Submitted:
07 May 2025
Posted:
08 May 2025
You are already at the latest version
Abstract
In this study, praseodymium-doped cobalt ferrite nanoparticles (CoFe₂₋yPryO₄, y = 0–0.2) were synthesized via sol-gel auto-combustion and systematically characterized to assess their structural, morphological, magnetic, and biological properties. X-ray diffraction (XRD) confirmed single-phase cubic cobalt ferrite formation for samples with y ≤ 0.05 and the emergence of a secondary orthorhombic PrFeO₃ phase at higher dopant concentrations. FTIR spectroscopy identified characteristic metal–oxygen vibrations and revealed a progressive shift of absorption bands with increasing Pr content. Vibrating sample magnetometry (VSM) demonstrated a gradual decline in saturation (Ms) and remanent (Mr) magnetization with praseodymium doping, an effect further intensified by cyclodextrin surface coating. TEM analyses revealed a particle size increase correlated with dopant level, while SEM images displayed a porous morphology typical of combustion-synthesized ferrites. In vitro cell viability assays showed minimal toxicity in normal human keratinocytes (HaCaT), while significant antiproliferative activity was observed against human cancer cell lines A375 (melanoma), MCF-7 (breast adenocarcinoma), and HT-29 (colorectal adenocarcinoma), particularly in Pr 6-CD and Pr 7-CD samples. These findings suggest that praseodymium substitution and cyclodextrin coating can effectively modulate the physicochemical and anticancer properties of cobalt ferrite nanoparticles, making them promising candidates for future biomedical applications.
Keywords:
praseodymium-doped magnetic nanoparticles
; cobalt ferrites
; cytotoxicity
; cell viability
; anticancer
1. Introduction
Introduced as concept in 1959, nanotechnology is now considered the most promising technology of the 21st century included in the medical field where nanoparticles may serve as novel diagnostic tools, targeted drug carriers or biomedical implants [1]. Among various types of nanoparticles, magnetic nanoparticles display unique properties such as diverse physicochemical parameters, easy synthesis, biocompatibility, and stability that render them suitable for biomedical purposes [2]. Magnetic nanoparticles are formed from metal elements or their oxides; superparamagnetic magnetite (Fe3O4) is the most commonly used given its high biocompatibility [2]. Partial substitution of iron with other metals such as Co, Ni, K, etc. produces ferrites that can be classified according to their crystal structure and magnetic properties; spinel ferrites display the general formula M2+Fe23+O4 and unique multifunctional properties such as strong magnetic behavior, high specific surface area, active sites at the surface allowing further functionalization, chemical stability as well as various shapes and sizes [3]. Unlike spinel ferrites where the divalent metal ion occupies tetrahedral sites while the iron ion Fe3+ can be found in octahedral sites, in cobalt ferrites (CoO·Fe2O3) the Co2+ ions are distributed in octahedral sites with the iron ions equally distributed in tetrahedral and octahedral sites which produces an inverse spinel structure [4] whose properties can be altered through composition and synthesis method.
Cobalt ferrite nanoparticles have been intensively studied due to their specific chemical, physical, electrical and mechanical properties that provide them with unique features which allow their application in various fields [5]. Unlike iron oxide nanoparticles that are prone to aggregation and oxidation and display renal and liver toxicity through oxidative cell damage following intraperitoneal administration, cobalt ferrite nanoparticles lack such toxic effects even at high doses necessary in hyperthermia [6]. Although frequently assessed as drug carriers, cobalt ferrites can act as anticancer agents themselves presumably due to their cytotoxic effects after cellular uptake as a result of pH-dependent release of Co2+ ions [7]. Moreover, when comparing Co2+ ions generated by cobalt chloride and cobalt ferrite, respectively, study showed that intrinsic cobalt toxicity prevents its use as monotherapy antitumor agent.
Cobalt ferrite nanoparticles were identified as antiproliferative agents against MCF7 breast cancer cells while lacking cytotoxic effects in HEK-293 human embryonic kidney cells thus displaying selective anticancer activity [8]. Similar results were reported in HCT-116 human colorectal cancer cells [9]. Studies show that at low concentrations the nanoparticles accumulate in the perinuclear region within cells; at increased concentrations, Co and Fe are present in the nuclear region triggering cell morphological alterations presumably due to an increased Co/Fe ratio as a result of biodegradation and Co accumulation in the cell nucleus [10]. However, more studies have been conducted on doped cobalt ferrites with different metal elements in the effort to enhance their properties and therefore their biomedical applications; doping with transitional metals leads to changes in the physical properties depending on the distribution of doping ions between the two interstitial sites of the cobalt ferrite spinel structure as well as on their valence [11]. Similarly, doping with large size rare earth metals produces significant adjustments in the physico-chemical features of cobalt ferrites such as specific surface area and particle size depending on the type and concentration of the dopant, synthesis method and cation distribution between the tetrahedral and octahedral sites [12]. Even more important, doping may increase the cytotoxicity of cobalt ferrite nanoparticles in cancer cells; substitution of Fe2+ with Mn2+ decreased MCF7 breast cancer cell viability presumably due to oxidative stress [13].
Rare earth elements or lanthanides exhibit biologic properties similar to those of calcium ions being involved in biomedical studies; they may be used as salts, coordination complexes, radioisotopes or oxides in cancer imagistic and therapy [14]. Rare earth metal-substituted cobalt ferrites can be easily synthesized and display a pure spinel phase, uniform and narrow particle size distribution; the substitution was able to decrease particle size and coercivity [15]. The molecular mechanism underlying the anticancer activity of such nanoparticles involves the pore formation within cell membrane, with strong selectivity versus healthy cells being reported; unlike healthy cells, cancer cell exhibits membrane depolarization that enables electroporation thus allowing delivery via induced pores [16]. Praseodymium (Pr)-doped nanorods decorated with poly-beta-cyclodextrin were synthesized as nanocarriers for the delivery of 5-fluorouracil; they display high-loading efficacy and a strong and selective anticancer activity in MCF7 breast cancer cells [17].
The aim of the current study was the synthesis of cobalt ferrite nanoparticles doped with praseodymium and their assessment as anticancer agents; the nanoparticles were physiochemically analyzed and tested thereafter on normal HaCaT cells and on A375, MCF-7 and HT-29 cancer cells. The biological assessment revealed that all compounds displayed a strong dose-dependent cytotoxic activity.
2. Materials and Methods
2.1. Chemicals
The raw materials used for synthetic purposes were iron nitrate nanohydrate (Fe(NO3)3·9H2O, Merck), cobalt nitrate hexahydrate (Co(NO3)2·6H2O, Sigma-Aldrich), dysprosium chloride x hydrate (PrCl3·xH2O, Sigma-Aldrich), glycine (C2H5NO2, Merck), (2-Hydroxypropyl)-γ-cyclodextrin (Sigma-Aldrich) and absolute ethanol (Merck).
2.2. Synthesis by Combustion Method
In order to synthesize 0.02 moles of cobalt ferrite, 0.04 moles iron nitrate, 0.02 moles cobalt nitrate and 0.09 mole of glycine were used; glycine acted as fuel while the nitrates functioned as oxidizing agent. The salts and the glycine were heated together at 60°C until the formation of a brown solution. Meanwhile, a porcelain capsule was preheated at around 350°C using a heating mantle. The mixture was then carefully poured in the capsule and the heating mantle was kept running at 350°C. After the entire water evaporated, the mixture became viscous and at some point, self-ignited. The combustion front propagated very fast in the entire mass, the reaction completing in around 9 seconds. During the reaction yellow flames indicating high temperature were observed; a black and crumbly powder formed as a result.
The praseodymium doped cobalt ferrite was synthesized following the same procedure but adding an extra step when praseodymium chloride was added into the mixture. The amount of cobalt nitrate (0.02 moles) and glycine (0.09 moles) remained unchanged while the praseodymium and iron nitrate quantities were varied. The Pr and Fe salts used are presented in Table 1. Regarding the reaction development, it is worth noted that white gases were released, the duration increased with 3-4 seconds and the final powder exhibited a more voluminous, spongy texture compared to the undoped sample.
The powders of both doped and undoped cobalt ferrite were washed several times using warm water, then dried and manually grinded in a mortar until a very fine powder is obtained.
2.3. Cyclodextrin Inclusion Complex
In order to enable biological evaluation, all samples were incorporated into (2-hydroxypropyl)-γ-cyclodextrin which has the ability to increase their water solubility; a molar ratio of 1:1 cyclodextrin:cobalt ferrite was used. Homogenization was accomplished by using a solvent composed of 0.9 ml distilled water and 2.1 ml absolute ethanol. Further, the mixture was stirred for 30 minutes and then dried at 70°C until full solvent evaporation. The resulting complex was then manually grinded into a fine powder that was later suspended in distilled water by using a UP200S ultrasonic homogenizer (Hielscher Ultrasonics GmbH, Teltow, Germany) for 2 hours at 50% amplitude and 0.8 cycles. After sonication, the particles remained suspended without the occurrence of sediments.
2.4. Characterization Methods
The investigation regarding sample phase composition was conducted through X-ray diffraction (XRD) analysis by means of a Rigaku Ultima IV device (Tokyo, Japan) operating at 40 kV and 40 mA; the XRD pattern was achieved by utilizing CuKα radiation. The FTIR spectra were obtained using KBr pellets on a Shimadzu IR Affinity-1S spectrophotometer (Shimadzu Scientific Instruments Inc., Columbia, MD, USA) operating between 400 and 4000 cm−1, with a resolution of 4 cm−1. Magnetic measurements were performed at room temperature on a LakeShore 8607 vibrating sample magnetometer (VSM, Shore Cryotronics, Westerville, OH, USA) at a magnetic field ranging between 30 and -30 KOe; prior to each test, samples were demagnetized in alternating field. Scanning electron microscopy (SEM) was performed on a Verios G4 UC Scanning Electron Microscope (Thermo Scientific, Czech Republic). The samples were coated prior to examination with 6 nm platinum using a Leica EM ACE200 Sputter coater in order to increase electrical conductivity and reduce charge buildup. SEM investigations were performed in High Vacuum mode using a detector for high-resolution images (Through Lens Detector, TLD) at an accelerating voltage of 10 kV. Morphological characterization was conducted in high contrast mode at 120 kV acceleration voltage on a Hitachi High-Tech HT7700 (Hitachi High-Technologies Corporation, Tokyo, Japan). The instrument is equipped with a STEM module, an energy dispersive X-ray (EDX) detector that allows elemental analysis and selected area electron diffraction (SAED) apertures that could be used to collect diffraction patterns. The samples were prepared by drop casting from their water suspension on 300 mesh carbon-coated copper grids (Ted Pella) and vacuum-dried at room temperature for 24 h.
