
Submitted:
07 May 2025
Posted:
07 May 2025
You are already at the latest version
Abstract
This study reports the rational design and systematic evaluation of a novel series of 2-substituted aniline pyrimidine derivatives as dual Mer/c-Met inhibitors. Among the synthesized compounds, 17c demonstrated potent dual kinase inhibition, with IC50 values of 6.4 ± 1.8 nM (Mer) and 26.1 ± 7.7 nM (c-Met). The compound exhibited significant antiproliferative activity across multiple cancer cell lines (HepG2, MDA-MB-231, and HCT116), while showing minimal hERG channel inhibition (IC50 > 40 μM), indicating favorable cardiac safety. Pharmacokinetic profiling revealed high metabolic stability in human liver microsomes (t1/2 = 53.1 min) and moderate oral bioavailability (F: 45.3%) with strong plasma protein binding affinity (>95%). Mechanistic studies further demonstrated 17c dose-dependently suppressed HCT116 cell migration and induced apoptosis. These integrated pharmacological properties position 17c as a promising therapeutic candidate for dual Mer/c-Met drive malignancies.
Keywords:
Mer kinase
; c-Met kinase
; Mer/c-Met dual inhibitor
; 2-substituted aniline pyrimidine
; Anti-cancer
1. Introduction
Mer kinase, a key member of TAM (Tyro3-Axl-Mer) receptor tyrosine kinase family [1,2,3], orchestrates multiple cellular processes critical to immune regulation, apoptosis resistance, and inflammatory signaling. Structurally, Mer is characterized by a conserved intracellular tyrosine kinase domain and extracellular ligand-binding region. Its expression is predominantly localized to immune cell (e.g., macrophages and dendritic cells), where it mediates efferocytosis (apoptotic cell clearance) and suppresses pro-inflammatory cytokine production, thereby maintaining immune tolerance [4]. Mechanistically, Mer activation occurs through Gas6 ligand binding, initiating downstream signaling cascades (e.g., PI3K/AKT, MAPK) that promote cell survival, chemotaxis, and tissue remodeling [4]. Importantly, Mer dysregulation has been pathologically linked to oncogenesis: overexpression in tumor cells enhances survival under chemotherapy (via BCL-2 upregulation), facilitates metastatic dissemination (through EMT activation), and fosters immunosuppressive microenvironments (by polarizing tumor-associated macrophages) [5,6,7,8]. These multifaceted roles established Mer as a high-value therapeutic target for precision oncology strategies targeting both tumor-intrinsic and immune-evasion mechanisms.
Current Mer kinase inhibitors are structurally categorized into two major chemotypes: aminopyrimidine-pyrazole (pyrrole) and aminopyrimidine derivatives (Figure 1). The first-generation inhibitor UNC569 [9,10], a pyrazole-based compound, selectively inhibits Mer kinase (IC50 = 1.2 nM) and suppresses downstream ERK/AKT signaling. However, its clinical translation is hindered by rapid metabolism (human microsomal t1/2< 15 min) and low oral bioavailability (F < 10%) [11]. Structural optimization of UNC569 yielded second-generation analogs. UNC2025: enhanced metabolic stability (t1/2 = 68 min in human hepatocytes) with maintained Mer potency (IC50 = 1.8 nM) [12,13]. MRX2843: dual Mer/Flt3 inhibitor (Mer IC50 = 1.2 nM) showing improved CNS penetration (brain/plasma ration = 0.4) [11], currently in phase II trails for relapsed/refractory acute lymphoblastic leukemia (NCT04872478) [14]. Notably, the aminopyrimidine-class inhibitor UNC2250 demonstrates exceptional Mer targeting (IC50 = 1.7 nM) and unique activity against Mer-EGFR fusion protein [15,16]. In xenograft models, UNC2250 induced tumor regression (~60% volume reduction vs. control) by dual blockade of Mer-mediated survival signaling and EGFR-driven proliferation [16].
The c-Met kinase, also known as the hepatocyte growth factor receptor (HGFR) [17,18], is a transmembrane receptor tyrosine kinase that plays a pivotal role in regulating critical cellular processes, including proliferation, survival, migration, and angiogenesis [19,20]. Activation of c-Met occurs through binding to its specific ligand HGF, which triggers receptor dimerization and autophosphorylation. This activation initiates downstream signaling cascades, such as the MAPK, PI3K/AKT, and STAT pathways. Aberrant c-Met signaling, caused by overexpression, mutations, or gene amplification, is strongly associated with tumorigenesis, promoting cancer progression, metastasis, and therapy resistance. Given its oncogenic significance, c-Met has become a promising therapeutic target. Current clinical investigations are actively exploring both small-molecule inhibitors and monoclonal antibodies targeting c-Met for the treatment of diverse malignancies.
Multiple c-Met inhibitors have entered clinical practice or are under clinical investigation (Figure 2). Notably, crizotinib, approved by the FDA in 2011, is primarily used for ALK-positive metastatic non-small cell lung cancer (NSCLC) but also exhibits c-Met inhibitory activity as part of its multi-target mechanism [21,22]. Cabozantinib, another tyrosine inhibitor targeting c-Met, VEGFR, Mer, and Kit, has been approved for multiple indications: metastatic medullary thyroid cancer, second-line treatment of advanced renal cell carcinoma post-antiangiogenic therapy, and first-line therapy for advanced renal cell carcinoma [23,24]. Additionally, savolitinib-a highly selective c-Met inhibitor-is currently in Phase III clinical trials for papillary renal cell carcinoma and other c-Met-driven malignancies [25]. Capmatinib, a selective c-Met inhibitor, received FDA approval specifically for metastatic NSCLC with MET exon 14 skipping mutations [26,27].
Mer and c-Met, both members of the receptor tyrosine kinase family, exhibit structurally homologous extracellular domains and share overlapping downstream signaling pathways. Notably, their functional convergence in regulating tumor proliferation, metastasis, and immune evasion establishes a compelling rationale for dual targeting of Mer and c-Met kinases. To date, cabozantinib and its derivatives remain the only clinical available agent with demonstrated inhibitory activity against both targets. However, cabozantinib’s clinical utility is predominantly attributed to its potent inhibition of c-Met and VEGFR-2, while the structural determinants governing its Mer kinase inhibitory activity remains poorly characterized. Furthermore, the emergence of cabozantinib resistance underscores the critical need to develop novel Mer/c-Met dual inhibitors with distinct chemical scaffolds. Importantly, the structure-activity relationship (SAR) of Mer/c-Met dual inhibitors remains underexplored, systematic exploration of these aspects through rational drug design is therefore essential to advance the development of next-generation dual-targeting agents.
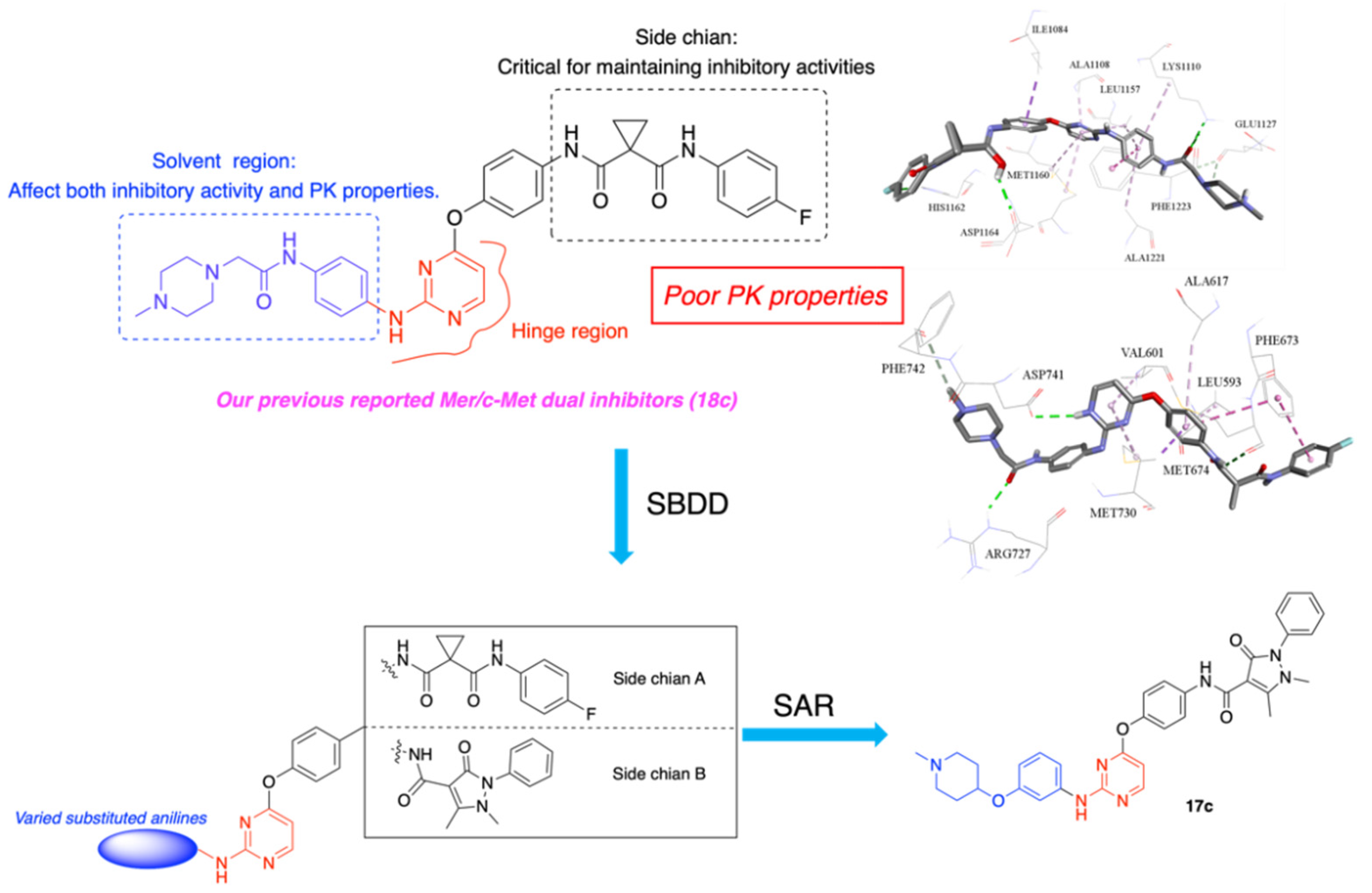
Previous study identified compound 18c as a dual Mer/c-Met dual inhibitor with potent dual-target inhibitory activity [28]. However, pharmacokinetic analysis revealed low oral bioavailability (F: 2.84%), promoting structural optimization. To elucidate key interaction for dual-target inhibition, we analyzed the binding modes of cabozantinib with Mer (PDB: 4M3Q) and c-Met (PDB: 3LQ8). Molecular docking showed that in Mer kinase, cabozantinib’s amide group forms hydrogen bonds with the hinge region (Pro672 and Met674), while the quinoline moiety occupies a hydrophobic cavity without direct hydrogen bonding (Figure 3). In c-Met kinase, both the quinoline core and the amide group participate in hydrogen bonding with residues in the ATP-binding pocket (Figure 4). Based on these observations and the binding mode of 18c [28], we propose modifying the 2-position of the pyrimidine ring to introduce hydrophilic groups. This strategy aims to enhance solubility and bioavailability while preserving dual-target activity.
Building upon our previous work (Figure 5), we designed and synthesized a series of 2-substituted aniline pyrimidine derivatives, with subsequent evaluation of their biological activities. Among these, compound 17c emerged as a lead candidate due to its superior metabolic stability in human liver microsomes and potent antiproliferative activities against three cancer cell lines (HepG2, MDA-MB-231, and HCT116). 17c also revealed favorable Pharmacokinetic properties, plasma protein binding, and favorable safety profile in hERG assays. Mechanistic studies further showed 17c induced dose-dependent apoptosis in HCT116 cells and effectively inhibited HCT116 cell migration. These results position 17c as a promising dual Mer/c-Met inhibitor warranting further development.
2. Results and Discussion
2.1. Chemistry
The synthesis of target compounds and key intermediates are described in Scheme 1 and Scheme 2, respectively. The structures of compounds were confirmed by 1H-NMR, 13C-NMR and HRMS spectroscopy.
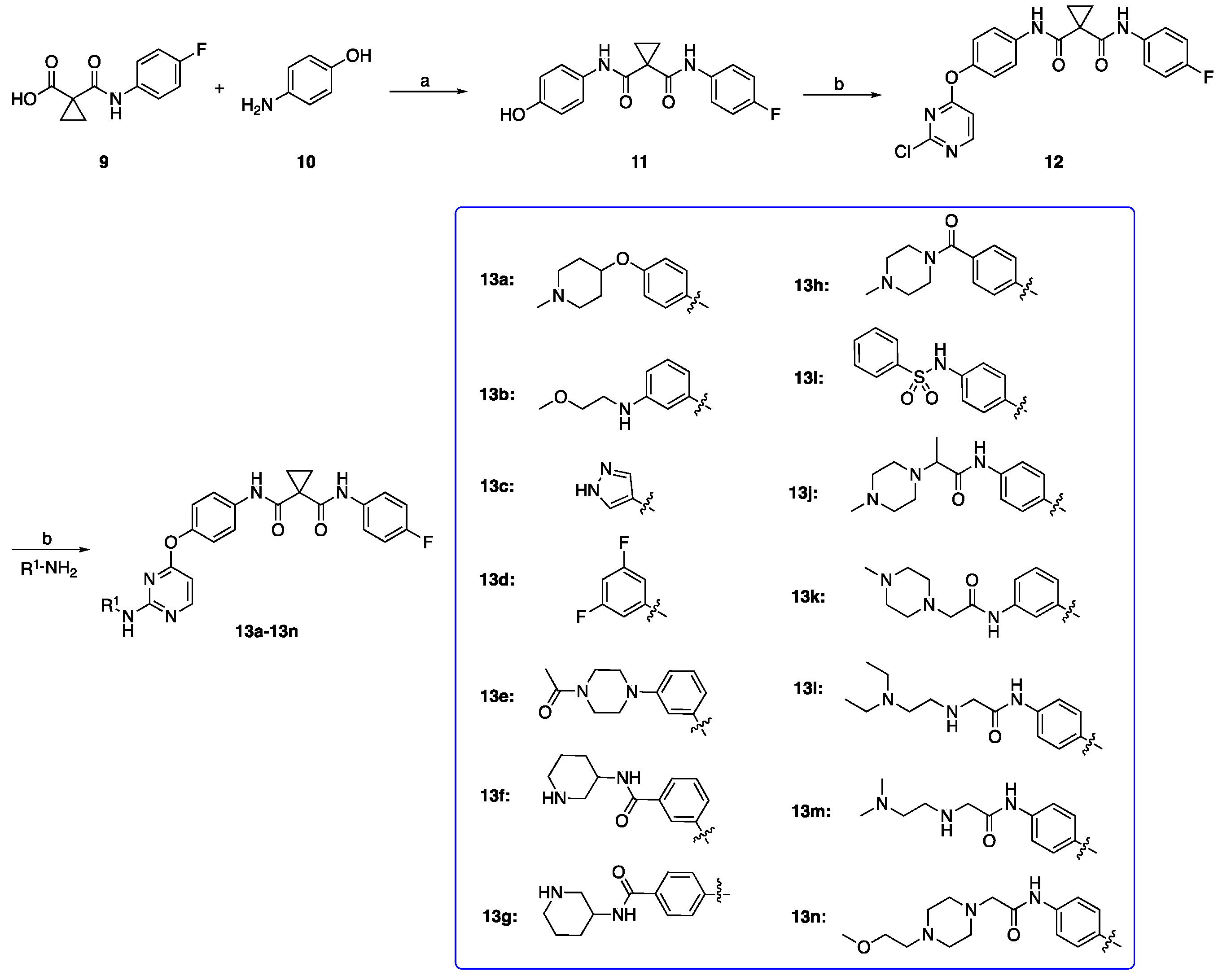
As depicted In scheme 1, a condensation reaction was employed where 1-((4-fluorophenyl)carbamoyl)cyclopropane-1-carboxylic acid (9) was combined with readily accessible 4-aminophenol (10) at ambient temperature, yielding the intermediate N-(4-fluorophenyl)-N-(4-hydroxyphenyl)cyclopropane-1,1-dicarboxamide (11) [29].Subsequently, the intermediate N-(4-((2-chloropyrimidin-4-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (12) was produced via an SN2 reaction between intermediate 11 with 2,4-dichloropyrimidine [30]. Finally, the desired compounds 13a–13h were obtained by reacting intermediate 12 with various substituted anilines, which were either commercial available or prepared in our prior research [31].
In Scheme 2, the preparation of the crucial intermediate N-(4-hydroxyphenyl)-1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide (15) commenced with a condensation process involving 1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxylic acid (14) and with 4-aminophenol (10) [32]. The intermediate 15 was then reacted with 2,4-dichloropyrimidine, yielding in the forming of the pivotal intermediate N-(4-((2-chloropyrimidin-4-yl)oxy)phenyl)-1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide (16). The target compounds 17a–17h were subsequently obtained by carrying out a nucleophilic substituted reaction between intermediate 16 and various substituted anilines.
2.2. Kinase Inhibitory Activities
All the target compounds (supplementary material, S1) were initially screened for their inhibitory activities on Mer and c-Met kinases compared to the lead compound (18c), and the results were depicted in Table 1 and Table 2. The corresponding curves were showed in Supplementary S3 and S4.
