Submitted:
09 October 2024
Posted:
12 October 2024
You are already at the latest version
Abstract
SPH5030 is developed to improve the selectivity of irreversible HER2 inhibitors, which shows excellent activity and better HER2 selectivity against four common HER2 mutants than neratinib and pyrotinib, and consumed to attenuate the off-target adverse events in patients. To comprehensively evaluate SPH5030, the preclinical pharmacokinetic studies of SPH5030 were systematically completed, including in vivo pharmacokinetic, tissue distribution and excretion in Sprague Dawley rats and Cynomolgus monkeys, as well as in vitro drug-drug interaction assessment and metabolite identification in different species. The results showed low clearance, reasonable bioavailability and widely tissues distributed properties, and mainly excreted in the feces in rats. In vitro studies SPH5030 showed stable metabolism and metabolized mainly by CYP3A4/5 and CYP2C8, with no or weak inhibitory potential on CYP450 enzymes and efflux transporters. In Phase I clinical trials, SPH5030 also showed well tolerated at doses ranging from 50 to 600mg and is expected to have an ideal clinical application prospect in the future.
Keywords:
SPH5030
; Selective and irreversible HER2 Inhibitor
; Pharmacokinetic
; Tissues distribution
; drug-drug interaction
1. Introduction
HER2 gene amplification occurs in about 20% of invasive breast cancer, 6% to 37% of gastric cancers and 5% of colorectal cancers. HER2 mutations are detected in about 2.5% of lung cancers, 15%-25% of breast cancers, 20% of gastric cancers, 20% of bile duct cancers, 27% of ovarian cancers and 18%~80% in endometrial cancer. As a diagnostic and prognostic biomarker for breast cancer, HER2 overexpression or HER2 oncogene amplification demonstrates increased tumor aggressiveness, recurrence and poorer survival [1,2]. While the members of human EGFR or HER receptor tyrosine kinase (RTK) subfamily, EGFR (HER1/erbB1), HER2 (neu/erbB2), HER3 (erbB3), and HER4 (erbB4) are structurally conserved with an extracellular ligand-binding domain, a transmembrane domain, and an intracellular kinase domain [3,4].
Currently, there are a variety of approved HER2-targeted therapies for HER2-positive cancer. Monoclonal antibody-based therapies significantly improving outcomes in HER2-positive breast cancer, but these treatments eventually become ineffective due to acquired resistance [6]. HER2-targeting ADC structurally composed of a humanized anti-HER2 antibody, provide valuable therapy with the potential to respond to cytotoxic agents-insensitive breast cancer and other HER2 expressing cancers in clinical practice [7], while interstitial lung disease and pneumonitis, Ocular toxicity, gastrointestinal and hematological toxicity are important risks requiring careful monitoring and prompt intervention [8]. As traditional chemotherapy modality, small molecule tyrosine kinase inhibitors (TKIs) inhibit tyrosine kinase phosphorylation by binding to HER2 intracellular tyrosine kinase and interfering with adenosine triphosphate (ATP) binding, thereby achieving anti-tumor effects through multiple pathways. In addition, small molecule TKIs are widely used in patients with HER2 positive ABC brain metastases and has the potential to hinder tumor penetration and accumulation due to their unique ability to cross the blood-brain tumor barrier or some other biological barrier.
SPH5030 is a novel tyrosine kinase inhibitor targeting HER2, with a modified structure from tucatinib and pyrotinib. It has advantages such as target selectivity of tucatinib and the irreversible inhibitory effect of pyrotinib [9]. SPH5030 can inhibit both constitutive and ligand-induced ErbB signaling, have shown non-cross-resistance with Trastuzumab in preclinical studies and also have promising activity in heavily pretreated metastatic HER2-positive cancer patients [10]. Irreversible inhibitors are covalently bound to cysteine residues in the receptor ATP site (cys797 for EGFR and cys805 for HER2) through an electrophilic structure in the molecule, and thus remain better inhibited against mutant HER2 receptors than reversible inhibitors [11,12,13]. Therefore, irreversible small-molecule tyrosine kinase inhibitors (TKIs) have potential advantages and attract more attention [14].
The objectives of the present work were to characterize the pharmacokinetic, tissue distribution and excretion properties after administration of SPH5030 in Sprague Dawley rats and Cynomolgus monkeys, as well as in vitro drug-drug interaction assessment and metabolite identification in different species. These preclinical studies may shed light on the indication selection and adverse reactions expectation in the future clinical trials.
2. Results
2.1. In Vivo Pharmacokinetic Properties
As shown in pharmacokinetic data, SPH5030 is a low clearance and widely distributed drug after intravenous administration based on plasma clearance of 5.83 and 6.95 L/min/kg and distribution volume of 1.66 and 2.67 L/kg for rats and monkeys, respectively. After oral administration, SPH5030 reached a Cmax at 2–4.5 h, and disappeared with a T1/2 of 4.61~9.14 h in rat and monkey plasma. The oral bioavailability of SPH5030 was about 56.4–64.3% in rats and 18.1–38.0% in monkeys calculated from the dose-corrected average AUC0–24h after iv and po dosing, which indicated that efflux transporters were involved. The main metabolite SPH6849 has similar T1/2 to SPH5030 and the plasma exposure AUC0-t is about 12.7-14.9% and 22-53% of SPH5030 for rats and monkeys. The detailed pharmacokinetic parameters of oral and intravenous administration in rats and monkeys were shown in Table 1.
2.2. Tissue Distribution and Excretion
After orally orally dosed 8 mg/kg in rats, SPH5030 were widely distributed in tissues, with higher exposure in most tissues than plasma exposure. AUC2-48 values of SPH5030 in adrenal gland, spleen, lung and liver tissues were more than 10 times that in plasma, and secondly in ovary, kidney, thymus, pancreas and fat tissues, about 3~8 times SPH5030 AUC2-48h in plasma. Notably, SPH5030 can get cross the blood-brain barrier with the brain/plasma distribution ratio of 0.10-0.12. The Cmax in tissues was 5 hours after SPH5030 administration, and almost disappeared 15 hours after administration, i.e., the concentration in most tissues was below 20% peak concentration. The main metabolite SPH6849 had less tissue-distributed tendency than SPH5030, only AUC0.25–12h values in liver, kidney, small intestine and adrenal gland higher than that in plasma. The blood-to-plasma concentration ratio of SPH5030 and SPH6849 was 0.627 and 0.565, respectively, which showed hardly enter into red blood cells in rats (Figure 1).
After a single oral dose of 8 mg/kg for rats, the cumulative excretion ratios of SPH5030 into feces and urine were 30.7% and 0.21% within 96 h, and the major metabolite SPH6849 accounted for 2.43% into feces, 0.01% into urine. The cumulative excretion of SPH5030 and SPH6849 into bile accounted for 0.30% within 48 h, much less than that in feces. The results indicated that SPH5030 and SPH6849 were mainly excreted through feces, but the possibility of omitting some other main metabolites in feces, urine and bile could not be excluded.
2.3. In Vivo and In Vivo Metabolic Profiles
The data of microsomal stability showed that SPH5030 were moderate hepatic clearance cross-species in mouse, rat, dog, monkey and human, meanwhile the residual rates were 40.6% ~ 59.7% after 60 minutes of incubation (Table 2). In the hepatocytes incubation systems, the tetrahydropyrrole cyclocarbonylation metabolite M8 (SPH6849) was mainly detected in human and monkey hepatocytes, and then there came M2, the amide hydrolysis metabolite. While for dogs, rats and mice, the amide hydrolysis metabolite M2 were mainly detected, only small amount of metabolite M8 were found.
Based on the results of enzymatic phenotype assay, there were two main metabolic pathways in SPH5030, i.e., the CYP450-dependent metabolism of tetrahydropyrrole cyclocarbonylation (M8) and dehydrogenation (M7), and the CYP450-independent metabolism of amide hydrolysis (M2) and monoxide oxidation (M9-2 and M9-3). Furthermore, M7 and M8 is mainly catalyzed by CYP3A4 and CYP3A5, to a lesser extent, catalyzed by CYP2C8. The results above were based on both of the human liver microsome chemical inhibitor method and human recombinant P450 enzyme method.
