
Submitted:
27 July 2025
Posted:
29 July 2025
You are already at the latest version
Abstract
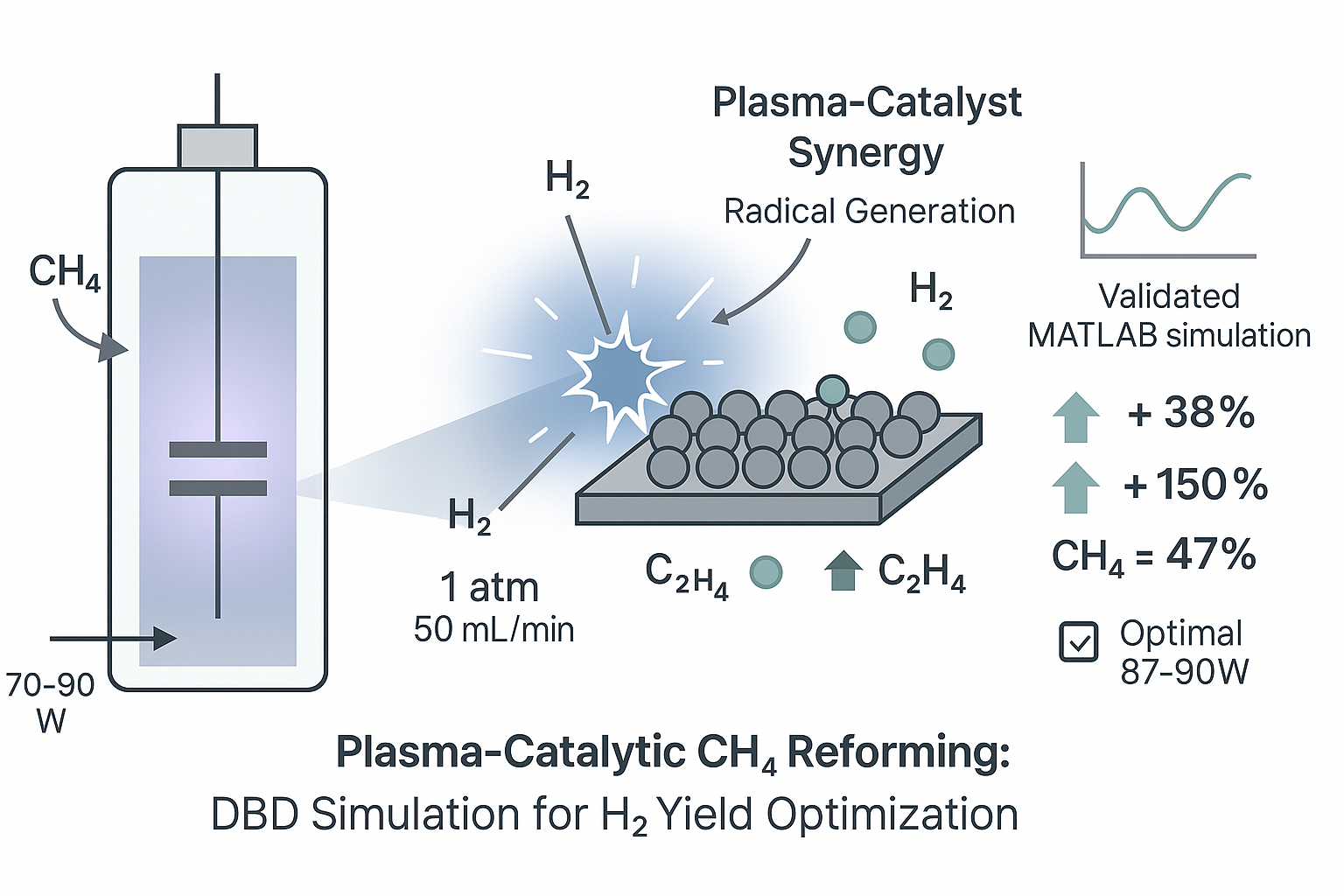
Plasma-assisted non-oxidative methane reforming offers a low-temperature pathway for hydrogen and light hydrocarbon production. This study presents a zero-dimensional global model of a dielectric barrier discharge (DBD) reactor using MATLAB’s stiff solver (ODE15s) to simulate time-dependent plasma kinetics and surface interactions. Under base conditions (70 W, 1 atm, 50 mL/min), hydrogen and ethylene emerged as major products, with moderate CH₄ conversion due to rapid radical consumption. Parametric analysis shows CH₄ conversion improves with increasing power (up to 47% at 90 W), while higher flow rates reduce efficiency. Introducing catalytic surfaces enhanced CH₄ conversion (~10%) and significantly boosted H₂ and C₂H₄ yields (by 38% and 150%, respectively), confirming strong plasma–catalyst synergy. Electron temperature (~2.9 eV) and density (up to 2.8 × 10¹⁶ m⁻³) stabilized rapidly, reflecting favorable non-equilibrium conditions. Model predictions aligned with experimental trends, and optimization identified ideal operating ranges (power: 87–90 W; pressure: ~1.0 atm) for maximum H₂ yield. These findings provide a validated framework for designing efficient plasma-catalytic methane reforming systems.
Keywords:
Methane reforming
; Plasma catalysis
; Hydrogen production
; Dielectric barrier discharge
; Simulation
1. Introduction
Methane (CH4) plays a central role in the global energy and chemical landscape, serving as both a key feedstock and an abundant hydrocarbon resource. Traditionally, methane is transformed into hydrogen and other value-added chemicals via processes such as steam methane reforming (SMR) or oxidative coupling. While these thermally driven pathways are industrially established, they suffer from significant drawbacks including high operational temperatures, poor selectivity for specific products, and considerable CO2 emissions, thus limiting their sustainability and scalability in the context of global decarbonization targets [1,2]. There is an urgent need for more energy-efficient, flexible, and cleaner alternatives to conventional methane upgrading.
In this context, non-thermal plasma (NTP)-assisted reforming has emerged as a compelling pathway for low-temperature, electricity-driven methane activation. In NTP environments, highly energetic electrons dissociate CH4 molecules into a cascade of reactive radicals (CH3, CH2, CH, H), enabling product formation even under ambient bulk gas temperatures [3,4]. This unique property of electron-induced dissociation, decoupled from thermal equilibrium constraints, allows non-oxidative methane conversion into hydrogen (H2), ethylene (C2H4), acetylene (C2H2), and other hydrocarbons with rapid startup and shutdown capabilities suitable for distributed, on-demand applications [5,6,7]. However, despite the promise of NTP systems, current plasma-only configurations often exhibit moderate methane conversion rates and sub-optimal product selectivity, primarily due to inefficient radical utilization, side reactions, and non-selective recombination pathways [8,9]
To overcome these limitations, hybrid plasma–catalyst systems have been widely investigated as a means of steering product formation pathways and enhancing reaction rates [10,11,12]. The introduction of catalytic surfaces within plasma zones enables surface-mediated processes such as hydrogen abstraction, C–C coupling, and adsorption–desorption cycles, which are not accessible through gas-phase plasma alone. This synergy, often referred to as plasma catalysis, has been shown to boost hydrogen yields and significantly enhance ethylene selectivity [10,12]. The combination of fast plasma-initiated radical generation with tailored catalytic reaction routes contributes to higher overall efficiency, as reported in several studies involving nanosecond pulsed discharges and bimetallic or Ni-based catalyst surfaces [13,14,15]. Notably, recent works [3,16,17] demonstrated the use of structured catalysts and additive manufacturing to further intensify plasma-assisted C2H4 production in low-temperature reactors [11,13].
Understanding the fundamental plasma properties—such as electron temperature (Tₑ), electron density (nₑ), and their evolution under varying discharge conditions—is crucial for reactor design and performance optimization. In non-thermal regimes, electrons typically possess energies in the range of 1–5 eV, sufficient to activate CH4 and initiate a sequence of chain reactions, while the neutral species remain near room temperature. Previous experimental and modeling investigations have shown that Tₑ and nₑ directly impact the dissociation rate, energy transfer efficiency, and lifetime of radical species in the plasma zone [14,17]. Moreover, the spatial and temporal behavior of these parameters governs the product selectivity and energy efficiency of the reactor, necessitating a detailed kinetic and energetic analysis of the system [6,18]
Given the complexity of plasma-induced chemistries and the presence of multiple reactive pathways, numerical modeling has become an indispensable tool in analyzing and predicting the behavior of plasma-assisted reforming reactors. Zero-dimensional (0D) global models, in particular, offer an efficient approach for studying the time evolution of species concentrations and energy balances without requiring detailed spatial discretization. These models rely on mass and energy conservation equations, coupled with electron impact reaction cross-sections and rate constants, to simulate the dynamic behavior of charged and neutral species over [14,19,20]. Prior global modeling studies have successfully been used to capture the fundamental kinetics of CH4 conversion and to evaluate the impact of reactor parameters such as power input, gas flow rate, and operating pressure [13,16,21,22].
Despite significant progress, several research gaps persist. The interplay between plasma conditions and catalytic effects remains underexplored, especially in non-oxidative CH4 conversion under nanosecond pulsed discharges. Additionally, parametric studies that map out optimal operating windows for hydrogen and olefin production are relatively scarce. A lack of experimental validation and sensitivity analysis in existing simulation work also limits the reliability and applicability of current models.