As summarized in Table 1, the substituents (R1) at the terminal pyrimidine amine group significantly modulate inhibitory potency against Mer and c-Met kinases. Compounds 13b, 13j, 13K, 13m, and 13n exhibited moderate to potent dual inhibitory, with Mer IC50 values ranging from 13.1 ± 2.1 ~ 37.3 ± 10.6 nM and c-Met IC50 values of 25.9 ± 7.7 ~ 48.6 ± 11.2 nM, compared to the lead compound 18c (Mer IC50:18.5 ± 2.3 nM, c-Met IC50: 33.6 ± 4.3 nM). Strikingly, compound 13e demonstrated the highest dual potency, achieving IC50 values of 7.5 ± 1.6 nM (Mer) and 7.5 ± 1.1 nM (c-Met), representing a 2.5-fold and 4.5-fold improvement over 18c, respectively. In contrast, compounds 13c and 13h showed reduced Mer inhibitory activity while maintaining c-Met inhibitory activity comparable to 18c. Conversely, compounds 13d, 13i, and 13l exhibited significantly reduced inhibitory activities against both Mer and c-Met kinase.
A novel series of pyrimidine derivatives were designed to optimize dual Mer/c-Met inhibiton. As shown in Table 2, the Mer kinase inhibitory activities varied significantly across the series. Compounds 17d, 17e, and 17g exhibited reduced inhibitory potency, with Mer IC50 values of 84.1 ± 13.9, 69.8 ± 12.5, and >1000 nM, respectively. In contrast, compounds 17a, 17b, and 17h retained activity comparable to the lead compound, featuring IC50 values of 26.2 ± 7.1, 12.1 ± 1.3, and 11.9 ± 1.7 nM. Notably, compounds 17c and 17f showcased superior inhibition, achieving Mer IC50 values of 6.4 ± 1.8 nM and 5.5 ± 0.8 nM.
For c-Met kinase inhibition, compounds 17c, 17f, and 17h, exhibited comparable IC50 values ranging from 25.0 ± 3.5 to 45.0 ± 6.8 nM. However, the potency of 17a, 17b, 17d, 17e, and 17g decreased (IC50 values from 100.7 ± 18.5 nM to 235.6 ± 25.6 nM), indicating that specific structural optimization at the pyrimidine scaffold detrimentally impact c-Met binding.
2.3. In Vitro Liver Microsomal Stability
Metabolic stability is a critical determinant of drug-like properties. To prioritize compounds for further development, we evaluate the human liver microsomal stability of selected candidates demonstrating potent dual Mer/c-Met dual inhibition in vitro. As shown in Table 3, compounds 13b, 13e, 13j, 13k, 13m, 13n, 17c, and 17f were subjected to microsome assays to determine their half-life and intrinsic clearance. Compound 17c demonstrated superior metabolic stability, with an extended t1/2 of 147.0 min and low CLint of 0.026 mL/min/mg, whereas 17f exhibited a shorter t1/2 of 51.0 min and higher CLint of 0.068 mL/min/mg. Notably, all other compounds showed rapid hepatic clearance (t1/2 < 40 min, CLint > 0.068 mL/min/mg), suggesting limited metabolic resistance. The results highlight 17c as the most metabolically stable candidate within this series.
2.4. Antiproliferation Assay In Vitro
Compound 17c was further evaluated for its antiproliferative activity against three cancer cell lines with high Mer and c-Met expression (HCT116 colon cancer, CAKI-1 renal cancer, and PC-3 prostate cancer) using CCK-8 assay [5,33,34,35]. As summarized in Table 4, 17c demonstrated superior antiproliferative activity compared to cabozantinib across all tested cell lines. Notably, 17c exhibited marked most potency with HCT116 cells, with an IC50 value of 0.46 ± 0.06 μmol/L—a 17.1-fold improvement over cabozantinib. In CAKI-1 cells, 17c showed an IC50 of 2.31 ± 0.67 μmol/L, representing a 2.0-fold enhancement compared to cabozantinib. Similarly, against PC-3 cells, 17c achieved an IC50 of 3.79 ± 1.09 μmol/L, which is 4.1 times more effective than cabozantinib. These results collectively establish compound 17c as a potent dual Mer/c-Met inhibitor with significant therapeutic potential. The dose-response curves are provided in Supplementary S5.
2.5. Molecular Docking Study of Compound 17c
Molecular docking was conducted to validate the binding mode of compound 17c with Mer kinase (PDB: 4M3Q) and c-Met (PDB: 3LQ8) kinases. As illustrated in Figure 8, the docking pose of 17c in Mer kinase revealed three critical interactions: (1) the aminopyrimidine moiety acts as hydrogen bond acceptor with Leu593; (2) the amide side chain forms a hydrogen bond with Lys619; (3) the benzene ring of side chain engages in a π-π stacking interaction. For c-Met kinase (Figure 9), 17c exhibited a similar binding mode: (1) the aminopyrimidine core forms a hydrogen bond with Met1160; (2) the amide group forms hydrogen bonds with Lys1110 and Phe1223; (3) the pyrimidine group participates in π-π interaction. These results confirm that the 2-substitutedaniline pyrimidine group effectively occupies the ATP-binding pockets of both kinases through conserved hydrogen bonding and hydrophobic interactions. The structural coherence between predicted binding modes and enzymes inhibitory data strongly supports the rational design strategy, validating the scaffold’s potential for dual Mer/c-Met inhibition.
2.6. Western Blot Assay
The inhibitory effects of compound 17c on Mer and c-Met kinase phosphorylation were assessed using Western blot analysis. As shown in Figure 10, increasing concentrations of 17c resulted in a reduction in the phosphorylation levels of both Mer and c-Met kinases. At a concentration of 2.5 μM, 17c significantly inhibited the phosphorylation of these kinases compared to the positive control, demonstrating its potential to effectively block Mer and c-Met kinase activation.
2.7. The hERG Test
To evaluate the potential cardiotoxicity risk associated with compound 17c, we assessed its inhibitory activity against the hERG potassium channel, a key determinant of cardiac safety. As summarized in Table 5, 17c demonstrated negligible hERG inhibition (IC50 > 40 μM), indicating a low risk of hERG activity was observed despite its potent dual inhibition of Mer and c-Met kinases.
2.8. Apoptosis Assay
To evaluate the apoptosis-inducing capability of compound 17c, the HCT116 colon cancer cell line was treated with varying concentrations of the compound. Apoptosis was evaluated using the TUNEL assay. DAPI staining (blue) was served as the positive control, while apoptotic cells were identified by TUNEL staining (green) after 48 h incubation with compound 17c, as shown in Figure 11. The quantity of apoptotic cells was measured at different concentrations of 17c, resulting in an ED50 value of 2.036 μM. These results demonstrate that compound 17c efficiently triggers apoptosis in HCT116 cells, thereby emphasizing its potential as a highly promising candidate for additional research and development.
2.9. Transwell Assay
The Transwell migration assay shown in Figure 12 was performed to evaluate the effect of compound 17c on the migration of HCT116 cells. A decrease in the number of cells migrating through the membrane was observed with increasing concentration of 17c. These results suggest that compound 17c effectively inhibits the migration capacity of HCT116 colon cancer cells in a concentration-dependent manner.
2.10. Plasma Protein Binding Affinity
The plasma protein binding (PPB) profile of compound 17c was assessed via equilibrium dialysis, with warfarin served as a positive control. As shown in Table 6, 17c exhibited high species-independent protein binding, with unbound fractions of 1.58% in human plasma and 1.78% in mouse plasma, corresponding to PPB values of 98.4% and 98.2%, respectively. Combined with its extended half-life and low clearance, the high PPB of 17c may contributed to sustained target engagement.
2.11. Pharmacokinetic Properties
The Pharmacokinetic (PK) profiles of compound 17c were evaluated in Sprague-Dawley (SD) rats following single-dose administration via oral (PO) and intravenous (iv) route. As summarized in Table 7, oral administration of 17c achieved a peak plasma concentration (Cmax) of 740 ng/mL, at Tmax = 4.0 h, with a half-time (T1/2) of 3.46 h and a mean residence time (MRT_inf) of 6.52 h. Compared to lead compound 18c, compound 17c demonstrated significantly improved PK properties, including prolonged oral half-time, reduced clearance, an extended mean residence time, and enhanced oral bioavailability (45.3%). These data indicate that 17c exhibits favorable PK properties, with sustained drug exposure and optimized absorption characteristics.
3. Materials and Methods
3.1. Chemical Part
All reagents were obtained from Titan Chemical Co., Ltd., and solvents were obtained from Shijiazhuang Kehai Huabo Instruments Co., Ltd., which were used without further purification unless otherwise indicated. All the Reactions were monitored by thin layer chromatography (TLC) using silica GF254 Plates. High Resolution Mass spectra (HRMS) were performed in electrospray ionization (ESI) mode on a Water Q-Tof mass spectrometer. Proton (1H) and carbon (13C) nuclear magnetic resonance (NMR) spectra were generated in DMSO-d6 on Bruker AM-400 and AM500 spectrometers (Bruker Bioscience, USA), with tetramethylsilane (TMS) as the internal reference, and chemical shift values were reported in ppm. The multiplicity of the signals is denoted as follows: s (singlet); d (doublet), t (triplet), q (quadruplet), qui (quintuplet), m (multiplet), dd (doublet of doublets), and dt (doublet of triplets). Coupling constants (J) are given in hertz (Hz). Flash chromatography was performed using silica gel (200-300 mesh) as the stationary phase, with a mobile phase consisting of a mixture of methanol (MeOH), ethyl acetate (EA), and petroleum ether (PE). The purity of all synthesized compounds was confirmed to be >95% by high-performance liquid chromatography (HPLC) using a Waters HPLC system equipped with a UV/visible detector, monitored at 254 nm. HPLC analysis was carried out on a 5 μm, 4.6 × 250 mm Hypersil ODS2 column, with a mobile phase consisting of a 3:7 (v/v) mixture of potassium dihydrogen phosphate solution and methanol, at a flow rate of 1.0 mL/min and an injection volume of 10 μL. All chemicals were of analytical grade and used without further purification.
3.1.1. Preparation of N-(4-Fluorophenyl)-N-(4-hydroxyphenyl)cyclopropane-1,1-dicarboxamide (Intermediate 11)
To a solution of 1-((4-fluorophenyl)carbamoyl)cyclopropane-1-carboxylic acid (2.02 g, 9.00 mmol) and 4-aminophenol (1.18 g, 10.8 mmol) in DMF (5 mL), EDC·HCl (2.07 g, 10.80 mmol) was added. The reaction solution was stirred at room temperature for 6 h and monitored by TLC. Ice water (125 mL) was added, and the precipitate was filtered off, washed, and dried in a vacuum to yield Intermediate 11 as a white solid (2.28 g, 80.6%), which can be used for the next step without any purification.
3.1.2. Preparation of N-(4-((2-Chloropyrimidin-4-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (Intermediate 12)
To a solution of intermediate 11 (2.01 g, 6.37 mmol) and 2,4-dichloropyrimidine (1.04 g, 7.01 mmol) in DMF (15 mL), K2CO3 (0.97 g, 7.01 mmol) was added under N2 atmosphere. The reaction solution was stirred at 80 °C for 6 h and monitored by TLC. The reaction mixture was poured into ice water (100 mL), and the precipitate was filtered off, washed, and dried in a vacuum to obtain the crude product, which was purified by silica gel chromatography using a mixture of (DCM: MeOH = 100:1~40:1) to afford the intermediate 12 as a white solid (2.34 g, 75.70 %).
3.1.3. General Procedure for Preparation of Target Compounds 13a–13n
To a mixture of the intermediate 12 (1.2 mmol), substituted aniline (1.0 mmol), and DMF (8 mL), p-toluenesulfonic acid (PTSA, 4.0 mmol) was added. The mixture was stirred at 90 °C for 4 h under N2 atmosphere. The reaction solution was cooled to room temperature, then poured into ice water (100 mL), and the precipitate was filtered off, washed, and dried in a vacuum to obtain the crude product, which was purified by silica gel chromatography using a mixture of (DCM: MeOH = 100:1-40:1) to afford the product 13a–13n.
N-(4-fluorophenyl)-N-(4-((2-((4-((1-methylpiperidin-4-yl)oxy)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)cyclopropane-1,1-dicarboxamide (compound 13a): White solid, yield: 23.27%. 1H NMR (500 MHz, DMSO-d6) δ 10.18 (s, 1H), 10.11 (s, 1H), 9.41 (s, 1H), 8.30 (d, J = 5.6 Hz, 1H), 7.70 (d, J = 8.9 Hz, 2H), 7.66 (dd, J = 9.1, 5.1 Hz, 2H), 7.43 (d, J = 7.5 Hz, 2H), 7.19–7.14 (m, 4H), 6.80 (d, J = 8.7 Hz, 2H), 6.35 (d, J = 5.6 Hz, 1H), 4.44 (s, 1H), 3.16–2.87 (m, 4H), 2.63 (s, 3H), 2.10–2.01 (m, 2H), 1.85 (s, 2H), 1.51 (d, J = 4.6 Hz, 4H). 13C NMR (126 MHz, DMSO-d6) δ 170.06, 168.73 (d, J = 4.3 Hz, 1C), 160.19, 159.70, 157.79, 151.65, 148.59, 136.63, 135.66, 134.42, 122.86 (d, J = 7.7 Hz, 1C), 122.43 (d, J = 10.0 Hz, 1C), 120.91,116.61, 115.62, 115.44, 98.07, 78.48, 50.54, 43.37, 32.01, 28.45, 15.91. HRMS: m/z C33H33FN6O4 [M+H]+ 597.2547, Found 597.2628. Purity: > 95% (HPLC).
N-(4-fluorophenyl)-N-(4-((2-((3-((2-methoxyethyl)amino)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)cyclopropane-1,1-dicarboxamide (compound 13b): White solid, yield: 20.08%. 1H NMR (500 MHz, DMSO-d6) δ 10.15 (s, 1H), 10.08 (s, 1H), 9.27 (s, 1H), 8.31 (d, J = 5.6 Hz, 1H), 7.71 (d, J = 9.0 Hz, 2H), 7.66 (dd, J = 9.0, 5.1 Hz, 2H), 7.21–7.14 (m, 4H), 6.86–6.79 (m, 3H), 6.34 (d, J = 5.6 Hz, 1H), 6.16 (d, J = 7.3 Hz, 1H), 5.21 (s, 1H), 3.44 (t, J = 5.9 Hz, 2H), 3.26 (s, 3H), 3.06 (t, J = 5.8 Hz, 2H), 1.49 (s, 4H). 13C NMR (126 MHz, DMSO-d6) δ 169.90, 168.67, 160.37, 160.22, 159.70, 157.79, 148.56, 141.27, 136.65, 135.67 (d, J = 2.0 Hz, 1C), 129.15, 122.82 (d, J = 7.7 Hz, 1C), 122.27, 115.61, 115.43, 107.93, 106.71, 103.23, 98.27, 70.93, 58.45, 42.93, 31.95, 15.86. HRMS: m/z C30H29FN6O4 [M+H]+ 557.2234, Found 557.2312. Purity: > 95% (HPLC).
N-(4-((2-((1H-pyrazol-4-yl)amino)pyrimidin-4-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (compound 13c): White solid, yield: 31.52%. 1H NMR (500 MHz, DMSO-d6) δ 12.31 (s, 1H), 10.17 (s, 1H), 10.08 (s, 1H), 9.65–9.18 (m, 1H), 8.29 (s, 1H), 7.72 (s, 2H), 7.65 (dd, J = 8.7, 5.0 Hz, 3H), 7.30–7.11 (m, 5H), 6.30 (d, J = 5.5 Hz, 1H), 1.50 (s, 4H). 13C NMR (126 MHz, DMSO-d6) δ 170.46, 168.68 (d, J = 11.7 Hz, 1C), 160.42, 159.84, 159.71, 157.80, 148.56, 136.67, 135.63, 130.12, 122.87 (d, J = 6.0 Hz, 1C), 122.40, 122.13, 115.62, 115.44, 98.04, 31.89, 15.97. HRMS: m/z C24H20FN7O3 [M+H]+ 474.1612, Found 474.1686. Purity: > 95% (HPLC).
N-(4-((2-((3,5-difluorophenyl)amino)pyrimidin-4-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (compound 13d): White solid, yield: 19.40%. 1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 10.07 (s, 1H), 10.01 (s, 1H), 8.47 (d, J = 5.6 Hz, 1H), 7.78 (d, J = 8.8 Hz, 2H), 7.69 (dd, J = 8.7, 5.1 Hz, 2H), 7.34 (d, J = 9.4 Hz, 2H), 7.28–7.17 (m, 4H), 6.67 (t, J = 9.1 Hz, 1H), 6.59 (d, J = 5.6 Hz, 1H), 1.53 (d, J = 4.1 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 170.09, 168.56, 160.40, 159.61, 157.59, 148.25, 143.31, 137.04, 135.59 (d, J = 2.6 Hz, 1C), 122.93 (d, J = 7.8 Hz, 1C), 122.29, 122.05, 115.62, 115.40, 101.74, 101.44, 99.97, 31.88, 15.93. HRMS: m/z C27H20F3N5O3 [M+H]+ 520.1518, Found 520.1594. Purity: > 95% (HPLC).
