
Submitted:
13 June 2025
Posted:
16 June 2025
You are already at the latest version
Abstract
Unlike other microbiomes, the breast microbiome remains stable across various factors, including pregnancy history, age and ethnicity. The breast is not a sterile organ, and its microbiota exhibits a distinct composition compared to other body sites. The breast microbiome is a community characterized by an abundance of Proteobacteria and Firmicutes, which represent the result of host microbial adaptation to the fatty acid environment in the tissue. The human milk microbiota (HMM) is a complex biological fluid enriched with immunomodulatory components and plays a pivotal role in shaping the neonatal gut ecosystem. Dysbiosis is strongly associated with mastitis.
Risk factors for BC include genetic mutations, late menopause, obesity, estrogen metabolism and alterations in gut microbial diversity. Gut microbiota can increase estrogen bioavailability by deconjugating estrogen-glucuronide moieties in a process called estrobolome. Perturbations of this process increases circulating estrogens and risk of BC. Fusobacterium nucleatum has recently been associated with BC. It moves from the oral cavity to other body sites hematogenously.
This review deals with the characteristics of the breast microbiome, with a focus on F. nucleatum, highlighting its dual role in promoting tumor growth and modulating immune responses.
F. nucleatum acts both on the Wnt/β-catenin pathway by positively regulating MYC expression, and on apoptosis by inhibiting caspase 8. Furthermore, F. nucleatum binds to TIGIT and CEACAM1, inhibiting T cell cytotoxic activity and protecting tumor cells from immune cell attack. F. nucleatum also inhibits T cell function through the recruitment of myeloid suppressor cells (MDSCs). These cells express PD-L1, which further reduces T cell activation.
A deeper understanding of F. nucleatum biology and its interactions with host cells and co-existing symbiotic microbiota could aid in the development of personalized anticancer therapy.
Keywords:
microbiome
; breast cancer
; breast feeding
; Fusobacterium nucleatum
; Wnt/b-catenin
; immune response
; Fap2
; FadA
; TIGIT
; estrobolome
; MDSC
1. Introduction
Unlike other microbiomes, the breast microbiome remains stable across various factors, including pregnancy history (previous births vs. nulliparous women), age of the individual, presence or absence of breast malignancy, geographic location and ethnicity, sample collection site within the breast and sequencing technologies used for analysis [1]. These findings confirm that the breast is not a sterile organ, and its microbiota exhibits a distinct composition compared to other body sites [1,2,3].
Determined bacteria found in breast tissue were also detected in other sites of the body like Lactobacillusiners and Prevotella (vagina), Enterobacteriaceae (gastrointestinal tract), Fusobacterium and Streptococcus (oral cavity), Propionibacterium and Micrococcus (skin), and Pseudomonas (respiratory tract). Both species with healthy properties such as Lactobacillus and Bifidobacterium and known pathogenic species like Enterobacteriaceae, Pseudomonas and Streptococcus agalactiae have been found [1].
The human breast harbors a unique and diverse microbiome that is distinct from microbial communities found in other body sites, including the gut, skin, and vagina [2]. A potential link between the breast microbiota and maternal health, infant development, and the pathogenesis of breast cancer has been hypothesized, although this remains an emerging and underexplored area of research [4,5]. For the first time in 2014 in two autonomy research it was detected the presence of native breast microbiome [1,2], evidence that the breast hosts a large ecosystem [2]. Since the breast consist mainly of adipose tissue with a large vascularization and lymphatic drainage, it is a supportive habitat for bacterial expansion, in particular Proteobacteria and Firmicutes [3] (Figure 1).
The breast microbiome in health is characterized by a balanced microbial community dominated by commensals such as Lactobacillus, Bifidobacterium, and Sphingomonas, supporting immune tolerance and neonatal gut colonization. In contrast, dysbiosis during inflammatory conditions such as mastitis or maternal obesity leads to an overgrowth of opportunistic pathogens (e.g., Staphylococcus aureus, E. coli), immune cell infiltration, and altered cytokine profiles, potentially disrupting lactation and compromising infant health outcomes as well as mum’s health in the long-term.
Bacteria like Bacillus sp., Enterobacteriaceae sp., and Staphylococcus sp. were detected in the breast tissue by growth experiments, which highlighted living bacterial species [2]. Other bacterial species were also identified after tissue stratification and regulation for known confounding factors and factors that may affect the microbiome (age of patient, race and hospital) [6,7]. The healthy controls and high-risk benign tissue samples show a comparable composition of microbiome, represented by great mean relative presence of 11 genera: Propionibacterium, Finegoldia, Granulicatella, Streptococcus, Anaerococcus, Ruminococcaceae, Corynebacterium 1, Alicyclobacillus, Odoribacter, Lactococcus, Esherichica/Shigella [8].
Thanks to the studies carried out, the possible generation of part of the mammary microbiome is now associated with translocation from the gastrointestinal tract and the skin, through the nipple-areola structure, through the orifices or through oral contact during breastfeeding or sexual intercourse [9]. One of the possible hypotheses of colonization could be explained by the movement from other body sites, such as the intestine or the oral cavity [9].
The microbiome could also play a defensive function towards the breast because the immune cells stimulate healthy tissue and the degradation of carcinogenic metabolites could be carried out by the activity of resident bacteria, always in a defensive view and maintenance of healthy tissue [1].
Both milk and tissue of the breast contain a large bacterial community that has an impact on the healthy development of children’s intestine and on maintaining of women health. The diversity of breast microbiota is comparable to that of the gut microbiome but significantly higher than that observed in the vaginal microbiome [1].
It was observed in women between 18 and 90 years old that the Proteobacteria are the most present in breast tissue with or without cancer (or around the cancer) as another common is represented by the Firmicutes [2].
2. Microbiota in Breast Feeding in Healthy and Inflammatory Conditions
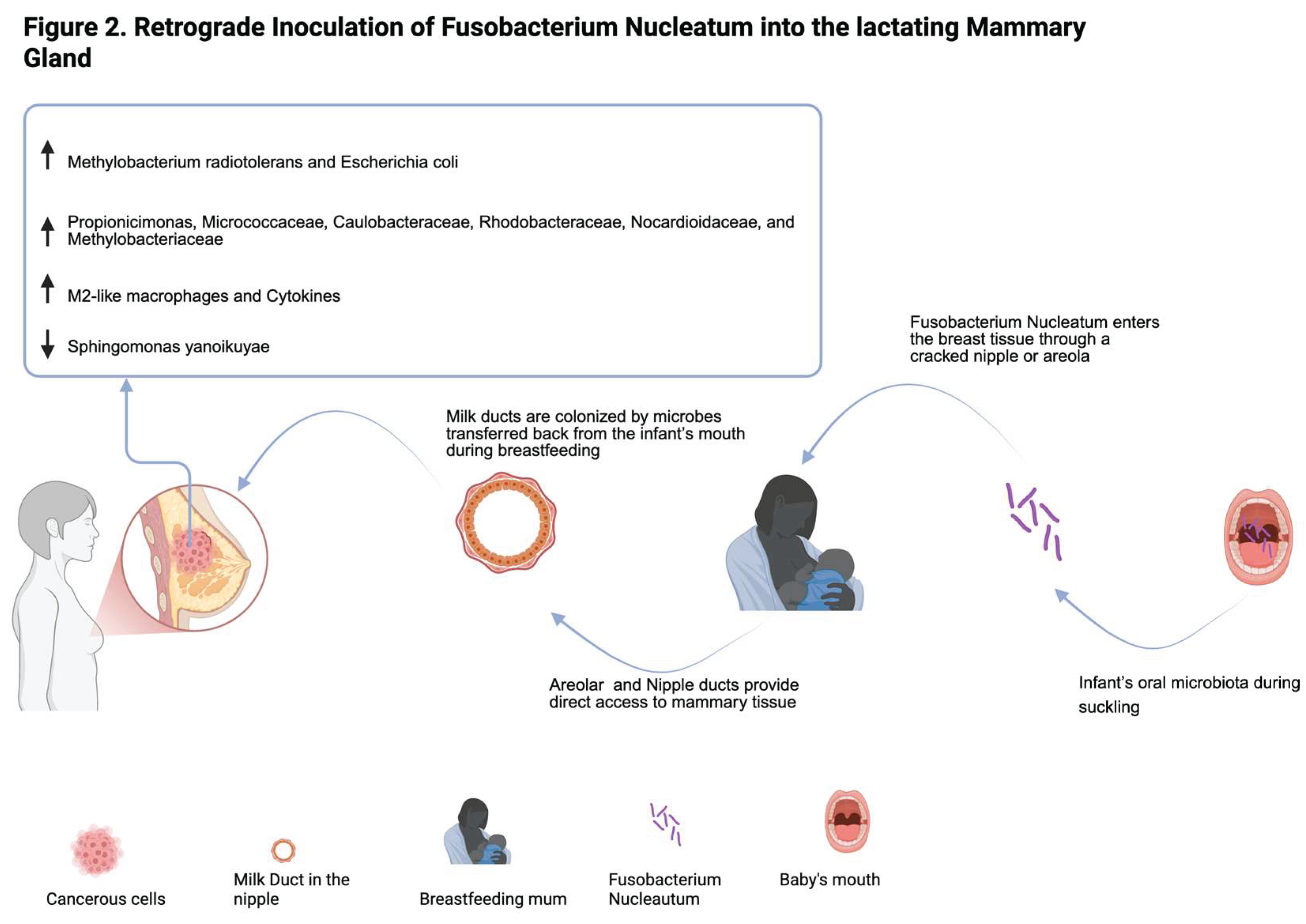
The breast microbiome plays a specific biological role rather than being a random accumulation of external microbes [10]. It is composed of a diverse array of bacteria, many of which are beneficial for infant health by supporting immune system maturation, metabolic programming, and protection against pathogens [11]. The bacterial communities in breast milk contribute significantly to the establishment and development of the neonatal gut microbiome. Research indicates that breast milk contains a dynamic and evolving microbiota, influenced by maternal factors such as diet, mode of delivery, and health status [5]. Key bacterial genera found in both breast milk and infant feces include Bifidobacterium (supports gut health and immune regulation); Lactobacillus (promotes gut barrier function); Streptococcus (plays a role in early microbial colonization); Staphylococcus (common but varies depending on maternal health) [12] (Figure 2). The human milk microbiota (HMM) also consists of a diverse range of bacterial species, as well as viruses, fungi, archaea, and protozoa that contribute to neonatal gut colonization [12]. The most prevalent bacteria belong to facultative anaerobic or strictly aerobic groups, which include: Firmicutes (Staphylococcus, Streptococcus, and Lactobacillus); Proteobacteria (Pseudomonas and Sphingomonas, dominant in early lactation); Actinobacteria (Bifidobacterium, a key player in gut health); Bacteroidetes (including Prevotella), found in lower abundance [13]. The presence of Proteobacteria and Firmicutes, particularly the class Bacilli, is greater than that of other taxonomic groups, and it might be due to a host microbial adaptation to the fatty acid environment in the tissue. Why potentially pathogenic bacteria that metabolize fat in breast tissue do not proliferate and cause infections is a question that remains largely speculative Of interest, Proteobacteria is the principal phylum in human milk with many of the same bacteria that we detected in tissue, raising the possibility that the tissue microbiota may contribute to the infant’s early gut colonization and may be a source of bacterial inocula for babies [14].