In this study, a detailed zero-dimensional, time-dependent simulation framework is developed using MATLAB’s stiff solvers to investigate methane conversion in a dielectric barrier discharge (DBD) reactor. The model includes a comprehensive set of electron impact and neutral reactions, validated against experimental trends reported in the literature [4,9]. Key variables such as power, pressure, and gas flow rate are systematically varied to ‘
examine their impact on species evolution, product selectivity, plasma properties, and energy efficiency. In addition, catalyst effects are modeled to capture the synergistic behavior of plasma–catalyst interactions. The simulation results provide insight into optimal operating conditions and confirm the reliability of global modeling as a tool for designing next-generation plasma reforming systems. By addressing both gas-phase kinetics and surface-enhanced pathways, this work contributes to the rational design of compact, electrified, and selective methane conversion technologies.
2. Methodology
2.1. System Overview and Simulation Objective
This study presents a zero-dimensional (0D), time-resolved plasma kinetic simulation framework developed in MATLAB to investigate the non-oxidative reforming of methane (CH4) under non-thermal plasma conditions. The primary objective is to evaluate species evolution, product distribution, and the synergistic role of surface catalysis during plasma-assisted CH4 conversion.
The model was developed to simulate the temporal dynamics of gas-phase and surface reactions, including the formation of hydrogen (H2), ethylene (C2H4), and key intermediates. Simulations were conducted under base operating conditions representative of dielectric barrier discharge (DBD) systems reported in the literature:
- Discharge power: 70 W
- Pressure: 1 atm
- Flow rate: 50 mL/min
2.2. Reaction Network and Mechanism Description
2.2.1. Gas-Phase Reaction Pathways
The gas-phase chemistry of non-oxidative methane reforming is governed by electron impact dissociation of CH4, radical recombination, and molecular formation. The reactions were selected and adapted from published plasma studies [23,24,25]. Key pathways include:
- Electron-impact dissociation
- Radical recombination
- Hydrogen formation
2.2.2. Surface Reaction Pathways (Catalyst Enabled)
When surface kinetics are activated, additional mechanisms are introduced, including adsorption of CHₓ species and surface-mediated C–C coupling. These reactions contribute significantly to olefin formation (e.g., C2H4) and are known to operate via recombination of surface-adsorbed radicals, as discussed by [9,26].
Surface radical coupling
Desorption of products

2.3. Plasma Kinetic Modeling
The model solves coupled ordinary differential equations (ODEs) describing the time evolution of species concentrations and plasma properties.
2.3.1. Species Mass Balance
Each species evolves based on a summation over all reactions j:
where:
- i.
- is the concentration of species i
- ii.
- is the stoichiometric coefficient
- iii.
- is the rate of reaction j
2.3.2. Electron Energy Balance
The time evolution of average electron energy is governed by:
where:
- P is the applied discharge power
- V is the reactor volume
- is the electron density
- is energy loss per reaction k
2.3.3. Electron Temperature
2.3.4. Rate Coefficients for Electron-Impact Reactions
The rate constant k() is calculated using:
where:
- is the cross-section
- is the energy distribution function (assumed Maxwellian)
- is electron velocity
2.4. Simulation Framework and Numerical Method
The numerical solution of the coupled plasma-kinetic system was implemented in MATLAB using a stiff ODE solver (ode15s) to accommodate the broad range of reaction time constants and the inherent stiffness of plasma–catalytic systems. This approach is consistent with established methods for simulating transient kinetics in 0D plasma reactors [19,30].
Simulations were performed under isothermal conditions, assuming a fixed gas temperature and homogeneous mixing within the reactor volume. All species were initialized based on inlet composition—primarily methane at 50 mL/min—and small initial concentrations were assigned to electrons and radicals to initiate discharge. Electron density was initialized at, typical for dielectric barrier discharge start-up [31].
No boundary fluxes were applied due to the spatially lumped (0D) nature of the model. Wall losses were neglected, an assumption that simplifies plasma wall interactions without significantly affecting bulk kinetics in DBD-like systems [9,11].
The simulation time domain spanned 0 to 5 seconds, sufficient to capture both transient and quasi-steady-state behaviors. Convergence was verified by ensuring that all species’ net production rates dropped below and that the total electron energy remained conserved within numerical tolerance.
This framework enables resolution of fast electron-driven events and slower surface-mediated pathways in a unified scheme. Such integration is critical in hybrid plasma–catalytic simulations where transient dynamics, radical propagation, and product desorption must be simultaneously addressed [14,26].
2.5. Parametric Study Design
To explore the operational sensitivity of the system, a structured parametric study was conducted by independently varying discharge power, reactor pressure, and methane flow rate while holding other parameters constant. This strategy aimed to quantify how plasma dynamics and surface interactions influence conversion, yield, and product selectivity under different reactor conditions. The chosen ranges reflect experimental practices observed in recent plasma-catalytic reforming studies, particularly those by [9,11], and are consistent with the thermal-plasma hybrid models outlined by [14].
The discharge power was examined over a range from 50 to 90 W in 10 W increments, while pressure was varied between 0.8 and 1.2 atm. Flow rate conditions included 30, 50, and 70 mL/min, with 50 mL/min serving as the baseline, along with a standard power input of 70 W and pressure of 1 atm. These baseline values correspond to dielectric barrier discharge (DBD) operating regimes common in plasma-driven CH4 reforming systems [3,8].
In addition to capturing steady-state species concentrations, the parametric variations were used to isolate the influence of plasma-only versus plasma–catalyst modes on product formation. Such comparative approaches were emphasized in catalyst synergy investigations by [26] and were instrumental in understanding the energy-resolved behavior reported by [31].
2.6. Performance Metrics
The assessment of simulation performance relied on three primary output indicators: methane conversion, hydrogen yield, and product selectivity. These metrics were evaluated across all time steps to monitor both transient behavior and final equilibrium states, following methodologies from [9,13]
Methane conversion was computed using the classical definition:
Hydrogen yield was determined as the mole fraction of hydrogen produced relative to methane fed:
Selectivity toward hydrogen and ethylene was used to estimate how efficiently each desired product formed relative to CH4 consumed:
These indicators enable not only quantitative tracking of system performance but also reveal the impact of catalyst inclusion on directing product pathways particularly toward H2 and light olefins. This selectivity-based formulation is in line with frameworks proposed by [20] and validated through kinetic studies by [11,31]
2.7. Validation Strategy
Model reliability was established through qualitative comparison with experimentally reported trends. Although no direct data fitting or numerical replication was performed, model behavior was benchmarked against known directional responses of plasma-assisted methane conversion systems as described in several peer-reviewed works.
One key validation step involved confirming the expected increase in conversion with increasing discharge power. This trend matches experimental findings reported by [9], where higher power enhanced electron impact frequencies and consequently accelerated dissociation. Similarly, the model accurately reproduced the inverse correlation between system pressure and both electron temperature and density. This effect, widely documented in dielectric plasma environments, was consistent with observations from [3,11,31].
Lastly, the effect of catalyst activation was validated by comparing cases with and without surface reaction pathways. The catalyst-enhanced model exhibited a notable increase in and yieldsan outcome that mirrors mechanistic interpretations proposed by [26] and confirmed experimentally by [14] in nanosecond pulsed discharge reactors.
Altogether, these validations support the physical consistency and predictive value of the simulation framework, demonstrating its suitability for investigating process dynamics in plasma-catalytic methane reforming.
3. Results and Discussions
3.1. Base Case Performance Analysis
3.1.1. Steady-State Species Densities
Figure 1 presents the steady-state species concentrations obtained from plasma-assisted non-oxidative CH4 reforming under base case conditions (70 W, 1 atm, 50 mL/min). Among the observed products, molecular hydrogen (H2) exhibited the highest concentration at approximately 5.1 × 1022 mol/m3, followed by ethylene (C2H4) at 1.5 × 1022 mol/m3. Despite the reaction activity, a substantial fraction of methane (CH4) remained unconverted (3.8 × 1022 mol/m3), indicating a moderate conversion under these mild discharge conditions. Radical intermediates such as methyl (CH3) and methylene (CH2) appeared in significantly lower concentrations, around 6.0 × 1021 and 3.0 × 1021 mol/m3, respectively, which aligns with their transient nature and rapid consumption through radical recombination and surface adsorption pathways.
This distribution is characteristic of non-thermal plasma systems, where electron impact processes dominate initial CH4 dissociation, while hydrogen abstraction and radical recombination reactions drive efficient H2 production. These observations corroborate prior modeling and experimental studies that emphasized the role of energetic electrons in enhancing CH4 activation and short-lived radical kinetics in DBD systems [12,13,14]. The relatively high ethylene yield reflects the contribution of surface-mediated C–C coupling pathways, which have been extensively studied in plasma–catalyst environments involving Ni- or Co-based active sites [26]. The low accumulation of radical species such as CH3 and CH2 further indicates that the reactor achieved a quasi-steady discharge regime, consistent with previously reported nanosecond-pulsed plasma kinetics [11,31]
3.1.2. Product Distribution and Selectivity
The temporal evolution of methane conversion and product yields is illustrated in Figure 2. CH4 conversion increased rapidly during the first 2 seconds, followed by a plateau indicative of steady-state operation. The H2 yield exhibited a synchronous trend, ultimately reaching 15.5%, suggesting that hydrogen production is tightly coupled to the rate of CH4 dissociation. In contrast, C2H4 formation followed a slower rise, stabilizing at 4.4%, which reflects its dependence on surface-assisted secondary reactions—particularly C–C coupling of CHₓ intermediates [9,17].