N-(4-((2-((3-(4-acetylpiperazin-1-yl)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (compound 13e): White solid, yield: 23.07%. 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 10.07 (s, 1H), 9.38 (s, 1H), 8.34 (d, J = 5.6 Hz, 1H), 7.70 (d, J = 9.0 Hz, 2H), 7.64 (dd, J = 9.1, 5.1 Hz, 2H), 7.20–7.13 (m, 5H), 7.08 (d, J = 8.1 Hz, 1H), 6.97 (t, J = 8.1 Hz, 1H), 6.47 (dd, J = 8.1, 1.7 Hz, 1H), 6.39 (d, J = 5.6 Hz, 1H), 3.56–3.47 (dd, J = 10.5, 7.0 Hz, 4H), 2.98 (t, J = 4.7 Hz, 2H), 2.93 (t, J = 4.9 Hz, 2H), 2.03 (s, 3H), 1.47 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 169.82, 168.71 (d, J = 4.3 Hz, 1C), 168.60, 160.30 (d, J = 7.60 Hz, 1C), 159.95, 157.56, 151.52, 148.56, 141.30, 136.59, 135.66 (d, J = 2.5 Hz, 1C), 129.26, 122.85 (d, J = 7.9 Hz, 1C), 122.10, 115.62, 115.40, 111.05, 110.13, 107.05, 98.72, 49.23, 45.87, 31.97, 21.64, 15.85. HRMS: m/z C33H32FN7O4 [M+H]+ 610.2500, Found 610.2576. Purity: > 95% (HPLC).
N-(4-fluorophenyl)-N-(4-((2-((3-(piperidin-3-ylcarbamoyl)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)cyclopropane-1,1-dicarboxamide (compound 13f): White solid, yield: 31.00%. 1H NMR (500 MHz, DMSO-d6) δ 10.11 (s, 1H), 10.07 (s, 1H), 8.23 (d, J = 5.6 Hz, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.67 (d, J = 8.9 Hz, 2H), 7.64 (dd, J = 8.9, 5.1 Hz, 2H), 7.19–7.12 (m, 4H), 7.07 (t, J = 7.8 Hz, 1H), 7.02 (s, 1H), 6.94 (d, J = 7.7 Hz, 1H), 6.69 (dd, J = 7.9, 1.5 Hz, 1H), 6.07 (d, J = 5.4 Hz, 1H), 5.27 (s, 1H), 4.59–4.20 (m, 2H), 3.83–3.74 (m, 1H), 2.88–2.76 (m, 2H), 1.93–1.85 (m, 1H), 1.75–1.68 (m, 1H), 1.64-1.54 (m, 1H), 1.46 (s, 4H), 1.44-1.34 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 169.81, 168.62 (d, J = 11.21 Hz, 1C), 167.20, 161.64, 160.43, 159.70, 157.79, 148.95, 148.31, 136.40, 136.11, 135.63 (d, J = 1.61 Hz, 1C), 129.02, 122.88 (d, J = 7.70 Hz, 1C), 121.96, 116.86, 115.58, 115.41, 115.05, 113.40, 95.84, 48.53, 46.15, 43.81, 31.91, 30.76, 23.97, 15.91. HRMS: m/z C24H20FN7O3 [M+H]+ 610.2500, Found 610.2576. Purity: > 95% (HPLC).
N-(4-fluorophenyl)-N-(4-((2-((4-(piperidin-3-ylcarbamoyl)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)cyclopropane-1,1-dicarboxamide (compound 13g): White solid, yield: 28.15%. 1H NMR (500 MHz, DMSO-d6) δ 10.10 (s, 1H), 10.08 (s, 1H), 8.22 (d, J = 5.5 Hz, 1H), 7.81 (d, J = 7.9 Hz, 1H), 7.69–7.61 (m, 4H), 7.58 (d, J = 8.4 Hz, 2H), 7.15 (t, J = 8.2 Hz, 4H), 6.54 (d, J = 8.4 Hz, 2H), 6.06 (d, J = 4.9 Hz, 1H), 5.64 (s, 1H), 4.62–4.15 (m, 2H), , 3.83–3.72 (m, 1H), 2.82 (t, J = 11.5 Hz, 2H), 1.88 (d, J = 9.6 Hz, 1H), 1.71 (d, J = 11.6 Hz, 1H), 1.61–1.51 (m, 1H), 1.46 (s, 4H), 1.44–1.33 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 169.81, 168.62 (d, J = 9.89 Hz, 1C), 166.27, 161.62, 160.39, 159.70, 157.79, 152.00, 148.32, 136.37, 135.62 (d, J = 1.39 Hz, 1C), 129.33, 122.88 (d, J = 7.70 Hz, 1C), 122.04, 121.94, 115.59, 115.41, 112.95, 95.78, 48.76, 45.99, 43.83, 31.88, 30.93, 24.04, 15.92. HRMS: m/z C24H20FN7O3 [M+H]+ 610.2500, Found 610.2567. Purity: > 95% (HPLC).
N-(4-fluorophenyl)-N-(4-((2-((4-(4-methylpiperazine-1-carbonyl)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)cyclopropane-1,1-dicarboxamide (compound 13h): White solid, yield: 18.28%. 1H NMR (400 MHz, DMSO-d6) δ 10.18 (s, 1H), 10.09 (s, 1H), 9.80 (s, 1H), 8.38 (d, J = 5.6 Hz, 1H), 7.71 (d, J = 9.0 Hz, 2H), 7.65 (dd, J = 9.1, 5.1 Hz, 2H), 7.58 (d, J = 8.5 Hz, 2H), 7.21 (d, J = 9.0 Hz, 2H), 7.19–7.13 (m, 4H), 6.48 (d, J = 5.6 Hz, 1H), 3.46 (s, 4H), 2.32 (s, 4H), 2.21 (s, 3H), 1.50 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 170.11, 169.51, 168.71, 160.41, 159.90, 157.56, 148.52, 141.96, 136.78, 135.62 (d, J = 2.5 Hz, 1C), 128.19, 122.89 (d, J = 7.9 Hz, 1C), 122.51, 122.33, 118.39, 115.62, 115.40, 99.15, 54.92, 45.97, 31.81, 31.62, 16.07. HRMS: m/z C33H32FN7O4 [M+H]+ 610.2500, Found 610.2568. Purity: > 95% (HPLC).
N-(4-fluorophenyl)-N-(4-((2-((4-(phenylsulfonamido)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)cyclopropane-1,1-dicarboxamide (compound 13i): White solid, yield: 21.35%. 1H NMR (500 MHz, DMSO-d6) δ 10.21 (s, 1H), 10.06 (s, 1H), 9.94 (s, 1H), 9.47 (s, 1H), 8.30 (d, J = 5.6 Hz, 1H), 7.70 (t, J = 9.6 Hz, 4H), 7.65 (dd, J = 8.9, 5.1 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.53 (t, J = 7.7 Hz, 2H), 7.41 (d, J = 8.3 Hz, 2H), 7.19–7.13 (m, 4H), 6.85 (d, J = 8.7 Hz, 2H), 6.36 (d, J = 5.6 Hz, 1H), 1.52 (d, J = 4.8 Hz, 4H).13C NMR (126 MHz, DMSO-d6) δ 169.99, 168.83, 168.65, 160.28, 160.03, 159.72, 157.81, 148.43, 139.99, 137.55, 136.77, 135.58 (d, J = 2.49 Hz, 1C), 133.15, 131.39, 129.57, 127.17, 122.94 (d, J = 7.88 Hz, 1C), 122.32, 122.08 (d, J = 10.53 Hz, 1C), 119.88, 115.60, 115.43, 98.61, 31.84, 16.02. HRMS: m/z C33H32FN7O4 [M+H]+ 639.1748, Found 639.1833. Purity: > 95% (HPLC).
N-(4-fluorophenyl)-N-(4-((2-((4-(2-(4-methylpiperazin-1-yl)propanamido)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)cyclopropane-1,1-dicarboxamide (compound 13j): White solid, yield: 23.75%. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 10.13 (s, 1H), 9.59 (s, 1H), 9.53 (s, 1H), 8.38 (d, J = 5.6 Hz, 1H), 7.77 (d, J = 8.9 Hz, 2H), 7.69 (dd, J = 9.1, 5.1 Hz, 2H), 7.53 (d, J = 8.7 Hz, 2H), 7.43 (d, J = 8.9 Hz, 2H), 7.26–7.18 (m, 4H), 6.42 (d, J = 5.6 Hz, 1H), 3.54–3.46 (m, 1H), 3.23 (q, J = 6.8 Hz, 1H), 2.61 (s, 4H), 2.47 (s, 4H), 2.26 (s, 3H), 1.56 (d, J = 10.5 Hz, 4H), 1.20 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.15, 170.05, 168.71 (d, J = 4.87 Hz, 1C), 160.15, 159.94, 157.55, 148.52, 136.74, 136.31, 135.63 (d, J = 2.57 Hz, 1C), 133.10, 122.86 (d, J = 7.87 Hz, 1C), 122.35, 122.12, 120.20, 119.52, 115.62, 115.40, 98.36, 63.74, 56.50, 55.27, 45.96, 31.89, 16.00, 13.25. HRMS: m/z C35H37FN8O4 [M+H]+ 653.2922 , Found 653.3001. Purity: > 95% (HPLC).
N-(4-fluorophenyl)-N-(4-((2-((3-(2-(4-methylpiperazin-1-yl)acetamido)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)cyclopropane-1,1-dicarboxamide (compound 13k): White solid, yield: 40.53%. 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 10.13 (s, 1H), 9.61 (s, 1H), 9.57 (s, 1H), 8.37 (d, J = 5.6 Hz, 1H), 7.76 (s, 1H), 7.74 (d, J = 9.0 Hz, 2H), 7.70 (dd, J = 9.1, 5.1 Hz, 2H), 7.36 (d, J = 8.1 Hz, 1H), 7.27–7.17 (m, 5H), 7.09 (t, J = 8.1 Hz, 1H), 6.41 (d, J = 5.6 Hz, 1H), 3.18 (s, 2H), 2.62 (s, 6H), 2.56 (s, 2H), 2.34 (s, 3H), 1.52 (s, 4H).13C NMR (101 MHz, DMSO-d6) δ 170.01, 168.65 (d, J = 6.48 Hz, 1C), 168.37, 160.27 (d, J = 6.39 Hz, 1C), 159.94, 157.55, 148.48, 140.90, 138.88, 136.71, 135.67 (d, J = 2.70 Hz, 1C), 128.95, 122.82 (d, J = 7.91 Hz, 1C), 122.31, 122.19, 115.63, 115.41, 113.65, 110.97, 98.72, 61.70, 54.63, 52.31, 45.38, 31.98, 15.87. HRMS: m/z C34H35FN8O4 [M+H]+ 639.2765, Found 639.2827. Purity: > 95% (HPLC).
N-(4-((2-((4-(2-((2-(diethylamino)ethyl)amino)acetamido)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (compound 13l): White solid, yield: 16.50%. 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 10.09 (s, 1H), 9.86 (s, 1H), 9.48 (s, 1H), 8.32 (d, J = 5.6 Hz, 1H), 7.71 (d, J = 8.9 Hz, 2H), 7.65 (dd, J = 9.0, 5.1 Hz, 2H), 7.51 (d, J = 8.7 Hz, 2H), 7.40 (d, J = 8.8 Hz, 2H), 7.20–7.13 (m, 4H), 6.36 (d, J = 5.6 Hz, 1H), 3.36 (s, 3H), 2.90 (s, 2H), 2.75 (s, 6H), 1.51 (d, J = 5.2 Hz, 4H), 1.06 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 170.02, 169.51, 168.74 (d, J = 2.1 Hz, 1C), 160.17, 159.94, 157.55, 148.51, 136.72, 136.35, 135.64 (d, J = 2.6 Hz, 1C), 133.07, 122.86 (d, J = 7.8 Hz, 1C), 122.25 (d, J = 12.7 Hz, 1C), 120.10, 119.64, 115.62, 115.40, 98.40, 52.32, 51.46, 46.90, 45.95, 31.92, 15.99, 10.73. HRMS: m/z C35H39FN8O4 [M+H]+ 655.3078, Found 655.3160. Purity: > 95% (HPLC).
N-(4-((2-((4-(2-((2-(dimethylamino)ethyl)amino)acetamido)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (compound 13m): White solid, yield: 30.11%. 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 10.12 (s, 1H), 9.91 (s, 1H), 9.48 (s, 1H), 8.32 (s, 1H), 7.71 (s, 2H), 7.65 (s, 2H), 7.49 (s, 2H), 7.42 (s, 2H), 7.18 (s, 4H), 6.36 (s, 1H), 3.32 (s, 3H), 2.71 (s, 2H), 2.55 (s, 2H), 2.32 (s, 6H), 1.51 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 170.03, 169.76, 168.76 (d, J = 3.0 Hz, 1C), 160.18, 157.57, 148.54, 136.70, 136.30, 135.61 (d, J = 2.6 Hz, 1C) 133.12, 122.89 (d, J = 7.9 Hz, 1C), 122.32, 122.19, 120.01, 119.65, 115.62, 115.40, 98.39, 58.06, 52.53, 46.03, 44.87, 31.88, 16.02. HRMS: m/z C33H35FN8O4 [M+H]+ 627.2765, Found 627.2841. Purity: > 95% (HPLC).
N-(4-fluorophenyl)-N-(4-((2-((4-(2-(4-(2-methoxyethyl)piperazin-1-yl)acetamido)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)cyclopropane-1,1-dicarboxamide (compound 13n): White solid, yield: 33.25%. 1H NMR (400 MHz, DMSO-d6) δ 10.14 (s, 1H), 10.09 (s, 1H), 9.48 (s, 1H), 9.46 (s, 1H), 8.32 (d, J = 5.6 Hz, 1H), 7.71 (d, J = 8.8 Hz, 2H), 7.64 (dd, J = 8.9, 5.1 Hz, 2H), 7.47 (d, J = 8.6 Hz, 2H), 7.37 (d, J = 8.8 Hz, 2H), 7.16 (q, J = 8.6 Hz, 4H), 6.36 (d, J = 5.6 Hz, 1H), 3.42 (t, J = 5.8 Hz, 2H), 3.22 (s, 3H), 3.04 (s, 2H), 2.50 (s, 2H), 2.47 (s, 8H), 1.51 (d, J = 8.2 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 170.05, 168.72, 168.18, 160.15, 157.56, 148.54, 136.71, 136.39, 135.63 (d, J = 2.5 Hz, 1C), 132.92, 122.87 (d, J = 7.9 Hz, 1C), 122.35, 122.17, 120.33, 119.54, 115.62, 115.40, 98.36, 70.34, 62.22, 58.46, 57.46, 53.42, 53.27, 31.89, 16.01. HRMS: m/z C36H39FN8O5 [M+H]+ 683.3027, Found 683.3107. Purity: > 95% (HPLC).
3.1.4. Preparation of N-(4-Hydroxyphenyl)-1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide (Intermediate 15)
To a solution of 4-antipyrine acid (14, 2.0 g, 8.61 mmol) and 4-aminophenol (10, 1.13 g, 10.33 mmol) in DMF (20 mL), HBTU (3.92 g, 10.33 mmol) and TEA (2.61 g, 25.84 mmol) were added. The reaction solution was stirred at room temperature for 8 h and monitored by TLC. The reaction solution was poured into ice water (200 mL), and the precipitate was filtered off, washed, and dried in a vacuum to yield intermediate 15 as a white solid (1.75 g, 63.0%), which can be used to the next step without any purification.
3.1.5. Preparation of N-(4-((2-Chloropyrimidin-4-yl)oxy)phenyl)-1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide (Intermediate 16)
To a solution of the intermediate 15 (1.75 g, 5.41 mmol) and 2,4-dichloropyrimidine (0.81 g, 5.41 mmol) in DMF (15 mL), K2CO3 (0.82 g, 5.95 mmol) was added. The mixture was stirred at 80 °C for 4.5 h. The reaction solution was poured into ice water (100 mL), and the precipitate was filtered off, washed, and dried in a vacuum to yield intermediate 16 as a white solid (1.85 g, 78.0%), which can be used directly without any further purification.
3.1.6. General Procedure for Preparation of Target Compounds 17a–17h
To a mixture of the intermediate 16 (1.2 mmol), substituted aniline (1.0 mmol) and DMF (8 mL), PTSA (4.00 mmol) was added. The mixture was stirred at 90 °C for 4 h under N2 atmosphere. The reaction solution was cooled to room temperature, then poured into ice water (100 mL), and the precipitate was filtered off, washed, and dried in a vacuum to obtain the crude product, which was purified by silica gel chromatography using a mixture of (DCM: MeOH = 100:1–30:1) to afford the product 17a–17h.
1,5-dimethyl-N-(4-((2-((4-((1-methylpiperidin-4-yl)oxy)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide (compound 17a): White solid, yield: 15.05%. 1H NMR (500 MHz, DMSO-d6) δ 10.82 (s, 1H), 9.41 (s, 1H), 8.29 (d, J = 5.6 Hz, 1H), 7.68 (d, J = 8.8 Hz, 2H), 7.60 (t, J = 7.7 Hz, 2H), 7.53 (d, J = 7.3 Hz, 1H), 7.45 (d, J = 7.5 Hz, 2H), 7.40–7.34 (m, 2H), 7.18 (d, J = 8.8 Hz, 2H), 6.71 (d, J = 7.7 Hz, 2H), 6.38 (d, J = 5.6 Hz, 1H), 4.25 (s, 1H), 3.38 (s, 3H), 2.77 (s, 2H), 2.74 (s, 3H), 2.45 (s, 2H), 2.31 (d, J = 13.7 Hz, 3H), 1.90 (s, 2H), 1.68 (s, 2H).13C NMR (126 MHz, DMSO-d6) δ 170.12, 163.52, 161.67, 160.36, 160.15, 154.26, 151.89, 148.17, 136.67, 134.09, 133.49, 129.97, 129.33, 127.59, 123.09, 120.95, 120.76, 116.33, 97.94, 97.60, 44.97, 33.83, 21.25, 11.98. HRMS: m/z C34H36N7O4 [M+H]+ 606.2751, Found 606.2832. Purity: > 95% (HPLC).