This schematic illustrates the retrograde inoculation route, whereby microbes from the infant’s oral cavity are transferred back into the mammary ducts during breastfeeding. This process occurs as negative pressure and peristaltic tongue movements during suckling allow milk and saliva to transiently flow back into the milk ducts. Microbial genera such as Streptococcus, Veillonella, Fusobacterium nucleatum, and Actinomyces have been identified as prominent oral-derived taxa colonizing breast milk via this mechanism (Pannaraj et al., 2017; Moossavi et al., 2019). This bidirectional exchange contributes to the shaping of both the milk microbiome and the infant gut microbiota, enhancing microbial diversity and potentially supporting immunological development. The figure also shows the changes in the microbiota of a cancerous breast environment.
In a recent cohort study, the presence of Bifidobacterium breve in the maternal rectum, breast milk, and infant stool further supports the entero-mammary pathway [15]. Human milk contains not only bacteria but also a complex virome and mycobiome, both of which may influence infant microbial colonization and immune development [13,16]. Human milk is increasingly recognized not only as a source of optimal nutrition but also as a critical contributor to the early microbial colonization of the infant’s gut and respiratory tract. Through this colonization, maternal milk plays a central role in shaping both immune system development and metabolic programming during the earliest stages of life [17]. A growing body of evidence supports the concept of a “gut-lung axis,” a bidirectional communication pathway between the gastrointestinal and respiratory microbiomes, further emphasizing the immunomodulatory potential of human milk in neonatal health [18].
Notably, microbial species found in maternal feces, breast milk, and the infant gut frequently overlap, suggesting the existence of multiple transmission pathways. These include the entero-mammary route, whereby maternal gut bacteria translocate across the intestinal barrier to the mammary gland via systemic circulation and lymphatic transport; retrograde inoculation from the infant’s oral microbiota during breastfeeding; and the transference of microbes from the maternal skin [19]. The identification of butyrate-producing bacteria such as Faecalibacterium, Roseburia, and Coprococcus spp. in both maternal fecal samples and breast milk further underscores the significance of gut-derived microbial seeding in establishing the infant’s early microbiome [20]. These taxa are known to play a role in the maturation of the gut epithelium and regulation of host immune responses.
The composition of the HMM is highly variable and influenced by a multitude of maternal and environmental factors. These include maternal age, parity, body mass index (BMI), mode of delivery, stage of lactation, dietary patterns, and antibiotic or probiotic use during pregnancy [19,21]. One key determinant is maternal BMI: obesity has been linked to increased levels of Lactobacillus in colostrum and Staphylococcus and Akkermansia in mature milk, along with reduced microbial diversity, particularly in Bacteroidetes and Firmicutes [19]. For example, maternal diet has been associated with shifts in microbial abundance, such as increased Bifidobacterium with polyunsaturated fatty acid intake, or elevated Staphylococcus levels with high vitamin C consumption.
Vitamin C intake is associated with more Staphylococcus spp., while polyunsaturated fatty acids support Bifidobacterium growth, essential for infant gut and immune health. High-calorie, high-fat diets are correlated with increased Firmicutes [13]. Collectively, these findings support the view that the HMM plays a fundamental role in establishing the infant microbiota, mediating immune tolerance, and potentially reducing the risk of chronic inflammatory and metabolic diseases later in life. Ongoing research into the ecology and function of HMM may pave the way for targeted maternal interventions aimed at optimizing infant health outcomes.
The mode of delivery significantly shapes the microbial composition of human milk. Infants born via Caesarean section (C-section) are exposed to different microbial communities than those born vaginally, and this extends to the milk they receive. Studies have found that milk from C-section births contains higher levels of Proteobacteria and lower levels of Bifidobacterium and Lactobacillus, potentially affecting the initial gut colonization in infants [11]. Interestingly, emergency C-sections tend to yield microbial profiles in breast milk that are more like vaginal deliveries, possibly due to the physiological onset of labor [11].
The composition of the HMM is significantly influenced by the mode of milk delivery-whether it is expressed and fed via a pump or delivered directly from the breast through breastfeeding. Direct breastfeeding facilitates dynamic microbial exchange between mother and infant, particularly through retrograde milk flow during suckling. This process appears to favor colonization by beneficial bacteria such as Bifidobacterium, which are less abundant in pumped milk. In contrast, expressed milk, due to its exposure to external surfaces, storage containers, and variable temperatures, tends to harbor more environmental or opportunistic microbes [22]. Such differences may have implications not only for the microbial profile of the milk but also for the immunological priming of the infant gut. Interestingly, the biological sex of the infant may also modulate HMM. Through retrograde flow and the intimate immunological dialogue that occurs during breastfeeding, male and female infants may exert different influences on the maternal breast microbiome.
Gestational age at birth is a key determinant of milk microbiota. Preterm milk typically contains reduced levels of Bifidobacterium, potentially contributing to the risk of gastrointestinal disorders such as necrotizing enterocolitis in premature infants [21]. Environmental factors also play a role: rural breast milk has been shown to contain greater microbial diversity than urban samples, likely due to broader environmental exposures including diet, hygiene, and contact with nature [23].
Beyond these influences, human milk is enriched with complex carbohydrates known as human milk oligosaccharides (HMOs), which play a central role in supporting a healthy infant microbiome. Although indigestible by the infant, HMOs selectively promote the growth of commensal bacteria, particularly Bifidobacterium spp., which in turn contribute to gut maturation and immune system development. The interplay between HMOs and milk microbiota fosters mucosal immunity, strengthens epithelial barrier function, and promotes the development of gut-associated lymphoid tissue mechanisms that are fundamental in protecting against infections and immune-related diseases [24,25]. The long-term implications of these interactions are increasingly evident in epidemiological studies. Azad et al. [26], in a comprehensive review, underscored the immunomodulatory potential of human milk, linking its microbial and bioactive components to reduced risks of allergic and autoimmune diseases. Taken together, these findings position the breast microbiome as a central actor in the developmental programming of infant immunity. Through its influence on microbial colonization, immune cell maturation, and the orchestration of diet–microbe–host interactions, the HMM serves not only as a conduit of maternal microbes but also as a dynamic and adaptive system that shapes the health trajectory of the infant well beyond the breastfeeding period.
Breast dysbiosis is strongly associated with mastitis. Research by Jiménez et al. [27] identified an overrepresentation of pathogenic species such as Staphylococcus aureus and Streptococcus spp. in women experiencing mastitis, suggesting that an imbalance in the local microbiome can trigger inflammatory cascades and compromise breastfeeding continuity (Figure 1). Beyond infection, alterations in breast microbiota have also been observed in women with obesity. Collado et al. [28] reported that obese mothers exhibit distinct microbial signatures in both colostrum and mature milk, with implications for milk composition, metabolic programming, and the vertical transmission of microbiota to the infant. In neonates, the implications of dysbiosis in early breast milk exposure are profound. Adequate microbial colonization in the first months of life is pivotal for the development of the gut microbiome, which in turn influences metabolism, immune regulation, and long-term health trajectories. An inadequate or imbalanced initial microbiota, especially one lacking key taxa such as Bifidobacterium or Lactobacillus, has been linked to the development of metabolic disorders, childhood obesity, and immune-related conditions including allergies and asthma [29]. Taken together, these findings underscore the importance of preserving the integrity of the breast microbiome through supportive maternal care, informed breastfeeding practices, and judicious use of medications such as antibiotics that may alter microbial balance. Further research into the dynamics of breast microbiota composition and its functional implications will be critical for the development of interventions aimed at optimizing maternal-infant health outcomes.Inizio modulo
Women living with autoimmune and chronic inflammatory diseases, such as inflammatory bowel disease (IBD), represent another population where the immunological and microbial composition of breast milk may be altered. Immunosuppressive therapies and systemic inflammation have the potential to modify breast milk’s bioactive profile, though data remain limited. Preliminary studies indicate that, despite these alterations, breastfeeding may continue to offer immunological benefits to the infant, including the transmission of protective antibodies and immune-modulating cytokines that may partially counteract the infant’s risk of inflammatory disorders [30].