In vivo studies, after 8mg/kg SPH5030 were dosed in rats, a total of 24 metabolites were identified in plasma, urine, fecal, and bile samples by UPLC-UV/Q-TOF MS, and no human specific metabolite was found. M0 was the main drug in plasma, and then M8 (SPH6849), the tetrahydropyrrole cyclocarbonylation metabolite. In the urine samples, primarily M0 (the parent drug), and then the amide hydrolytic metabolite M2, finally the monoxide metabolites M9-2 and M9-3. In fecal samples, primarily M0, secondly M2, then the tetrahydropyrrole cyclocarbonylation and O-deethylated metabolite M5, finally M8. In bile samples, the primary drug M0, M8 and O-deethylated metabolite M5 were the main substances. In cynomolgus monkey, after single oral dose of 8mg/kg, M0 was the main substance in plasma, secondly, the M8 (SPH6849). The major metabolites of SPH5030 and the mainly metabolic pathways are shown in Figure 2.
2.4. In Vivo Drug-Drug Interaction Assay
The in vitro CYP inhibition study only found weak inhibitory effects with IC50 of 16.0 μM, 17.1 μM and 20.7 μM for CYP2C8, CYP3A4/5 (1′-hydroxyl-Midazolam) and CYP3A4/5 (6β-hydroxy-Testosterone), respectively, which were much higher than human plasma Cmax even at the recommended clinical dose of 400 mg (Table 3).
In vitro CYP induction study detected both of enzyme activities and mRNA expression in human primary hepatocytes. There was no concentration-dependent increase in the enzyme activities of CYP1A2, CYP2B6, and CYP3A4 after treated by SPH5030, but showed the increase of CYP3A4 enzyme mRNA, i.e., more than 2-fold of the vector control and greater than 20% of the positive control rifampicin. So it is deemed a possible induction of CYP3A4 enzyme after incubation with SPH5030 (Table 4). The concentration of SPH5030 did not decrease after multiple consecutive doses in rats and monkeys. Therefore, the risk of CYP3A4 enzyme induction of SPH5030 remains low till now and need further validation in the clinical trials.
Numerous studies showed that the transporter might affect the drug uptake and in vivo disposition process, thus affecting the absorption and excretion of the drug. The Papp values of SPH5030 on different modality cells are demonstrated in Table 5, which indicated the low permeability of SPH5030 (10.0, 50.0 µM) from the apical to basal side based on the criteria of lower Papp, A → B than acyclovir (the positive control for low permeability compounds, 0.77 × 10-6 cm/s). SPH5030 is also an efflux transporter substrate, as the significantly decreased efflux ratio (from 12.37 to 0.66) after co-administered with GF120918 (the inhibitor of P-gp and BCRP).
In the MDR1/BCRP-transfected MDCK cell models, after co-administered with GF120918 or Ko143, the efflux ratio of SPH5030 decreased, further indicating that SPH5030 is the minor substrate of both P-gp and BCRP, with efflux ratio of 7 and 2.72 (Table 5). SPH5030 is also the weak inhibitor of efflux transporters P-gp and BCRP, when digoxin (P-gp) and imatinib (BCRP) were efflux transporter substrates in MDCK-MDR1 and MDCK-BCRP cells.
In the uptake transporter studies, we found a concentration-dependent inhibition of MATE1 and MATE2-K by SPH5030 with IC50 of 1.68 and 0.496 µM, respectively. SPH5030 also has been characterized as a weak inhibitor of OATP1B1 and OATP1B3 with IC50 values > 50 μM, and not a substrate of OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1 and MATE2-K. It is clinically recommended to further evaluate the possibility of drug-drug interactions due to inhibition of MATE1 and MATE2-K based on plasma exposure.
2.5. Interspecies Simple Allometric Scaling of CL and Vdss
Assuming a one-compartment model for the prediction of danicamtiv human pharmacokinetics, the estimated plasma pharmacokinetic parameters of CL obtained from preclinical intravenous pharmacokinetic studies were used to predict human CL using simple allometric interspecies scaling. Simple allometric scaling of unbound plasma CL from rat and cynomolgus monkey led to a predicted human plasma clearance (total) of 0.75 mL/min/kg with an allometric exponent of 1.0763 (Figure 3). When using two species for the allometric growth model method, the model fitting is not ideal, resulting in significant uncertainty in the allometric amplification model. Therefore, a single genus allometric growth model method was adopted, and based on the average results of the two genera, the CL of the human body was calculated to be 8.71 L/h. In addition, the in vivo-in vitro extrapolation (IVIVE) method was used to predict a human clearance rate of 16.67 L/h based on human liver microsomes metabolism data. Therefore, the predicted human clearance rate is 12.69 L/h.
The predicted apparent clearance rate (CL/F) of SPH5030 is 21.15 L/h, the apparent volume of distribution (V/F) is 216.5 L, and the absorption rate constant Ka is 0.5011. Based on the predicted results of human PK parameters, the human drug duration curve after oral administration of 100mg SPH5030 in a simulated human body is shown in Figure 5. At this dose, the peak concentration of the first administration is about 310 ng/mL, the steady-state valley concentration of Cmin, ss is about 55 ng/mL.
3. Discussion
SPH5030 is a novel tyrosine kinase inhibitor targeting HER2, which has advantages of better target selectivity and the irreversible inhibitory effect. SPH5030 inhibits the kinase activity of HER2 and EGFR at IC50 values of 3.51 nM and 8.13 nM and is an irreversible HER2 inhibitors with better HER2 selectivity, of which the IC50 values of HER2 inhibitory activity is 23 times smaller than Neratinib and 21.1 times than Pyrotinib [9]. Furthermore, SPH5030 shows better cell proliferation inhibitory activities in cell-based assays with IC50 NCI-N87 of 1.09 nM and IC50BT-474 of 2.01 nM, and assumed to attenuate the off-target adverse events in patients.
In vitro and in vivo preclinical pharmacokinetics studies show good druggability and less drug-drug interactions in SPH5030, which is expected a good clinical application prospects in the future. In rats and monkeys, SPH5030 exhibits a low clearance and widely tissue distributed drug characteristics under intravenous and oral administration. Besides, SPH5030 was the major circulating entity in rats and monkeys and no gender differences in drug plasma exposure were observed. The oral bioavailability of SPH5030 is about 56.4–64.3% in rats and 18.1–38.0% in monkeys, and the elimination half-life (T1/2) 4.61~9.14 h in rats and monkeys. M8 is detected in plasma about 12.7-14.9% and 22-53% of SPH5030 for rats and monkeys, respectively. As a low solubility and low permeability compound, as well as the weak efflux transporter substrate and inhibitor, SPH5030 can be corroborated by non-linear oral exposure across some ranges of the dose. As is reported in 2024 ACSO annual meeting abstracts [20], in Phase I clinical trials, SPH5030 plasma exposures from single-dose and steady-state showed a proportional dose increase, but absorption of SPH5030 was found to reach a plateau from 400 to 600mg.
As to the tissue distribution, SPH5030 is widely distributed and maintain a high concentration in the target organ with peak tissue concentration of about 5 hours after dosing. AUC2-48 values of SPH5030 in lung and liver tissues were more than 10 times, and in ovary, kidney, pancreas and fat tissues, about 3~8 times that of plasma. These pharmacokinetic properties of SPH5030 allow for a reasonable dosing frequency and reduction in the dose administered. Therefore, SPH5030 can be considered for treatment of lung and ovarian cancers related to HER2-positive or HER2 mutations. Furthermore, SPH5030 can cross the blood-brain barrier and is distributed in brain tissue, which also has the potential to be used in the patients with brain metastases. Meanwhile, SPH5030 exhibited good efficacy effects in the clinical trials [20]. Of the 28 evaluable patients for efficacy, 6 pts (21.4%) achieved partial response (PR, breast cancer (BC) = 5, colorectal cancer (CRC) = 1), while 16 patients (57.1%) achieved stable disease (4 patients SD≥ 24 weeks,14.3%). Objective response rate (ORR) was 21.4%, disease control rate was 78.6%, and clinical benefit rate was 35.7%. Three patients in the 600 mg dose group achieved PR, with ORR of 50.0%.
