
Submitted:
29 July 2025
Posted:
30 July 2025
You are already at the latest version
Abstract
H5N1 Influenza A virus continues to pose a significant zoonotic threat, with increasing evidence of interspecies transmission and genetic adaptation. Previous studies primarily focused on avian or human isolates, with limited comprehensive analysis of H5N1 evolution across multiple mammalian hosts. Existing molecular surveillance often lags behind viral evolution; this study underscores the necessity for real-time monitoring of ongoing mutations affecting pathogenicity and transmissibility. Our goals are (1) to retrieve and analyse HA, NP, and NA gene sequences of H5N1 Influenza A virus from diverse hosts, including humans, poultry, and multiple mammalian species, to assess genetic diversity and evolutionary patterns. (2) To evaluate positive selection sites across the three major genes (HA, NP, NA) to determine adaptive mutations linked to host adaptation and viral survival. To achieve these goals, in this study, we analysed 78 HA, 62 NP, and 61 NA gene sequences from diverse hosts, including humans, poultry, and multiple mammalian species, retrieved from the NCBI database. The phylogenetic analysis revealed distinct clade formations, indicating regional spread and cross-species transmission events, particularly from avian sources to mammals and humans. Selection pressure analysis identified positive selection across all three genes, suggesting adaptive mutations contributing to host adaptation and viral survival. Homology modeling and molecular dynamics simulations were performed to generate high-quality structural models of HA, NP, and NA proteins, which were subsequently validated using multiple stereochemical parameters. Domain analysis confirmed conserved functional motifs, while protein-ligand docking demonstrated stable interactions at conserved binding sites, despite observed residue substitutions in recent isolates. Earlier research concentrated on HA alone; this study integrates HA, NP, and NA genes for a broader understanding of viral evolution and adaptation. These findings highlight ongoing evolutionary changes in H5N1 genes that may enhance viral adaptability and pathogenicity, underscoring the need for continuous molecular surveillance and updated antiviral strategies.
Keywords:
H5N1
; Influenza-A virus
; Clade 2.3.4.4b
; hemagglutinin
; nucleoprotein
; neuraminidase
; phylogenetics
; selection pressure
; molecular dynamics
; protein-ligand docking
1. Introduction
Influenza A viruses (IAV), belonging to the family Orthomyxoviridae, are among the most significant pathogens affecting humans and animals, causing seasonal epidemics and occasional pandemics that result in substantial morbidity, mortality, and economic losses worldwide [1,2,3,4]. Among these, highly pathogenic avian influenza (HPAI) H5N1 viruses pose a particularly grave zoonotic threat. Since their first detection in domestic poultry in the late 1990s, H5N1 viruses have spread across Asia, Europe, Africa, and the Americas, causing devastating outbreaks in poultry populations and sporadic but severe infections in humans [5]. The World Health Organization (WHO) reports a case fatality rate exceeding 50% in humans, underscoring the pandemic potential of these viruses should sustained human-to-human transmission occur [6]. The evolution of H5N1 viruses is driven by their segmented RNA genome, which facilitates frequent genetic reassortment and accumulation of point mutations through antigenic drift. These mechanisms enable the virus to evade host immune responses, adapt to new hosts, and develop resistance to antivirals [7]. While wild aquatic birds serve as the primary reservoir of IAV, recent years have witnessed an alarming increase in spillover events to domestic mammals (such as cats, dogs, pigs, and cattle) and wildlife, highlighting the virus’s expanding host range [8]. Notably, the emergence and global dissemination of H5N1 clade 2.3.4.4b since 2020 have raised concerns due to its unprecedented spread across continents and its association with widespread infections in avian and mammalian hosts, including recent reports of infections in sea lions, mink, and dairy cattle [9]. These events amplify fears of viral adaptation toward efficient mammalian transmission, which could pave the way for the next influenza pandemic [10]. At the molecular level, three viral proteins—hemagglutinin (HA), nucleoprotein (NP), and neuraminidase (NA) play critical roles in host adaptation and viral fitness. HA mediates receptor binding and membrane fusion, thereby determining host specificity and pathogenicity [11]. The NA protein facilitates viral release from infected cells by cleaving sialic acid residues, influencing transmission dynamics and drug susceptibility. NP forms the ribonucleoprotein complex essential for genome packaging and replication, and mutations in NP have been implicated in host adaptation and immune evasion [12]. Several studies have analysed HA due to its central role in receptor binding and antigenicity; however, there is limited integrated analysis of HA alongside NP and NA in the context of genetic evolution, structural variation, and functional consequences across diverse hosts [13]. Existing literature also exhibits several gaps that impede a comprehensive understanding of H5N1 evolution and its zoonotic potential: Limited cross-host evolutionary insight: Most studies have focused on either avian or human isolates, neglecting sequences from other mammalian hosts that increasingly act as intermediate or spillover reservoirs [14]. Inadequate multi-gene approach: Research on HA is abundant, but NP and NA, which influence replication efficiency and drug resistance, are often overlooked in combined analyses [15]. Lack of structural-functional correlation: While sequence-based analyses of H5N1 genes are available, few studies incorporate structural modelling, molecular dynamics (MD) simulations, and ligand-binding assessments to evaluate the functional implications of adaptive mutations [16]. Insufficient data on contemporary isolates: rapid viral evolution necessitates real-time surveillance of recent isolates from multiple species yet integrated molecular and structural studies using updated datasets remain scarce. Addressing these gaps is crucial for predicting the evolutionary trajectory of H5N1, guiding the development of antiviral drugs, and informing vaccine design strategies. The present study aims to provide a comprehensive evolutionary and structural characterization of H5N1 clade 2.3.4.4b isolates using three key viral genes, HA, NP, and NA, retrieved from multiple species, including humans, poultry, and various domestic mammals [17]. Specifically, we: conduct phylogenetic analyses to elucidate genetic relationships and identify patterns of interspecies transmission. Assess selection pressures acting on HA, NP, and NA genes to detect signatures of adaptive evolution. Predict and validate three-dimensional structures of these proteins using homology modelling and molecular dynamics simulations to understand the structural consequences of mutations [18]. Analyse conserved domains and evaluate protein-ligand interactions through molecular docking to determine the impact of residue substitutions on receptor binding and antiviral susceptibility. By integrating evolutionary, structural, and functional analyses, this study provides valuable insights into the mechanisms underlying H5N1 adaptability, thereby enhancing preparedness for future outbreaks and informing the development of effective countermeasures.
2. Materials and Methods
2.1. Retrieval of Gene Sequences and Analysis
A total of 78, 62 and 61 gene sequences of HA, NP and NA proteins, respectively from various isolates of H5N1 strain of Influenza A virus clade 2.4.4.4b from varied hosts such as human, chicken, dog, horse, cattle, cat, pig, and goat (NP gene sequences from horse were not available) were retrieved from NCBI nr-nucleotide database. These retrieved gene sequences were renamed with their accession ID, host name, country of isolation, and isolate ID. Further, sequence similarity and coverage were assessed for all sequences.
