroonen, J.; Poulet, C.; Martin, D.; et al. Expression pattern of synaptic vesicle protein 2 (SV2) isoforms in patients with temporal lobe epilepsy and hippocampal sclerosis. Neuropathol. Appl. Neurobiol. 2014, 40, 191–204. [CrossRef]
- Pavinato, L.; Stanic, J.; Barzasi, M.; Gurgone, A.; Chiantia, G.; Cipriani, V.; Eberini, I.; Palazzolo, L.; Di Luca, M.; Costa, A.; et al. Missense variants in RPH3A cause defects in excitatory synaptic function and are associated with a clinically variable neurodevelopmental disorder. Anesthesia Analg. 2023, 25, 100922. [CrossRef]
- Lipstein, N.; Verhoeven-Duif, N.M.; Michelassi, F.E.; Calloway, N.; van Hasselt, P.M.; Pienkowska, K.; van Haaften, G.; van Haelst, M.M.; van Empelen, R.; Cuppen, I.; et al. Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder. J. Clin. Investig. 2017, 127, 1005–1018. [CrossRef]
- Kontaxi, C.; Ivanova, D.; Davenport, E.C.; Kind, P.C.; Cousin, M.A. Epilepsy-Related CDKL5 Deficiency Slows Synaptic Vesicle Endocytosis in Central Nerve Terminals. J. Neurosci. 2023, 43, 2002–2020. [CrossRef]
- D'Adamo, M.C.; Liantonio, A.; Conte, E.; Pessia, M.; Imbrici, P. Ion Channels Involvement in Neurodevelopmental Disorders. Neuroscience 2020, 440, 337–359. [CrossRef]
- Hü Bner, C. A. & Jentsch, T. J. Ion channel diseases.
- Rodan, L.H.; Spillmann, R.C.; Kurata, H.T.; Lamothe, S.M.; Maghera, J.; Jamra, R.A.; Alkelai, A.; Antonarakis, S.E.; Atallah, I.; Bar-Yosef, O.; et al. Phenotypic expansion of CACNA1C-associated disorders to include isolated neurological manifestations. Anesthesia Analg. 2021, 23, 1922–1932. [CrossRef]
- Jiang, X. et al. Both gain-of-function and loss-of-function de novo CACNA1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox-Gastaut syndrome. Epilepsia 60, 1881–1894 (2019).
- Pinggera, A.; Lieb, A.; Benedetti, B.; Lampert, M.; Monteleone, S.; Liedl, K.R.; Tuluc, P.; Striessnig, J. CACNA1D De Novo Mutations in Autism Spectrum Disorders Activate Cav1.3 L-Type Calcium Channels. Biol. Psychiatry 2015, 77, 816–822. [CrossRef]
- Sampedro-Castañeda, M.; Baltussen, L.L.; Lopes, A.T.; Qiu, Y.; Sirvio, L.; Mihaylov, S.R.; Claxton, S.; Richardson, J.C.; Lignani, G.; Ultanir, S.K. Epilepsy-linked kinase CDKL5 phosphorylates voltage-gated calcium channel Cav2.3, altering inactivation kinetics and neuronal excitability. Nat. Commun. 2023, 14, 1–18. [CrossRef]
- Chen, C.; Ziobro, J.; Robinson-Cooper, L.; Hodges, S.L.; Chen, Y.; Edokobi, N.; Lopez-Santiago, L.; Habig, K.; Moore, C.; Minton, J.; et al. Epilepsy and sudden unexpected death in epilepsy in a mouse model of human SCN1B-linked developmental and epileptic encephalopathy. Brain Commun. 2023, 5, fcad283. [CrossRef]
- Gardella, E. & Møller, R. S. Phenotypic and genetic spectrum of SCN8A-related disorders, treatment options, and outcomes. Epilepsia 60, S77–S85 (2019).
- Wagnon, J.L.; Barker, B.S.; Hounshell, J.A.; Haaxma, C.A.; Shealy, A.; Moss, T.; Parikh, S.; Messer, R.D.; Patel, M.K.; Meisler, M.H. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann. Clin. Transl. Neurol. 2015, 3, 114–123. [CrossRef]
- Zhu, Z.; Bolt, E.; Newmaster, K.; Osei-Bonsu, W.; Cohen, S.; Cuddapah, V.A.; Gupta, S.; Paudel, S.; Samanta, D.; Dang, L.T.; et al. SCN1B Genetic Variants: A Review of the Spectrum of Clinical Phenotypes and a Report of Early Myoclonic Encephalopathy. Children 2022, 9, 1507. [CrossRef]
- Kruger, L.C.; O'Malley, H.A.; Hull, J.M.; Kleeman, A.; Patino, G.A.; Isom, L.L. β1-C121W Is Down But Not Out: Epilepsy-AssociatedScn1b-C121WResults in a Deleterious Gain-of-Function. J. Neurosci. 2016, 36, 6213–6224. [CrossRef]
- Aledo-Serrano, Á.; Gómez-Iglesias, P.; Toledano, R.; Garcia-Peñas, J.J.; Garcia-Morales, I.; Anciones, C.; Soto-Insuga, V.; Benke, T.A.; del Pino, I.; Gil-Nagel, A. Sodium channel blockers for the treatment of epilepsy in CDKL5 deficiency disorder: Findings from a multicenter cohort. Epilepsy Behav. 2021, 118, 107946. [CrossRef]
- Kalm, T.; Schob, C.; Völler, H.; Gardeitchik, T.; Gilissen, C.; Pfundt, R.; Klöckner, C.; Platzer, K.; Klabunde-Cherwon, A.; Ries, M.; et al. Etiological involvement of KCND1 variants in an X-linked neurodevelopmental disorder with variable expressivity. Am. J. Hum. Genet. 2024, 111, 1206–1221. [CrossRef]
- Miceli, F. et al. Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. Journal of Neuroscience 35, 3782–3793 (2015).
- Ambrosino, P. et al. A novel KCNC1 gain-of-function variant causing developmental and epileptic encephalopathy: “Precision medicine” approach with fluoxetine. Epilepsia 64, e148–e155 (2023).
- Miceli, F. et al. Distinct epilepsy phenotypes and response to drugs in KCNA1 gain- and loss-of function variants. Epilepsia 63, e7–e14 (2022).
- Veale, E.L.; Golluscio, A.; Grand, K.; Graham, J.M.; Mathie, A. A KCNB1 gain of function variant causes developmental delay and speech apraxia but not seizures. Front. Pharmacol. 2022, 13, 1093313. [CrossRef]
- Zhao, T.; Wang, L.; Chen, F. Potassium channel-related epilepsy: Pathogenesis and clinical features. Epilepsia Open 2024, 9, 891–905. [CrossRef]
- Wu, W.; Yao, H.; Negraes, P.D.; Wang, J.; Trujillo, C.A.; de Souza, J.S.; Muotri, A.R.; Haddad, G.G. Neuronal hyperexcitability and ion channel dysfunction in CDKL5-deficiency patient iPSC-derived cortical organoids. Neurobiol. Dis. 2022, 174. [CrossRef]
- Nelson, S.B.; Valakh, V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87, 684–698. [CrossRef]
- Kan, A. S. H. et al. Understanding paralogous epilepsy-associated GABAA receptor variants: Clinical implications, mechanisms, and potential pitfalls. Proc Natl Acad Sci U S A 121, e2413011121 (2024).