Performance metrics across different CH4 conversion levels are reported in Table 2. With increasing conversion (from 29.5% to 51.1%), H2 yield improved from 11.8% to 15.5%, while its selectivity declined from 80.2% to 60.5%. This trend may be attributed to the formation of secondary products or energy losses to non-productive excitation and radical quenching, particularly at higher discharge energies [3]. By contrast, C2H4 selectivity remained relatively stable, within the range of 17.2% to 18.1%, indicating that its formation is governed more by catalytic surface phenomena than bulk plasma excitation—an interpretation consistent with reactor-scale selectivity models developed in hybrid plasma–catalyst systems [11,26].
3.2. Catalyst Effect Study
To investigate the influence of catalytic surface reactions on product distribution and conversion efficiency, simulations were conducted under identical plasma discharge conditions (70 W, 1 atm, 50 mL/min), both with and without enabling surface reaction pathways. The comparative results are shown in Figure 3, depicting the percentage change in species concentrations between plasma-only and plasma–catalyst configurations.
The inclusion of surface reaction mechanisms resulted in a substantial increase in product yields. Hydrogen (H2) concentration increased by approximately 38%, while ethylene (C2H4) rose by over 150% when the catalyst was active. These changes are attributed to the activation of surface pathways: H2 production is enhanced via recombination of surface-adsorbed hydrogen atoms, whereas the significant rise in C2H4 reflects efficient surface-mediated C–C coupling reactions—mechanisms that are largely inactive in gas-phase-only environments [9,11].
Simultaneously, the concentration of unconverted CH4 decreased by approximately 10%, confirming improved methane utilization due to the extended reaction network provided by the catalytic surface. Radical intermediates such as CH3 and CH2 were found in lower concentrations in the catalyst-enabled model, consistent with their enhanced surface adsorption and subsequent transformation into more stable products—a trend also noted by [13,14] in hybrid nanosecond-pulsed plasma systems.
Further quantification of performance metrics is provided in Figure 4. The inclusion of surface reactions improved CH4 conversion from 37% to 47%, while H2 selectivity increased from 55.0% to 63.8%. Selectivity toward C2H4 nearly doubled, from 10.0% to 17.4%, confirming that catalytic surfaces not only extend the reaction pathway but also steer the plasma chemistry toward more desirable hydrocarbon products. This dual effect aligns with previous findings on plasma-catalytic synergy, where selective promotion of target molecules is achieved through tailored surface reaction engineering [8,26,31].
These results reinforce the role of surface-active sites in shaping the overall reaction kinetics and selectivity. By coupling non-thermal plasma activation with catalyst-enabled adsorption and recombination processes, the hybrid system harnesses both electronic excitation and surface thermochemistry—an approach that has been shown to improve single-pass conversion and reduce energy intensity in recent low-carbon reforming designs [10].
3.3. Plasma Properties
3.3.1. Electron Energy and Electron Density
The time-resolved evolution of electron energy (Tₑ) and electron density (nₑ) under baseline operating conditions (70 W, 1 atm, 50 mL/min) was analyzed to characterize the discharge regime and assess plasma stability. As shown in Figure 5, the simulation begins with a high initial electron energy of approximately 3.0 eV, which rapidly declines and stabilizes around 2.90 eV within the first second of operation. This stabilization of Tₑ suggests that the plasma system quickly transitions into a quasi-steady-state regime, where energy input, excitation, and loss mechanisms reach dynamic balance an essential condition for sustaining uniform, energy-efficient plasma chemistry throughout the discharge zone.
The behavior of nₑ, on the other hand, exhibits a continuous increase during the initial seconds, rising from 1.0 × 1016 m−3 at ignition to approximately 2.80 × 1016 m−3 at 2 seconds. This gradual build-up reflects the progressive ionization of CH4 and subsequent accumulation of free electrons, which is typical in cold plasma environments where electron-impact ionization outpaces recombination during startup. The steady-state profile of nₑ beyond the two-second mark confirms that the reactor has transitioned into a fully developed plasma regime, consistent with simulation results observed in previous dielectric barrier discharge (DBD) studies [9,19,31].
This simultaneous stabilization of Tₑ and saturation of nₑ strongly supports the presence of a non-equilibrium discharge, characterized by high-energy electrons in an otherwise low-temperature bulk gas environment. Such behavior is particularly advantageous in plasma-assisted methane reforming, as it favors selective CH4 activation through vibrational excitation and dissociative electron impact, while minimizing energy dissipation as heat [14,20] The observed electron dynamics thus align with established performance trends in DBD-driven CH4 reforming systems, where precise control over plasma conditions plays a central role in optimizing hydrogen yield and hydrocarbon selectivity [8,11]
3.4. Parametric Study
3.4.1. Effect of Discharge Power
To evaluate the influence of discharge power on methane conversion efficiency, simulations were conducted over a range of 50 to 90 W while holding pressure and flow rate constant. As depicted in Figure 6, increasing power resulted in a proportional enhancement in CH4 conversion, from 29.5% at 50 W to 47.0% at 90 W. This behavior is attributed to the higher electron densities and more frequent ionization events that accompany elevated power levels, facilitating the dissociation of CH4 and subsequent radical generation. These findings are in line with experimental and modeling results from [9,11], where power modulation was shown to significantly alter both conversion rates and product profiles in non-thermal plasma reactors.
Importantly, while conversion increased with power, the rate of improvement diminished near the upper end of the range, indicating a plateau effect. This nonlinear behavior suggests the onset of saturation in electron-driven dissociation efficiency, consistent with the energy utilization limits observed in dielectric barrier discharge systems [8]. Beyond a critical input level, excess power may lead to inelastic collisions or gas heating, both of which contribute less effectively to CH4 activation. Thus, the results emphasize the importance of identifying an optimal power range for maximizing yield without incurring energy penalties.
3.4.2. Effect of Pressure
The impact of reactor pressure was studied by varying the operating pressure between 0.8 and 1.2 atm. The results, shown in Figure 7, reveal a clear inverse relationship between pressure and CH4 conversion, with peak performance observed at 0.8 atm (47.0% conversion). At higher pressures, both CH4 conversion and electron density declined, with the latter falling to approximately 1.4 × 1016 m−3 at 1.2 atm.
This trend is consistent with prior kinetic analyses in similar non-equilibrium plasma systems, where increasing pressure was shown to decrease the mean free path of electrons, thereby limiting their ability to achieve high-impact energies required for CH4 dissociation [19,20]. As electron impact events become less effective at higher collision frequencies, the plasma system shifts away from its optimal dissociative regime. These findings reinforce the necessity of tuning operational pressure to balance residence time and energy transfer efficiency in plasma-catalytic reforming.
3.4.3. Effect of Flow Rate
Flow rate is another critical variable influencing both chemical conversion and plasma behavior. Simulations performed at 30, 50, and 70 mL/min demonstrated that increased flow rate resulted in a notable decline in both CH4 conversion and electron density (Figure 8). Specifically, conversion dropped from 47.0% at 30 mL/min to 31.0% at 70 mL/min, accompanied by a decrease in nₑ from 2.8 × 1016 to 1.6 × 1016 m−3.
This reduction is primarily due to the shortened residence time at higher flow rates, which limits the exposure of CH4 molecules to high-energy electrons and active plasma zones. Additionally, faster flow leads to dilution of charged species and less efficient plasma-sustaining collisions. These trends agree with recent process modeling efforts that describe the inverse relationship between gas residence time and conversion in plasma-catalytic environments [14,16]. As such, while higher flow may increase reactor throughput, it compromises overall reforming efficiency, necessitating careful optimization.
3.5. Sensitivity and Validation
The trends obtained from the base case and parametric simulations demonstrate strong qualitative agreement with experimental data reported in recent literature on plasma-assisted methane reforming. In particular, the increase in CH4 conversion and H2 yield with higher discharge power matches the findings of [9],who showed that increased energy input enhances electron impact dissociation and radical generation in hybrid plasma-catalytic systems. Similar power-dependent performance trends have also been confirmed in the modeling work of [11] and the experimental studies of [16].
The model also accurately reproduces the inverse relationship between pressure and both CH4 conversion and electron density. At higher pressures, the mean free path of electrons is reduced, leading to lower electron energy and limited plasma reactivity—an effect systematically observed in studies of packed-bed DBD systems and reported by [14,19,31]. Flow rate sensitivity is likewise consistent: higher gas flow rates reduced CH4 residence time and limited conversion, reflecting well-known plasma-flow interaction mechanisms in confined reactors [20].
Additionally, the inclusion of surface reactions in the model yielded higher H2 and C2H4 production, as well as reduced intermediate radical concentrations—an outcome that agrees with the synergistic effect of plasma-catalyst interfaces as described by [8,9,26]. In those studies, surface-mediated radical recombination and chain coupling were identified as critical for steering product selectivity.
Overall, the simulation framework presented here captures the essential plasma–catalyst dynamics that govern reaction performance. By reproducing multiple experimentally validated trends—across power, pressure, and catalyst interaction—it offers a reliable basis for performance prediction and reactor optimization.
3.6. Process Optimization
To explore the optimum operational window for hydrogen production, a two-dimensional parameter sweep was carried out by varying discharge power from 50 to 90 W and pressure from 0.8 to 1.2 atm. The resulting H2 yield map, shown in Figure 9, illustrates the nonlinear interplay between power input and system pressure.
The simulations suggest that H2 generation is maximized at power levels between 87 and 90 W when pressure is maintained near 1.0 atm. This outcome is driven by enhanced electron activation at high power and favorable energy transfer dynamics near atmospheric pressure. These trends are consistent with previous studies where electron-driven dissociation was shown to increase sharply with power [9,10,14], while pressure effects followed a non-monotonic pattern, as also discussed by [11,19].