2.2. Phylogenetic and Selection Pressure Analyses of HA, NP, and NA Genes
The multiple sequence alignments were performed for sequences of HA, NP, and NA genes using Clustal Omega (Sievers and Higgins, 2021). Then, phylogenetic analysis was performed using PHYLIP (Felsenstein, 1981) with the Neighbor-Joining algorithm. Then, phylogenetic trees were visualized for gene sequences of HA, NP, and NA proteins using MEGA11 [19]. Further, selection pressure analysis was done for both HA, NP, and NA gene sequences at gene and codon levels using HYPHY/Datamonkey at the threshold of p <=0.1 [20].
2.3. Protein Sequence Retrieval and Phylogenetic Analysis of HA, NP, and NA Proteins
Protein coding sequences of all sequences of HA, NP, and NA genes were retrieved from NCBI for all the isolates from hosts such as horse (NP protein sequences from horse were not available), dog, cat, human, chicken, goat, pig, and cattle. Further, multiple sequence alignment was performed for protein sequences of HA, NP, and NA genes using Clustal Omega (Sievers and Higgins, 2021). Then, phylogenetic analysis was performed using PHYLIP (Felsenstein, 1981) with the Neighbour-Joining algorithm. Then, phylogenetic trees were visualized for protein sequences of HA, NP, and NA genes using MEGA11 (Tamura et al., 2021).
2.4. Structure Prediction and Assessment of HA, NP, and NA Proteins
The structures of the (HA, NP, and NA) proteins were predicted using the protein MODELLER software (Protein Modeling with Modeller: A Comprehensive Guide - Omics tutorials) [21], after template structure selection for HA, NP, and NA proteins using BLASTp [22]. Further, the quality of the structure was assessed using ERRAT [22], ProSA [23], and PROCHECK [24] for each protein. Further, domains were also assessed from the predicted structures using InterPro [25].
2.5. MD-Simulation of Predicted Structures of HA, NP, and NA Proteins
The GROMACS server (https://www.gromacs.org/) was used for MD-simulation to get the more stable optimized structures of HA, NP, and NA proteins within the cellular environment. First, the OPLS force field was selected for all the atoms of the proteins, along with the SPC/E water model with a 1000 kJ mol-1 nm-2 force constant (kpr) to mimic the cellular aqueous medium. To simulate the aqueous medium, each protein was placed in the centre of a cubic box filled with the water molecules at least 1.0 nm away from the box edge, along with the solvent configuration spc216.gro. The volume of the cubic box was optimized to ensure the complete coverage of the solvent over each protein. Further, 7 Na+, 12 Cl-, and 6 Na+ ions were added by replacing a similar number of solvent molecules to neutralize the -7, 12, and -6 charges of the (HA, NP, and NA) proteins, respectively. Before going for dynamics, energy minimization of the protein-solvent system was performed to avoid steric clashes or inappropriate geometry using steepest descent minimization up to a maximum force < 1000.0 kJ/mol/nm for 50000 maximum of minimization steps. Further, to begin real dynamics, equilibration of the solvent and ions around each protein was done in two phases. The first phase was conducted under a constant NVT (number of particles, volume, and temperature) ensemble to stabilize the temperature of the protein-solvent system using a modified Berendsen thermostat for 100 ps. The second phase was conducted under a constant NPT (number of particles, pressure, and temperature) ensemble to stabilize the pressure of the system using the Parrinello-Rahman barostat and modified Berendsen thermostat for 100 ps. After achieving the desired temperature and pressure of the well-equilibrated protein-solvent system, MD simulation was performed for each protein for 1000 ps. The final optimized structure of each protein was utilized for further protein-ligand docking.
2.6. Docking of the H5N1 Clade 2.3.4.4b (HA, NP, and NA) Proteins with Their Ligand-Receptors
Information on natural ligands of HA, NP, and NA proteins was taken from the published literature, such as the sialated glycan receptor (NAG-GAL-SIA) of the HA protein [13,26,27], RNA sequence of the NP protein [28], and sialic acid (SIA) of the NA protein [29]. Structures of ligands were drawn using SwissADME online tools (SwissADME), and further, hydrogen atoms were added, and charges were assigned to optimized structures of HA, NP, and NA proteins, and hydrogen atoms were added to their respective ligands using PyMol 3.1 (https://pymol.org/). Further, molecular docking was performed for each protein with its respective ligand to get the information of the binding site in terms of least binding score and binding site residues using the CD-Dock2 server (https://cadd.labshare.cn/cb-dock2/) [30] Utilizing a template-based bling docking approach. CD-Dock2 follows a procedure that combines curvature-based cavity detection with Auto Dock Vina-based molecular docking.
3. Results and Discussion
3.1. Sequence Analysis of HA, NP, and NA Genes
Details of a sum of 78, 62, and 61 sequences retrieved from NCBI for HA, NP, and NA genes, respectively, are provided in Supplementary Tables S1–S3, respectively. With respect to PP755366.1/goat/Minnesota/24-007234-006/2024 HA gene, other 77 HA gene sequences have sequence similarity up to 85% and coverage up to 99.8%. Out of 78 gene sequences of HA protein, 10, 18, 11, 06, 04, 09, 03, and 17 sequences were extracted from cattle, human, cat, goat, horse, swine, dog, and chicken hosts, respectively from USA, Japan, South Korea, Thailand, Indonesia, Viet Nam, China, Nigeria, Germany, Hong Kong and Bangladesh between 2001 to 2025. Similarly, out of 62 gene sequences of NP protein, 04, 11, 06, 02, 02, 04, 01, and 32 sequences were extracted from cattle, human, cat, goat, horse, swine, dog, and chicken hosts, respectively from Germany, Sudan, Burkina Faso, China, USA, Indonesia, Thailand, Cambodia, Kuwait, Poland, Russia, Ivory Coast, Israel, Scotland, Cameroon, Bangladesh, Bhutan, Nepal, Pakistan, Afghanistan, South Korea, Malaysia, Nigeria, Laos, Ghana, Philippines, India, Egypt, England, Viet Nam, France, Chile, Canada and Hong Kong. There was sequence similarity up to 82% and coverage up to 95.5% among NP gene sequences with respect to the NP gene sequence of PV124897.1/cattle/ Hong Kong/RG-DelNS1-p10/2024. Similarly, there was sequence similarity up to 77.4% and coverage up to 94.8% among NA gene sequences with respect to the NA gene sequence of PV649629.1/chicken/NY/25-010935-008-original/2025. Out of 61 gene sequences of NA protein, 08, 14, 06, 03, 03, 12, 02, and 13 sequences were extracted from cattle, human, cat, goat, horse, swine, dog, and chicken hosts, respectively.