Finally, the in vitro drug-drug interaction studies of SHP5030 were preliminary assessed, due to the fact that there are DDI problems in most of the HER2-targeting inhibitors, such as Tucatinib, Pyrotinib and Rapatinib. Once orally absorbed, SPH5030 was eliminated through extensive oxidative metabolism and amide hydrolysis metabolism, and to a lesser extent as an unchanged drug excreted through feces, urine and hepatobiliary routes, which accounted for approximately 30.91% of the dose in rats. SPH5030 undergoes oxidative metabolism by P450 isoenzymes, of which M7 and M8 is mainly catalyzed by CYP3A4 and CYP3A5, to a lesser extent, CYP2C8. In vitro study showed weak inhibitory effects with IC50 of 16.0 μM, 17.1 μM and 20.7 μM for CYP2C8, CYP3A4/5 (1’-hydroxyl-Midazolam) and CYP3A4/5 (6β-hydroxy-Testosterone). At the recommended clinical dose of 400 mg, the safety margin of CYP3A4 inhibition is at least 5~7 times higher than human plasma Cmax (Table 3). Although in vitro induction study showed increased tendency in CYP3A4 enzyme mRNA level after treatment of SPH5030, no concentration-dependent increase of the enzyme activities of CYP3A4 was found in vitro study. Furthermore, the concentration of SPH5030 did not decrease after multiple consecutive dosing both in rats and monkeys. Therefore, the risk of CYP3A4 enzyme induction of SPH5030 remains low and need further validation in the clinical trials. These characteristics may exclude most of the possible drug-drug interaction in clinical trails.
Phenotyping experiments demonstrated that CYP3A4 is the most active enzyme responsible for the biotransforma tion of SPH5030. Considering that CYP3A4 has a broad substrate specificity and a number of multiple-medication drugs were mainly metabolized by CYP3A4, it is worth monitoring the effects of co-administrated CYP3A4 inhibitors, inducers or substrates in future clinical trials [21]. Besides, SPH5030 is the inhibitor of uptake transporters MATE1 and MATE2-K, and not the substrate for OAT1, OAT3. As MATE1 and MATE2-K transporters are always involved in cationic drug renal tubular secretion in concert with OAT1, which partially explains the reason for less excretion through urine of SPH5030.
Allometric scaling for the prediction of human CL and Vdss of SPH5030 was based on interspecies simple allometric scaling of rat and cynomolgus monkey intravenous pharmacokinetic parameters [17]. Since the distribution phase rates of SPH5030 far exceeded the terminal elimination rates for each of the preclinical species tested, we assumed a one-compartment model for the prediction human pharmacokinetics [22,23]. Prediction of human CL was performed by extrapolating preclinical species unbound plasma intravenous clearance, calculated based on experimental determination of plasma protein binding fraction unbound (fu,p) for each species including human. Results from this simple allometric scaling method provided a CLp of 0.75 mL/min/kg with an allometric exponent of 1.0763. To eliminate the significant uncertainty in the allometric amplification model, a single genus allometric growth model method was adopted. Based on the average results of the two genera, the CL of the human body was calculated to be 8.71 L/h. In addition, the in vivo-in vitro extrapolation (IVIVE) method was used to predict a human clearance rate of 16.67 L/h based on human liver cell metabolism data. Therefore, the predicted human clearance rate is 21.15 L/h, and the apparent volume of distribution (V/F) is 216.5 L, and the absorption rate constant Ka is 0.5011. Based on the predicted results of human PK parameters, after oral administration of 100mg SPH5030, the peak concentration of the first administration was about 310 ng/mL, the steady-state valley concentration of Cmin, ss were about 55 ng/mL.
Based on the report in clinical trials, SPH5030 was well tolerated at doses ranging from 50 to 600mg.Treatment-emerged adverse events (TEAEs) and ≥grade (G) 3 TEAEs occurred in 30 pts (100.0%) and 10 pts (33.3%), respectively. The most common TRAEs (≥20%) were diarrhea (66.7%, 16.7% ≥ G3), creatinine increased (30.0%, 0% ≥ G3), hypokalemia (23.3%, 3.3% ≥ G3), fatigue (23.3%, 3.3% ≥ G3), AST increased (20.0%, 0% ≥ G3), and bilirubin increased (20.0%, 0%≥ G3). One in 6 pts receiving 600 mg dose experienced a DLT (diarrhea, G3). Overall, as an orally irreversible HER2 inhibitor, SPH5030 demonstrated desirable pharmacokinetic properties and safety profiles, which is expected to have an ideal clinical application prospect in the future.
4. Materials and Methods
4.1. Drugs, Chemicals and Reagents
SPH5030 maleate and its major metabolite (SPH6849) were synthesized by Shanghai Pharmaceutical Co., Ltd. (Shanghai, China). HPLC grade acetonitrile and methanol were obtained from Merck (Darmstadt, Germany), formic acid was obtained from Fluka (Munich, Germany), and ammonium acetate was obtained from ROE (ROE, USA). Purified water was generated by Milli-Q gradient water purification system (Millipore, Molsheim, France).
4.2. Animals
Sprague Dawley (SD) rats (170-280g, 6–8 weeks old) were purchased from Shanghai Sipur-Bikai Laboratory Animal Co., Ltd and Shanghai JSJ Laboratory Animal Co., Ltd. Cynomolgus monkeys (3-5 kg, 3–6 years old) were purchased from Hainan Xinzhengyuan Biotechnology Co., Ltd. All animals were treated in accordance with Institutional Guide for the Care and Use of Laboratory Animals.
4.3. Analytical Method and Method Validation
LC-MS/MS consists of an HPLC system (Shimadzu, Japan) and a Sciex Triple QuadTM 6500+ triple quadrupole mass spectrometer (AB SCIEX) with electrospray ionization (ESI) source. MS detection was performed in positive ESI mode (scan mode MRM) with the source temperature maintaining at 550 °C. Data acquisition and analysis were performed using Analyst software (AB SCIEX). A Eclipse Plus C18(100 × 4.6 mm, 3.5 μm; Agilent) thermostatted at 40 °C. The mobile phases were 2 mM NH4OAc & 0.2% FA in H2O (A) and ACN (B) at a flow rate of 0.600 mL/min. The UPLC-MS/MS method for all animal plasma, tissue, feces, bile, and urine samples were validated for selectivity, lower limit of quantification, linearity, accuracy and precision, dilution reliability, matrix effects, and stability.
4.4. Intravenous and Intragastric PK Studies of SPH5030
SPH5030 was administrated at a dose of 4 mg/kg (intravenous, i.v.), 4 mg/kg, 8 mg/kg, and 16 mg/kg (intragastric, i.g.) for both rats and Cynomolgus monkeys. Blood samples were collected from the retrobulbar venous plexus at 0 and 5 min, and at 0.25, 0.5, 1, 2, 3, 5, 7, 10, 24, 36 and 48 h for rats (0.2 mL for each time point), and from a limb vein at 0, 5, and 15 min and at 0.5, 1, 2, 4, 5, 6, 7, 8, 10, 24, 36 and 48 h for monkeys (0.2 mL for each time point) post-dose. The collected blood samples were placed in EDTA-K2 anticoagulation test tubes, centrifuged at 3500 rpm for 10 min (4 °C). The plasma was separated within 2 h and stored at 70 °C for testing. The concentrations of SPH5030 and its metabolite SPH6849 in the plasma of rats and cynomolgus monkeys at different time points were determined by validated LC-MS/MS method. The non-compartmental model of Phoenix WinNonlin 7.0 software (Pharsight, USA) was used to calculate the pharmacokinetic parameters of rats and cynomolgus monkeys after administration.
