
Submitted:
02 June 2025
Posted:
02 June 2025
You are already at the latest version
Abstract
After spinal cord injury (SCI), axons become severed, and glial cells undergo changes in response to environmental cues, contributing to glial scar formation. Chondroitin Sulfate Proteoglycans (CSPGs) are released during glial scarring and are an underlying mechanism for the inhibitory effects on axonal regeneration in the central nervous system (CNS). This review initially focuses on CSPGs and provides a comprehensive overview of the glial scarring process. In addition to established therapeutic agents, such as chondroitinase ABC (chABC), this review addresses therapeutic approaches that target the interactions between CSPG and its transmembrane receptors on growth cones. These receptors belong to the leukocyte common antigen-related (LAR) phosphatase subfamily and the Nogo family, including protein tyrosine phosphatase sigma (PTPσ), LAR receptors, and Nogo receptor variants. This review then shifts to the efficacy of other therapeutic peptides, such as intracellular sigma peptide (ISP) and intracellular LAR peptide (ILP), on the interactions between CSPG and PTPσ and LAR receptors. Likewise, it also evaluates decoys for their effectiveness in obstructing interactions between CSPG and Nogo receptors. Additionally, it extensively discusses the impact of deleting these receptors or utilizing peptides and decoys to counteract the inhibitory effects of CSPG and its transmembrane receptors on axonal regeneration, while acknowledging the limitations of these therapeutics and extending future applications. This review aims to highlight these potential therapeutic avenues targeting CSPG to promote axonal regeneration in the CNS, elucidating their use for future treatment.
Keywords:
axonal regeneration
; chondroitin sulfate proteoglycans (CSPG)
; central nervous system (CNS)
; glial scar
; chondroitinase ABC (chABC)
; protein tyrosine phosphatase sigma (PTPσ)
; leukocyte common antigen-related (LAR)
; nogo
; intracellular sigma peptide (ISP)
; intracellular LAR peptide (ILP)
1. Introduction
Nearly a century ago, Santiago Ramon y Cajal, a pioneer in the history of modern neuroscience, proposed that nerve pathways were fixed and immutable. This proposal stemmed from the prevailing dogma that there was a lack of neuroplasticity in the adult central nervous system (CNS), making them incapable of regeneration. However, Ramon y Cajal hoped that future science would be able to alter that perception [1]. Axonal regeneration in the peripheral nervous system (PNS) occurs spontaneously with the assistance of regeneration-associated genes, glial cells, and macrophages. Yet in the CNS, axonal regeneration is minimal and does not occur spontaneously after spinal cord injury (SCI) or other CNS injuries [2].
Chondroitin sulfate proteoglycans (CSPGs) are expressed predominantly in the CNS extracellular matrix (ECM). CSPGs are well-established as crucial inhibitors for axonal regeneration and neural plasticity after SCI. Among these CSPGs, various subtypes, including the lectican family consisting of aggrecan, brevican, neurocan, and versican, are morphologically similar but differ in their core proteins. Other CSPGs, such as neuron-glial antigen 2 (NG2) associated with oligodendrocyte precursor cells (OPCs), discussed later in the review, and phosphacan, are slightly distinct; NG2 is a transmembrane proteoglycan, whereas phosphacan is a secreted splice variant of a transmembrane receptor. CSPGs of the lectican family consist of a globular domain at the N-terminus (G1) and a globular domain at the C-terminus (G3) connected by a core protein that provides the backbone for the structure of CSPGs. These domains have unique functions, as the G1 domain attaches to proteins but mainly to hyaluronan (HA), identifying them as HA-binding domains. In contrast, the G3 domain interacts primarily with tenascin, making it a tenascin-binding domain. However, NG2 and phosphacan do not contain these binding domains, distinguishing them from lecticans [3,4].
The source of the inhibitory properties attributed to CSPGs is from glycosaminoglycan (GAG) side chains, a collection of unbranched sugar chains consisting of alternating pairs of glucuronic acid and N-acetyl-galactosamine, which are responsible for exacerbating the CSPG-mediated inhibition and are a shared component of all CSPGs [3,4,5]. A specific sulfated GAG chain, the chondroitin sulfate (CS)-GAG chain, can be found in CSPG with varying CS sulfation patterns in CS chains and is integral for contributing to CSPGs’ binding affinity to signaling receptors and molecules [4]. CSPG core proteins are constructed of various amino acids; the core proteins are specifically constructed of repeating serine and glycine residues in regions where CS-GAG chains are attached. These regions are often referred to as Ser-Gly sites, which serve as the attachment sites to the CS-GAG chains [6]. These sites, anchored on the core protein through a common tetrasaccharide linker, consisting of glucuronic acid, galactose, galactose, and xylose moieties, are covalently linked to CS-GAG chains, allowing for the structural composition of CSPGs [6,7]. Figure 1 depicts the common CSPG structure among all variations constructed with a core protein and CS-GAG chains and the classification of CSPGs with subtypes and their further constituents.
CSPGs are molecules synthesized and secreted primarily by glial cells, such as astrocytes, OPCs, and microglia, along with fibroblasts and immune cells, like macrophages, in the CNS following SCI. CSPGs play critical roles in the CNS, establishing structural support and catalyzing the versatile properties of the ECM. CSPGs interact with receptors on the growth cone of regenerating neurons, influencing neuronal function through their CS-GAG chains and molecular structure. This influences the organizational function and structure of the growth cone, inducing intracellular responses that affect regeneration and regulate cellular functions related to repair. During neurite regrowth, CSPG can hinder the development of growth cones via inhibitory mechanisms following injury [8]. When injury occurs in the CNS, CSPG levels become more abundant, constructing an inhibitory barrier that hinders axonal regeneration. As reinforced previously, the distinct CS patterns of the CS-GAG chains strongly influence function, as chondroitin 6-sulfates are more permissive towards neurite outgrowth, whereas chondroitin 4-sulfates exert more inhibitory effects [9].
The purpose of this review article is to comprehensively elucidate the inhibitory behavior and effects of CSPG on the CNS after injury. Through examining current research, this article aims to explore the complex interplay of CSPGs in the formation of glial scarring, hindrance of axonal regeneration, and analysis of therapeutics targeting CSPGs to suppress them and guide axonal growth. This review delves deeper into the specific effects of protein tyrosine phosphatase sigma (PTPσ), leukocyte common antigen-related (LAR), and Nogo receptors by analyzing the cellular and molecular mechanisms underlying CSPG-mediated inhibition and their interactions with CSPGs, all of which are known to downregulate axonal regeneration. This article also examines the potential therapeutic strategies that target CSPGs to stimulate axonal extension. By providing a detailed analysis, this review seeks to spotlight the therapeutic avenues associated with modulating CSPG to overcome CNS regeneration challenges and offer efficacious therapeutic approaches to catalyze axonal regeneration.
2. The Formation of Glial Scarring and CSPGs
The glial scar remains a crucial extrinsic factor, limiting axonal regeneration capacity after SCI. Following SCI, a complex cellular response known as glial scarring is initiated, which plays a dual role in blocking nerve regeneration and preventing damaged tissue from spreading. Besides blocking axonal regrowth, glial scarring also provides several beneficial functions, such as repairing the blood-brain barrier (BBB) while limiting scar tissue spread and surplus inflammation [10]. Immediately after injury, acute response events occur as neural tissue is damaged, releasing several distress signals, including cellular debris and adenosine triphosphate, which bind to purinergic receptors on glial cells. This induces a chain of activation from certain glial cells, including microglia, astrocytes, and macrophages, to their activated forms by switching from a monitoring mode to an active response state throughout the glial scarring process [11].
Reactive astrocytes and activated macrophages are commonly categorized into two primary phenotypes: A1 and A2 for astrocytes, and M1 and M2 for macrophages; these activated states are present following SCI. A1 astrocytes and M1 macrophages are predominantly pro-inflammatory, causing synaptic dysfunction and neural apoptosis. In contrast, A2 astrocytes and M2 macrophages are associated with more anti-inflammatory and neuroprotective roles, upregulating neurotrophic factors and catalyzing nerve repair. However, this classification oversimplifies the heterogeneity of both reactive astrocytes and macrophages, as their responses are highly dependent on the situation and are more nuanced in their activation states, existing on a spectrum, rather than being completely binary [12].
Initially, narrowing attention immediately after SCI occurs, the inflammatory cascade is initiated by the release of alarmins, damage-associated molecular patterns (DAMPs), and other cytokines from severed axons. This recruits both native and invading immune cells, like macrophages and microglia, through a cascade. However, the specific functions of microglia and macrophages have been challenging because of the difficulties in distinguishing the two cell variants due to their shared morphology and protein markers. Gene expression profiling and recent advancements in microglial markers have improved their ability to differentiate their functions [12,13,14]. Following SCI, microglia secrete cytokines and chemokines which recruit immune cells, mediate inflammation, and engulf cellular debris via phagocytosis [14]. In the chronic phase of SCI with CSPG, microglia form a physical barrier between astrocytes and infiltrating leukocytes, working with astrocytes to conceal fibroblasts and immune cells by inducing further inflammation [12,15]. These microglia further release cytokines, such as insulin-like growth factor 1 (IGF-1), triggering astrocytic activation and further proliferation, contributing to the scarring process [11].
Activated astrocytes also participate in the scarring cascade by recruiting monocytes from the peripheral bloodstream via the release of cytokines and chemokines. These monocytes differentiate into macrophages as they enter the lesion site, and macrophages are distinguishable from microglia in lesion sites; they induce the retraction of dystrophic growth cones. Macrophages also phagocytose cellular debris and remove ineffective neutrophils [11,12]. These microglia-derived and monocyte-derived macrophages initially adopt a mixed phenotype after SCI of both M1 and M2 anti-inflammatory subtypes. M1 macrophages amplify astrocyte activation, promoting scar formation, whereas M2 macrophages enhance the polarization of reactive astrocytes and contribute to scar organization. However, chronically after injury with CSPG presence, macrophages tend to adopt an M1 phenotype [13,14,16]. SCI induces significant changes in glial cells, including microglia, astrocytes, and OPCs, which augment glial scar formation, hindering axonal outgrowth [17,18].
Focusing on a non-glial component of the scar in response to inflammatory cues, fibroblasts also play a role in the glial scar, worth mentioning. Fibroblasts are present in the basal laminae and parenchyma that circulate along the spinal cord, whereas fibroblast-like cells only contribute to the basal laminae. However, following SCI, these fibroblasts are activated, leading to proliferation and migration towards the site of the lesion [13,14]. The fibroblast density in the environment is also upregulated as a subset of residing pericytes surrounding the lesion area, which proliferate and differentiate into fibroblast-like cells, assisting fibrotic scar construction after injury. In response to injury, these fibroblasts produce stromal ECM components composed of various molecules such as collagen, laminin, and fibronectin [19,20]. This further amplifies glial scarring by constructing the fibrotic scar of fibroblasts and other stromal ECM components.