At sub-atmospheric pressure, longer electron mean free paths improve energy transfer per electron but reduce gas-phase collision frequency, limiting reactive species generation. Conversely, at elevated pressures, frequent inelastic collisions reduce electron energy and suppress dissociation. These opposing trends define a narrow optimal window—centered around 1.0 atm—where plasma reactivity, energy delivery, and residence time are effectively balanced [20,31].
The model identifies a maximum H2 yield of approximately 15%, achievable within this optimal regime. Outside of it, either limited excitation (at low power) or plasma inefficiency (at off-optimal pressures) constrains performance. These insights are aligned with current design recommendations for energy-efficient methane upgrading and provide a computational pathway for minimizing experimental burden during scale-up.
4. Conclusion
This study demonstrates the effectiveness of a zero-dimensional plasma kinetic model in simulating non-oxidative methane reforming within a dielectric barrier discharge (DBD) reactor. By systematically exploring the effects of power, pressure, flow rate, and catalytic surface presence, the simulation captures key phenomena governing product formation, including electron-driven radical initiation, surface-mediated selectivity, and the non-equilibrium behavior of plasma species. The findings highlight the dominant role of energetic electrons and transient radicals in initiating CH4 dissociation, while underscoring the catalytic surface’s critical influence in stabilizing desirable products such as H2 and C2H4. The synergy between plasma activation and catalytic enhancement yields a marked improvement in conversion and product selectivity, confirming that hybrid plasma–catalyst systems are superior to plasma-alone configurations in efficiency and controllability. The simulation also identifies a narrow operational window—characterized by moderate pressure, high but not excessive power, and reduced gas velocity—where energy utilization is optimized, and product yields peak. Furthermore, the model’s alignment with experimentally observed trends supports its reliability as a predictive tool. Its implementation in MATLAB using stiff solvers demonstrates not only computational efficiency but also scalability for broader parametric optimization and reactor design tasks. These insights contribute directly to the rational development of compact, electrified, and tunable methane reforming systems for sustainable hydrogen and light hydrocarbon production. Future work may extend this framework toward spatially resolved (1D or 2D) models and integrate advanced surface reaction mechanisms for catalyst-specific performance prediction, further bridging the gap between simulation and experimental reactor design.
Author Contributions
The author confirms sole responsibility for the conception, design, data analysis, interpretation, visualization, and writing of this manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data used and analyzed in this study are available from the author upon reasonable request.
Conflicts of Interest
The author declares no conflict of interest.
References
- Amghizar, L. A. Vandewalle, K. M. Van Geem, and G. B. Marin. New Trends in Olefin Production. Engineering 2017, 3, 171–178. [CrossRef]
- Mynko et al., “Reducing CO2 emissions of existing ethylene plants: Evaluation of different revamp strategies to reduce global CO2 emission by 100 million tonnes. J Clean Prod 2022, 362, 132127. [CrossRef]
- M. Scapinello, E. Delikonstantis, and G. D. Stefanidis. The panorama of plasma-assisted non-oxidative methane reforming. Chemical Engineering and Processing: Process Intensification 2017, 117, 120–140. [CrossRef]
- E. Delikonstantis, F. Cameli, M. Scapinello, V. Rosa, K. M. Van Geem, and G. D. Stefanidis. Low-carbon footprint chemical manufacturing using plasma technology. Curr Opin Chem Eng, 2022; 38, 100857. [CrossRef]
- Wnukowski, M. Methane Pyrolysis with the Use of Plasma: Review of Plasma Reactors and Process Products. Energies 2023, 16, 6441. [Google Scholar] [CrossRef]
- P. Lamichhane et al. Critical review: ‘Green’ ethylene production through emerging technologies, with a focus on plasma catalysis. Renewable and Sustainable Energy Reviews, 2024; 189, 114044. [CrossRef]
- C. Shen, D. Sun, and H. Yang. Methane coupling in microwave plasma under atmospheric pressure. Journal of Natural Gas Chemistry, 2011; 20, 449–456. [CrossRef]
- S. Heijkers, M. Aghaei, and A. Bogaerts. Plasma-Based CH4 Conversion into Higher Hydrocarbons and H2: Modeling to Reveal the Reaction Mechanisms of Different Plasma Sources. The Journal of Physical Chemistry C, 2020; 124, 7016–7030. [CrossRef]
- R. Liu et al. Hybrid plasma catalysis-thermal system for non-oxidative coupling of methane to ethylene and hydrogen. Chemical Engineering Journal, 2024; 498, 155733. [CrossRef]
- E. Morais, E. Delikonstantis, M. Scapinello, G. Smith, G. D. Stefanidis, and A. Bogaerts. Methane coupling in nanosecond pulsed plasmas: Correlation between temperature and pressure and effects on product selectivity. Chemical Engineering Journal, 2023; 462, 142227. [CrossRef]
- F. Cameli, M. Scapinello, E. Delikonstantis, and G. D. Stefanidis. Electrified methane upgrading via non-thermal plasma: Intensified single-pass ethylene yield through structured bimetallic catalyst. Chemical Engineering and Processing - Process Intensification, 2024; 204, 109946. [CrossRef]
- M. Scapinello, E. Delikonstantis, and G. D. Stefanidis. Direct methane-to-ethylene conversion in a nanosecond pulsed discharge. Fuel, 2018; 222, 705–710. [CrossRef]
- E. Delikonstantis et al. Valorizing the Steel Industry Off-Gases: Proof of Concept and Plantwide Design of an Electrified and Catalyst-Free Reverse Water–Gas-Shift-Based Route to Methanol. Environmental Science & Technology, 2023; 57, 14961–14972. [CrossRef]
- E. Morais, F. Cameli, G. D. Stefanidis, and A. Bogaerts. Selective catalytic hydrogenation of C2H2 from plasma-driven CH4 coupling without extra heat: mechanistic insights from micro-kinetic modelling and reactor performance. EES Catalysis, 2025; 3, 475–487. [CrossRef]
- S. Ravasio and C. Cavallotti. Analysis of reactivity and energy efficiency of methane conversion through non thermal plasmas. Chem Eng Sci. 2012; 84, 58–590. [CrossRef]
- E. Delikonstantis, M. Scapinello, O. Van Geenhoven, and G. D. Stefanidis. Nanosecond pulsed discharge-driven non-oxidative methane coupling in a plate-to-plate electrode configuration plasma reactor. Chemical Engineering Journal, 2020; 380, 122477. [CrossRef]
- E. Delikonstantis, M. Scapinello, and G. D. Stefanidis. Low energy cost conversion of methane to ethylene in a hybrid plasma-catalytic reactor system. Fuel Processing Technology, 2018; 176, 33–42. [CrossRef]
- P.-A. Maitre, M. S. Bieniek, and P. N. Kechagiopoulos. Modelling excited species and their role on kinetic pathways in the non-oxidative coupling of methane by dielectric barrier discharge. Chem Eng Sci, 2021; 234, 116399. [CrossRef]
- K. van ‘t Veer, F. Reniers, and A. Bogaerts. Zero-dimensional modeling of unpacked and packed bed dielectric barrier discharges: the role of vibrational kinetics in ammonia synthesis. Plasma Sources Sci Technol, 2020; 29, 45020. [CrossRef]
- B. Wanten, R. Vertongen, R. De Meyer, and A. Bogaerts. Plasma-based CO2 conversion: How to correctly analyze the performance? 2023; 86, 180–196. [CrossRef]
- N. Budhraja, A. Pal, and R. S. Mishra. Plasma reforming for hydrogen production: Pathways, reactors and storage. Int J Hydrogen Energy, 2023; 48, 2467–2482. [CrossRef]
- J. Medford et al. CatMAP: A Software Package for Descriptor-Based Microkinetic Mapping of Catalytic Trends. Catal Letters 2015, 145, 794–807. [CrossRef]
- Y. Gao, S. Zhang, H. Sun, R. Wang, X. Tu, and T. Shao. Highly efficient conversion of methane using microsecond and nanosecond pulsed spark discharges. Appl Energy 2018, 226, 534–545. [CrossRef]
- S. Zhang, Y. Gao, H. Sun, H. Bai, R. Wang, and T. Shao. Time-resolved characteristics and chemical kinetics of non-oxidative methane conversion in repetitively pulsed dielectric barrier discharge plasmas. J Phys D Appl Phys 2018, 51, 274005. [Google Scholar] [CrossRef]
- E. C. Neyts, K. (Ken) Ostrikov, M. K. Sunkara, and A. Bogaerts. Plasma Catalysis: Synergistic Effects at the Nanoscale. Chem Rev 2015, 115, 13408–13446. [Google Scholar] [CrossRef] [PubMed]
- Y. Engelmann, P. Mehta, E. C. Neyts, W. F. Schneider, and A. Bogaerts. Predicted Influence of Plasma Activation on Nonoxidative Coupling of Methane on Transition Metal Catalysts. ACS Sustainable Chemistry & Engineering 2020, 8, 6043–6054. [Google Scholar] [CrossRef]
- R. Horn and R. Schlögl. Methane Activation by Heterogeneous Catalysis. Catal Letters 2014, 145, 23–39. [Google Scholar] [CrossRef]
- R. J. Carman and R. P. Mildren. Electron energy distribution functions for modelling the plasma kinetics in dielectric barrier discharges. J Phys D Appl Phys 2000, 33, L99–L103. [Google Scholar] [CrossRef]
- M. A. Lieberman and S. Ashida. Global models of pulse-power-modulated high-density, low-pressure discharges. Plasma Sources Sci Technol 1996, 5, 145–158. [Google Scholar] [CrossRef]
- Hurlbatt et al. Concepts, Capabilities, and Limitations of Global Models: A Review. Plasma Processes and Polymers 2016, 14. [CrossRef]
- T. Butterworth et al. Plasma induced vibrational excitation of CH4—a window to its mode selective processing. Plasma Sources Sci Technol, 2020; 29, 95007. [CrossRef]
Figure 1.