4.5. Tissue Distribution and Excretion Test in SD Rats
SD rats were divided into four time groups (3/sex/group) and fasted for 12 h before drug administration. 8 mg/kg SPH5030 was dosed by gavage and were anesthetized and bled through the abdominal aorta at 2 h, 5 h, 15 h and 48 h after administration, respectively, and dissected immediately. The tissues were collected and washed with cold saline, labeled and stored at 70 °C for testing.
In excretion test, SPH5030 was administered to SD rats by gavage at a dose of 8 mg/kg after fasted overnight. Urine and faecal samples were collected from rats (3M/3F) from 0-8, 8-24, 24-48 h, 48-72 h and 72-96 h after administration. SD rats were placed in metabolic cages, fed and watered freely, and blank urine and fecal samples were collected. The volume of the urine samples was recorded and the fecal samples were weighed. Another six SD rats (3M/3F) were anesthetized and bile duct cannulation was performed to allow bile collection at 0 - 4, 4 - 8, 8 - 24 and 24 - 48 h post dosing and the volumes were recorded. All samples were stored at -70ºC for testing. The concentrations of SPH5030 and SPH6849 in tissues, feces, urine and bile were determined by the validated liquid chromatography-tandem mass spectrometry method.
4.6. In Vivo and In Vitro Metabolite Identification
The plasma samples collected from rats and monkeys of the same sex at 1 h, 5 h, 10 h and 24 h after drug administration were mixed in equal volumes to obtain combined plasma samples, respectively. The combined biological samples were taken separately, added with approximately twice the volume of acetonitrile. The supernatant was taken, blown dry at 40℃ under nitrogen flow, and re-dissolved with acetonitrile/water (1: 9, v/v) for UPLC-UV/Q-TOF MS analysis. PeakView ® V1.2 and MetabolitePilot V1.5 software from AB Sciex were used for data analysis.
The hepatocytes incubation system (100 µL) contained William’s Medium E medium (pH 7.4), hepatocytes at a cell density of 1.0×106 cells/mL and SPH5030 at a final concentration of 3.0 µM was incubated at 37 °C. After 3 h of reaction, 100 µL of ice-cold acetonitrile was added to stop the reaction. The incubated samples were diluted with acetonitrile, vortexed for 1 min, and centrifuged for 5 min (14 000 rpm). The supernatant was collected, dried under nitrogen flow at 40ºC, and then reconstituted with acetonitrile/water (1:9, v/v) for UPLC-Q/TOF MS analysis. PeakView ® V1.2 and MetabolitePilot V1.5 software from AB Sciex were used for data analysis.
4.7. In Vitro Metabolic Profiles Analysis
Pooled human liver microsomes with selective inhibitors and recombinant human CYP450 enzymes were used to identify the CYP450 isozymes responsible for the metabolism of SPH5030 in vitro [15]. Human liver microsomal chemical inhibition assay: incubation system (100 µL) contained 0.5 mg/mL human liver microsome, 1.0 mM NADPH and various concentrations of inhibitors. After 10 min of pre-incubation, 3.0 µM SPH5030 was added to the mixture to initiate the reaction. Meanwhile, recombinant CYP450 enzyme assay were also employed to identify the metabolizing CYP450 isozymes: incubation system (100 µL) contained 3.0 µM SPH5030 and 1.0 mM NADPH was incubated at 37 ℃. After 3 min of pre-incubation, the human recombinant P450 enzymes (BD Biosciences, Woburn, MA, USA) was added to the mixture to initiate the reaction. The final concentration of each recombinant enzyme protein was 50 pmol/mL. After incubation for 60 min, both of the incubation system reaction above were stopped by adding into the same volume of ice-cold acetonitrile. After appropriate pretreatment, the supernatant was taken for UPLC-Q/TOF MS analysis.
SPH5030 was also tested for its CYP enzyme inhibition effects on marker reactions specific for CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4/5. For the reversible CYP450 inhibition assay, 5 min pre-incubation at 37 °C was needed and the reaction was initiated by the addition of NADPH (1.00 mM). For the time-dependent CYP450 inhibition assay, before probe substrates were added, 30 min of pre-incubation in the presence or absence of NADPH (1μM) was processed. The final concentration of the probe substrates and incubation time corresponding to each CYP450 isoenzyme are shown in Table 1-2. The metabolites generated from probe substrates were detected and IC50 was calculated by sigmoidal (logistic) fitting in Graphpad Prism (v8.0, GraphPad Software, San Diego, CA). For time-dependent Inhibition assay, IC50 shift was determined by the ratio of IC50 values between the 30 min pre-incubation minus and plus NADPH.
For CYP450 induction assay, cryopreserved primary human hepatocytes were thawed and seeded in collagen coated 48-well cell culture plates at a density of 0.7 million viable cells/mL and incubated overnight at 37 °C with 5% CO2. In the following two days, the cells were treated with SPH5030 or vehicle (0.1% DMSO) and incubated at 37℃ and 5% CO2, with medium change and replenishment every 24 h. After treatment, probe substrates were incubated with the cells at 37℃ for 30 min. and the metabolites formation s was detected by LC/MS/MS. Total RNA from the hepatocytes was isolated and cDNA was synthesized. The expression levels of CYP1A2, CYP2B6, CYP3A4 and 18S (internal reference) were detected by real-time fluorescence quantitative PCR.2.5 [16].
4.8. Transporter Mediated Interaction
Caco-2 cells purchased from the American Type Culture Collection (Manassas, VA, USA) were cultured for 21 days in an incubator at 37 °C in a 5% CO2 and 90% relative humidity environment. Cell layers with a transepithelial electrical resistance value of > 300 Ω cm2 were used. SPH5030 (2.00, 10.0 and 50.0 μM) in HBSS containing 0.5% BSA was added to the apical side to assess permeability in the A → B direction and in the basolateral side to assess permeability in the B → A direction. After incubation at 37 °C for 2 h, samples were collected from both the donor and receiver sides. All incubations were performed in duplicate. The concentration of SPH5030 was measured by the LC-MS/MS method described above. The apparent permeability coefficient (Papp) was calculated using the equation, Papp (cm/s) = [dq/dt/C0 × A]. The efflux ratio (ER) was estimated as ER = Papp, B → A/Papp, A → B.
MDCK-MDR1 and MDCK-BCRP cells were seeded into 96-Multiwell Insert Systems at 5×105/cm2 and cultured for 7 days for confluent cell monolayer formation. After washed twice, blank or inhibitor-containing transport buffer was pre-incubated with cells for 30 min. For the substrate determination, transport buffer containing SPH5030 at 10 μM was treated bidirectionally of the cell monolayer with or without selective inhibitors. For the inhibitor determination, transport buffer containing probe substrateswas treated bidirectionally of the cell monolayer with or without selective inhibitor. After 120 min incubation at 37 °C with 5% CO2, Samples collected from the donor and receiver side were analyzed by LC/MS/MS together with the initial dosing samples.
HEK293-OATP1B1, HEK293-OATP1B3, HEK293-OAT1, HEK293-OAT3, HEK293-OCT2, HEK293-Mock, HEK293-MATE1, HEK293-MATE2-K and HEK293-Wildtype cells were seeded into poly-D-lysine coated 96 well plate at 5×104 cells/well and cultured for 48 hours. For the substrate investigation, SPH5030 (1.00/ 10.0 µM, with or without selective inhibitors) were incubated on the transporter transfected cells for 10 minutes after the cells were washed and preincubated with transport buffer for 10 min. For the inhibitor investigation, the cells were washed and preincubated with transport buffer containing SPH5030 (0.3-50.0 µM) for 10 min (the pre-incubation time of OATP1B1 and OATP1B3 was 30 min). Following this, different concentrations of SPH5030 and probe substrates were co-incubated on the transfected cells for a certain time. All of the cells samples from substrate and inhibitor assay were then washed, lysed and taken out for LC/MS/MS analysis. Protein concentrations of the cell lysate were quantified using a BCA assay kit according to the manufacturer’s instructions.