3.2. Phylogenetic Analysis of HA, NP, and NA Genes
The phylogenetic analysis of the HA gene (Figure 1) shows that the genes from different isolates of H5N1 Influenza virus could be grouped mainly in four clades. It can be seen from clade I that the HA gene from American isolates of H5N1 Influenza virus from different hosts such as goat, cattle, human, cat, and chicken, are more closely related to each other, which may suggest that H5N1 Influenza virus might have spread through humans to other domestic host species living with the human population. A similar pattern is seen in clade II, and it also suggests that the H5N1 isolates might have reached the USA via an affected human from H5N1-infected chickens in Europe, and H5N1 isolates might have reached Europe through affected chickens from Asia. Clade III and clade IV suggest that in Asia, the H5N1 influenza virus might have reached human and other domestic animal species populations through chickens. It can also be seen that HA genes from most of the isolates from China are similar to each other and might have transgressed from Chicken to other mammalian species. It is also evident from the phylogenetic tree of the HA gene that the HA gene from most of the isolates of different hosts from Asian countries other than China might have spread from China and are more similar to each other. Another important observation from Figure 1 is that the HA gene sequences of isolates in clades I and II were recently isolated and sequenced, and were similar to each other, regardless of the host of the isolates. However, the HA gene sequences of isolates in clades III and IV were older. This suggests that older isolates were somewhat distinct from recent isolates.
The phylogenetic analysis of the NP gene (Figure 2) shows that NP genes from different isolates of H5N1 Influenza virus could be grouped mainly in seven clades. Clade I is the largest, suggesting that H5N1 isolates may have migrated from Asia to the Middle East and via Russia to Europe. It also suggests the migration of H5N1 infection could have occurred from Chicken to different domestic host species in Asia and other countries. Similarly, a conclusion could be derived from clades II and III that the migration of H5N1 infection could have occurred from Chicken to different domestic host species in Asia. Clade III also suggests the migration of H5N1 from Asia to Africa via infected chickens. Clade IV suggests migration of H5N1 from Asia to Europe via Egypt. It can be seen from the clade VI that the NP gene from American isolates of H5N1 Influenza virus from different hosts such as goat, cattle, human, cat, swine and chicken are more closely related to each other, which may suggest that H5N1 Influenza virus might have spread through human to other domestic host species living with human population. Clades V and VII consist of only one isolate of H5N1 each from chicken and cattle from Scotland and Hong Kong, respectively. It can also be seen in Figure 1 that NP gene sequences of isolates in clades IV, VI, and VII were isolated and sequenced recently except KX364460.1/swine/Shandong/SD1/2014 and were similar to each other irrespective of the host of the isolates; however, the NP gene sequences of isolates of other clades were older. This also suggests that older isolates were somewhat distinct from recent isolates.
Phylogenetic analysis of the NA gene (Figure 3) found that the NA gene sequences of various isolates of H5N1 Influenza A virus could be grouped into three clades. Clade I is the largest, and Clade II is the smallest. Clade I suggests that most of the Asian isolates are similar to each other and are older, irrespective of the host, in comparison to isolates in clades II and III. It also suggests that in Asia, the H5N1 influenza virus might have spread from chicken to human and from human to other domestic species, though in Middle East countries like Iraq and Egypt, it might have been transmitted by an infected human and spread to other domestic species reared along with the human population. It can be seen from clade II that the NA gene from various isolates from chicken spread to human and pig populations. Clade III suggests that different isolates of H5N1 Influenza A virus might have been introduced to North and South America from infected chickens from Viet Nam. It is also very interesting to see from clade III that there must have been a fresh spread of new strains/isolates of H5N1 Influenza virus from Asia to Europe, the Middle East, North America, and South America, which could be concluded based on the similarity of the NA genes from recent isolates, irrespective of host and country of isolation.
3.3. Selection Pressure Analysis of HA, NP, and NA Genes
Selection pressure analysis (Table 1) suggested that there was positive selection pressure on all three HA, NP, and NA genes, though a sum of 03, 09, and 13 codons of HA, NP, and NA genes, respectively (see Table 2 for details) were under negative selection pressure. Overall positive selection on the HA, NP and NA genes suggests that new modifications occurred over the time have been favoured by the nature for the better survival of H5N1 Influenza virus, which helped it to spread from avian to human and other mammal species which were not the natural host of Influenza virus but over the time it succeed to increase its pathogenicity and invaded the all domestic species reared within human population.
3.4. Phylogenetic Analysis of the (HA, NP, and NA) Proteins of the H5N1 Clade 2.3.4.4b
Phylogenetic tree (Figure 4) of HA protein sequences of various isolates of H5N1 Influenza virus shows that there are mainly 04 clades. Clade I is the largest clade and has HA protein sequences of isolates from the USA from cat, chicken, human, goat, and cattle. It shows high sequence similarity among HA protein sequences of isolates from the USA, irrespective of the host. Clade II has sequences of isolates from Asia (South Korea, Japan, Bangladesh), Europe (Germany), and the USA from swine, cat, cattle, chicken, and human, which suggests that the H5N1 Influenza virus might have reached the USA from Asia via Europe. Clade III contained HA sequences of isolates from East Asia (China, Hong Kong, and Viet Nam) extracted from human, equine, chicken, and swine. Clade IV also contained HA sequences of isolates from East Asia (China, Hong Kong, Indonesia, Thailand, and Viet Nam) extracted from human, chicken, dog, cat, swine, and human, chicken, except one sequence from Nigeria, which suggests that the H5N1 influenza virus might have reached Africa via Asia. All HA sequences of all isolates of H5N1 Influenza virus in clades I and II are from isolates recently extracted in comparison to isolates in clade III and IV, which suggests that the new isolates prevailing in the USA might have reached the USA from new variants of Asia, which might be more adaptable in invading the hosts in new environmental conditions.
The phylogenetic analysis of the NP protein (Figure 5) shows that NP genes from different isolates of H5N1 Influenza virus could be grouped mainly into eleven clades. Clade VI X, and XI included only one sequence each from Scotland and India extracted from chicken, and from Hong Kong extracted from cattle. Clade I has sequences of H5N1 isolates from East Asia (Pakistan, Bangladesh, Afghanistan, Russia, Japan, China), the Middle East (Kuwait, Israel, Egypt), Africa (Sudan, Burkina Faso, Ivory Coast), and Europe (Poland) extracted mainly from chicken and a few from humans. It suggests that H5N1 isolates may have infected humans from chickens and migrated from East Asia to the Middle East and Africa. Clade II included sequences from East Asia (China, Cambodia, Japan, Thailand) from chicken, cat, dog, human, and equine. Clade III included sequences of isolates from Asia (Nepal, Bhutan, Indonesia) extracted from chicken, cat, and swine. Similarly, clade IV has sequences of isolates from Asia (China, Malaysia, Laos, Viet Nam) extracted from equine, human, and chicken. It could be derived from clades II, III, and IV that the migration of H5N1 infection could have occurred from Chicken to different domestic host species in Asia. Clade V included sequences of isolates from Asia (China, South Korea, Indonesia) and Africa (Ghana, Nigeria, Cameroon) extracted from chicken and swine. It might suggest the migration of the H5N1 influenza virus from Asia to Africa via infected chickens. Clade VII included sequences of isolates from Asia (China, Bangladesh, South Korea, Philippines) and Europe (Germany) extracted from cat and chicken. Clade VIII included sequences from Asia (Viet Nam), the Middle East (Egypt), and Europe (England, France). It suggests that the migration of the H5N1 Influenza virus from Asia to Europe and the Middle East. Clade IX includes sequences of NP protein isolates from North America (USA, Canada) and South America (Chile, Colombia) from different hosts such as goat, cattle, human, cat, swine, and chicken. It can be seen that the sequences of clades VII, VIII, and IX are more recent and closely related to each other than the older sequences of clades I, II, III, IV, and V.