2.5. Cell Culture
Immortalized human keratinocytes HaCaT (CLS Cell Lines Service GmbH, Eppelheim, Germany), human melanoma cells A375, human breast adenocarcinoma cells MCF-7 and human colorectal adenocarcinoma HT-29 cells (American Type Culture Collection ATTC, Lomianki, Poland) were used in the current study. HaCaT and A375 cells were cultured on Dulbecco’s Modified Eagle Medium (DMEM) high glucose supplemented with 10% fetal bovine serum (FBS) and 1% mixture of penicillin/streptomycin (100 U/mL). McCoy’s 5A supplemented with 10% FBS and 1% antibiotic mixture was employed for culturing the HT-29 cell line, while MCF-7 cells were cultured in Eagle’s Minimum Essential Medium (EMEM) containing the same concentrations of FBS, antibiotic mixture and 0.01 mg/mL human recombinant insulin. All cells were incubated at 37 °C and 5% CO2.
2.6. Cell Viability Assessment
The Alamar blue colorimetric assay was employed to determine the cell viability percentages of HaCaT, A375, MCF-7 and HT-29 cells after stimulation with increasing concentrations (0.025, 0.05, 0.1, 0.25 and 0.5 mg/mL) of the newly synthesized compounds for 48h. The cells (1 x 104) were seeded onto 96-well plates and incubated until reaching 85-90% confluence. The old medium was removed using an aspiration station and then replaced with fresh medium containing the tested concentrations of each compound. After 48h, 20 μL Alamar blue 0.01% was added to each well and the cells were further incubated for another 3h. To determine cell population, the absorbance of the wells was determined at two wavelengths, 570 nm and 600 nm, using a microplate reader (xMarkTM Microplate, Bio-Rad Laboratories, Hercules, CA, USA). All experiments were performed in triplicates.
2.7. Statistical Analysis
One-way ANOVA followed by Dunnett’s post-hoc test (GraphPad Prism version 6.0.0, GraphPad Software, San Diego, CA, USA) were used in order to assess statistical significance; the differences between the groups were considered statistically significant if p< 0.05, as follows: * p < 0.05, ** p < 0.01, and *** p < 0.001.
3. Results
3.1. X-Ray Diffraction (XRD)
All the X-ray diffraction patterns are well defined, exhibiting intense and narrow diffraction peaks that confirm the high sample crystallinity (Figure 1); the undoped sample Pr 1 shows specific diffraction peaks that characterize single-phase cobalt ferrite. The absence of a secondary phase in the spectra of doped samples Pr 2 and Pr 3 suggests the successful incorporation of praseodymium in the cobalt ferrite lattice and the definite formation of praseodymium substituted cobalt ferrite. The identification of the single-phase was made with the JCPDS card 22-1086. The diffraction peaks were reported at the 2θ angles 18.33°, 30.20°, 35.59°, 37.20°, 43.19°, 53.54°, 57.09°, and 62.68°, that correlate with the crystallographic planes (111), (220), (311), (222), (400), (422), (511), and (440), indicating the presence of cubic cobalt ferrite.
For doped samples Pr 4, 5, 6 and 7, other diffraction peaks were recorded which indicate the occurrence of a secondary phase identified as perovskite-type crystalline PrFeO3 by using JCPDS card 047-0065. The diffraction peaks located at 2θ angles of 22.78°, 32.55°, 46.61° and 58.14° correspond to the crystallographic planes (110), (112), (004) and (204) that confirm the formation of orthorhombic PrFeO3. However, in sample Pr 4, the presence of PrFeO3 is signaled by one very small diffraction peak that indicates its presence in trace amounts; in samples Pr 5, Pr 6 and Pr 7, an increased intensity is noticed for the diffraction peaks assigned to the secondary phase. Moreover, a direct correlation between the praseodymium content of the sample and the amount of PrFeO3 can be established.
3.2. Fourier-Transform Infrared Spectroscopy (FTIR)
The FTIR spectra for all samples are displayed in Figure 2; one can notice that all samples show an intense absorption band and a smaller one within the 400-800 cm-1 domain, characteristic for the metal-oxygen vibration; also, a small shift was noticed for the absorption maximum due to the praseodymium presence in the sample.
3.3. Investigation of Magnetic Properties (VSM)
The magnetic properties of the samples were analyzed and the hysteresis are presented in Figure 3.
One can notice that all samples display a similar behavior as indicated by resembling characteristics of the hysteresis, with variations in the recorded magnitude. The first two samples have a comparable saturation mass magnetization (Ms) value of approximatively 72 emu/g which decreases with the increase of the praseodymium content reaching 61.47 emu/g in sample Pr 7. In terms of remnant mass magnetization (Mr), samples Pr 1 and Pr 3 exhibit a value of 30 emu/g that decreased towards sample Pr 7 (28 emu/g), in a similar trend with the previously reported Ms. Interestingly enough, in samples Pr2 and Pr5 out-of-line Mr values of 34 and 29 emu/g, respectively, were recorded.
In cyclodextrin coated samples, the values of Ms and Mr suffer a significant decrease by comparison to the naked samples; as such, in Pr 1-CD (cobalt ferrite-CD) the respective values dropped to 8.8 emu/g (Ms) and 3.6 emu/g (Mr). In naked samples, the decrease of both Ms and Mr follows a linear trend while in cyclodextrin coated samples the same parameters decrease in a more irregular manner; however, in both cases, the strong reduction of Ms/Mr values with increasing amounts of praseodymium is maintained, with Ms values ranging between 8.8 and 7.6 emu/g and Mr between 4 and 3.2 emu/g. Of note, the presence of cyclodextrin greatly influences the magnetic properties of the sample.
3.4. Scanning Electron Microscopy (SEM) and Transmitted Electron Microscopy (TEM)
The SEM images of samples Pr 1 - Pr 7 display a spongious appearance with clustered particles. The aspect of the undoped sample is similar with the one of doped sample (Figure 4).
The TEM images present a roughly estimate of the particle size range for the obtained doped and undoped cobalt ferrite nanoparticles. For the sample that contains undoped cobalt ferrite, the particle diameters are located between 27 and 64 nm. Some particle conglomerates that reach up to 96 nm are also observable (Figure 5). In the case of doped samples, the range of the particles is within the following intervals: Pr 2 – 45 - 100 nm; Pr 3 – 41 -140 nm; Pr 4 – 54-140 nm; Pr 5 – 82-146 nm; Pr 6 – 75-210 nm; Pr 7 – 119-246 nm. An apparent dopant quantity-dependent diameter increase can also be observed (Figure 5).
3.5. Cell Viability
The viability of human keratinocytes HaCaT, human melanoma A375, human breast adenocarcinoma MCF-7 and human colorectal adenocarcinoma HT-29 cells was evaluated 48h post-treatment with the newly synthesized compounds (0.025, 0.05, 0.1, 0.25 and 0.5 mg/mL) by means of Alamar blue assay. The incubation of HaCaT cells revealed that Pr 1- CD did not altered cell viability at any of the concentrations tested, whereas Pr 2-CD, Pr 3-CD, Pr 4-CD, Pr 5-CD, Pr 6-CD and Pr 7-CD altered cell proliferation only when used at 0.5 mg/ml, the highest concentration tested (Figure 6).
In melanoma cells, Pr 2-CD, Pr 3-CD, Pr 4-CD, Pr 5-CD, Pr 6-CD and Pr 7-CD decreased cell viability in the most aggressive manner, expressing the lowest IC50 values (Table 2). Moreover, the other compound, Pr 1-CD also exerted significant inhibitory effects against A375 cells (Figure 7), particularly in high concentrations, as follows: 56.63% ± 11.91 (0.25 mg/mL) and 50.21% ± 11.29 (0.5 mg/mL).
When tested against colorectal adenocarcinoma HT-29 cells, Pr 5-CD (IC50 0.13 mg/mL), Pr 6-CD (IC50 0.17 mg/mL), and Pr 7-CD (IC50 0.049 mg/mL) induced the strongest antiproliferative effects, particularly when used in high concentration (Figure 8). Pr 1-CD, Pr 2-CD, Pr 3-CD and Pr 4-CD also decreased significantly cell viability, as follows: 72.95% ± 10.53 (Pr 1-CD 0.5 mg/mL), 57.45% ± 15.57 (Pr 2-CD 0.5 mg/mL), 59.48% ± 21.43 (Pr 3-CD 0.5 mg/mL) and 52.82% ± 29.23 (Pr 4-CD 0.5 mg/mL).
The stimulation of breast adenocarcinoma MCF-7 cells for 48h with the tested samples showed that all compounds reduced cell viability in a dose-dependent manner, with Pr 6-CD and Pr 7-CD exerting the strongest anticancer effect as indicated by an IC50 value of 0.008 mg/mL. All the newly synthesized compounds decreased MCF-7 cell viability (Figure 9) at an accelerated rate when the highest tested concentrations were applied, as follows: 37.34% ± 8.09 (Pr 1-CD 0.5 mg/mL), 37.7% ± 3.29 (Pr 2-CD 0.5 mg/mL), 38.77% ± 4.72 (Pr 3-CD 0.5 mg/mL), 31.53% ± 7.95 (Pr 4-CD 0.5 mg/mL), 42.97% ± 6.70 (Pr 5-CD 0.5 mg/mL), 37.06% ± 1.87 (Pr 6-CD 0.5 mg/mL), 33.79% ± 10.49 (Pr 7-CD 0.5 mg/mL).