4.9. Simple Allometric Scaling of Preclinical In Vivo Clearance and Volume of Distribution for Human Pharmacokinetics Prediction
Assuming a one-compartment model, simple allometric scaling [17] was applied using mean pharmacokinetic parameters of unbound CL and unbound Vdss for danicamtiv from intravenous pharmacokinetic studies in mouse, rat and cynomolgus monkey, which were correlated to corresponding body weight (BW) using the power equation Y = a × BWb, where a and b are coefficient and exponent, respectively. Least square fitting of log BW versus log Y allowed for estimates of exponents and coefficients. Rat, cynomolgus monkey and human body weights of 0.30, 3 and 60 kg, respectively, were used [18]. Assuming a one-compartment model, the predicted t1/2 of danicamtiv in human was calculated using the predicted pharmacokinetic parameters (t1/2 = Vss × 0.693/CL) [19].
Author Contributions
Conceptualization, Xia G.; methodology, Xu J.; software, Gong X.; validation, Zhang Z. and Li L.; formal analysis, Xu J.; investigation, , Gu Y., Xu J. and Ding S.; resources, Li D. and Gong W.; data curation, Gu Y. and Xu J.; writing—original draft preparation, Gu Y.; writing—review and editing, Gong X. and Xia G.; visualization, Gong X.; supervision, Xia G. and Zhang Z.; project administration, Gong W.. All authors have read and agreed to the published version of the manuscript.”
Funding
This research received no external funding.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board (Animal Care Committee) of of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences. All of the studies has been reviewed and approved by the Committee on the Use and Management of Experimental Animals (IACUC).
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Cronin K.A.; Harlan L.C.; Dodd K.W.; Abrams J.S.; Ballard B.R. Population-based estimate of the prevalence of HER-2 positive breast cancer tumors for early stage patients in the US. Cancer Invest. 2010, 28, 963-968. [CrossRef]
- Li X.; Yang C.; Wan H.; Zhang G.; Feng J.; Zhang L.; Chen X.; Zhong D.; Lou L.; Tao W.; Zhang L. Discovery and development of pyrotinib: A novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor with favorable safety profiles for the treatment of breast cancer. European journal of pharmaceutical sciences. 2017, 110, 51–61. [CrossRef]
- Franklin, M.C.; Carey K.D.; Vajdos F.F.; Leahy D.J.; de Vos A.M.; Sliwkowski M.X. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004, 5, 317-328. [CrossRef]
- Blume J.P.; Hunter T. Oncogenetic Kinase Signaling. Nature. 2001, 411, 355-365. [CrossRef]
- Olayioye M.A. Update on HER-2 as a target for cancer therapy: intracellular signaling pathways of ErbB2/HER-2 and family members. Breast Cancer Res. 2001, 3, 385–389. [CrossRef]
- Iida M.; Brand T.M.; Starr M.M.; Huppert E.J.; Luthar N.; Bahrar H.; Coan J.P.; Pearson H.E.; Salgia R.; Wheeler D.L. Overcoming acquired resistance to cetuximab by dual targeting HER family receptors with antibody-based therapy. Mol Cancer. 2014, 13, 1-16. [CrossRef]
- Kondrashov A.; Sapkota S.; Sharma A.; Riano I.; Kurzrock R.; Adashek J.J. Antibody-Drug Conjugates in Solid Tumor Oncology: An Effectiveness Payday with a Targeted Payload. Pharmaceutics. 2023, 15, 2160-2187. [CrossRef]
- Nakada T.; Sugihara K.; Jikoh T.; Abe Y.; Agatsuma T. The Latest Research and Development into the Antibody-Drug Conjugate, [fam-] Trastuzumab Deruxtecan (DS-8201a), for HER2 Cancer Therapy. Chem Pharm Bull (Tokyo). 2019, 67, 173-185. [CrossRef]
- Li D.; Tu Y.; Jin K.; Duan L.; Hong Y.; Xu J.; Chen N.; Zhang Z.; Zuo H.; Gong W.; Zhang J.; Wang Q.; Qian H.; Wang X.; Ke Y.; Xia G. Discovery of SPH5030, a Selective, Potent, and Irreversible Tyrosine Kinase Inhibitor for HER2-Amplified and HER2-Mutant Cancer Treatment. J Med Chem. 2022, 65, 5334–5354. [CrossRef]
- Mckertish C.M.; Kayser V. A Novel Dual-Payload ADC for the Treatment of HER2+ Breast and Colon Cancer. Pharmaceutics. 2023, 15, 2020-2017. [CrossRef]
- Nguyen K.S.; Kobayashi S.; Costa D.B. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clinical Lung Cancer. 2009, 10, 281-289. [CrossRef]
- Yun C.H.; Mengwasser K.E.; Toms A.V.; Woo M.S.; Greulich H.; Wong K.K.; Meyerson M.; Eck M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008, 105, 2070-2075. [CrossRef]
- Kulukian A.; Lee P.; Taylor J.; Rosler R.; de Vries P.; Watson D.; Forero T.A.; Peterson S. Pre-clinical activity of HER2-selective tyrosine kinase inhibitor tucatinib as a single agentor in combination with trastuzumab or docetaxel in solid tumor models. Molecular Cancer Therapeutics. 2020, 19, 976–987. [CrossRef]
- Vu T.; Claret F.X. Trastuzumab: updated mechanisms of action and resistance in breast cancer. Front Oncol. 2012, 2, 1-6. [CrossRef]
- Wrighton S.A.; Stevens J.C. The human hepatic cytochromes P450 involved in drug metabolism. Crit Rev Toxicol. 1992, 22, 1-21. [CrossRef]
- Mattinen L.; Kublbeck J.; Rechardt O.; Honkakoski P.; Rautio J. Direct and rapid transcript analysis assay for CYP mRNA expression and inducibility in human primary hepatocytes. Drug Metab Lett. 2014, 8, 77-87. [CrossRef]
- Boxenbaum H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J Pharmacokinet Biopharm. 1982, 10:201-227. [CrossRef]
- Davies B.; Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993, 10, 1093-1095. [CrossRef]
- Grillo M.P.; Markova S.; Evanchik M.; Trellu M.; Moliner P.; Brun P.; Perreard-Dumaine A.; Vicat P.; Driscoll JP.; Carlson TJ. Preclinical in vitro and in vivo pharmacokinetic properties of danicamtiv, a new targeted myosin activator for the treatment of dilated cardiomyopathy. Xenobiotica. 2021, 51, 222-238. [CrossRef]
- J Clin Oncol 42, 2024 (suppl 16; abstr 3117). https://conferences.asco.org/am/embargo-abstract-release.
- Czyrski A.; Resztak M.; Świderski P.; Brylak J.; Główka F.K. The Overview on the Pharmacokinetic and Pharmacodynamic Interactions of Triazoles. Pharmaceutics. 2021, 13, 1961-1987. [CrossRef]
- Smith DA.; Beaumont K.; Maurer TS.; Di L. Relevance of half-life in drug design. J Med Chem. 2018, 24, 61, 4273-4282. [CrossRef]
- Westra N, Touw D, Lub-de Hooge M, Kosterink J, Oude Munnink T. Pharmacokinetic Boosting of Kinase Inhibitors. 2023, 15, 1149-1174. [CrossRef]
Figure 1.
The tissue distribution of SPH5030 and SPH6849.