The psychosocial state of the lactating parent is increasingly recognized as a modulator of the HMM [31]. Disruptions to the hypothalamic-pituitary-adrenal (HPA) axis can impair epithelial barrier integrity within the breast, facilitating shifts in microbial colonization. Reduced microbial diversity and altered immune signaling in the milk of stressed mothers could have downstream effects on infant development, including immune function, stress reactivity, and even gut-brain axis programming [32].
These insights collectively underscore the dynamic interplay between maternal health and the microbial constitution of human milk. Understanding the influence of inflammatory states on HMM not only aids in optimizing breastfeeding support strategies but also contributes to more targeted interventions that safeguard maternal and infant health.
Looking ahead, emerging research is uncovering the complex interplay between the human milk virome and mycobiome that is, the communities of viruses (including bacteriophages) and fungi (e.g., Malassezia, Candida) present in breast milk. These non-bacterial components contribute not only to the microbial richness of milk but also to its immunomodulatory properties. Bacteriophages, for example, may help regulate bacterial populations in the infant gut, while fungal elements might influence immune responses and barrier function. This broader ecological view challenges the traditionally bacteria-centric model of microbiome science and suggests that human milk serves as a rich, biologically active fluid-delivering not just nutrition, but also a sophisticated toolkit of microbial agents and regulatory factors.
2. Molecular Alterations in Breast Cancer
Breast cancer (BC) has emerged as the most commonly diagnosed female neoplasm in all age groups and the leading cause of cancer death among women worldwide [33] The etiology of BC is multifactorial, with both endogenous and environmental factors potentially interacting in its pathogenesis. Risk factors for BC include genetic mutations, late menopause, obesity and reproductive factors [34,35,36].
BC is a complex molecular disease involving alterations to several cellular pathways that control cell growth and proliferation. Affected pathways include MAPK, RB/E2F, PI3K/AKT/mTOR, and TP53 [37]. The complex molecular pathways involved in BC progression originate from deregulated crosstalk controlling cell growth and renewal. These pathways include HER2, c-MYC, the estrogen receptor (ER), cyclins D1 and E, TP53, and the phosphatase and tensin homolog (PTEN). They are also affected by the failure to repair DNA damage that occurs during physiological processes, such as those involving the BRCA1 molecular network [38,39].
The presence of hereditary factors significantly increases the likelihood of developing BC. The risk of developing BC due to pathogenic mutations in high-penetrance genes, such as BRCA1 [OMIM 113705] and BRCA2 [OMIM 600185], has been recognised for several decades [40,41]. Some of the genetic risk can be explained by pathogenic variants in other BC susceptibility genes, including TP53, CDH1, PALB2 and PTEN and various rare gene variants have also been reported to increase the risk of developing BC [42,43]. However, the presence of multiple polygenic susceptibility alleles, resulting in a cumulative risk from low-penetrance mutations, is also a widely recognised risk factor [44,45]. Moreover, several related genes to metabolism, oxidative stress and inflammation have been linked to an increased risk of BC, including those involved in one-carbon metabolism, estrogen metabolism, and lipid metabolism [46,47,48,49]. Therapeutic strategies have been proposed to benefit patients with mutations in BRCA1/2 and other genes required for homologous recombination (HR). Around 22% of BCs are estimated to exhibit a BRCA1/2 alteration, whereas a functional deficit in the HR system overseen by BRCA1 has been identified in 69% of triple-negative breast cancers. These cancers tend to be more aggressive, carry a higher risk of recurrence and have a poorer prognosis [50,51]. The estimated lifetime risk of developing BC for carriers of the BRCA1 and BRCA2 mutations is 69% and 72%, respectively. Twenty years after an initial BC diagnosis, the respective risks of developing contralateral BC are 40% and 26% [52]. Estrogens have been associated with an increased risk of BC. However, emerging clinical and experimental evidence suggests that progesterone, whether produced naturally or synthetically, is the primary hormonal factor underlying this risk. In fact, estrogens may indirectly contribute to BC risk by inducing progesterone receptors and amplifying progesterone signalling [53].
In clinical settings, methods based on immunohistochemistry using four surrogate markers, estrogen receptor [ER], progesterone receptor [PR], HER2, and Ki67 are commonly used to identify BC subtypes [54,55]. According to the World Health Organization (WHO) classification system, breast carcinoma is classified as either invasive or non-invasive [56]. Invasive carcinoma accounts for 70%-75% of cases and is grouped into four categories based on hormone receptor expression. These categories are estrogen receptor positive (ER+), progesterone receptor positive (PR+), human epidermal growth factor receptor positive (HER2+), and triple negative (TNBC). TNBC is characterized by the absence of expression of any of these receptors. Lobular carcinoma, which accounts for 12%-15% of cases, is also characterized by deregulation of E-cadherin/β-catenin [57]. There are also eighteen other uncommon subtypes, accounting for 0.5–5% of cases. Pathological descriptions include the tumour’s histological type and grade, as well as an immunohistochemical assessment of its hormone receptor status (ER and PR), HER2 expression, and Ki67 expression [58].
BC is a heterogeneous group of diseases that respond differently to various personalized treatment modalities [59,60]. The most common method used in clinical practice is a molecular classification approach using gene expression profiling: luminal A, luminal B, enriched HER-2 and basal-like or triple-negative breast cancer (TNBC) [61]. Because each subtype has unique responses and prognoses to therapeutic interventions, a subtype-specific treatment approach is necessary [61].
The progression of BC is determined by a number of factors, including the characteristics of tumour cells, elements of the tumour microenvironment (TME) (both cellular and non-cellular), and the characteristics of the surrounding tissue. Significant advances have been made in the treatment and diagnosis of primary breast cancer in recent years [62]. Alongside traditional imaging techniques and pathological diagnostic methods, liquid biopsies, multiple immunofluorescence tests and digital pathology approaches are being increasingly adopted in clinical practice. Various treatment options are available for BC, and recent clinical studies emphasise the importance of personalised, targeted therapies [39,63]. The long-term follow-up management of BC patients is also crucial as this can impact therapeutic outcomes and improve quality of life [64]. Whether or not the tumour has spread to the sentinel lymph nodes (SLNs) is a key factor in predicting the outcome of BC. The SLNs are usually the first place to which the cancer spreads, and patients with lymph node metastases (LNM) have lower overall and disease-free survival rates, regardless of the type of BC [65,66,67]. The spread of tumour cells to the draining lymph node can lead to lymphangiogenesis and the trafficking of cytokines and chemokines. This can result in immune evasion and alterations to the TME, leading to the expansion of cancer cells [68]. Several studies are currently investigating the factors in tumour and lymph node microenvironments that drive metastatic capacity [69]. Systemic treatments that prevent the development of distant metastases are ineffective for certain types of BC, such as triple-negative BC. Metastatic disease is the leading cause of death for most BC patients. The prolonged latency period between initial treatment and recurrence suggests that tumor cells adapt to and interact with molecular signals from the primary tissue microenvironment and the host systemic environment. This process facilitates disease progression and the invasion of other organs, such as the liver, lung, brain, and bone [70].
Recent studies suggest that exposure to environmental chemicals with endocrine-disrupting properties may increase a woman’s risk of developing BC at certain stages in her life. Significant structural and functional changes occur in the mammary gland during critical periods, including prenatal development, puberty, pregnancy, and menopause. Furthermore, alterations in the mammary microenvironment and hormone signaling can enhance BC susceptibility [71]. Alterations in gut microbial diversity that cause dysbiosis have been linked to BC development. These alterations modulate host immune responses and inflammatory pathways, thereby promoting tumorigenesis and progression. Additionally, differences in gut microbiota populations have been observed between women with and without BC, further implicating the role of gut microbiota in cancer development [72]. Changes in estrogen metabolism, a key factor in BC development, have also been linked to the composition of the gut microbiota [73]. Gut microbiota expressing the enzyme β-glucuronidase (GUS) can increase estrogen bioavailability by deconjugating estrogen-glucuronide moieties [74]. This process enables estrogen to be reabsorbed into the bloodstream. Increased circulating estrogens can induce estrogen receptor positivity in BC cells. Furthermore, GUS-expressing microbiota can affect the effectiveness and toxicity of anticancer therapies by altering glucuronide-conjugated drug metabolites. Consequently, GUS inhibitors have emerged as a potential anticancer treatment [75]. Further studies are needed to determine how carcinogenesis, tumor marker expression, and endocrine receptor-targeted therapies affect the microbiota and vice versa.
3. The Microbiota in Breast Cancer
BC is the second cause of death related to cancer in the women worldwide [76]. It is clinically heterogeneous, with a variety of subtypes identified according to the molecular nature. Although other clinical parameters also have an impact, the microbiota in particular appears to be an increasingly emerging risk factor. It was shown that the gut microbiota influences the onset and development of BC through different processes; these mechanisms can be represented by the control of the immunity activity, the modification of estrogen levels and the production of bacterial metabolites, which may affect cancer cells themselves and their habitat [77]. The cancer progression is driven by an increase in inflammatory mediators, generated by disruption of host and microbiota balance, leading to dysbiosis [78,79]. Dysbiosis induced by antibiotics has been shown to cause an important rise of myeloid cells greatly expressing arginase-1 and IL- 6, known as suppressive/inflammatory molecules, at the cancer site [80].
Among all immunity cells, the M2-like macrophages are the most present in the BC microenvironment and are related to lower survival in HR+ BC patients [51]. These cells have been observed to infiltrate into BC and in health close mammary gland during the initial and late stages of cancer progression. Interestingly, a rich and various microbiome has been detected in BC [77]. There are many analyses which indicated a deep different microbioma of the breast between cancerous and health tissue and between benign and malignant cancer [4,9,81], demonstrating that changing the microbial community can influence BC progression [9].
