
Submitted:
04 June 2025
Posted:
06 June 2025
You are already at the latest version
Abstract
(1) Background: The highly pathogenic avian influenzas virus H5N1 clade 2.3.4.4b is an emerging threat that poses great risk to the poultry industry. Few human cases have been linked to the infection with this clade in many parts of the world, including the USA. Unfortunately, there are no specific vaccines or antiviral drugs that could help prevent and treat the infection of this virus in birds. Our major objective is to identify/repurpose some (novel/known) antiviral compounds that may inhibit viral replication by targeting some key viral proteins. (2) Methods: We used state-of-the-art machine learning tools such as molecular docking and MD-simulation methods from Bio-via Discovery Studio (v24.1.0.321712). The target proteins homology models were validated through structural assessment via DOPE scores, Ramachandran plots, and Ver-ify-3D metrics, ensuring reliable structural representations, confirming their reliability for subsequent in silico approaches, including molecular docking and molecular dynamics simulation for 50 ns highlighted the structural stability and compactness of the docked complexes. (3) Results: Molecular docking revealed strong binding affinities of both compounds, particularly the GS441524, which demonstrated superior interaction energy and hydrogen bonding with critical functional residues of (HA, NA, M2, and PB2/CBD). The MM-GBSA binding free energy calculations supported these findings, with GS441524 showing more favorable energies than several known standard inhibitors for HA (F0045S), NA (Zanamivir), M2 (Rimantadine and Amantadine) and PB2/CBD (Pb2-39), especially in its interaction with NA and PB2/CBD. Molecular dynamics simulations over 50 ns highlighted the structural stability and compactness of the GS441524 complex with PB2/CBD. Hydrogen bond analysis further confirmed persistent and specific interactions, suggesting that GS441524 may effectively disrupt PB2-mediated RNA synthesis. (4) Conclusions: Our findings are consistent with previous evidence supporting the antiviral activity of certain nucleoside analog inhibitors, including GS441524, against various coronaviruses. These results further support the potential repurposing of GS441524 as a promising therapeutic candidate against H5N1 avian influenza clade 2.3.4.4b. However, additional functional studies are necessary to validate these in silico predictions of its anti-H5N1 activity.
Keywords:
highly pathogenic avian influenza virus
; H5N1
; Clade 2.3.4.4b
; drug design
; molecular docking
; homology modeling
; antiviral compounds
1. Introduction
Influenza virus Type A (IVA) is an RNA, negative sense, and has a segmented genome. The viral genome consists of 7-8 segments [1]. Each viral segment encodes at least one protein. These viral proteins include the hemagglutinin (HA), neuraminidase (NA), the matrix protein (M), the viral nucleoprotein (NP), the viral polymerase called (RNA dependent RNA polymerase (RdRp), which consists of three subunits (PA, PB1, and PB2), as well as the nonstructural protein-1 (NSP-1) [1]. Influenza viruses have a unique feature during their transcription. The viral genome is translocated into the host cell nucleus to acquire the cap from the host cell DNA polymerase to prime the viral transcription [2]. The avian influenza virus (AIV) belongs to the Influenza virus type-A. They have been classified based on their virulence into the low pathogenic (LPAIV) and the high pathogenic (HPAIV) viruses [3]. The influenza viruses are experiencing dynamic changes at the genomic level. There are several reasons for the emergence/reemergence of new viruses, strains, clades, or subclades of avian influenza viruses, including (1) Some point mutations across different proteins called antigenic drift, the exchange of some viral genome segments in case of double infection with more than one type of viruses called antigenic shift, and (3) the poor proofreading capabilities of the viral RdRp [4,5]. The IVA is classified based on their key proteins into H1-H16 and N1-N9 [6]. This pattern of frequent changes in the IVA on the genomic levels. These frequent changes of the viruses on the genomic levels may generate new viral isolates that are resistant to the currently used drugs and vaccines. This is hammering the presently used vaccine and antiviral therapy that is prepared against the previously seeded viruses. Thus, there is an urgent need to monitor these viruses on the genomic levels through active, vigilant molecular surveillance as well as frequent upgrading of the vaccine and drugs that match the most recently circulating strains of the virus. The highly pathogenic avian influenza virus continues to pose a great risk to human health and the poultry industry worldwide. During 2021, the H5N1 clade 2.3.4.4b started to cause several outbreaks in poultry farms across the globe. It spread rapidly to different parts of the world [7]. In early 2024, this clade was reported in cattle and in the milk in many cattle herds across the USA [8]. Human infections due to the same clade were reported in many countries across the world [9]. Unfortunately, there is no current vaccine or even antiviral drugs specific to prevent/treat the infected hosts with this newly emerged clade of the HPAIV. Several antiviral drugs were used in the past to treat many IVA in humans. There are several strategies to use for these antiviral drugs by targeting various key IAV proteins [10,11,12,13]. Most of these strategies are mainly targeting some key viral proteins involved in the viral replication and maturation in addition to some other host proteins that play important roles during the viral replication in the host cells [14]. Here, are focusing on some antiviral strategies that involve the AIV proteins. These strategies mainly focus on the inhibition of the viral entry, release, or the processing and maturation of the viral virions [14]. Some of these strategies mainly depend on the generation of some monoclonal antibodies that mainly neutralize the activities of the viral expressed proteins on the surface of the viral envelope,, particularly the HA and NA [15,16]. In this study, we are mainly focusing on the synthetic antiviral compounds that target various key proteins of the virus. These strategies include (1) inhibitors of the HA maturation, (2) inhibitors of the NA protein, (3) inhibitors of the RdRp, (4) the ion channel (M2) blockers, and (5) inhibitors of the NSP1 [14]. There are some compounds that inhibit the maturation of the HA protein of the IAV, such as Nitazoxanide [17]. The NA protein plays some crucial roles in AIV replication, including the degradation of the mucous layers in the respiratory tracts, thus granting access to the virus to be in touch with the epithelial cells of the respiratory tracts. The transmembrane protein NA also plays a critical role in the removal of the sialic acid from the HA protein. Thus, targeting the NA protein of the AIV is one of the most promising antiviral strategies. Several drugs are currently FDA-approved targeting the NA protein of the AIV, such as (Zanamivir, Oseltamivir, Peramivir, and Laninamivir) [18,19,20]. The viral polymerase RdRp is an attractive target for some antiviral drugs. Several antiviral compounds were shown to inhibit various subunits of this viral polymerase [21,22]. Several compounds targeting the cap-binding domain of the PB2 subdomain (PB2/CBD) have shown some inhibitory effects on the viral replication, such as the Pimodivir [23,24]. The nucleoside analogue antiviral drugs such as Ribavirin showed a broad-spectrum antiviral drug against many RNA viruses, including IAV [25]. The GS-441524, as a nucleoside analog, has been recently used as a powerful inhibitor of the feline infectious peritonitis virus main protease (Mpro) but was never tested as an anti-influenza virus potential drug [26,27]. In the current study, we are comparing the potential impacts of the currently known anti-AIV drugs with some new potential drugs using the molecular docking and simulation approach. We believe these AI tools will pave the way for screening a large number of synthetic and natural compounds to be used as new drugs against many viral diseases, including the currently circulating H5N1 clade 2.3.4.4b. However, further laboratory validations are required to confirm this prediction and to identify the mechanism of actions of these potential drugs.
2. Results
2.1. Homology Modeling of Some Key Proteins of the Avian H5N1 clade 2.3.4.4b (HA, NA, PB2/CBD, and M2)
To perform the homology modeling, we searched for experimentally determined template reference sequences closely related to the query sequences and shared at least 35-65% sequence identity and similarity. The multiple sequence alignment of HA, NA, and PB2/CBD with the templates is shown in (Figure S1-3). The sequences of the target proteins from H5N1 as receptors are HA (804 AA.), NA (797 AA), PB2/CBD (173 AA) of viral RdRp, and four chain M2 (24 AA each chain) channels.
Results of the homology models of the target proteins of the H5N1 clade 2.3.4.4b (HA, NA, PB2, and M2) based on their DOPE scores and energy scores are listed in (Figure S4). We selected the top-ranked models per protein based on their PDF total energy and the lowest DOPE scores (Table 1). Details of the ranked model per protein were selected to predict the 3D model per protein are listed in (Table 1).
The Ramachandran plot results indicate that the majority of the amino acid residues per each protein were located in the most favored regions. The remaining amino acid residues were located in the proper locations. Our results showed high accuracy of all the predicted homology models for the target proteins (HA, NA, PB2, and M2) of the H5N1 clade 2.3.4.4b (Figure 1). Results of both the Ramachandran plot and Verify-3D scores suggest the generated models per each protein are very stable and have the highest scores of the top-quality homology models (Table 1).
2.2. Results of the Binding Affinity of the Antiviral Compounds/Ligands with the Avian H5N1 Clade 2.2.4.4b HA Protein Using the Molecular Docking Approach
Our molecular docking studies revealed a high binding affinity of nearly all the selected compounds. The CDOCKER interaction energy scores suggested binding affinity score (-CDOCK scores) as listed in (Table 2). The GS445214 compound interacted with the active site residues of the same binding pocket as reference standard F0045(S) (Figure 2). The 2D interaction patterns of top hits of F0045(S) (standard) and GS441524 are illustrated in Figure S5.
The highest -CDOCKER interaction energy score (positive value) referred to the most favorable binding of ligand to the protein. The HA protein was docked with GS445214 and standard compound F0045(S), with the -CDOCKER Score of HA- GS441524 complex is 26.62 is higher as compared to the HA complex with a reference standard F0045(S), which is 22.92. The sofosbuvir was not docked in the same binding pocket of HA where the Standard F0045(S) and GS441524 were docked. The standard ligand F0045(S) does not make any hydrogen bond but makes hydrophobic interactions with interacting residues HIS24, TRP366, ILE390, and VAL393 of HA protein. However, the best hit of GS441524 with HA established four hydrogen bonds with residues ALA45, THR331, ILE390, and THR394 in the active site of the target HA protein. The docking results of NA also showed that the CDOCK Scores of standard reference compounds Zanamivir and Oseltamivir with NA are 49.26 and -65.74. However, The -CDOCK scores of sofosbuvir and GS445214 with NA are 53.43 and 32.34 (Table 2). The binding affinity of reference standard Oseltamivir-NA complex is exhibited good as its -CDOCK score is in negative values as compared to sofosbuvir-NA, GS445214, and Reference standard-Zanamivir which are showing positive -CDOCK score (Table 2). However, the binding affinity score (-CDOCK score) of NA-Sofosbuvir is higher than that of the NA-Zanamivir complex. The NA- GS441524 also showed good binding affinity as compared to the Standard Zanamivir-NA docking complex. Due to the interaction of NA-Zanamivir, seven hydrogen bonds were established, and zanamivir interacted with NA binding site residues TYR344, ARG368 (four hydrogen bonds), ASP151, and SER367. However, the top hit of ligand sofosbuvir established eight hydrogen bonds through NA protein interacting residues TYR344, ARG368, TYR402 (two hydrogen bonds), ARG225, LYS432, ASP151, and GLU278 in the active site. On the other hand, GS441524 established 10 hydrogen bonds with interacting residues ARG368 (four hydrogen bonds), VAL149, ASP151 (three hydrogen bonds), SER367, PRO431 (three hydrogen bonds) and LYS432. The GS445214 and Sofosbuvir interacted with active site residues of the same binding pocket as reference standard Zanamivir (Figure 3). The 2D interaction patterns of top hits of Zanamivir (standard), sofosbuvir and GS441524 are illustrated in Figure S6.
Further, the docking of compound GS445214 and Sofosbuvir with PB2/CBD protein of H5N1 also showed promising results as compared to reference standard compound PB2-39 and MGT (CAP) for protein PB2-CBD. The binding affinity scores of sofosbuvir-PB2/CBD and GS441524 docked with PB2/CBD exhibited by -CDOCK score are 50.88 and 34.23. However, the -CDOCK score of reference standard PB2-39 compound and MGT (CAP) for PB2 is 54.55 and 80.67, slightly higher than the binding affinity score of PB2-Sofosbuvir and PB2/CBD-GS441524 docking complex (Table 2). The standard ligand PB2-39 makes only two hydrogen bonds with interacting residues ARG355 and ASN429 of PB2/CBD protein, while MGT (CAP) interacted with residues LYS339, ARG335 (three hydrogen bonds), PHE404 (two hydrogen bonds), GLU361, HIS357 (two hydrogen bonds) and PHE323 established 10 hydrogen bonds with PB2 protein. The interaction of sofosbuvir with PB2/CBD established three hydrogen bonds with residues LYS339, LYS376, and HIS357 in the active site. However, the top hit of GS441524 with PB2/CBD suggested that six hydrogen bonds were established due to interaction of GS441524 with residues ARG355, LYS376, PHE404, GLN406, GLU361 and SER324 in the active site of PB2/CBD protein (Figure 4). The 2D interaction patterns of top hits of MGT and PB2-39 (standard), Sofosbuvir and GS441524 are illustrated in Figure S7. The presence of several hydrogen bonds, along with their favorable interaction score inside the binding cavity of the HA, NA, and PB2/CBD, predicts the validity of the GS441524 as a potential potent inhibitor.
The transmembrane domain of the M2 protein is a tetrameric channel, the target of the anti-influenza drugs Amantadine and its methyl derivative Rimantadine. The M2 protein was docked with Rimantadine and Amantadine as the standard compounds. The CDOCK Score of the M2- Rimantadine complex is 33.02, and that of the M2-amantadine complex is 27.55 (Table 2).
The sofosbuvir showed a good binding affinity with the M2 at the same binding pocket with the -CDOCKER Score of 62.05, which is higher as compared to the M2 complex with the Amantadine and Rimantadine compounds (Table 2).