N-(4-((2-((3-((2-methoxyethyl)amino)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)-1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide (compound 17b): White solid, yield: 5.80%. 1H NMR (500 MHz, DMSO-d6) δ 10.82 (s, 1H), 9.28 (s, 1H), 8.31 (d, J = 5.4 Hz, 1H), 7.69 (d, J = 8.5 Hz, 2H), 7.60 (t, J = 7.4 Hz, 3H), 7.52 (t, J = 7.2 Hz, 1H), 7.45 (d, J = 7.4 Hz, 2H), 7.19 (d, J = 8.6 Hz, 2H), 6.87–6.77 (m, 3H), 6.36 (d, J = 5.4 Hz, 1H), 6.16 (d, J = 6.8 Hz, 1H), 3.42 (t, J = 5.5 Hz, 2H), 3.37 (s, 3H), 3.24 (s, 3H), 3.05 (s, 2H), 2.72 (s, 3H).13C NMR (126 MHz, DMSO-d6) δ 169.92, 163.53, 161.65, 160.35, 160.23, 154.29, 149.19, 148.09, 141.27, 136.65, 133.52, 129.95, 129.32, 129.04, 127.58, 122.77, 120.61, 107.94, 106.65, 103.26, 98.29, 97.57, 70.93, 58.42, 42.94, 33.79, 11.93. HRMS: m/z C31H21N7O4Na [M+Na]+ 588.2438, Found 588.2337. Purity: > 95% (HPLC).
1,5-dimethyl-N-(4-((2-((3-((1-methylpiperidin-4-yl)oxy)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide (compound 17c): White solid, yield: 7.01%. 1H NMR (500 MHz, DMSO-d6) δ 10.81 (s, 1H), 9.51 (s, 1H), 8.35 (d, J = 5.5 Hz, 1H), 7.68 (d, J = 8.7 Hz, 2H), 7.60 (t, J = 7.5 Hz, 2H), 7.52 (t, J = 7.4 Hz, 1H), 7.45 (d, J = 7.5 Hz, 2H), 7.27 (s, 1H), 7.20 (d, J = 8.7 Hz, 2H), 7.16 (d, J = 7.8 Hz, 1H), 7.00 (t, J = 8.0 Hz, 1H), 6.51 (d, J = 7.7 Hz, 1H), 6.42 (d, J = 5.5 Hz, 1H), 4.30 (s, 1H), 3.34 (s, 2H), 2.93 (s, 2H), 2.72 (s, 3H), 2.45 (s, 3H), 2.00 (s, 2H), 1.76 (s, 2H).13C NMR (126 MHz, DMSO-d6) δ 169.95, 163.51, 161.64, 160.33, 160.13, 157.35, 154.28, 147.98, 141.90, 136.69, 133.50, 129.96, 129.49, 129.33, 127.59, 122.70, 120.68, 112.14, 108.92, 107.25, 98.85, 97.57, 51.75, 44.37, 33.80, 31.62, 30.30, 11.94. HRMS: m/z C34H36N7O4 [M+H]+ 606.2751, Found 606.2823. Purity: > 95% (HPLC).
1,5-dimethyl-N-(4-((2-((4-(4-methylpiperazine-1-carbonyl)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide (compound 17d): White solid, yield: 63.0%. 1H NMR (500 MHz, DMSO-d6) δ 10.85 (s, 1H), 9.82 (s, 1H), 8.38 (d, J = 5.6 Hz, 1H), 7.71 (d, J = 8.8 Hz, 2H), 7.60 (t, J = 7.6 Hz, 2H), 7.49-7.56 (m, 3H), 7.45 (d, J = 7.3 Hz, 2H), 7.21 (d, J = 8.8 Hz, 2H), 7.13 (d, J = 8.3 Hz, 2H), 6.51 (d, J = 5.6 Hz, 1H), 3.37 (s, 4H), 3.33 (s, 3H), 2.73 (s, 3H), 2.27 (s, 4H), 2.15 (s, 3H).13C NMR (126 MHz, DMSO-d6) δ 170.17, 169.40, 163.51, 161.67, 160.46, 159.84, 154.35, 148.05, 141.99, 136.85, 133.49, 129.95, 129.30, 128.45, 128.17, 127.57, 123.12, 120.68, 118.37, 99.11, 97.65, 54.90, 33.80, 11.98. HRMS: m/z C34H34N8O4Na [M+Na]+ 641.2703, Found 641.2603. Purity: > 95% (HPLC).
N-(4-((2-((4-(4-acetylpiperazin-1-yl)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)-1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide (compound 17e): White solid, yield: 15.79%. 1H NMR (500 MHz, DMSO-d6) δ 10.85 (s, 1H), 9.36 (s, 1H), 8.28 (d, J = 5.5 Hz, 1H), 7.69 (d, J = 8.7 Hz, 2H), 7.60 (t, J = 7.6 Hz, 2H), 7.52 (t, J = 7.4 Hz, 1H), 7.47 (d, J = 7.4 Hz, 2H), 7.26 (s, 2H), 7.16 (d, J = 8.8 Hz, 2H), 6.68 (d, J = 6.9 Hz, 2H), 6.38 (d, J = 5.5 Hz, 1H), 3.48 (s, 4H), 3.40 (s, 3H), 2.99 (s, 2H), 2.87 (s, 2H), 2.74 (s, 3H), 2.00 (s, 3H).13C NMR (126 MHz, DMSO-d6) δ 170.26, 168.66, 163.48, 161.69, 160.29, 160.03, 154.04, 148.29, 145.98, 136.72, 133.47, 133.25, 129.97, 129.38, 127.69, 123.28, 120.82, 120.48, 116.55, 97.44, 50.18, 49.24, 45.98, 33.75, 21.60, 11.88. HRMS: m/z C34H34N8O4Na [M+Na]+ 641.2703, Found 641.2607. Purity: > 95% (HPLC).
N-(4-((2-((3-(4-acetylpiperazin-1-yl)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)-1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide (compound 17f): White solid, yield: 20.0%. 1H NMR (500 MHz, DMSO-d6) δ 10.82 (s, 1H), 9.40 (s, 1H), 8.34 (d, J = 5.5 Hz, 1H), 7.68 (d, J = 8.8 Hz, 2H), 7.60 (t, J = 7.6 Hz, 2H), 7.52 (t, J = 7.4 Hz, 1H), 7.45 (d, J = 7.4 Hz, 2H), 7.19 (d, J = 8.5 Hz, 3H), 7.09 (d, J = 7.3 Hz, 1H), 6.96 (t, J = 8.0 Hz, 1H), 6.51 (d, J = 7.5 Hz, 1H), 6.41 (d, J = 5.5 Hz, 1H), 3.51 (t, J = 5.7 Hz, 4H), 3.37 (s, 4H), 2.96 (s, 4H), 2.72 (s, 3H), 2.01 (s, 3H).13C NMR (126 MHz, DMSO-d6) δ 169.84, 168.70, 163.53, 161.64, 160.33, 160.26, 154.27, 151.57, 148.05, 141.31, 136.61, 133.51, 129.95, 129.32, 129.14, 127.60, 122.59, 120.57, 111.11, 110.15, 107.18, 98.73, 97.56, 49.29, 48.88, 45.86, 33.78, 21.63, 11.92. HRMS: m/z C34H34N8O4Na [M+Na]+ 641.2703, Found 641.2599. Purity: > 95% (HPLC).
1,5-dimethyl-N-(4-((2-((6-(2-(4-methylpiperazin-1-yl)acetamido)pyridin-3-yl)amino)pyrimidin-4-yl)oxy)phenyl)-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide (compound 17g): White solid, yield: 18.6%. 1H NMR (600 MHz, DMSO-d6) δ 10.81 (s, 1H), 9.72 (s, 1H), 9.67 (s, 1H), 8.49 (s, 1H), 8.35 (d, J = 5.6 Hz, 1H), 7.98 (s, 1H), 7.88 (d, J = 8.6 Hz, 1H), 7.69 (d, J = 8.9 Hz, 2H), 7.60 (t, J = 7.7 Hz, 2H), 7.52 (t, J = 7.5 Hz, 1H), 7.48–7.43 (m, 2H), 7.20 (d, J = 8.9 Hz, 2H), 6.43 (d, J = 5.6 Hz, 1H), 3.37 (s, 5H), 3.11 (s, 2H), 2.72 (s, 3H), 2.52–2.50 (m, 2H), 2.38 (s, 4H), 2.18 (s, 3H).13C NMR (126 MHz, DMSO-d6) δ 170.10, 168.61, 163.52, 161.66, 161.66, 160.40, 160.08, 154.31, 147.90, 145.69, 139.23, 136.78, 133.75, 133.50, 129.95, 129.32, 128.77, 127.57, 122.68, 120.68, 113.01, 99.15, 97.62, 61.50, 55.13, 52.97, 46.02, 33.78, 11.94. HRMS: m/z C34H36N10O4Na [M+Na]+ 671.2921, Found 671.2823. Purity: > 95% (HPLC).
1,5-dimethyl-N-(4-((2-((4-(2-(4-methylpiperazin-1-yl)propanamido)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide (compound 17h): White solid, yield: 33.8%. 1H NMR (600 MHz, DMSO-d6) δ 10.81 (s, 1H), 9.55 (s, 1H), 9.49 (s, 1H), 8.32 (d, J = 5.6 Hz, 1H), 7.71–7.67 (m, 2H), 7.60 (t, J = 7.7 Hz, 2H), 7.52 (dt, J = 8.3, 1.5 Hz, 1H), 7.51–7.47 (m, 2H), 7.45–7.42 (m, 2H), 7.37 (d, J = 8.6 Hz, 2H), 7.22–7.18 (m, 2H), 6.37 (d, J = 5.6 Hz, 1H), 3.37 (s, 3H), 3.15 (q, J = 6.8 Hz, 1H), 2.72 (s, 3H), 2.52–2.50 (m, 2H), 2.40 (d, J = 71.7 Hz, 6H), 2.13 (s, 3H), 1.13 (d, J = 6.9 Hz, 3H).13C NMR (126 MHz, DMSO-d6) δ 171.20, 170.01, 163.52, 161.65, 160.31, 160.17, 154.32, 148.03, 136.71, 133.49, 133.13, 129.95, 129.31, 127.53, 122.77, 120.64, 120.16, 119.56, 98.41, 97.65, 63.77, 55.38, 49.39, 46.16, 33.78, 13.29, 11.94. HRMS: m/z C36H40N9O4 [M+H]+ 662.3125, Found 662.3207. Purity: > 95% (HPLC).
3.2. Mer and c-Met Inhibitory Activity Assay
The in vitro kinase inhibition assays for all compounds against Mer and c-Met were conducted by Shanghai Bioduro Biological Technology Co., Ltd. (China). The detailed procedures are as follows: Prepare 1x buffer: Assay buffer 1X, MgCl2 5mM, DTT 1mM. Compound dilution with DMSO. For test compounds, make 100× final concentration solution. Transfer 100 nL compounds to 384-well plate by using the automated liquid handler. The finial DMSO% in the assay is 1%. Dilute enzyme stock solutions with 1X assay buffer to concentration of 0.6nM to make a 2X working solution. Add 5μL to the assay plate manually (final 1nM) with multichannel pipette, Spin down at 1000rpm and centrifuge for 30sec and Incubate 15min at 25°C temperatures. Dilute Substrate solutions with 1X assay buffer. Add 5μL mix or buffer to the assay plate manually (ATP final 3.5uM and TK-Sub-Biotin final 0.5uM) with multichannel pipette, Spin down at 1000rpm and centrifuge for 30sec. After 30°C 60 min, add 10μl of Detection solution (TK-antidody-cryptate final 0.25X and streptavidine-XL 665 final 0.3125 uM) to each well of the assay. Mix briefly with Centrifuge, equilibrate for 60 min. Record luminescence on Envision.
3.3. Antiproliferation Assay
The HepG2, MDA-MB-231, and HCT116 cell lines are derived from our own laboratory. The antiproliferative activities were assessed using the CCK-8 assay. Cell viability was measured according to the manufacturer’s instructions for the Cell Counting Kit-8 (Meilunbio, Dalian, China). In this assay, 2.5 × 10³ cells per well were seeded into 96-well plates.
After an initial 12-hour incubation, cancer cells were exposed to varying concentrations of compound 17c for 72 hours. Following this, CCK-8 solution was added to each well and incubated for an additional 2 hours. The absorbance at 450 nm was then recorded using the BioTek Synergy HTX microplate reader. The experiment was performed in triplicate and repeated three times. IC50 values for compound 17c were calculated using GraphPad PRISM 9.5.
3.4. hERG Potassium Currents Assay
CHO cells, purchased from Cell Bank of Chinese Academy of Sciences (Cat. No.: SCSP-507), were examined using the QPatch system (Sophion) and the automated whole-cell patch clamp method. The detail procedures are as follows: CHO cells stably expressing the transcript of hERG were investigated by the automated whole-cell patch clamp technique, using the QPatch system (Sophion). Cells were grown in 175 cm2 flasks in serum-supplemented F12 medium in 37 oC 5% CO2 incubator. Two days after plating (at 70-80% confluence), culture medium was removed and cells were washed with 7 mL PBS (Phosphate Buffered Saline), then cells were treated with 3 mL Detachin for 2 min at 37 oC, followed by the addition of 7 mL serum-free medium, cells were resuspended in the growth medium at a density of 2-5*106 cells/mL. The cell suspension was then transferred to the QPatch system, where it was centrifuged; the cells were washed and resuspended in extracellular solution (in mM): 140 NaCl, 5 KCl, 1 CaCl2, 1.25 MgCl2, 10 HEPES and 10 Glucose, pH 7.4 with NaOH. The composition of the intracellular solution was (in mM): 140 KCl, 1 MgCl2, 1 CaCl2, 10 EGTA and 10 HEPES, pH 7.2 with KOH. The cells were held at -80 mV and activated by a +40 mV pre-pulse of 5 s duration followed by a step to -50 mV for 5 s. The voltage protocol is to be repeated every 15 s, the peak of the tail current evoked by the −50 mV step was recorded and measured as test parameter. The solutions containing the compounds were applied to the cells for 2.5 min following a 5 min baseline recording in extracellular solution. Each cell was received from six escalating concentrations. Each concentration was tested on at least 3 cells (n≥3). The reference compound cisapride was applied at the end of the test compound addition. Data were retrieved and analyzed using Assay Software v5.6.4 provided by Sophion, GraphPad Prism 8 and Excel. The concentrations of compounds to yield 50 % block of the hERG currents (IC50) were obtained by fitting normalized concentration-inhibition relationships to the equation in Prism 8 software.
3.5. Liver Microsome Stability Assay
Each test compound was initially dissolved in DMSO to prepare a 10 mM stock solution, which was then diluted to 200 μM using acetonitrile. Incubation mixtures were set up in a total volume of 200 μL, containing 0.1 M PBS (pH 7.4), 2 mM NADPH, 0.2 mg/mL liver microsomes, and 1 μM of the test compound or positive control. After a 5-minute preincubation of all components (except NADPH) at 37 °C, NADPH was added. The mixture was pipette-mixed thoroughly, and 20 μL was immediately transferred to a “Quenching” plate as a 0-minute sample, followed by pipette-mixing. At subsequent time points (5, 15, 30, and 60 minutes), 20 μL aliquots were transferred from the incubate to the “Quenching” plate after pipette-mixing. To each well of the plate, 200 μL of acetonitrile containing the internal standard was added. The plate was then centrifuged at 4000 rpm for 10 minutes. Finally, 50 μL of the supernatant was mixed with 50 μL of deionized water and analyzed by LC-MS/MS.
3.6. Docking Study
Molecular docking simulations were performed in our lab. The binding interactions of the compounds were investigated with the aid of Discovery Studio 2019. For this analysis, the X-ray crystallographic structures of MerTK (PDB: 4M3Q) and c-Met (PDB: 3LQ8), both resolved at 1.84 Å resolution, were utilized. The protein-ligand interactions were visualized and analyzed through the interaction mode diagram generated by Discovery Studio 2019.
3.7. Western Blot Assay
Western blot assay was performed according to established protocols. HCT colon cancer cells were exposed to the specified concentration of compound 17c for 24 h, after which whole cell lysate were prepared using RAPI cell lysis buffer (Beyotime Institute of Biotechnology, China). Primary antibodies, purchased from Beyotime Institute of Biotechnology, were diluted at a ratio of 1:2000. The protein band on polyvinylidene fluoride membranes were visualized and analyzed using Glyco Band-Scan Software Version 4.50. The antibody details are as follows:
Name: Phospho-MER/TYRO3 (Tyr753/Tyr685) antibody, Species: Rabit, Sequence: synthesized peptide derived from human MER/TYRO3 around the Phosphorylation site of Tyr753/Tyr685, Dilution: 1:2000, RRID number: AB_2840500, Catalogue number: AF8443, Supplier: Affinity Biosciences.
Name: Phospho-c-Met (Tyr1234) antibody, Species: Rabit, Sequence: synthesized peptide derived from human c-Met around the Phosphorylation site of Tyr1234, Dilution: 1:2000, RRID number: AB_2834564, Catalogue number: AF3129, Supplier: Affinity Biosciences.