In recent years, there has been a great interest in the characterisation of the microbiota of various areas of the body and under different health conditions, this is because different studies have shown that bacterial communities are different between areas of the body and that complex interactions between host and bacteria are established [82,83].
Xuan et al. [1] detected a different presence of the genera Methylbacterium and Sphingomonas across paired healthy and/or normal adjacent tissue and cancerous tissue; this may propose that they could have a role in cancer progression. Sphingomonas yanoikuyae is present in health breast tissue, while it reduces dramatically in the tumour tissue, instead Methylobacterium radiotolerans is the most enriched bacterium in the cancerous tissue [84]. In tumor tissue Methylbacterium radiotolerans was relatively abundant and predominant (found in 100% of samples), whereas in paired normal tissue Sphingomonas yanoikuyae was relatively abundant and predominant [1]. The inverse correlation of these two bacteria in normal and tumour breast tissues is evidence that dysbiosis is associated with breast cancer [1].
In women with cancer, there is a higher amount of Escherichia coli than in healthy controls, which is known for its ability to promote cancer [85]. A study by Constantini et al.l. [86] shows that the genus Ralstonia is present in breast tissue, including in tumors and adjacent normal tissue. A significant abundace of Propionicimonas, Micrococcaceae, Caulobacteraceae, Rhodobacteraceae, Nocardioidaceae, and Methylobacteriaceae has been highlighted in an Asian cohort of breast cancer patients [84]. In another study it was underlined a reduction of Bacteroidaceae, while an increase of Agrococcus has been observed in parallel with the progression of cancer. Additionally, the presence of Fusobacterium, Atopobium, Gluconacetobacter, Hydrogenophaga, and Lactobacillus genera has been associated with cancer progression [87]. A recent study on a cohort of triple negative Breast Cancer (TNBC) patients has showed an important presence of Clostridiales in cancerous tissue related to a triggered immune microenvironment [88].
The evidence of a metabolically active tissue-resident microbiota is supported by the presence of bacteria correlated with the production of the metabolite trimethylamine N-oxide (TMAO), capable to stimulate antitumor immunity mediated by CD8+ T cells and M1 macrophages [89].
The gut microbiome involved in several processes: it modulates inflammation, influences cell genomic stability through the dysregulation of different pathways, but it is also linked to the progression of cancer by acting on estrogens metabolism through enterohepatic circulation [90].
It has been suggested that some microbes may play a role in breast carcinogenesis by promoting anti-cancer immunity and immune surveillance and/or modulating systemic estrogens levels [1,91].
The associations between BC and estrogen levels could reflect differences within people’s intestinal microbial communities [92], as demonstrated by Adlercreutz and collaborators 50 years ago, who demonstrated the vital role of the gut microbiota [93]. Even more recent is the Fuhrman and co-workers’ study which shows that postmenopausal estrogen metabolism is associated with microbial diversity [94]. The investigation of the connection between breast cancer and gut microbiota are still are still ongoing.
The so-called ‘estrobolome’, comprised of enteric bacterial genes that produce estrogen and its metabolites, was extensively discussed by Plottel and Blaser [95] in 2011. Elevated levels of circulating estrogens and their metabolites can result from perturbations in the microbiota/estrobolome, which can increase the risk of breast cancer. Clinical research has proven that the gut microbiota and urinary estrogens and estrogen metabolites are associated in a positive way [94,96]. Estrogens are metabolized in the liver, where they are conjugated and expelled with the bile into the lumen of gastrointestinal tract; there, they are de-conjugated by bacterial β-glucuronidase, and then they are reabsorbed as free estrogens through enterohepatic circulation, reaching different organs like the breast [90]. These metabolic products are generated through estrogen metabolism made by some bacteria inside the Clostridia Ruminococcaceae families [90]. Additionally, there are other metabolites similar to estrogen that are produced by gut oxidative and reductive reaction and by induced synthesis of estrogen-inducible growth factors, which could play a carcinogenic role.
Furthermore, bacterial β-glucuronidase may be involved in the deconjugation of xenobiotics and/or xenoestrogens, resulting in their reuptake through the enterohepatic pathway and consequently extending the duration of their stay in the body [97].
The Firmicutes phylum contains two dominant subgroups, the Clostridium leptum cluster and the Clostridium coccoides cluster, where a lot of β-glucuronidase bacteria can be found. The Escherichia/Shigella bacterial group, which is a proteobacteria phylum, also has enzymes that metabolize β -glucuronidase [98].
The relationship between gut microbiome and breast cancer risk through estrogen-independent pathways has been investigated in other studies [95,99,100]. Postmenopausal BC patients had less diverse gut bacteria and a significantly altered microbiota composition, as evidenced by a case-control study comparing the fecal microbiota of BC patients with paired controls [100]. The case patients show in the gut microbiome higher levels of Clostridiaceae, Faecalibacterium, and Ruminococcaceae but lower of Dorea and Lachnospiraceae. Furthermore, in cancer patients the fecal microbiota was less various (α-diversity). Total estrogens are connected to α-diversity only in control patients, but not in case patients and it was surprising to discover that cancer patients had higher, but not statistically significant, levels of systemic estrogens. A possible reason could be found in the other known risk factors for breast cancer like adiposity and obesity, because in these situations the microbiota appears to be less diversified [101].
It is interesting to note that bacteria like Listeria fleischmannii were highly associated with genes responsible for epithelial-to-mesenchymal transition, while Haemophilus influenza was linked to pathways involving tumor growth, cell cycle progression, E2F signaling, and mitotic spindle assembly. These discoveries indicate that the microbiome composition of tumors can be linked to certain intrinsic traits. Despite this, further investigation is necessary for this type of investigation. It is not clear if there is a connection between certain bacteria and mutations that breast cancer cells harbor. The fact that Escherichia coli, Staphylococcus, and Bacterioides fragilis isolated from breast tumors have been described as having clear genotoxic activity is what makes this topic particularly fascinating [4].
In their research, Urbaniak et al. examined breast tissue collected from different parts of the breast in women aged 18 to 90, with or without cancer, and some subjects did not have a history of lactation. The population of bacteria was found to be diverse, but Proteobacteria was the main phylum. Viable bacteria were found in certain samples through culture, despite the absence of signs or symptoms among the subjects [2].
From the analogy of normal adjacent tissue of BC woman and tissue of non-cancer women was found a significant increase, in healthy patients, of Prevotella, Lactococcus, Streptococcus, Corynebacterium, and Micrococcus, whereas in patients with BC of Bacillus, Staphylococcus, Enterobacteriaceae, Comamondaceae, and Bacteroidetes [102].
These bacteria can cause damage to DNA in vitro. In addition, it was detected a reduction of some lactic acid bacteria, which are well known for their beneficial health results, like anti-carcinogenic qualities [102]. According to these findings, the bacteria or their components may impact the local immune microenvironment, and there may be an unsuspected link between dysbiosis and breast cancer [103].
The Cancer Genome Atlas provided Thompson and co-workers [104] with the information needed to characterize the breast microbiota in 668 breast tumor tissues and 72 non-cancerous adjacent tissues. They also reported potential microbial compositional shifts among the various disease subtypes. Proteobacteria was the most prevalent phylum found in tumor sites (48%), while in the vagina, oral cavity, skin, gastrointestinal tract and bladder the Proteobacteria phylum represents only a small percentage of the global bacterial community, unlike the breast where it is the most abundant phylum [82,105,106,107]. Actinobacteria and Firmicutes were less represented, at 26.3% and 16.2% respectively. Results coherent with past outcome from Hieken et al. [9] and Urbaniak et al. [4]. In tumor samples there was a difference in the abundance of two prevalent species, Mycobacterium fortuitum and Mycobacterium phlei. The presence of Proteobacteria in tumor tissue samples increased, while Actinobacteria was also observed in non-cancerous adjacent tissue samples [104].
A significant number of viruses have been found in breast tumor tissue and/or TME, along with bacteria, parasites, and fungi. Certain authors have suggested that some of these viral signatures could be linked to specific breast cancer subtypes [108].
According to Banerjee and co-workers, the analysis of 100 triple negative breast cancer samples revealed predominant viral, bacterial, fungal, and parasitic genomic sequence signatures, which were significantly present in the analysis of 17 matched and 20 non-matched control samples.
Actinomycetaceae, Caulobacteriaceae, Sphingobacteriaceae, Enterobacteriaceae, Prevotellaceae, Brucellaceae, Bacillaceae, Peptostreptococcaceae and Flavobacteriaceae were among the many families linked to cancer [109].
4. Fusobacterium Nucleatum and Its Onco-Immunomodulatory Role in Breast Cancer
The presence of Fusobacterium nucleatum (F. nucleatum) in human breast cancer tissue has been established undoubtedly in recent years, and its capacity to accelerate breast cancer progression has been proven in murine models [81]. The involvement of F. nucleaum in the development of BC as a risk factor was inferred by Hieken et al. through experiments and risk meta-analysis [110]. To understand how F. nucleatum colonizes BC, it is essential to recognize that BC creates a TME favorable for F. nucleatum to grow [111].
F. nucleatum is a common oral gram-negative anaerobe associated with a wide spectrum of human diseases as periodontal disease [112,113,114], adverse pregnancy outcomes [115,116,117], rheumatoid arthritis [118], inflammatory bowel disease [119,120]. Its association with cancer was later established. It was first identified in colorectal cancer [121,122,123,124,125], then was also associated with oral, pancreatic, esophageal, gastric, cervical and breast cancers [126,127,128,129,130,131,132]. Hence the term of oncobacterium.