The GS441524 compound docked with M2 exhibited good binding affinity with the CDOCK Score of 42.59, which is comparatively higher than the binding affinity with the known drugs (amantadien and Ramantadien). However, this binding affinity score is slightly lower than that of sofosbuvir with M2. The M2 interacting with standard ligands and the test ligands established hydrogen bond and hydrophobic bond interactions are listed in (Table 2). The top hit of the sofosbuvir and the GS441524 compounds with the M2 created six and four hydrogen bonds, respectively (Figure 5). The 2D interaction patterns of the top hits of the Rimantadine, the Amantadine, sofosbuvir, and the GS441524 compounds are presented in (Figure S8).
2.3. Results of the Calculation of the Ligands- H5N1 Clade 2.3.4.4b Proteins Binding Energy – (MM-GBSA)
The MM-GBSA calculations for the compounds GS441524 and Sofosbuvir were performed using data from the top 10 docking hits, along with the respective reference standard compounds: F0045(s) for HA, PB2-39 for PB2/CBD, and Zanamivir for the NA protein.
Based in the interaction with the active site of the HA protein, GS441524 exhibited a binding energy of −61.43 kcal/mol, compared to −48.86 kcal/mol for the reference standard. In the case of NA protein interactions, the top docking hits of Sofosbuvir and GS441524 showed binding free energies of −138.23 kcal/mol and −83.10 kcal/mol, respectively. These values were compared to the −201 kcal/mol binding energy of the top hit for the reference compound Zanamivir, indicating the complexes’ relative stability and supporting the docking results’ validity.
The interaction of PB2/CBD with the standard compounds PB2-39 and MGT (CAP) resulted in binding free energies of −236.79 kcal/mol and −236.00 kcal/mol, respectively, which are significantly more negative compared to the binding energies observed for PB2/CBD-Sofosbuvir (−90.00 kcal/mol) and PB2/CBD-GS441524 (−99.00 kcal/mol). These findings suggest greater complex stability for the standard compounds and support the reliability of the docking results (Table 2). For the M2 ion channel protein, the reference compounds Rimantadine and Amantadine showed binding energies of −112.23 kcal/mol and −104.46 kcal/mol, respectively. In contrast, the test compounds Sofosbuvir and GS441524 demonstrated more favorable (i.e., more negative) binding energies of −116.11 kcal/mol and −120.65 kcal/mol, indicating potentially stronger interactions with the M2 protein than the reference compounds.
2.4. Molecular Dynamics Simulation Results for Avian H5N1 Clade 2.2.4.4b (HA, NA, PB2, and M2) Proteins and Some Selected Compounds/Drugs
Proteins play essential structural and functional roles in biological systems, including microbial pathogenesis, by mediating receptor-based internalization and replication. While molecular docking offers insights into the stability of protein-ligand complexes, it lacks the ability to capture their dynamic behavior over time. To address this limitation, molecular dynamics (MD) simulations were performed for 50 nanoseconds (ns) to evaluate the interactions of the GS441524 compound with the HA, NA, and PB2/CBD proteins of avian H5N1 clade 2.2.4.4b. Reference standard complexes—HA-F0045(S), NA-Zanamivir, and PB2/CBD-PB2-39 were also simulated under the same conditions for comparative analysis.
The MD simulations provided detailed insights into the molecular behavior of each complex by capturing time-dependent structural deviations and flexibility. Root Mean Square Deviation (RMSD) was used to assess conformational stability, while Root Mean Square Fluctuation (RMSF) and Radius of Gyration were calculated to evaluate residue-level mobility and compactness of the complexes, respectively. These parameters are critical for understanding the ligand-bound protein systems’ overall dynamics, structural stability, and flexibility. Additionally, hydrogen bond monitoring and hydrogen bond distance analyses were conducted to further explore each complex’s dynamic interactions and stability. The comparative trajectory analysis of GS441524-bound complexes (HA, NA, PB2/CBD) with their respective reference standards is presented and discussed in this section.
2.4.1. Analysis of the RMSD Calculations Conferring Stability of Ligand-Protein Complexes
The RMSD plot analysis showed that all complexes, including the reference compounds, remained relatively stable when RMSD values were within the 3 Å threshold, as lower RMSD values typically indicate greater structural stability of protein-ligand complexes. For the HA-GS441524 complex, however, the RMSD values exhibited significant fluctuations compared to the HA-F0045(S) complex, suggesting lower dynamic stability of the GS441524-bound HA complex during the simulation (Figure 6A). The RMSD of the backbone atoms for HA-GS441524 and HA-F0045(S) ranged from approximately 1.8 Å to 5.5 Å. The average RMSD values were 3.2 Å for HA-GS441524 and 4.0 Å for HA-F0045(S), both exceeding the 3 Å stability threshold (Figure 6A). These results indicate that neither GS441524 nor the reference compound F0045(S) maintained consistent structural stability over the 50 ns simulation period.
The RMSD analysis of the neuraminidase (NA) complex with its standard inhibitor Zanamivir (NA–Zanamivir) revealed minimal structural fluctuations, with an average backbone RMSD of 3.13 Å—slightly exceeding the commonly accepted 3 Å stability threshold. In comparison, the NA–GS441524 complex showed a lower average RMSD of 2.5 Å (Figure 6B), suggesting enhanced structural stability. Thus, over the 50 ns simulation period, GS441524 exhibited superior dynamic stability when bound to the NA relative to the NA–Zanamivir complex.
RMSD analysis of the PB2/CBD complexes with PB2-39 and GS441524 indicated that the PB2/CBD–GS441524 complex exhibited the highest stability, maintaining RMSD values below 3 Å throughout the simulation. A slight increase to approximately 3 Å was noted after 30 ns; however, the RMSD remained steady between 2.7 Å and 3 Å from 30 to 50 ns without significant fluctuations (Figure 6C). In contrast, the reference complex PB2/CBD–F0045(S) displayed an average RMSD around 3 Å but underwent noticeable fluctuations between 17–21 ns and 32–35 ns. These findings suggest that GS441524 forms a more stable complex with PB2/CBD than the reference compound. Overall, the PB2 protein maintained a stable dynamic profile upon ligand binding, supporting the potential of GS441524 as an effective PB2 inhibitor.
2.4.2. Analysis of the Stability and Mobility of the Interaction Between the H5N1 Clade 2.3.4.4b Proteins and the Selected Drugs/Compounds -RMSF
The RMSF (Root Mean Square Fluctuation) plots for the C-α atoms of all complexes were generated from the MD simulation trajectories. In both the HA–F0045(S) and HA–GS441524 complexes, residues within the 337–393 region exhibited the highest flexibility. The HA–F0045(S) complex showed a peak fluctuation of approximately 6 Å, whereas the HA–GS441524 complex displayed slightly reduced fluctuations, peaking around 4.5 Å. Outside this region, most residues in both complexes fluctuated within a 0.5–2.5 Å range throughout the simulation. The overall RMSF profiles of the HA–GS441524 complex were comparable to those of the reference HA–F0045(S) complex, suggesting similar residue mobility and dynamic behavior (Figure 7A).
RMSF analysis of the NA–Zanamivir and NA–GS441524 complexes revealed that residues spanning positions 169 to 270 exhibited the most prominent fluctuations during the simulation. In this region, both complexes reached a maximum fluctuation of approximately 2.5 Å. Outside this range, most residues fluctuated between 0.5 and 1.5 Å over the 50 ns simulation period. The overall RMSF values for the NA–GS441524 complex were slightly lower but remained comparable to those of the NA–Zanamivir complex, suggesting similar residue flexibility and dynamic behavior in both complexes (Figure 7B).
The RMSF analysis of the PB2/CBD-PB2-39 and PB2/CBD-GS441524 complexes revealed that residues between positions 85 and 110 underwent the highest fluctuations during the simulation. The reference complex PB2/CBD-PB2-39 showed a peak fluctuation of approximately 6 Å, whereas the PB2/CBD-GS441524 complex exhibited a slightly lower peak of around 5 Å. Outside this region, residue fluctuations remained within 0.5–2.5 Å over the 50 ns trajectory. Notably, the PB2/CBD-GS441524 complex demonstrated lower overall RMSF values compared to the reference, suggesting reduced residue mobility and improved structural stability. Both complexes exhibited comparable fluctuation patterns around 3 Å within the same residue range, indicating that the PB2 protein preserved structural integrity upon ligand binding. These findings, reinforced by consistently low Radius of Gyration (Rg) values, highlight the compactness and stability of both the reference and test complexes (Figure 7C).
2.4.3. Assessment of the Compactness and Structural Integrity of the H5N1 Clade 2.3.4.4b Proteins-Ligands Complex over Time-Based on the Radius of Gyration (Rg) Using the Molecular Dynamics (MD) Simulations Approach
The average Rg value for the HA-F0045(S) complex was approximately 38 Å, while the HA-GS441524 complex exhibited a slightly lower average Rg of about 37.5 Å (Figure 8A). These similar values indicate that HA-GS441524 maintains a comparable level of compactness to the reference complex. Throughout the simulation, the Rg of HA-GS441524 remained steady around or above 37.5 Å, with fewer fluctuations than the HA-F0045(S) complex, suggesting a relatively stable structural conformation. Although both complexes showed minor fluctuations, the HA-GS441524 complex appeared slightly less compact. Overall, all analyzed complexes demonstrated acceptable levels of compactness, with some conformational changes attributed to ligand binding.
In the case of the NA protein, the average Radius of Gyration (Rg) values for the NA-Zanamivir and NA-GS441524 complexes were 20.1 Å and 20.2 Å, respectively (Figure 8B). These closely aligned values suggest that both complexes preserved structural integrity throughout the simulation, exhibiting only minor fluctuations. Similarly, for the PB2/CBD protein, the PB2/CBD-PB2-39 complex had an average Rg of approximately 16.4 Å, whereas the PB2/CBD-GS441524 complex showed a slightly lower average Rg of 16.2 Å (Figure 8C). The lower Rg value and minimal variation in the PB2/CBD-GS441524 complex indicate a more compact and structurally stable configuration compared to the reference. Overall, the Rg analysis supports that all protein-ligand complexes presented in this study maintained satisfactory compactness during the 50 ns simulation, with no significant conformational changes induced by ligand binding.
2.4.4. Assessment of H5N1 Clade 2.3.4.4b Proteins–Ligands Interactions Based on the Hydrogen Bonds Analysis of the Molecular Dynamics Simulations
The hydrogen bond (HB) formation in the complex during the simulation showed the strength of the complex and dynamic behavior with each bond formed during the time frame. To investigate the stability of the ligand interaction with protein, the measurement of intermolecular hydrogen bond development and hydrogen bond distance between ligand and interacting residues of proteins were evaluated.
The hydrogen bond monitor of complex HA-F0045(S) suggested that around two hydrogen bonds formed throughout the simulation time frame. However, two other hydrogen bonds were also established in various frames during the simulation (Figure 9A). The complex HA-GS441524 formed four intermolecular hydrogen bonds throughout the simulation frames and two hydrogen bonds in various frames during the complete 50 ns simulation time (Figure 9B).
In the case of NA protein hydrogen bond monitoring, the complex NA-zanamivir (standard) maintained four hydrogen bonds throughout the entire simulation time (Figure 9C). However, the complex NA-GS441524 formed three stable hydrogen bonds. (Figure 9D).
The hydrogen bond analysis of the PB2/CBD complex with the standard ligand PB2-39 (PB2/CBD-PB2-39) revealed the formation of two stable hydrogen bonds throughout the 50 ns simulation (Figure 9E), with two additional hydrogen bonds forming intermittently. In contrast, the PB2/CBD-GS441524 complex consistently maintained five stable hydrogen bonds, along with two more that appeared intermittently during the simulation (Figure 9F). These results indicate that the PB2/CBD-GS441524 complex exhibited greater stability compared to the reference PB2/CBD-PB2-39 complex.
2.4.5. Results of the Estimation of the Hydrogen Bond Distances as a Measure of the Complex Stability Between the H5N1 Clade 2.3.4.4b with the Selected Legends
To evaluate the stability of hydrogen bonds, changes in the distances between the compounds and key amino acid residues involved in hydrogen bonding were analyzed throughout the MD simulations. This was done by monitoring the variation in distance between the Cα atoms of the binding pocket and the ligands. For stable hydrogen bonds, the average distance should ideally remain below 3 Å.
In the HA-F0045(S) complex, the standard ligand F0045(S) formed three hydrogen bonds with residues GLN46, THR331, and HIS44 of the HA protein. The average bond distances were approximately 3.0 Å, 3.1 Å, and 5.0 Å, respectively (Figure 10A). However, these interactions showed fluctuations ranging from 2.2 Å to 7.0 Å over the course of the simulation, indicating relatively unstable bonding, particularly in the case of HIS44.
In contrast, the HA-GS441524 complex displayed interactions between GS441524 and four HA residues: THR331, HIS44, GLN46, and HIS465. The average hydrogen bond distances were about 3.2 Å, 3.3 Å, 4.1 Å, and 4.2 Å, respectively, with fluctuations ranging from 2.9 Å to 12.0 Å (Figure 10B). These larger variations further suggest that the interactions between GS441524 and the HA protein were also relatively unstable during the simulation.
For the NA-zanamivir complex (standard ligand), three hydrogen bonds were observed with residues TYR344, ARG368, and LYS432. The average hydrogen bond distances were 2.0 Å, 2.1 Å, and 2.7 Å, respectively, indicating stable interactions between zanamivir and the NA protein (Figure 10C).
In comparison, the NA-GS441524 complex formed hydrogen bonds with four residues: LYS432, ARG368, ARG430, and ARG18. The average bond distances were 2.7 Å, 3.0 Å, 3.2 Å, and 3.5 Å, respectively. These values suggest less stable interactions compared to the standard zanamivir complex (Figure 10D).
For the PB2/CBD-PB2-39 complex (standard ligand), three hydrogen bonds were established with residues ARG355, ASN429, and GLN406, with average bond distances of 3.0 Å, 3.1 Å and 3.3 Å, respectively (Figure 10E).