- Sun, J.-H.; Chen, J.; Valenzuela, F.E.A.; Brown, C.; Masser-Frye, D.; Jones, M.; Romero, L.P.; Rinaldi, B.; Li, W.L.; Li, Q.-Q.; et al. X-linked neonatal-onset epileptic encephalopathy associated with a gain-of-function variant p.R660T in GRIA3. PLOS Genet. 2021, 17, e1009608. [CrossRef]
- Ragnarsson, L.; Zhang, Z.; Das, S.S.; Tran, P.; Andersson, Å.; Portes, V.D.; Altuzarra, C.D.; Remerand, G.; Labalme, A.; Chatron, N.; et al. GRIN1 variants associated with neurodevelopmental disorders reveal channel gating pathomechanisms. Epilepsia 2023, 64, 3377–3388. [CrossRef]
- Rinaldi, B.; Bayat, A.; Zachariassen, L.G.; Sun, J.-H.; Ge, Y.-H.; Zhao, D.; Bonde, K.; Madsen, L.H.; Awad, I.A.A.; Bagiran, D.; et al. Gain-of-function and loss-of-function variants in GRIA3 lead to distinct neurodevelopmental phenotypes. Brain 2023, 147, 1837–1855. [CrossRef]
- Hamanaka, K.; Miyoshi, K.; Sun, J.-H.; Hamada, K.; Komatsubara, T.; Saida, K.; Tsuchida, N.; Uchiyama, Y.; Fujita, A.; Mizuguchi, T.; et al. Amelioration of a neurodevelopmental disorder by carbamazepine in a case having a gain-of-function GRIA3 variant. Hum. Genet. 2022, 141, 283–293. [CrossRef]
- Ahring, P.K.; Liao, V.W.Y.; Gardella, E.; Johannesen, K.M.; Krey, I.; Selmer, K.K.; Stadheim, B.F.; Davis, H.; Peinhardt, C.; Koko, M.; et al. Gain-of-function variants in GABRD reveal a novel pathway for neurodevelopmental disorders and epilepsy. Brain 2021, 145, 1299–1309. [CrossRef]
- Musto, E. et al. GABRA1-related disorders: From genetic to functional pathways. Ann Neurol 95, 27–41 (2024).
- Yennawar, M.; White, R.S.; Jensen, F.E. AMPA Receptor Dysregulation and Therapeutic Interventions in a Mouse Model of CDKL5 Deficiency Disorder. J. Neurosci. 2019, 39, 4814–4828. [CrossRef]
- Gennaccaro, L.; Fuchs, C.; Loi, M.; Roncacè, V.; Trazzi, S.; Ait-Bali, Y.; Galvani, G.; Berardi, A.C.; Medici, G.; Tassinari, M.; et al. A GABAB receptor antagonist rescues functional and structural impairments in the perirhinal cortex of a mouse model of CDKL5 deficiency disorder. Neurobiol. Dis. 2021, 153, 105304. [CrossRef]
- Lefebvre, V.; Smits, P. Transcriptional control of chondrocyte fate and differentiation. Birth Defects Res. Part C: Embryo Today: Rev. 2005, 75, 200–212. [CrossRef]
- Lefebvre, V. & Bhattaram, P. Vertebrate skeletogenesis. Curr Top Dev Biol 90, 291–317 (2010).
- Hernández-García, F.; Fernández-Iglesias, Á.; Suárez, J.R.; Gil Peña, H.; López, J.M.; Pérez, R.F. The Crosstalk Between Cartilage and Bone in Skeletal Growth. Biomedicines 2024, 12, 2662. [CrossRef]
- Tonelli, F.; Bek, J.W.; Besio, R.; De Clercq, A.; Leoni, L.; Salmon, P.; Coucke, P.J.; Willaert, A.; Forlino, A. Zebrafish: A Resourceful Vertebrate Model to Investigate Skeletal Disorders. Front. Endocrinol. 2020, 11, 489. [CrossRef]
- Lefebvre, V.; Huang, W.; Harley, V.R.; Goodfellow, P.N.; de Crombrugghe, B. SOX9 Is a Potent Activator of the Chondrocyte-Specific Enhancer of the Proα1(II) Collagen Gene. Mol. Cell. Biol. 1997, 17, 2336–2346. [CrossRef]
- Liu, Y.; Li, H.; Tanaka, K.; Tsumaki, N.; Yamada, Y. Identification of an Enhancer Sequence within the First Intron Required for Cartilage-specific Transcription of the α2(XI) Collagen Gene. J. Biol. Chem. 2000, 275, 12712–12718. [CrossRef]
- Genzer, M.A.; Bridgewater, L.C. A Col9a1 enhancer element activated by two interdependent SOX9 dimers. Nucleic Acids Res. 2007, 35, 1178–1186. [CrossRef]
- Lamandé, S.R.; Bateman, J.F. Genetic Disorders of the Extracellular Matrix. Anat. Rec. 2019, 303, 1527–1542. [CrossRef]
- Saltarelli, M.A.; Quarta, A.; Chiarelli, F. Growth plate extracellular matrix defects and short stature in children. Ann. Pediatr. Endocrinol. Metab. 2022, 27, 247–255. [CrossRef]
- Chen, N.; Wu, R.W.; Lam, Y.; Chan, W.C.; Chan, D. Hypertrophic chondrocytes at the junction of musculoskeletal structures. Bone Rep. 2023, 19, 101698. [CrossRef]
- Ortega, N.; Behonick, D.J.; Werb, Z. Matrix remodeling during endochondral ossification. Trends Cell Biol. 2004, 14, 86–93. [CrossRef]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.-S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [CrossRef]
- Komori, T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res. 2010, 339, 189–195. [CrossRef]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med. 2013, 19, 179–192. [CrossRef]
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. Int. J. Mol. Sci. 2019, 20, 5525. [CrossRef]
- Huybrechts, Y.; Mortier, G.; Boudin, E.; Van Hul, W. WNT Signaling and Bone: Lessons From Skeletal Dysplasias and Disorders. Front. Endocrinol. 2020, 11, 165. [CrossRef]
- Wang, Y.; Li, Y.-P.; Paulson, C.; Shao, J.-Z.; Zhang, X.; Wu, M.; Chen, W. Wnt and the Wnt signaling pathway in bone development and disease. Front. Biosci. 2014, 19, 379–407. [CrossRef]
Figure 1.
- Comparison of gene expression levels between cdkl5-/- mutant zebrafish and WT siblings at 5 dpf (A, C, E, G) and 35 dpf (B, D, F, H). (A, B) Box plots of the distribution of gene expression levels based on log2 transformed FPKM (Fragments Per Kilobase of transcript per Million mapped reads) values for each sample. The abscissa axis is the sample name, and the ordinate axis represents the log2 (FPKM + 1) values. (C, D) Heatmap of the square of Pearson correlation coefficients (R2) among the ten sequenced samples. (E, F) Principal Component Analysis (PCA) maps based on the gene expression value (FPKM) of samples. The abscissa axis is the first principal component (PC1) representing the most variation in the data, and the ordinate axis is the second principal component (PC2), representing the second most variation in the data. (G, H) Venn diagram of gene expression indicating the number of common and uniquely expressed genes in the WT siblings and cdkl5-/- groups.
Figure 1.