It could be seen from the phylogenetic tree (Figure 6) of sequences of NA protein of isolates of H5N1 Influenza virus that all sequences could be grouped mainly into two clades. Clade I included all the older sequences of isolates from Asia (China, Indonesia, Thailand, Indonesia, Cambodia, Viet Nam), Middle East (Egypt, Iraq) and Europe (Germany) extracted from hosts chicken, human, equine, cat, dog, and swine, except one sequence of isolate from Bangladesh extracted in 2024. Clade II included all sequences of newer isolates extracted recently from North America, South America (Peru, Uruguay, Atacama), Asia (India, Viet Nam, South Korea, Philippines, Hong Kong), the Middle East (Egypt) extracted from cat, chicken, pig, goat, human, and cattle. It suggests that newer isolates of H5N1 Influenza virus might have developed the adaptation for newer climatic conditions (because they are different from the older isolates, as both fall in different clades) and spreading to North America and South America from Asia.
3.5. Protein Structure Prediction, MD-Simulation, and Domain Analysis of the (HA, NP and NA) Proteins
First, 6pcx.1.A protein structure was selected as a template structure with 91.87% sequence identity and 89% coverage with the target sequence of the HA protein for the structure prediction using homology modelling. Similarly, 2q06.A was selected as a template for the structure prediction, which has a sequence identity of 97.39% and coverage of 100% with the target sequence of the NP protein. 5hug.1.A protein sequence was selected as a template structure for structure prediction, which had 96.12% sequence identity and 88% coverage with the target sequence of the NA protein. The best model obtained from homology modelling was selected for each protein. Predicted structures of HA, NP, and NA proteins are shown in Figure 7a, Figure 7b, and Figure 7c, respectively. Further, quality assessment was performed for all proteins by PROCHECK (Ramachandran score), ERRAT, VERIFY3D, QMEAN, Z-score, and assessed as good predicted structures with higher % of allowed region, higher ERRAT score, higher VERIFY3D score, QMEAN score close to 0, and z-score within range, respectively (details of pre-MD-simulation scores are given in Table 3). Further, predicted structures were optimized using MD-simulation and assessed as mentioned above for pre-MD-simulation structure assessment using Ramachandran score, ERRAT, VERIFY3D, QMEAN, Z-score, etc., as results are shown in Table 4 as post-MD-simulation scores, which shows the significant improvement in structure post-MD-simulation reflected from the improvement in different scores.
Additionally, domains in all three proteins were analysed using InterPro. Hemagglutinin domain belonging to viral capsid protein domain superfamily and HA1 A/B domain belonging to Hemagglutn_HA1_a/b domain superfamily were found in HA protein as reported in HA protein of Influenza A virus isolates. However, no specific domain was found in the NP protein, and it contained only a small, low-complexity region in positions 177 to 187. The Sialidase Influenza A/B (IPR033654) domain, belonging to the Sialidase domain superfamily, was identified in the NA protein, consistent with other Influenza A virus isolates.
3.6. Molecular Docking of HA, NP, and NA Proteins with Their Ligands
Template-based blind molecular docking was performed for all three proteins with their respective Ligands, the results of which are provided in Table 5, including information on ligand, template used, docking score, and ligand-binding site residues. Figure 8 shows the ligand-binding site in the protein (shown in a cartoon model) in full view and in a closer view, along with the ligand (shown in a stick-ball model). Ligand-binding site residues derived using molecular docking were found to be conserved throughout 78 sequences of HA protein from different isolates, irrespective of varied hosts such as human, chicken, dog, horse, cattle, cat, pig, and goat, except S149, A150, D199, which were found substituted with A149, S150/V150, N199. In HA protein sequences of recent isolates, as grouped in clade I and II (Figure 4), the binding site residues A149, N199 (S199 in PV388362.1/chicken/Bangladesh/CE-115-09-CA-21-BR-O/2024), Q234 substituted S149, D199, and K234, respectively, were found in older isolates grouped in clade III and IV. These substitutions might have helped the H5N1 Influenza virus adapt to new environmental conditions and help it spread to newer geographical locations like North and South America. Binding site residue A150 was found substituted with S150, V150 in HA protein sequence of HM440059.1/swine/Banten/UT3062/2005, and CY098641.1/human/Hubei/1/2006, respectively. The binding site residue Q238 was found substituted with L238 in the HA protein sequence of CY116662.1/cat/Indonesia/5-F2/2005. Ligand-binding site residues derived using molecular docking were found to be conserved throughout 62 sequences of the NP protein from different isolates, irrespective of varied hosts such as human, chicken, dog, cattle, cat, pig, and goat, except S68, H82, and L136, which were substituted with Y68, R82, and I136, respectively. In the NP sequence of PV410637.1/chicken/Viet Nam/LBFecal247MC/2023, binding site residue H82 was found substituted by R82, and in the NP protein sequence of KX215429.1/chicken/Bhutan/1029/2012, binding site residue S68 was found substituted by Y68. In the NP protein sequence of KX215429.1/chicken/Bhutan/1029/2012, binding site residue L136 was found substituted by I136. Ligand-binding site residues derived using molecular docking were found to be conserved throughout 61 sequences of NA protein from different isolates, irrespective of varied hosts such as human, chicken, dog, horse, cattle, cat, pig, and goat.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
KS, methodology, investigation, data analysis, funding, writing first draft, writing final manuscript: YSM, methodology, investigation, data analysis, writing first draft, writing final manuscript: MGH, methodology, conceptualization, investigation, data analysis, supervision, funding, writing first draft, writing final manuscript.
Funding
This study was supported by Long Island University (LIU) (Grant No. 40262), with financing provided by Helaine Lerner and Joan Rechnitz, as part of the Heilbrunn Family College of Veterinary Medicine Research and Scholarship Fund (Research Project #1) at LIU.
Institutional Review Board Statement
NA.
Informed Consent Statement
NA.
Data Availability Statement
All data will be available upon request from the corresponding author.