Bar chart of concentrations at steady state.
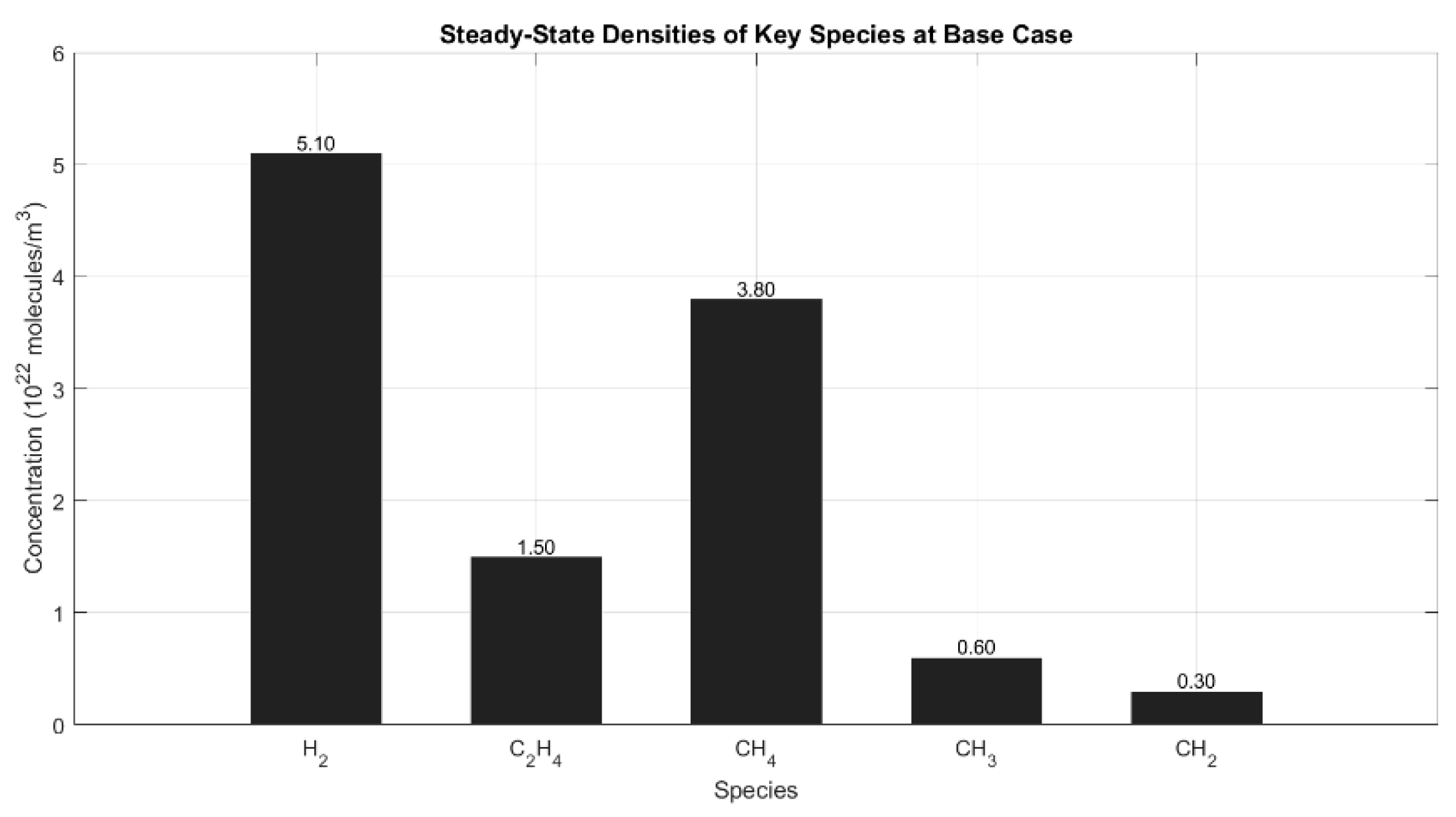
Figure 2.
Time evolution of CH4 conversion & yields (line plot).
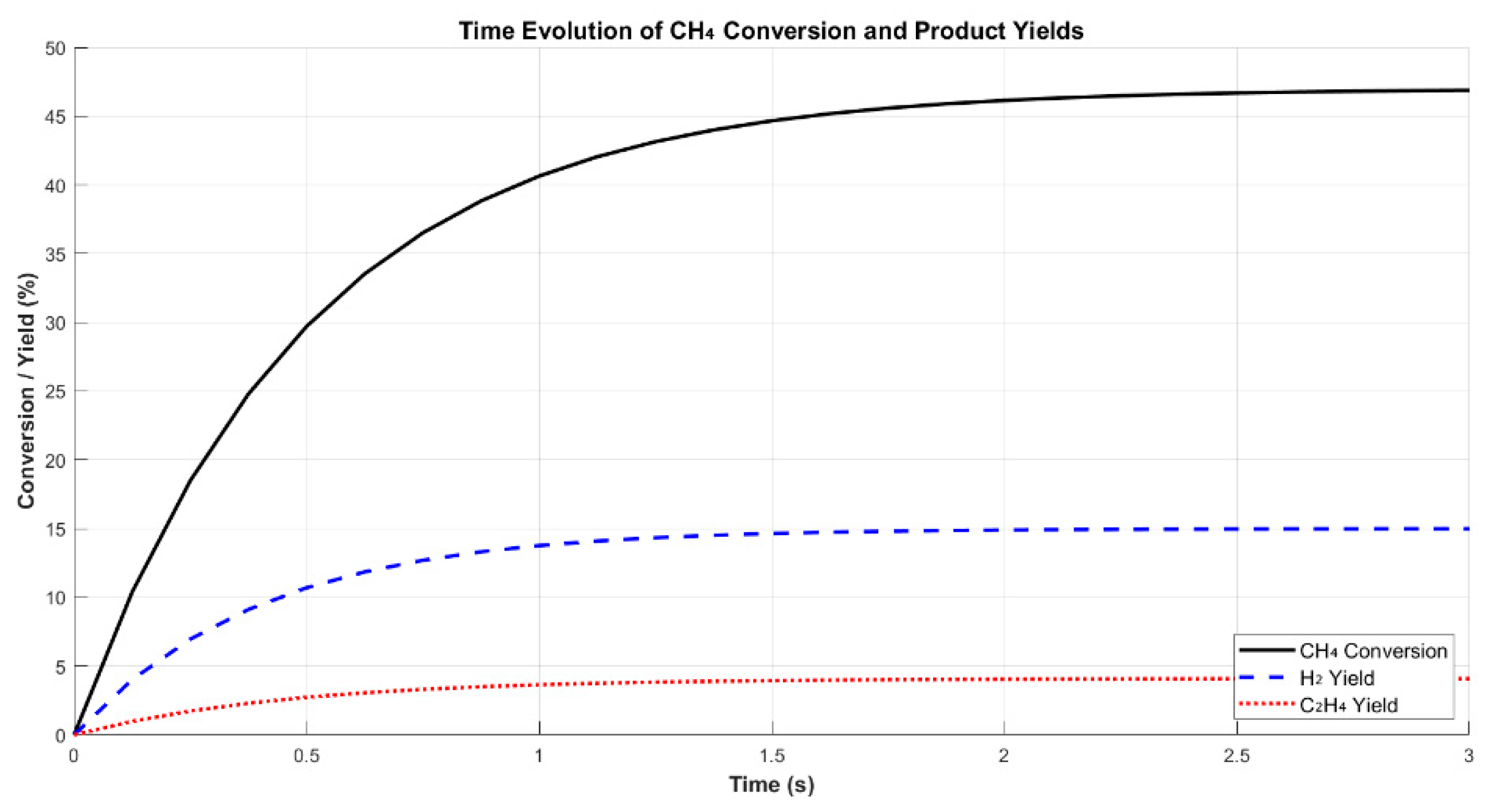
Figure 3.
Percentage change in key species concentrations with catalyst vs. plasma-only configuration.
Figure 3.
Percentage change in key species concentrations with catalyst vs. plasma-only configuration.