- Comparison of gene expression levels between cdkl5-/- mutant zebrafish and WT siblings at 5 dpf (A, C, E, G) and 35 dpf (B, D, F, H). (A, B) Box plots of the distribution of gene expression levels based on log2 transformed FPKM (Fragments Per Kilobase of transcript per Million mapped reads) values for each sample. The abscissa axis is the sample name, and the ordinate axis represents the log2 (FPKM + 1) values. (C, D) Heatmap of the square of Pearson correlation coefficients (R2) among the ten sequenced samples. (E, F) Principal Component Analysis (PCA) maps based on the gene expression value (FPKM) of samples. The abscissa axis is the first principal component (PC1) representing the most variation in the data, and the ordinate axis is the second principal component (PC2), representing the second most variation in the data. (G, H) Venn diagram of gene expression indicating the number of common and uniquely expressed genes in the WT siblings and cdkl5-/- groups.
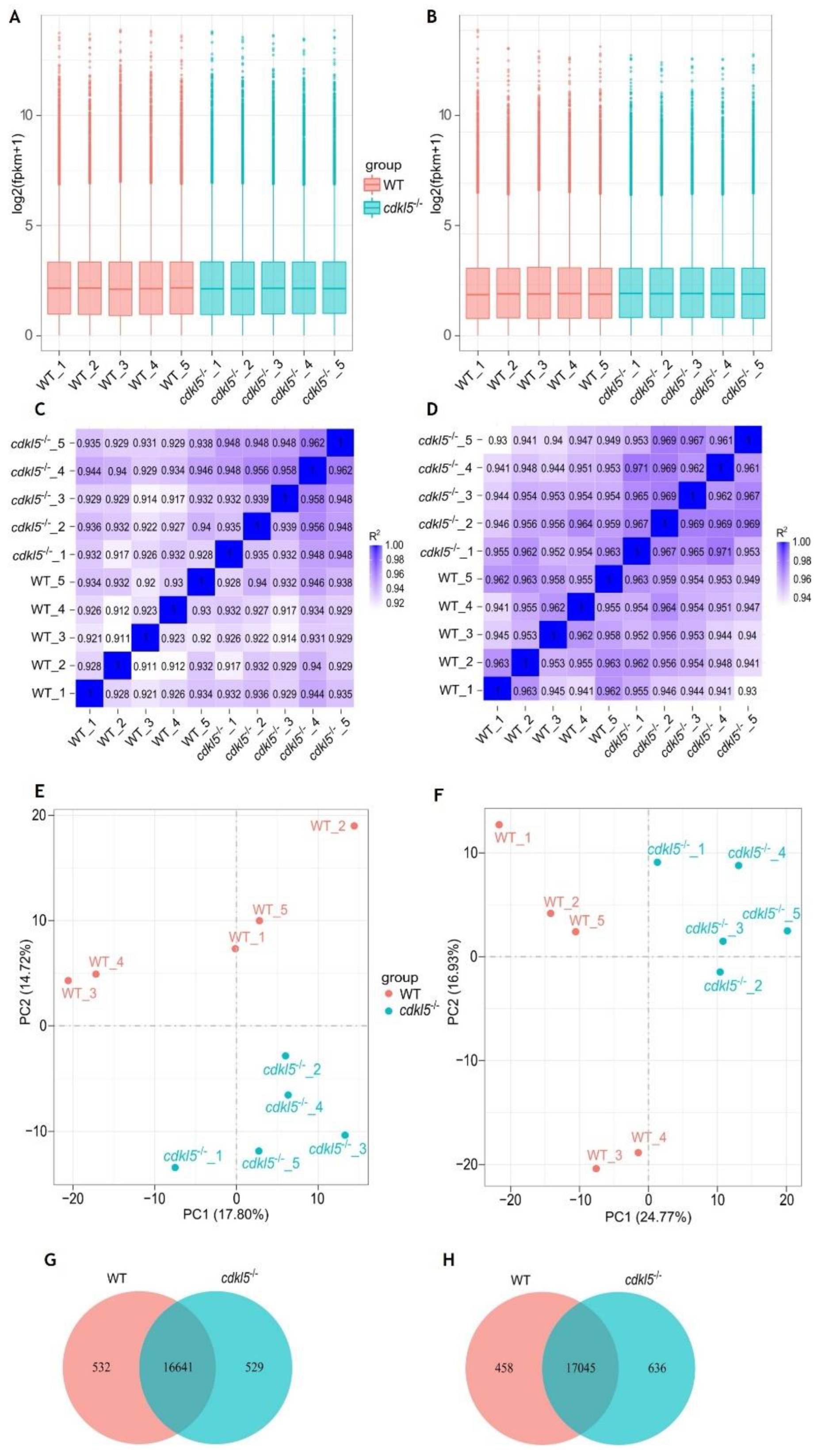
Figure 2.
Differential gene expression analysis between cdkl5-/- mutant zebrafish and WT siblings at 5 dpf (A, C, E) and 35 dpf (B, D, F). (A, B) Heatmap of hierarchical clustering analysis of differentially expressed genes (DEGs), based on log2(FPKM+1) values. Each column corresponds to a different sample, while each row represents a distinct gene. Red and green colors indicate genes with higher and lower expression levels, respectively. (C, D) The number of differentially expressed genes in cdkl5-/- vs WT groups. (E, F) Volcano plots show the distribution of the differentially expressed genes. The abscissa axis represents the value of log2FoldChange, and the ordinate axis represents the statistical significance value (-log10 (p value)) of gene expression between the groups. Red and green dots indicate significantly up-regulated (up) and down-regulated (down) genes, respectively. Blue dots represent genes with no statistically significant differences in expression (no) between the two groups.
Figure 2.
Differential gene expression analysis between cdkl5-/- mutant zebrafish and WT siblings at 5 dpf (A, C, E) and 35 dpf (B, D, F). (A, B) Heatmap of hierarchical clustering analysis of differentially expressed genes (DEGs), based on log2(FPKM+1) values. Each column corresponds to a different sample, while each row represents a distinct gene. Red and green colors indicate genes with higher and lower expression levels, respectively. (C, D) The number of differentially expressed genes in cdkl5-/- vs WT groups. (E, F) Volcano plots show the distribution of the differentially expressed genes. The abscissa axis represents the value of log2FoldChange, and the ordinate axis represents the statistical significance value (-log10 (p value)) of gene expression between the groups. Red and green dots indicate significantly up-regulated (up) and down-regulated (down) genes, respectively. Blue dots represent genes with no statistically significant differences in expression (no) between the two groups.