4. Discussion
In the current study we aimed to achieve the synthesis of cobalt ferrite nanoparticles doped with praseodymium in the effort to identify therapeutic alternatives to fight melanoma, colorectal adenocarcinoma and breast cancer. Although conventional therapies that combine cancer staging with a multitude of strategies (chemotherapy, radiation, surgery) can produce satisfying outcomes for a limited period of time, they also come with severe side effects due to lack of selectivity and may also induce drug resistance. Additionally, conventional anticancer drugs may exhibit poor pharmacokinetic profiles that may be associated with partial lack of efficacy or toxicity. Such challenges can be overcome by using nanoformulations that provide benefits in terms of bioavailability, stability and administration [18]. Among the huge number of metallic nanoparticles, cobalt ferrites have gained increased interest due to their high coercivity at room temperature combined with moderate magnetization [19]. Cobalt ferrites are spinel nanoparticles that have been tested as therapeutic agents both as drug carriers as well as hyperthermia agents [20]; however, their intrinsic anticancer properties were poorly investigated so far. One such study revealed their ability to reduce viability in MCF7 breast cancer cells in a dose-dependent manner with stronger effects compared to magnesium-doped cobalt ferrites [21]. Such effects could be triggered by the intrinsic toxicity of cobalt ions that may act as cytotoxic agents following their release as a result of lysosome degradation of the cobalt ferrite nanoparticle in an acid environment [21]. The use of cobalt ferrites as anticancer agents is therefore worthy of investigation particularly considering their ability to combine heat and cytotoxic effects into one single platform. Moreover, the introduction of small amounts of rare earth metals into the lattice of cobalt ferrites can tune their magnetic, electronic and cytotoxic properties presumably due to the complex interactions between the 3d cation orbitals and 2p oxygen orbitals as a result of metal distribution over the tetragonal and octahedral positions [22]. Based on previous promising results with dysprosium doping [4], we chose to prepare praseodymium-doped cobalt ferrite nanoparticles whose physicochemical and biological analysis was conducted; their low water solubility introduced the challenge of facilitating their uptake by the aqueous biological medium which was solved by adding the hydrophilic HPGCD.
A multitude of synthetic approaches have been designed to achieve cobalt ferrite nanoparticle, including sol-gel emulsion or auto-combustion, co-precipitation, gamma-irradiation, microwave mediated, hydrothermal, thermal decomposition [23], displaying various challenges such as the use of toxic reagents or uncontrollable properties of the resulting nanoparticles. The combustion method used in the current study provides undisputable advantages in terms of feasibility (single-step method, easily applicable and not expensive) but also quality of the final product: high crystallinity, homogenous ferrites where the intrinsic energy of the components enable the occurrence of metastable phases in one step [24]. Additionally, through controlling the reaction parameters, one can modulate the properties of the final nanoparticles [25].
The X-ray diffraction analysis revealed the formation of cubic cobalt ferrite and the successful incorporation of praseodymium in the ferrite structure. Dheeraj Yadav et al. [26] investigated the influence of the annealing temperature on cobalt ferrite formation. They identified the cobalt ferrite using the JCPDS file 22-1086; on the XRD spectra, the most intense maximum diffractions were located at 2θ values similar to the ones observed in the current work (e.g., 35.55° vs. 35.59°). Also, the allocated crystallographic planes were identical to our case. The same JCPDS file was employed in other studies [27,28,29,30] for the identification of the spinel structure.
In our study, the synthesis of CoFe2-yPryO4 led to the formation of single phase for samples Pr 1, Pr 2, Pr 3 and Pr 4 (y=0; 0.01; 0.03; 0.05, respectively), and to the occurrence of a secondary phase for samples Pr 5, Pr 6 and Pr 7 (y=0.1; 0.15; 0.2, respectively). Of note, the formation of the secondary phase did not produce a visible decrease of the peak intensity in praseodymium-doped cobalt ferrites. The secondary phase, orthorhombic perovskite PrFeO3 was identified using the JCPDS card 047-0065 which was also used in other studies for similar purposes [31,32].
Our results are in agreement with those reported by Pachpinde et al. [33] who synthetized PrxCoFe2-xO4 (x=0 – 0.1) by sol-gel auto combustion method using citric acid and nitrates as precursors; results showed that for x˂0.05 praseodymium was successfully incorporated into the ferrite lattice while for values above 0.05, a secondary phase of perovskite PrFeO3was found alongside the cubic cobalt ferrite. An explanation for the occurrence of the secondary phase was provided by Nikmanesh et al. [28] who investigated the effect of praseodymium doping on the cobalt ferrite structure; a sol-gel auto combustion method was used for the synthesis of CoFe2−xPrxO4 (x = 0, 0.02, 0.04, 0.06). The research focused on the synthesis and thorough investigation of the impact provided by iron substitution with a lanthanide upon the spinel lattice; a secondary phase of PrFeO3 occurred for x=0.06 as revealed by the two diffraction peaks located at 32.56° and 46.68° in the XRD spectra, values similar to the ones recorded in our study. The authors hypothesized that such a secondary phase can be a result of the difference between the ionic radius of iron and substituting praseodymium; similar conclusions were drawn in other studies where various lanthanides were doped in ferrite structures [34,35,36].
In the next step, the FTIR spectra were built, revealing a strong absorption band around 570 cm-1 in each investigated sample, with a shoulder present around 400 cm-1, characteristic for the stretching vibrations attributed to the tetrahedral vibration (metal-oxygen in site A) and the octahedral vibration (metal-oxygen in site B), respectively [37]. In a series of trivalent ions of rare earth metals (Gd, Sm, Ho, Er, Yb, Y) used as dopants for cobalt ferrite, Jing et al. [35] emphasized that the absorption band around 570 cm-1 shifts to higher wavenumbers for samarium, holmium, erbium, ytterbium and yttrium while for gadolinium the shift occurs towards lower wavenumbers. Moreover, they assigned the 570 cm-1 band to the tetrahedral stretching vibration between metal and oxygen, and the second band to the octahedral stretching vibration between metal and oxygen. Additionally, the samples that contain praseodymium exhibited a shift towards lower wavenumbers, e.g., 567 cm-1 in Pr 7. A similar observation was previously reported in praseodymium-doped cobalt ferrite [33] as well as in dysprosium-doped cobalt ferrite [4], were the wavenumber decreased with the increase of the rare earth metal substitution. The replacement of iron by praseodymium, with larger ionic radius, results in an increased unit cell dimensions and alters the iron-oxygen vibrations thus leading to change in band positions. The peak intensity also changes with an increased Pr3+ substitution; the change in the frequency of the second stretching band points to the preferred orientation of Pr3+ ions towards octahedral sites. Also, the slight decrease of the second band frequency may be attributed to the presence of the secondary phase in samples with higher Pr3+ substitution level [33].
The VSM analysis revealed that the presence of praseodymium in the ferrite lattice reduced the Ms and Mr values, particularly in samples where cyclodextrin was used as coating material; such intense magnetization reduction can be attributed to the large amount of cyclodextrin that lacks magnetic behavior and is able to shield the intrinsic magnetic properties of the included ferrite. Similar data was reported in samples of CoFe2−xPrxO4 (x = 0, 0.02, 0.04, 0.06) [28] where the Ms values decreased with the increase of praseodymium content; as an example, for x=0, Ms was around 80 emu/g and reached 60 emu/g for x=0.06. In our study, the Ms value for x=0 was around 73 emu/g and dropped to 66 emu/g for x=0.05. Vani et al. [30] studied the effect of terbium ions (Tb3+) on the cobalt ferrite structure and properties; they reached a similar conclusion in terms of magnetic properties, reporting that the value of Ms decreases with the increase of terbium concentration in the sample presumably due to the substitution of the highly magnetic iron with the less magnetic terbium. Such conclusions were supported by other studies on rare earth metal-doped cobalt ferrites where the values of both Ms and Mr could be clearly correlated with the content of the doping material [4,38,39].
The aspect of the samples illustrated in the SEM images, a sponge, foam-like cavernous structure is typical for the cobalt ferrite obtained by combustion synthesis. Similar morphological features were previously presented by Qing et al. [40] in the case of gadolinium doped cobalt ferrite and Abbas et al. [41] for aluminum doped cobalt ferrite, both prepared by sol-gel autocombustion method. Although, the SEM images are comparable for the undoped and doped samples, in the case of TEM images can be clearly noticed that the presence of the dopant influences the particle dimensions and shape. In addition, the images present a tendency of particle agglomeration or even superposition. In our case, was observed an increase of particle size with the increase of dopant quantity, starting from a range of 24-64 nm for the pure sample going towards 120-245 nm for sample Pr 7, that has the highest amount of dopant. Similar findings were presented by Yadav et al. [42] in the study regarding the synthesis of CoFe2−xPrxO4 (x = 0.0, 0.025, 0.05, 0.075, 0.1) using starch-assisted sol–gel autocombustion. Starting with a range of 5-10 nm for the undoped sample, the particles dimension doubled in size for a content of praseodymium of 0.075. Manikandan et al. [43] synthesized by chemical oxidation method pure and doped cobalt ferrite, using praseodymium in different ratios between 0% - 5%. The FESEM results indicated spherical particles with a range between 30 to 160 nm in the case of pure sample. When praseodymium was added into the sample, and the doping percentage increased up to 5%, was remarked an increase of particle size ranging from 70 to 200 nm. Furthermore, in the case of zinc-cobalt ferrite [44] was observed an increase in particle dimension and altering of particle shape after praseodymium doping. Comparable findings were presented in the case of gadolinium doped cobalt ferrite CoFe2−xGdxO4 (x=0-0.30) [45].
The biological evaluation of the newly synthetized Pr-doped cobalt ferrite nanoparticles as cyclodextrin inclusion complexes was conducted both on normal human keratinocytes cell line and three cancer cell lines, namely human melanoma, human breast adenocarcinoma and human colorectal adenocarcinoma cell lines. This strategy provides insights on the efficacy of the tested samples as anticancer agents while investigating their selectivity. Reference to previously reported data is challenging since, to the best of our knowledge, this is the first biological evaluation of Pr-doped CoFe2O4 nanoparticles. The biological assessment in HaCaT normal human keratinocytes revealed that the tested compounds exhibited low cytotoxic, dose-dependent activity; thus, an antiproliferative activity was only displayed when the highest concentration (0.5 mg/mL) of the tested compounds was applied. Moreover, taken together with the high IC50 values (> 0.5 mg/mL) obtained, the results indicate a high biocompatibility of tested compounds as well as their potential selective anticancer activity which stands as an important parameter in the development of new anticancer agents. The high biocompatibility of our compounds when tested in HaCat cells are also in line with previous data on cobalt ferrite nanoparticles that reported low cytotoxicity even at concentrations of 2 mg/mL [46], with significant reductions in cell viability only at concentrations as high as 4 mg/mL combined with 72 and 96 h incubation time [46].