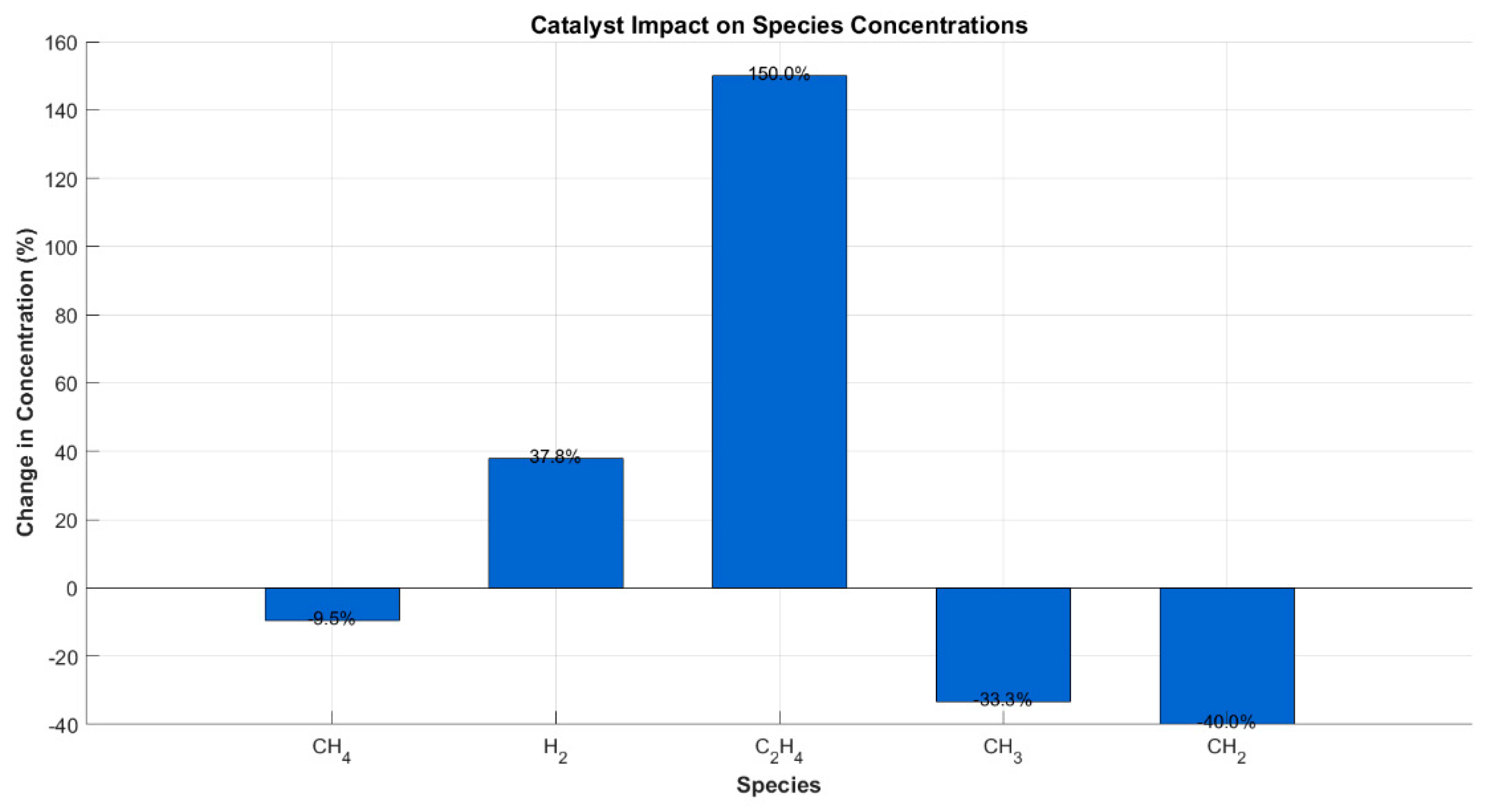
Figure 4.
CH4 conversion and product selectivity with and without catalyst. Surface chemistry boosts both conversion and selectivity toward H2 and light olefins.
Figure 4.
CH4 conversion and product selectivity with and without catalyst. Surface chemistry boosts both conversion and selectivity toward H2 and light olefins.
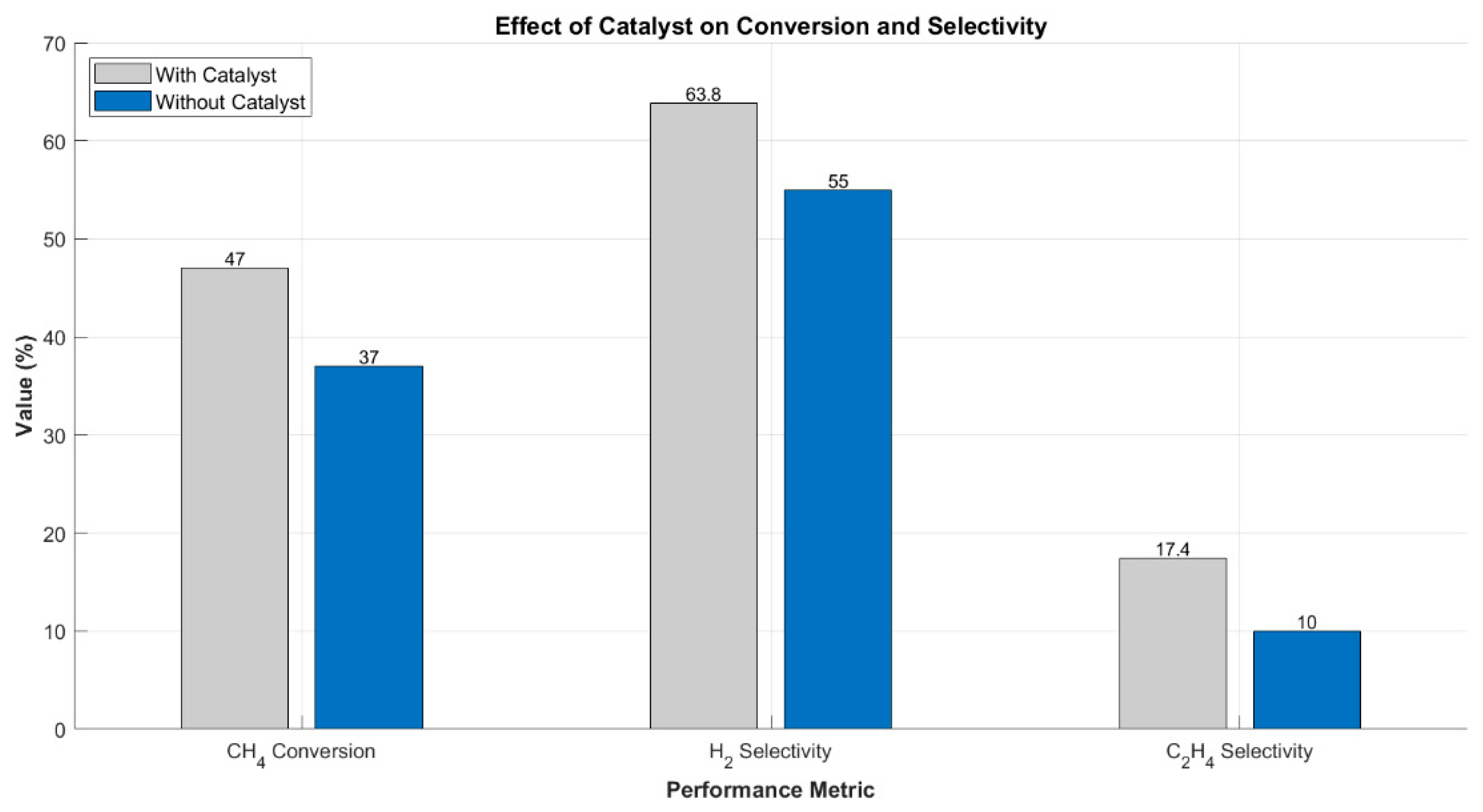
Figure 5.
Time evolution of electron energy (Tₑ) and electron density (nₑ) under base case discharge conditions. Tₑ stabilizes at ~2.90 eV, while nₑ increases progressively, reaching a steady-state around 2.80 × 1016 m−3.
Figure 5.
Time evolution of electron energy (Tₑ) and electron density (nₑ) under base case discharge conditions. Tₑ stabilizes at ~2.90 eV, while nₑ increases progressively, reaching a steady-state around 2.80 × 1016 m−3.
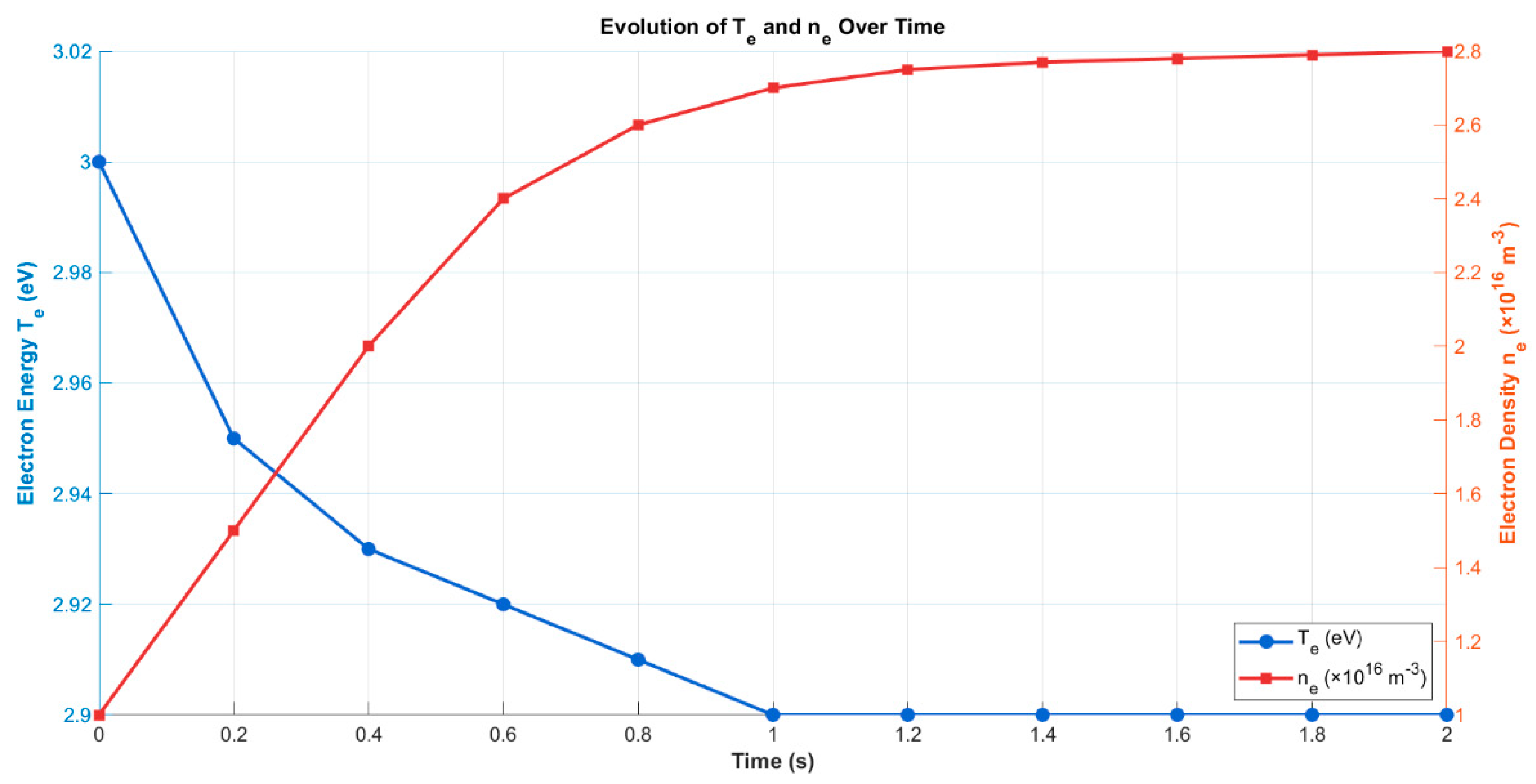
Figure 6.