When astrocytes are activated by various environmental cues, such as DAMPs, IGF-1, and other inflammatory cytokines, they undergo phenotypic alterations to become reactive astrocytes outlined by cellular hypertrophy and the upregulation of intermediate filament proteins like glial fibrillary acidic protein (GFAP), nestin, and vimentin [13,21]. This process, known as reactive gliosis or astrogliosis, produces reactive astrocytes, distinguishable from regular or quiescent astrocytes by their morphology, increased hypertrophy, and upregulation of intermediate filament protein markers [13]. Several studies have investigated astrogliosis to better understand the process after SCI. For instance, Wanner et al. experimented using crush SCI in transgenic mice to examine the process of astroglial proliferation after SCI, administering GFAP fluorescence immunostaining to label proliferating astroglial cells, yielding quantitative results that the presence of astroglial cell density in the glial scar border was nearly twice that in regular uninjured tissue. These proliferating astroglia around the lesion were also discovered to morphologically exhibit elongated shapes of themselves, expressing progenitor cell-associated markers. Astroglial proliferation triggered by SCI consists of higher percentages of newly proliferated cells surrounding the lesion area, with a direct relationship through declining percentages as the distance from the lesion core increases [22].
These reactive astrocytes accumulate around the lesion core, elongate perpendicularly towards the core, and are transformed into scar-forming reactive astrocytes. These scar-forming reactive astrocytes then proliferate and accumulate to stabilize the local environment of the scar, which increases astrocytic density. Over time, the scar-forming reactive astrocytes complete their phenotypic changes, causing them to develop in parallel with the lesion core rather than in a perpendicular orientation, forming a dense, mature glial scar that impairs neural repair influenced by STAT-3 signaling [11,13,22]. This suggests that the location of astrocytes plays an essential role in their function. For instance, reactive astrocytes specifically surrounding the lesion core in proximity alter the orientation and density, elongating these astrocytes and forming scar-forming reactive astrocytes with the extension of overlapping processes, constructing the astrocytic scar [12,23].
These scar-forming reactive astrocytes secrete CSPG, which is repulsive and inhibits repair. Reactive astrocytes secrete CSPG into the environment, elevating their secretion into the ECM and interacting with other stromal ECM components to further inhibit regrowth around the glial scar. Zhang et al. conducted SCI injury in lamprey, highlighting that after astrocytes had switched to their reactive state, CSPG levels were upregulated immediately after injury, while CSPG levels appeared to surge over time in the process of glial scarring [24]. The collection of scar-forming reactive astrocytes around the lesion site forms a physical and biochemical barrier between the lesion site and the external environment, enclosing the fibrotic tissue, macrophages, and stromal ECM from enlarging the lesion, The secretion of CSPG from primarily reactive astrocytes serves as a repellent to external molecules from entering or passing the astrocytic scar [13,25]. The formation of glial scars poses both physical and chemical barriers. In the acute stages following injury, the glial scar hinders diffusion of the induced lesion cavity and appears to be protective. However, it also has chronic effects that exacerbate regeneration-blocking growth cones and upregulate CSPGs, causing proteoglycans to act as repellents and ultimately impede functional recovery [12].
Cells that express NG2, a subtype of the CSPG family, are referred to as (NG2+) cells. These cells present in glial scars are recognized for their inhibitory role in regrowth and serve as key markers for OPCs in such lesions. OPCs, a type of glial cell, are precursors to oligodendrocytes, which generate myelin sheaths and facilitate remyelination. NG2-expressing OPCs (NG2-OPCs) express NG2, which makes them, alongside reactive astrocytes, another recognizable and critical producer of CSPG. After an injury, OPCs have dual effects on the glial scar, seemingly conflicting with each other as OPCs have contributed to remyelination via differentiation into oligodendrocytes or myelinating Schwann cells, fostering a pro-regenerative environment [8,11,26]. However, these OPCs undergo hypertrophy and upregulate NG2 expression, classifying them as NG2+ cells that are detrimental to repair [11,13]. After SCI, macrophages retract dystrophic axons to the caudal end of the lesion, where they interact with NG2-OPCs. These NG2-OPCs produce inhibitory NG2, which mediates synaptic-like connections with dystrophic axons and stabilizes but entraps them, thereby hindering regeneration [27]. Further research on NG2-OPCs is necessary to gain insight into their more complex contribution to scar injury and approaches that could be utilized to modulate glial scarring. Figure 2 portrays the lesion environment with glial scarring and the presence of CSPG in the spinal cord after SCI, illustrating the primary components present at the time of CSPG presence in glial scarring.
3. Delving into the Transmembrane Receptors of CSPG
CSPGs bind to transmembrane receptors on growth cones, allowing them to inhibit axonal regeneration following SCI. The receptors discussed in this review encompass interactions between CSPG and its transmembrane receptors. Two of these receptors, PTPσ and LAR, are part of the LAR phosphatase subfamily of receptor-type protein tyrosine phosphatases (RPTPs), transmembrane receptors that mediate the detrimental impact of CSPGs on axonal growth [3,28]. Focusing on the morphology of both PTPσ and LAR receptors, these transmembrane receptors have contents that influence their high affinity for CSPGs. Extracellularly, both receptors are composed of immunoglobulin-like (Ig-like) domains and fibronectin type-III (FN-III) domains, which are composed of three single Ig-like domains at the N-terminus, below which are around four to eight FN-III repeats connected to the transmembrane, which are responsible for the complex binding to the inhibitory CS-GAG chains from CSPGs [29]. After crossing the transmembrane and analyzing the structure from an intracellular perspective, both receptors contained two cytoplasmic phosphatase domains, with the first domain (D1) being catalytically active with phosphatase activity. Conversely, the second domain (D2) has been associated with being catalytically inactive, lacking any form of phosphatase activity in the C-terminus. Various factors can alter signaling and function based on the morphological changes these receptors can undergo. When alternative splicing occurs in FN-III domains, it causes variability in the number of FN-III units connecting the extracellular Ig-like domains to the transmembrane domain [30,31]. After traveling past the transmembrane domain, the inhibitory response signal from the binding of the CS-GAG chains and these receptors triggers the activation of the D1 and D2 domains, which emit intracellular signals that inhibit axonal regrowth through these two RPTPs.
Although these two receptors belong to the same subfamily of RPTPS, there are some slight differences in the molecular structures of both PTPσ and LAR receptors. Extracellularly, the difference in the conformation of the Ig-like domains and the arrangement or quantitative value of the FN-III domains helps binding molecules, such as CSPG, to identify which receptor is PTPσ or LAR. Remarkably, in their extracellular and cytosolic domains, differences in the binding specificity of ligands and residues have also been found to have the ability to regulate their roles in these functions [30,32]. PTPσ and LAR are recognized as receptors that contribute to CSPG-induced axonal degeneration; however, these receptors can have multifaceted effects on axonal growth influenced by the binding of various extracellular ligands. For instance, heparan sulfate proteoglycans (HSPGs), a different proteoglycan with heparan sulfate (HS) chains instead of CS chains, have been able to promote neurite extension from growth cones after binding to PTPσ or LAR. Between both receptors, molecular structure variance and proteoglycan dependence can have varying effects on axonal regeneration but serve as a receptor between the extracellular and intracellular environments [28,33].
Focusing on PTPσ, this receptor is primarily recognized for its role in mediating inhibitory effects on CSPGs. However, there are more factors, such as its morphology and behavior toward CS chains, that influence its affinity for CSPG. PTPσ regulates neural development by mediating axonal growth, and its ligand binding significantly influences its function [32]. A study conducted by Coles et al. explored in vitro cultures and the inhibitory role of CS chains in mediating PTPσ and influencing neurite growth. They found that when CS chains from CSPG were present in glial scars, they could not prompt PTPσ oligomerization or clustering to form an oligomer and became a competitor for PTPσ with HS chains in HSPG, exacerbating axonal regrowth. This study suggests that not only the individual effects on CS or HS but also the relative ratio levels of CS and HS are critical in influencing axonal regrowth. All three Ig-like domains, more specifically the first Ig-like domain (Ig1), are known to be crucially influential in PTPσ interactions with CS-GAG chains. They also established the receptor as a switch model for HS-modulated clustering and CS with a high affinity for binding to Ig1, blocking PTPσ dimerization [33]. PTPσ interaction with HS chain ligands incited PTPσ dimer conformation, which inactivates phosphatase activity in the D1 and D2 domains, enhancing axonal extension. Conversely, CS chain ligands as a part of CSPG, interacting with PTPσ, have detrimentally induced PTPσ monomer formation, allowing for high phosphatase activity from the two intracellular domains of PTPσ, inhibiting axonal regeneration on growth cones [34]. Based on these studies, PTPσ can be portrayed as a nuanced receptor with its ligand playing a pivotal role in its effects on axonal regrowth.
Shifting focus to the LAR receptor, like PTPσ, mediates CSPG-inhibition on axonal regeneration. They are also molecules that modulate neural development and cytoskeletal dynamics. Like PTPσ, the LAR receptor functions with a highly extracellularly dependent framework on its binding ligand. LAR has also been found to significantly influence intracellular signaling pathways [35]. However, the extracellular structure of LAR has been discovered to be of a different conformation than PTPσ, as a study by Biersmith et al., while analyzing the molecular structure using the same method of crystal structure analysis, unveiled that the Ig-like domains of Ig1 and Ig2 of LAR receptors were in a horseshoe-like conformation. This was significant because PTPσ receptors are known to have either linear or slightly curved Ig-like domains. This discovery provides more insight into how this structural difference in Ig-like confirmation enables CSPG to recognize each of the two receptors with distinct conformations [36]. Xie et al. utilized liprin-α proteins, a ligand that organizes synapses and intracellularly binds to LAR. They found that as this protein binds intracellularly to the cytoplasmic D1 and D2 domains of LAR, along with homophilic liprin-α oligomerization, it induces LAR clustering. This clustering suppressed phosphatase activity. When they disrupted the intracellular D1 domain, they exhibited tyrosine dephosphorylation and suppression of LAR clustering, which elevated phosphatase levels, suggesting inhibition of axonal regeneration [34,37]. However, further research evaluating its effects on axonal regeneration unlocks a new pathway for promoting regeneration via the LAR receptor. LAR appears to be a receptor with a multifaceted function, with some structural differences between PTPσ, which affect its functions but still play a role, like PTPσ mediating CSPG-inhibition on neural regrowth.
Centering attention on Nogo receptors, several variants of receptors are classified under Nogo receptors. This classification includes three main categories of receptors: Nogo receptor 1 (NgR1), Nogo receptor 2 (NgR2), and Nogo receptor 3 (NgR3). NgR1 receptors have been shown to bind to myelin-associated inhibitors (MAIs), including Nogo-A, myelin-associated glycoprotein (MAG), and oligodendrocyte-myelin glycoprotein (OMgp), which are all present within myelin sheaths of the CNS that modulate neural extension with high affinity [38]. However, NgR2 is slightly more selective in binding to MAIs with only attaching to MAG, while both NgR1 and NgR3 were two receptors that were found to bind to CSPG, suggesting that these receptors can mediate CSPG inhibition and modulate CSPG-mediated inhibitory impacts on diminishing axonal regrowth. These Nogo-receptor bindings further activate the RhoA/ROCK pathway, which is detrimental to axonal growth [38,39]. The molecular structure of Nogo receptors is somewhat distinct from the similar morphology of PTPσ and LAR, as the intracellular domains of PTPσ and LAR tend to be pivotal for their function, whereas Nogo receptors lack any form of intracellular structure. The morphology of NgR1, NgR2, and NgR3 seems to be quite alike, differentiated extracellularly at the N-terminus, and an accumulation of leucine-rich repeat (LRR) proteins flanked N-terminally (LRRNT). This aggregation constructs the LRRNT capping domain, followed by a tandem of LRR proteins varying in size, and is terminated with another cluster of LRRs flanked C-terminally (LRRCT), constructing the LRRCT capping domain. All LRRs with both capping domains are linked through a stalk region to a glycosylphosphatidylinositol (GPI) anchor. These GPI anchor proteins are attached to the transmembrane, permitting CSPG to attach to it and LRRCT, which allows CSPG inhibitory properties to impede growth via the NgR1 and NgR3 receptors [40].