3.8. Apotosis Study
Apoptosis was analyzed in a dose-dependent manner using the One Step TUNEL Apoptosis Assay Kit (Meilunbio, Dalian, China). HCT116 cells (1.5 × 10⁴ cells/well) were seeded onto TC-treated glass coverslips in 24-well plates and incubated for 24 hours. Following this, the cells were exposed to varying concentrations of compound 17c for 48 hours. After treatment, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.3% Triton-X-100. TUNEL staining was performed as per the manufacturer’s guidelines, with DAPI used for nuclear counterstaining. Fluorescent images were obtained using an Olympus microscope. The ED50 value for compound 17c was calculated using GraphPad PRISM 9.5. Each assay was performed in triplicate and repeated three times for consistency.
3.9. Transwell Assay
Migration assays were conducted using Transwell® inserts (Costar, Cambridge, MA) with an 8 μm pore size polycarbonate membrane. HCT116 cells (1.5 × 10⁵) were seeded into the upper chamber with serum-free medium containing 0.2% BSA, while the lower chamber contained medium supplemented with 15% FBS. Compound 17c was applied at the specified concentrations to both sides of the membrane. After 24 hours of incubation at 37 °C, the cells were fixed in methanol and stained with 0.1% crystal violet. Non-migrated cells on the upper surface of the membrane were removed using a cotton swab. Five random fields per well were imaged under brightfield microscopy, and the number of migrated cells was quantified using ImageJ software. Each experiment was performed in triplicate.
3.10. Plasma Protein Binding in Plasma
Stock solution (20 mM) of compound 17c was prepared in DMSO. The stock solution was then diluted into 100 μM with acetonitrile. The HT Dialysis apparatus was assembled according to the manufacturer’s instructions. 150 μL of the plasma spiked with 1 μM final compound concentrations was added to one site of well and 150 μL of PBS was added to the opposite side of well (n=3). The plate was equilibrated for 6 hours at 37 °C in a 5% CO2 incubator. For T0 samples: 200 μL of spiked plasma was stored at –20 °C. For T6 samples: 200 μL of spiked plasma was incubated at 37 °C in a 5% CO2 incubator for 6 hours. After incubation, 50 μL of the samples (plasma or PBS) was collected into a vessel containing 50 μL of the opposite matrix. 300 μL of acetonitrile with IS were added into the vessels. The 96-well plate was centrifuged at 4000 rpm for 10 min. 50 μL of supernatant was mixed with 50 μL of ddH2O and then injected onto the LC-MS/MS system for analysis.
3.11. Pharmacokinetic Properties
Male and female SD rats (SLRC Laboratory Animal Inc., Shanghai, China) were used for the study. In the oral (PO) administration, 3 rats per group were given a single 10 mg/kg dose. Blood samples (100 μL per time point) were collected via jugular vein cannula and taken at 5, 15, and 30 minutes, and 1, 2, 4, 8, and 24 h post-dose. For the intravenous (IV) administration, 3 rats per group received 2.0 mg/kg, and blood samples were collected at 5, 15, and 30 minutes, and 1, 2, and 4 hours after dosing. Rats were fasted overnight prior to dosing and remained fasted for 6 h post-dose. Plasma was separated by centrifugation and stored at –40 °C until analysis. Compound concentrations were determined by LC/MS/MS (Shimadzu LC-30AD), and pharmacokinetic parameters, including Cmax, Tmax, T1/2, and AUC0-inf, were calculated.
4. Conclusions
We report a novel series of dual Mer/c-Met inhibitors, with 17c emerging as the lead candidate. Molecular docking revealed that 17c forms hydrogen bonds with Mer (Leu593/Lys619) and c-Met (Met1160/Lys1110/Phe1223), complemented by π-π interaction. 17c exhibited potent enzymatic inhibition and superior antiproliferative activity over cabozantinib against three cancer cell lines. Pharmacokinetically, 17c showed 45.3% oral bioavailability (16-fold higher than 18c), extended half-life (3.46 h), and low hepatic clearance (0.026 mL/min/mg). Safety profiling indicated minimal hERG inhibition. Mechanistically, 17c induced apoptosis and suppressed HCT116 migration. These data position 17c as a promising dual Mer/c-Met inhibitor warranting further development.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
Conceptualization, Investigation, Project administration, Writing—original draft, and Writing—review & editing, J.X.; Investigation, Methodology, Project administration, Software, Writing—original draft, and Writing—review & editing, D.W.; Data curation, Investigation, and Methodology, R.J.; Formal analysis, Investigation, and Software, P.X.; Investigation, Methodology, and Validation, R.R.; Data curation, Investigation, Methodology, Project administration, and Writing—review & editing, D.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially funded by Science Research Project of Hebei Education Department (BJK2024144), College Students’ Innovation and Entrepreneurship Training Program (202314432007), Special Project for Basic Research Business Expenses of Provincial Universities (JCYJ2023004), Doctoral Research Fund Project (BSZ2022004), and the National Natural Science Foundation of China (No.82003601).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Structures of all compounds, Raw materials of biology, Mer inhibitory results, c-Met inhibitory results, and 1H-NMR, 13C-NMR, HRMS, and HPLC spectra of all target compounds are listed in Supplementary Materials. All data supporting this study are available from the corresponding authors upon reasonable request.
Acknowledgments
Thank the Instrumental Analysis Center of Hebei University of Science and Technology for providing HRMS and NMR tests.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
References
- Wang, X.; Frye, S. Chapter Nineteen—Mer Receptor Tyrosine Kinase: Therapeutic Opportunities in Oncology, Virology, and Cardiovascular Indications. Annu. Rep. Med. Chem. 2014, 49, 301–314. [Google Scholar] [CrossRef]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yi, M.; Qin, S.; Wu, K.M. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol. Cancer. 2019, 18, 125. [Google Scholar] [CrossRef] [PubMed]
- Smart, S.; Vasileiadi, E.; Wang, X. The Emerging Role of TYRO3 as a Therapeutic Target in Cancer. Cancers (Basel) 2018, 10, 474. [Google Scholar] [CrossRef]
- Baladi, T.; Abet, V.; Piguel, S. State-of-the-art of small molecule inhibitors of the TAM family: the point of view of the chemist. Eur. J. Med. Chem. 2015, 105, 220–237. [Google Scholar] [CrossRef]
- Davra, V.; Kumar, S.; Geng, K.; Calianese, D.; Mehta, D.; Gadiyar, V.; Kasikara, C.; Lahey, K.C.; Chang, Y.J.; Wichroski, M.; Gao, C.; Lorenzo, M.S.D.; Kotenko, S.V.; Bergsbaken, T.; Mishra, P.; Gause, W.C.; Quigley, M.; Spires, T.E.; Birge, R.B. Axl and Mertk Receptors Cooperate to Promote Breast Cancer Progression by Combined Oncogenic Signaling and Evasion of Host Antitumor Immunity. Cancer Res. 2021, 81, 698–712. [Google Scholar] [CrossRef]
- Park, H.J.; Baen, J.Y.; Lee, Y.J.; Choi, Y.H.; Kang, J.L. The TAM-family receptor Mer mediates production of HGF through the RhoA-dependent pathway in response to apoptotic cells. Mol. Biol. Cell 2012, 23, 3254–3265. [Google Scholar] [CrossRef]
- Nguyen, K.Q.; Tsou, W.I.; Kotenko, S.; Birge, R.B. TAM receptors in apoptotic cell clearance, autoimmunity, and cancer. Autoimmunity 2013, 46, 294–297. [Google Scholar] [CrossRef]
- Sayama, A.; Okado, K.; Yamaguchi, M.; Samata, N.; Imaoka, M.; Kai, K.; Mori, K. The impact of the timing of dosing on the severity of UNC569-induced ultrastructural changes in the mouse retina. Toxicol. Pathol. 2020, 48(5), 669–676. [Google Scholar] [CrossRef]
- Koda, Y.; Itoh, M.; Tohda, S. Effects of MERTK inhibitors UNC569 and UNC1062 on the growth of acute myeloid Leukaemia cells. Anticancer Res. 2018, 38(1), 199–204. [Google Scholar] [CrossRef]
- DeRyckere, D.; Lee-Sherick, A.B.; Huey, M.G.; Hill, A.A.; Tyner, J.W.; Jacobsen, K.M.; Page, L.S.; Kirkpatrick, G.G.; Eryildiz, F.; Montgomery, S.A.; Zhang, W.; Wang, X.D.; Fry, S.V.; Earp, H.S.; Graham, D.K. UNC2025, a MERTK small-molecule inhibitor, is therapeutically effective alone and in combination with methotrexate in leukemia models. Clin. Cancer Res. 2017, 23(6), 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Huelse, J.M.; Kireev, D.; Tan, Z.; Chen, L.; Goyal, S.; Wang, X.; Frye, S.V.; Behera, M.; Schneider, F.; Ramalingam, S.S. MERTK activation drives osimertinib resistance in EGFR-mutant non–small cell lung cancer. J. Clin. Invest. 2022, 132(15). [CrossRef]
- Zhang, W.; DeRyckere, D.; Hunter, D.; Liu, J.; Stashko, M.A.; Minson, K.A.; Cummings, C.T.; Lee, M.; Glaros, T.G.; Newton, D.L.; Sather, S. UNC2025, a potent and orally bioavailable MER/FLT3 dual inhibitor. J. Med. Chem. 2014, 57(16), 7031–7041. [Google Scholar] [CrossRef] [PubMed]
- Lee-Sherick, A.B.; Jacobsen, K.M.; Henry, C.J.; Huey, M.G.; Parker, R.E.; Page, L.S.; Hill, A.A.; Wang, X.; Frye, S.V.; Earp, H.S.; Jordan, C.T.; Deryckere, D.; Hraham, D.K. MERTK inhibition alters the PD-1 axis and promotes anti-leukemia immunity. JCI insight 2018, 3(21). [CrossRef]
- Zhang, W.; Zhang, D.; Stashko, M.A.; DeRyckere, D.; Hunter, D.; Kireev, D.; Miley, M.J.; Cummings, C.; Lee, M.; Norris-Drouin, J.; Stewart, W.M. Pseudo-cyclization through intramolecular hydrogen bond enables discovery of pyridine substituted pyrimidines as new Mer kinase inhibitors. J. Med. Chem. 2013, 56(23), 9683–9692. [Google Scholar] [CrossRef]
- Shi, C.; Li, X.; Wang, X.; Ding, N.; Ping, Y.; Shi, Y.; Mi, L.; Lai, Y.; Song, Y.; Zhu, J. The proto-oncogene Mer tyrosine kinase is a novel therapeutic target in mantle cell lymphoma. J. Hematol. Oncol. 2018, 11, 1–13. [Google Scholar] [CrossRef]
- Hu, L.; Wang, X.; Song, Z.; Chen, F.; Wu, B. Leveraging CAR macrophages targeting c-Met for precision immunotherapy in pancreatic cancer: insights from single-cell multi-omics. Mol. Med. 2024, 30(1), 231. [Google Scholar] [CrossRef]
- Barbosa-Matos, C.; Borges-Pereira, C.; Libório-Ramos, S.; Fernandes, R.; Oliveira, M.; Mendes-Frias, A.; Silvestre, R.; Osório, N.S.; Bastos, H.N.; Santos, R.F.; Guimarães, S. Deregulated immune cell recruitment orchestrated by c-MET impairs pulmonary inflammation and fibrosis. Resp. Res. 2024, 25(1), 1–18. [Google Scholar] [CrossRef]
- Faiella, A.; Riccardi, F.; Cartenì, G.; Chiurazzi, M.; Onofrio, L. The emerging role of c-Met in carcinogenesis and clinical implications as a possible therapeutic target. J. Oncol. 2022, 2022(1), 5179182. [Google Scholar] [CrossRef]
- Lee, M.; Jain, P.; Wang, F.; Ma, P.C.; Borczuk, A.; Halmos, B. MET alterations and their impact on the future of non-small cell lung cancer (NSCLC) targeted therapies. Expert Opin. Ther. Tar. 2021, 25(4), 249–268. [Google Scholar] [CrossRef]
- Gou, Q.; Gou, Q.; Gan, X.; Xie, Y. Novel therapeutic strategies for rare mutations in non-small cell lung cancer. Sci. Rep. 2024, 14(1), 10317. [Google Scholar] [CrossRef]
- Cen, S.Y.; Lin, F.; Li, X.; Hu, Y.; Liu, J.P.; Xue, Z.; Gao, Y.; Sun, Y.P.; Zhu, S.; Dang, Y.; Zhao, Y. Crizotinib and its enantiomer suppress ferroptosis by decreasing PE-O-PUFA content. Cell Death Discov. 2024, 10(1), 360. [Google Scholar] [CrossRef]
- Elghawy, O.; Barsouk, A.; Xu, J.; Chen, S.; Cohen, R.B.; Sun, L. Real word outcomes of cabozantinib therapy in poorly differentiated thyroid carcinoma. Eur. Thyroid J. 2024, 13(6). [CrossRef]
- Patil, J.; Bhattacharya, S.; Saoji, S.D.; Dande, P. Cabozantinib-phospholipid complex for enhanced solubility, bioavailability, and reduced toxicity in liver cancer. Ther. Deliv. 2025, 16(1), 25–41. [Google Scholar] [CrossRef] [PubMed]
- Miao, K.; Zhang, X.; Wang, H.; Si, X.; Zhang, L. Savolitinib versus crizotinib for treating MET positive non-small cell lung cancer. Thorac. Cancer 2023, 14(13), 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Li, J.; Lin, S.; Wu, S.; Chai, K.; Jiang, X.; Qian, J.; Jiang, C. A real-world pharmacovigilance study of FDA adverse event reporting system events for Capmatinib. Sci. Rep. 2024, 14(1), 11388. [Google Scholar] [CrossRef]
- Dhillon, S. Capmatinib: first approval. Drugs, 2020, 80, 1125–1131. [CrossRef]
- Huang, D.W.; Chen, Y.; Yang, J.X.; Zhao, B.Y.; Wang, S.Y.; Chai, T.T.; Cui, J.; Zhou, X. L.; Shang, Z. H. Design, Synthesis, and Biological Evaluation of 2-Substituted Aniline Pyrimidine Derivatives as Potent Dual Mer/c-Met Inhibitors. Molecules, 2024, 29(2), 475. [CrossRef]
- Wei, D.S.; Fan, H.; Zheng, K.; Qin, X.; Yang, L.; Yang, Y.; Duan, Y.; Zhang, Q.; Zeng, C.; Hu, L. Synthesis and anti-tumor activity of [1,4] dioxino [2, 3-f] quinazoline derivatives as dual inhibitors of c-Met and VEGFR-2. Bioorg. Chem. 2019, 88, 102916. [Google Scholar] [CrossRef]
- Gao, G.R.; Li, M.Y.; Lv, Y.C.; Cao, S.F.; Tong, L.J.; Wei, L.X.; Ding, J.; Xie, H.; Duan, W.H. Design, synthesis and biological evaluation of biphenylurea derivatives as VEGFR-2 kinase inhibitors (II). Chinese Chem. Lett. 2016, 27(2), 200–204. [Google Scholar] [CrossRef]
- Park, H.; Jung, H.Y.; Mah, S.; Hong, S. Systematic computational design and identification of low Picomolar inhibitors of Aurora kinase A. J. chem. Inf. Model. 2018, 58(3), 700–709. [Google Scholar] [CrossRef]
- Huang, D.W.; Yang, J.X.; Zhang, Q.W.; Wang, G.; Zhang, Z.X.; Zhang, Y.; Li, J.Q. Structure-guided design and development of novel N-phenylpyrimidin-2-amine derivatives as potential c-Met inhibitors. Eur. J. Med. Chem. 2021, 223, 113648. [Google Scholar] [CrossRef]
- Hwang, S.T.; Um, J.Y.; Chinnathambi, A.; Alharbi, S.A.; Narula, A.S.; Namjoshi, O.A.; Blough, B.E.; Ahn, K.S. Evodiamine mitigates cellular growth and promotes apoptosis by targeting the c-Met pathway in prostate cancer cells. Molecules 2020, 25(6), 1320. [Google Scholar] [CrossRef]
- Li, J.; Tan, G.S.; Cai, Y.B.; Liu, R.H.; Xiong, X.L.; Gu, B.H.; He, W.; Liu, B.; Ren, Q.Y.; Wu, J.P.; Chi, B.; Zhao, Y.H.; Xu, Y.R.; Zou, Z.X.; Kang, F.H.; Xu, K.P. A novel Apigenin derivative suppresses renal cell carcinoma via directly inhibiting wild-type and mutant MET. Biochem. Pharmacol. 2021, 190, 114620. [Google Scholar] [CrossRef]
- Gu, Y.; Chen, Y.; Wei, L.; Wu, S.; Shen, K.; Liu, C.; Dong, Y.; Zhao, Y.; Zhang, Y.; Zhang, C.; Zheng, W. ABHD5 inhibits YAP-induced c-Met overexpression and colon cancer cell stemness via suppressing YAP methylation. Nat. Commun. 2021, 12(1), 6711. [Google Scholar] [CrossRef]
Figure 1.
The representative Mer inhibitors.