CH4 conversion as a function of time under varying discharge power (50–90 W). Higher power enhances both conversion rate and final efficiency.
Figure 6.
CH4 conversion as a function of time under varying discharge power (50–90 W). Higher power enhances both conversion rate and final efficiency.
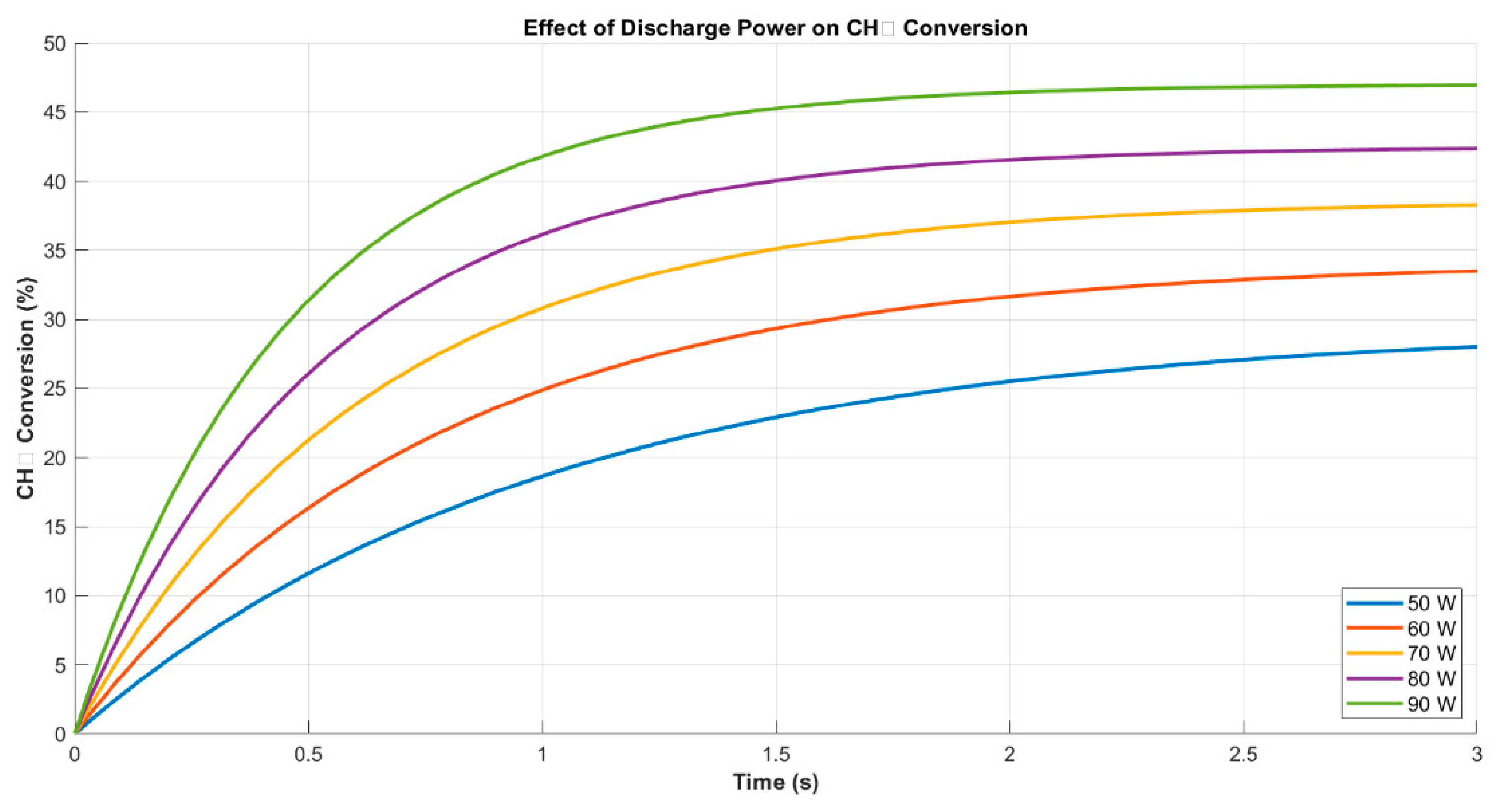
Figure 7.
CH4 conversion and electron density vs. time across pressures from 0.8 to 1.2 atm. Elevated pressure suppresses both dissociation efficiency and plasma reactivity.
Figure 7.
CH4 conversion and electron density vs. time across pressures from 0.8 to 1.2 atm. Elevated pressure suppresses both dissociation efficiency and plasma reactivity.
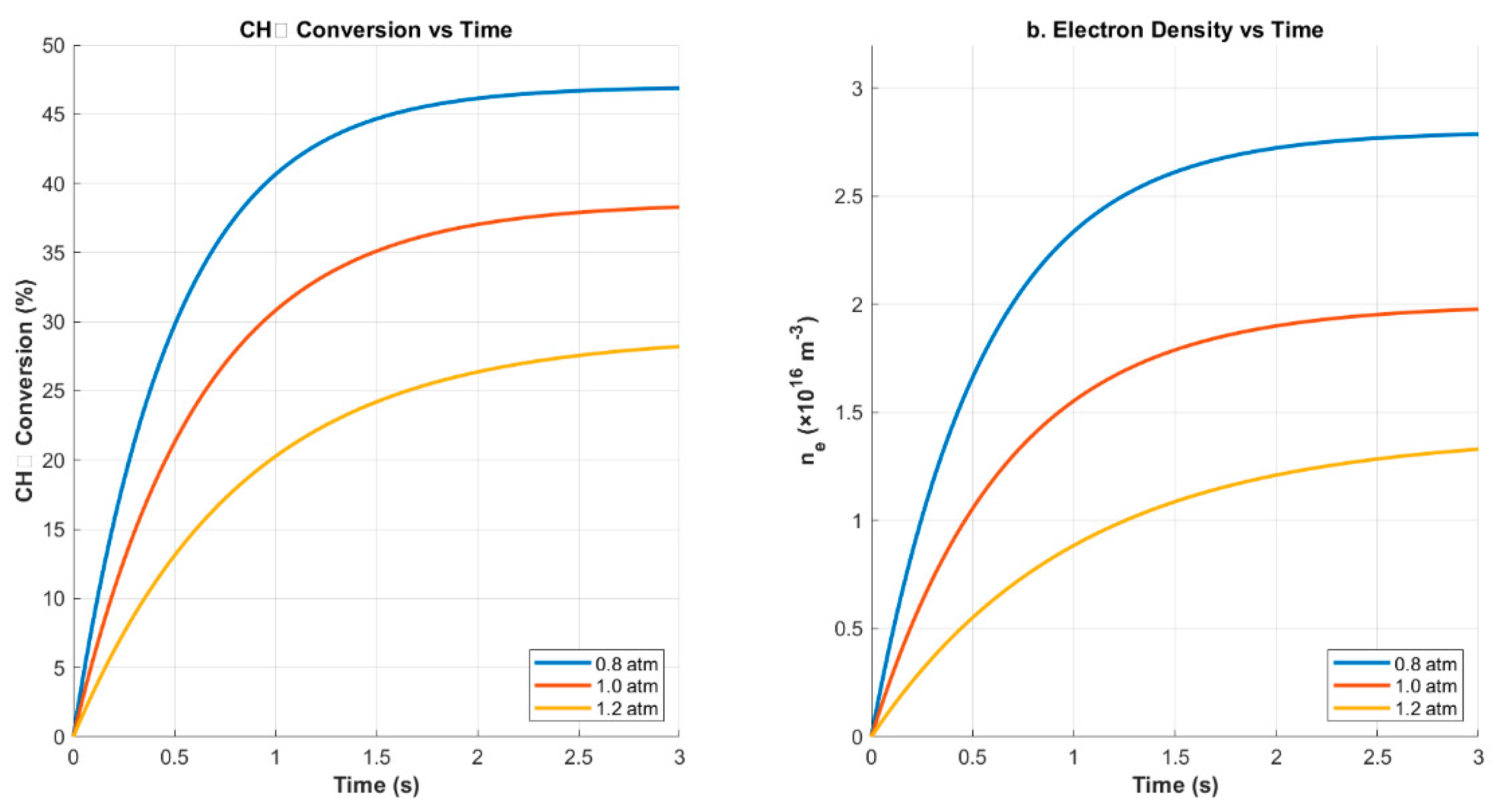
Figure 8.