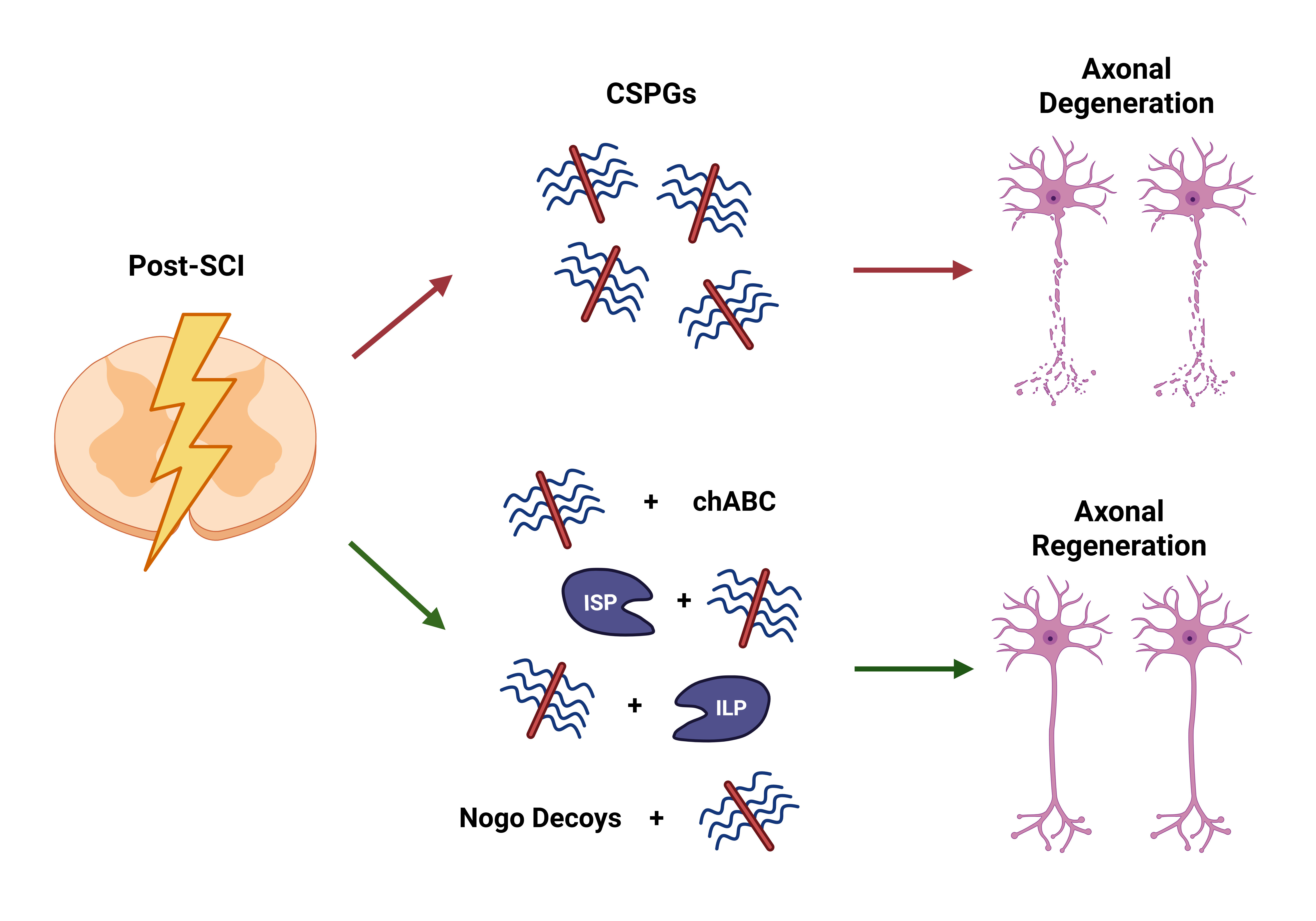
4. Overcoming CSPG-Mediated Inhibition with Therapeutic Strategies
4.1. Therapeutic with ChABC and Analyzing Its Development
Over the past few decades, it has been established that CSPG elicits an inhibitory role in axonal regeneration; however, in the last few years, scientists have utilized CSPG inhibitors to focus on axonal regeneration. Exploring and utilizing therapeutics to combat CSPG-mediated inhibition is a promising approach to axonal regeneration. Chondroitinase ABC (chABC) is the most familiar therapeutic that utilizes an enzymatic approach derived from bacteria, which targets CSPG via its CS-GAG chains. These GAGs have been associated with being responsible for the growth-diminishing properties of CSPG, hampering axonal regeneration [41]. Therapy with chABC enzymatically digested the GAG chains associated with CSPG, removing the regrowth inhibitory properties and reducing the activity of CSPG expression. Using chABC, the CS-GAG chains of CSPGs were further proven to be the culprit for the inhibitory properties of CSPGs, as they left the core remaining and unaffected neural regrowth. This utilization resulted in significantly improved axonal sprouting and regeneration while enhancing synaptic plasticity in axons with contusive SCI [41,42].
Several earlier studies have incorporated chABC to digest these CS-GAG chains from CSPG, perpetuating its effects and promoting axonal regeneration throughout injuries in multiple spinal systems, constituting injured corticospinal regions along with intact serotonergic descending neural pathways and intact sensory afferents [43]. In this scientific journey of chABC treatment, Bradbury et al. discovered that administering chABC to rats with dorsal column lesions upregulated growth-associated protein 43 and suppressed CSPG levels, prompting the regeneration of ascending sensory neurons and descending corticospinal tract (CST) axons associated with motor neurons. It ultimately yielded functional recovery of locomotor systems by eradicating inhibitory GAG chains [42]. García-Alías et al. incorporated chABC into rats with induced dorsal column lesions and similarly observed enhanced sprouting of CST axons. However, they also combined chABC treatment with rehabilitative strategies, which elicited improvements in specific motor skills [44]. With similar findings, Starkey et al. performed unilateral pyramidology in mice, discovering enhanced axonal sprouting in the CST compared to controls [45]. Likewise, chABC treatments have been sweeping with the removal of the inhibitory properties of CSPG, but discovering ways through further advances in treatment and studies to enhance their effectiveness can provide a more effective therapeutic strategy [46]. For instance, Bartus et al. discovered the use of gene therapy for chABC, with a treatment method of utilizing a lentiviral vector to deliver chABC intraspinal, leading to larger-scale CSPG digestion, enhancing the efficacy of chABC treatment and preventing the inhibitory effects of CSPG more extensively [47]. These studies have identified chABC as a major possible therapeutic contributor by focusing on the inhibitory section of CSPG to promote axonal regeneration in the CNS after SCI.
The development of chABC therapeutic strategies has led to more discoveries and higher treatment efficacies. For instance, a study conducted a unilateral cervical rhizotomy, inducing injury to the dorsal roots of the spinal nerves. In SCI models, rhizotomy successfully impaired sensory afferent fibers. Then, with the administration of chABC to digest CSPG chains, the chABC-treated mice were compared to wild-type (WT) mice with no application. This study revealed that after undergoing spinal cord unilateral cervical rhizotomy, the application of chABC permitted reorganization of the remaining seventh cervical vertebra primary afferent terminals, which restored post-synapses, leading to functional sensory recovery [43,48]. Based on these observations, with the assistance of transgenic mice, Carter et al. performed contusive SCI and intracerebroventricular injection of chABC and found that ChABC-treated mice exhibited a significant reduction in neuronal loss compared with WT mice. This indicates that ChABC has neuroprotective effects, preserves neuronal structures along with successful regeneration in the CST, and enhances sensory recovery. Interestingly, four weeks after chABC delivery, researchers noticed an observable activation and upregulation of intracellular signaling pathways, including the Erk and Akt pathways, in the chABC-treated site of SCI [49]. Based on studies, it is evident that chABC remains a revolutionary therapeutic agent that degrades the CS-GAG chain from CSPG, promoting axonal sprouting and regeneration in the CNS.
4.2. Utilizing PTPσ and LAR Deletion to Target CSPG-Inhibition
The interactions between CSPG and its receptors (PTPσ, LAR, and Nogo), posing inhibitory effects, have been well established and discussed [28,32]. The binding of CSPGs to their receptors can have inhibitory effects on axonal regeneration by inducing the release of inhibitory molecules for axonal regeneration via intracellular signaling pathways. Narrowing attention to the PTPσ receptor and removing the inhibitory receptor seemed promising for regeneration, so studies such as McLean et al. and Shen et al. examined the impact of PTPσ knockout (PTPσ (−/−)) on nerve regeneration after inducing SCI injury in mice in the ECM. The results were quite remarkable as the study yielded that PTPσ (−/−) mice exhibited an enhanced rate of nerve regeneration and neurite outgrowth compared to WT controls. This suggests that removing the PTPσ receptor can accelerate the regeneration of injured nerves and axons [50,51]. These findings were reinforced later by Fry et al., who conducted a study with PTPσ (−/−) mice and WT controls. Both groups underwent induced SCI, revealing that mouse with PTPσ (−/−) displayed via the hemisection of the spinal cord, a significant increase in regeneration of the CST with sixty percent of PTPσ (−/−) neurites that could cross to the inhibitory CSPG side, whereas WT mice could only regenerate less than ten percent of injured neurites. This significant increase in axonal growth across the lesion site signifies that with the removal of PTPσ, CSPG cannot bind to it, suppressing inhibitory cascade signaling and increasing neural regeneration [52].
Incorporating this technique into the LAR receptor, another key receptor like PTPσ, is vital for serving as an inhibitory receptor that binds to CSPG, eliciting a similar response to PTPσ in the ECM and triggering intracellular cascades that prevent axonal regeneration. Incorporating the knockout of the LAR receptor (LAR (−/−)) through in vitro experimentation with dorsal root ganglions (DRGs) from mice resulted in the LAR (−/−) group exhibiting significant neurite regrowth on CSPG substrates when compared with the WT group cultures. Deleting the LAR receptor obstructed the CSPG-LAR interaction, affecting the intracellular cascade, enhancing neurite regeneration after CNS injury [53]. Xu et al. reinforced these findings by utilizing LAR (−/−) in vivo in mice to increase the chances of axonal regeneration. This leads to findings showing a significant improvement in axonal regeneration with neurite outgrowth. This can be reinforced as fewer receptors for CSPG to bind with, also reducing additional intracellular pathways with reduced activation of RhoA, preventing the activation of the inhibitory RhoA/ROCK pathway, which is associated with axon retraction and growth cone collapse [54]. These studies confirm that LAR (−/−) is highly effective in preventing CSPG function and promoting axonal regrowth.