In contrast, the PB2/CBD-GS441524 complex formed five hydrogen bonds with GLU361, PHE404, LYS367, ARG355, and GLN406. The average hydrogen bond distances were 2.0 Å, 2.2 Å, 2.3 Å, 2.5 Å, and 3.0 Å, respectively, indicating more stable interactions than those seen in the PB2/CBD-PB2-39 complex (Figure 10F).
3. Discussion
This study utilized an integrated computational strategy—including homology modeling, molecular docking, molecular dynamics (MD) simulations, and MM-GBSA binding free energy calculations—to evaluate the structural stability and inhibitory potential of GS441524 and sofosbuvir against key viral proteins of the currently circulating H5N1 clade 2.3.4.4b, namely hemagglutinin (HA), neuraminidase (NA), polymerase basic protein 2 C-terminal domain (PB2/CBD), and matrix protein 2 (M2). These proteins are critical for viral entry, replication, and host adaptation, making them promising therapeutic targets for combating avian influenza [28,29,30,31].
The high-quality homology models of HA, NA, and PB2/CBD were validated using multiple structural assessment parameters, including favorable DOPE scores, low PDF total energy values, and Ramachandran plot analyses. The majority of amino acid residues were located within favored regions of the Ramachandran plots, confirming appropriate backbone dihedral angles and overall model stability. Additionally, the Verify-3D scores for all models exceeded the minimum acceptable thresholds and approached or surpassed expected high-quality benchmarks, further supporting the structural reliability of the predicted protein models [32]. These findings confirmed the reliability of the models for use in subsequent docking and simulation analyses.
Molecular docking results revealed that GS441524 and sofosbuvir exhibit strong binding affinities toward the HA, NA, and PB2/CBD proteins, often matching or surpassing those of established reference ligands (Table 2). Notably, GS441524 showed higher -CDOCKER interaction energy scores than F0045(S) when docked with HA and formed multiple hydrogen bonds with critical residues like ILE390 and THR394, which play key roles in the viral fusion process [33]. Similarly, both sofosbuvir and GS441524 exhibited strong interactions with the NA protein, forming up to 10 hydrogen bonds with key catalytic residues such as ARG368 and ASP151, which are essential for neuraminidase enzymatic function [34].
The PB2-39 is among the identified PB2/CBD inhibitors and has been recognized as a potent agent against the replication of various influenza A virus subtypes. When used in combination with the viral release inhibitor zanamivir, PB2-39 demonstrated a synergistic antiviral effect, enhancing the overall efficacy of the treatment [30].
In the PB2/CBD docking analysis, GS441524 showed extensive interactions with catalytically and structurally significant residues, such as ARG355 and PHE404, forming multiple hydrogen bonds. Its binding energy was comparable to, or slightly lower than, that of the reference compound PB2-39. MM-GBSA calculations supported these findings, revealing favorable binding energies for GS441524 with HA (−61.43 kcal/mol), NA (−83.10 kcal/mol), and PB2/CBD (−99.08 kcal/mol) (Table 2). Notably, the binding energy of GS441524 with NA exceeded that of Zanamivir, a clinically approved neuraminidase inhibitor. [35]. These results highlight GS441524’s potential as a viable anti-influenza agent. Although its binding energies with PB2/CBD were slightly less favorable compared to MGT or PB2-39, the overall interaction profile and energy scores reinforce its promise as a strong candidate for inhibiting influenza virus replication.
The M2 tetrameric channel protein of the influenza A virus serves as a crucial membrane-bound proton channel that acidifies virions enclosed within endosomes, thereby mediating the viral uncoating process and enabling the release of viral RNA into the host cell cytoplasm [36]. In our molecular docking analysis, the binding affinities of standard antiviral agents (amantadine and rimantadine) and test compounds (sofosbuvir and GS441524) were assessed against the M2 transmembrane domain of influenza A virus. Rimantadine and amantadine exhibited moderate binding affinities, with CDOCKER interaction scores of 33.02 and 27.55, respectively, and corresponding binding energies of −112.23 kcal/mol and −104.46 kcal/mol. However, known mutations within the M2 membrane-spanning domain that confer resistance to amantadine have been shown to generate ion channel activity that is no longer inhibited by the drug [37].
The binding sites for amantadine and rimantadine within the M2 ion channel are located near the histidine gate (His37) and pore-lining residues (Leu26, Ala30, and Ser31), where these inhibitors function as channel blockers. Binding interaction analysis showed that both standard and test compounds formed key hydrogen bonds and hydrophobic interactions with conserved residues of the H5N1 clade 2.3.4.4b M2 channel (Table 2). Notably, the test compound sofosbuvir demonstrated a substantially higher CDOCKER score (62.05) and more favorable binding energy (−116.11 kcal/mol) than the standard inhibitors, suggesting a stronger and more stable interaction with the M2 channel. Likewise, GS441524—the parent nucleoside of remdesivir, resulted in a solid CDOCKER score of 42.59 and the most negative binding energy (−120.65 kcal/mol) among all tested compounds. This indicates a potentially higher binding affinity and robust interaction with the M2 protein, particularly through engagement with key residues such as VAL27, ALA30, and HIS37. These findings, closely aligned with known interaction patterns of standard drugs, support the potential of GS441524 as a promising M2 channel inhibitor and affirm the reliability of the docking analysis [38,39,40].
The molecular dynamics simulations conducted over a 50 ns period revealed varying levels of stability across the protein-ligand complexes. In the case of HA, both GS441524 and the reference ligand F0045(S) showed elevated RMSD values exceeding 3 Å, indicating moderate conformational fluctuations and reduced stability. In contrast, the GS441524 complexes with NA and PB2/CBD displayed consistent RMSD values at or below 3 Å throughout the simulation, reflecting strong dynamic stability and limited structural deviations [41,42]. The RMSF analysis revealed minimal fluctuations at the residue level across all complexes, with PB2/CBD-GS441524 exhibiting slightly greater residue stability compared to the reference PB2-39, indicating tighter binding and decreased flexibility. Additionally, the radius of gyration (Rg) measurements confirmed the overall structural compactness of the ligand-protein complexes. Notably, PB2/CBD-GS441524 displayed a more compact and stable conformation than the standard reference, further supporting its enhanced binding affinity and stability [43,44].
The hydrogen bonding analysis of GS441524 with NA demonstrated favorable interaction patterns, although these were slightly less stable compared to those observed with PB2/CBD. GS441524 formed more hydrogen bonds with HA than the reference ligand F0045(S), but the dynamic stability of this complex was comparatively weaker, indicating that further optimization may be needed to improve binding efficacy with both HA and NA. Conversely, hydrogen bond analysis strongly supported the stability of GS441524’s interaction with PB2/CBD. In the PB2/CBD-GS441524 complex, up to five persistent hydrogen bonds were maintained, with bond lengths consistently below 3 Å, reflecting stable and specific binding. This suggests GS441524 is a promising inhibitor of PB2/CBD, potentially outperforming the standard inhibitor PB2-39. These findings are especially important given the critical role of the PB2 protein in viral polymerase function and host adaptation, where its inhibition could significantly disrupt viral replication [45,46].
These computational results are consistent with previous studies that have demonstrated the broad-spectrum antiviral efficacy of GS441524 against various RNA viruses, such as SARS-CoV-2 and feline infectious peritonitis virus (FIPV) [27,47,48,49]. Its ability to target highly conserved functional domains in influenza virus proteins indicates that GS441524 holds promise as a novel therapeutic candidate against avian influenza strains like H5N1, which remain significant zoonotic and pandemic threats. In summary, this study provides important insights into the drug repurposing potential of GS441524 and highlights the effectiveness of computational methods in accelerating the discovery of broad-spectrum antiviral agents against emerging and re-emerging viral pathogens.
4. Materials and Methods
4.1. The H5N1 Clade 2.3.4.4b Sequences Retrieval from the GenBank
In this study, we retrieved the full-length genome sequences of 279 isolates representing the H5N1 clade 2.3.4.4b from the GenBank (NCBI) (https://www.ncbi.nlm.nih.gov/protein) database. The sequence of each protein was downloaded in a fasta format. The sequences of these isolates included (115 from chickens, 40 from ducks, 30 from migratory birds, and 51 from the Canadian geese). We extracted the nucleotides and protein sequences of the (HA, NA, PB2, and M2) proteins out of these sequences.
4.2. Multiple Sequence Alignment (MSA) and Generation of the Consensus Sequences
We performed the multiple protein sequence alignments using Geneiuos software ((https://www.geneious.com/). The consensus sequences per protein were generated based on the individual’s MSA, which was one per protein. The generated consensus sequences will be used for each protein’s downstream prediction and homology modeling.
4.3. The Homology Modeling of the H5N1 Clade 2.3.4.4b (HA, NA, PB2 abd M2) Proteins
We used the MODELLER tool of the Biovia Discovery Studio (v24.1.0.321712) and applied the comparative modeling approach to build the homology model per each target protein. This approach enabled the creation of the 3D model structure for the query protein sequences, as previously described [50]. Briefly, the sequences of the target viral proteins, HA, NA, and the cap-binding domain of PB2 (PB2/CBD) of the viral RNA polymerase complex RdRp were retrieved and downloaded in FASTA format from the NCBI database (https://www.ncbi.nlm.nih.gov/). The homology modeling procedure requires the alignment of the query sequences with template protein sequences from the BLAST search tool. The query protein sequence aligned with 100 most identical template sequences with a maximum similarity of 99% and a minimum of 30% in BLAST with BLOSSUM62 algorithm protocol. Five identical template sequences (similarity: maximum 99-minimum 35%) were chosen to align sequences to the templates. Further, the 3D-build homology model protocol was run to align template structures and predict query sequence 3D confirmation based on template proteins. Discrete Optimized Protein Energy (DOPE) score was used to assess the quality of the generated different 3D protein models to predict five different homology models per query sequence. The DOPE Score and the PDF total energy score are used to evaluate the accuracy of each protein model. The more negative values of the DOPE score and the more positive PDF total energy values, the more stable and accurate the predicted protein model. The best model having the lowest DOPE score was selected for further in silico computational studies as previously described [51]. All the predicted 3D structure models were verified through the Ramachandran plot and the Verify 3D model tool of Biovia Discovery Studio. The Ramachandran plot suggested the stability of structure based on amino acid residues located in the most favored, highly allowed, and allowed region or site of the plot. If the verified score results from the Verify 3D method of the modeled protein is higher than the verified expected low Score value, then the model is considered of acceptable quality. The closer the verified score result is to the verified expected high score value, the better the quality of the predicted protein structure model. The prediction of structure is further visualized using the Biovia Discovery Studio v24.1.0.321712.
4.4. Protein and Ligand Preparation
The prepared proteins, through homology modeling, are used as receptors for ligand interaction. Compounds that are used as ligands in this study are retrieved in 3D-SDF file format from PubChem database: https://pubchem.ncbi.nlm.nih.gov. The compound IDs of all the ligands are listed in (Table 2). For energy minimization of the modeled protein, the smart minimizer at 10000 steps was run to minimize the energy of the prepared protein through the CHARMm forcefield. Further, the preparation of the ligand was done following the energy minimization through fill minimization protocol through smart Minimizer at 10000 steps with CHARMm forcefield using the Biovia Discovery Studio v24.1.0.321712 as previously described [50].
4.5. Molecular Docking of Compounds with Target Proteins
The molecular docking approach can be used to model the interaction between HA, NA, and PB2/CBD protein with some selected and reference standard anti-influenza virus compounds. The interactions between these small molecules and proteins at the atomic level, molecular docking, help us understand how these molecules bind and function within biological processes [52,53]. In the CDOCKER tool of the Biovia Discovery Studio docking protocol, we typically use 10 docking poses per ligand. For each dock/ligand pose, the higher the positive values of the CDOCKER interaction energy score (-CDOCK score), the more strongly the binding affinity is expected [53,54,55].The computational tool of the Biovia Discovery Studio also enables the visualization of the ligand-target interaction (molecular docking) and the identification of the compounds that bind more efficiently with the target [53]. This analysis typically involves examining the docking scores, ligand-protein interactions, and the visualization of the docked complexes. To describe the defining binding site in protein, -CDOCK score through Biovia Discovery Studio v24.1.0.321712. The docking figures were also created with the BIOVIA Discovery Studio, and the docking scores were ranked, with the most positive scores for each complex chosen.
4.6. Calculations of the Molecular Mechanics-Generalize-Born Surface Area (MM-GBSA)
The binding free energy of the protein-ligand complexes in this study was measured using the MM-GBSA approach, which integrates the molecular mechanics (MM) force fields with a generalized Born and surface area continuum with a none-implicit solvation model. The MM-GBSA calculation incorporates the CHARMm force field, with partial charge estimation using Memory Rone. The Di-electric constant was set to be one, and the minimum non-bond higher and lower cutoff distance was set to be 12 and 10 Å. The MM-GBSA was determined in this study using the equation (ΔG = E_complex (minimized)– [E_ligand (minimized) + E_receptor (minimized)) using the Biovia Discovery Studio v24.1.0.321712. The default setting employed to compute MM-GBSA involved rendering all protein atoms rigid while the ligand atoms are relaxed through the Biovia Discovery Studio software.
4.7. Molecular Dynamics (MD) Simulation
Based on in vitro analysis, docking energies, and conformational pose analysis, the top four complexes were selected for MD simulation as previously described [56]. To further explore the dynamic behavior of ligand-protein complexes in this study, the best predicted top hits of compounds and reference standards with respect to HA, NA, and PB2/CBD shown in (Table 2) were selected to perform 50,000 picoseconds (ps) through standard dynamics cascade simulation. To generate the molecular topology files for the protein complex and to create the topology of ligands, the CHARMm36 force field was used. The simulation system consists of an explicit boundary solvent model, orthorhombic box with a minimal distance of 7 nm between the protein surface and the edge of the box neutralized with the inclusion of cation type sodium (Na) and anion type Chloride (Cl) counter ions.