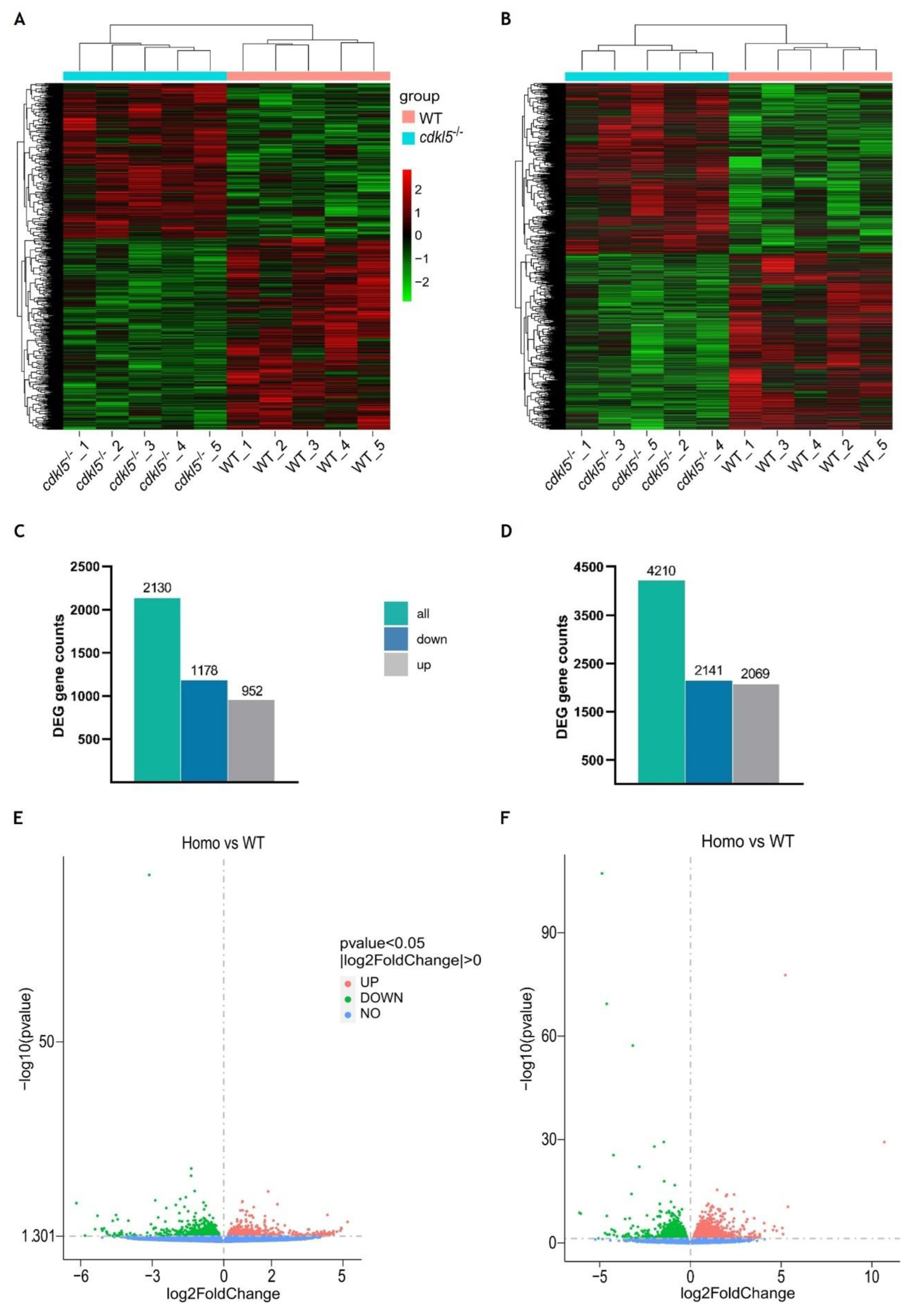
Figure 3.
Enriched gene ontology (GO) terms for all differentially expressed genes between cdkl5-/- mutant zebrafish and WT siblings. The top 10 significantly enriched GO terms in biological process (BP), cellular component (CC), and molecular function (MF) for all DEGs at 5 dpf (A) and 35 dpf (B) are displayed. The ordinate axis represents the GO term while the abscissa axis represents the level of significance of GO term's enrichment, expressed as -log10(padj). Different colors represent different functional categories. The numbers above each bar indicate the number of enriched DEGs for each GO term.
Figure 3.
Enriched gene ontology (GO) terms for all differentially expressed genes between cdkl5-/- mutant zebrafish and WT siblings. The top 10 significantly enriched GO terms in biological process (BP), cellular component (CC), and molecular function (MF) for all DEGs at 5 dpf (A) and 35 dpf (B) are displayed. The ordinate axis represents the GO term while the abscissa axis represents the level of significance of GO term's enrichment, expressed as -log10(padj). Different colors represent different functional categories. The numbers above each bar indicate the number of enriched DEGs for each GO term.
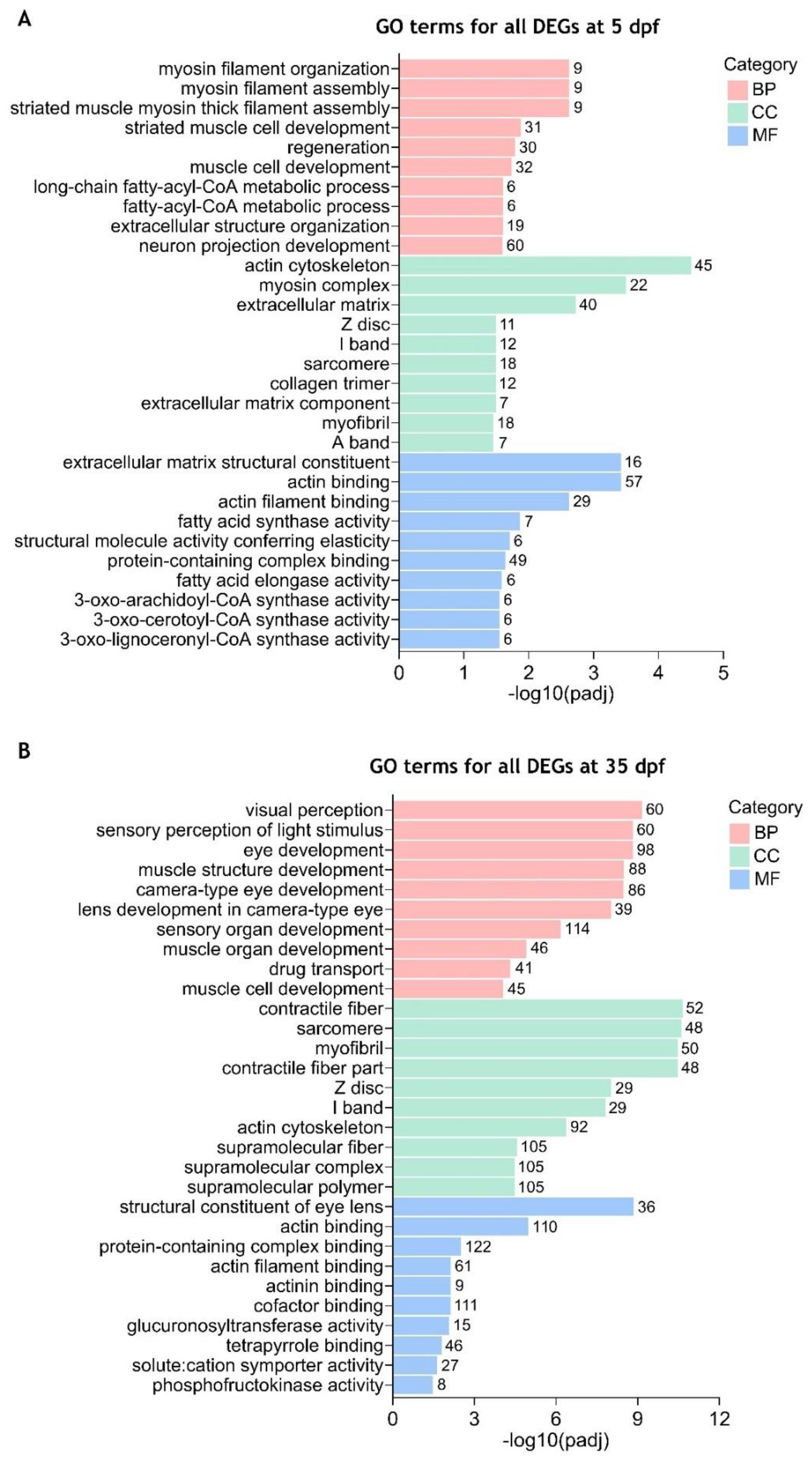
Figure 4.