4.3. Targeting the Effects of CSPG-PTPσ Interactions with ISP
Lang et al. targeted the PTPσ receptor with an alternative approach, exploring ways of inhibiting CSPG-PTPσ interactions with the experimentation of intracellular sigma peptide (ISP). This peptide binds to PTPσ as it mimics the wedge of PTPσ, facilitating its penetration into neural membranes and binding to PTPσ in its intracellular domains. This binding impedes the signaling that is activated because of CSPG binding to PTPσ as an ISP, allowing ISP to serve as a barrier to the inhibitory effects of CSPG-PTPσ interactions from affecting axonal regeneration by blocking any inhibitory intracellular signaling pathways. After contusive SCI was administered to a group of ISP-treated and WT mice, the ISP-treated mice displayed noticeable improvements in regeneration, especially serotonergic fibers, beyond the site of injury; they further promoted functional recovery of sensory and motor neural systems [55,56]. Moreover, Luo et al. also experimented with ISP, performing induced ischemic injury in the spinal cord of mice in an ISP-treated group and a WT group. This led to the finding that, in the ISP-treated group, there were quantitatively greater amounts of neuroblasts, which are precursors in the generation of neurons, suggesting the high efficacy of ISP in hindering the inhibitory effects of CSPG-PTPσ on neural growth. ISP treatment of neural stem cells further upregulated phosphorylated Erk levels, reversing inhibition of the Erk signaling pathway and promoting its regeneration. There was also an increase in axonal density at the injury site in the peri-infarct region. Incorporating ISP further stimulated neuroblast migration to the injury site, suggesting that, as neuroblast migration is pivotal in adult neurogenesis, the inhibitory behavior of CSPG with PTPσ was blocked with increased axonal sprouting and neuroblast migration deep into scar regions in the ISP-treated group with functional recovery [55,57]. Another study conducted by Milton et al. discovered that cervical spinal hemi-lesions led to paralyzed forelimb and paw function in rats. When exposed to ISP treatment, the treated group exhibited forearm and digit function recovery, suggesting that ISP blocked the inhibitory effects of CSPG-PTPσ interactions after injury [58]. It has also been found that ISP intercepting CSPG-PTPσ effects are associated with the upregulation of autophagy flux and lysosomal function. These processes and functions stimulate synaptic and axonal regeneration [59].
4.4. Targeting the Effects of CSPG-LAR Interactions with ILP
A highly promising peptide for axonal regeneration, extending from the deletion of the LAR receptor and targeting CSPG-LAR interactions, is the intracellular LAR peptide (ILP). ILP peptides, like ISP peptides, can bind to the intracellular domains of LAR receptors, penetrate the membrane, and mimic the wedge domain of LAR, which diminishes the inhibitory effects of CSPG-LAR interactions by blocking intracellular signaling from CSPG-LAR interactions and preventing axonal degeneration. Fisher et al. attempted to alleviate this degeneration by incorporating ILP into their study in mice, as they found that ILP-treated mice exhibited increased axonal regrowth and decreased CSPG-LAR receptor signaling activity, indicating successful receptor modulation of CSPG-LAR effects. When both DRGs and cerebellar granule neurons (CGNs) were cultured in vitro on CSPG substrates, ILP selectively blocked LAR activity and elicited significant recovery of neurite growth. Furthermore, in mice, ILP administration after modulated dorsal transection injury significantly recovered the length of neurite regrowth and enhanced descending serotonergic fibers within the lesion site and caudal spinal cord, while promoting locomotor recovery with the growth of CST axons. More extensively, it was discovered that ILP has extensive efficacy in limiting downstream regulation by enhancing Akt phosphorylation, promoting growth via the Akt signaling pathway, and suppressing the RhoA/ROCK pathway [53,54]. Expanding from ILP, a study conducted by Cheng et al. further explored LAR peptides from an extracellular perspective, where LAR peptides were intrathecally injected into mice with prompted cervical SCI. Compared to the WT group, peptide-treated mice were found to have enhanced synaptic plasticity and axonal sprouting [35]. This reinforced the efficacy of ILP and other LAR peptides as a powerful therapeutic, achieving regrowth from SCI by obstructing the signaling caused by CSPG-LAR interactions.
4.5. Using ISP + ILP to Mediate CSPG-Inhibition
Since the use of ISP for PTPσ or ILP for LAR resulted in improvements in neural regeneration, providing both ISP and ILP (ISP + ILP) seemed quite sensible and promising for further axonal regeneration after SCI with greater efficacy. Dyck et al. did just this, experimenting using both ISP + ILP, where both peptides were administered intrathecally into mice of a WT group and a group with induced SCI. Blocking the effects of both PTPσ and LAR receptors with ISP and ILP, respectively, resulted in a reduction in pro-inflammatory M1 microglia and macrophages, replacing them with the expression of anti-inflammatory M2 microglia and macrophages, reducing the inflammatory response in the process of glial scar formation, even in the presence of CSPGs. This administration also found that axonal lengths had grown significantly, and that axonal density increased after ISP + ILP treatment, suggesting that the treatment is a promising reparative for modulating the inflammatory response following SCI in the CNS [53,57,60]. Furthermore, ISP + ILP usage performed by Hosseini et al. measured its efficacy through ISP + ILP treatment in vitro with neurons derived from engrafted human directly reprogrammed caudalized neural precursor cells (NPCs) and found that ISP or ILP application individually had minimal effect; however, with ISP + ILP co-treatment, the PTPσ and LAR receptors were co-blockaded, significantly reversing the inhibitory effects of CSPGs. Furthermore, the neurites derived from human-engrafted NPCs transplanted into rats with SCI were found to be more efficacious with ISP + ILP co-treatment, exhibiting increased neurite length after transplantation. It also significantly suppressed the inhibitory properties of the interactions of CSPG between PTPσ and LAR, with the co-inhibition of these two receptors, promoting synapse formation and increasing neuronal replacement with significant locomotor recovery post-SCI in treated rats, with the regeneration of CST axons. The study further revealed that ISP + ILP co-treatment created a more permissive environment for axonal regeneration by suppressing astrocyte differentiation while enhancing neuron density and morphological complexity in the lesion site after SCI [61]. In essence, by examining these studies, it can be concluded that ISP + ILP overcomes inhibitory signals from CSPGs, facilitating neuronal regrowth and synaptic connectivity with more efficiency than a single peptide of ISP or ILP alone, emerging as a promising therapeutic for SCI.
4.6. Targeting the Interactions of CSPG and Nogo
Shifting the focus to the interactions between CSPGs and Nogo receptors, it has been previously established that NgR1 and NgR3 have a high affinity for CS-GAG chains of CSPG. Some studies have deleted NgR1 (NgR1 (−/−)) and NgR3 (NgR3 (−/−)), which has prompted evidence of regrowth after injury. Some studies have incorporated soluble Nogo receptor decoys, which are fused proteins constructed of fragment crystallizable (Fc) regions from Ig, merged with the ligand-binding domains of NgR1 (NgR1-Fc) and NgR3 (NgR3-Fc) [62]. Dickendesher et al. also investigated the behavior of NgR1-Fc and NgR3-Fc to better understand the relationship between CSPG and Nogo receptors. They experimented with induced retro-orbital optic nerve crush (ONC) within the retinal ganglion cell (RGC) axons of rodents. This resulted in the upregulation of CSPGs in the injured optic nerve section, where soluble NgR1-Fc and NgR3-Fc exhibited a high affinity for binding with CSPGs. They also discovered that the GAG-binding motif m2, a cluster of basic amino acids and residues located in the stalk region of both NgR1 and NgR3, binds to CSPG. NgR1-Fc and NgR3-Fc mimic this motif to bind to the CS-GAG chains of CSPG; hence, their presence is a crucial factor in the soluble NgR1-Fc and NgR3-Fc binding. When chABC was applied to this environment, binding was significantly reduced, confirming that these Nogo decoys interact with CSPG by retaining the GAG-binding motif m2 on both NgR1 and NgR3. It was also observed that the RGC of mice after induced nerve crush resulted in a surge in CSPG levels; in comparison with WT, NgR1 (−/−) or NgR3 (−/−) alone did not completely suppress CSPG inhibition; however, co-treatment utilizing both NgR1 (−/−) and NgR3 (−/−) or the deletion of all three Nogo-receptors (NgR1, NgR2, and NgR3) significantly neutralized CSPG inhibition and enhanced RGC axonal regeneration [39,62].
Wang et al. incorporated the NgR(310)ecto-Fc decoy or a type of NgR1-Fc decoy after inducing a dorsal column injury in rat groups which as administered had elicited regeneration of primary afferent fibers into the injury environment. However, utilizing other therapies such as chABC and a preconditioning peripheral sciatic nerve axotomy along with the NgR1-Fc produced more robust neural repair and regeneration, allowing for regrowth past the injury site [62]. Wang et al. attempted to utilize the NgR1-Fc decoy also referred to as AXER-204 as the therapeutic for axonal regeneration. They investigated whether NgR1-Fc could target the ligands that bind to Nogo receptors, which are predominantly made up of MAIs (NogoA, MAG, and OMgp) and CSPG, as these ligands are associated with interacting with Nogo receptors and activating RhoA/ROCK signaling pathway, contributing to axonal degeneration. Using contusive SCI with an NgR1-Fc treated group and a WT control group in macaques, results exhibited that the NgR1-Fc treated macaques significantly promoted locomotor recovery by enhancing the growth of axons in the CST beyond the lesion site [39,63,64]. A more recent clinical study incorporated this decoy into human patients with chronic cervical SCI discussing that as a soluble decoy, AXER-204 could bind to all NgR1 inhibitory ligands and obstruct their inhibitory effects, suggesting that AXER-204 or NgR1-Fc could be utilized to bind to CSPG and neutralize CSPG-Nogo interactions; the study also found the NgR1-Fc decoy successfully reached targeted cerebrospinal fluid (CSF) concentrations for patients [65]. This provides promise for intercepting interactions between NgR1 and CSPGs with NgR1-Fc to inhibit RhoA/ROCK signaling and promote axonal regeneration; however, much more research is necessary to investigate these effects and extend applications such as to NgR3. Improving the research on these decoys, which have come to clinical trials, can yield a promising reparative for prompting neural regrowth after SCI.
Figure 3 showcases how a subset of therapeutic approaches reviewed thus far, including chABC, ISP, ILP, and NgR1-Fc, interact with and impact CSPGs, PTPσ, LAR, and Nogo receptors, influencing intrinsic pathways and promoting axonal regeneration. It also acknowledges the limited understanding of NgR3-Fc and the need for more research to better understand its interactions and how it influences the association between CSPGs and NgR3, affecting regrowth.
5. Discussion
5.1. Addressing Challenges and Opportunities in Current Therapeutic Research
Fundamentally, in the adult CNS, axonal regeneration does not occur spontaneously, primarily because of glial scar formation and the lack of regeneration-associated genes. Glial scars contain a dual role in preventing the spread of damaged tissue or lesions after injury but also mitigate the growth of injured axons by posing both a physical and chemical barrier with scar-forming reactive astrocytes and CSPGs [12,66]. As previously discussed, CSPG tends to be highly expressed during scar formation and has several inhibitory properties that affect axonal outgrowth via transmembrane receptors on growth cones with multiple scar-associated receptors (PTPσ, LAR, and Nogo) [67]. Understanding the challenges and further exploration of these extrinsic treatments could provide strategies for improving the efficacy of these therapeutics by interacting with transmembrane receptors and promoting axonal regeneration. Table 1 summarizes the therapeutics discussed in this review and provides the findings on their influence on axonal regeneration from the discussed studies.