Conflicts of Interest
All authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| HA | Hemagglutinin |
| NA | Neuraminidase |
| NP | Nucleoprotein |
| IAV | Influenza A Virus |
| HPAI | Highly Pathogenic Avian Influenza |
| WHO | World Health Organization |
| MD | Molecular Dynamics |
| NCBI | National Center for Biotechnology Information |
| NVT | Constant Number of Particles, Volume, and Temperature |
| NPT | Constant Number of Particles, Pressure, and Temperature |
| OPLS | Optimized Potentials for Liquid Simulations |
| SPC/E | Extended Simple Point Charge (water model) |
| BLASTp | Basic Local Alignment Search Tool for Proteins |
| ProSA | Protein Structure Analysis |
| QMEAN | Qualitative Model Energy Analysis |
| CD-Dock2 | Curvature Detection-based Docking Tool |
| NAG-GAL-SIA | N-acetyl-alpha-neuraminic acid-(2-3)-beta-D-galactopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose |
| SIA | N-acetylneuraminic acid (Sialic acid) |
References
- Alnaeem, A.A.; Al-Shabeb, A.; Hemida, M.G. Evaluation of the immune status of birds and domestic and companion animals for the influenza A virus in Eastern Saudi Arabia. Vet World 2020, 13, 1966–1969. [Google Scholar] [CrossRef]
- Dadonaite, B.; Gilbertson, B.; Knight, M.L.; Trifkovic, S.; Rockman, S.; Laederach, A.; Brown, L.E.; Fodor, E.; Bauer, D.L.V. The structure of the influenza A virus genome. Nat Microbiol 2019, 4, 1781–1789. [Google Scholar] [CrossRef]
- Hemida, M.G.; Chu, D.; Abdelaziz, A.; Alnaeem, A.; Chan, S.M.S.; Peiris, M. Molecular characterisation of an avian influenza (H5N8) outbreak in backyard flocks in Al Ahsa, Eastern Saudi Arabia, 2017-2018. Vet Rec Open 2019, 6, e000362. [Google Scholar] [CrossRef]
- Hemida, M.G.; Perera, R.; Chu, D.K.W.; Alnaeem, A.A.; Peiris, M. Evidence of equine influenza A (H3N8) activity in horses from Eastern and Central Saudi Arabia: 2013-2015. Equine Vet J 2019, 51, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F.; Hermann, E.; Rasmussen, A.L. Highly pathogenic avian influenza H5N1: history, current situation, and outlook. J Virol 2025, 99, e0220924. [Google Scholar] [CrossRef] [PubMed]
- Charostad, J.; Rezaei Zadeh Rukerd, M.; Mahmoudvand, S.; Bashash, D.; Hashemi, S.M.A.; Nakhaie, M.; Zandi, K. A comprehensive review of highly pathogenic avian influenza (HPAI) H5N1: An imminent threat at doorstep. Travel Med Infect Dis 2023, 55, 102638. [Google Scholar] [CrossRef]
- Skeik, N.; Jabr, F.I. Influenza viruses and the evolution of avian influenza virus H5N1. Int J Infect Dis 2008, 12, 233–238. [Google Scholar] [CrossRef]
- Diskin, E.R.; Friedman, K.; Krauss, S.; Nolting, J.M.; Poulson, R.L.; Slemons, R.D.; Stallknecht, D.E.; Webster, R.G.; Bowman, A.S. Subtype Diversity of Influenza A Virus in North American Waterfowl: a Multidecade Study. J Virol 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Cui, P.; Shi, J.; Wang, C.; Zhang, Y.; Xing, X.; Kong, H.; Yan, C.; Zeng, X.; Liu, L.; Tian, G.; et al. Global dissemination of H5N1 influenza viruses bearing the clade 2.3.4.4b HA gene and biologic analysis of the ones detected in China. Emerg Microbes Infect 2022, 11, 1693–1704. [Google Scholar] [CrossRef]
- Pardo-Roa, C.; Nelson, M.I.; Ariyama, N.; Aguayo, C.; Almonacid, L.I.; Munoz, G.; Navarro, C.; Avila, C.; Ulloa, M.; Reyes, R.; et al. Cross-species transmission and PB2 mammalian adaptations of highly pathogenic avian influenza A/H5N1 viruses in Chile. bioRxiv 2023. [Google Scholar] [CrossRef]
- Sriwilaijaroen, N.; Suzuki, Y. Molecular basis of the structure and function of H1 hemagglutinin of influenza virus. Proc Jpn Acad Ser B Phys Biol Sci 2012, 88, 226–249. [Google Scholar] [CrossRef]
- Cohen, M.; Zhang, X.Q.; Senaati, H.P.; Chen, H.W.; Varki, N.M.; Schooley, R.T.; Gagneux, P. Influenza A penetrates host mucus by cleaving sialic acids with neuraminidase. Virol J 2013, 10, 321. [Google Scholar] [CrossRef]
- Wu, N.C.; Wilson, I.A. Structural Biology of Influenza Hemagglutinin: An Amaranthine Adventure. Viruses 2020, 12. [Google Scholar] [CrossRef]
- Tan, C.C.S.; van Dorp, L.; Balloux, F. The evolutionary drivers and correlates of viral host jumps. Nat Ecol Evol 2024, 8, 960–971. [Google Scholar] [CrossRef]
- Prachanronarong, K.L.; Canale, A.S.; Liu, P.; Somasundaran, M.; Hou, S.; Poh, Y.P.; Han, T.; Zhu, Q.; Renzette, N.; Zeldovich, K.B.; et al. Mutations in Influenza A Virus Neuraminidase and Hemagglutinin Confer Resistance against a Broadly Neutralizing Hemagglutinin Stem Antibody. J Virol 2019, 93. [Google Scholar] [CrossRef]
- Malbranke, C.; Bikard, D.; Cocco, S.; Monasson, R. Improving sequence-based modeling of protein families using secondary-structure quality assessment. Bioinformatics 2021, 37, 4083–4090. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Shi, J.; Cui, P.; Yan, C.; Zhang, Y.; Zhang, Y.; Wang, C.; Chen, Y.; Zeng, X.; Tian, G.; et al. Evolution and biological characterization of H5N1 influenza viruses bearing the clade 2.3.2.1 hemagglutinin gene. Emerg Microbes Infect 2024, 13, 2284294. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Bergner, A.; Schwede, T. Modelling three-dimensional protein structures for applications in drug design. Drug Discov Today 2014, 19, 890–897. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.B.; Dushoff, J. Codon bias and frequency-dependent selection on the hemagglutinin epitopes of influenza A virus. Proc Natl Acad Sci U S A 2003, 100, 7152–7157. [Google Scholar] [CrossRef]
- Fiser, A.; Sali, A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol 2003, 374, 461–491. [Google Scholar] [CrossRef]
- Lipman, D.J.; Pearson, W.R. Rapid and sensitive protein similarity searches. Science 1985, 227, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Tejero, R.; Montelione, G.T. Evaluating protein structures determined by structural genomics consortia. Proteins 2007, 66, 778–795. [Google Scholar] [CrossRef]
- Blum, M.; Andreeva, A.; Florentino, L.C.; Chuguransky, S.R.; Grego, T.; Hobbs, E.; Pinto, B.L.; Orr, A.; Paysan-Lafosse, T.; Ponamareva, I.; et al. InterPro: the protein sequence classification resource in 2025. Nucleic Acids Res 2025, 53, D444–D456. [Google Scholar] [CrossRef]
- Gamblin, S.J.; Vachieri, S.G.; Xiong, X.; Zhang, J.; Martin, S.R.; Skehel, J.J. Hemagglutinin Structure and Activities. Cold Spring Harb Perspect Med 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.K.; Guttman, M.; Ebner, J.L.; Lee, K.K. Dynamic changes during acid-induced activation of influenza hemagglutinin. Structure 2015, 23, 665–676. [Google Scholar] [CrossRef]
- Hosaka, H.; Nakagawa, A.; Tanaka, I.; Harada, N.; Sano, K.; Kimura, M.; Yao, M.; Wakatsuki, S. Ribosomal protein S7: a new RNA-binding motif with structural similarities to a DNA architectural factor. Structure 1997, 5, 1199–1208. [Google Scholar] [CrossRef]
- Chauhan, R.P.; Gordon, M.L. An overview of influenza A virus genes, protein functions, and replication cycle highlighting important updates. Virus Genes 2022, 58, 255–269. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. CB-Dock2: improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res 2022, 50, W159–W164. [Google Scholar] [CrossRef]
Figure 1.