Effect of varying flow rate on CH4 conversion and electron density. Lower flow rates support better plasma-molecule interaction and improved conversion.
Figure 8.
Effect of varying flow rate on CH4 conversion and electron density. Lower flow rates support better plasma-molecule interaction and improved conversion.
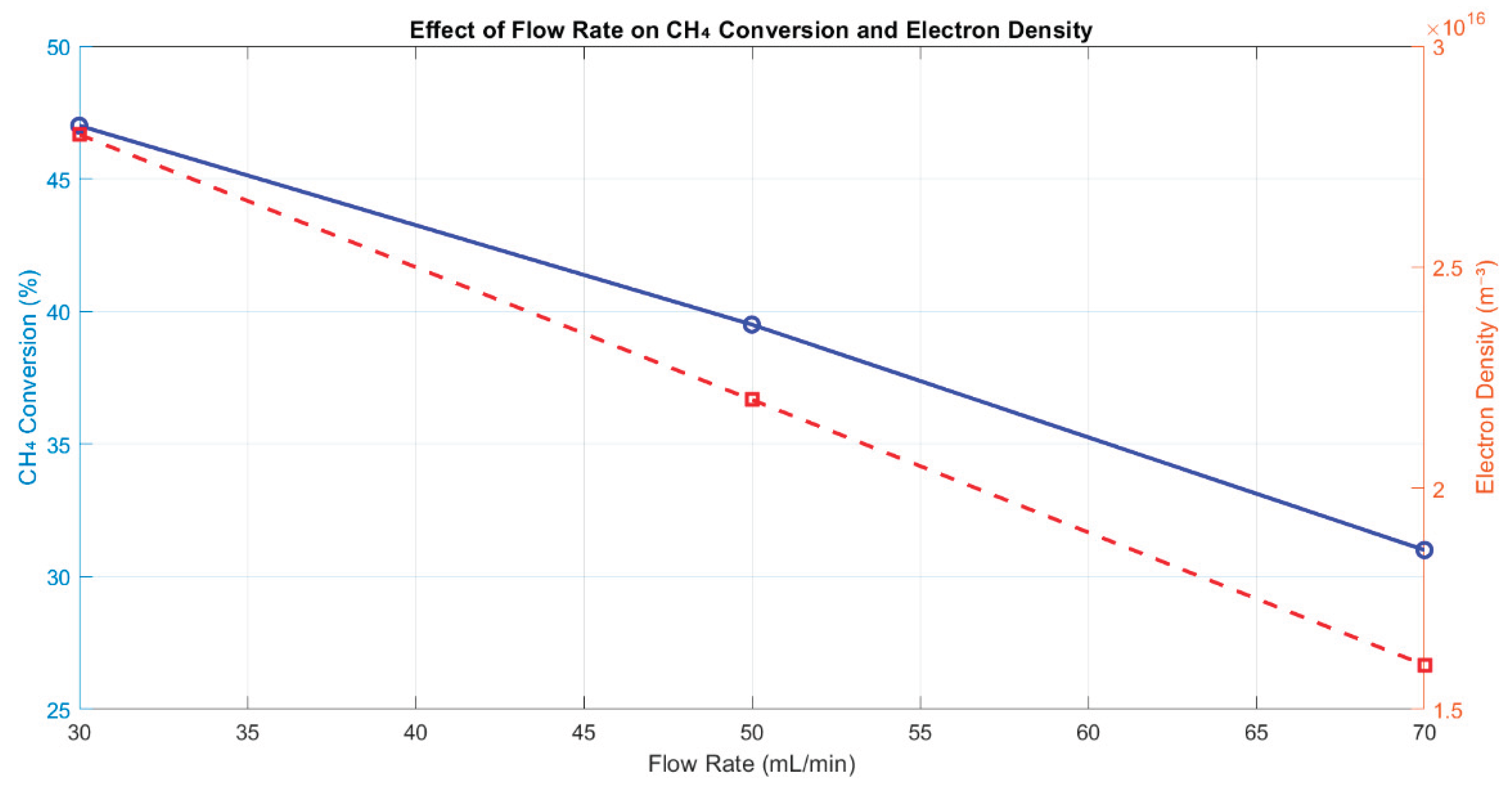
Figure 9.
Simulated H2 yield surface plot as a function of discharge power and pressure. Maximum yield occurs near 87–90 W and ~1.0 atm.
Figure 9.
Simulated H2 yield surface plot as a function of discharge power and pressure. Maximum yield occurs near 87–90 W and ~1.0 atm.
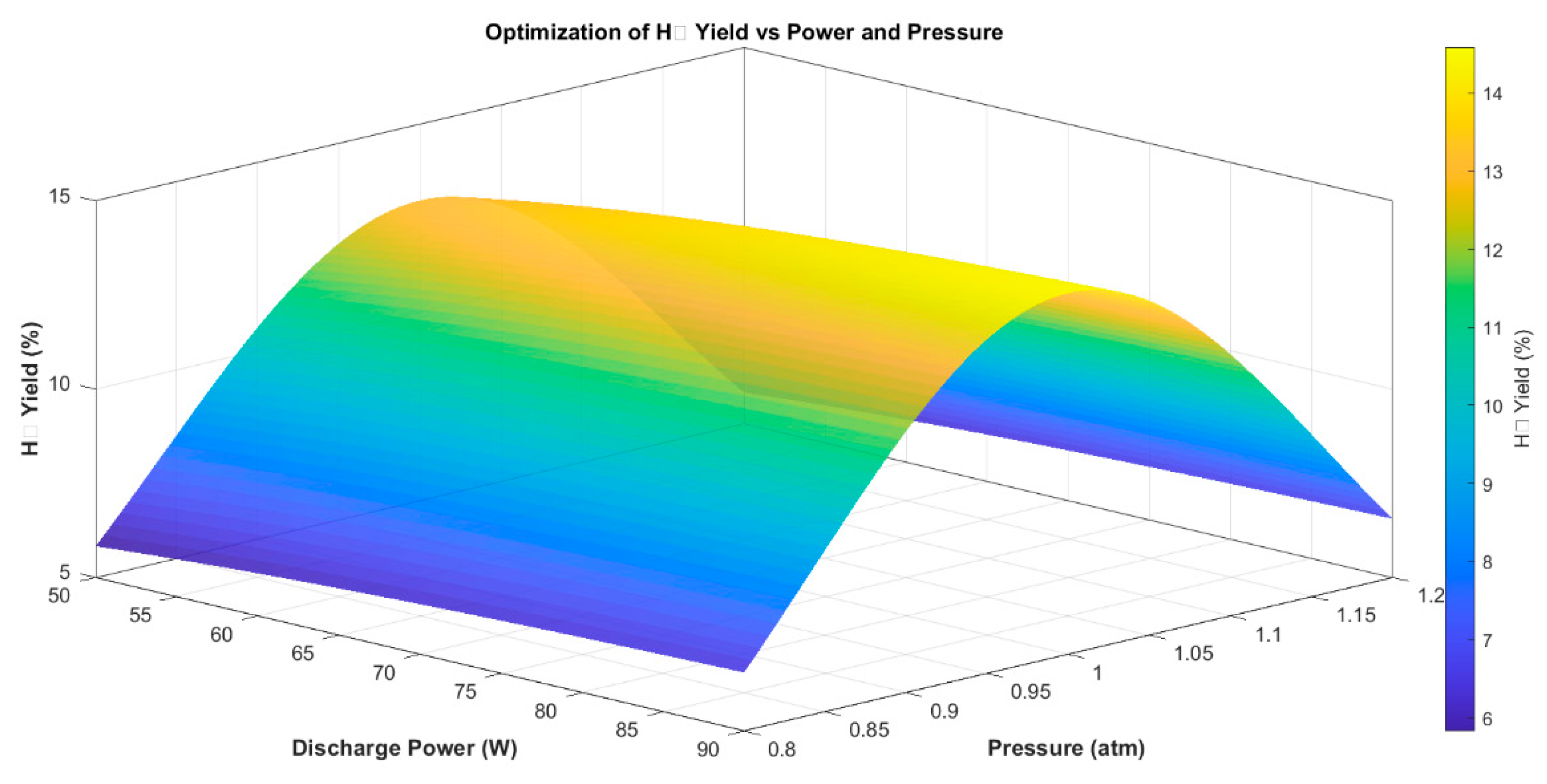
Table 2.
CH4 Conversion, Product Yields, and Selectivity Values under Base Case Conditions.
| CH4 Conversion (%) | H2 Yield (%) | C2H4 Yield (%) | H2 Selectivity (%) | C2H4 Selectivity (%) |
|---|---|---|---|---|
| 29.5 | 11.8 | 2.6 | 80.2 | 17.5 |
| 34.2 | 12.9 | 3.1 | 75.4 | 18.1 |
| 38.6 | 13.7 | 3.5 | 70.9 | 18.0 |
| 42.5 | 14.3 | 3.8 | 67.2 | 17.8 |
| 47.0 | 15.0 | 4.1 | 63.8 | 17.4 |
| 51.1 | 15.5 | 4.4 | 60.5 | 17.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