The evaluation of Pr-doped cobalt ferrite nanoparticles cytotoxic effects in A375 human melanoma cells disclosed a dose-dependent antiproliferative activity notably after treatment with the highest concentration of each compound (0.5 mg/mL). The intensity of the cell viability inhibitory activity correlates with the amount of Pr in the tested cobalt ferrite nanoparticles; the cytotoxic activity of the tested compounds increased exponentially with the amount of Pr. The role of Pr in exerting the inhibitory effect is obvious when comparing the doped cobalt ferrites to the undoped ones, in all tested concentrations (e.g., Pr 1-CD vs. Pr 2-CD). To the best of our knowledge, this is the first assessment of Pr-doped cobalt ferrites as anticancer agents; however, we previously conducted similar studies on Dy-doped CoFe2O4 nanoparticles that revealed a dose-dependent cytotoxic activity against the A375 cell line which was also correlated to the amount of dysprosium used as substitution for the iron ions [4]. Cobalt ferrite nanoparticles were also previously investigated for magnetic hyperthermia and light-based treatments in the eradication of cancer stem cells in A375 cell line, displaying effective cytotoxicity with IC50 values of 0.025 mg/ml [47].
The antiproliferative activity of the tested compounds was confirmed in HT-29 colorectal adenocarcinoma cell line. When tested in the highest concentration, all compounds displayed a strong dose-dependent cytotoxic activity. Compounds Pr 5-CD and notably Pr 6-CD exhibited exponential cytotoxicity in correlation to an increased praseodymium substitution, with cell viability values of 29.54% and 25.28% respectively. Pr 7-CD exhibited the strongest antiproliferative activity in all tested concentrations, decreasing HT-29 cell viability to 9.28% when tested in the highest concentration, presumably due to its high amount of Pr. As we stated previously, we were not able to find another attempt to assess the anticancer activity of Pr-doped cobalt ferrite nanoparticles; however, in a similar study, Pr-substituted Ni-Co nano-spinel ferrites exhibited efficient cell viability inhibition in HCT-116 colorectal carcinoma cells, displaying an average inhibitory activity of 50% [16]. Moreover, Nd3+ and Ce3+ co-substituted Co ferrite nanoparticles were tested on the same colorectal carcinoma cell line, results indicating significantly reduced cell viability through dose-dependent apoptotic effects [48].
When tested on the MCF-7 breast adenocarcinoma cell line, all compounds displayed a dose-dependent antiproliferative activity, with IC50 values ranging from 0.08 to 0.32 (Table 2). The cytotoxic activity in MCF-7 cells is more evident when compared to results on the HaCaT cell line, where an IC50 value >0.5 was obtained for all compounds, thus emphasizing the selectivity of the tested compounds against MCF-7 cells. An overview of cell viability inhibition in all tested cancer cell lines, with respect to their IC50 values (Table 2), revealed that the MCF-7 cell line was the most responsive to the antiproliferative effect exerted by the Pr-doped CoFe2O4 nanoparticles. When compared to our previous results on Dy-doped CoFe2O4 nanoparticles, the Pr-doped CoFe2O4 nanoparticles displayed higher cytotoxicity, reducing MCF-7 cell viability to less than 50% when using half the concentration [4]. Pr-metal nanorods coated with poly-CD polymer were synthesized as carrier for 5-fluorouracil and investigated in terms of their anticancer potential against the MCF-7 cell line; they were revealed as a suitable vehicle while providing sustained release for the incorporated drug [17]. Also, cobalt ferrites were investigated as drug carriers in MCF-7 cells; one study involved the use of cobalt ferrites for the delivery of docetaxel where an intrinsic cytotoxicity of the bare nanoparticles was reported [49]. Conversely, another study where the cobalt ferrite nanoparticles were used as carriers for letrozole revealed the absence of cytotoxicity of the bare nanoparticles in breast cancer cells; however, an additional coating with methionine was applied in this case which might have increased their physiological acceptance [50]. In addition, cobalt ferrite nanoparticles demonstrated a mild anti-proliferative activity against MCF-7 cancer cell line promoting a mean viability of 74-85% viability in all tested concentrations [51]. It is important to note however that the enhanced inhibitory activity of Pr-doped cobalt ferrites could be previewed if considering the promising results reported by Andiappan et al. when investigating the anticancer effects of Pr-doped Schiff bases in several cancer cells; they revealed that the metal ions (Pr3+) exhibited strong affinity for the cell walls thus leading to DNA fragmentation and arresting cell proliferation [52]. Additionally, a polymeric quadrivalent praseodymium complex was tested in seven cancer cell lines and, although weaker than conventional anticancer agents, they induced cytotoxic effects in all types of cancer cells but particularly in A375 melanoma cells [53]. Exposure of cancer cells to low concentrations of a small coordinated complex of Pr and pyrithione significantly diminishes cell survival regardless of drug-resistant phenotype by affecting the cell waste clearing mechanisms as well as mitochondrial metabolism [54]. Collectively, our data corroborate with previously published results in terms of the anticancer effects of both Pr and cobalt ferrites thus emphasizing the potential of Pr-doped nanoparticles to act as an effective alternative to conventional treatments.
5. Conclusions
The successful synthesis of praseodymium-substituted cobalt ferrites was confirmed through comprehensive structural and spectroscopic characterization. In vitro studies revealed high tolerability on normal HaCaT cells and notable antiproliferative effects on multiple cancer cell lines (A375, MCF-7, HT-29), particularly in the case of samples Pr 6-CD and Pr 7-CD, which exhibited the lowest IC₅₀ values. Overall, the study underscores the potential of praseodymium-doped cobalt ferrite nanoparticles, particularly those surface-modified with cyclodextrin, as multifunctional platforms for targeted biomedical applications.
Author Contributions
Conceptualization, S.R. and C.Ș.; methodology, R.R. and A.G.; software, M.M.; validation, A.G and C.Ș.; formal analysis, M.M., A.Mil., A.Mio., R.N.G., and N.L.M; investigation, S.R., R.R. A.Mil., A.Mio., R.N.G, C.T. and N.L.M; data curation, C.T..; writing—original draft preparation, S.R. and R.R., .; writing—review and editing, A.G and C.Ș.; visualization, A.G and C.Ș.; supervision, C.Ș. All authors have read and agreed to the published version of the manuscript.
Funding
We would like to acknowledge the “Victor Babes” University of Medicine and Pharmacy Timisoara for their support in covering the costs of publication for this research paper.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Haleem, A.; Javaid, M.; Singh, R.P.; Rab, S.; Suman, R. Applications of nanotechnology in medical field: a brief review. Glob. Heal. J. 2023, 7, 70–77. [CrossRef]
- Ali, A.; Shah, T.; Ullah, R.; Zhou, P.; Guo, M.; Ovais, M.; Tan, Z.; Rui, Y.K. Review on Recent Progress in Magnetic Nanoparticles: Synthesis, Characterization, and Diverse Applications. Front. Chem. 2021, 9, 629054.
- Amiri, M.; Salavati-Niasari, M.; Akbari, A. Magnetic nanocarriers: Evolution of spinel ferrites for medical applications. Adv. Colloid Interface Sci. 2019, 265, 29–44.
- Rotunjanu, S.; Racoviceanu, R.; Mioc, A.; Milan, A.; Negrea-Ghiulai, R.; Mioc, M.; Marangoci, N.L.; Şoica, C. Newly Synthesized CoFe2−xDyxO4 (x = 0; 0.1; 0.2; 0.4) Nanoparticles Reveal Promising Anticancer Activity against Melanoma (A375) and Breast Cancer (MCF-7) Cells. Int. J. Mol. Sci. 2023, 24. [CrossRef]
- Tamboli, Q.Y.; Patange, S.M.; Mohanta, Y.K.; Sharma, R.; Zakde, K.R. Green Synthesis of Cobalt Ferrite Nanoparticles: An Emerging Material for Environmental and Biomedical Applications. J. Nanomater. 2023, 2023, 9770212. [CrossRef]
- Garanina, A.S.; Nikitin, A.A.; Abakumova, T.O.; Semkina, A.S.; Prelovskaya, A.O.; Naumenko, V.A.; Erofeev, A.S.; Gorelkin, P. V; Majouga, A.G.; Abakumov, M.A.; et al. Cobalt Ferrite Nanoparticles for Tumor Therapy: Effective Heating versus Possible Toxicity. Nanomaterials 2022, 12. [CrossRef]
- Balakrishnan, P.B.; Silvestri, N.; Fernandez-Cabada, T.; Marinaro, F.; Fernandes, S.; Fiorito, S.; Miscuglio, M.; Serantes, D.; Ruta, S.; Livesey, K.; et al. Exploiting Unique Alignment of Cobalt Ferrite Nanoparticles, Mild Hyperthermia, and Controlled Intrinsic Cobalt Toxicity for Cancer Therapy. Adv. Mater. 2020, 32, 2003712. [CrossRef]
- Panda, J.; Das, S.; Kumar, S.; Tudu, B.; Sarkar, R. Investigation of antibacterial, antioxidant, and anticancer properties of hydrothermally synthesized cobalt ferrite nanoparticles. Appl. Phys. A Mater. Sci. Process. 2022, 128, 1–11. [CrossRef]
- Alfareed, T.M.; Slimani, Y.; Almessiere, M.A.; Nawaz, M.; Khan, F.A.; Baykal, A.; Al-Suhaimi, E.A. Biocompatibility and colorectal anti-cancer activity study of nanosized BaTiO3 coated spinel ferrites. Sci. Rep. 2022, 12, 14127. [CrossRef]
- Marmorato, P.; Ceccone, G.; Gianoncelli, A.; Pascolo, L.; Ponti, J.; Rossi, F.; Salomé, M.; Kaulich, B.; Kiskinova, M. Cellular distribution and degradation of cobalt ferrite nanoparticles in Balb/3T3 mouse fibroblasts. Toxicol. Lett. 2011, 207, 128–136. [CrossRef]
- Chakrabarty, S.; Dutta, A.; Pal, M. Enhanced magnetic properties of doped cobalt ferrite nanoparticles by virtue of cation distribution. J. Alloys Compd. 2015, 625, 216–223. [CrossRef]
- Salih, S.J.; Mahmood, W.M. Review on magnetic spinel ferrite (MFe(2)O(4)) nanoparticles: From synthesis to application. Heliyon 2023, 9, e16601. [CrossRef]
- Fiaz, S.; Ahmed, M.N.; Haq, I. ul; Shah, S.W.A.; Waseem, M. Green synthesis of cobalt ferrite and Mn doped cobalt ferrite nanoparticles: Anticancer, antidiabetic and antibacterial studies. J. Trace Elem. Med. Biol. 2023, 80, 127292. [CrossRef]
- Wang, J.; Li, S. Applications of rare earth elements in cancer: Evidence mapping and scientometric analysis. Front. Med. 2022, 9, 946100. [CrossRef]
- Wu, X.; Ding, Z.; Song, N.; Li, L.; Wang, W. Effect of the rare-earth substitution on the structural, magnetic and adsorption properties in cobalt ferrite nanoparticles. Ceram. Int. 2016, 42, 4246–4255. [CrossRef]
- Rehman, S.; Jermy, B.R.; Rather, I.A.; Sabir, J.S.M.; Aljameel, S.S.; Almessiere, M.A.; Slimani, Y.; Khan, F.A.; Baykal, A. Pr(3+) Ion-Substituted Ni-Co Nano-Spinel Ferrites: Their Synthesis, Characterization, and Biocompatibility for Colorectal Cancer and Candidaemia. Pharmaceuticals (Basel). 2023, 16. [CrossRef]
- Manikantan, V.; Sri Varalakshmi, G.; Ashapak Tamboli, U.; Sumohan Pillai, A.; Alexander, A.; Lucas, A.; Akash, B.A.; Enoch, I.V.M. V Praseodymium metal nanorods as a 5-fluorouracil carrier. J. Rare Earths 2024, 42, 1328–1336. [CrossRef]
- Gelperina, S.; Kisich, K.; Iseman, M.D.; Heifets, L. The potential advantages of nanoparticle drug delivery systems in chemotherapy of tuberculosis. Am. J. Respir. Crit. Care Med. 2005, 172, 1487–1490. [CrossRef]
- Maaz, K.; Mumtaz, A.; Hasanain, S.K.; Ceylan, A. Synthesis and magnetic properties of cobalt ferrite (CoFe2O4) nanoparticles prepared by wet chemical route. J. Magn. Magn. Mater. 2007, 308, 289–295. [CrossRef]
- Srinivasan, S.Y.; Paknikar, K.M.; Bodas, D.; Gajbhiye, V. Applications of cobalt ferrite nanoparticles in biomedical nanotechnology. Nanomedicine 2018, 13, 1221–1238.