The initial treatment with chABC was repeated, and this therapeutic administration resulted in significant regeneration of injured axons. However, there are some limitations to the chABC application in humans, as chABC is thermally unstable, thus having the chance of suppressing the efficacy of this treatment. Several delivery challenges are associated with chABC administration in humans, as they could pose a larger obstacle in providing accurate delivery [46,68]. Experimenting administration techniques that modulate delivery with synthetic scaffolds or nanoparticles has induced more pathways to improve chABC treatment [66]. However, further research is needed to determine its efficacy in facilitating chABC treatment. Therefore, chABC is a bacterial enzyme that can harm the immune system. This issue was remarkably resolved by utilizing a doxycycline-inducible switch, which was found to modulate chABC delivery as an immune evasive [69]. Although this chABC treatment has barely been immersed in clinical trials to amplify regeneration after SCI, chABC has been utilized for lumbar disc herniation through a phase III clinical trial in Japan, suggesting a possible therapeutic significance of chABC application for SCI in the future [46].
The peptide-based therapeutics discussed, including ISP, ILP, and NgR1-Fc (AXER-204), have been shown to improve axonal regeneration in preclinical models. However, their clinical translation into human treatments faces significant obstacles, specifically when delivering these molecules across the BBB. The BBB’s tight and adherens junctions are drastic contributors to the BBB’s selective permeability, often impeding the efficacy of administered peptide therapeutics. This requires other routes of delivery, such as intrathecal administration, which can be considered more invasive for clinical application [70]. While peptide-based therapeutics are promising with their low immunogenicity, their application is limited by factors such as short in vivo half-life and susceptibility to enzymatic degradation due to dosing issues without optimal therapeutic concentrations. Remarkably, a discussed clinical trial of AXER-204 demonstrated its ability to reach target concentrations in the CSF yet also emphasized the need for additional optimized dosing delivery approaches to enhance peptide efficacy [65,71]. Therefore, further research is needed to enhance the efficacy and safety profiles of peptide therapeutics when being translated to treatment across the BBB for minimal invasiveness and achieving optimized dosing regimens to prevent adverse effects [70,71].
Considering that either PTPσ (−/−) or LAR (−/−) prompted neuronal regrowth, experimenting with the knockout of both PTPσ and LAR receptors seemed to be a reparative process with higher efficacy. Ohtake et al. incorporated this into their study by deleting either of these receptors in the presence of CSPG, yielding augmented neurite regrowth in adult DRG cultures. In another group, the dual knockout of PTPσ and LAR receptors elicited robust axonal regeneration to a significantly greater extent than the regrowth of knocking out one of the receptors [32]. However, this specific dual knockout strategy involving PTPσ (−/−) and LAR (−/−) has not been widely reported in the literature. More studies focusing on deleting both PTPσ and LAR receptors are necessary to develop a more comprehensive understanding of therapeutics to target the synergistic effects of both PTPσ and LAR receptors in mediating CSPG inhibition. Bridging to generalized dual receptor targeting, there are existing gaps in the therapeutic potential of combined inhibition of PTPσ or LAR with NgR1 or NgR3, as it remains largely unexplored. Addressing these gaps with more research on investigating the effects of dual receptor targeting with dual knockout or co-inhibition studies focusing on CSPG-mediated receptors could yield valuable insights to enhance regrowth following injury.
Furthermore, as discussed previously, the NgR1-Fc decoy binds to NgR1 ligands and blocks the interactions between NgR1 and its inhibitory ligands, such as CSPGs and MAIs, enhancing regrowth [62]. This may also bind to ligands that interact with NgR3, which could indirectly assist in inhibiting CSPG-NgR3 interactions. However, further research on this is necessary, along with more research on the NgR3-Fc decoy, to develop a greater understanding of the potential of the NgR3-Fc decoy in the relationship between CSPGs and NgR3 to promote regeneration.
The clinical advancement of peptide-based and other protein therapeutics discussed to inhibit CSPG and promote regeneration mandates rigorous adherence to regulatory guidelines to ensure their safety and efficacy of the treatment. Regulatory bodies establish comprehensive frameworks with guidelines that emphasize the significance of thorough characterization of the biological therapeutics, including assessments of analysis, stability, and quality [72]. These regulatory and safety considerations are pivotal to the successful translation of peptide-based and protein therapeutics, ensuring that they meet the rigorous standards required for clinical application.
5.2. Alternative Direction of Treatments for Regeneration
This review examines potential therapeutics that mediate CSPG inhibition via its receptors to enhance axonal regeneration in the CNS. However, researchers are also developing therapeutics to amplify axonal regrowth by altering the expression of intracellular pathways to favor regrowth mechanisms. This review previously highlighted that, as CSPG attaches to one of its transmembrane receptors, it induces distinct intracellular pathway signaling to be upregulated or suppressed for each receptor. The binding of PTPσ and LAR to CSPGs activates the RhoA/ROCK pathway while inhibiting the Erk pathway and PI3K/Akt/mTOR pathways. CSPG and Nogo receptor binding activate RhoA/ROCK and other signaling pathways, inhibiting axonal growth via other downregulating cascades of these three primary pathways [3].
CSPG intrinsically activates Rho and its downstream ROCK, thereby impeding axonal regrowth. A treatment utilizing C3 transferase (C3), a bacterial exoenzyme, suppresses and disables Rho into an inactive state through adenosine diphosphate-ribosylation of the Rho effector domain, intercepting Rho activation [73,74]. The pharmacological inhibitor Y27632 has also been used as an emerging therapeutic target for ROCK in the RhoA/ROCK pathway [75]. This suggests that C3 and Y27632 could be potential therapeutics that could be advanced to target the RhoA/ROCK signaling pathway intracellularly and catalyze axonal regeneration after SCI.
The PI3K/Akt/mTOR signaling pathway is inhibited intracellularly by CSPGs via receptors that exacerbate axonal regrowth after SCI. PTEN is a negative PI3K/Akt/mTOR pathway regulator that inhibits its activity and plays a detrimental role in regeneration [76], and the knockout of PTEN in vitro in neuronal cultures containing cortical neurons with CSPG-treated substrates still unveiled a noticeable enhancement in neurite outgrowth compared with controls [77]. Previously, mTOR was hypothesized to reside in mature neurons; however, it was discovered to be functionally integrated with growth cones, suggesting that restoring the mTOR pathway to its growth state can induce repair [78]. PTEN deletion and mTOR seem to hold substantial promise for upregulating the PI3K/Akt/mTOR signaling pathway and promoting neural regrowth by utilizing an intracellular pathway [79].
Additional research to investigate the relationship between these receptors and the Erk pathway, along with efficacious therapeutics, is necessary. Further studies have unraveled the use of new technologies in prompting regeneration with optogenetic systems. Utilizing systems such as optoRAF and optoAKT in Drosophila, they induce the activation of both the Akt and Erk pathways, prompting axonal regrowth [80]. These treatments diverge from the therapeutics mentioned in this review by targeting the interaction of CSPG and its receptors. However, these therapeutics elucidate their target of intracellular signaling pathways to promote axonal regeneration. Essentially, although from a different perspective, these therapeutics appear promising and applicable to the broader scope of axonal regeneration treatment.
6. Conclusions
In the dynamic landscape of axonal regeneration research, the inhibitory effects of CSPG on glial scarring and axonal regeneration after SCI have been well-established as impediments to regeneration. This review discussed CSPG and its transmembrane receptors while highlighting studies of therapeutic options that inhibit CSPG extracellularly to promote axonal regrowth. It has been outlined that a widely known therapeutic established to inhibit CSPG in SCI has been chABC; however, it has been established that there have been limitations to this treatment. Therefore, other treatments that were discussed targeting transmembrane receptors were shown to be promising for regeneration. Certain possible therapeutics and combining certain therapeutic strategies were identified, however, more research is necessary to validate their efficacy. Modifying the intracellular pathway from a different therapeutic approach has also been mentioned as a possible regeneration approach with more investigation advised. Acknowledging the role of CSPG in SCI can address challenges, and elucidating these extracellular therapeutic strategies to overcome CSPG-mediated inhibition can harness the potential of these therapeutics to enhance axonal regeneration in the CNS and revolutionize SCI treatment.