For the energy minimization, the steepest descent (minimization 1) with RMS gradient one and conjugate gradient (minimization 2) with RMS gradient 0.1. Both minimization algorithms were used for 50,00 steps. The heating phase was performed using simulation time 10 ps with time step 2 femtosecond (fs); for immersion, the initial temperature is 50 and the target temperature 300 K with save results interval 2 ps.
The equilibration of complexes was carried out for a 10000 ps simulation run with a two fs (femtosecond) time step, and the save result interval is two ps. The Particle-Mesh-Ewald (PME) algorithm was used for long-range electrostatic interactions with fourth-order cubic interpolation and kappa 0.34 Å grid spacing. The advanced dynamic integrator used Leapfrog Verlet algorithm with applied shake constraint. The implicit solvent model was used with dielectric constant 1, Nonbond list radius cutoff 14 in which nonbond higher cutoff distance is 12 and nonbond lower cutoff distance is 10. The production step of a standard dynamic cascade of MD simulation was carried out for 50 ns (50000 picoseconds). Trajectory analysis was done to confirm the number of hydrogen bonds established per each confirmation, hydrogen bond distance variation of ligand and interacted residues per confirmation, Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), and Radius of Gyration (RG) of each system [57]. The stability of the complex is indicated by the highest potential inhibitor from the stable complex protein-ligand through Biovia Discovery Studio software.
5. Conclusions
This comprehensive in silico study provides strong evidence that GS441524, the active metabolite of remdesivir, is a promising multi-target antiviral agent against H5N1 avian influenza virus clade 2.3.4.4v. Using structural modeling, molecular docking, MM-GBSA binding energy calculations, and 50 ns molecular dynamics simulations, GS441524 demonstrated stable and high-affinity binding to four key viral proteins: hemagglutinin (HA), neuraminidase (NA), and the cap-binding domain of PB2 (PB2/CBD). Among these, GS441524 showed especially notable dynamic stability and binding strength with NA and PB2/CBD, supported by favorable -CDOCKER scores, robust hydrogen bonding, consistent RMSD/RMSF values, lower radius of gyration, and shorter hydrogen bond distances. These results suggest GS441524’s potential dual inhibition of viral entry and replication. Consistent with prior reports of its broad-spectrum antiviral activity against multiple RNA viruses, GS441524’s targeting of conserved influenza protein domains positions it as a viable candidate for repurposing against H5N1, a significant zoonotic and pandemic threat. However, in silico findings require experimental validation. Future work should include in vitro antiviral testing, in vivo efficacy and toxicity studies, pharmacokinetics, and exploration of resistance mechanisms. Evaluating combination therapies with existing antivirals like PB2-39, Zanamivir, oseltamivir, or Baloxavir could further advance GS441524’s clinical development.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
MK: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Writing – original draft, Writing – review & editing. AS: Data curation, Investigation, Methodology, Software, Validation, Writing – original draft, Writing – review & editing, Resources. ND: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Writing – original draft, Writing – review & editing. MC: Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing, Conceptualization, Funding acquisition. MH: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
This study was funded by a seed grant (PI: MGH) from Long Island University (Grant no: 36524) and funds from the USDA-NIFA Animal Health and Disease Research grant (NI24AHDRXXXXG066).
Institutional Review Board Statement
Not Applicable
Informed Consent Statement
Not Applicable
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors upon request.
Acknowledgments
We thank Dylan Feldman and Muddapuram Deeksha Goud for their technical assistance in retrieving the protein sequences from GenBank
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Dou, D.; Revol, R.; Ostbye, H.; Wang, H.; Daniels, R. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front Immunol 2018, 9, 1581. [Google Scholar] [CrossRef] [PubMed]
- De Vlugt, C.; Sikora, D.; Pelchat, M. Insight into Influenza: A Virus Cap-Snatching. Viruses 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Sims, L.D. Avian influenza: past, present and future. Rev Sci Tech 2024, Special Edition, 83–88. [Google Scholar] [CrossRef]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza Virus: Dealing with a Drifting and Shifting Pathogen. Viral Immunol 2018, 31, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y. Pathogenicity and virulence of influenza. Virulence 2023, 14, 2223057. [Google Scholar] [CrossRef]
- Humayun, F.; Khan, F.; Fawad, N.; Shamas, S.; Fazal, S.; Khan, A.; Ali, A.; Farhan, A.; Wei, D.Q. Computational Method for Classification of Avian Influenza A Virus Using DNA Sequence Information and Physicochemical Properties. Front Genet 2021, 12, 599321. [Google Scholar] [CrossRef]
- Webby, R.J.; Uyeki, T.M. An Update on Highly Pathogenic Avian Influenza A(H5N1) Virus, Clade 2.3.4.4b. J Infect Dis 2024, 230, 533–542. [Google Scholar] [CrossRef]
- Burrough, E.R.; Magstadt, D.R.; Petersen, B.; Timmermans, S.J.; Gauger, P.C.; Zhang, J.; Siepker, C.; Mainenti, M.; Li, G.; Thompson, A.C.; et al. Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4b Virus Infection in Domestic Dairy Cattle and Cats, United States, 2024. Emerg Infect Dis 2024, 30, 1335–1343. [Google Scholar] [CrossRef]
- Pulit-Penaloza, J.A.; Brock, N.; Belser, J.A.; Sun, X.; Pappas, C.; Kieran, T.J.; Basu Thakur, P.; Zeng, H.; Cui, D.; Frederick, J.; et al. Highly pathogenic avian influenza A(H5N1) virus of clade 2.3.4.4b isolated from a human case in Chile causes fatal disease and transmits between co-housed ferrets. Emerg Microbes Infect 2024, 13, 2332667. [Google Scholar] [CrossRef]
- Loregian, A.; Mercorelli, B.; Nannetti, G.; Compagnin, C.; Palu, G. Antiviral strategies against influenza virus: towards new therapeutic approaches. Cell Mol Life Sci 2014, 71, 3659–3683. [Google Scholar] [CrossRef]
- Sarker, A.; Gu, Z.; Mao, L.; Ge, Y.; Hou, D.; Fang, J.; Wei, Z.; Wang, Z. Influenza-existing drugs and treatment prospects. Eur J Med Chem 2022, 232, 114189. [Google Scholar] [CrossRef] [PubMed]
- van der Vries, E.; Schutten, M.; Fraaij, P.; Boucher, C.; Osterhaus, A. Influenza virus resistance to antiviral therapy. Adv Pharmacol 2013, 67, 217–246. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Y.; Liu, S. Emerging antiviral therapies and drugs for the treatment of influenza. Expert Opin Emerg Drugs 2022, 27, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Bonomini, A.; Mercorelli, B.; Loregian, A. Antiviral strategies against influenza virus: an update on approved and innovative therapeutic approaches. Cell Mol Life Sci 2025, 82, 75. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Wohlbold, T.J.; Zheng, N.Y.; Huang, M.; Huang, Y.; Neu, K.E.; Lee, J.; Wan, H.; Rojas, K.T.; Kirkpatrick, E.; et al. Influenza Infection in Humans Induces Broadly Cross-Reactive and Protective Neuraminidase-Reactive Antibodies. Cell 2018, 173, 417–429.e10. [Google Scholar] [CrossRef]
- Sun, X.; Ma, H.; Wang, X.; Bao, Z.; Tang, S.; Yi, C.; Sun, B. Broadly neutralizing antibodies to combat influenza virus infection. Antiviral Res 2024, 221, 105785. [Google Scholar] [CrossRef]
- Li, F.; Ma, C.; Wang, J. Inhibitors targeting the influenza virus hemagglutinin. Curr Med Chem 2015, 22, 1361–1382. [Google Scholar] [CrossRef]
- Hayden, F.G.; Atmar, R.L.; Schilling, M.; Johnson, C.; Poretz, D.; Paar, D.; Huson, L.; Ward, P.; Mills, R.G. Use of the selective oral neuraminidase inhibitor oseltamivir to prevent influenza. N Engl J Med 1999, 341, 1336–1343. [Google Scholar] [CrossRef]
- Read, R.C. Treating influenza with zanamivir. Lancet 1998, 352, 1872–1873. [Google Scholar] [CrossRef]
- Slain, D. Intravenous Zanamivir: A Viable Option for Critically Ill Patients With Influenza. Ann Pharmacother 2021, 55, 760–771. [Google Scholar] [CrossRef]
- Massari, S.; Desantis, J.; Nizi, M.G.; Cecchetti, V.; Tabarrini, O. Inhibition of Influenza Virus Polymerase by Interfering with Its Protein-Protein Interactions. ACS Infect Dis 2021, 7, 1332–1350. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, T.; Zhang, Y.; Zhang, N.; Guo, Y.; Gao, X.; Peng, W.; Shu, S.; Zhao, C.; Cui, D.; et al. BAG6 inhibits influenza A virus replication by inducing viral polymerase subunit PB2 degradation and perturbing RdRp complex assembly. PLoS Pathog 2024, 20, e1012110. [Google Scholar] [CrossRef] [PubMed]
- Finberg, R.W.; Lanno, R.; Anderson, D.; Fleischhackl, R.; van Duijnhoven, W.; Kauffman, R.S.; Kosoglou, T.; Vingerhoets, J.; Leopold, L. Phase 2b Study of Pimodivir (JNJ-63623872) as Monotherapy or in Combination With Oseltamivir for Treatment of Acute Uncomplicated Seasonal Influenza A: TOPAZ Trial. J Infect Dis 2019, 219, 1026–1034. [Google Scholar] [CrossRef]
- O'Neil, B.; Ison, M.G.; Hallouin-Bernard, M.C.; Nilsson, A.C.; Torres, A.; Wilburn, J.M.; van Duijnhoven, W.; Van Dromme, I.; Anderson, D.; Deleu, S.; et al. A Phase 2 Study of Pimodivir (JNJ-63623872) in Combination With Oseltamivir in Elderly and Nonelderly Adults Hospitalized With Influenza A Infection: OPAL Study. J Infect Dis 2022, 226, 109–118. [Google Scholar] [CrossRef]