- Ahmad, N.; Alomar, S.Y.; Albalawi, F.; Khan, M.R.; Farshori, N.N.; Wahab, R.; Shaik, M.R. Anticancer potentiality of green synthesized Mg-Co ferrites nanoparticles against human breast cancer MCF-7 cells. J. King Saud Univ. - Sci. 2023, 35, 102708. [CrossRef]
- Pęczkowski, P.; Szostak, E.; Pocheć, E.; Michalik, J.M.; Piętosa, J.; Tahraoui, T.; Łuszczek, M.; Gondek, Ł. Biocompatibility and potential functionality of lanthanum-substituted cobalt ferrite spinels. J. Alloys Compd. 2023, 966, 171433. [CrossRef]
- Kashid, P.; Suresh, H.; Mathad, S.; Shedam, R.; Shedam, M. A Review on Synthesis, Properties and Applications on Cobalt Ferrite. Int. J. Adv. Sci. Eng. 2022, 9, 2567. [CrossRef]
- Prabhakaran, T.; Hemalatha, J. Combustion synthesis and characterization of cobalt ferrite nanoparticles. Ceram. Int. 2016, 42, 14113–14120. [CrossRef]
- Pop, D.; Buzatu, R.; Moacă, E.A.; Watz, C.G.; Cîntă-Pînzaru, S.; Tudoran, L.B.; Nekvapil, F.; Avram, Ștefana; Dehelean, C.A.; Crețu, M.O.; et al. Development and characterization of Fe3O4@carbon nanoparticles and their biological screening related to oral administration. Materials (Basel). 2021, 14, 3556. [CrossRef]
- Yadav, D.; Gahlawat, R.; Shukla, R. A comprehensive analysis of the impact of annealing temperature variation on the structural, optical, morphological, magnetic, and photocatalytic properties of CoFe2O4 nanoparticles. Ionics (Kiel). 2024, 30, 6559–6574. [CrossRef]
- Hadela, A.; Lakić, M.; Potočnik, M.; Košak, A.; Gutmaher, A.; Lobnik, A. Novel reusable functionalized magnetic cobalt ferrite nanoparticles as oil adsorbents. Adsorpt. Sci. Technol. 2020, 38, 168–190. [CrossRef]
- Nikmanesh, H.; Jaberolansar, E.; Kameli, P.; Varzaneh, A.G. Effect of praseodymium in cation distribution, and temperature-dependent magnetic response of cobalt spinel ferrite nanoparticles. Nanotechnology 2022, 33. [CrossRef]
- Khan Asghar, H.M.N. ul H.; Nawaz, M.K.; Hussain, R.; Gilani, Z.A. Synthesis and Characterization of Praseodymium Doped Nickel Zinc Ferrites using Microemulsion Method. J. Mater. Phys. Sci. 2020, 1, 98–108. [CrossRef]
- Vani, K.; Hashim, M.; Rana, G.; Ismail, M.M.; Batoo, K.M.; Hadi, M.; Kumar, N.P.; Naveena, G.; Sathish, B.; Sriramulu, G.; et al. Impact of rare earth Tb3+ substitution in cobalt ferrites: Tuning structural, dielectric, magnetic properties and photocatalytic activity. Ceram. Int. 2025, 51, 240–251. [CrossRef]
- Nguyen, A.; Nguyen, N.; Mittova, I.; Perov, N.; Mittova, V.; Cam, T.; Hoang, C.; Nguyen, M.; Nguyen, V.; Pham, V.; et al. Crystal structure, optical and magnetic properties of PrFeO 3 nanoparticles prepared by modified co-precipitation method. 2021, 355–361. [CrossRef]
- Ni, P.; Ma, S.; Ma, N.; Xu, C.; Fan, G.; Guo, J.; Wei, J.; Liu, J. Porous hollow sphere structure PrFeO3 as an efficient sensing material for n-butanol detection. J. Alloys Compd. 2024, 1002, 175392. [CrossRef]
- Pachpinde, A.M.; Langade, M.M.; Lohar, K.S.; Patange, S.M.; Shirsath, S.E. Impact of larger rare earth Pr3+ ions on the physical properties of chemically derived PrxCoFe2−xO4 nanoparticles. Chem. Phys. 2014, 429, 20–26. [CrossRef]
- Tahir Farid, H.M.; Ahmad, I.; Bhatti, K.A.; Ali, I.; Ramay, S.M.; Mahmood, A. The effect of praseodymium on Cobalt-Zinc spinel ferrites. Ceram. Int. 2017, 43, 7253–7260. [CrossRef]
- Jing, X.; Guo, M.; Li, Z.; Qin, C.; Chen, Z.; Li, Z.; Gong, H. Study on structure and magnetic properties of rare earth doped cobalt ferrite: The influence mechanism of different substitution positions. Ceram. Int. 2023, 49, 14046–14056. [CrossRef]
- Pachpinde, A.M.; Langade, M.M.; Mandle, U.M.; Shinde, B.L.; Lohar, K.S. Effect of rare earth sustituents Pr3+ and Ho3+ on structural and magnetic properties of cobalt ferrites. Rasayan J. Chem 2023, 16.