In the colon model, it has been hypothesized that the F. nucleatum reaches the organ through the hematogenous route rather than the gastrointestinal route [133]. This transient bacteremia is recurrent in patients with periodontal disease. But it is also likely that oral fusobacteria can enter the circulatory system even during trivial injuries during daily hygiene.
At first F. nucleatum binds to vascular endothelial cadherin (VE-cadherin) through the surface adhesin A (FadA) that promotes the penetration of F. nucleatum into endothelial cells [134,135]. Upon reaching the host cell, F. nucleatum binds via its autotransporter protein 2 (Fap2). This protein specifically recognizes Gal-N-acetylgalactosamine (Gal-GalNAc), a sugar abundantly exposed by colorectal cells [133]. It is very likely that F. nucleatum may also reach Gal-GalNAc-displaying tumors in other organs via this route. In breast cancer F. nucleatum has been reported to be overabundant as well as Gal-GalNAc. This suggests a colonization mechanism of F. nucleatum like colon cancer [9,136,137]. F. nucleatum enrichment in breast cancer is largely due to Fap2 as demonstrated in Fap2-deficient mutant mice that exhibited a decrease in bacterial colonization compared to Fap2 WT mice [132].
There are three possible routes through which Fusobacterium translocates to the mammary gland contributing to carcinogenesis [138]. 1) Due to bleeding gums during periodontal disease, F. nucleatum via transient bacteriemia penetrates VE-cadherin using its surface FadA which disrupts tight junctions, increasing permeability. Then the surface lectin Fap2 recognizes GalNAc overexpressed in tumor cells and it enters BC cells through rich blood supply. Furthermore, F. nucleatum activates p38 inducing the production of MMPs by tumor cells, which in turn activates the invasion, angiogenesis, and metastasis pathways [139]. 2) Breast-gut axis. Oral F. nucleatum arrives the gut through circulation or the lymphatic system. It opens the tight junctions between intestinal epithelial cells, dendritic cells and CD18+ cells facilitating their translocation from the intestinal lumen to the breast, especially during pregnancy and lactation [9]. In breast tissue F. nucleatum Fap2 interacts with Gal-GalNAc to enter breast cancer cells. 3) Direct contact with nipple/areola during breastfeeding or sexual activity, oral F. nucleatum gets access to enter mammary tissue. The nipple-areolar openings represent a potential opportunity for bacteria from the skin and oral cavity to populate the breast tissue during breastfeeding or sexual intercourse [9].
F. nucleatum contributes to carcinogenesis by increasing tumor cell proliferation and inhibiting tumor cell apoptosis [96,138,140,141].
In details, F. nucleatum induces a proinflammatory microenvironment that becomes a TME after its binding with E-cadherin. It makes FadA activating the Wnt/β-catenin signaling pathway, thus increasing transcription of nuclear factor-κβ (NF-κβ), oncogenes cyclin D1 and c-Myc [121,122]. F. nucleatum promotes tumor proliferation not only by activating the Wnt/ β-catenin pathway but also by inhibiting apoptosis. F. nucleatum also activates β-catenin signaling through the TLR4/PAK1 cascade [142]. The binding of F. nucleatum lipopolysaccharide (LPS) to TLR4 activates NF-κβ which upregulates the expression of genes that inhibit caspase 8. This represses apoptosis [138]. Since TLR4 is highly expressed in BC cells, it is conceivable that F. nucleatum may also promote BC progression through a similar, TLR4-dependent mechanism [143].
In addition to contributing to carcinogenesis by activating the oncogenetic pathways described above F. nucleatum modulates immunity against mammary tumors by manipulating antitumor immunity. Several studies have found that F. nucleatum mediates the development and immune evasion in BC although specific connection of the gut-mammary in the immune system awaits further investigation. Is it known that in colon cancer cell, Fap2 of F. nucleatum binds the human immune receptor named “T cell immunoreceptor with Ig and ITIM domains “(TIGIT), present on Tumor Infiltrating Lymphocytes (TILs) [144]. TIGIT regulates T-cell mediated immunity. This binding inhibits T cell cytotoxic activity on tumor cells, protecting them from immune cell attack.
Another way by which F. nucleatum inhibits T cell function is through the recruitment of myeloid suppressor cells (MDSCs) [143]. This may be due to increased expression of immune checkpoint inhibitors such as CD47 (cluster of differentiation 47, that inhibits phagocytosis and cytotoxicity of phagocytes on host and cancer cells) and matrix metalloproteinase 9 (MMP-9). Increased infiltration of MDSCs into the tumor environment is associated with cancer progression as MDSCs have potent immune inhibitory effects. These cells deplete the extracellular environment of amino acids such as arginine and cysteine required for T cell proliferation. MDSCs express PD-L1, which further reduces T cell activation. Upregulated PD-L1 binds to PD-1 on T cells and inhibits their proliferation.
In addition, the MYC upregulation by F.nucleatum positively stimulates the PD-L1 and CD47 expression in cancer cells [145]. Finally, F. nucleatum recruitment of MDSCs into the TME has dual outcome, leading to immune suppression and tumor progression [143].
Furthermore, F. nucleatum also binds a carcinoembryonic antigen-related cell adhesion molecules (CEACAM1) expressed by T and natural killer (NK) cells, and thus downregulating the antitumor immune response [144]. This binding is mediated by a trimeric auto-transmitting adhesive called CEACAM binding protein of Fusobacterium (CbpF) [144]. By means of these mechanisms F. nucleatum creates an escaping of tumor cell and thus a TME with immunosuppressive activity.
In breast cancer tissues, TIGIT and CEACAM1 expressions resulted upregulated and downregulated, respectively [146,147]. In early-stage breast cancer, TIGIT was highly co-expressed with other immune checkpoint receptors, like PD-1, cytotoxic T lymphocyte–associated antigen 4 (CTLA-4), LAG-3, and TIM-3. High levels of TIGIT and CTLA-4 are associated with favorable prognosis with longer overall survival and relapse-free survival. Yet, the therapeutic use of antibodies against these receptors has not shown good efficacy but rather high toxicity [113,146].
In breast cancer, F. nucleatum promotes expression of both PD-L1 and CD47 [148]. PD-L1 binds to PD-1 expressed on T cells, thereby inhibiting T cell proliferation, and reducing the secretion of cytokine. Thus, the PD-1/PD-L1 axis is thought to be responsible for cancer immune escape [148,149]. Furthermore, F. nucleatum helps breast cancer cells to evade killing by CD8+ T lymphocytes through the NFkb/PD-L1 pathway [140]. By this way, F. nucleatum inhibits immune responses and promotes self-tolerance.
5. Conclusions and Future Perspectives
F.nucleatum promotes breast carcinogenesis by a dual mechanism. On the one hand, it promotes cell proliferation and inhibits apoptosis. On the other hand, it inhibits the immune response.
In conclusion, F.nucleatum might modulate breast tumor immune microenvironment (TIME) through activating the human inhibitory receptors TIGIT and CEACAM1, upregulating CD47 and PD-L1 expression to promoting immune tolerance, recruiting myeloid-derived suppressor Cells (MDSCs) for immune migration. F. nucleatum recruits MDSCs to the TME which leads to immune suppression and tumour progression.
Considering these results, which demonstrate the presence of F. nucleatum in human breast cancer, targeting F. nucleatum may hold potential as a therapeutic strategy to enhance cancer treatment.
The experimental therapeutic use of antibodies against TIGIT and CTLA-4 receptors has not given good results, while di use of PD-L1 and CD-47 antagonists might also be more effective in combination with the eradication of F. nucleatum. However, at present, no clinical research has been conducted on the possible role of the bacterium in the treatment outcome of human breast cancer. Therefore, further investigation of the biology of F. nucleatum and its interactions with TME and with breast cancer cells harboring mutations is needed.
Author Contributions
Conceptualization and supervision, M.C.C.; methodology, A.Z., M.M.; investigation, A.P.; writing review and editing, A.D., G.M.A., M.C.C. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Xuan, C., et al., Microbial dysbiosis is associated with human breast cancer. PLoS One, 2014. 9(1): p. e83744.
- Urbaniak, C., et al., Microbiota of human breast tissue. Appl Environ Microbiol, 2014. 80(10): p. 3007-14. [CrossRef]
- O’Connor, H., et al., Resident bacteria in breast cancer tissue: pathogenic agents or harmless commensals? Discov Med, 2018. 26(142): p. 93-102.
- Urbaniak, C., et al., The Microbiota of Breast Tissue and Its Association with Breast Cancer. Appl Environ Microbiol, 2016. 82(16): p. 5039-48.
- Fernandez, L., et al., The human milk microbiota: origin and potential roles in health and disease. Pharmacol Res, 2013. 69(1): p. 1-10. [CrossRef]
- Smith, A., et al., Distinct microbial communities that differ by race, stage, or breast-tumor subtype in breast tissues of non-Hispanic Black and non-Hispanic White women. Sci Rep, 2019. 9(1): p. 11940.
- Yatsunenko, T., et al., Human gut microbiome viewed across age and geography. Nature, 2012. 486(7402): p. 222-7. [CrossRef]
- Tzeng, A., et al., Human breast microbiome correlates with prognostic features and immunological signatures in breast cancer. Genome Med, 2021. 13(1): p. 60. [CrossRef]
- Hieken, T.J., et al., The Microbiome of Aseptically Collected Human Breast Tissue in Benign and Malignant Disease. Sci Rep, 2016. 6: p. 30751.