- Ayari, M.G.; Favetta, P.; Warszycki, D.; Vasseur, V.; Herve, V.; Degardin, P.; Carbonnier, B.; Si-Tahar, M.; Agrofoglio, L.A. Molecularly Imprinted Hydrogels Selective to Ribavirin as New Drug Delivery Systems to Improve Efficiency of Antiviral Nucleoside Analogue: A Proof-of-Concept Study with Influenza A Virus. Macromol Biosci 2022, 22, e2100291. [Google Scholar] [CrossRef]
- Cosaro, E.; Pires, J.; Castillo, D.; Murphy, B.G.; Reagan, K.L. Efficacy of Oral Remdesivir Compared to GS-441524 for Treatment of Cats with Naturally Occurring Effusive Feline Infectious Peritonitis: A Blinded, Non-Inferiority Study. Viruses 2023, 15. [Google Scholar] [CrossRef]
- Murphy, B.G.; Perron, M.; Murakami, E.; Bauer, K.; Park, Y.; Eckstrand, C.; Liepnieks, M.; Pedersen, N.C. The nucleoside analog GS-441524 strongly inhibits feline infectious peritonitis (FIP) virus in tissue culture and experimental cat infection studies. Vet Microbiol 2018, 219, 226–233. [Google Scholar] [CrossRef]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog 2013, 9, e1003657. [Google Scholar] [CrossRef]
- Hussein, A.F.A.; Cheng, H.; Tundup, S.; Antanasijevic, A.; Varhegyi, E.; Perez, J.; AbdulRahman, E.M.; Elenany, M.G.; Helal, S.; Caffrey, M.; et al. Identification of entry inhibitors with 4-aminopiperidine scaffold targeting group 1 influenza A virus. Antiviral Res 2020, 177, 104782. [Google Scholar] [CrossRef]
- Yuan, S.; Chu, H.; Zhang, K.; Ye, J.; Singh, K.; Kao, R.Y.; Chow, B.K.; Zhou, J.; Zheng, B.J. A novel small-molecule compound disrupts influenza A virus PB2 cap-binding and inhibits viral replication. J Antimicrob Chemother 2016, 71, 2489–2497. [Google Scholar] [CrossRef]
- Charostad, J.; Rezaei Zadeh Rukerd, M.; Mahmoudvand, S.; Bashash, D.; Hashemi, S.M.A.; Nakhaie, M.; Zandi, K. A comprehensive review of highly pathogenic avian influenza (HPAI) H5N1: An imminent threat at doorstep. Travel Med Infect Dis 2023, 55, 102638. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Luthy, R.; Bowie, J.U. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol 1997, 277, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Weis, W.I.; Cusack, S.C.; Brown, J.H.; Daniels, R.S.; Skehel, J.J.; Wiley, D.C. The structure of a membrane fusion mutant of the influenza virus haemagglutinin. EMBO J 1990, 9, 17–24. [Google Scholar] [CrossRef]
- Doyle, T.M.; Jaentschke, B.; Van Domselaar, G.; Hashem, A.M.; Farnsworth, A.; Forbes, N.E.; Li, C.; Wang, J.; He, R.; Brown, E.G.; et al. The universal epitope of influenza A viral neuraminidase fundamentally contributes to enzyme activity and viral replication. J Biol Chem 2013, 288, 18283–18289. [Google Scholar] [CrossRef]
- Colman, P.M. Zanamivir: an influenza virus neuraminidase inhibitor. Expert Rev Anti Infect Ther 2005, 3, 191–199. [Google Scholar] [CrossRef]
- Cady, S.D.; Hong, M. Amantadine-induced conformational and dynamical changes of the influenza M2 transmembrane proton channel. Proc Natl Acad Sci U S A 2008, 105, 1483–1488. [Google Scholar] [CrossRef]
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528. [Google Scholar] [CrossRef]
- Kozakov, D.; Chuang, G.Y.; Beglov, D.; Vajda, S. Where does amantadine bind to the influenza virus M2 proton channel? Trends Biochem Sci 2010, 35, 471–475. [Google Scholar] [CrossRef]
- Thomaston, J.L.; Samways, M.L.; Konstantinidi, A.; Ma, C.; Hu, Y.; Bruce Macdonald, H.E.; Wang, J.; Essex, J.W.; DeGrado, W.F.; Kolocouris, A. Rimantadine Binds to and Inhibits the Influenza A M2 Proton Channel without Enantiomeric Specificity. Biochemistry 2021. [Google Scholar] [CrossRef]
- Intharathep, P.; Laohpongspaisan, C.; Rungrotmongkol, T.; Loisruangsin, A.; Malaisree, M.; Decha, P.; Aruksakunwong, O.; Chuenpennit, K.; Kaiyawet, N.; Sompornpisut, P.; et al. How amantadine and rimantadine inhibit proton transport in the M2 protein channel. J Mol Graph Model 2008, 27, 342–348. [Google Scholar] [CrossRef]
- Abouzied, A.S.; Alqarni, S.; Younes, K.M.; Alanazi, S.M.; Alrsheed, D.M.; Alhathal, R.K.; Huwaimel, B.; Elkashlan, A.M. Structural and free energy landscape analysis for the discovery of antiviral compounds targeting the cap-binding domain of influenza polymerase PB2. Sci Rep 2024, 14, 25441. [Google Scholar] [CrossRef] [PubMed]
- Abouzied, A.S.; Alqarni, S.; Younes, K.M.; Alanazi, S.M.; Alrsheed, D.M.; Alhathal, R.K.; Huwaimel, B.; Elkashlan, A.M. Author Correction: Structural and free energy landscape analysis for the discovery of antiviral compounds targeting the cap-binding domain of influenza polymerase PB2. Sci Rep 2025, 15, 3691. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, L.; Amichai, S.; Zandi, K.; Cox, B.; Schinazi, R.; Amblard, F. Novel influenza polymerase PB2 inhibitors for the treatment of influenza A infection. Bioorg Med Chem Lett 2019, 29, 126639. [Google Scholar] [CrossRef]
- Omoto, S.; Speranzini, V.; Hashimoto, T.; Noshi, T.; Yamaguchi, H.; Kawai, M.; Kawaguchi, K.; Uehara, T.; Shishido, T.; Naito, A.; et al. Characterization of influenza virus variants induced by treatment with the endonuclease inhibitor baloxavir marboxil. Sci Rep 2018, 8, 9633. [Google Scholar] [CrossRef]
- Graef, K.M.; Vreede, F.T.; Lau, Y.F.; McCall, A.W.; Carr, S.M.; Subbarao, K.; Fodor, E. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J Virol 2010, 84, 8433–8445. [Google Scholar] [CrossRef]
- Kumosani, T.A.; Abbas, A.T.; Basheer, B.; Hassan, A.M.; Yaghmoor, S.S.; Alyahiby, A.H.; Asseri, A.H.; Dwivedi, V.D.; Azhar, E.I. Investigating Pb2 CAP-binding domain inhibitors from marine bacteria for targeting the influenza A H5N1. PLoS One 2025, 20, e0310836. [Google Scholar] [CrossRef]
- Pitts, J.; Babusis, D.; Vermillion, M.S.; Subramanian, R.; Barrett, K.; Lye, D.; Ma, B.; Zhao, X.; Riola, N.; Xie, X.; et al. Intravenous delivery of GS-441524 is efficacious in the African green monkey model of SARS-CoV-2 infection. Antiviral Res 2022, 203, 105329. [Google Scholar] [CrossRef]
- Mohseni, N.; Royster, A.; Ren, S.; Ma, Y.; Pintado, M.; Mir, M.; Mir, S. A novel compound targets the feline infectious peritonitis virus nucleocapsid protein and inhibits viral replication in cell culture. J Biol Chem 2023, 299, 102976. [Google Scholar] [CrossRef]
- Cox, R.M.; Wolf, J.D.; Lieber, C.M.; Sourimant, J.; Lin, M.J.; Babusis, D.; DuPont, V.; Chan, J.; Barrett, K.T.; Lye, D.; et al. Oral prodrug of remdesivir parent GS-441524 is efficacious against SARS-CoV-2 in ferrets. Nat Commun 2021, 12, 6415. [Google Scholar] [CrossRef]
- Khan, M.Y.; Shah, A.U.; Duraisamy, N.; ElAlaoui, R.N.; Cherkaoui, M.; Hemida, M.G. Leveraging Artificial Intelligence and Gene Expression Analysis to Identify Some Potential Bovine Coronavirus (BCoV) Receptors and Host Cell Enzymes Potentially Involved in the Viral Replication and Tissue Tropism. Int J Mol Sci 2025, 26. [Google Scholar] [CrossRef]
- Takeda-Shitaka, M.; Nojima, H.; Takaya, D.; Kanou, K.; Iwadate, M.; Umeyama, H. Evaluation of homology modeling of the severe acute respiratory syndrome (SARS) coronavirus main protease for structure based drug design. Chem Pharm Bull (Tokyo) 2004, 52, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Agu, P.C.; Afiukwa, C.A.; Orji, O.U.; Ezeh, E.M.; Ofoke, I.H.; Ogbu, C.O.; Ugwuja, E.I.; Aja, P.M. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci Rep 2023, 13, 13398. [Google Scholar] [CrossRef]
- Gagnon, J.K.; Law, S.M.; Brooks, C.L. , 3rd. Flexible CDOCKER: Development and application of a pseudo-explicit structure-based docking method within CHARMM. J Comput Chem 2016, 37, 753–762. [Google Scholar] [CrossRef]
- Ding, X.; Wu, Y.; Wang, Y.; Vilseck, J.Z.; Brooks, C.L. , 3rd. Accelerated CDOCKER with GPUs, Parallel Simulated Annealing, and Fast Fourier Transforms. J Chem Theory Comput 2020, 16, 3910–3919. [Google Scholar] [CrossRef]
- Ahmad, S.; Nabi, R.; Alvi, S.S.; Khan, M.; Khan, S.; Khan, M.Y.; Hussain, I.; Shahanawaz, S.D.; Khan, M.S. Carvacrol protects against carbonyl osmolyte-induced structural modifications and aggregation to serum albumin: Insights from physicochemical and molecular interaction studies. Int J Biol Macromol 2022, 213, 663–674. [Google Scholar] [CrossRef]
- Jin, Z.; Wang, Y.; Yu, X.F.; Tan, Q.Q.; Liang, S.S.; Li, T.; Zhang, H.; Shaw, P.C.; Wang, J.; Hu, C. Structure-based virtual screening of influenza virus RNA polymerase inhibitors from natural compounds: Molecular dynamics simulation and MM-GBSA calculation. Comput Biol Chem 2020, 85, 107241. [Google Scholar] [CrossRef]
Figure 1.
Results of the Ramachandran plot analysis of the predicted homology models of the H5N1 clade 2.2.4.4b proteins (A) HA, (B) NA, (C) PB2/CBD, and (D) M2.
Figure 1.
Results of the Ramachandran plot analysis of the predicted homology models of the H5N1 clade 2.2.4.4b proteins (A) HA, (B) NA, (C) PB2/CBD, and (D) M2.
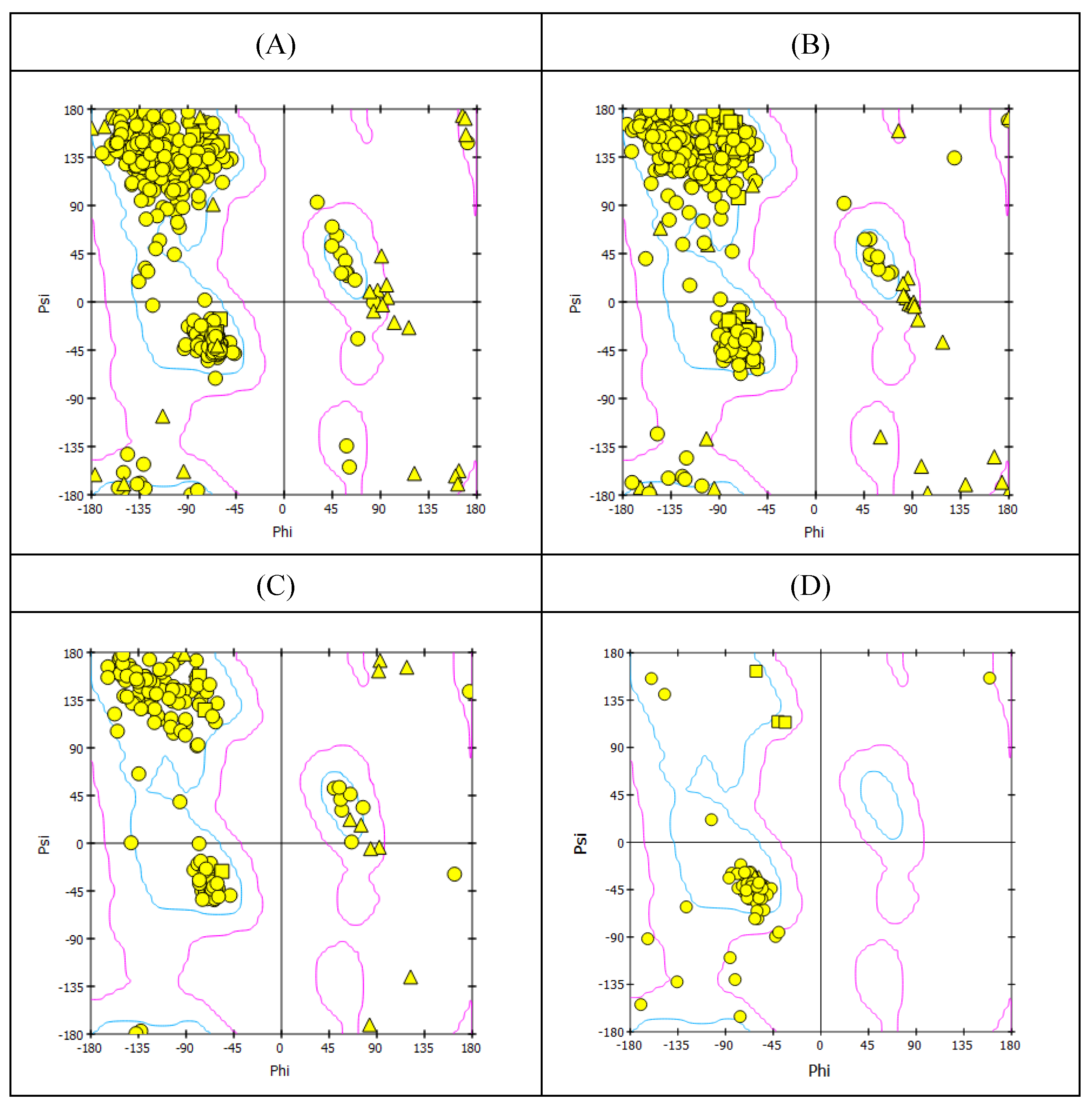
Figure 2.
Representation of the molecular interaction of ligands in the binding sites of the structure of HA protein. (A) F0045(S) interacted through a hydrogen bond with HIS24 of the HA1 chain, VAL 393, and ILE390 of the HA2 chain of HA protein. (B) GS441524 interacted through hydrogen bonds with ALA45 and THR331 of the HA1 chain, VAL 393, and ILE390 of the HA2 chain of HA protein.
Figure 2.
Representation of the molecular interaction of ligands in the binding sites of the structure of HA protein. (A) F0045(S) interacted through a hydrogen bond with HIS24 of the HA1 chain, VAL 393, and ILE390 of the HA2 chain of HA protein. (B) GS441524 interacted through hydrogen bonds with ALA45 and THR331 of the HA1 chain, VAL 393, and ILE390 of the HA2 chain of HA protein.
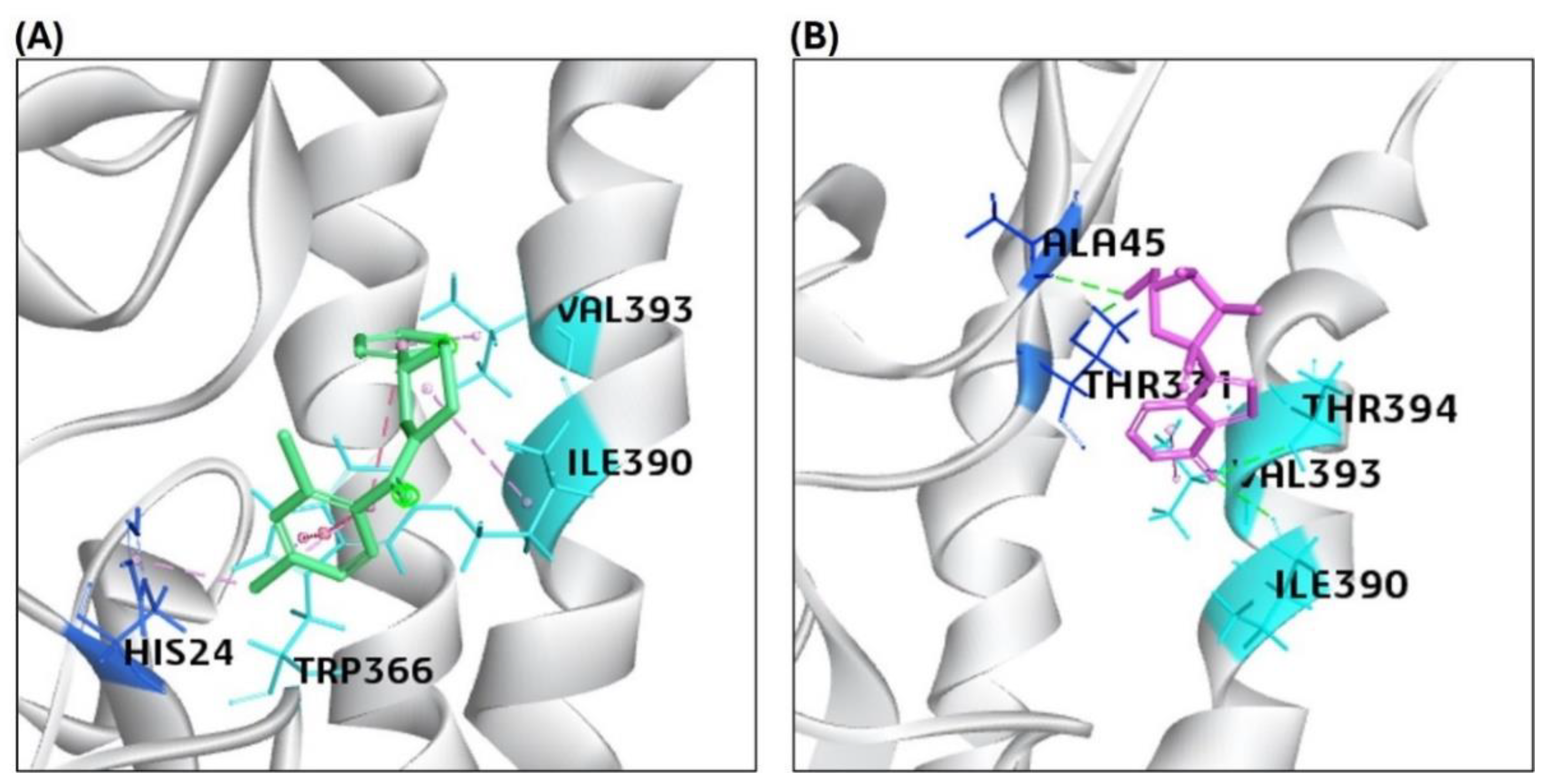
Figure 3.