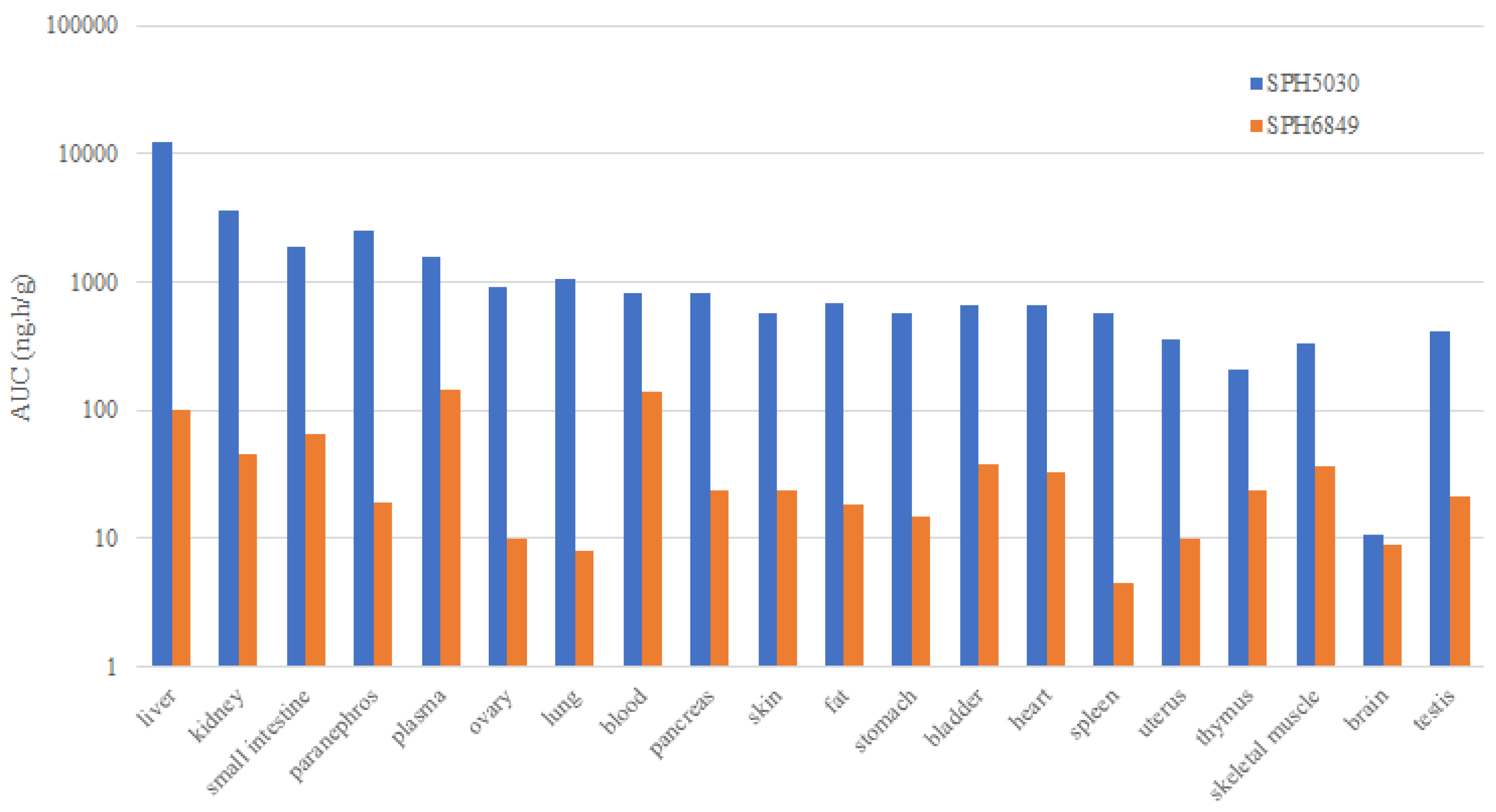
Figure 2.
The major metabolism pathway of SPH5030.
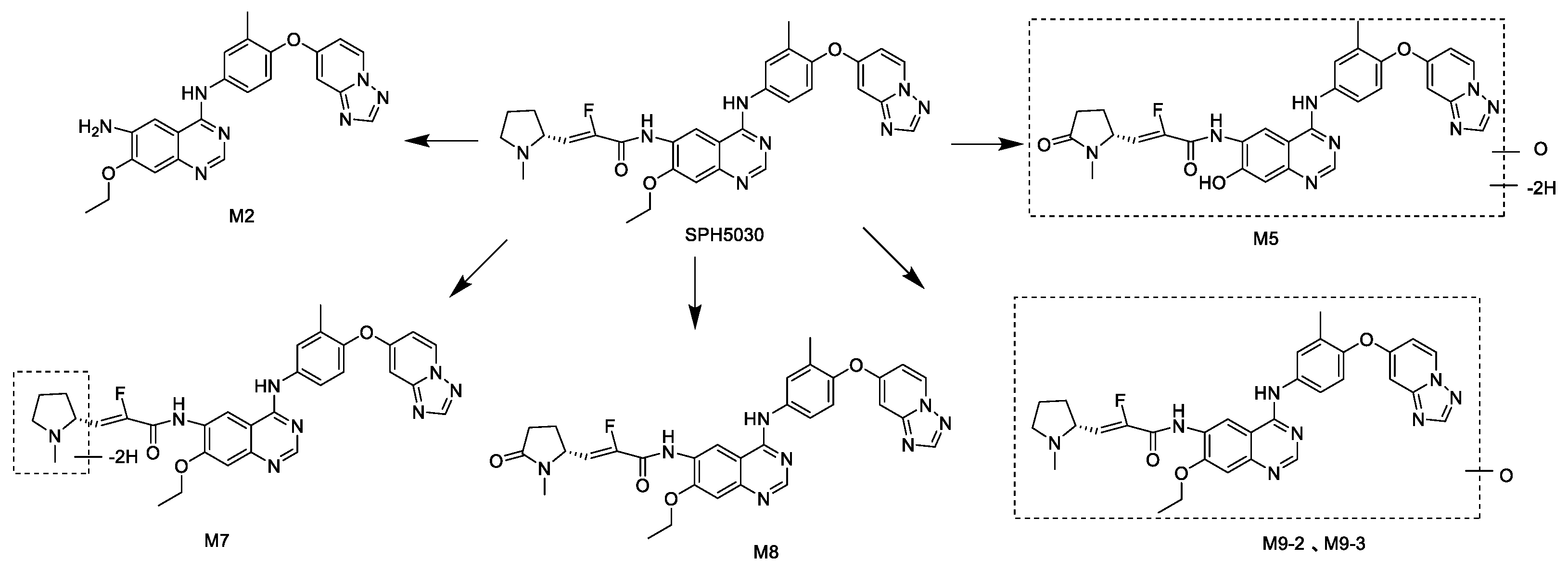
Figure 3.
Human clearance extrapolated by allometric scaling.