- Kumar, H.; Singh, J.P.; Srivastava, R.C.; Negi, P.; Agrawal, H.M.; Asokan, K. FTIR and Electrical Study of Dysprosium Doped Cobalt Ferrite Nanoparticles. J. Nanosci. 2014, 2014, 1–10. [CrossRef]
- Zhao, L.; Yang, H.; Zhao, X.; Yu, L.; Cui, Y.; Feng, S. Magnetic properties of CoFe2O4 ferrite doped with rare earth ion. Mater. Lett. 2006, 60, 1–6. [CrossRef]
- Javed, F.; Abbas, M.A.; Asad, M.I.; Ahmed, N.; Naseer, N.; Saleem, H.; Errachid, A.; Lebaz, N.; Elaissari, A.; Ahmad, N.M. Gd3+ doped cofe2o4 nanoparticles for targeted drug delivery and magnetic resonance imaging. Magnetochemistry 2021, 7, 1–16. [CrossRef]
- Lin, Q.; Lin, J.; He, Y.; Wang, R.; Dong, J. The Structural and Magnetic Properties of Gadolinium Doped CoFe2O4 Nanoferrites. J. Nanomater. 2015, 2015, 294239. [CrossRef]
- Abbas, N.; Rubab, N.; Sadiq, N.; Manzoor, S.; Khan, M.I.; Garcia, J.F.; Aragao, I.B.; Tariq, M.; Akhtar, Z.; Yasmin, G. Aluminum-doped cobalt ferrite as an efficient photocatalyst for the abatement of methylene blue. Water (Switzerland) 2020, 12, 2285. [CrossRef]
- Yadav, R.S.; Havlica, J.; Kuřitka, I.; Kozakova, Z.; Masilko, J.; Hajdúchová, M.; Enev, V.; Wasserbauer, J. Effect of Pr3+Substitution on Structural and Magnetic Properties of CoFe2O4Spinel Ferrite Nanoparticles. J. Supercond. Nov. Magn. 2015, 28, 241–248. [CrossRef]
- Manikandan, V.S.; Kavin Kumar, T.; Poobalan, R.K.; Sakthivel, P.; Chidhambaram, N.; Dineshbabu, N.; Dhanabalan, S.S.; Abarzúa, C.V.; Morel, M.J.; Thirumurugan, A. Exploring the magnetic and supercapacitor characteristics of praseodymium-doped CoFe2O4 magnetic nanoparticles. J. Mater. Sci. Mater. Electron. 2024, 35, 46. [CrossRef]
- Khan, N.-H.; Gilani, Z.A.; Abid, M.; Samiullah; Hussain, G.; Khalid, M.; Noor Huda Khan Asghar, H.M.; Nawaz, M.Z.; Ali, S.M.; Khan, M.A.; et al. Structural, dielectric and magnetic characteristics of praseodymium doped Cobalt-Zinc spinel ferrites for communication and microwave frequency applications. Appl. Phys. A 2024, 130, 829. [CrossRef]
- Peng, J.; Hojamberdiev, M.; Xu, Y.; Cao, B.; Wang, J.; Wu, H. Hydrothermal synthesis and magnetic properties of gadolinium-doped CoFe2O4 nanoparticles. J. Magn. Magn. Mater. 2011, 323, 133–137. [CrossRef]
- Oliveira, A.B.B.; De Moraes, F.R.; Candido, N.M.; Sampaio, I.; Paula, A.S.; De Vasconcellos, A.; Silva, T.C.; Miller, A.H.; Rahal, P.; Nery, J.G.; et al. Metabolic Effects of Cobalt Ferrite Nanoparticles on Cervical Carcinoma Cells and Nontumorigenic Keratinocytes. J. Proteome Res. 2016, 15, 4337–4348. [CrossRef]
- Khalili Najafabad, B.; Attaran, N.; Barati, M.; Mohammadi, Z.; Mahmoudi, M.; Sazgarnia, A. Cobalt ferrite nanoparticle for the elimination of CD133+CD44+ and CD44+CD24−, in breast and skin cancer stem cells, using non-ionizing treatments. Heliyon 2023, 9. [CrossRef]
- Almessiere, M.A.; Slimani, Y.; Sertkol, M.; Khan, F.A.; Nawaz, M.; Tombuloglu, H.; Al-Suhaimi, E.A.; Baykal, A. Ce–Nd Co-substituted nanospinel cobalt ferrites: An investigation of their structural, magnetic, optical, and apoptotic properties. Ceram. Int. 2019, 45, 16147–16156. [CrossRef]
- Panda, J.; Satapathy, B.S.; Mandal, B.; Sen, R.; Mukherjee, B.; Sarkar, R.; Tudu, B. Anticancer potential of docetaxel-loaded cobalt ferrite nanocarrier: an in vitro study on MCF-7 and MDA-MB-231 cell lines. J. Microencapsul. 2021, 38, 36–46. [CrossRef]
- Eshrati Yeganeh, F.; Eshrati Yeganeh, A.; Fatemizadeh, M.; Farasati Far, B.; Quazi, S.; Safdar, M. In vitro cytotoxicity and anti-cancer drug release behavior of methionine-coated magnetite nanoparticles as carriers. Med. Oncol. 2022, 39. [CrossRef]
- Ansari, S.M.; Bhor, R.D.; Pai, K.R.; Mazumder, S.; Sen, D.; Kolekar, Y.D.; Ramana, C. V. Size and Chemistry Controlled Cobalt-Ferrite Nanoparticles and Their Anti-proliferative Effect against the MCF-7 Breast Cancer Cells. ACS Biomater. Sci. Eng. 2016, 2, 2139–2152. [CrossRef]
- Andiappan, K.; Sanmugam, A.; Deivanayagam, E.; Karuppasamy, K.; Kim, H.-S.; Vikraman, D. Detailed investigations of rare earth (Yb, Er and Pr) based inorganic metal-ion complexes for antibacterial and anticancer applications. Inorg. Chem. Commun. 2023, 150, 110510. [CrossRef]
- Aly, A.A.M.; Ibrahim, A.B.M.; Zidan, A.S.A.; Mosbah, H.K.; Atta, S.A.; Schicht, I.; Villinger, A. Isolation and crystal structure of the first Pr(IV) coordination polymer and the complex anti-proliferative activity evaluation against seven cancer cell lines. J. Mol. Struct. 2022, 1256, 132508. [CrossRef]
- Bellot, G.L.; Liu, D.; Fivaz, M.; Yadav, S.K.; Kaur, C.; Pervaiz, S. Lanthanide conjugate Pr-MPO elicits anti-cancer activity by targeting lysosomal machinery and inducing zinc-dependent cataplerosis. Cell Commun. Signal. 2024, 22, 509. [CrossRef]
Figure 1.
XRD patterns of the samples.
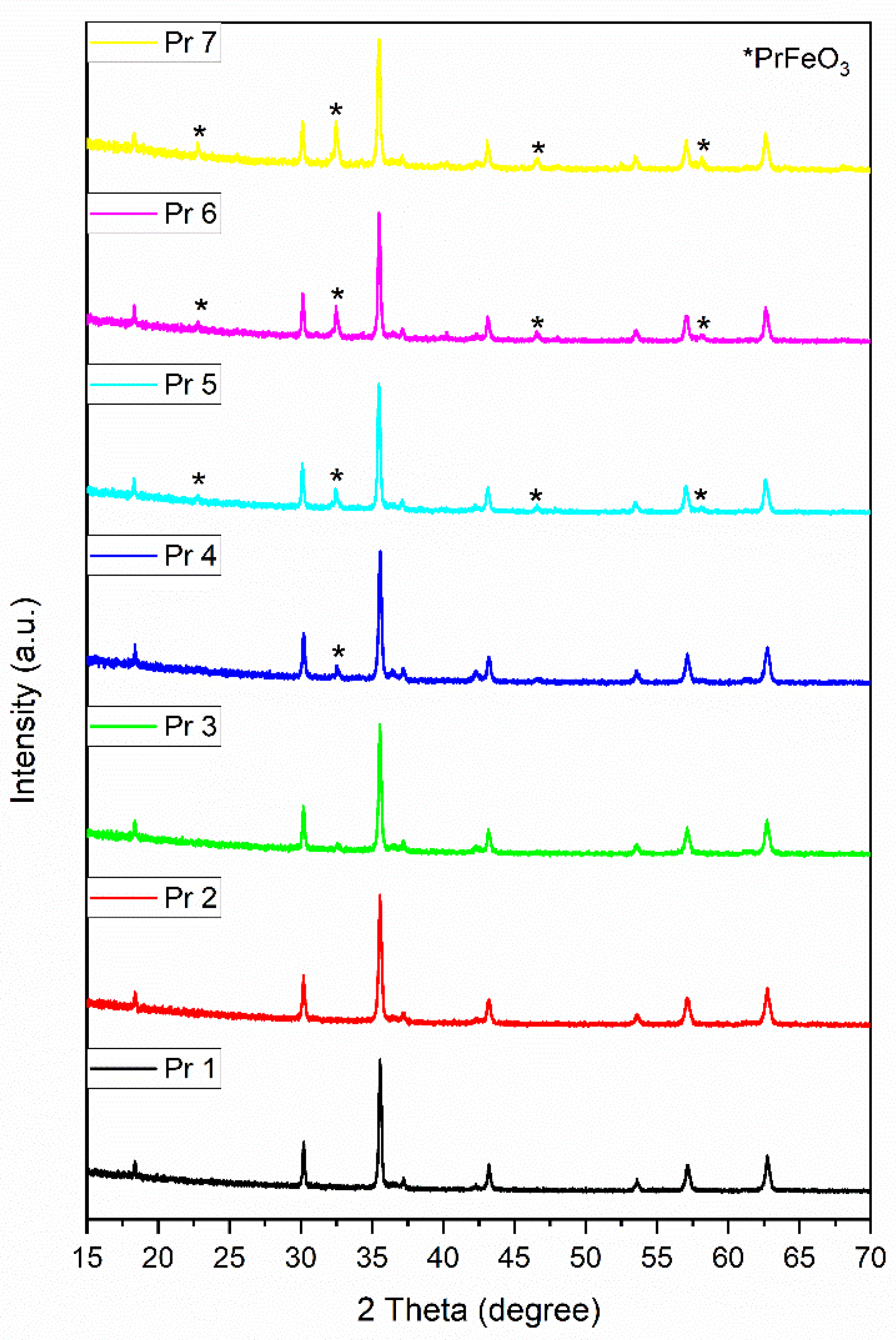
Figure 2.
FTIR spectra of the samples.
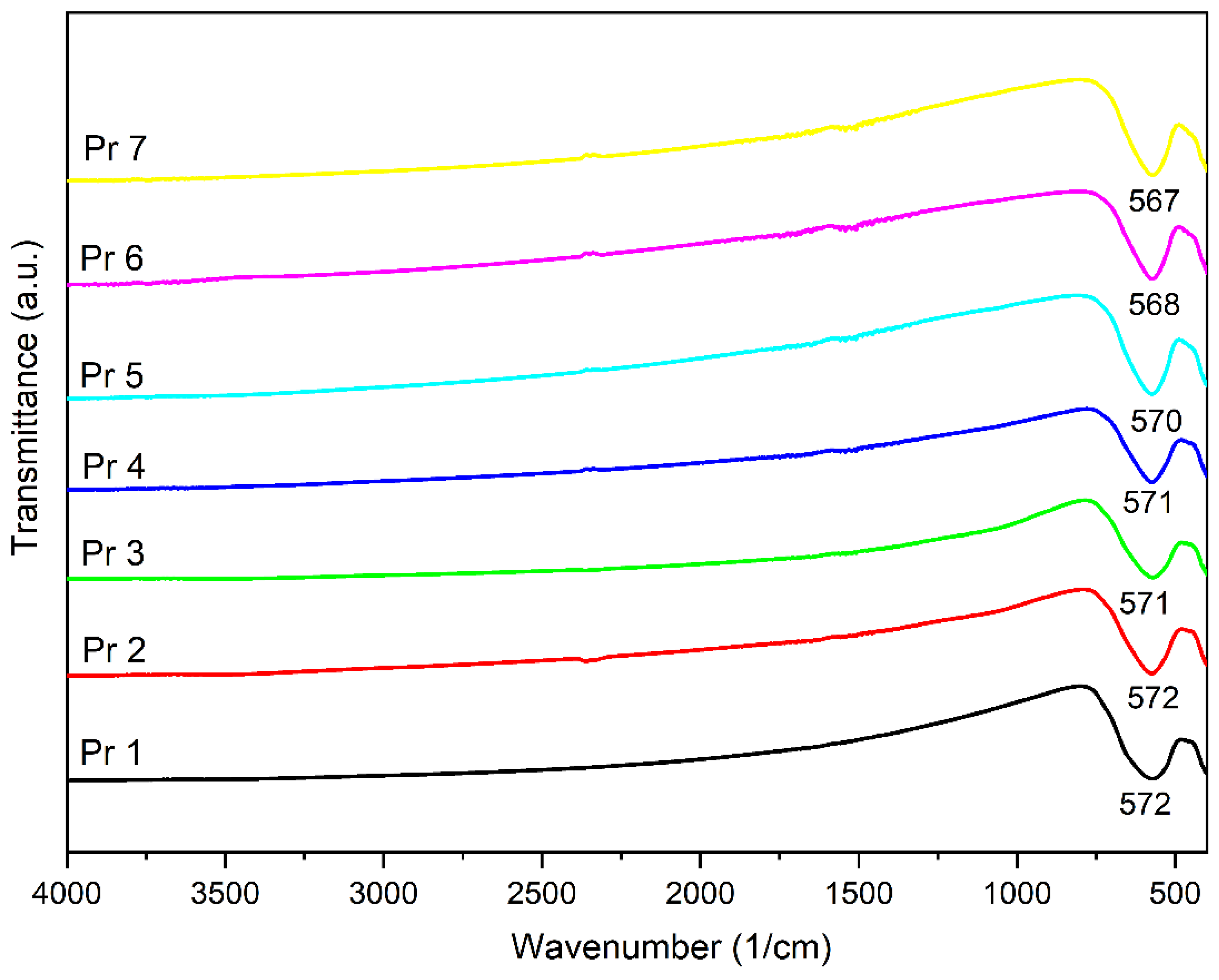
Figure 3.