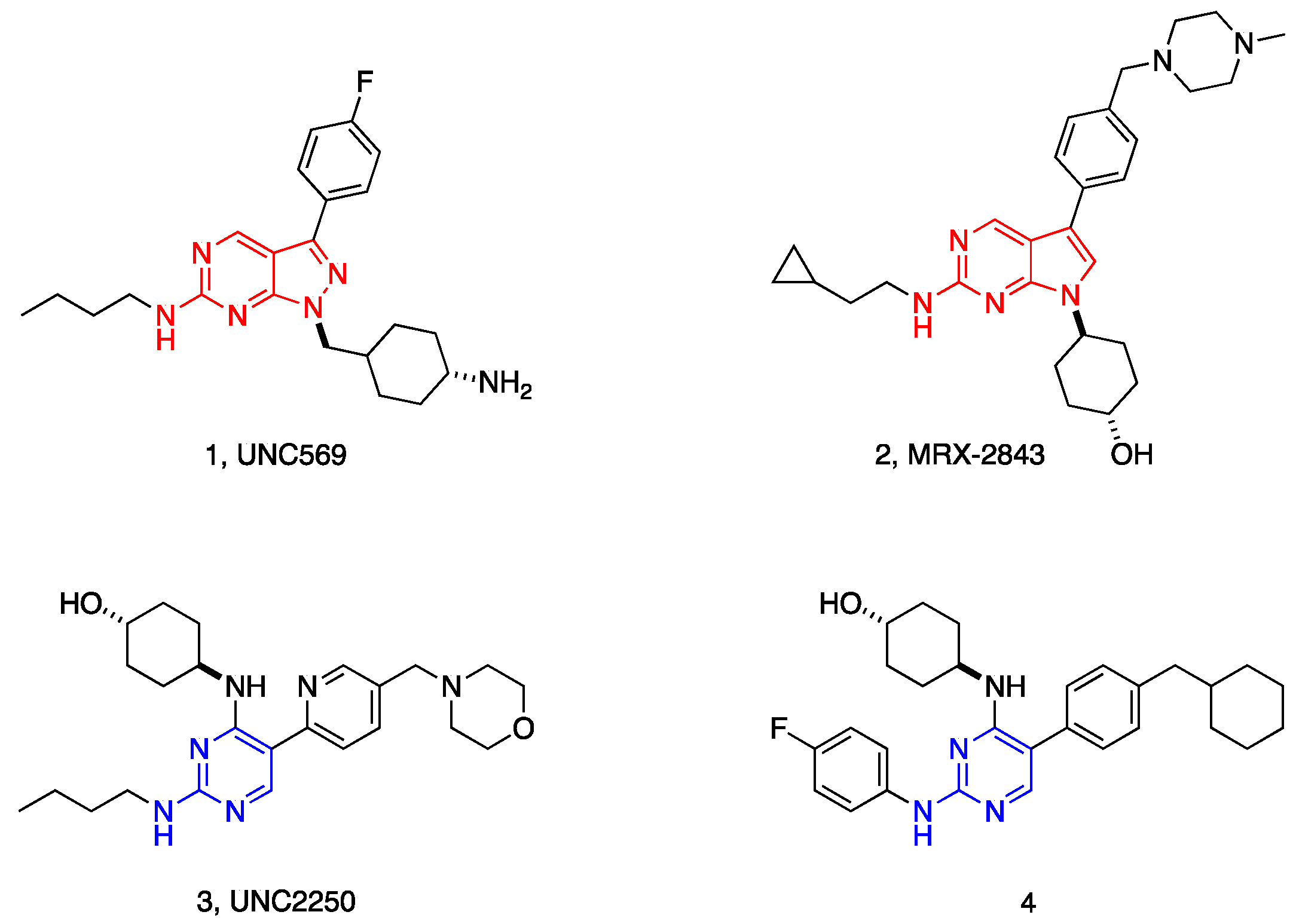
Figure 2.
The representative c-Met inhibitors.
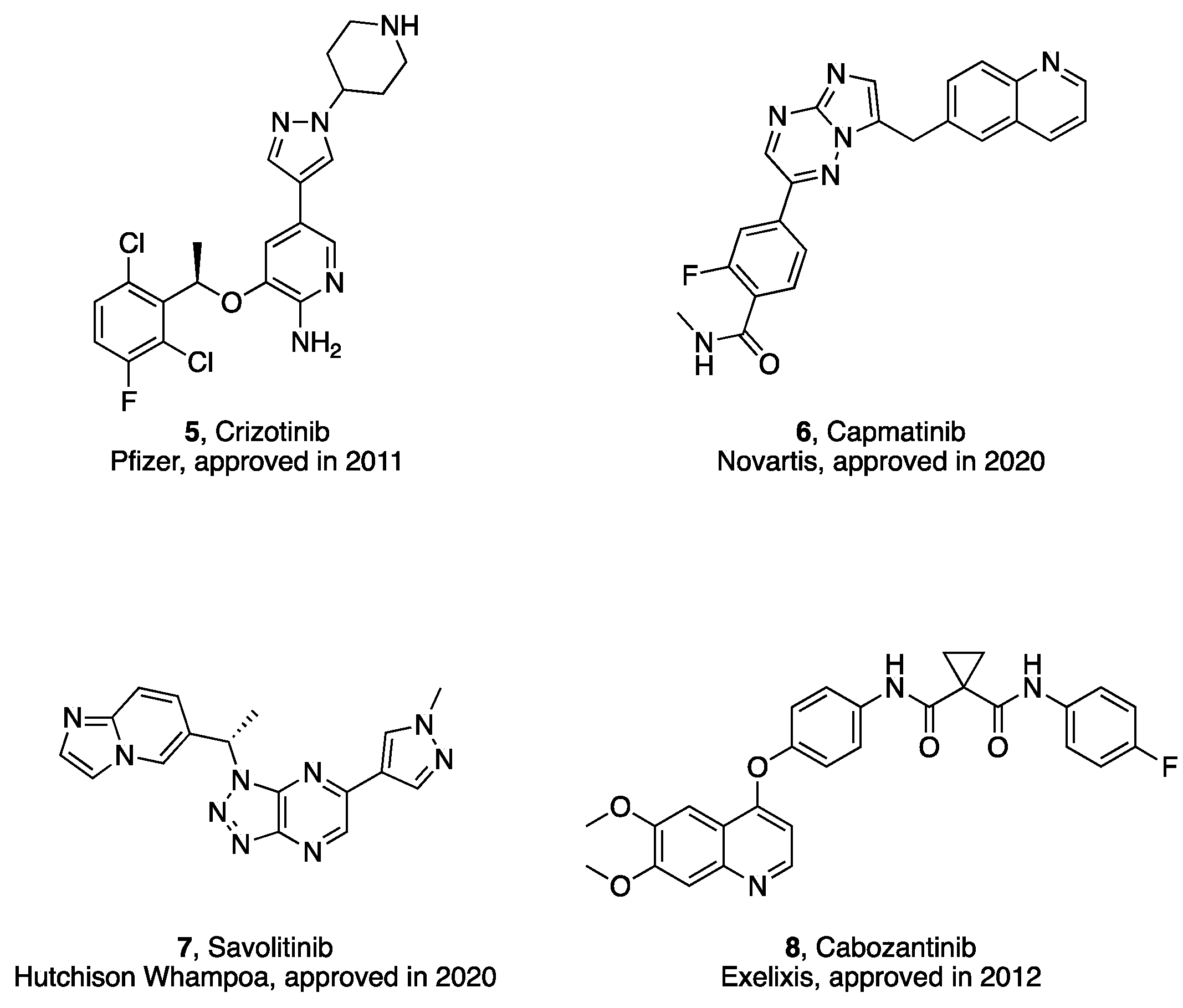
Figure 3.
The docking mode of cabozantinib with Mer kinase (PDB: 4M3Q), A) 3D binding mode; B) 2D binding mode.
Figure 3.
The docking mode of cabozantinib with Mer kinase (PDB: 4M3Q), A) 3D binding mode; B) 2D binding mode.
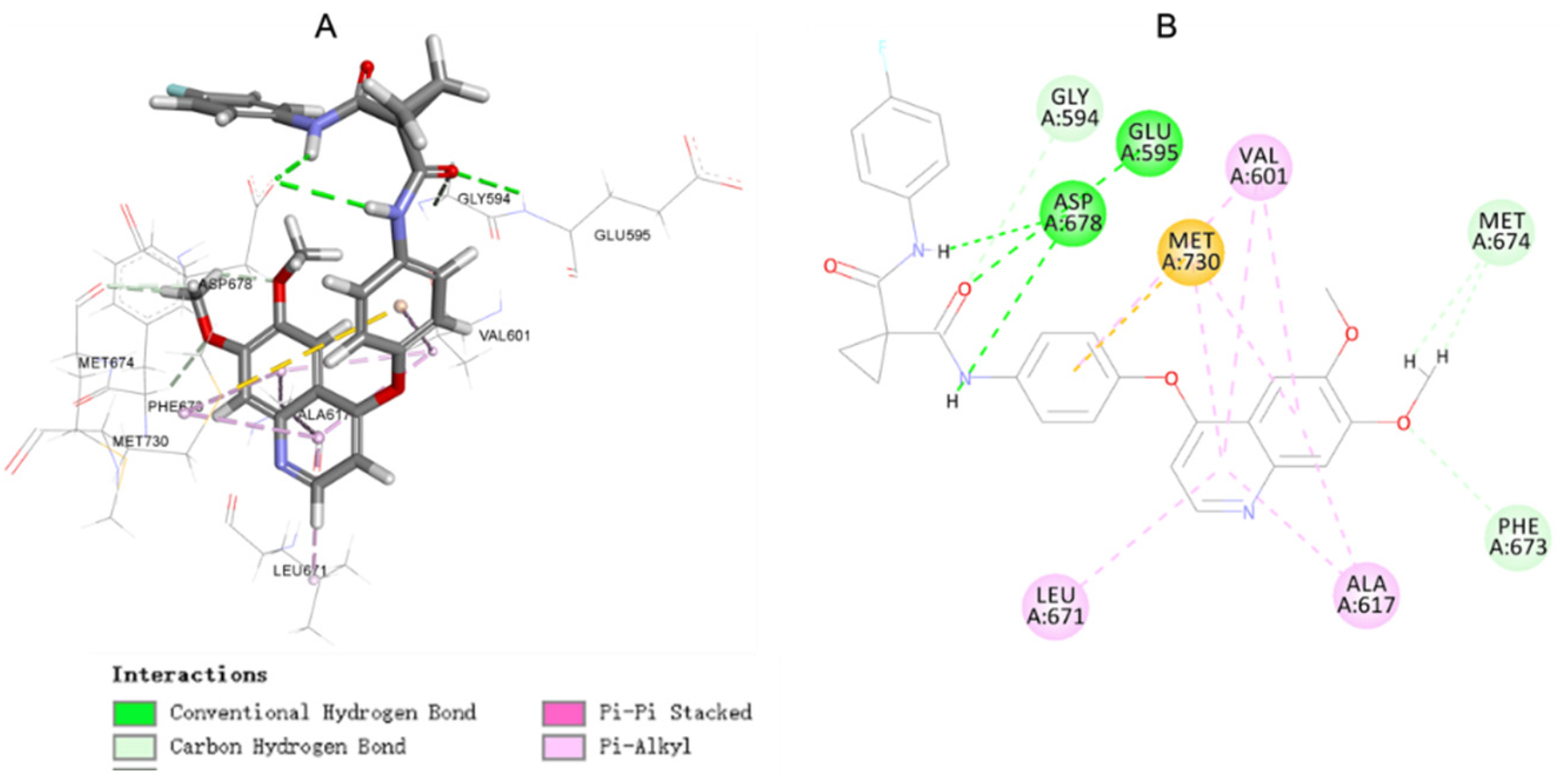
Figure 4.
The docking mode of cabozantinib with c-Met kinase (PDB: 3LQ8), A) 3D binding mode; B) 2D binding mode.
Figure 4.
The docking mode of cabozantinib with c-Met kinase (PDB: 3LQ8), A) 3D binding mode; B) 2D binding mode.
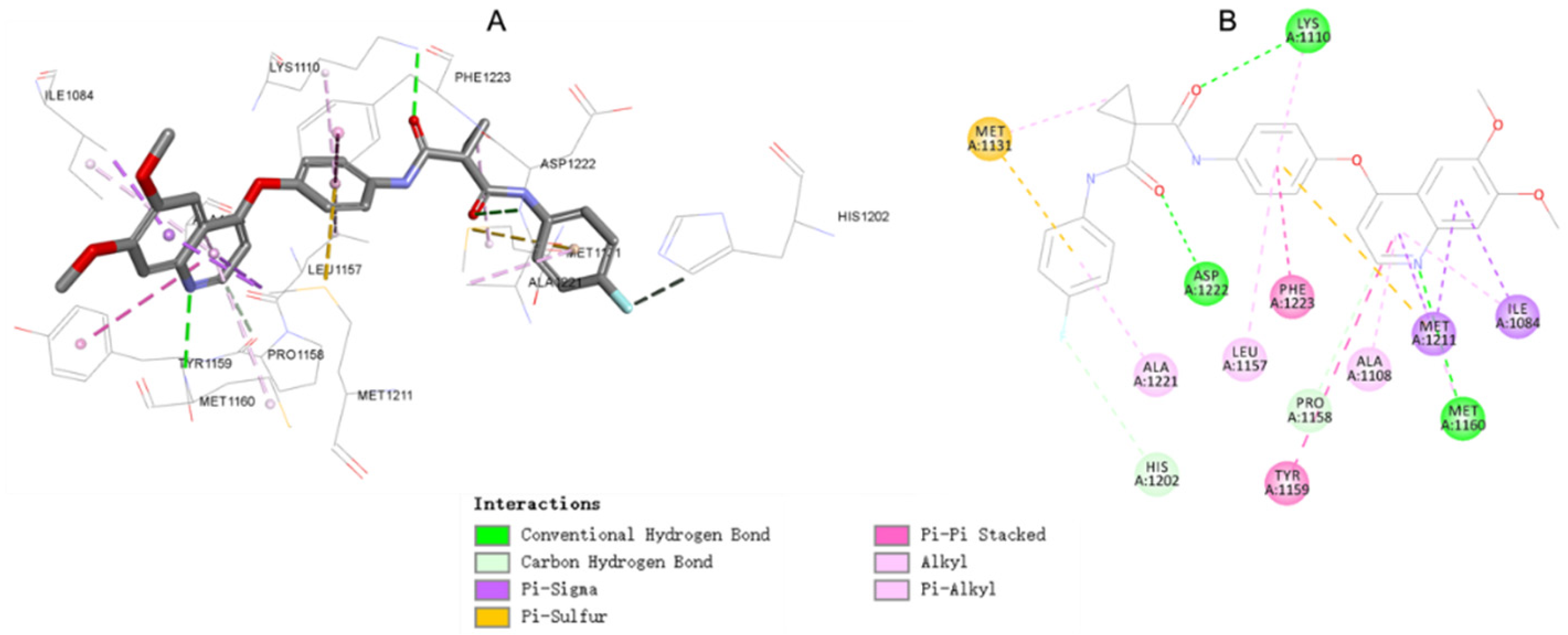
Figure 5.
Design strategy of target compounds.
Scheme 1.
Reagents and conditions: (a) EDC·HCl/DMF, r.t., 6 h; (b) K2CO3/DMF, 80 °C, 6 h; (c) PTSA/DMF, 90 °C, 4 h.
Scheme 1.
Reagents and conditions: (a) EDC·HCl/DMF, r.t., 6 h; (b) K2CO3/DMF, 80 °C, 6 h; (c) PTSA/DMF, 90 °C, 4 h.
Scheme 2.
Reagents and conditions: (a) HBTU/TEA/DMF, r.t., 8 h; (b) K2CO3/DMF, 80 °C, 4.5 h; (c) PTSA/DMF, 90 °C, 4.0 h.
Scheme 2.
Reagents and conditions: (a) HBTU/TEA/DMF, r.t., 8 h; (b) K2CO3/DMF, 80 °C, 4.5 h; (c) PTSA/DMF, 90 °C, 4.0 h.