Phylogenetic tree for the HA gene of H5N1 Influenza A virus clade 2.3.4.4b from various hosts.
Figure 1.
Phylogenetic tree for the HA gene of H5N1 Influenza A virus clade 2.3.4.4b from various hosts.
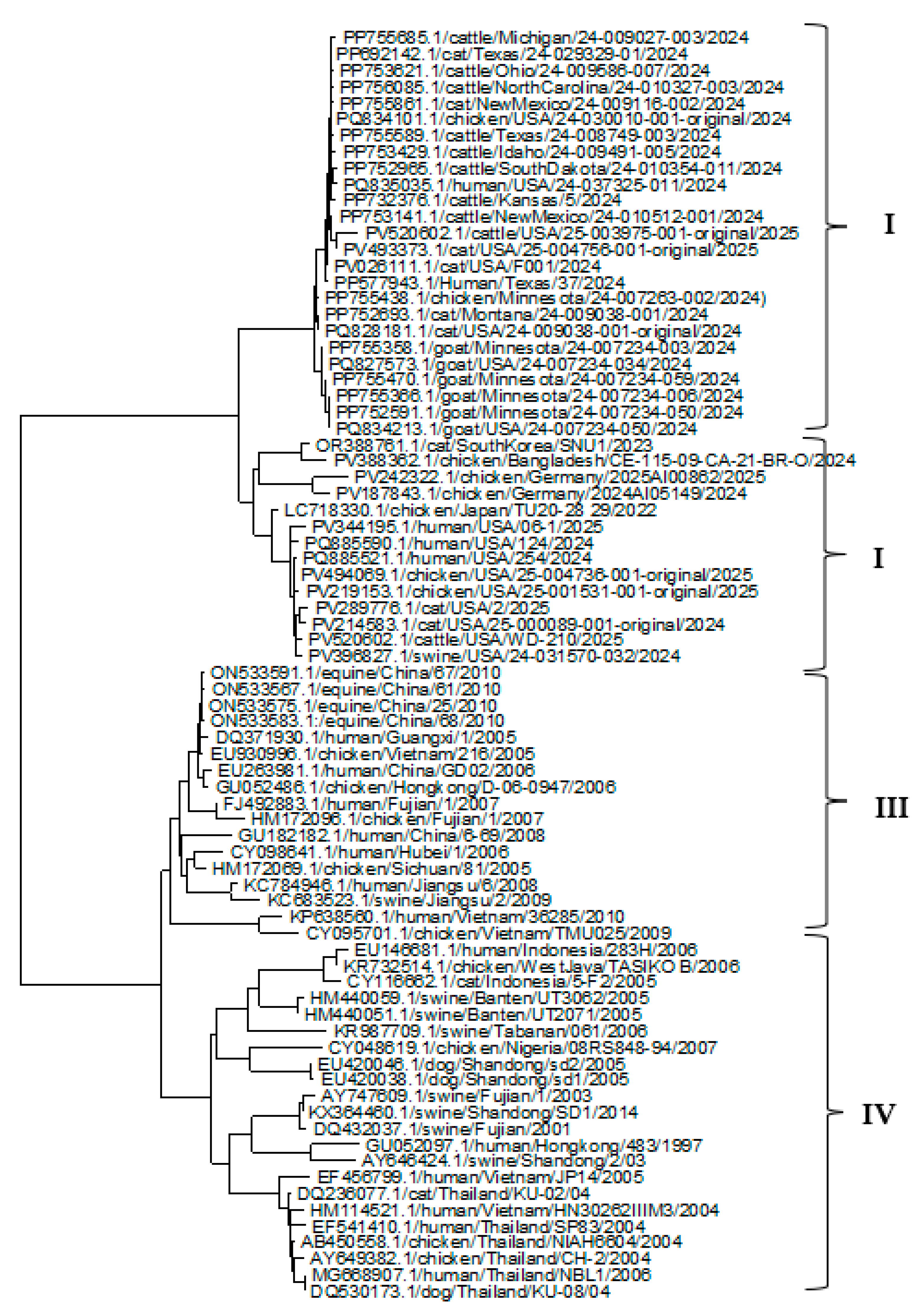
Figure 2.
Phylogenetic tree for the NP gene of the H5N1 Influenza A virus clade 2.3.4.4b from various hosts.
Figure 2.
Phylogenetic tree for the NP gene of the H5N1 Influenza A virus clade 2.3.4.4b from various hosts.