Gene ontology (GO) terms enrichment analysis for the separated downregulated and upregulated DEGs between cdkl5-/- mutant zebrafish and WT siblings. The top 10 enriched GO terms in biological process (BP), cellular component (CC), and molecular function (MF) for the downregulated (A, B) and upregulated (C, D) DEGs at 5 dpf (A, C) and 35 dpf (B, D). Stripped bars indicate the GO terms that were not statistically significant. The numbers in front of each bar indicate the number of enriched DEGs for each GO term.
Figure 4.
Gene ontology (GO) terms enrichment analysis for the separated downregulated and upregulated DEGs between cdkl5-/- mutant zebrafish and WT siblings. The top 10 enriched GO terms in biological process (BP), cellular component (CC), and molecular function (MF) for the downregulated (A, B) and upregulated (C, D) DEGs at 5 dpf (A, C) and 35 dpf (B, D). Stripped bars indicate the GO terms that were not statistically significant. The numbers in front of each bar indicate the number of enriched DEGs for each GO term.
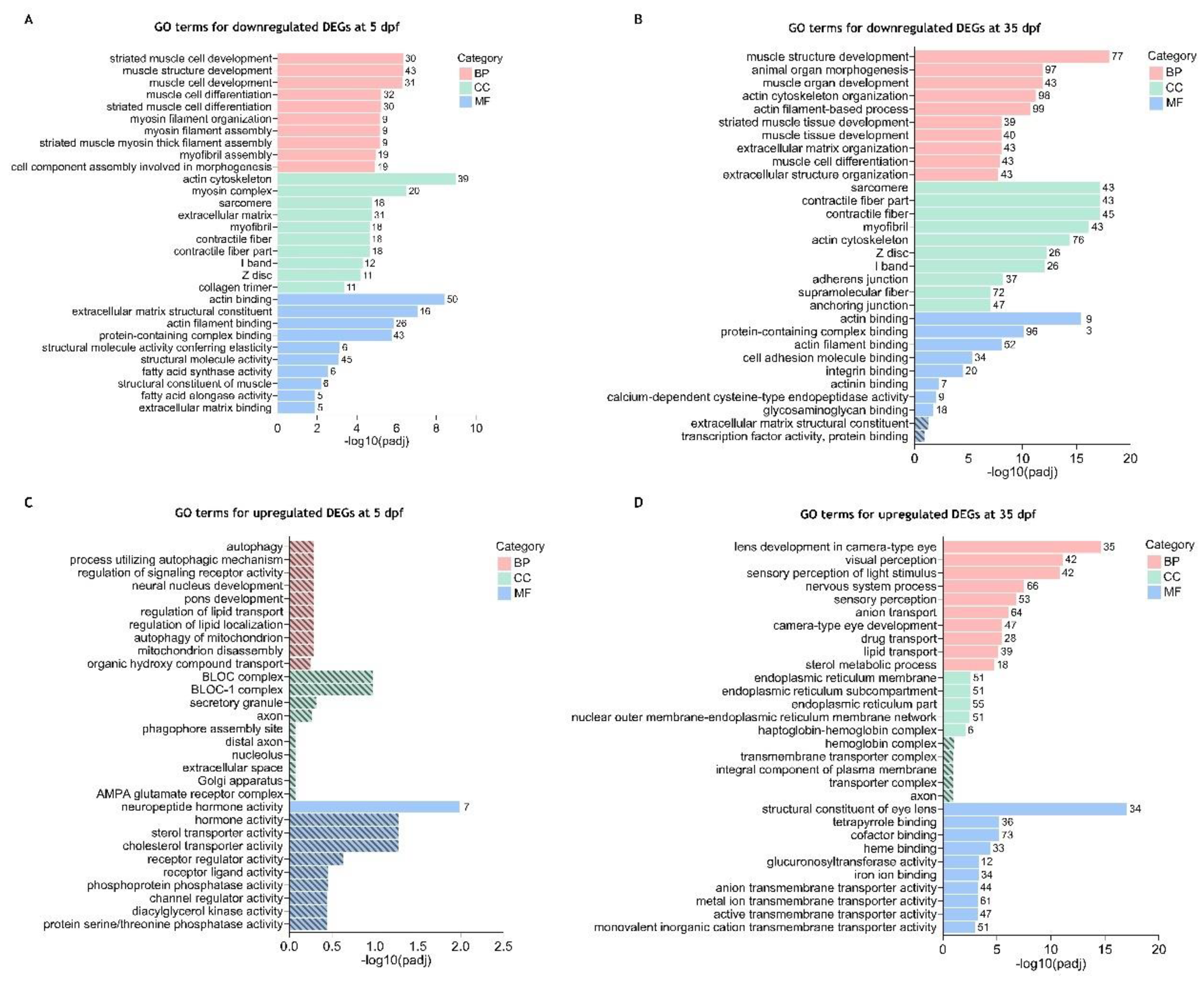
Figure 5.
Enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in zebrafish cdkl5-/- mutants compared to WT siblings. (A) Significantly enriched KEGG pathways for the downregulated DEGs at 5 dpf. (B) Significantly enriched KEGG pathways for the downregulated DEGs at 35 dpf. (C) Significantly enriched KEGG pathways for the upregulated DEGs at 35 dpf. The number of DEGs contributing to each pathway is shown in front of the corresponding bar.
Figure 5.
Enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in zebrafish cdkl5-/- mutants compared to WT siblings. (A) Significantly enriched KEGG pathways for the downregulated DEGs at 5 dpf. (B) Significantly enriched KEGG pathways for the downregulated DEGs at 35 dpf. (C) Significantly enriched KEGG pathways for the upregulated DEGs at 35 dpf. The number of DEGs contributing to each pathway is shown in front of the corresponding bar.