Magnetic hysteresis of samples.

Figure 4.
SEM of the samples.

Figure 5.
TEM of the samples.

Figure 6.
HaCaT cell viability 48h post-treatment with Pr 1-CD, Pr 2-CD, Pr 3-CD, Pr 4-CD, Pr 5-CD, Pr 6-CD and Pr 7-CD (0.025, 0.05, 0.1, 0.25 and 0.5 mg/mL). The results are expressed as viability percentages compared to the control group (100%) (* p < 0.05, ** p < 0.01). The data represents the mean values ± SD of three independent experiments performed in triplicate.
Figure 6.
HaCaT cell viability 48h post-treatment with Pr 1-CD, Pr 2-CD, Pr 3-CD, Pr 4-CD, Pr 5-CD, Pr 6-CD and Pr 7-CD (0.025, 0.05, 0.1, 0.25 and 0.5 mg/mL). The results are expressed as viability percentages compared to the control group (100%) (* p < 0.05, ** p < 0.01). The data represents the mean values ± SD of three independent experiments performed in triplicate.
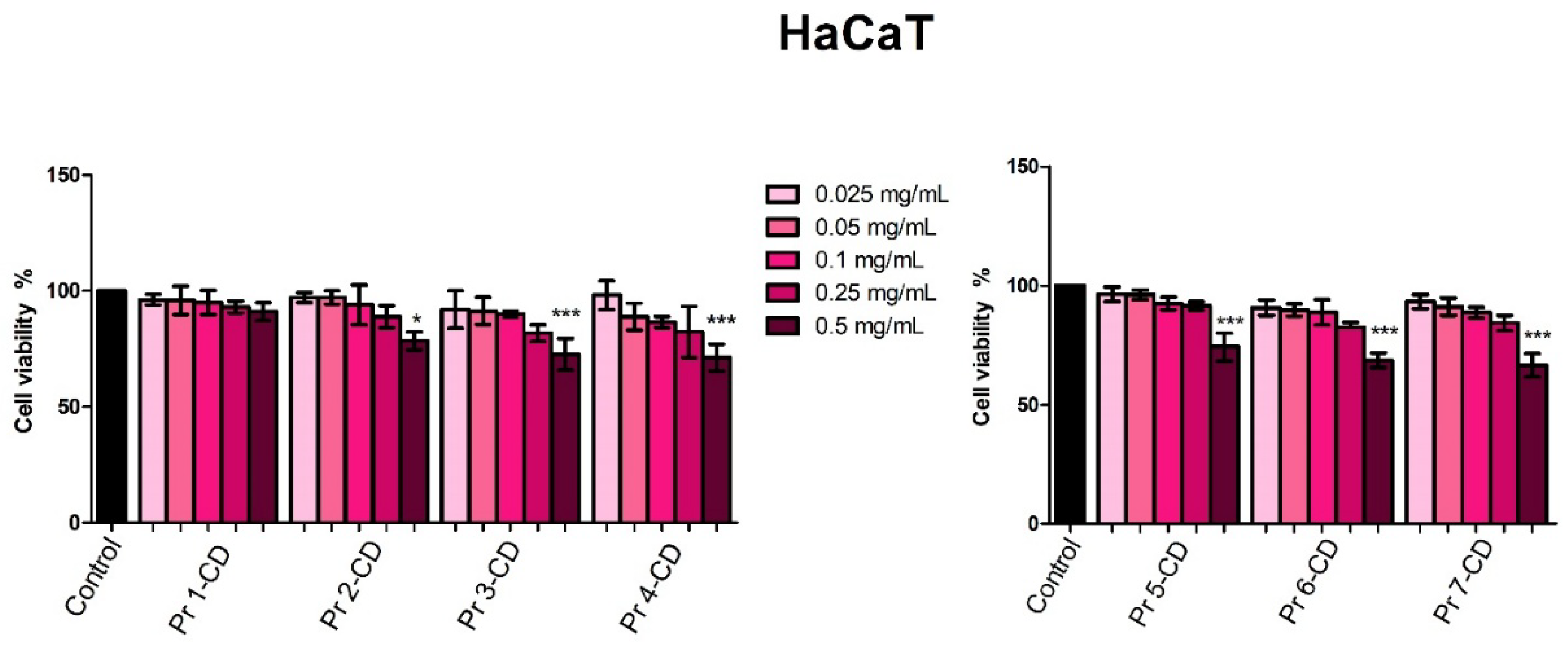
Figure 7.
A375 cell viability 48h post-treatment with Pr 1-CD, Pr 2-CD, Pr 3-CD, Pr 4-CD, Pr 5-CD, Pr 6-CD and Pr 7-CD (0.025, 0.05, 0.1, 0.25 and 0.5 mg/mL). The results are expressed as viability percentages compared to the control group (100%) (* p < 0.05, ** p < 0.01, *** p < 0.001). The data represents the mean values ± SD of three independent experiments performed in triplicate.
Figure 7.
A375 cell viability 48h post-treatment with Pr 1-CD, Pr 2-CD, Pr 3-CD, Pr 4-CD, Pr 5-CD, Pr 6-CD and Pr 7-CD (0.025, 0.05, 0.1, 0.25 and 0.5 mg/mL). The results are expressed as viability percentages compared to the control group (100%) (* p < 0.05, ** p < 0.01, *** p < 0.001). The data represents the mean values ± SD of three independent experiments performed in triplicate.
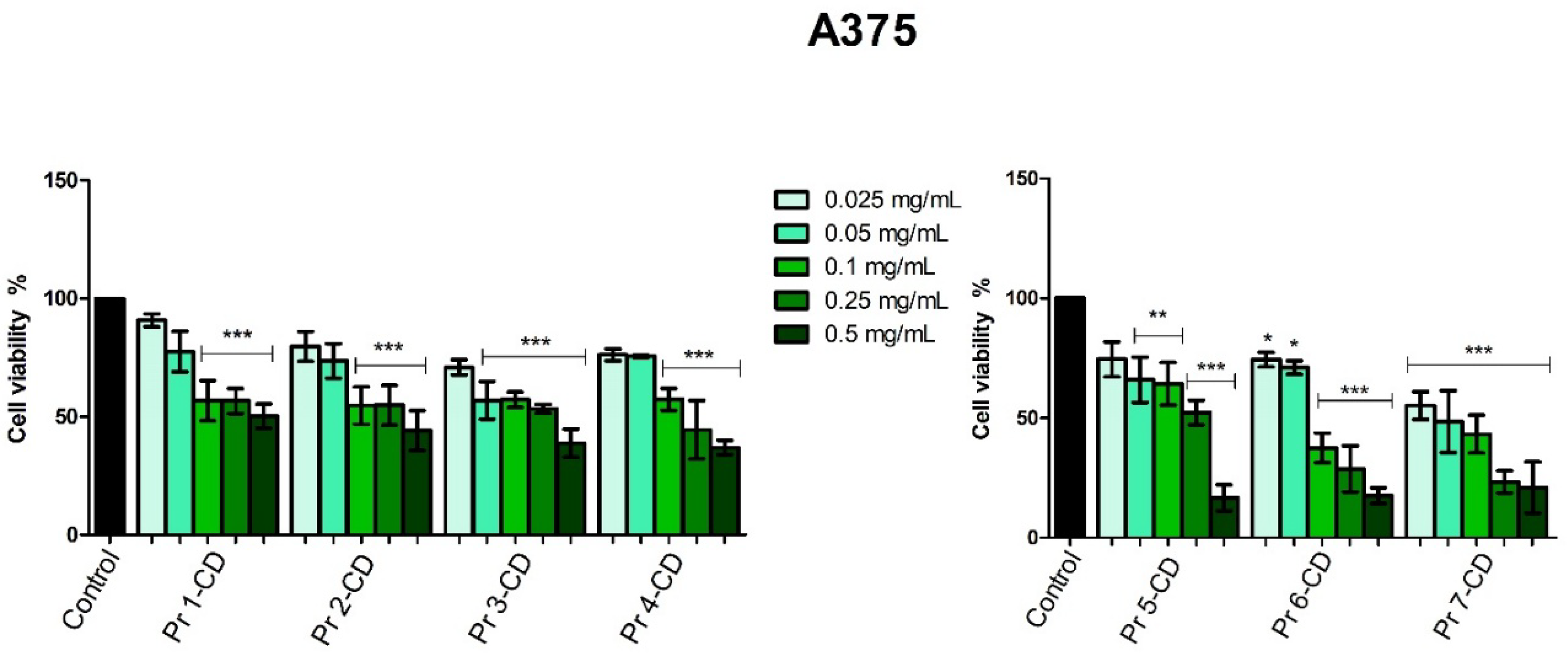
Figure 8.