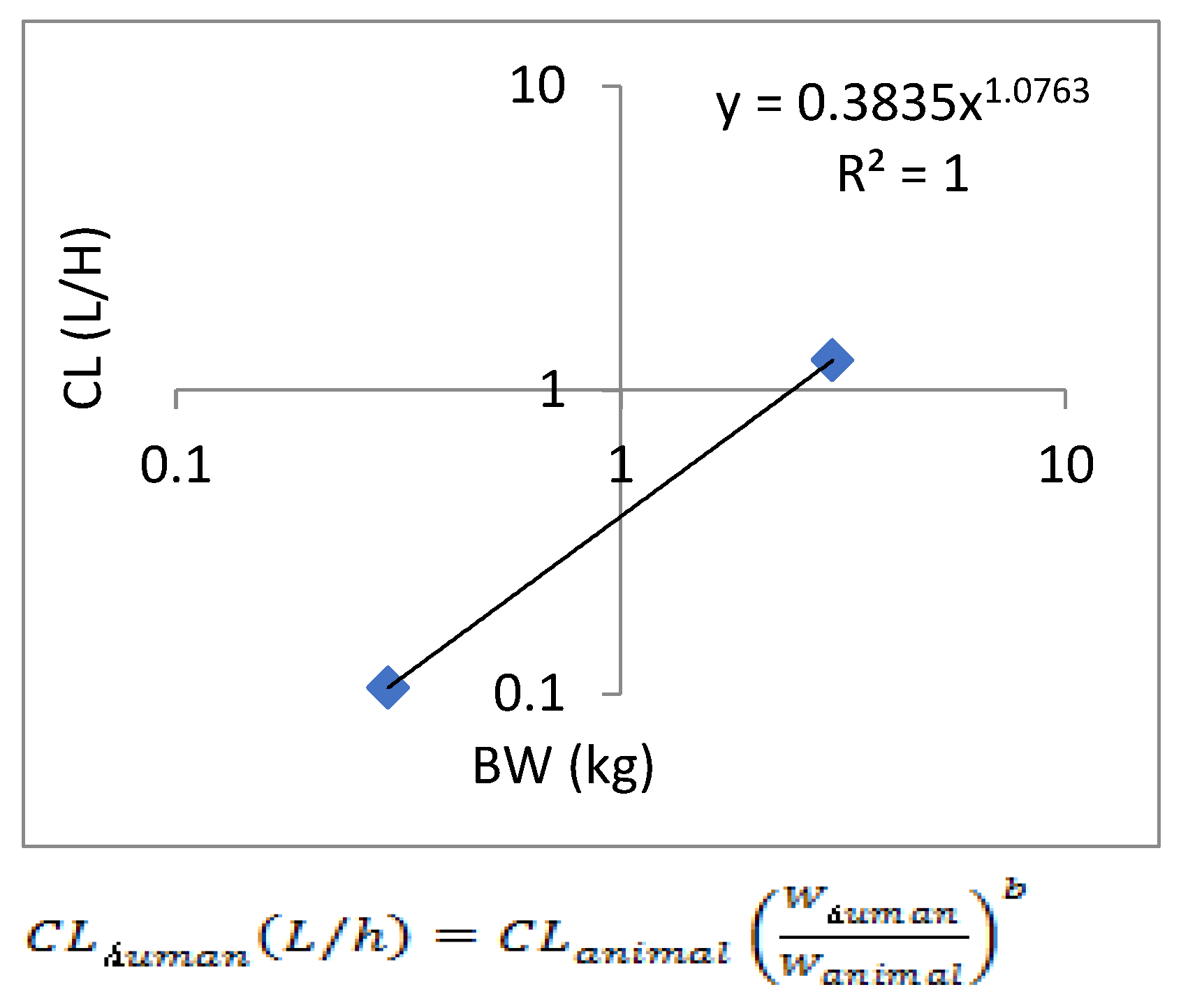
Figure 4.
Human volume of distribution Vss extrapolated by allometric scaling.
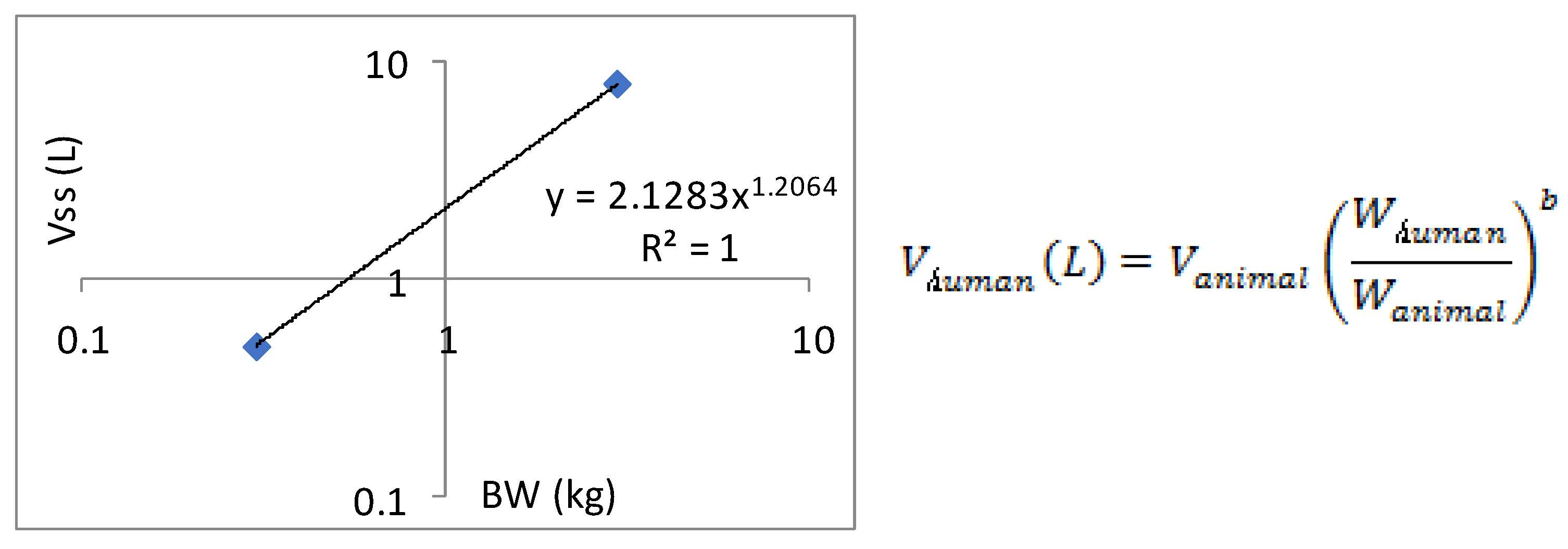
Figure 5.
Prediction of blood concentration and time of SPH5030 after oral administration of 100 mg in human.
Figure 5.
Prediction of blood concentration and time of SPH5030 after oral administration of 100 mg in human.
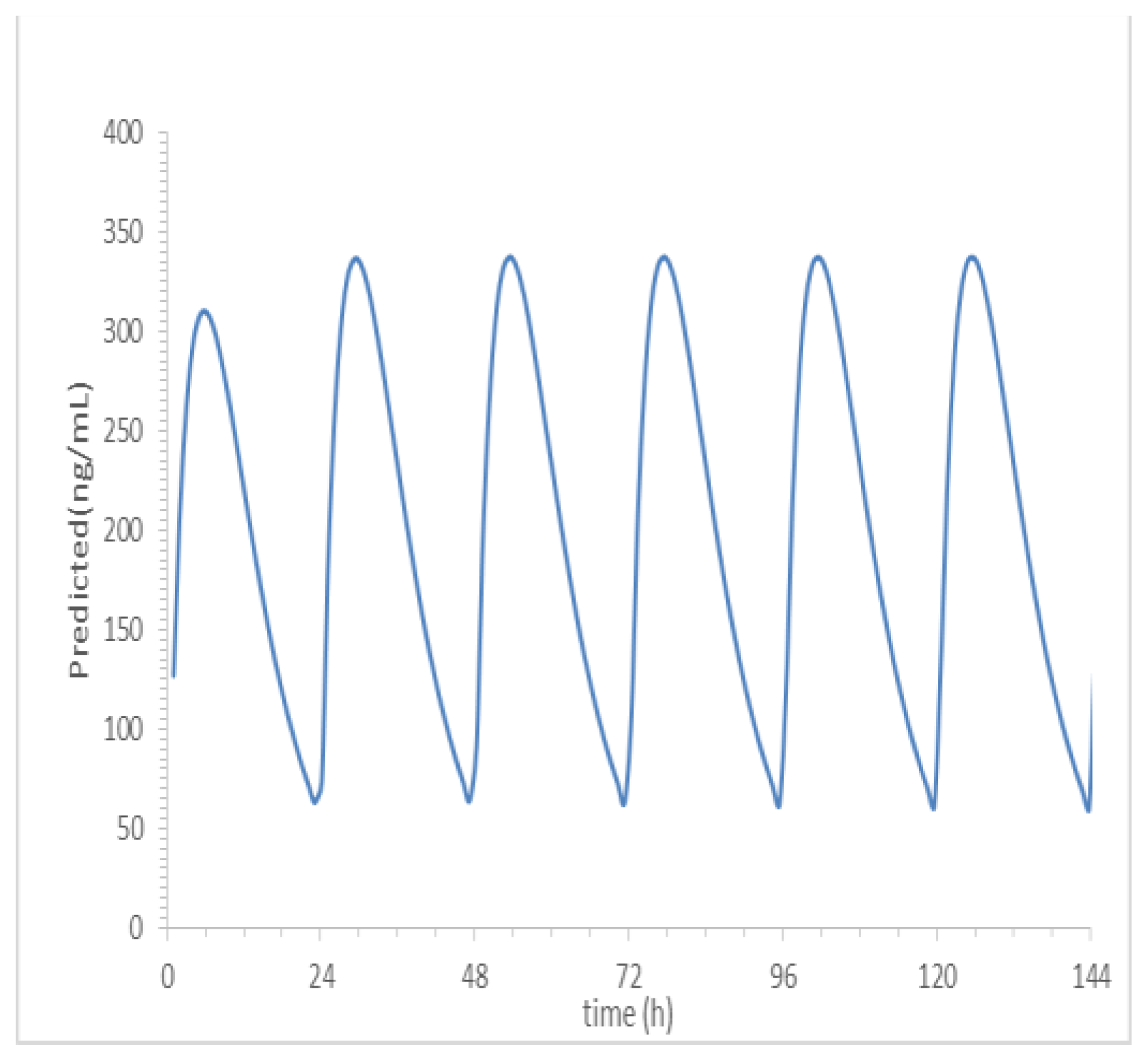

Table 1.