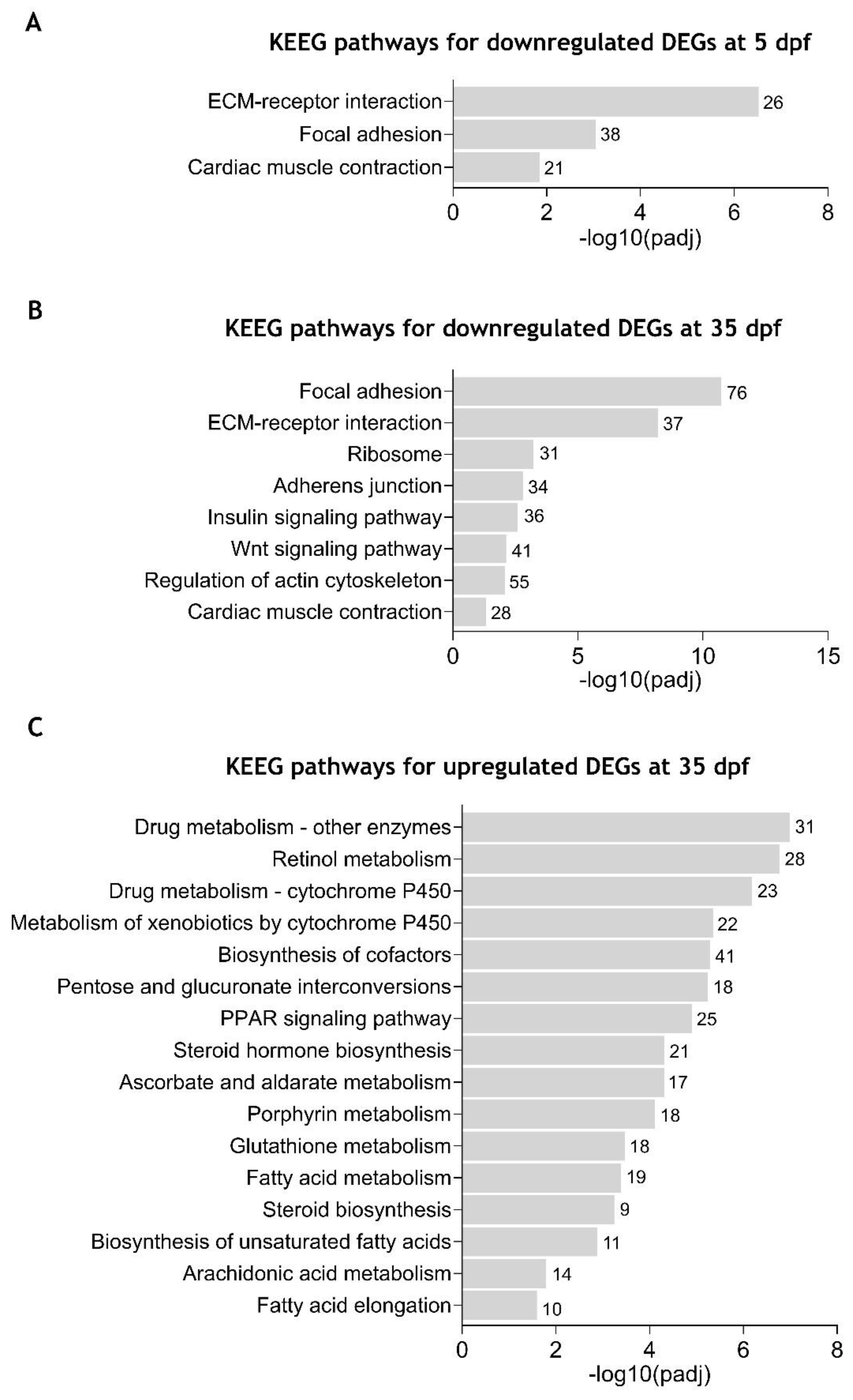
Figure 6.
RT-qPCR analysis of a randomly selected set of DEG between cdkl5-/- mutant zebrafish and WT siblings at 5 dpf (A) and 35 dpf (B). Relative expression values are presented as mean±SD and statistical analysis was performed using Student's t-test with Welch correction. *, **, ***, and **** indicate p < 0.05, p < 0.01, p < 0.001 and p < 0.0001, respectively.
Figure 6.
RT-qPCR analysis of a randomly selected set of DEG between cdkl5-/- mutant zebrafish and WT siblings at 5 dpf (A) and 35 dpf (B). Relative expression values are presented as mean±SD and statistical analysis was performed using Student's t-test with Welch correction. *, **, ***, and **** indicate p < 0.05, p < 0.01, p < 0.001 and p < 0.0001, respectively.
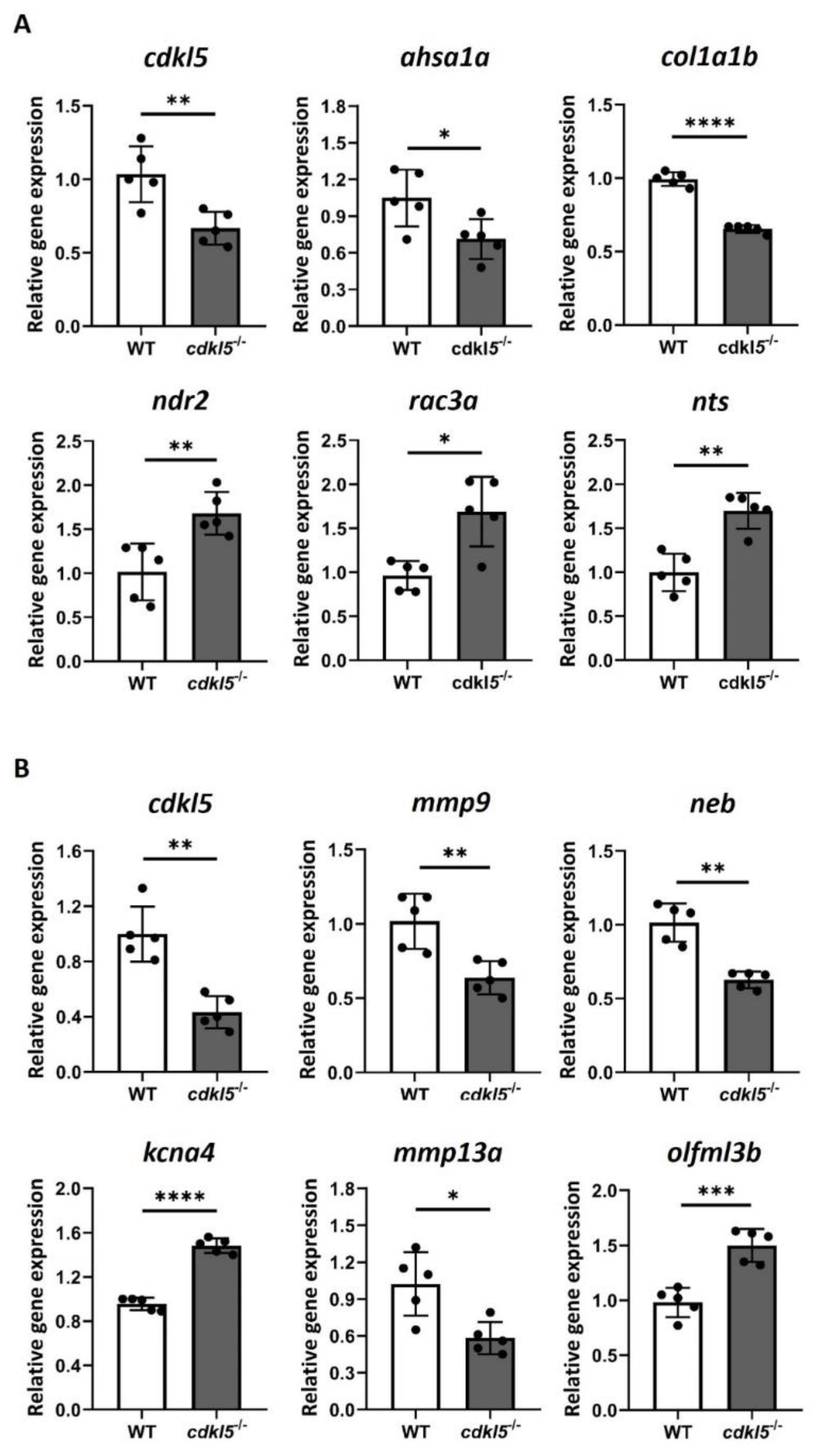
Figure 7.
Motor neuron alterations in cdkl5-/- zebrafish at 3 dpf. (A) Representative fluorescence images of the WT and cdkl5−/− motor neurons, labelled by Tg(hb9:GFP). (B) Number of motor neurons per hemisegment. (C) Axonal length of CaP motoneurons. Values are presented as mean±SD and statistical analysis was performed using Student's t-test with Welch correction. ** and **** indicate and p < 0.01 and p < 0.0001, respectively.
Figure 7.
Motor neuron alterations in cdkl5-/- zebrafish at 3 dpf. (A) Representative fluorescence images of the WT and cdkl5−/− motor neurons, labelled by Tg(hb9:GFP). (B) Number of motor neurons per hemisegment. (C) Axonal length of CaP motoneurons. Values are presented as mean±SD and statistical analysis was performed using Student's t-test with Welch correction. ** and **** indicate and p < 0.01 and p < 0.0001, respectively.