HT-29 cell viability 48h post-treatment with Pr 1-CD, Pr 2-CD, Pr 3-CD, Pr 4-CD, Pr 5-CD, Pr 6-CD and Pr 7-CD (0.025, 0.05, 0.1, 0.25 and 0.5 mg/mL). The results are expressed as viability percentages compared to the control group (100%) (* p < 0.05, ** p < 0.01, *** p < 0.001). The data represents the mean values ± SD of three independent experiments performed in triplicate.
Figure 8.
HT-29 cell viability 48h post-treatment with Pr 1-CD, Pr 2-CD, Pr 3-CD, Pr 4-CD, Pr 5-CD, Pr 6-CD and Pr 7-CD (0.025, 0.05, 0.1, 0.25 and 0.5 mg/mL). The results are expressed as viability percentages compared to the control group (100%) (* p < 0.05, ** p < 0.01, *** p < 0.001). The data represents the mean values ± SD of three independent experiments performed in triplicate.
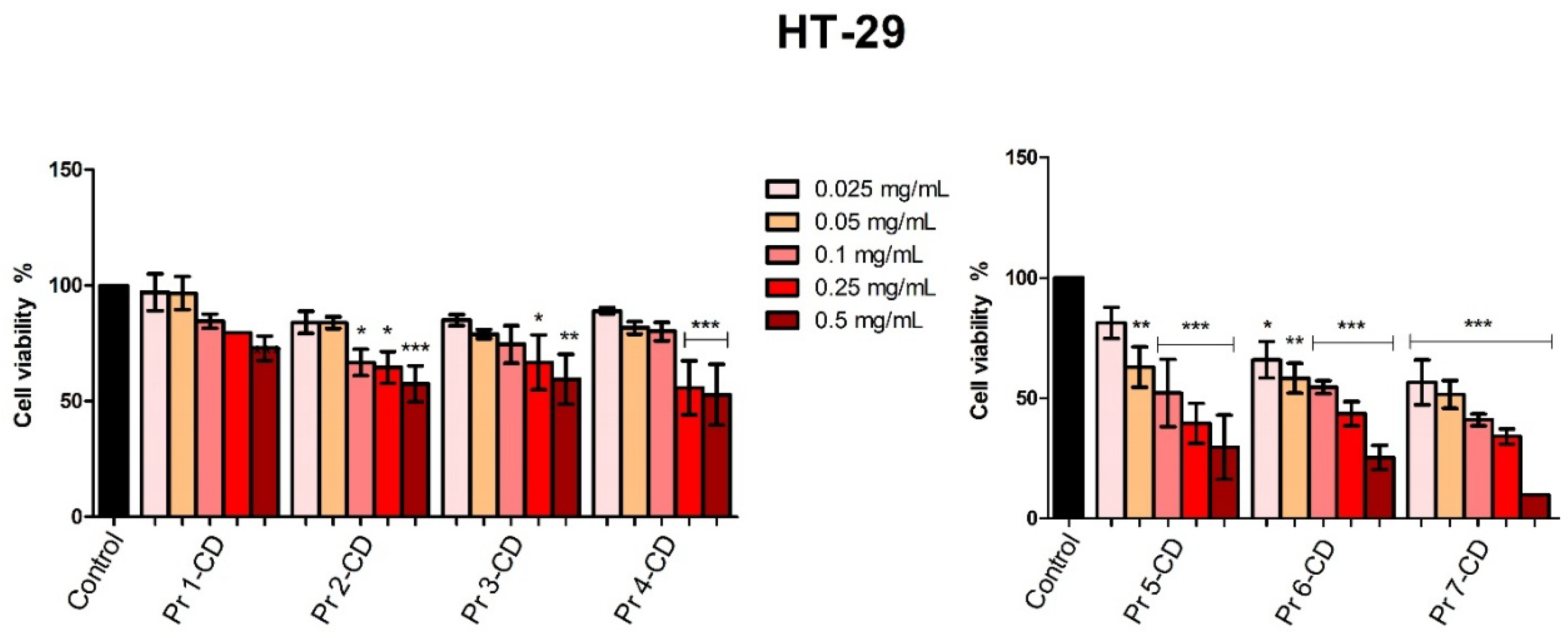
Figure 9.
MCF-7 cell viability 48h post-treatment with Pr 1-CD, Pr 2-CD, Pr 3-CD, Pr 4-CD, Pr 5-CD, Pr 6-CD and Pr 7-CD (0.025, 0.05, 0.1, 0.25 and 0.5 mg/mL). The results are expressed as viability percentages compared to the control group (100%) (* p < 0.05, *** p < 0.001). The data represents the mean values ± SD of three independent experiments performed in triplicate.
Figure 9.
MCF-7 cell viability 48h post-treatment with Pr 1-CD, Pr 2-CD, Pr 3-CD, Pr 4-CD, Pr 5-CD, Pr 6-CD and Pr 7-CD (0.025, 0.05, 0.1, 0.25 and 0.5 mg/mL). The results are expressed as viability percentages compared to the control group (100%) (* p < 0.05, *** p < 0.001). The data represents the mean values ± SD of three independent experiments performed in triplicate.
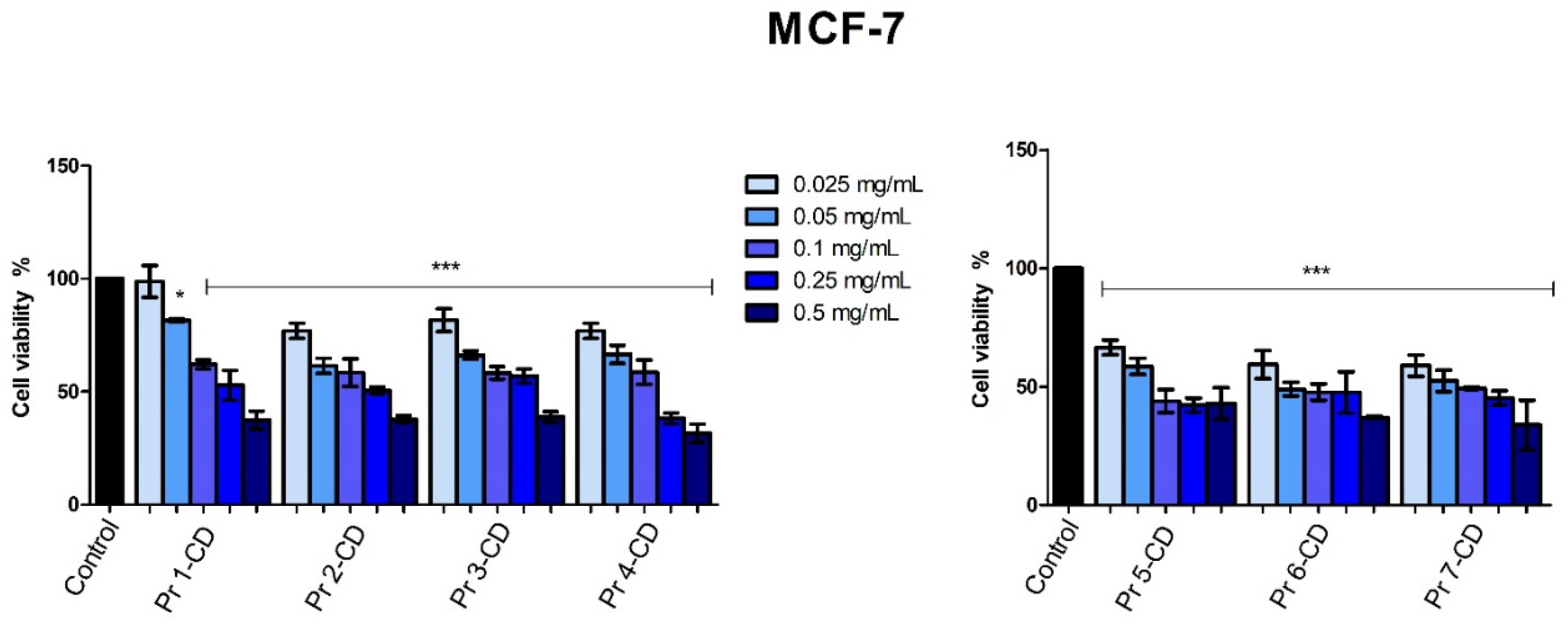

Table 1.
Sample composition.
| Sample | PrCl3·xH2O (moles) | Fe(NO3)3·9H2O (moles) |
| Pr1 | - | 0.0400 |
| Pr2 | 0.0002 | 0.0398 |
| Pr3 | 0.0006 | 0.0394 |
| Pr4 | 0.0010 | 0.0390 |
| Pr5 | 0.0020 | 0.0380 |
| Pr6 | 0.0030 | 0.0370 |
| Pr7 | 0.0040 | 0.0360 |
Table 2.
The calculated IC50 values (mg/mL) of Pr 1-CD, Pr 2-CD, Pr 3-CD, Pr 4-CD, Pr 5-CD, Pr 6-CD and Pr 7-CD on HaCaT, A375, HT-29 and MCF-7 cell lines 48h post-stimulation.
Table 2.
The calculated IC50 values (mg/mL) of Pr 1-CD, Pr 2-CD, Pr 3-CD, Pr 4-CD, Pr 5-CD, Pr 6-CD and Pr 7-CD on HaCaT, A375, HT-29 and MCF-7 cell lines 48h post-stimulation.
| Compound | HaCaT | A375 | HT-29 | MCF-7 |
| Pr 1-CD | > 0.5 | > 0.5 | > 0.5 | 0.25 |
| Pr 2-CD | > 0.5 | 0.28 | > 0.5 | 0.19 |
| Pr 3-CD | > 0.5 | 0.20 | > 0.5 | 0.32 |
| Pr 4-CD | > 0.5 | 0.18 | > 0.5 | 0.14 |
| Pr 5-CD | > 0.5 | 0.22 | 0.13 | 0.12 |
| Pr 6-CD | > 0.5 | 0.08 | 0.17 | 0.08 |
| Pr 7-CD | > 0.5 | 0.05 | 0.049 | 0.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