Representation of the molecular interaction of ligands in the binding sites of the structure of NA protein. (A) Zanamivir interacted through hydrogen bonds with residues TYR344, ARG368, ASP151 and SER367 (B) Sofosbuvir established hydrogen bonds with residues TYR344, ARG368, TYR402, ARG225, LYS432, ASP151 and GLU278. (C) GS441524 interacted through hydrogen bonds with ARG368, VAL149, ASP151, SER367, PRO431 (three hydrogen bonds), and LYS432 residues of NA protein.
Figure 3.
Representation of the molecular interaction of ligands in the binding sites of the structure of NA protein. (A) Zanamivir interacted through hydrogen bonds with residues TYR344, ARG368, ASP151 and SER367 (B) Sofosbuvir established hydrogen bonds with residues TYR344, ARG368, TYR402, ARG225, LYS432, ASP151 and GLU278. (C) GS441524 interacted through hydrogen bonds with ARG368, VAL149, ASP151, SER367, PRO431 (three hydrogen bonds), and LYS432 residues of NA protein.
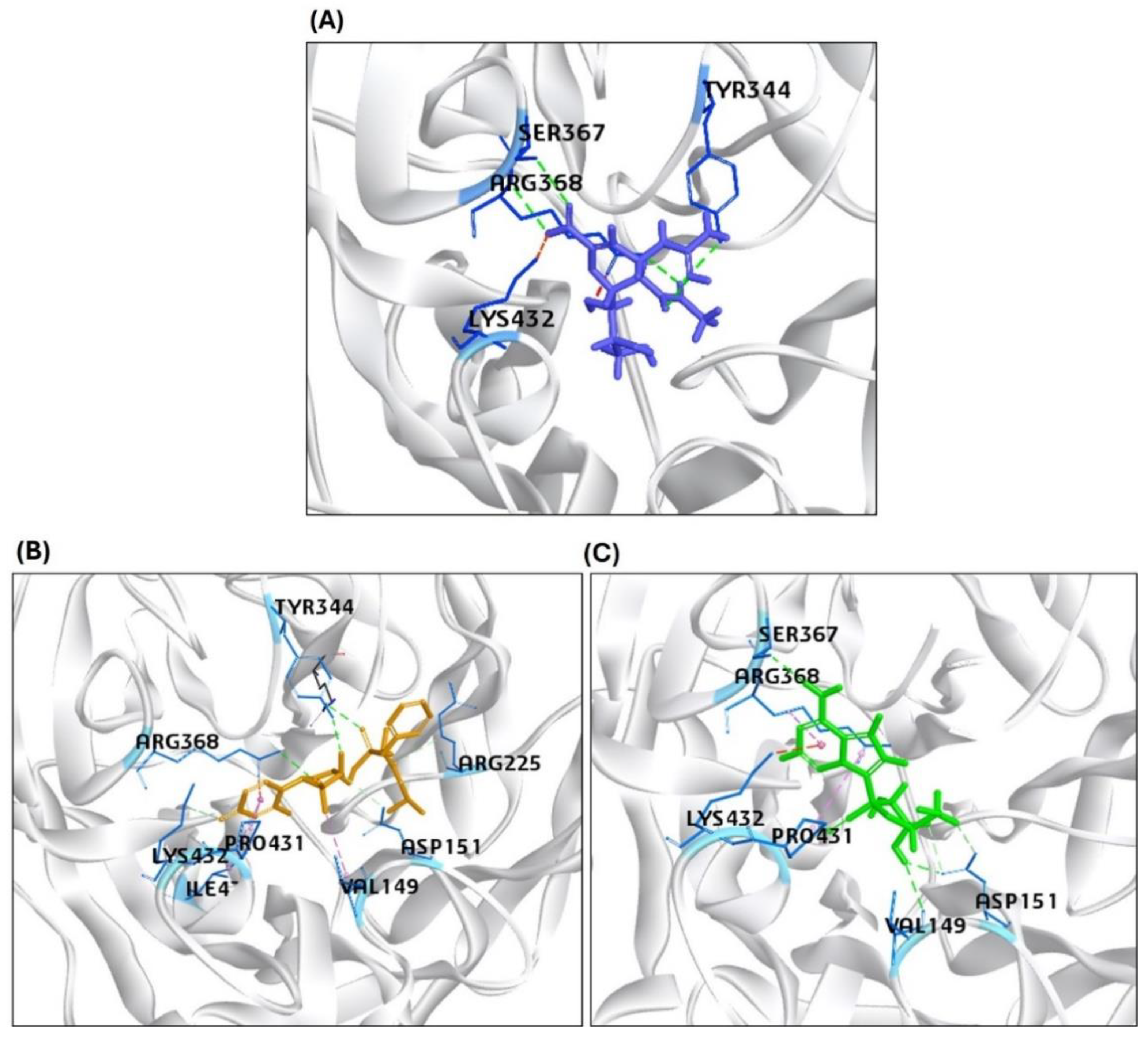
Figure 4.
Results of the molecular interaction model of the tested compounds and the binding sites of the H5N1 clade 2.3.4.4b B2/CBD protein. (A) MGT interacted through hydrogen bonds with LYS339, ARG335, PHE404, GLU361, HIS357, and PHE323 residues of PB2/CBD. (B) PB2-39 interacted through hydrogen bonds with only ARG355 and ASN429 residues of PB2/CBD (C) Sofosbuvir interacted through hydrogen bonds with LYS339, LYS376 and HIS357 and (D) GS441524 interacted through hydrogen bonds with ARG355, LYS376, PHE404, GLN406, GLU361 and SER324 residues in the binding site of the structure of PB2/CBD protein.
Figure 4.
Results of the molecular interaction model of the tested compounds and the binding sites of the H5N1 clade 2.3.4.4b B2/CBD protein. (A) MGT interacted through hydrogen bonds with LYS339, ARG335, PHE404, GLU361, HIS357, and PHE323 residues of PB2/CBD. (B) PB2-39 interacted through hydrogen bonds with only ARG355 and ASN429 residues of PB2/CBD (C) Sofosbuvir interacted through hydrogen bonds with LYS339, LYS376 and HIS357 and (D) GS441524 interacted through hydrogen bonds with ARG355, LYS376, PHE404, GLN406, GLU361 and SER324 residues in the binding site of the structure of PB2/CBD protein.
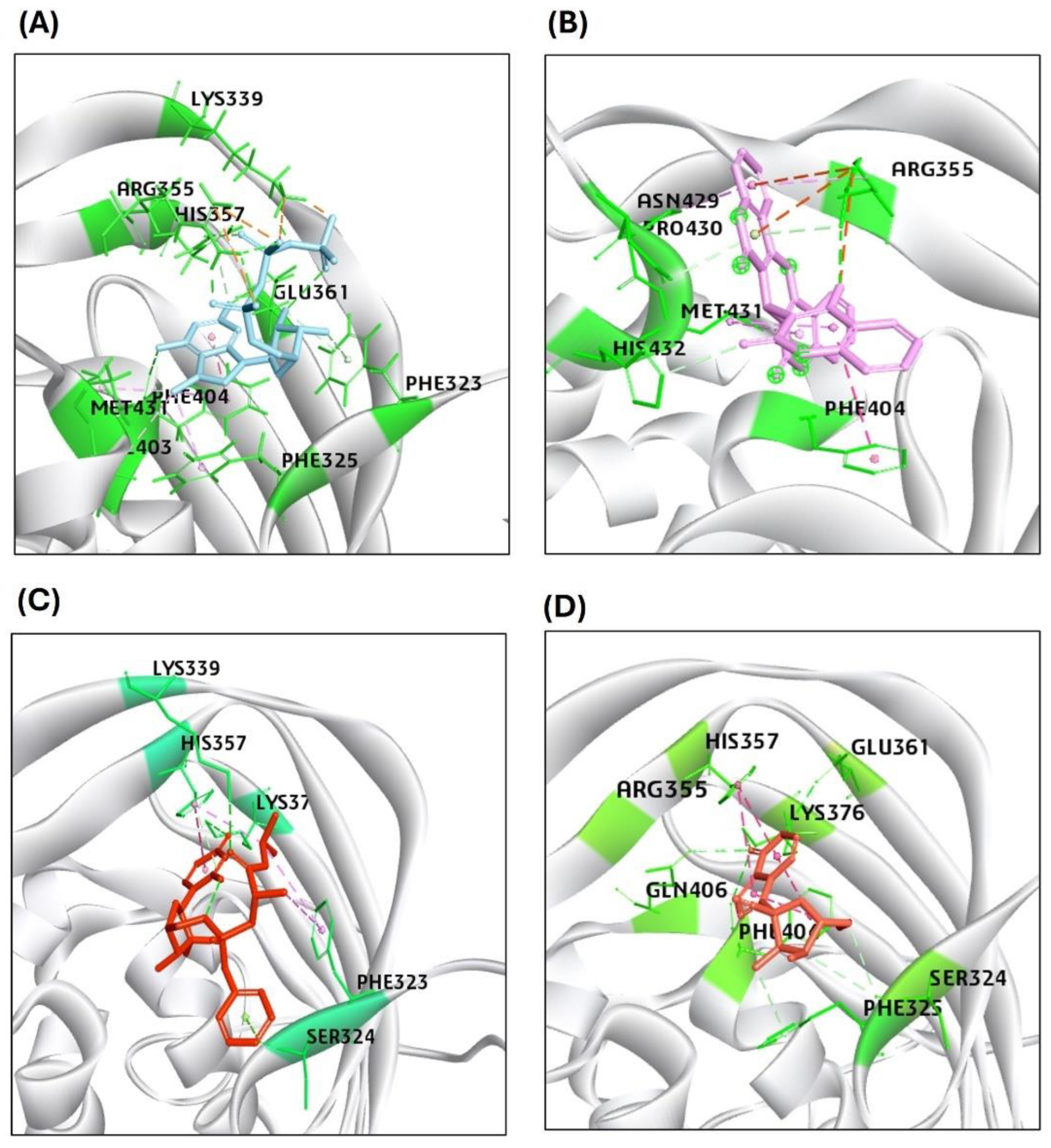
Figure 5.
Results of the molecular interaction model of the tested compounds and the binding sites of the H5N1 clade 2.3.4.4b M2 tetrameric channel protein. (A) The Rimantadine interacted with the M2 protein residues of Chain-1 through hydrogen bonds: (AlA30; Chain 2: VAL27, ALA30; Chain 3: ALA30; Chain 4: VAL27, ALA30). (B) The Amantadine interacted with the M2 protein residues of Chain-2 through hydrogen bonds: : (VAL27,ALA30); (Chain 3: ALA30); (Chain 4: ALA30, GLY34) amino acid residues of the M2 protein (C) The Sofosbuvir interacted through hydrogen bonds with M2-Chain 1: through (VAL27, AlA30, GLY34, HIS37); Chain 2 through: (ALA30, SER31, HIS37, LEU38); Chain 3 through: (VAL27, ALA30, HIS37); and Chain 4 through: (VAL27, ALA30, GLY34, HIS37) amino acid residues of the M2 protein (D) The GS441524 compound interacted with hydrogen bonds with the M2-Chain 1: (AlA30, GLY34); Chain 2: (SER22, VAL27, ALA30); Chain 3: (ALA30, and HIS37) amno acid residues of the M2 channel protein.
Figure 5.
Results of the molecular interaction model of the tested compounds and the binding sites of the H5N1 clade 2.3.4.4b M2 tetrameric channel protein. (A) The Rimantadine interacted with the M2 protein residues of Chain-1 through hydrogen bonds: (AlA30; Chain 2: VAL27, ALA30; Chain 3: ALA30; Chain 4: VAL27, ALA30). (B) The Amantadine interacted with the M2 protein residues of Chain-2 through hydrogen bonds: : (VAL27,ALA30); (Chain 3: ALA30); (Chain 4: ALA30, GLY34) amino acid residues of the M2 protein (C) The Sofosbuvir interacted through hydrogen bonds with M2-Chain 1: through (VAL27, AlA30, GLY34, HIS37); Chain 2 through: (ALA30, SER31, HIS37, LEU38); Chain 3 through: (VAL27, ALA30, HIS37); and Chain 4 through: (VAL27, ALA30, GLY34, HIS37) amino acid residues of the M2 protein (D) The GS441524 compound interacted with hydrogen bonds with the M2-Chain 1: (AlA30, GLY34); Chain 2: (SER22, VAL27, ALA30); Chain 3: (ALA30, and HIS37) amno acid residues of the M2 channel protein.