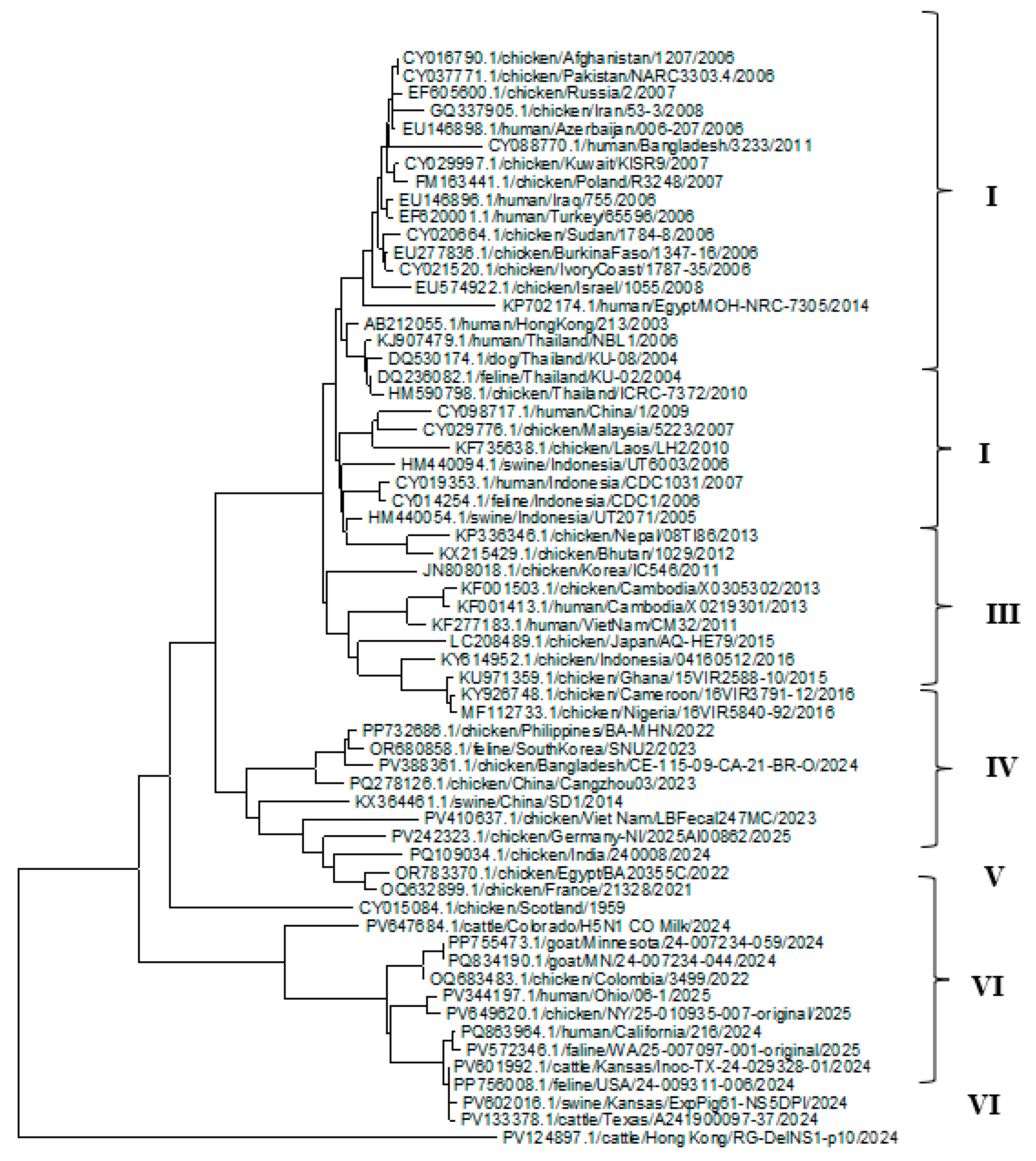
Figure 3.
Phylogenetic tree for the NA gene of the H5N1 Influenza A virus clade 2.3.4.4b from various hosts.
Figure 3.
Phylogenetic tree for the NA gene of the H5N1 Influenza A virus clade 2.3.4.4b from various hosts.

Figure 4.
Phylogenetic tree for the HA Protein of the H5N1 Influenza A virus clade 2.3.4.4b from various hosts.
Figure 4.
Phylogenetic tree for the HA Protein of the H5N1 Influenza A virus clade 2.3.4.4b from various hosts.
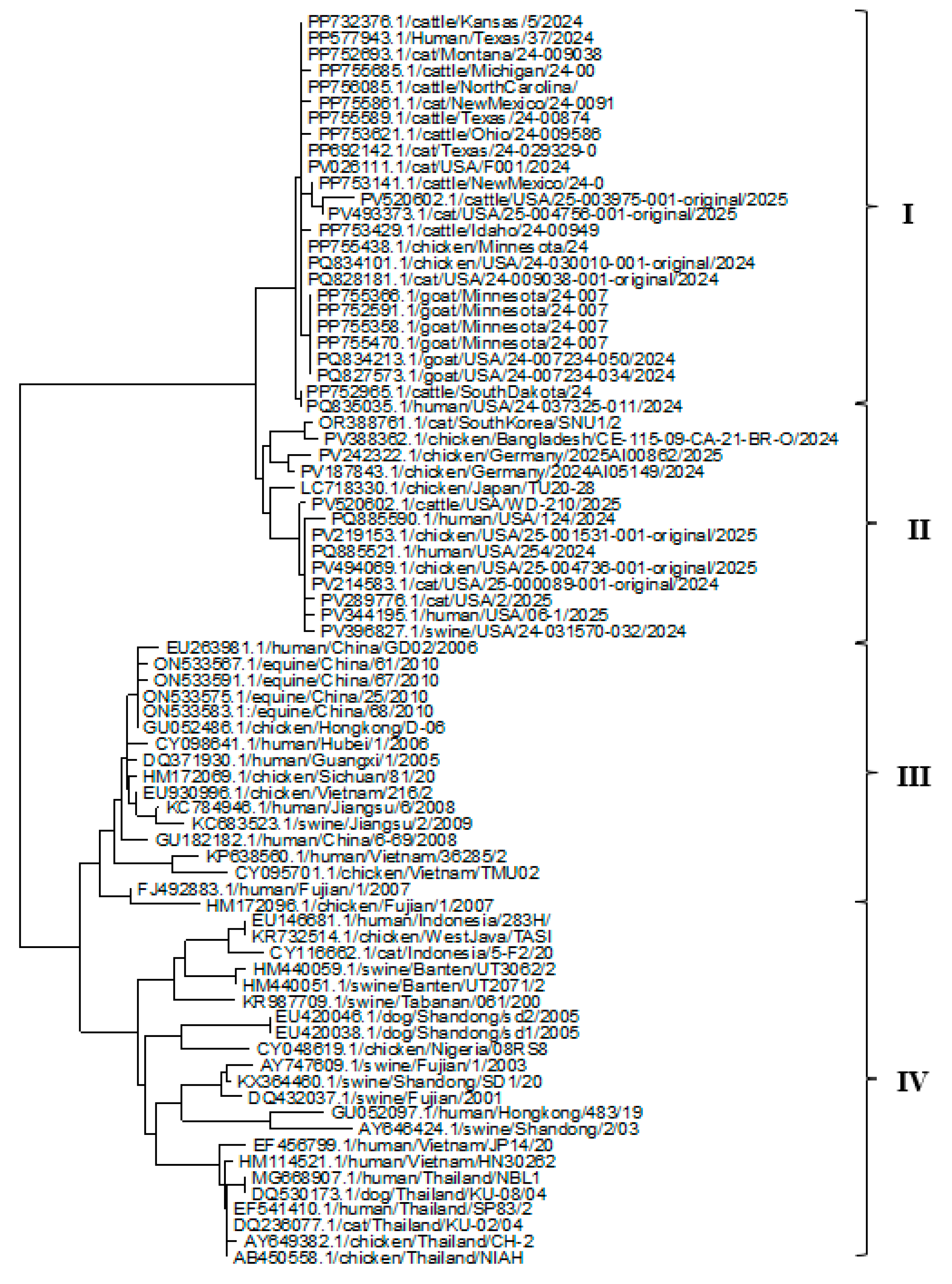
Figure 5.