- Fitzstevens, J.L., et al., Systematic Review of the Human Milk Microbiota. Nutr Clin Pract, 2017. 32(3): p. 354-364. [CrossRef]
- Pannaraj, P.S., et al., Association Between Breast Milk Bacterial Communities and Establishment and Development of the Infant Gut Microbiome. JAMA Pediatr, 2017. 171(7): p. 647-654. [CrossRef]
- Gomez-Gallego, C., et al., The human milk microbiome and factors influencing its composition and activity. Semin Fetal Neonatal Med, 2016. 21(6): p. 400-405. [CrossRef]
- Milani, C., et al., The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol Mol Biol Rev, 2017. 81(4). [CrossRef]
- Ward, T.L., et al., Human milk metagenome: a functional capacity analysis. BMC Microbiol, 2013. 13: p. 116. [CrossRef]
- Makino, H., et al., Transmission of intestinal Bifidobacterium longum subsp. longum strains from mother to infant, determined by multilocus sequencing typing and amplified fragment length polymorphism. Appl Environ Microbiol, 2011. 77(19): p. 6788-93. [CrossRef]
- Boix-Amoros, A., M.C. Collado, and A. Mira, Relationship between Milk Microbiota, Bacterial Load, Macronutrients, and Human Cells during Lactation. Front Microbiol, 2016. 7: p. 492.
- Rodriguez, J.M., The origin of human milk bacteria: is there a bacterial entero-mammary pathway during late pregnancy and lactation? Adv Nutr, 2014. 5(6): p. 779-84. [CrossRef]
- Enaud, R., et al., The Gut-Lung Axis in Health and Respiratory Diseases: A Place for Inter-Organ and Inter-Kingdom Crosstalks. Front Cell Infect Microbiol, 2020. 10: p. 9. [CrossRef]
- Ferretti, P., et al., Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe, 2018. 24(1): p. 133-145 e5. [CrossRef]
- Arrieta, M.C., et al., The intestinal microbiome in early life: health and disease. Front Immunol, 2014. 5: p. 427. [CrossRef]
- Cabrera-Rubio, R., et al., The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am J Clin Nutr, 2012. 96(3): p. 544-51. [CrossRef]
- Tsuji, H., et al., Molecular monitoring of the development of intestinal microbiota in Japanese infants. Benef Microbes, 2012. 3(2): p. 113-25. [CrossRef]
- Kumar, H., et al., Distinct Patterns in Human Milk Microbiota and Fatty Acid Profiles Across Specific Geographic Locations. Front Microbiol, 2016. 7: p. 1619. [CrossRef]
- Plaza-Diaz, J., L. Fontana, and A. Gil, Human Milk Oligosaccharides and Immune System Development. Nutrients, 2018. 10(8). [CrossRef]
- Kirmiz, N., et al., Milk Glycans and Their Interaction with the Infant-Gut Microbiota. Annu Rev Food Sci Technol, 2018. 9: p. 429-450. [CrossRef]
- Azad, M.B., et al., Infant gut microbiota and food sensitization: associations in the first year of life. Clin Exp Allergy, 2015. 45(3): p. 632-43. [CrossRef]
- Jimenez, E., et al., Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Curr Microbiol, 2005. 51(4): p. 270-4. [CrossRef]
- Collado, M.C., et al., Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci Rep, 2016. 6: p. 23129. [CrossRef]
- Davis, E.C., et al., Gut microbiome in the first 1000 days and risk for childhood food allergy. Ann Allergy Asthma Immunol, 2024. 133(3): p. 252-261. [CrossRef]
- Forbes, J.D., et al., A comparative study of the gut microbiota in immune-mediated inflammatory diseases-does a common dysbiosis exist? Microbiome, 2018. 6(1): p. 221. [CrossRef]
- Moossavi, S., et al., Composition and Variation of the Human Milk Microbiota Are Influenced by Maternal and Early-Life Factors. Cell Host Microbe, 2019. 25(2): p. 324-335 e4. [CrossRef]
- Rodriguez, J.M., L. Fernandez, and V. Verhasselt, The Gut‒Breast Axis: Programming Health for Life. Nutrients, 2021. 13(2). [CrossRef]
- Sung, H., et al., Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin, 2021. 71(3): p. 209-249. [CrossRef]
- Friebel, T.M., S.M. Domchek, and T.R. Rebbeck, Modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: systematic review and meta-analysis. J Natl Cancer Inst, 2014. 106(6): p. dju091. [CrossRef]
- Mavaddat, N., et al., Distribution of age at natural menopause, age at menarche, menstrual cycle length, height and BMI in BRCA1 and BRCA2 pathogenic variant carriers and non-carriers: results from EMBRACE. Breast Cancer Res, 2025. 27(1): p. 87. [CrossRef]
- Aceto, G.M., et al., Germline TP53 mutation spectrum in Sudanese premenopausal breast cancer patients: correlations with reproductive factors. Breast Cancer Res Treat, 2019. 175(2): p. 479-485. [CrossRef]
- Loibl, S., et al., Breast cancer. Lancet, 2021. 397(10286): p. 1750-1769.
- Toss, A. and M. Cristofanilli, Molecular characterization and targeted therapeutic approaches in breast cancer. Breast Cancer Res, 2015. 17(1): p. 60. [CrossRef]
- Xiong, X., et al., Breast cancer: pathogenesis and treatments. Signal Transduct Target Ther, 2025. 10(1): p. 49.
- Rebbeck, T.R., et al., Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum Mutat, 2018. 39(5): p. 593-620. [CrossRef]
- Veschi, S., et al., High prevalence of BRCA1 deletions in BRCAPRO-positive patients with high carrier probability. Ann Oncol, 2007. 18 Suppl 6: p. vi86-92. [CrossRef]
- Newman, L., US Preventive Services Task Force Breast Cancer Recommendation Statement on Risk Assessment, Genetic Counseling, and Genetic Testing for BRCA-Related Cancer. JAMA Surg, 2019. 154(10): p. 895-896. [CrossRef]
- Anaclerio, F., et al., Clinical usefulness of NGS multi-gene panel testing in hereditary cancer analysis. Front Genet, 2023. 14: p. 1060504. [CrossRef]
- Rizzolo, P., et al., Contribution of MUTYH Variants to Male Breast Cancer Risk: Results From a Multicenter Study in Italy. Front Oncol, 2018. 8: p. 583. [CrossRef]
- Wang, Y., et al., Association Between Polymorphisms in DNA Damage Repair Pathway Genes and Female Breast Cancer Risk. DNA Cell Biol, 2024. 43(5): p. 219-231. [CrossRef]
- Bouras, E., et al., Circulating inflammatory cytokines and risk of five cancers: a Mendelian randomization analysis. BMC Med, 2022. 20(1): p. 3. [CrossRef]
- Moscatello, C., et al., Relationship between MUTYH, OGG1 and BRCA1 mutations and mRNA expression in breast and ovarian cancer predisposition. Mol Clin Oncol, 2021. 14(1): p. 15.
- Donovan, M.G., et al., Dietary fat and obesity as modulators of breast cancer risk: Focus on DNA methylation. Br J Pharmacol, 2020. 177(6): p. 1331-1350. [CrossRef]
- Ming, R., et al., Causal effects and metabolites mediators between immune cell and risk of breast cancer: a Mendelian randomization study. Front Genet, 2024. 15: p. 1380249. [CrossRef]
- Davies, H., et al., HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med, 2017. 23(4): p. 517-525. [CrossRef]
- Chopra, N., et al., Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat Commun, 2020. 11(1): p. 2662. [CrossRef]
- Kuchenbaecker, K.B., et al., Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA, 2017. 317(23): p. 2402-2416.
- Kim, J. and P.N. Munster, Estrogens and breast cancer. Ann Oncol, 2025. 36(2): p. 134-148.
- Sorlie, T., et al., Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A, 2001. 98(19): p. 10869-74. [CrossRef]
- Goldhirsch, A., et al., Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann Oncol, 2013. 24(9): p. 2206-23. [CrossRef]
- Tan, P.H., et al., The 2019 World Health Organization classification of tumours of the breast. Histopathology, 2020. 77(2): p. 181-185. [CrossRef]
- De Schepper, M., et al., Results of a worldwide survey on the currently used histopathological diagnostic criteria for invasive lobular breast cancer. Mod Pathol, 2022. 35(12): p. 1812-1820.
- Risom, T., et al., Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell, 2022. 185(2): p. 299-310 e18.
- Yersal, O. and S. Barutca, Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J Clin Oncol, 2014. 5(3): p. 412-24. [CrossRef]
- Lim, S.K., et al., Impact of Molecular Subtype Conversion of Breast Cancers after Neoadjuvant Chemotherapy on Clinical Outcome. Cancer Res Treat, 2016. 48(1): p. 133-41. [CrossRef]
- Perou, C.M., et al., Molecular portraits of human breast tumours. Nature, 2000. 406(6797): p. 747-52. [CrossRef]
- Fico, F. and A. Santamaria-Martinez, The Tumor Microenvironment as a Driving Force of Breast Cancer Stem Cell Plasticity. Cancers (Basel), 2020. 12(12). [CrossRef]
- Gupta, T., S. Vinayak, and M. Telli, Emerging strategies: PARP inhibitors in combination with immune checkpoint blockade in BRCA1 and BRCA2 mutation-associated and triple-negative breast cancer. Breast Cancer Res Treat, 2023. 197(1): p. 51-56. [CrossRef]
- Bottosso, M., et al., Moving toward precision medicine to predict drug sensitivity in patients with metastatic breast cancer. ESMO Open, 2024. 9(3): p. 102247. [CrossRef]
- Lukasiewicz, S., et al., Breast Cancer-Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies-An Updated Review. Cancers (Basel), 2021. 13(17).