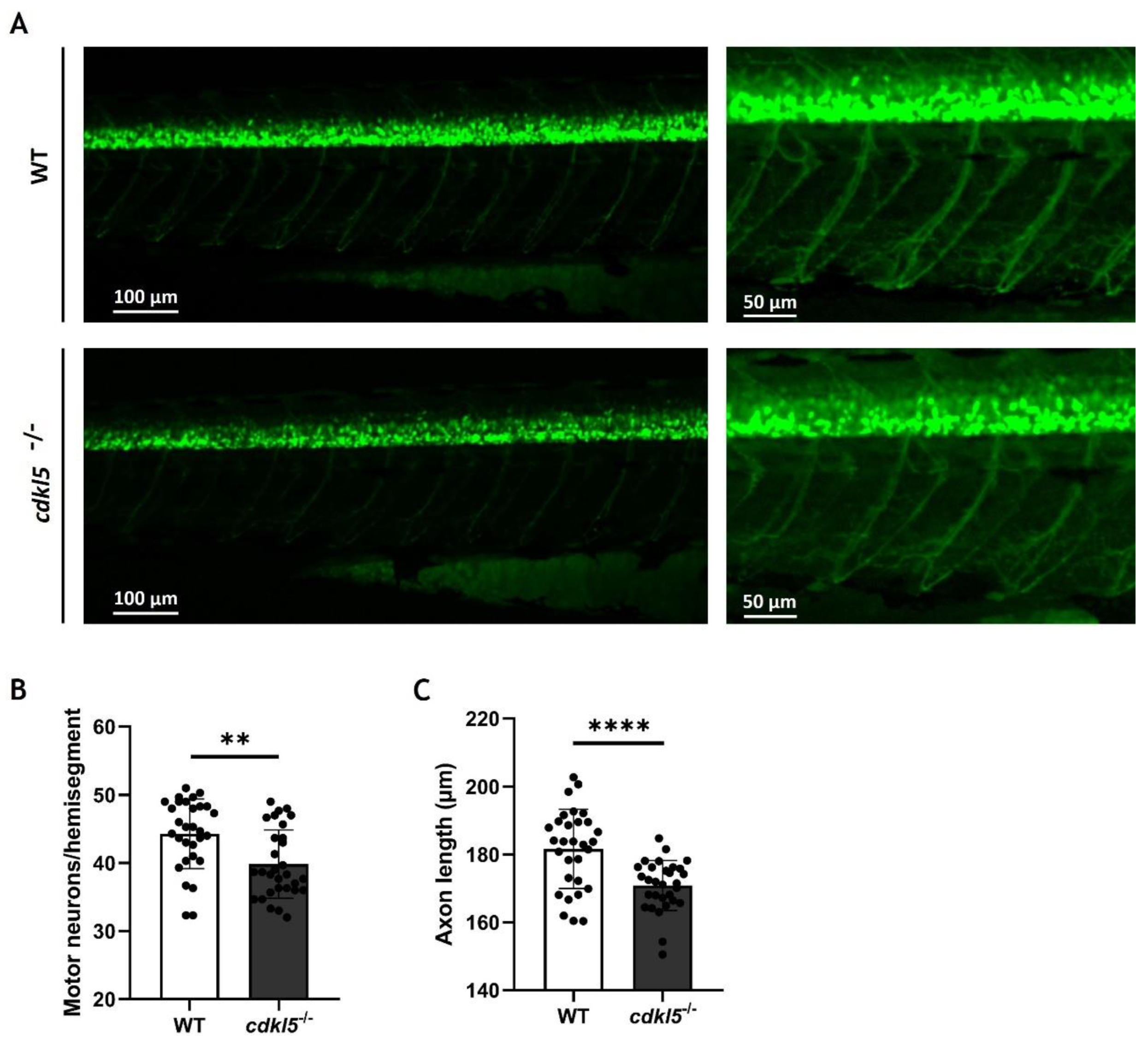

Table 2.
The top 20 known up-regulated and down-regulated differentially expressed genes between cdkl5-/- mutant and WT siblings’ zebrafish at 5 and 35 days post-fertilization. For each gene is reported the log2(fold change) of cdkl5-/- over WT, the p-value and corrected p-value (padj) calculated by DESeq2.
Table 2.
The top 20 known up-regulated and down-regulated differentially expressed genes between cdkl5-/- mutant and WT siblings’ zebrafish at 5 and 35 days post-fertilization. For each gene is reported the log2(fold change) of cdkl5-/- over WT, the p-value and corrected p-value (padj) calculated by DESeq2.
| Gene symbol | Protein name | Log2(fold change) | p-value | padj |
|---|---|---|---|---|
| 5 days post-fertilization embryos | ||||
| sort1a | Sortilin 1a | 1.8794 | 2.9996e-13 | 1.1657e-09 |
| tdo2b | Tryptophan 2,3-Dioxygenase 2b | 0.8033 | 9.0703e-11 | 1.9583e-07 |
| cflara | CASP8 and FADD-like apoptosis regulator a | 0.7984 | 1.3040e-10 | 2.5339e-07 |
| ube2l3a | Ubiquitin-conjugating enzyme E2L 3a | 2.2685 | 4.5907e-10 | 6.8616e-07 |
| pdap1a | pdgfa associated protein 1a | 0.8095 | 1.3519e-08 | 1.3135e-05 |
| csrp2 | Cysteine and glycine-rich protein 2 | 0.7129 | 1.4895e-08 | 1.3782e-05 |
| igfbp1b | Insulin-like growth factor binding protein 1b | 0.9922 | 2.3254e-08 | 2.0442e-05 |
| guca1c | Guanylate cyclase activator 1C | 0.9678 | 2.9697e-08 | 2.4044e-05 |
| nts | Neurotensin | 4.3662 | 1.9960e-07 | 0.0001 |
| tmem240b | Transmembrane protein 240b | 1.1948 | 2.8351e-07 | 0.0002 |
| ndufs7 | NADH Dehydrogenase [Ubiquinone] Iron-Sulfur Protein 7 | 1.0968 | 3.1092e-07 | 0.0002 |
| c7b | Complement component 7b | 1.7532 | 3.7864e-07 | 0.0002 |
| mt2 | Metallothionein 2 | 1.2515 | 9.1100e-07 | 0.0005 |
| phf13 | PHD finger protein 13 | 0.5807 | 2.5537e-06 | 0.0012 |
| krt15 | Keratin 15 | 0.9521 | 2.9615e-06 | 0.0013 |
| cspg4bb | chondroitin sulfate proteoglycan 4bb | 1.7450 | 4.0039e-06 | 0.0017 |
| ccdc149a | coiled-coil domain containing 149a | 2.0997 | 5.2690e-06 | 0.0021 |
| arr3b | Arrestin 3b | 0.7502 | 5.3098e-06 | 0.0021 |
| tcnba | Transcobalamin beta a | 0.7376 | 7.0446e-06 | 0.0024 |
| cel.2 | Carboxyl ester lipase, tandem duplicate 2 | 0.5815 | 8.2700e-06 | 0.0028 |
| gamt | Guanidinoacetate N-methyltransferase | -3.1079 | 1.1615e-92 | 2.2569e-88 |
| nucks1a | Nuclear casein kinase and cyclin-dependent kinase substrate 1a | -1.3425 | 5.4136e-19 | 5.2596e-15 |
| pir | Pirin | -1.3499 | 3.3308e-17 | 2.1574e-13 |
| ctsl.1 | Cathepsin L.1 | -1.2024 | 2.3532e-13 | 1.1431e-09 |
| card19 | Caspase recruitment domain family member 19 | -1.5736 | 8.6568e-12 | 2.8035e-08 |
| ugt1b5 | UDP glucuronosyltransferase 1 family, polypeptide B5 | -1.8045 | 2.4384e-11 | 6.7686e-08 |
| dynlt3 | Dynein Light Chain Tctex-Type 3 | -0.8301 | 1.6882e-10 | 2.9821e-07 |
| klhdc8a | Kelch domain containing 8A | -6.1579 | 2.4233e-10 | 3.9239e-07 |
| ccdc120 | Coiled-coil domain containing 120 | -1.9919 | 6.3624e-10 | 8.8305e-07 |
| foxj3 | Forkhead box J3 | -0.7362 | 9.5553e-10 | 1.2378e-06 |
| slc15a2 | Solute carrier family 15, member 2 | -0.8038 | 8.1421e-09 | 8.3268e-06 |
| slc12a10.1 | Solute carrier family 12, member 10, tandem duplicate 1 | -1.8439 | 2.4197e-08 | 2.0442e-05 |
| cishb | Cytokine inducible SH2-containing protein b | -1.8439 | 7.1115e-08 | 5.4873e-05 |
| mbd1b | Methyl-CpG binding domain protein 1b | -1.1586 | 7.3423e-08 | 5.4873e-05 |
| slc41a1 | Solute carrier family 41, member 1 | -4.4974 | 2.1192e-07 | 0.0001 |
| rs1a | Retinoschisin 1a | -0.5785 | 2.2400e-07 | 0.0001 |