Phylogenetic tree for the NP protein of the H5N1 Influenza A virus clade 2.3.4.4b from various hosts.
Figure 5.
Phylogenetic tree for the NP protein of the H5N1 Influenza A virus clade 2.3.4.4b from various hosts.
Figure 6.
Phylogenetic tree for the NA protein of H5N1 Influenza A virus from various hosts.

Figure 7.
Predicted structures of HA (a), NP (b), and NA proteins (c).
Figure 8.
Ligand (shown in stick-ball model)-binding site of HA protein (shown in cartoon model) in full (a) and closer (b) views; Ligand (stick-ball model)-binding site of NP protein (cartoon model) in full (c) and closer (d) views; Ligand (stick-ball model)-binding site of NA protein (cartoon model) in full (e) and closer (f) views.
Figure 8.
Ligand (shown in stick-ball model)-binding site of HA protein (shown in cartoon model) in full (a) and closer (b) views; Ligand (stick-ball model)-binding site of NP protein (cartoon model) in full (c) and closer (d) views; Ligand (stick-ball model)-binding site of NA protein (cartoon model) in full (e) and closer (f) views.

Table 1.
List of the non-synonymous/synonymous rate ratio for the (HA, NP, and NA) genes.
| Gene | No. of sequences | No. of codons | No. of codons under selection | Non-synonymous/ synonymous rate ratio | Selection pressure |
|---|---|---|---|---|---|
| HA gene | 78 | 530 | 3 | 2.7856 | Positive |
| NP gene | 61 | 439 | 2 | 2.4435 | Positive |
| NA gene | 62 | 435 | 13 | 2.4312 | Positive |
Table 2.
Selection pressure on codons of HA, NP and NA genes.
| Protein | Codon no. | S | N | dS | dN | Selection pressure |
|---|---|---|---|---|---|---|
| HA | 151 | 0.000 | 6.000 | 0.000 | 3.000 | Positive |
| 191 | 3.000 | 1.000 | 3.752 | 0.454 | Negative | |
| 487 | 2.000 | 0.000 | 2.499 | 0.000 | Negative | |
| NP | 10 | 0 | 4 | 0 | 2.507 | Positive |
| 138 | 1 | 9 | 0 | 9 | Negative | |
| NA | 17 | 1 | 5 | 0.678 | 3.538 | Positive |
| 45 | 6 | 5 | 7.237 | 2.313 | Negative | |
| 91 | 0 | 6 | 0 | 2.979 | Positive | |
| 105 | 0 | 6 | 0 | 3.02 | Positive | |
| 112 | 0 | 7 | 0 | 3.464 | Positive | |
| 175 | 3 | 0 | 3 | 0 | Negative | |
| 181 | 0 | 6 | 0 | 2.986 | Positive | |
| 235 | 0 | 8 | 0 | 4.102 | Positive | |
| 271 | 3 | 1 | 4.117 | 0.44 | Negative | |
| 347 | 1.5 | 5.5 | 0.917 | 4.03 | Positive | |
| 348 | 0 | 9 | 0 | 4.287 | Positive | |
| 352 | 0 | 6 | 0 | 2.997 | Positive | |
| 402 | 0 | 6 | 0 | 3 | Positive |
Table 3.
Various quality assessment scores of predicted structures of HA, NP and NA proteins: pre and post MD-simulation.
Table 3.
Various quality assessment scores of predicted structures of HA, NP and NA proteins: pre and post MD-simulation.
| Proteins | Assessment scores | Pre MD-simulation | Post MD-simulation |
|---|---|---|---|
| HA | Procheck | 92.06 (Ramachandran score) | 94.79 (Ramachandran score) |
| 79.1% most favoured core | 88.7% most favoured core | ||
| 18.7% additional allowed region | 9.7% additional allowed region | ||
| 1.6% generously allowed region | 0.9% generously allowed region | ||
| 0.7% disallowed region | 0.7% disallowed region | ||
| ERRAT | 91.9913 | 92.5926 | |
| Verify3D | 78.64% | 79.44% | |
| QMEAN | 0.71 | 0.78 | |
| ProSA | -9.8 | -9.5 | |
| NP | Procheck | 89.02 (Ramachandran score) | 91.75 (Ramachandran score) |
| 85.4% most favoured core | 86.1% most favoured core | ||
| 12.9% additional allowed region | 12.4% additional allowed region | ||
| 1.4% generously allowed region | 1.2% generously allowed region | ||
| 0.2% disallowed region | 0.2% disallowed region | ||
| ERRAT | 90.9548 | 96.1276 | |
| Verify3D | 84.21% | 82.32% | |
| QMEAN | 0.69 | 0.79 | |
| ProSA | -9.7 | -9.36 | |
| NA | Procheck | 91.53 (Ramachandran score) | 94.82 (Ramachandran score) |
| 76.9% most favoured core | 86.7% most favoured core | ||
| 20.4% additional allowed region | 13.0% additional allowed region | ||
| 2.8% generously allowed region | 0.3% generously allowed region | ||
| 0.0% disallowed region | 0.0% disallowed region | ||
| ERRAT | 82.9913 | 89.0957 | |
| Verify3D | 78.64% | 84.75% | |
| QMEAN | 0.88 | 0.93 | |
| ProSA | -5.4 | -5.15 |
Table 4.
Details and results of molecular docking of HA, NP, and NA proteins with their respective ligands.
Table 4.
Details and results of molecular docking of HA, NP, and NA proteins with their respective ligands.
| Protein | Ligand | Template | Docking score | Binding site residues |
|---|---|---|---|---|
| HA | NAG-GAL-SIA (N-acetyl-alpha-neuraminic acid-(2-3)-beta-D-galactopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose) | 1hge | -2.8 | V147 S148 A149 A150 P157 W165 N198 N199 E202 L206 Q234 G237 Q238 |
| NP | RNA | 7dxp | -7.5 | Q58 I61 T62 R65 M66 L68 S69 A70 D72 E73 R74 R75 N76 H82 G86 K87 D88 P89 K90 K91 T92 G93 G94 P95 L108 L110 K113 R117 L136 M137 H140 T171 L172 P173 R174 R175 S367 |
| NA | SIA (N-ACETYL-ALPHA-NEURAMINIC ACID) | 4gzq | -5.7 | R118 E119 D151 R152 R156 W179 S180 R225 E228 S247 E277 E278 R293 N295 Y344 G345 R368 Y402 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