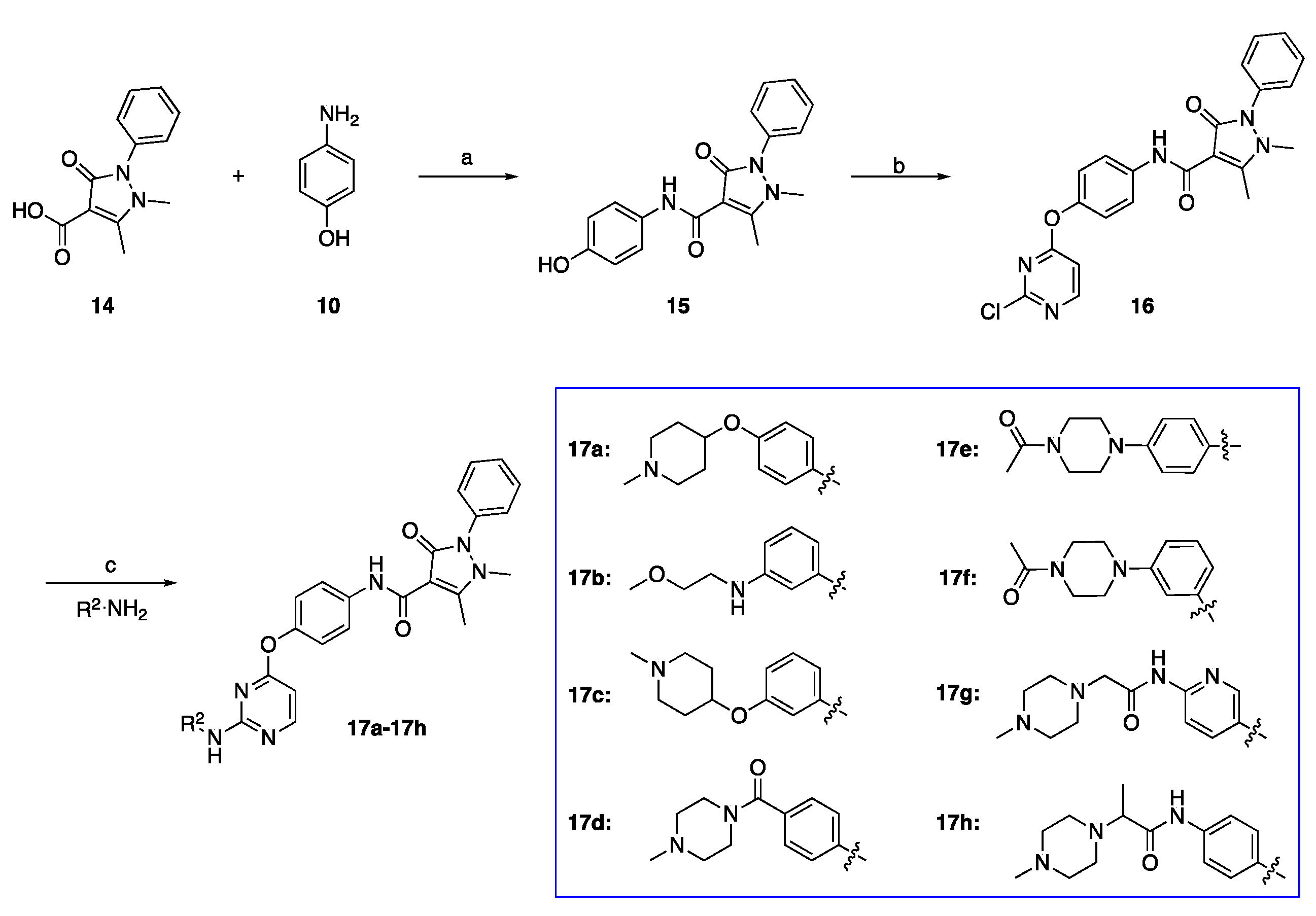
Figure 6.
SAR study of the designed compounds with side chain A.
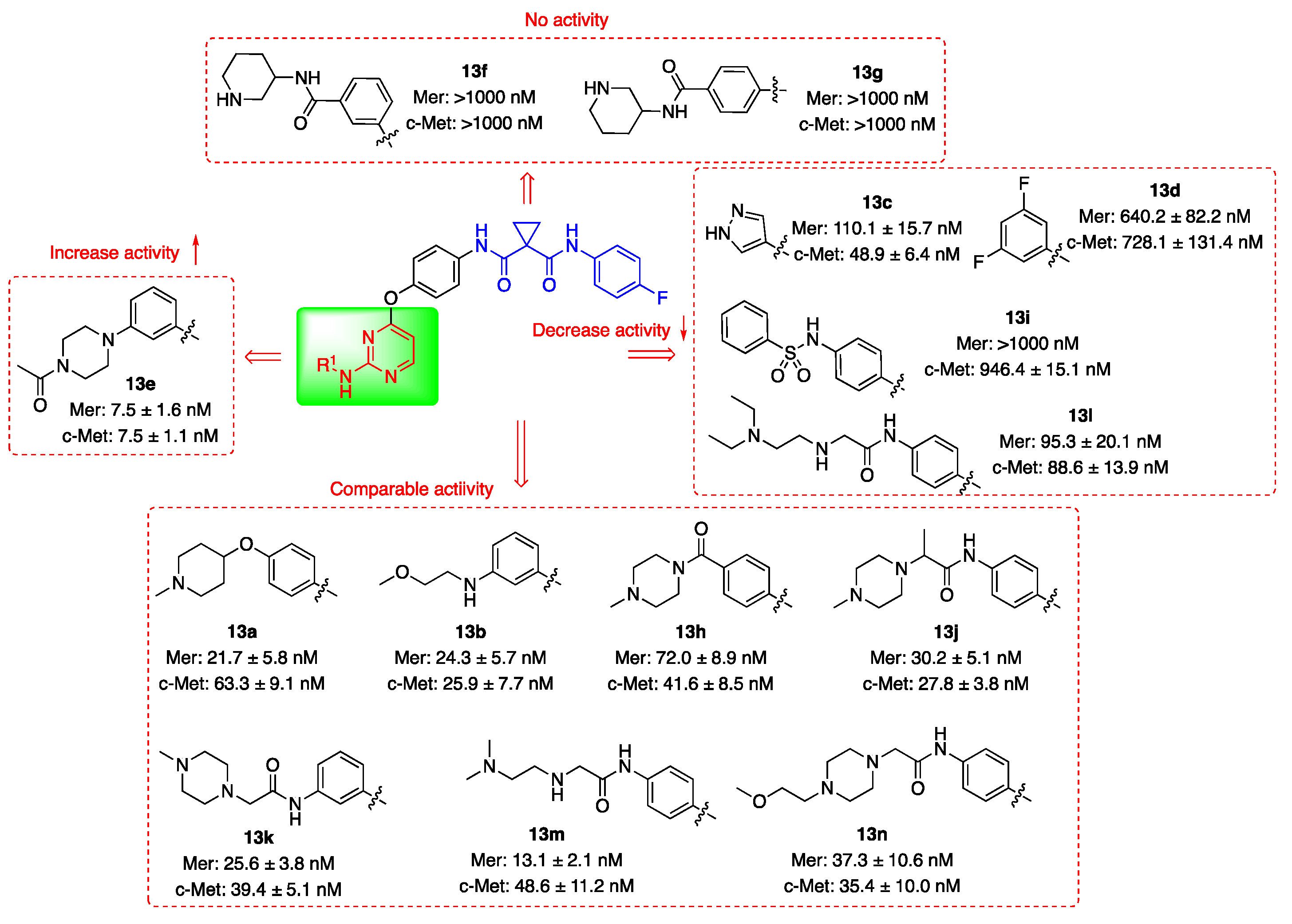
Figure 7.
SAR study of the designed compounds with side chain B.
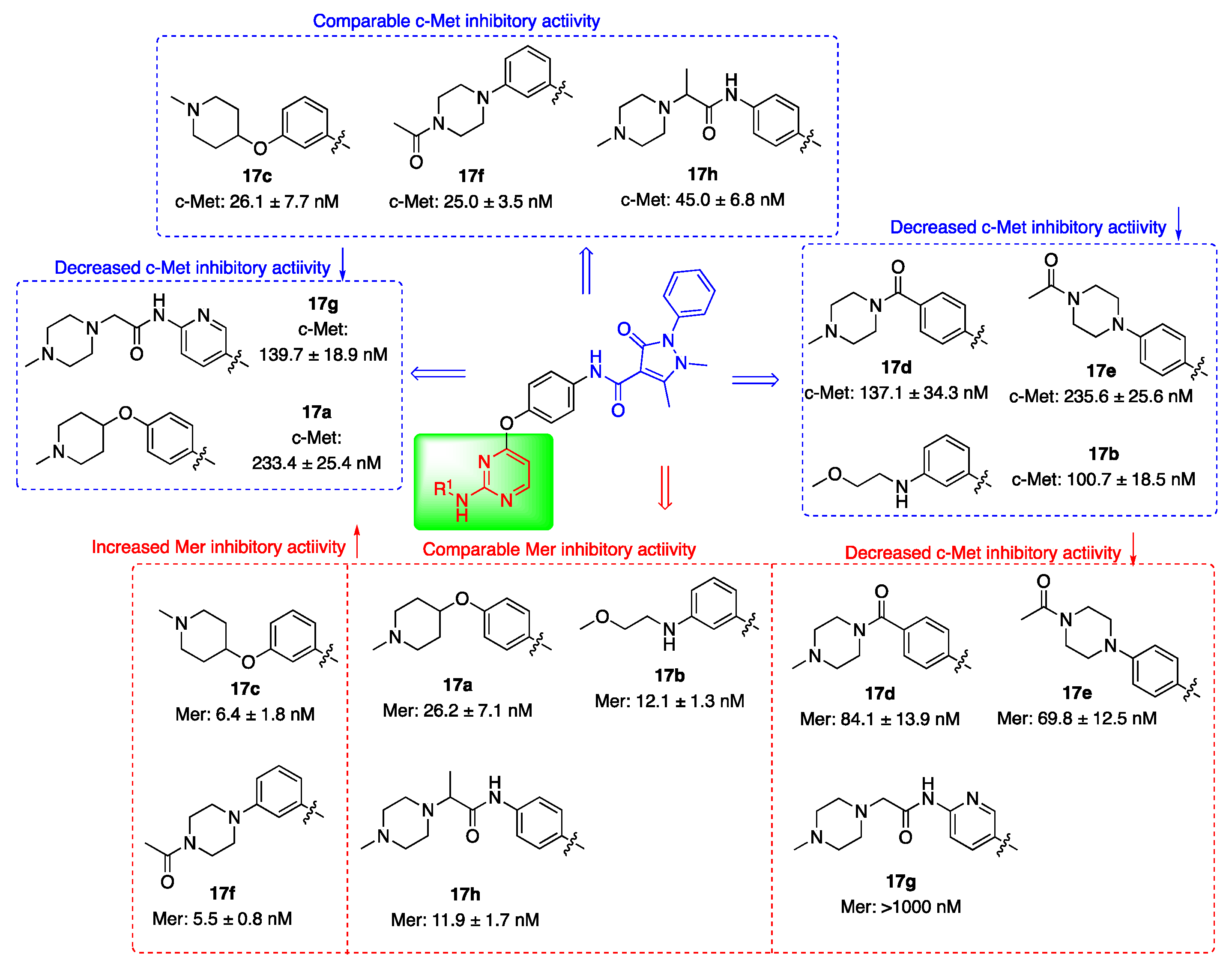
Figure 8.
The docking mode of 17c with Mer kinase (PDB: 4M3Q). A) 3D binding mode; B) 2D binding mode.
Figure 8.
The docking mode of 17c with Mer kinase (PDB: 4M3Q). A) 3D binding mode; B) 2D binding mode.
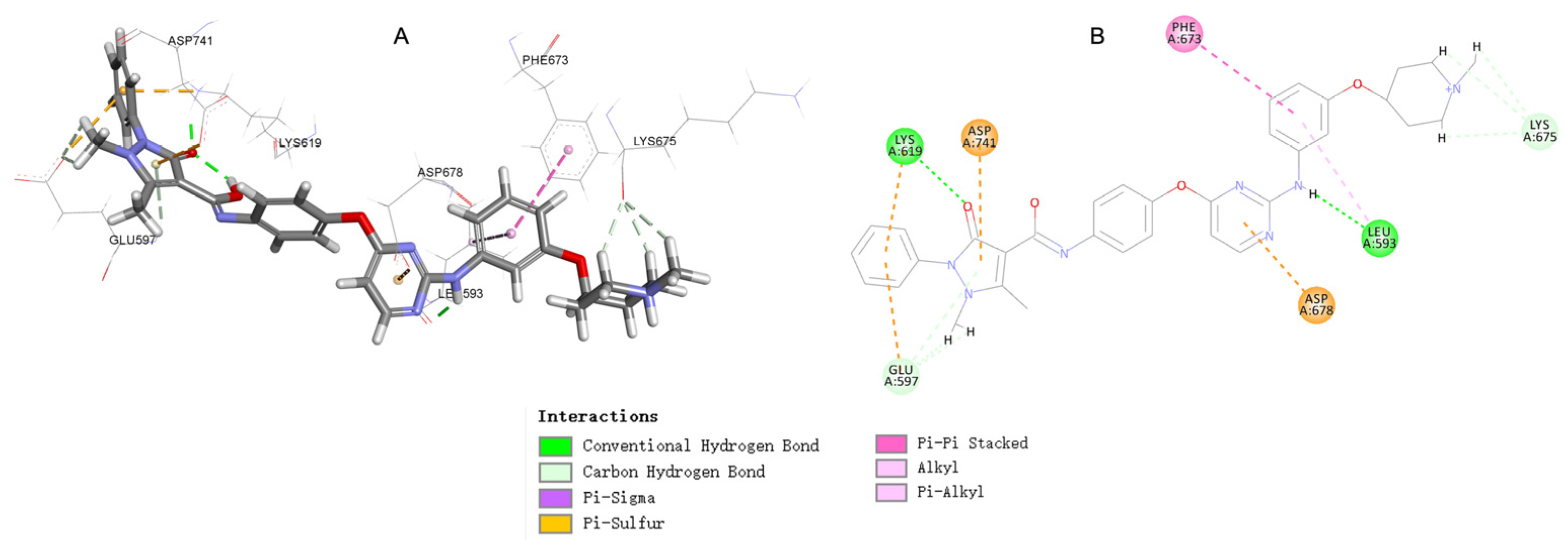
Figure 9.
The docking mode of 17c with c-Met kinase (PDB: 3LQ8), A) 3D binding mode; B) 2D binding mode.
Figure 9.
The docking mode of 17c with c-Met kinase (PDB: 3LQ8), A) 3D binding mode; B) 2D binding mode.
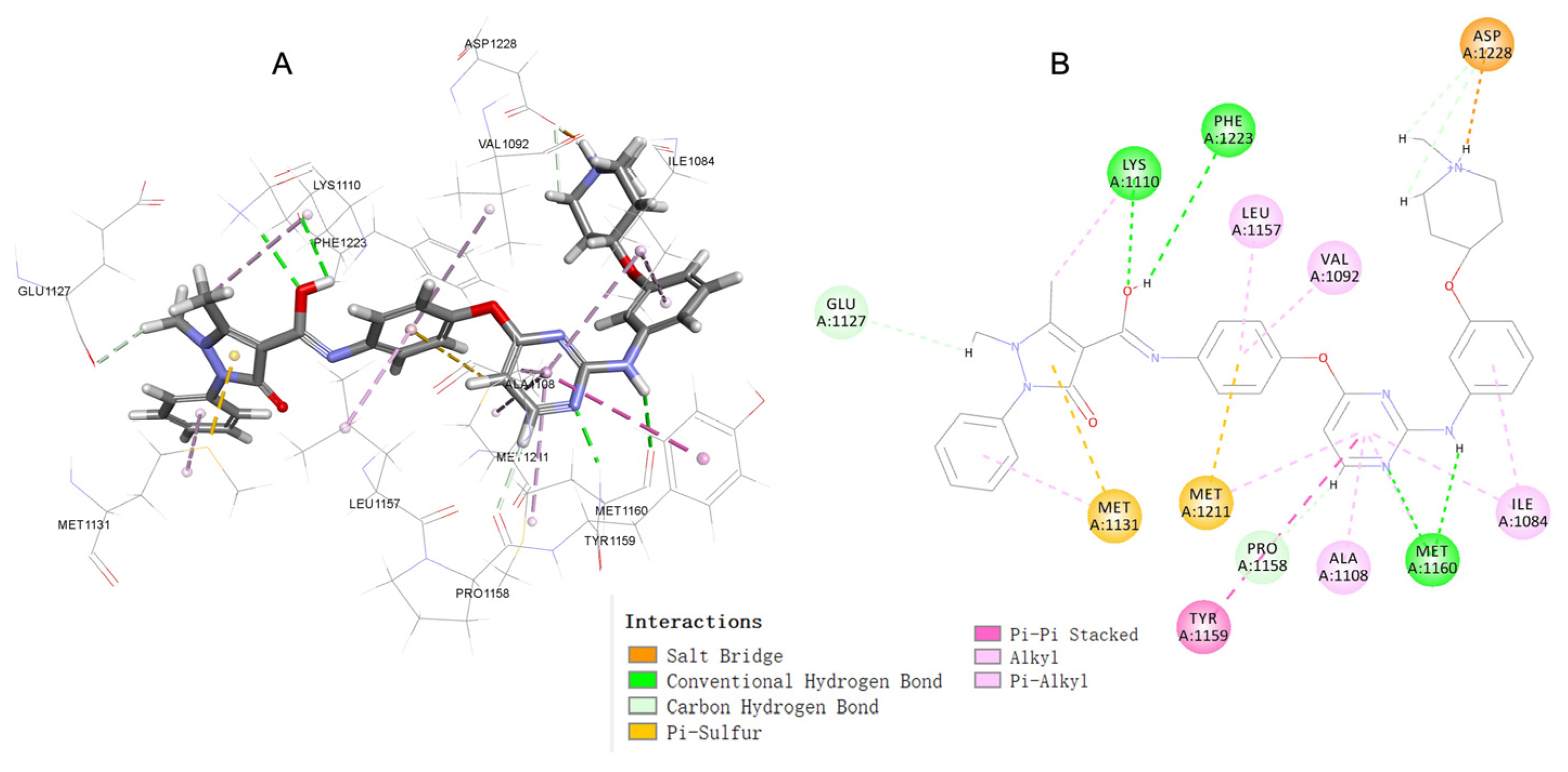
Figure 10.
Western blot analysis was applied to analyze the expression and phosphorylation of c-Met and MERTK in HCT116 cells with staurosporine or increasing doses of compound 17c. All experiments were performed at least three times.
Figure 10.
Western blot analysis was applied to analyze the expression and phosphorylation of c-Met and MERTK in HCT116 cells with staurosporine or increasing doses of compound 17c. All experiments were performed at least three times.
Figure 11.
Compound 17c induced cell apoptosis in HCT116 cancer cell. (A) Quantitative analysis of TUNEL staining. (B) Representative TUNEL and DAPI-stained images. Magnification, x100. HCT116 cells grown on cover slip, treated with various indicated dose of 17c for 48 h, and stained for TUNEL (green). The number of TUNEL-positive cells were counted from 5 non-overlap random fields per group. DAPI (blue). Data are representative of three independent experiments.