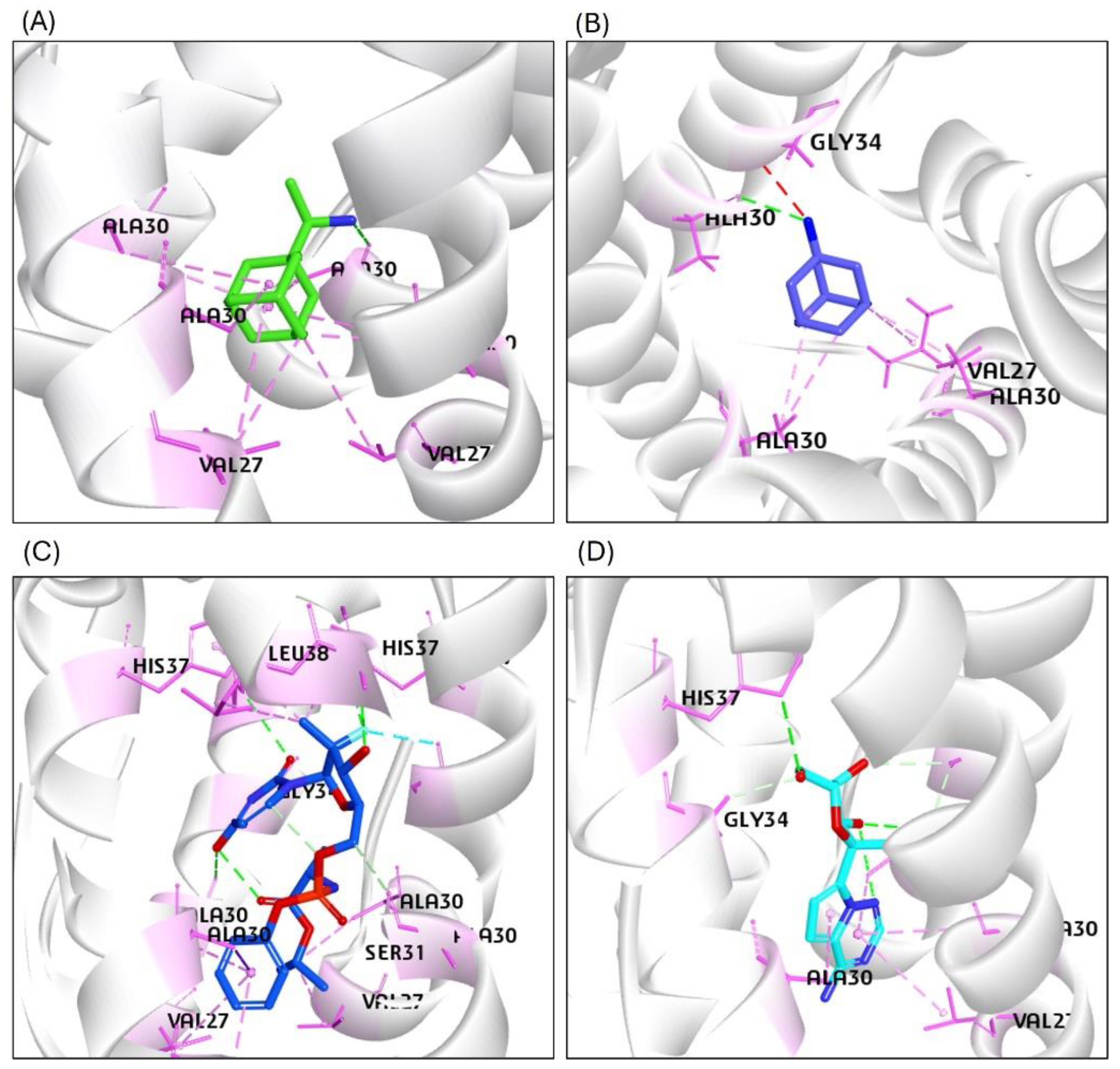
Figure 6.
The RMSD plots based on the 50 ns molecular dynamics simulation trajectories for the H5N1 clade 2.3.4.4b proteins/ligands complexes: (A) HA in complex with the reference compound F0045(S) and GS441524; (B) NA in complex with the standard inhibitor Zanamivir and GS441524; (C) PB2/CBD in complex with the reference ligand PB2-39 and GS441524.
Figure 6.
The RMSD plots based on the 50 ns molecular dynamics simulation trajectories for the H5N1 clade 2.3.4.4b proteins/ligands complexes: (A) HA in complex with the reference compound F0045(S) and GS441524; (B) NA in complex with the standard inhibitor Zanamivir and GS441524; (C) PB2/CBD in complex with the reference ligand PB2-39 and GS441524.
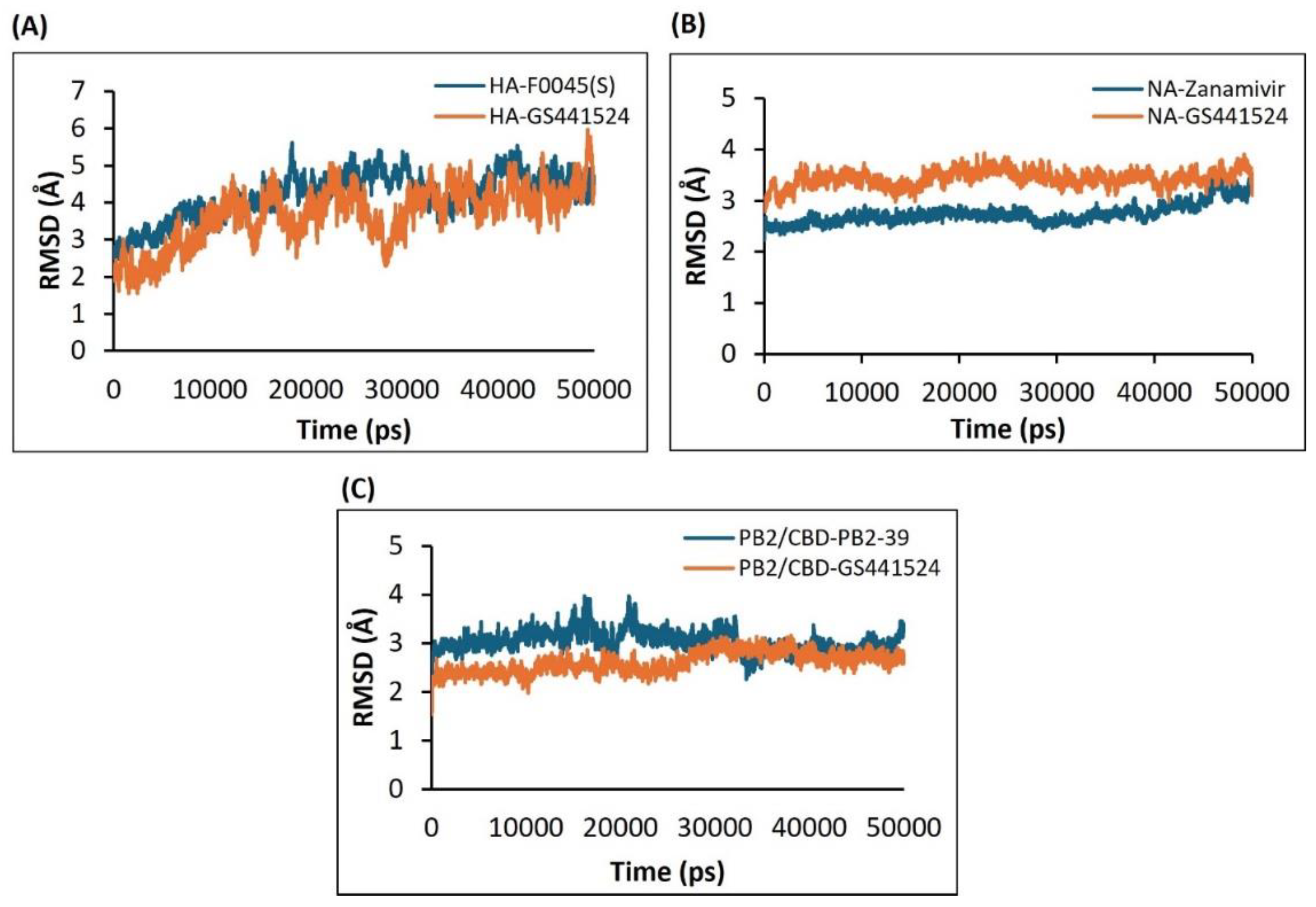
Figure 7.
RMSF plots generated from the 50 ns MD simulation trajectories for the following protein-ligand complexes: Models for the HA in complex with F0045(S) (reference compound) or GS441524. (B) Model for the NA in complex with Zanamivir (reference compound) or the GS441524 compound (C) Model for the PB2/CBD complex with the PB2-39 (reference compound) or the GS441524 compound.
Figure 7.
RMSF plots generated from the 50 ns MD simulation trajectories for the following protein-ligand complexes: Models for the HA in complex with F0045(S) (reference compound) or GS441524. (B) Model for the NA in complex with Zanamivir (reference compound) or the GS441524 compound (C) Model for the PB2/CBD complex with the PB2-39 (reference compound) or the GS441524 compound.
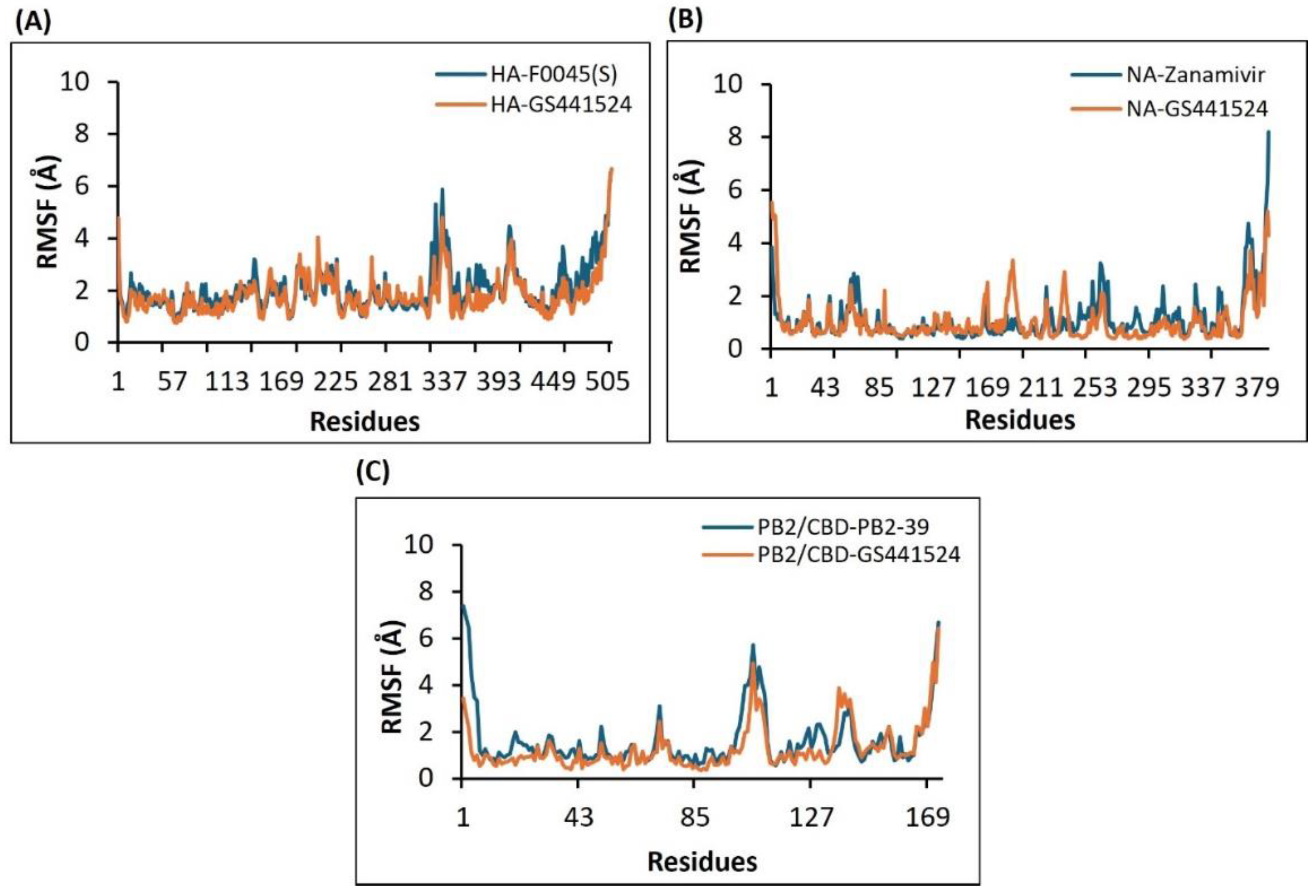
Figure 8.
The Radius of gyration (Rg) plots derived from the 50 ns molecular dynamics simulation trajectories for the following H5N1 clade 2.3.4.4b protein-ligand complexes: (A) HA-F0045(S) (reference standard) and HA-GS441524; (B) NA-Zanamivir (reference standard) and NA-GS441524; and (C) PB2/CBD-PB2-39 (reference standard) and PB2/CBD-GS441524.
Figure 8.
The Radius of gyration (Rg) plots derived from the 50 ns molecular dynamics simulation trajectories for the following H5N1 clade 2.3.4.4b protein-ligand complexes: (A) HA-F0045(S) (reference standard) and HA-GS441524; (B) NA-Zanamivir (reference standard) and NA-GS441524; and (C) PB2/CBD-PB2-39 (reference standard) and PB2/CBD-GS441524.