- Rossing, M., et al., Clinical implications of intrinsic molecular subtypes of breast cancer for sentinel node status. Sci Rep, 2021. 11(1): p. 2259. [CrossRef]
- Liu, J., et al., The prognostic role of lymph node ratio in breast cancer patients received neoadjuvant chemotherapy: A dose-response meta-analysis. Front Surg, 2022. 9: p. 971030. [CrossRef]
- Lei, P.J., et al., Cancer cell plasticity and MHC-II-mediated immune tolerance promote breast cancer metastasis to lymph nodes. J Exp Med, 2023. 220(9). [CrossRef]
- Reticker-Flynn, N.E., et al., Lymph node colonization induces tumor-immune tolerance to promote distant metastasis. Cell, 2022. 185(11): p. 1924-1942 e23. [CrossRef]
- Si, H., et al., The covert symphony: cellular and molecular accomplices in breast cancer metastasis. Front Cell Dev Biol, 2023. 11: p. 1221784. [CrossRef]
- Terry, M.B., et al., Environmental exposures during windows of susceptibility for breast cancer: a framework for prevention research. Breast Cancer Res, 2019. 21(1): p. 96. [CrossRef]
- Arnone, A.A., et al., Diet Modulates the Gut Microbiome, Metabolism, and Mammary Gland Inflammation to Influence Breast Cancer Risk. Cancer Prev Res (Phila), 2024. 17(9): p. 415-428. [CrossRef]
- Alpuim Costa, D., et al., Human Microbiota and Breast Cancer-Is There Any Relevant Link?-A Literature Review and New Horizons Toward Personalised Medicine. Front Microbiol, 2021. 12: p. 584332. [CrossRef]
- Hu, S., et al., Gut microbial beta-glucuronidase: a vital regulator in female estrogen metabolism. Gut Microbes, 2023. 15(1): p. 2236749. [CrossRef]
- Arnone, A.A. and K.L. Cook, Gut and Breast Microbiota as Endocrine Regulators of Hormone Receptor-positive Breast Cancer Risk and Therapy Response. Endocrinology, 2022. 164(1). [CrossRef]
- Menon, G., F.M. Alkabban, and T. Ferguson, Breast Cancer, in StatPearls. 2025: Treasure Island (FL).
- Dethlefsen, L., M. McFall-Ngai, and D.A. Relman, An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature, 2007. 449(7164): p. 811-8. [CrossRef]
- Willing, B.P., S.L. Russell, and B.B. Finlay, Shifting the balance: antibiotic effects on host-microbiota mutualism. Nat Rev Microbiol, 2011. 9(4): p. 233-43.
- Arendt, L.M., et al., Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res, 2013. 73(19): p. 6080-93.
- Buchta Rosean, C., et al., Preexisting Commensal Dysbiosis Is a Host-Intrinsic Regulator of Tissue Inflammation and Tumor Cell Dissemination in Hormone Receptor-Positive Breast Cancer. Cancer Res, 2019. 79(14): p. 3662-3675. [CrossRef]
- Nejman, D., et al., The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science, 2020. 368(6494): p. 973-980. [CrossRef]
- Human Microbiome Project, C., Structure, function and diversity of the healthy human microbiome. Nature, 2012. 486(7402): p. 207-14.
- Costello, E.K., et al., Bacterial community variation in human body habitats across space and time. Science, 2009. 326(5960): p. 1694-7. [CrossRef]
- Meng, Z., et al., New Developments and Opportunities of Microbiota in Treating Breast Cancers. Front Microbiol, 2022. 13: p. 818793.
- Davis, C.P., et al., Urothelial hyperplasia and neoplasia. III. Detection of nitrosamine production with different bacterial genera in chronic urinary tract infections of rats. J Urol, 1991. 145(4): p. 875-80. [CrossRef]
- Costantini, L., et al., Characterization of human breast tissue microbiota from core needle biopsies through the analysis of multi hypervariable 16S-rRNA gene regions. Sci Rep, 2018. 8(1): p. 16893. [CrossRef]
- Thu, M.S., et al., Human gut, breast, and oral microbiome in breast cancer: A systematic review and meta-analysis. Front Oncol, 2023. 13: p. 1144021.
- Wang, H., et al., The microbial metabolite trimethylamine N-oxide promotes antitumor immunity in triple-negative breast cancer. Cell Metab, 2022. 34(4): p. 581-594 e8. [CrossRef]
- Hix, L.M., et al., CD1d-expressing breast cancer cells modulate NKT cell-mediated antitumor immunity in a murine model of breast cancer metastasis. PLoS One, 2011. 6(6): p. e20702.
- Rea, D., et al., Microbiota effects on cancer: from risks to therapies. Oncotarget, 2018. 9(25): p. 17915-17927. [CrossRef]
- Wang, H., et al., Breast tissue, oral and urinary microbiomes in breast cancer. Oncotarget, 2017. 8(50): p. 88122-88138. [CrossRef]
- Kwa, M., et al., The Intestinal Microbiome and Estrogen Receptor-Positive Female Breast Cancer. J Natl Cancer Inst, 2016. 108(8).
- Adlercreutz, H. and F. Martin, Biliary excretion and intestinal metabolism of progesterone and estrogens in man. J Steroid Biochem, 1980. 13(2): p. 231-44. [CrossRef]
- Fuhrman, B.J., et al., Associations of the fecal microbiome with urinary estrogens and estrogen metabolites in postmenopausal women. J Clin Endocrinol Metab, 2014. 99(12): p. 4632-40. [CrossRef]
- Plottel, C.S. and M.J. Blaser, Microbiome and malignancy. Cell Host Microbe, 2011. 10(4): p. 324-35. [CrossRef]
- D’Antonio, D.L., et al., The Oncobiome in Gastroenteric and Genitourinary Cancers. Int J Mol Sci, 2022. 23(17). [CrossRef]
- Yang, J., et al., Gastrointestinal microbiome and breast cancer: correlations, mechanisms and potential clinical implications. Breast Cancer, 2017. 24(2): p. 220-228. [CrossRef]
- Dabek, M., et al., Distribution of beta-glucosidase and beta-glucuronidase activity and of beta-glucuronidase gene gus in human colonic bacteria. FEMS Microbiol Ecol, 2008. 66(3): p. 487-95.
- Goedert, J.J., et al., Postmenopausal breast cancer and oestrogen associations with the IgA-coated and IgA-noncoated faecal microbiota. Br J Cancer, 2018. 118(4): p. 471-479. [CrossRef]
- Goedert, J.J., et al., Investigation of the association between the fecal microbiota and breast cancer in postmenopausal women: a population-based case-control pilot study. J Natl Cancer Inst, 2015. 107(8). [CrossRef]
- Backhed, F., et al., The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A, 2004. 101(44): p. 15718-23. [CrossRef]
- Koller, V.J., et al., Impact of lactic acid bacteria on oxidative DNA damage in human derived colon cells. Food Chem Toxicol, 2008. 46(4): p. 1221-9. [CrossRef]
- Zhang, J., Y. Xia, and J. Sun, Breast and gut microbiome in health and cancer. Genes Dis, 2021. 8(5): p. 581-589. [CrossRef]
- Thompson, K.J., et al., A comprehensive analysis of breast cancer microbiota and host gene expression. PLoS One, 2017. 12(11): p. e0188873.
- Wolfe, A.J., et al., Evidence of uncultivated bacteria in the adult female bladder. J Clin Microbiol, 2012. 50(4): p. 1376-83. [CrossRef]
- Hummelen, R., et al., Deep sequencing of the vaginal microbiota of women with HIV. PLoS One, 2010. 5(8): p. e12078. [CrossRef]
- Grice, E.A., et al., Topographical and temporal diversity of the human skin microbiome. Science, 2009. 324(5931): p. 1190-2. [CrossRef]
- Banerjee, S., et al., Distinct Microbial Signatures Associated With Different Breast Cancer Types. Front Microbiol, 2018. 9: p. 951.
- Banerjee, S., et al., Distinct microbiological signatures associated with triple negative breast cancer. Sci Rep, 2015. 5: p. 15162.
- Gaba, F.I., R.C. Gonzalez, and R.G. Martinez, The Role of Oral Fusobacterium nucleatum in Female Breast Cancer: A Systematic Review and Meta-Analysis. Int J Dent, 2022. 2022: p. 1876275. [CrossRef]
- Wei, M.Q., et al., Facultative or obligate anaerobic bacteria have the potential for multimodality therapy of solid tumours. Eur J Cancer, 2007. 43(3): p. 490-6. [CrossRef]
- Saygun, I., et al., Salivary infectious agents and periodontal disease status. J Periodontal Res, 2011. 46(2): p. 235-9. [CrossRef]
- Liu, P., et al., Detection of fusobacterium nucleatum and fadA adhesin gene in patients with orthodontic gingivitis and non-orthodontic periodontal inflammation. PLoS One, 2014. 9(1): p. e85280. [CrossRef]
- Yang, N.Y., et al., Progression of periodontal inflammation in adolescents is associated with increased number of Porphyromonas gingivalis, Prevotella intermedia, Tannerella forsythensis, and Fusobacterium nucleatum. Int J Paediatr Dent, 2014. 24(3): p. 226-33.
- Han, Y.W., et al., Term stillbirth caused by oral Fusobacterium nucleatum. Obstet Gynecol, 2010. 115(2 Pt 2): p. 442-445. [CrossRef]
- Gauthier, S., et al., The origin of Fusobacterium nucleatum involved in intra-amniotic infection and preterm birth. J Matern Fetal Neonatal Med, 2011. 24(11): p. 1329-32.