Author Contributions
Conceptualization, K.E.; methodology, K.E.; writing—original draft preparation, K.E.; writing—review and editing, K.E.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
The author declares no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CNS | Central Nervous System |
| PNS | Peripheral Nervous System |
| SCI | Spinal Cord Injury |
| CSPG | Chondroitin sulfate proteoglycan |
| ECM | Extracellular matrix |
| NG2 | Neuron-glial antigen 2 |
| OPC | Oligodendrocyte precursor cell |
| G1 | Globular domain at the N-terminus |
| G3 | Globular domain at the C-terminus |
| HA | Hyaluronan |
| GAG | Glycosaminoglycan |
| CS | Chondroitin sulfate |
| PTPσ | Protein tyrosine phosphatase sigma |
| LAR | Leukocyte common antigen-related |
| BBB | Blood-brain barrier |
| DAMP | Damage-associated molecular pattern |
| IGF-1 | Insulin-like growth factor 1 |
| GFAP | Glial fibrillary acidic protein |
| RPTPs | Receptor-type protein tyrosine phosphatases |
| Ig-like | Immunoglobulin-like |
| FN-III | Fibronectin type-III |
| HSPG | Heparan sulfate proteoglycan |
| HS | Heparan sulfate |
| NgR1 | Nogo receptor 1 |
| NgR2 | Nogo receptor 2 |
| NgR3 | Nogo receptor 3 |
| MAIs | Myelin-associated inhibitors |
| MAG | Myelin-associated glycoprotein |
| OMgp | Oligodendrocyte-myelin glycoprotein |
| LRR | Leucine-rich repeat |
| LRRNT | LRR proteins flanked N-terminally |
| LRRCT | LRR proteins flanked C-terminally |
| GPI | Glycosylphosphatidylinositol |
| ChABC | Chondroitinase ABC |
| CST | Corticospinal tract |
| WT | Wild-type |
| PTPσ (−/−) | PTPσ knockout |
| LAR (−/−) | LAR knockout |
| DRG | Dorsal root ganglion |
| ISP | Intracellular sigma peptide |
| ILP | Intracellular LAR peptide |
| CGN | Cerebellar granule neuron |
| NPC | Neural precursor cell |
| NgR1 (−/−) | NgR1 knockout |
| NgR3 (−/−) | NgR3 knockout |
| Fc | Fragment crystallizable |
| NgR1-Fc | NgR1 decoy merged with Fc region proteins |
| NgR3-Fc | NgR3 decoy merged with Fc region proteins |
| ONC | Optic nerve crush |
| RGC | Retinal ganglion cell |
| CSF | Cerebrospinal fluid |
| C3 | C3 transferase |
References
- Cajal, S.R. Degeneration and Regeneration of the Nervous System; Oxford University Press: New York, NY, USA, 1928. [Google Scholar]
- Huebner, E.A.; Strittmatter, S.M. Axon regeneration in the peripheral and central nervous systems. Results Probl. Cell Differ. 2009, 48, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Sami, A.; Selzer, M.E.; Li, S. Advances in the signaling pathways downstream of glial-scar axon growth inhibitors. Front. Cell Neurosci. 2020, 14, 174. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, K.E.; Fawcett, J.W. Chondroitin sulphate proteoglycans: preventing plasticity or protecting the CNS? J. Anat. 2004, 204, 33–48. [Google Scholar] [CrossRef]
- Avram, S.; Shaposhnikov, S.; Buiu, C.; Mernea, M. Chondroitin sulfate proteoglycans: structure-function relationship with implication in neural development and brain disorders. Biomed. Res. Int. 2014, 2014, 642798. [Google Scholar] [CrossRef] [PubMed]
- Kokenyesi, R.; Bernfield, M. Core protein structure and sequence determine the site and presence of heparan sulfate and chondroitin sulfate on syndecan-1. J. Biol. Chem. 1994, 269, 12304–12309. [Google Scholar] [CrossRef]
- Bourgeais, M.; Fouladkar, F.; Weber, M.; Boeri-Erba, E.; Wild, R. Chemo-enzymatic synthesis of tetrasaccharide linker peptides to study the divergent step in glycosaminoglycan biosynthesis. Glycobiology 2024, 34, cwae016. [Google Scholar] [CrossRef]
- Ohtake, Y.; Li, S. Molecular mechanisms of scar-sourced axon growth inhibitors. Brain Res. 2015, 1619, 22–35. [Google Scholar] [CrossRef]
- Yang, S.; Gigout, S.; Molinaro, A.; et al. Chondroitin 6-sulphate is required for neuroplasticity and memory in ageing. Mol. Psychiatry 2021, 26, 5658–5668. [Google Scholar] [CrossRef]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef]
- Yang, T.; Dai, Y.; Chen, G.; Cui, S. Dissecting the dual role of the glial scar and scar-forming astrocytes in spinal cord injury. Front. Cell Neurosci. 2020, 14, 78. [Google Scholar] [CrossRef]
- Tran, A.P.; Warren, P.M.; Silver, J. New insights into glial scar formation after spinal cord injury. Cell Tissue Res. 2022, 387, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, E.J.; Burnside, E.R. Moving beyond the glial scar for spinal cord repair. Nat. Commun. 2019, 10, 3879. [Google Scholar] [CrossRef] [PubMed]
- Clifford, T.; Finkel, Z.; Rodriguez, B.; Joseph, A.; Cai, L. Current advancements in spinal cord injury research—glial scar formation and neural regeneration. Cells 2023, 12, 853. [Google Scholar] [CrossRef] [PubMed]
- Bellver-Landete, V.; Bretheau, F.; Mailhot, B.; et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat. Commun. 2019, 10, 518. [Google Scholar] [CrossRef]
- Gensel, J.C.; Zhang, B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 2015, 1619, 1–11. [Google Scholar] [CrossRef]
- Shibuya, S.; Yamamoto, T.; Itano, T. Glial and axonal regeneration following spinal cord injury. Cell Adhes. Migr. 2009, 3, 99–106. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Fonken, L.K. Glial cells shape pathology and repair after spinal cord injury. Neurotherapeutics 2018, 15, 554–577. [Google Scholar] [CrossRef]
- Dias, D.O.; Göritz, C. Fibrotic scarring following lesions to the central nervous system. Matrix Biol. 2018, 68–69, 561–570. [Google Scholar] [CrossRef]
- Li, Z.; Yu, S.; Hu, X.; et al. Fibrotic scar after spinal cord injury: crosstalk with other cells, cellular origin, function, and mechanism. Front. Cell Neurosci. 2021, 15, 720938. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, H.; Qin, X.; Tian, M.; Zhang, H. PET imaging of reactive astrocytes in neurological disorders. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 1275–1287. [Google Scholar] [CrossRef]
- Wanner, I.B.; Anderson, M.A.; Song, B.; et al. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci. 2013, 33, 12870–12886. [Google Scholar] [CrossRef]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef]
- Zhang, G.; Jin, L.Q.; Rodemer, W.; Hu, J.; Root, Z.D.; Medeiros, D.M.; Selzer, M.E. The composition and cellular sources of CSPGs in the glial scar after spinal cord injury in the lamprey. Front. Mol. Neurosci. 2022, 15, 918871. [Google Scholar] [CrossRef]
- Karimi-Abdolrezaee, S.; Billakanti, R. Reactive astrogliosis after spinal cord injury—Beneficial and detrimental effects. Mol. Neurobiol. 2012, 46, 251–264. [Google Scholar] [CrossRef]
- Assinck, P.; Duncan, G.J.; Plemel, J.R.; et al. Myelinogenic plasticity of oligodendrocyte precursor cells following spinal cord contusion injury. J. Neurosci. 2017, 37, 8635–8654. [Google Scholar] [CrossRef]
- Filous, A.R.; Tran, A.; Howell, C.J.; et al. Entrapment via synaptic-like connections between NG2 proteoglycan+ cells and dystrophic axons in the lesion plays a role in regeneration failure after spinal cord injury. J. Neurosci. 2014, 34, 16369–16384. [Google Scholar] [CrossRef]
- Sharma, K.; Selzer, M.E.; Li, S. Scar-mediated inhibition and CSPG receptors in the CNS. Exp. Neurol. 2012, 237, 370–378. [Google Scholar] [CrossRef]
- Sclip, A.; Südhof, T.C. LAR receptor phospho-tyrosine phosphatases regulate NMDA-receptor responses. Elife 2020, 9, e53406. [Google Scholar] [CrossRef]
- Takahashi, H.; Craig, A.M. Protein tyrosine phosphatases PTPδ, PTPσ, and LAR: Presynaptic hubs for synapse organization. Trends Neurosci. 2013, 36, 522–534. [Google Scholar] [CrossRef]
- Xu, Y.; Fisher, G.J. Receptor type protein tyrosine phosphatases (RPTPs)—Roles in signal transduction and human disease. J. Cell Commun. Signal. 2012, 6, 125–138. [Google Scholar] [CrossRef]
- Ohtake, Y.; Wong, D.; Abdul-Muneer, P.M.; Selzer, M.E.; Li, S. Two PTP receptors mediate CSPG inhibition by convergent and divergent signaling pathways in neurons. Sci. Rep. 2016, 6, 37152. [Google Scholar] [CrossRef]
- Coles, C.H.; Shen, Y.; Tenney, A.P.; et al. Proteoglycan-specific molecular switch for RPTPσ clustering and neuronal extension. Science 2011, 332, 484–488. [Google Scholar] [CrossRef]
- Cornejo, F.; Cortés, B.I.; Findlay, G.M.; Cancino, G.I. LAR receptor tyrosine phosphatase family in healthy and diseased brain. Front. Cell Dev. Biol. 2021, 9, 659951. [Google Scholar] [CrossRef]
- Cheng, L.; Sami, A.; Ghosh, B.; et al. LAR inhibitory peptide promotes recovery of diaphragm function and multiple forms of respiratory neural circuit plasticity after cervical spinal cord injury. Neurobiol. Dis. 2021, 147, 105153. [Google Scholar] [CrossRef]
- Biersmith, B.H.; Hammel, M.; Geisbrecht, E.R.; Bouyain, S. The immunoglobulin-like domains 1 and 2 of the protein tyrosine phosphatase LAR adopt an unusual horseshoe-like conformation. J. Mol. Biol. 2011, 408, 616–627. [Google Scholar] [CrossRef]
- Xie, X.; Luo, L.; Liang, M.; Zhang, W.; Zhang, T.; Yu, C.; Wei, Z. Structural basis of liprin-α-promoted LAR-RPTP clustering for modulation of phosphatase activity. Nat. Commun. 2020, 11, 169. [Google Scholar] [CrossRef]
- Costa, G.; Ribeiro, F.F.; Sebastião, A.M.; Muir, E.M.; Vaz, S.H. Bridging the gap of axonal regeneration in the central nervous system: A state of the art review on central axonal regeneration. Front. Neurosci. 2022, 16, 1003145. [Google Scholar] [CrossRef]
- Dickendesher, T.L.; Baldwin, K.T.; Mironova, Y.A.; et al. NgR1 and NgR3 are receptors for chondroitin sulfate proteoglycans. Nat. Neurosci. 2012, 15, 703–712. [Google Scholar] [CrossRef]