| etnk2 | Ethanolamine kinase 2 | -5.2767 | 3.2897e-07 | 0.0002 |
| lrp2b | Low density lipoprotein receptor-related protein 2b | -1.2138 | 5.5833e-07 | 0.0003 |
| gnl3l | G protein nucleolar 3 like | -2.7313 | 8.3341e-07 | 0.0004 |
| myh7ba | Myosin heavy chain 7B | -0.8244 | 1.2271e-06 | 0.0006 |
| col5a1 | Collagen type V alpha 1 chain | -0.4288 | 1.2532e-06 | 0.0006 |
| 35 days post-fertilization juveniles | ||||
| olfml3b | Olfactomedin-like 3b | 5.2512 | 1.5620e-78 | 1.6759e-74 |
| kdm5ba | Lysine Demethylase 5Ba | 1.5014 | 3.4759e-16 | 6.2161e-13 |
| crygm2d18 | crystallin, gamma M2d18 | 2.4337 | 7.8911e-15 | 1.2096e-11 |
| ipo9 | Importin 9 | 2.0313 | 8.7711e-15 | 1.2549e-11 |
| rlbp1b | Retinaldehyde binding protein 1b | 1.9842 | 2.3868e-14 | 3.2013e-11 |
| ggctb | Gamma-glutamylcyclotransferase b | 1.0173 | 3.8752e-12 | 4.3769e-09 |
| lim2.4 | Lens intrinsic membrane protein 2.4 | 1.2252 | 1.4216e-11 | 1.4528e-08 |
| tfcp2l1 | Transcription factor CP2-like 1 | 1.1479 | 4.2758e-11 | 3.6703e-08 |
| krt15 | Keratin 15 | 1.5442 | 1.1659e-10 | 9.0605e-08 |
| crygm2d15 | Crystallin gamma M2d15 | 2.2803 | 1.2171e-10 | 9.0605e-08 |
| crygm2d16 | Crystallin gamma M2d16 | 2.3116 | 1.2224e-10 | 9.0605e-08 |
| crygm2d11 | Crystallin gamma M2d11 | 1.5472 | 1.2244e-10 | 9.0605e-08 |
| rrp9 | Ribosomal RNA processing 9 | 1.6369 | 1.4287e-10 | 9.8906e-08 |
| cryba2a | Crystallin beta a2a | 1.1551 | 2.3574e-10 | 1.5809e-07 |
| crygm2d13 | Crystallin gamma M2d13 | 1.6363 | 4.4261e-10 | 2.7139e-07 |
| parapinopsinb | Parapinopsin b | 2.9711 | 6.3436e-10 | 3.5825e-07 |
| foxq1a | Forkhead box Q1a | 0.6750 | 7.2768e-10 | 3.9040e-07 |
| dynlrb1 | Dynein light chain roadblock-type 1 | 0.8483 | 8.6657e-10 | 4.5358e-07 |
| timm17a | Translocase of inner mitochondrial membrane 17A | 1.4058 | 2.2971e-09 | 1.0060e-06 |
| crygm2d14 | Crystallin, gamma M2d14 | 1.8210 | 2.5513e-09 | 1.0950e-06 |
| opn1lw2 | Opsin 1, long-wave-sensitive, 2 | -4.8460 | 4.6970e-108 | 1.0080e-103 |
| opn1lw1 | Opsin 1, long-wave-sensitive, 1 | -4.5892 | 3.4828e-70 | 2.4914e-66 |
| gnl3l | G protein nucleolar 3 like | -3.1493 | 4.3148e-58 | 2.3149e-54 |
| nucks1a | Nuclear casein kinase and cyclin-dependent kinase substrate 1a | -1.4428 | 4.5028e-30 | 1.7153e-26 |
| pir | Pirin | -1.9600 | 9.5388e-29 | 2.9243e-25 |
| slc41a1 | Solute carrier family 41, member 1 | -4.2114 | 2.8678e-26 | 7.6930e-23 |
| ccdc120 | Coiled-coil domain containing 120 | -2.7935 | 6.8052e-23 | 1.6227e-19 |
| ugt1b5 | UDP glucuronosyltransferase 1 family, polypeptide B5 | -1.4197 | 1.1478e-18 | 2.4631e-15 |
| dynlt3 | Dynein Light Chain Tctex-Type 3 | -0.8398 | 1.6501e-17 | 3.2191e-14 |
| nfasca | Neurofascin a | -1.0500 | 1.0572e-12 | 1.3345e-09 |
| mbd1b | Methyl-CpG binding domain protein 1b | -1.6633 | 3.8504e-12 | 4.3769e-09 |
| myh7ba | Myosin heavy chain 7B | -0.8647 | 3.0928e-11 | 2.8019e-08 |
| ehd2b | EH-domain containing 2b | -0.4425 | 1.2915e-10 | 9.2386e-08 |
| igfn1.1 | immunoglobulin like and fibronectin type III domain containing 1, tandem duplicate 1 | -1.1561 | 4.2629e-10 | 2.7139e-07 |
| cishb | Cytokine inducible SH2-containing protein b | -1.6556 | 4.3495e-10 | 2.7139e-07 |
| htra1b | HtrA serine peptidase 1b | -0.6314 | 4.9847e-10 | 2.8959e-07 |
| myhc4 | Myosin heavy chain 4 | -1.5783 | 6.6454e-10 | 3.6570e-07 |
| vgll2b | Vestigial-like family member 2b | -1.2804 | 1.3015e-09 | 6.3616e-07 |
| myom2a | Myomesin 2a | -1.1321 | 1.3043e-09 | 6.3616e-07 |
| vwa1 | von Willebrand factor A domain containing 1 | -0.5727 | 1.4088e-09 | 6.7183e-07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