- Barak, S., et al., Evidence of periopathogenic microorganisms in placentas of women with preeclampsia. J Periodontol, 2007. 78(4): p. 670-6. [CrossRef]
- Temoin, S., et al., Identification of oral bacterial DNA in synovial fluid of patients with arthritis with native and failed prosthetic joints. J Clin Rheumatol, 2012. 18(3): p. 117-21. [CrossRef]
- Tahara, T., et al., Fusobacterium detected in colonic biopsy and clinicopathological features of ulcerative colitis in Japan. Dig Dis Sci, 2015. 60(1): p. 205-10. [CrossRef]
- Strauss, J., et al., Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis, 2011. 17(9): p. 1971-8. [CrossRef]
- Rubinstein, M.R., et al., Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe, 2013. 14(2): p. 195-206. [CrossRef]
- Rubinstein, M.R., et al., Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/beta-catenin modulator Annexin A1. EMBO Rep, 2019. 20(4).
- Castellarin, M., et al., Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res, 2012. 22(2): p. 299-306. [CrossRef]
- Kostic, A.D., et al., Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res, 2012. 22(2): p. 292-8. [CrossRef]
- Pignatelli, P., et al., The Potential of Colonic Tumor Tissue Fusobacterium nucleatum to Predict Staging and Its Interplay with Oral Abundance in Colon Cancer Patients. Cancers (Basel), 2021. 13(5). [CrossRef]
- Pignatelli, P., et al., The Role of Fusobacterium nucleatum in Oral and Colorectal Carcinogenesis. Microorganisms, 2023. 11(9). [CrossRef]
- D’Antonio, D.L., et al., Intratumoral Fusobacterium nucleatum in Pancreatic Cancer: Current and Future Perspectives. Pathogens, 2024. 14(1). [CrossRef]
- Hayashi, M., et al., Intratumor Fusobacterium nucleatum promotes the progression of pancreatic cancer via the CXCL1-CXCR2 axis. Cancer Sci, 2023. 114(9): p. 3666-3678. [CrossRef]
- Yamamura, K., et al., Human Microbiome Fusobacterium Nucleatum in Esophageal Cancer Tissue Is Associated with Prognosis. Clin Cancer Res, 2016. 22(22): p. 5574-5581. [CrossRef]
- Chen, W.D., et al., Salivary Fusobacterium nucleatum serves as a potential diagnostic biomarker for gastric cancer. World J Gastroenterol, 2022. 28(30): p. 4120-4132. [CrossRef]
- Audirac-Chalifour, A., et al., Cervical Microbiome and Cytokine Profile at Various Stages of Cervical Cancer: A Pilot Study. PLoS One, 2016. 11(4): p. e0153274. [CrossRef]
- Parhi, L., et al., Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat Commun, 2020. 11(1): p. 3259.
- Abed, J., et al., Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe, 2016. 20(2): p. 215-25. [CrossRef]
- Fardini, Y., et al., Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol Microbiol, 2011. 82(6): p. 1468-80. [CrossRef]
- Fan, Z., et al., Fusobacterium nucleatum and its associated systemic diseases: epidemiologic studies and possible mechanisms. J Oral Microbiol, 2023. 15(1): p. 2145729. [CrossRef]
- Kolbl, A.C., et al., The role of TF- and Tn-antigens in breast cancer metastasis. Histol Histopathol, 2016. 31(6): p. 613-21.
- Abed, J., et al., Tumor Targeting by Fusobacterium nucleatum: A Pilot Study and Future Perspectives. Front Cell Infect Microbiol, 2017. 7: p. 295. [CrossRef]
- Guo, X., K. Yu, and R. Huang, The ways Fusobacterium nucleatum translocate to breast tissue and contribute to breast cancer development. Mol Oral Microbiol, 2024. 39(1): p. 1-11. [CrossRef]
- Mehner, C., et al., Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget, 2014. 5(9): p. 2736-49. [CrossRef]
- Guo, J., et al., Fusobacterium nucleatum promotes PD-L1 expression in cancer cells to evade CD8(+) T cell killing in breast cancer. Hum Immunol, 2024. 85(6): p. 111168. [CrossRef]
- Wang, N., et al., Intratumoral microbiome: implications for immune modulation and innovative therapeutic strategies in cancer. J Biomed Sci, 2025. 32(1): p. 23. [CrossRef]
- Chen, Y., et al., Invasive Fusobacterium nucleatum activates beta-catenin signaling in colorectal cancer via a TLR4/P-PAK1 cascade. Oncotarget, 2017. 8(19): p. 31802-31814. [CrossRef]
- Van der Merwe, M., et al., The onco-immunological implications of Fusobacterium nucleatum in breast cancer. Immunol Lett, 2021. 232: p. 60-66.
- Gur, C., et al., Fusobacterium nucleatum supresses anti-tumor immunity by activating CEACAM1. Oncoimmunology, 2019. 8(6): p. e1581531. [CrossRef]
- Casey, S.C., et al., MYC regulates the antitumor immune response through CD47 and PD-L1. Science, 2016. 352(6282): p. 227-31. [CrossRef]
- Fang, J., et al., Prognostic value of immune checkpoint molecules in breast cancer. Biosci Rep, 2020. 40(7). [CrossRef]
- Mollavelioglu, B., et al., High co-expression of immune checkpoint receptors PD-1, CTLA-4, LAG-3, TIM-3, and TIGIT on tumor-infiltrating lymphocytes in early-stage breast cancer. World J Surg Oncol, 2022. 20(1): p. 349. [CrossRef]
- Matlung, H.L., et al., The CD47-SIRPalpha signaling axis as an innate immune checkpoint in cancer. Immunol Rev, 2017. 276(1): p. 145-164.
- Han, Y., D. Liu, and L. Li, PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res, 2020. 10(3): p. 727-742.
Figure 1.
Healthy breast and inflammatory condition.
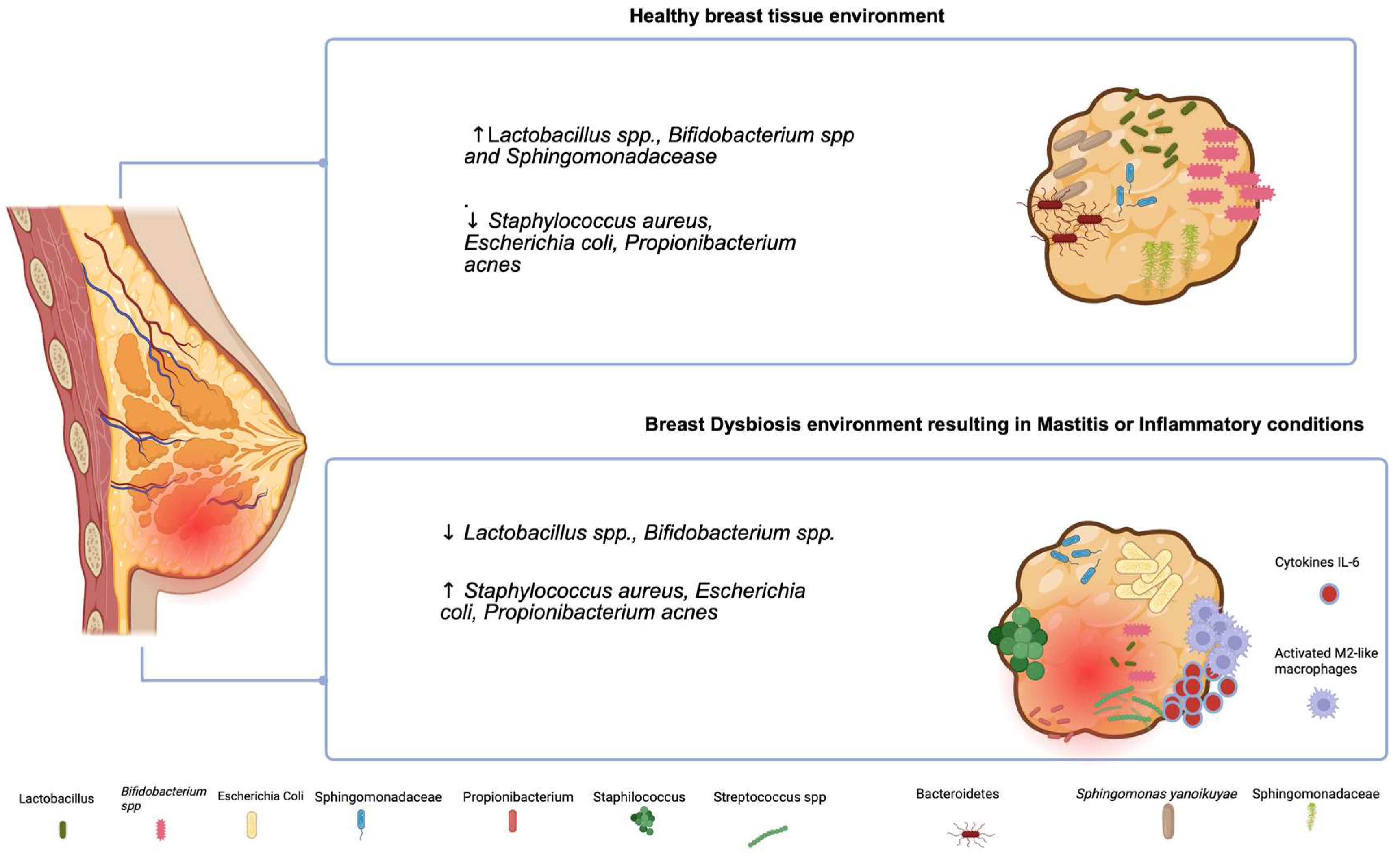
Figure 2.
Retrograde Inoculation of Fusobacterium nucleatum into the lactating Mammary Gland.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