- Giger, R.J.; Venkatesh, K.; Chivatakarn, O.; et al. Mechanisms of CNS myelin inhibition: Evidence for distinct and neuronal cell type specific receptor systems. Restor. Neurol. Neurosci. 2008, 26, 97–115. [Google Scholar] [CrossRef]
- Mukherjee, N.; Nandi, S.; Garg, S.; Ghosh, S.; Ghosh, S.; Samat, R.; Ghosh, S. Targeting chondroitin sulfate proteoglycans: An emerging therapeutic strategy to treat CNS injury. ACS Chem. Neurosci. 2020, 11, 231–232. [Google Scholar] [CrossRef]
- Bradbury, E.J.; Moon, L.D.; Popat, R.J.; et al. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 2002, 416, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, E.J.; Carter, L.M. Manipulating the glial scar: chondroitinase ABC as a therapy for spinal cord injury. Brain Res. Bull. 2011, 84, 306–316. [Google Scholar] [CrossRef] [PubMed]
- García-Alías, G.; Barkhuysen, S.; Buckle, M.; Fawcett, J.W. Chondroitinase ABC treatment opens a window of opportunity for task-specific rehabilitation. Nat. Neurosci. 2009, 12, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Starkey, M.L.; Bartus, K.; Barritt, A.W.; Bradbury, E.J. Chondroitinase ABC promotes compensatory sprouting of the intact corticospinal tract and recovery of forelimb function following unilateral pyramidotomy in adult mice. Eur. J. Neurosci. 2012, 36, 3665–3678. [Google Scholar] [CrossRef]
- Hu, J.; Jin, L.Q.; Selzer, M.E. Inhibition of central axon regeneration: perspective from chondroitin sulfate proteoglycans in lamprey spinal cord injury. Neural Regen. Res. 2022, 17, 1955–1956. [Google Scholar] [CrossRef]
- Bartus, K.; James, N.D.; Didangelos, A.; et al. Large-scale chondroitin sulfate proteoglycan digestion with chondroitinase gene therapy leads to reduced pathology and modulates macrophage phenotype following spinal cord contusion injury. J. Neurosci. 2014, 34, 4822–4836. [Google Scholar] [CrossRef]
- Cafferty, W.B.; Yang, S.H.; Duffy, P.J.; Li, S.; Strittmatter, S.M. Functional axonal regeneration through astrocytic scar genetically modified to digest chondroitin sulfate proteoglycans. J. Neurosci. 2007, 27, 2176–2185. [Google Scholar] [CrossRef]
- Carter, L.M.; Starkey, M.L.; Akrimi, S.F.; Davies, M.; McMahon, S.B.; Bradbury, E.J. The yellow fluorescent protein (YFP-H) mouse reveals neuroprotection as a novel mechanism underlying chondroitinase ABC-mediated repair after spinal cord injury. J. Neurosci. 2008, 28, 14107–14120. [Google Scholar] [CrossRef]
- McLean, J.; Batt, J.; Doering, L.C.; Rotin, D.; Bain, J.R. Enhanced rate of nerve regeneration and directional errors after sciatic nerve injury in receptor protein tyrosine phosphatase sigma knock-out mice. J. Neurosci. 2002, 22, 5481–5491. [Google Scholar] [CrossRef]
- Shen, Y.; Tenney, A.P.; Busch, S.A.; et al. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science 2009, 326, 592–596. [Google Scholar] [CrossRef]
- Fry, E.J.; Chagnon, M.J.; López-Vales, R.; Tremblay, M.L.; David, S. Corticospinal tract regeneration after spinal cord injury in receptor protein tyrosine phosphatase sigma deficient mice. Glia 2010, 58, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.; Xing, B.; Dill, J.; et al. Leukocyte common antigen-related phosphatase is a functional receptor for chondroitin sulfate proteoglycan axon growth inhibitors. J. Neurosci. 2011, 31, 14051–14066. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Park, D.; Ohtake, Y.; et al. Role of CSPG receptor LAR phosphatase in restricting axon regeneration after CNS injury. Neurobiol. Dis. 2015, 73, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.T.; Cregg, J.M.; DePaul, M.A.; et al. Modulation of the proteoglycan receptor PTPσ promotes recovery after spinal cord injury. Nature 2015, 518, 404–408. [Google Scholar] [CrossRef]
- Rink, S.; Arnold, D.; Wöhler, A.; et al. Recovery after spinal cord injury by modulation of the proteoglycan receptor PTPσ. Exp. Neurol. 2018, 309, 148–159. [Google Scholar] [CrossRef]
- Luo, F.; Wang, J.; Zhang, Z.; et al. Inhibition of CSPG receptor PTPσ promotes migration of newly born neuroblasts, axonal sprouting, and recovery from stroke. Cell Rep. 2022, 40, 111137. [Google Scholar] [CrossRef]
- Milton, A.J.; Kwok, J.C.; McClellan, J.; et al. Recovery of Forearm and Fine Digit Function After Chronic Spinal Cord Injury by Simultaneous Blockade of Inhibitory Matrix Chondroitin Sulfate Proteoglycan Production and the Receptor PTPσ. J. Neurotrauma 2023, 40, 2500–2521. [Google Scholar] [CrossRef]
- Wang, H.; Feng, N.; Liu, C.; et al. Inhibition of CSPG-PTPσ Activates Autophagy Flux and Lysosome Fusion, Aids Axon and Synaptic Reorganization in Spinal Cord Injury. Mol. Neurobiol. 2025, 62, 773–785. [Google Scholar] [CrossRef]
- Dyck, S.; Kataria, H.; Alizadeh, A.; Santhosh, K.T.; Lang, B.; Silver, J.; Karimi-Abdolrezaee, S. Perturbing chondroitin sulfate proteoglycan signaling through LAR and PTPσ receptors promotes a beneficial inflammatory response following spinal cord injury. J. Neuroinflammation 2018, 15, 90. [Google Scholar] [CrossRef]
- Hosseini, S.M.; Alizadeh, A.; Shahsavani, N.; Chopek, J.; Ahlfors, J.E.; Karimi-Abdolrezaee, S. Suppressing CSPG/LAR/PTPσ axis facilitates neuronal replacement and synaptogenesis by human neural precursor grafts and improves recovery after spinal cord injury. J. Neurosci. 2022, 42, 3096–3121. [Google Scholar] [CrossRef]
- Wang, X.; Hasan, O.; Arzeno, A.; Benowitz, L.I.; Cafferty, W.B.; Strittmatter, S.M. Axonal regeneration induced by blockade of glial inhibitors coupled with activation of intrinsic neuronal growth pathways. Exp. Neurol. 2012, 237, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Ji, S.J.; Jaffrey, S.R. Intra-axonal translation of RhoA promotes axon growth inhibition by CSPG. J. Neurosci. 2012, 32, 14442–14447. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, T.; Maynard, G.D.; Terse, P.S.; Cafferty, W.B.; Kocsis, J.D.; Strittmatter, S.M. Nogo receptor decoy promotes recovery and corticospinal growth in non-human primate spinal cord injury. Brain 2020, 143, 1697–1713. [Google Scholar] [CrossRef] [PubMed]
- Maynard, G.; Kannan, R.; Liu, J.; et al. Soluble Nogo-receptor-Fc decoy (AXER-204) in patients with chronic cervical spinal cord injury in the USA: A first-in-human and randomised clinical trial. Lancet Neurol. 2023, 22, 672–684. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, S.; Liu, C.; Han, X.; Gu, X.; Zhou, S. Deciphering glial scar after spinal cord injury. Burns Trauma 2021, 9, tkab035. [Google Scholar] [CrossRef]
- Mencio, C.P.; Hussein, R.K.; Yu, P.; Geller, H.M. The role of chondroitin sulfate proteoglycans in nervous system development. J. Histochem. Cytochem. 2021, 69, 61–80. [Google Scholar] [CrossRef]
- Muir, E.; De Winter, F.; Verhaagen, J.; Fawcett, J. Recent advances in the therapeutic uses of chondroitinase ABC. Exp. Neurol. 2019, 321, 113032. [Google Scholar] [CrossRef]
- Burnside, E.R.; De Winter, F.; Didangelos, A.; et al. Immune-evasive gene switch enables regulated delivery of chondroitinase after spinal cord injury. Brain 2018, 141, 2362–2381. [Google Scholar] [CrossRef]
- Parrasia, S.; Szabò, I.; Zoratti, M.; Biasutto, L. Peptides as pharmacological carriers to the brain: Promises, shortcomings and challenges. Mol. Pharm. 2022, 19, 3700–3729. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; et al. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Elsayed, Y.Y.; Kühl, T.; Imhof, D. Regulatory guidelines for the analysis of therapeutic peptides and proteins. J. Pept. Sci. 2025, 31, e70001. [Google Scholar] [CrossRef] [PubMed]
- Monnier, P.P.; Sierra, A.; Schwab, J.M.; et al. The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol. Cell. Neurosci. 2003, 22, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Yamashita, T. Axon growth inhibition by RhoA/ROCK in the central nervous system. Front. Neurosci. 2014, 8, 338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Jiang, C.; et al. Rho kinase inhibitor Y27632 improves recovery after spinal cord injury by shifting astrocyte phenotype and morphology via the ROCK/NF-κB/C3 pathway. Neurochem. Res. 2022, 47, 3733–3744. [Google Scholar] [CrossRef]
- Geoffroy, C.G.; Lorenzana, A.O.; Kwan, J.P.; et al. Effects of PTEN and Nogo codeletion on corticospinal axon sprouting and regeneration in mice. J. Neurosci. 2015, 35, 6413–6428. [Google Scholar] [CrossRef]
- Liu, S.; Jia, J.; Zhou, H.; et al. PTEN modulates neurites outgrowth and neuron apoptosis involving the PI3K/Akt/mTOR signaling pathway. Mol. Med. Rep. 2019, 20, 4059–4066. [Google Scholar] [CrossRef]
- Poulopoulos, A.; Murphy, A.J.; Ozkan, A.; Davis, P.; Hatch, J.; Kirchner, R.; Macklis, J.D. Subcellular transcriptomes and proteomes of developing axon projections in the cerebral cortex. Nature 2019, 565, 356–360. [Google Scholar] [CrossRef]
- Huang, H.; Miao, L.; Yang, L.; et al. AKT-dependent and -independent pathways mediate PTEN deletion-induced CNS axon regeneration. Cell Death Dis. 2019, 10, 203. [Google Scholar] [CrossRef]
- Wang, Q.; Fan, H.; Li, F.; Skeeters, S.S.; Krishnamurthy, V.V.; Song, Y.; Zhang, K. Optical control of ERK and AKT signaling promotes axon regeneration and functional recovery of PNS and CNS in Drosophila. Elife 2020, 9, e57395. [Google Scholar] [CrossRef]
Figure 1.
Structure of CSPGs and classification of CSPG molecules. This figure was illustrated referencing [3,5]. Image created with BioRender.com.
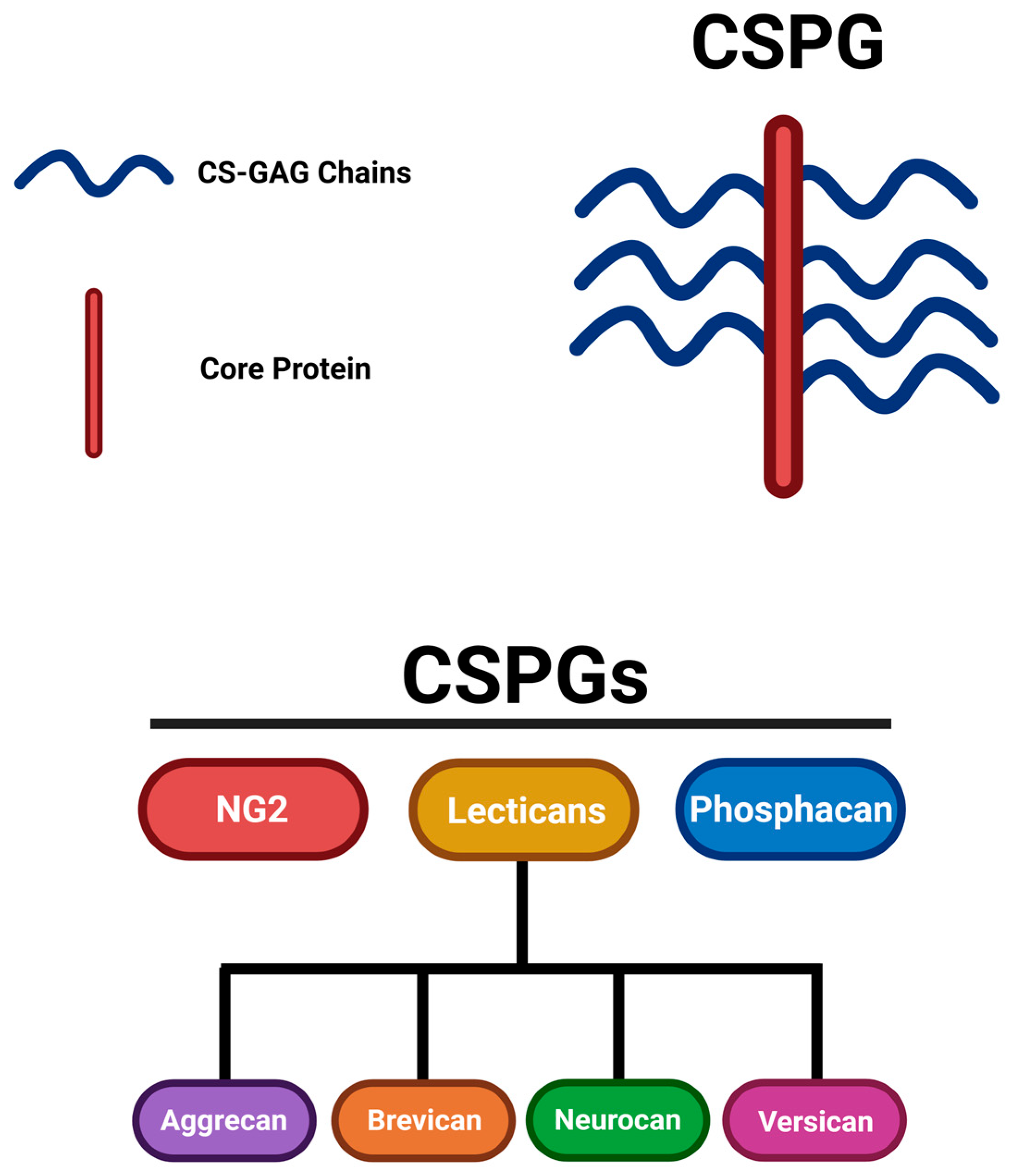
Figure 2.