Figure 11.
Compound 17c induced cell apoptosis in HCT116 cancer cell. (A) Quantitative analysis of TUNEL staining. (B) Representative TUNEL and DAPI-stained images. Magnification, x100. HCT116 cells grown on cover slip, treated with various indicated dose of 17c for 48 h, and stained for TUNEL (green). The number of TUNEL-positive cells were counted from 5 non-overlap random fields per group. DAPI (blue). Data are representative of three independent experiments.
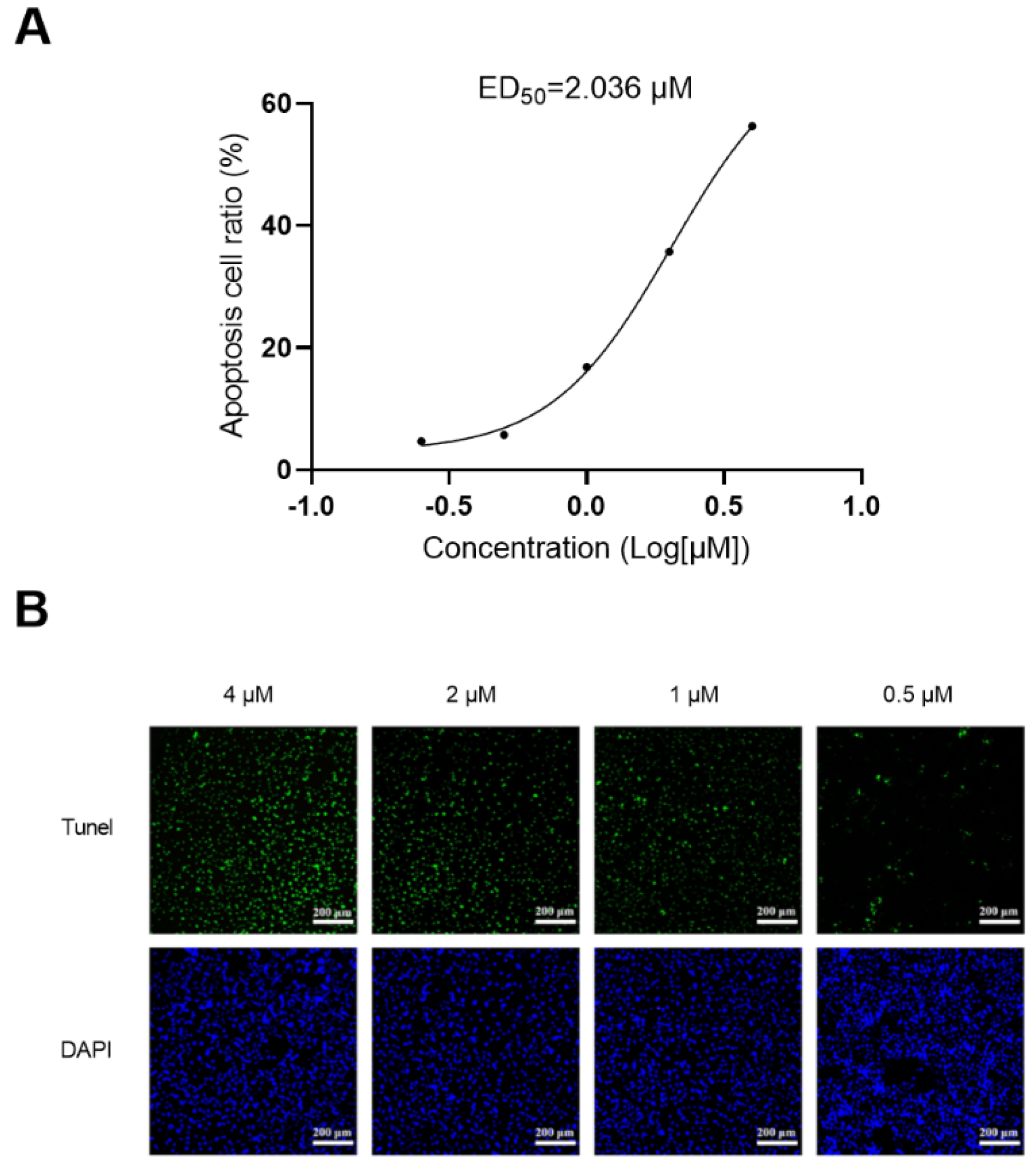
Figure 12.
Compound 17c inhibited cell migration in HCT116 cancer cell. (A) Representative images of transwell assay of HCT116 cells treated with compound 17c for 24 h. (B) Quantification of transwell assays.
Figure 12.
Compound 17c inhibited cell migration in HCT116 cancer cell. (A) Representative images of transwell assay of HCT116 cells treated with compound 17c for 24 h. (B) Quantification of transwell assays.
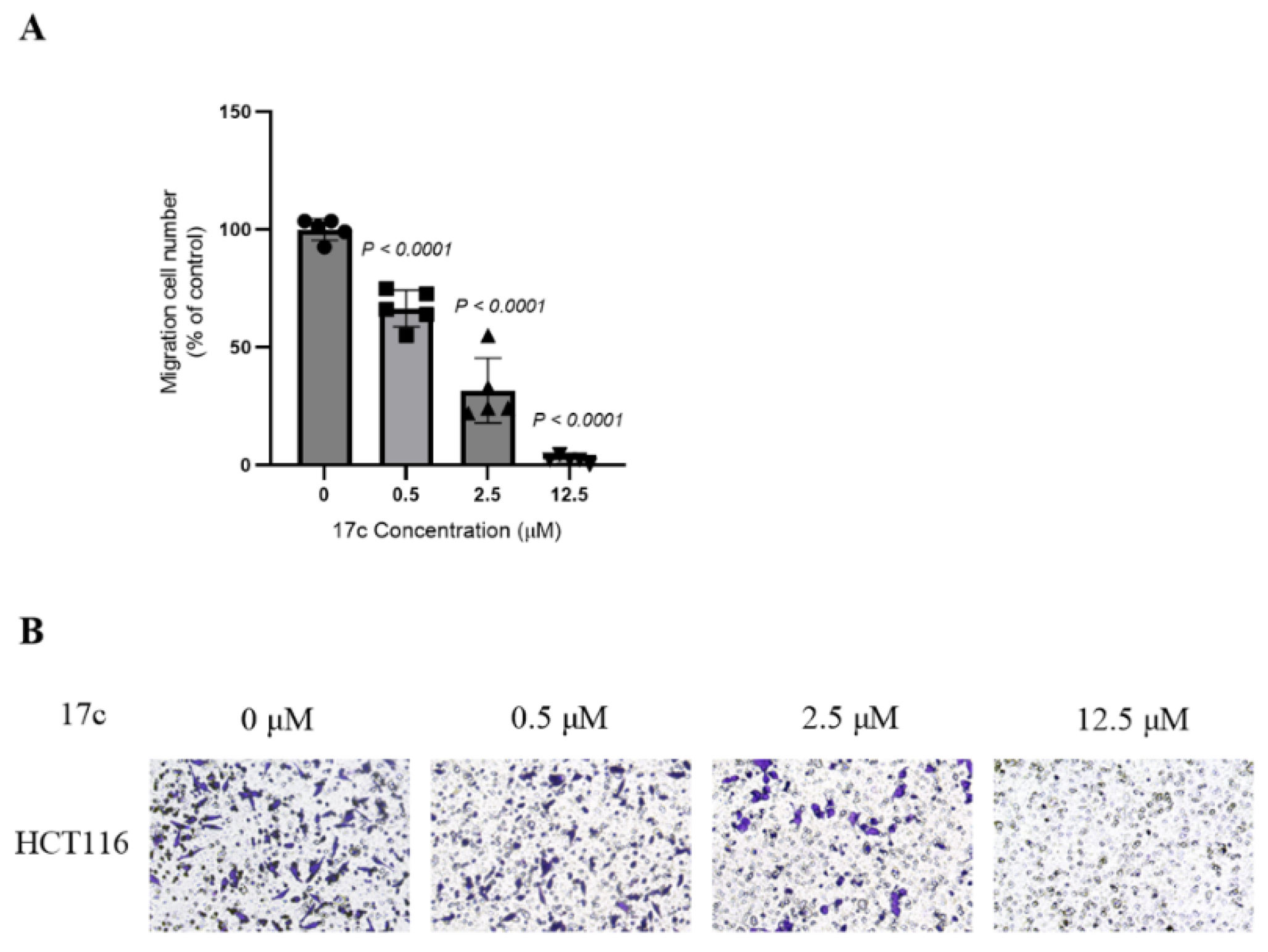

Table 1.
Inhibitory activities of compound 13a–13n against Mer and c-Met kinase.
| Compd. | R1 | IC50 (nM)1 | |
| Mer | c-Met | ||
| 13a | 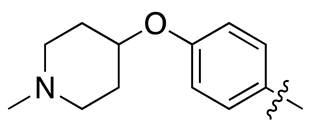 |
21.7 ± 5.8 | 63.3 ± 9.1 |
| 13b | 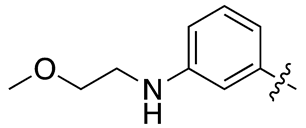 |
24.3 ± 5.7 | 25.9 ± 7.7 |
| 13c | 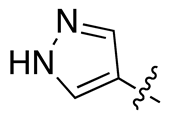 |
110.1 ± 15.7 | 48.9 ± 6.4 |
| 13d | 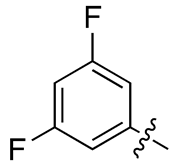 |
640.2 ± 82.2 | 728.1 ± 131.4 |
| 13e | 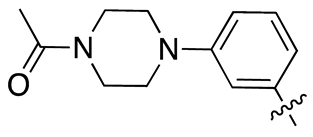 |
7.5 ± 1.6 | 7.5 ± 1.1 |
| 13f | 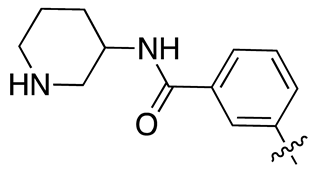 |
>1000 | >1000 |
| 13g | 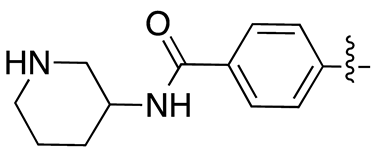 |
>1000 | >1000 |
| 13h | 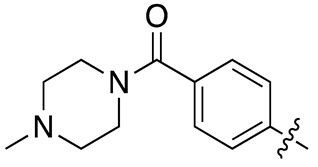 |
72.0 ± 8.9 | 41.6 ± 8.5 |
| 13i | 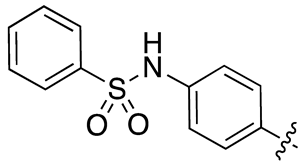 |
>1000 | 946.4 ± 15.1 |
| 13j | 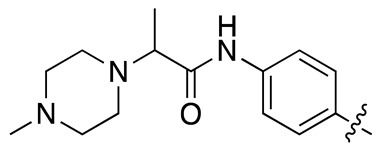 |
30.2 ± 5.1 | 27.8 ± 3.8 |
| 13k | 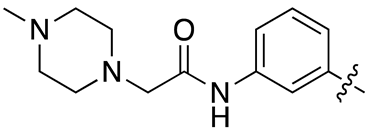 |
25.6 ± 3.8 | 39.4 ± 5.1 |
| 13l | 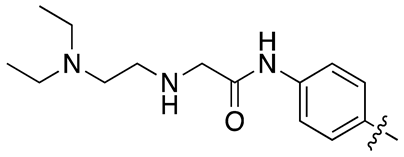 |
95.3 ± 20.1 | 88.6 ± 13.9 |
| 13m | 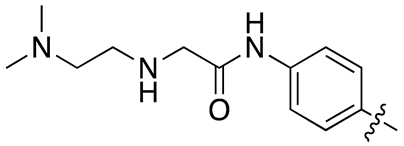 |
13.1 ± 2.1 | 48.6 ± 11.2 |
| 13n | 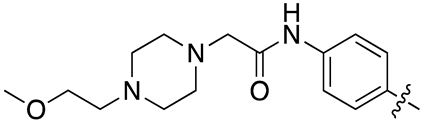 |
37.3 ± 10.6 | 35.4 ± 10.0 |
| 18c | 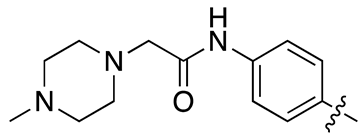 |
18.5 ± 2.3 | 33.6 ± 4.3 |
1 Data are means from three independent experiments.
Table 2.
Inhibitory activities of compound 17a–17h against Mer and c-Met kinase.
| Compd. | R2 | IC50 (nM)1 | |
| Mer | c-Met | ||
| 17a | 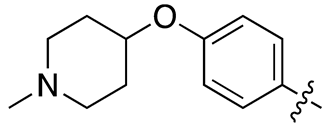 |
26.2 ± 7.1 | 233.4 ± 25.4 |
| 17b | 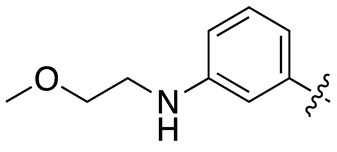 |
12.1 ± 1.3 | 100.7 ± 18.5 |
| 17c | 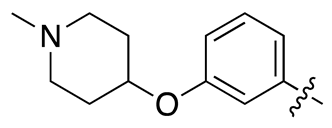 |
6.4 ± 1.8 | 26.1 ± 7.7 |
| 17d | 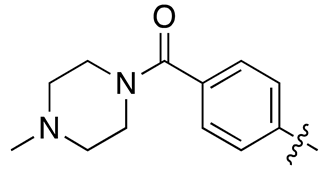 |
84.1 ± 13.9 | 137.1 ± 34.3 |
| 17e | 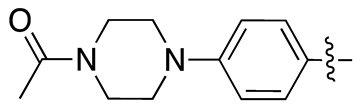 |
69.8 ± 12.5 | 235.6 ± 25.6 |
| 17f | 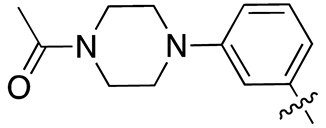 |
5.5 ± 0.8 | 25 ± 3.5 |
| 17g | 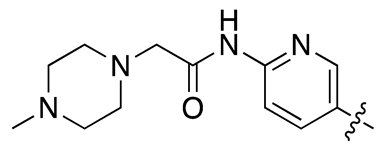 |
>1000 | 139.7 ± 18.9 |
| 17h | 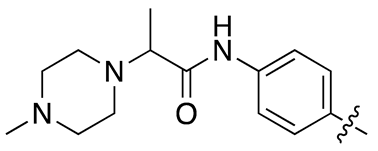 |
11.9 ± 1.7 | 45.0 ± 6.8 |
| 18c | 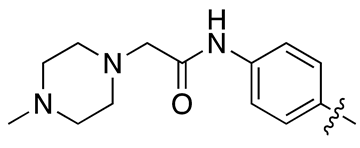 |
18.5 ± 2.3 | 33.6 ± 4.3 |
1 Data are means from three independent experiments.
Table 3.
Microsome stability study of compound 13b, 13e, 13j, 13k, 13m, 13n, 17c, and 17f.
| Cpd. | HUMAN | |
|---|---|---|
| T1/2 (min) | CL (mL/min/mg) | |
| 13b | 38.3 | 0.0905 |
| 13e | 30.4 | 0.1142 |
| 13j | 11.9 | 0.2912 |
| 13k | 8.6 | 0.4046 |
| 13m | 27.9 | 0.1521 |
| 13n | 38.4 | 0.1243 |
| 17c | 147.0 | 0.0236 |
| 17f | 51.0 | 0.0680 |
| Testosterone | 32.1 | 0.11 |
Table 4.
Cytotoxic activities of compound 17c on cancer cell lines in vitro.
| Cpds | IC50(μM)1 of 3 cell lines | |||
|---|---|---|---|---|
| HepG2 | MDA-MB0231 | HCT116 | ||
| 17c | 0.46 ± 0.06 | 2.31 ± 0.67 | 3.79 ± 1.09 | |
| Cabozatinib | 7.87 ± 2.11 | 4.62 ± 0.54 | 15.57 ± 4.39 | |
1 Data are means from three independent experiments.
Table 5.
Activity on hERG potassium currents of compound 17c.
| Cpd. | IC50 (μM) |
| 17c | >40 |
| Cisapride | 0.04 |
Table 6.
Stability recovery of compound 17c and positive control in plasma.
| Cpd. |
Con. ( μM) |
Species | Mean Fu | Mean Fb |
Stability (%) |
| 17c | 1 | Human | 0.0158 | 98.4% | 99.3% |
| 17c | 1 | Rat | 0.0178 | 98.2% | 102.2% |
| Warfarin | 1 | Human | 0.00877 | 99.1% | 99.8% |
| Warfarin | 1 | Rat | 0.0102 | 99.0% | 98.2% |
Fu=Area ratio of compound in buffer/Area ratio of compound in plasma. Fb=100×(1–Fu).
Table 7.
Pharmacokinetic parameters of compound 17c.
| Adm. | Dose (mg/Kg) |
AUClast (h*ng/mL) |
T1/2 (h) |
Tmax (h) |
Cmax (ng/mL) |
CL_obs (mL/min/kg) | MRTINF_obs(h) | F (%) |
| p.o. | 10 | 4626 | 3.46 | 4.00 | 740 | – | 6.52 | 45.3 |
| i.v. | 2 | 2154 | 3.33 | – | – | 16.4 | 3.23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