Figure 9.
Analysis of the number of hydrogen bonds formed during interactions between H5N1 clade 2.3.4.4b proteins and ligands, as illustrated in the plots derived from the 50 ns molecular dynamics (MD) simulation trajectory data. (A) Results of the HA-F0045(S) complex (B) Results of the HA-GS4415524 complex (C) Results of the NA-Zanamivir (reference standard) complex, (D) Results of the NA-GS441524 complex, (E) Results of the PB2/CBD-PB2-39 complex (reference standard) and (F) Results of the PB2/CBD-GS441524 complex.
Figure 9.
Analysis of the number of hydrogen bonds formed during interactions between H5N1 clade 2.3.4.4b proteins and ligands, as illustrated in the plots derived from the 50 ns molecular dynamics (MD) simulation trajectory data. (A) Results of the HA-F0045(S) complex (B) Results of the HA-GS4415524 complex (C) Results of the NA-Zanamivir (reference standard) complex, (D) Results of the NA-GS441524 complex, (E) Results of the PB2/CBD-PB2-39 complex (reference standard) and (F) Results of the PB2/CBD-GS441524 complex.
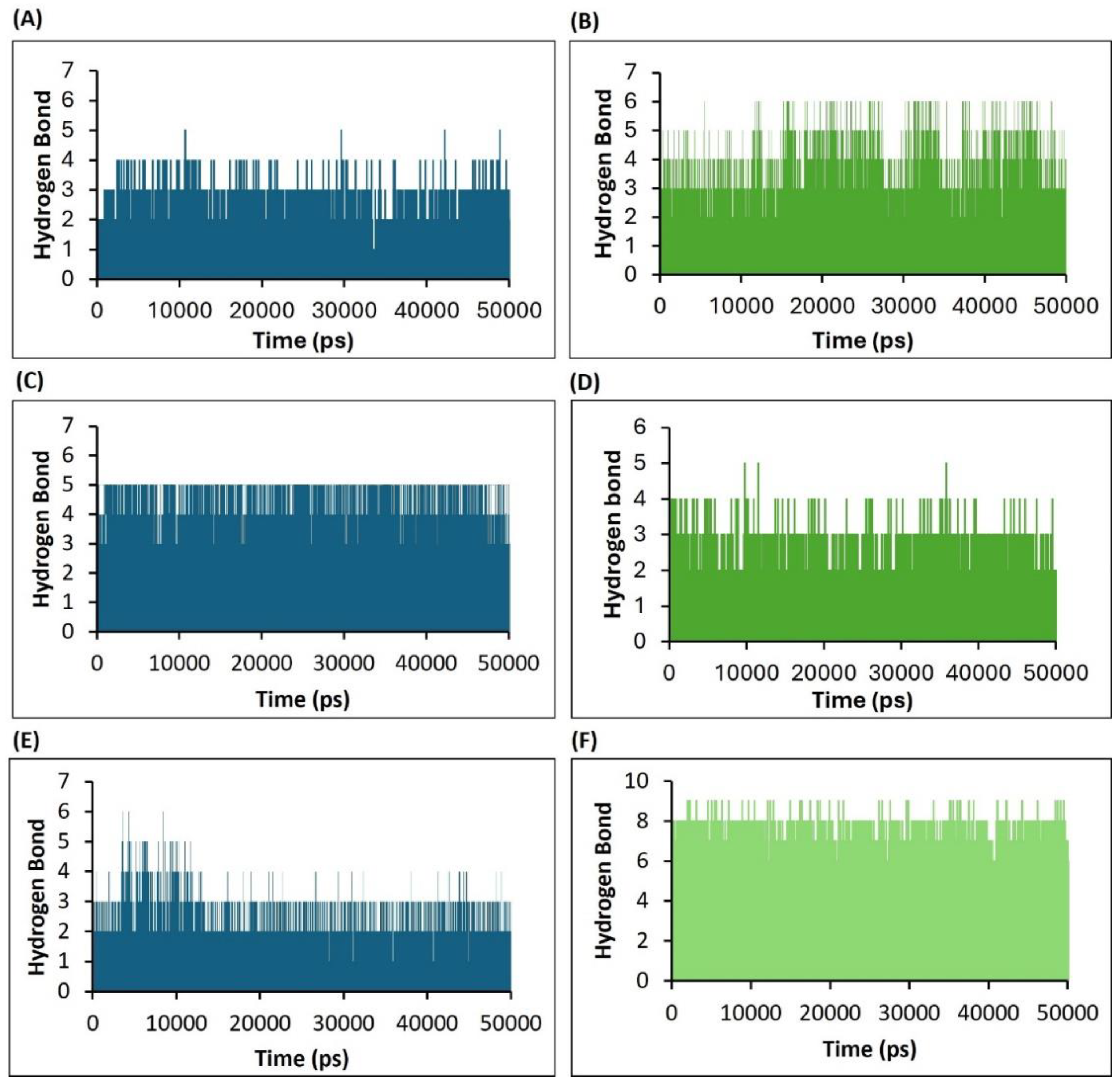
Figure 10.
Plots depicting the variation in hydrogen bond distances between ligands and interacting protein residues, based on the 50 ns MD simulation trajectories for the following complexes. (A) HA-F0045(S) (reference standard) (B) HA-GS4415524 (C) NA-Zanamivir (reference standard) (D) NA-GS441524 (E) PB2/CBD-PB2-39 (reference standard) and (F) PB2/CBD-GS441524.
Figure 10.
Plots depicting the variation in hydrogen bond distances between ligands and interacting protein residues, based on the 50 ns MD simulation trajectories for the following complexes. (A) HA-F0045(S) (reference standard) (B) HA-GS4415524 (C) NA-Zanamivir (reference standard) (D) NA-GS441524 (E) PB2/CBD-PB2-39 (reference standard) and (F) PB2/CBD-GS441524.
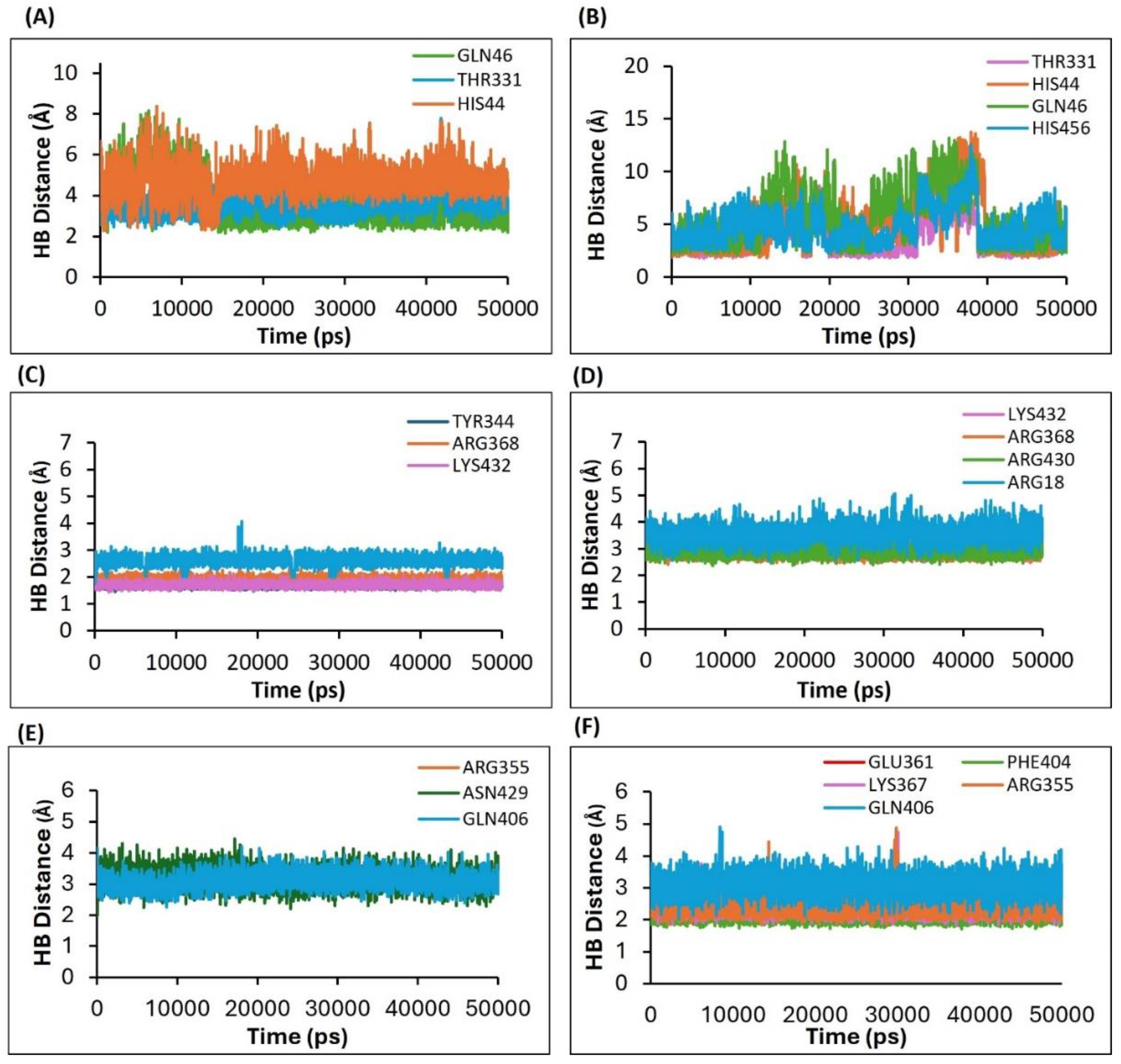

Table 1.
List of the predicted homology models of the major proteins of the H5N1 clade 2.3.4.4b and their verification parameters.
Table 1.
List of the predicted homology models of the major proteins of the H5N1 clade 2.3.4.4b and their verification parameters.
| N | Predicted Homology Model | PDF total energy | DOPE Score | Verify Score | Verify Expected High Score | Expected Low Score |
| 1 | HA | 26755 | -54478 | 205 | 232 | 104 |
| 2 | NA | 18118 | -43552 | 180 | 176 | 79 |
| 3 | PB2/CBD | 1926 | -79787 | 277 | 333 | 154 |
| 4 | M2 | 5702 | -10588 | 31 | 45 | 20 |
Table 2.
Summary of the binding affinity scores, the binding free energies, and the molecular interactions between the avian H5N1 Clade 2.2.4.4b (HA, NA, PB2/CBD, and M2) proteins and their respective ligands.
Table 2.
Summary of the binding affinity scores, the binding free energies, and the molecular interactions between the avian H5N1 Clade 2.2.4.4b (HA, NA, PB2/CBD, and M2) proteins and their respective ligands.
| N | Protein | Ligands/ Compound-ID |
MM-GBSA Binding energy |
-CDOCK Score (Binding affinity score) |
Interacting amino acid residues |
| 1 | HA | *F0045(S) (CID: 25281297) |
-48.864 | 22.9215 | HIS24, TRP366, ILE390, VAL393 |
| 2 | HA | GS441524 (CID: 44468216) |
-61.432 | 26.672 | ALA45, THR331, ILE390, VAL393, THR394 |
| 3 | PB2/CBD | *MGT (CID: 175837) |
-486.373 | 80.6777 | PHE323, PHE325, LYS339, ARG355, HIS357, GLU361, VAL403, PHE404, MET431 |
| 4 | PB2/CBD | *PB2-39 (CID: 54694510) |
-236.799 | 54.5527 | PHE404, ASN429, PRO430, HIS432, ARG355 |
| 5 | PB2/CBD | Sofosbuvir (CID: 45375808) |
-110.053 | 50.8859 | PHE323, SER324, LYS339, HIS357, LYS367 |
| 6 | PB2/CBD | GS441524 (CID: 44468216) |
-99.0791 | 34.2346 | ARG324, ARG355, HIS357, PHE404, GLN406, GLU361, LYS376 |
| 7 | NA | *Zanamivir (CID: 60855) |
-201 | 49.26 | ASP151, TYR344, SER367, ARG368, LYS432 |
| 8 | NA | *Oseltamivir (CID: 65028) |
28 | -65.74 | SER101, ASP103, ILE117, ILE122, THR131, PRO162, VAL163, GLU165, PRO167, SER441 |
| 9 | NA | GS441524 (CID: 44468216) |
-83.10 | 32.34 | VAL149, ASP151, SER367, ARG368, PRO431, LYS432 |
| 10 | NA | Sofosbuvir (CID: 45375808) |
-138.23 | 53.43 | VAL149, ASP151, ARG225, GLU278, TYR344, ARG368, TYR402, ILE427, PRO431, LYS432 |
| 11 | M2 | Rimantadine (CID: 5071) |
-112.23 | 33.02 | Chain 1: AlA30; Chain 2: VAL27,ALA30 Chain 3: ALA30; Chain 4: VAL27, ALA30 |
| 12 | M2 | Amantadine (CID: 5071) |
-104.46 | 27.55 | Chain 2: VAL27,ALA30; Chain 3: ALA30; Chain 4: ALA30, GLY34 |
| 13 | M2 | Sofosbuvir (CID: 45375808) |
-116.11 | 62.05 | Chain 1: VAL27, AlA30, GLY34, HIS37; Chain 2: ALA30, SER31, HIS37, LEU38 Chain 3: VAL27, ALA30, HIS37 Chain 4: VAL27, ALA30, GLY34, HIS37 |
| 14 | M2 | GS441524 (CID: 44468216) |
-120.65 | 42.59 | Chain 1: AlA30, GLY34; Chain 2: SER22, VAL27, ALA30 Chain 3: ALA30, HIS37 |
* Represents the reference standard ligands for HA, NA, and PB2. CDOCK Score refers to the CDOCKER interaction energy.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