CSPGs in glial scarring inhibiting regeneration following SCI. This figure was illustrated referencing [11,13,14]. Image created with BioRender.com.
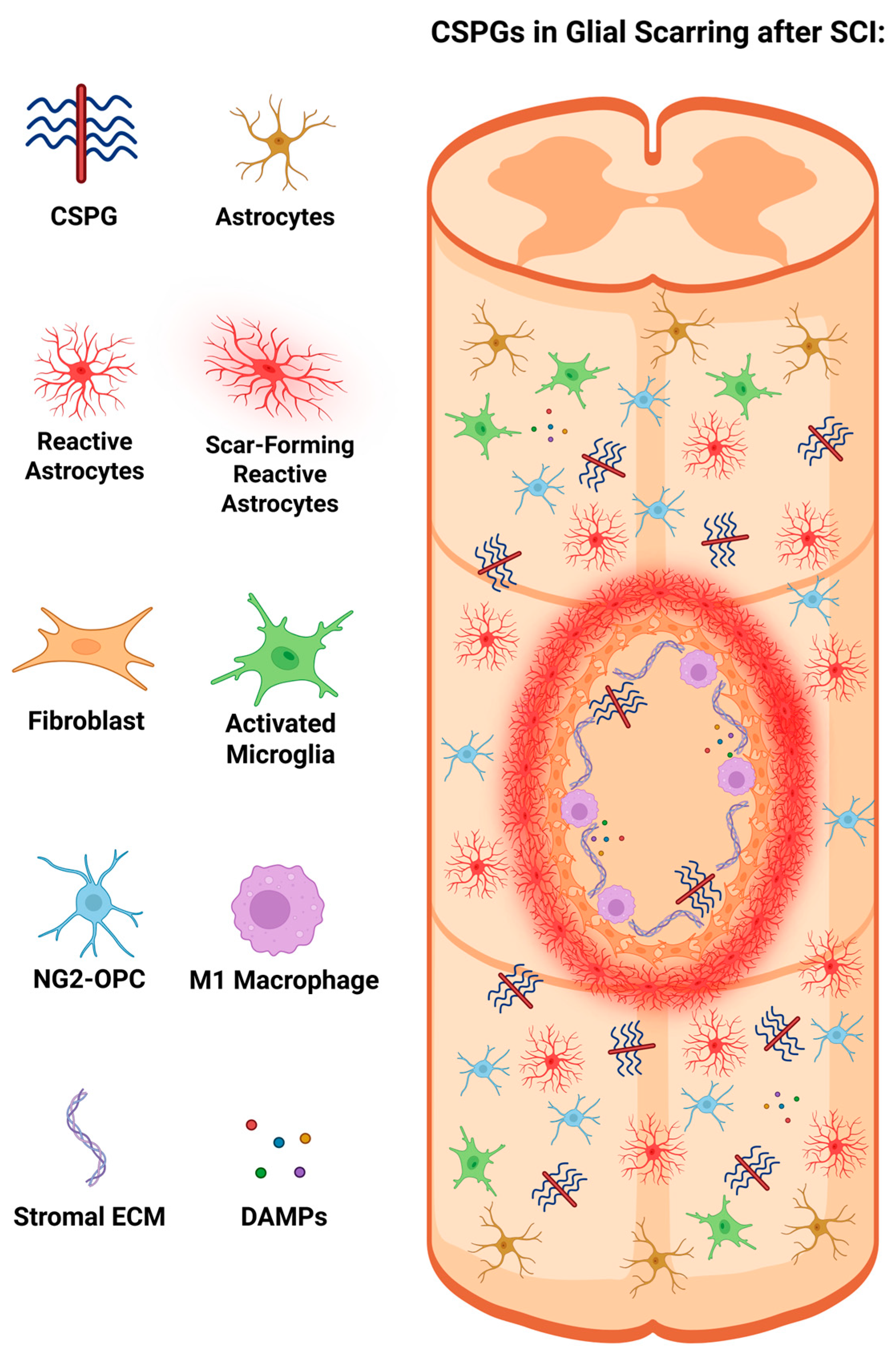
Figure 3.
Therapeutics targeting CSPGs and its transmembrane receptors to enhance axonal regeneration in the CNS. This figure was illustrated referencing [3,28,32]. Image created with BioRender.com.
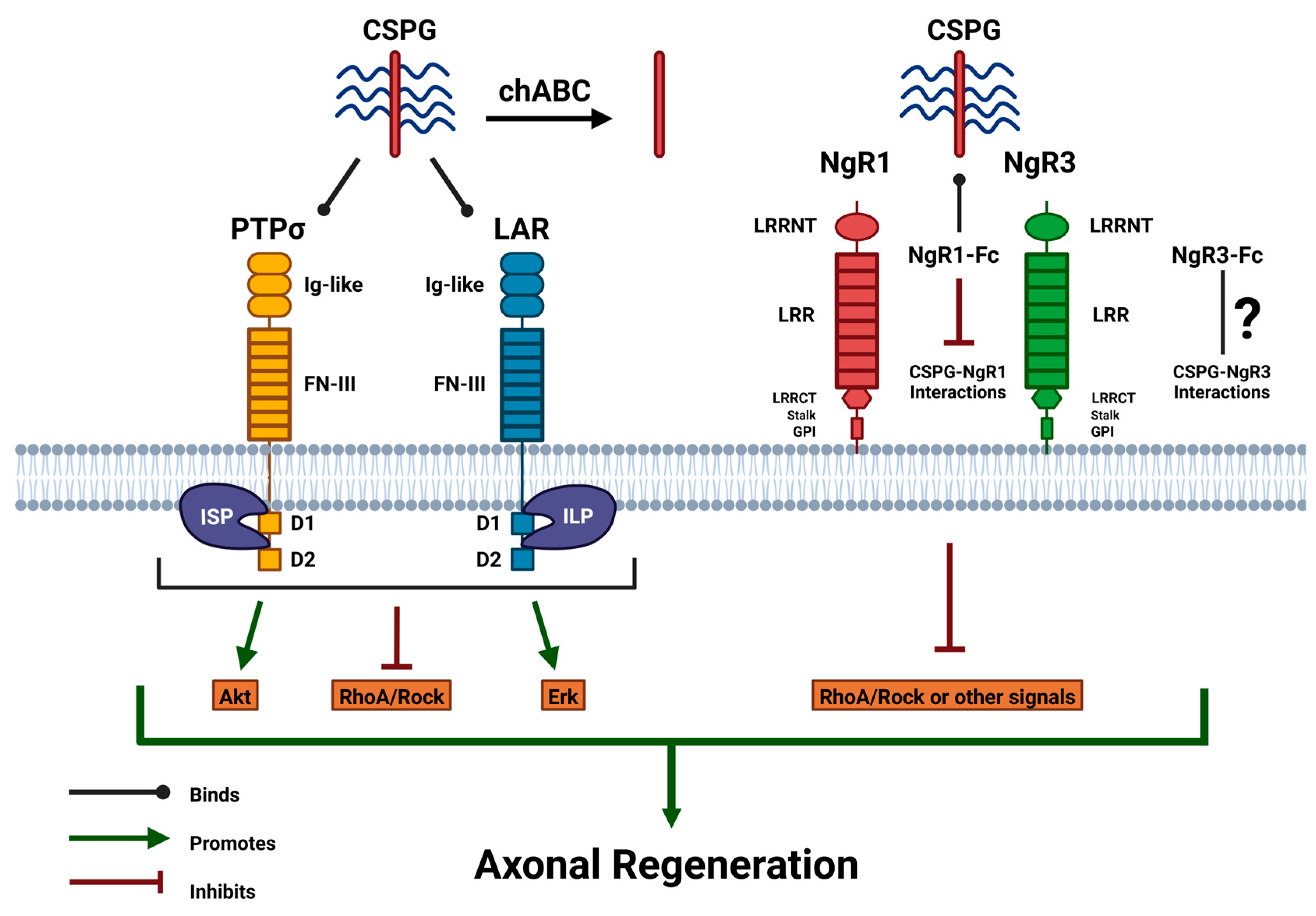

Table 1.
Summary of studies utilizing treatments to suppress the inhibitory effects of CSPGs.
| Therapeutic Name | Primary Target(s) | Mechanism of Action | Findings | References |
| ChABC | CSPG | Degrades the CS-GAG chains on CSPGs, removing the inhibitory properties of CSPG. | Promotes axonal regeneration, enhances neuroplasticity, and provides functional recovery in various SCI models. | [42,44,45,47,48,49] |
| PTPσ (−/−) | PTPσ Receptor | Knockout of PTPσ to inhibit CSPG-PTPσ interactions. | Enhances neural outgrowth to the lesion area and regeneration in CST axons. | [50,51,52] |
| LAR (−/−) | LAR Receptor | Knockout of LAR to inhibit CSPG-LAR interactions. | Augmented neurite regrowth with improving locomotor function and mitigating the activation of the RhoA/ROCK pathway. | [53,54] |
| ISP | PTPσ Receptor | Targets the intracellular domains of PTPσ to prevent inhibitory effects of CSPG-PTPσ interactions. | Regrowth of serotonergic fibers with functional recovery, along with axonal sprouting and neuroblast migration. | [55,56,57,58,59] |
| ILP | LAR Receptor | Targets the intracellular domains of LAR to prevent inhibitory effects of CSPG-LAR interactions. | Improved regeneration of neurite growth with enhanced axonal sprouting, Akt phosphorylation, and RhoA/ROCK reduction. | [53,54] |
| ISP + ILP | PTPσ and LAR Receptors | Combined treatment inhibits the effects of both CSPG-PTPσ and CSPG-LAR interactions. | Enhanced axonal density and neurite length, along with suppression of astrocyte differentiation. | [60,61] |
| NgR1 (−/−) & NgR3 (−/−) | NgR1 and NgR3 Receptors | Knockout of Nogo Receptors that mediate CSPG properties (NgR1 & NgR3) to inhibit CSPG-Nogo interactions. | Attenuated CSPG levels and promoted regeneration of RGC axons after induced ONC in models. | [39,62] |
| NgR1-Fc (AXER-204) | NgR1 Receptor Ligands | NgR1-Fc is a decoy that can bind to MAIs and CSPGs to inhibit their inhibitory properties. | Permitting growth beyond the lesion site with amplifying growth in CST axons and catalyzing neural repair. | [62,64,65] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