Overall pharmacokinetic properties of SPH5030 in animal species and human.
| Species |
Dose mg/kg |
Route |
Tmax (h) |
Cmax (µg/mL) |
AUCtot (µg·h/mL) |
MRT0-∞ (h) |
T1/2 (h) |
CL (mL/min/kg) |
Vss (L/kg) |
F (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Rat | 4 | iv | - | - | 13.5 | 5.18 | 4.13 | 5.83 | 1.66 | |
| 4 | ig | 4 | 0.642 | 7.62 | 7.77 | 4.64 | - | - | 56.4 | |
| 8 | ig | 5 | 1.3 | 15.4 | 8.4 | 8.27 | - | - | 57 | |
| 16 | ig | 4 | 2.49 | 34.7 | 8.9 | 6.75 | - | - | 64.3 | |
| Monkey | 4 | iv | - | - | 10.0 | 6.45 | 5.92 | 6.95 | 2.67 | - |
| 4 | ig | 2 | 0.304 | 2.39 | 7.6 | 5.19 | - | - | 23.9 | |
| 8 | ig | 4 | 0.363 | 3.62 | 7.44 | 4.95 | - | - | 18.1 | |
| 16 | ig | 4.5 | 0.713 | 15.2 | 15.9 | 9.14 | - | - | 38 |
Tmax, time to peak; Cmax, maximum plasma concentration; AUCtot, the total area under the plasma concentration–time curve; MRT, mean residence time; T1/2, half-life; CL, clearance after iv; Vss, volume of distribution at steady state after iv; F, absolute oral bioavailability; iv, intrravenous; ig, intragastric.
Table 2.
In vitro metabolic stability assay of SPH5030 in liver microsomes and plasma protein binding rate assay.
Table 2.
In vitro metabolic stability assay of SPH5030 in liver microsomes and plasma protein binding rate assay.
| Species | T1/2 (min) |
CLint# (mL/min/kg) |
CLhb* (mL/min/kg) |
BRPP**(%) | ||
|---|---|---|---|---|---|---|
| SPH5030 | SPH6849 | |||||
| Human | 73.7 | 23.6 | 11.0 | 99.2 | 98.8 | |
| Monkey | 46.5 | 43.6 | 21.9 | 99.4 | 99.1 | |
| Dog | 56.4 | 61.3 | 20.5 | 99.0 | 98.0 | |
| Rat | 54.2 | 45.9 | 25.1 | 99.9 | 99.9 | |
| Mouse | 86.6 | 63.0 | 37.1 | 99.8 | 99.8 | |
# CLint, inherent clearance; * CLhb, hepatic clearance; ** BRPP, plasma protein binding rate.
Table 3.
Estimated safety margins of CYP and hERG inhibition based on total human Cmax.
| Dose (mg) | Cmax (µM) | CYP 1A2, 2B6, 2C9, 2C19, 2D6 (>50 µM) | CYP3A4 (Midazolam 6β-Hydroxylation) (17.1 µM) | CYP3A4 (Testosterone 6 β- Hydroxylation) (20.7 µM) | CYP2C8 (16 µM) | hERG (>100 µM) |
|---|---|---|---|---|---|---|
| 25 | 0.110 | > 454.0 × | > 155.0 × | > 18.0 × | > 145.0 × | > 909.0 × |
| 50 | 0.413 | > 121.0 × | > 41.0 × | > 50.0 × | > 38.0 × | > 242.0 × |
| 100 | 0.620 | > 80.0 × | > 27.0 × | > 33.0 × | > 25.0 × | > 161.0 × |
| 200 | 1.474 | > 33.0 × | > 11.0 × | > 14.0 × | > 10.0 × | > 67.0 × |
| 400 | 2.914 | > 17.0 × | > 5.0 × | > 7.0 × | > 5.0 × | > 34.0 × |
| 800 | 6.848 | > 7.0 × | > 2.0 × | > 3.0 × | > 2.0 × | > 14.0 × |
CYP cytochrome P450, Cmax maximum plasma concentration, hERG human−ether-a-go-go-related gene.
Table 4.
CYP450 induction of the enzyme activity and mRNA level in hepatocytes.
| Hepatocyte Batch |
SPH5030 (µM) |
CYP Enzyme Activity | mRNA Result | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Induction Multiple | % of PC | Induction Multiple | % of PC | |||||||||||
| 1A2 | 2B6 | 3A4 | 1A2 | 2B6 | 3A4 | 1A2 | 2B6 | 3A4 | 1A2 | 2B6 | 3A4 | |||
| 0.10 | 0.91 | 0.68 | 1.50 | -0.56 | -4.46 | 7.24 | 0.54 | 1.00 | 1.56 | -0.42 | -0.01 | 3.19 | ||
| QBU | 1.00 | 0.71 | 0.60 | 1.67 | -1.69 | -5.49 | 9.81 | 0.51 | 1.43 | 3.80 | -0.44 | 1.21 | 16.1 | |
| 10.0 | 0.44 | 0.41 | 1.50 | -3.30 | -8.07 | 7.30 | 0.84 | 1.90 | 11.1 | -0.15 | 2.50 | 57.7 | ||
| 0.10 | 1.06 | 1.12 | 1.44 | 0.16 | 0.80 | 3.66 | 0.41 | 0.37 | 0.54 | -0.40 | -3.85 | -0.96 | ||
| GKL | 1.00 | 1.00 | 1.32 | 1.64 | 0.01 | 2.14 | 5.27 | 0.35 | 0.60 | 0.92 | -0.44 | -2.46 | -0.16 | |
| 10.0 | 0.31 | 0.56 | 0.48 | -1.76 | -2.96 | -4.29 | 0.56 | 1.16 | 2.96 | -0.30 | 0.99 | 4.09 | ||
| 0.10 | 0.99 | 1.05 | 1.31 | -0.03 | 0.67 | 4.15 | 0.54 | 0.89 | 1.84 | -1.79 | -1.27 | 24.2 | ||
| HJK | 1.00 | 0.78 | 1.06 | 1.50 | -1.06 | 0.78 | 6.86 | 0.39 | 1.07 | 2.20 | -2.37 | 0.84 | 34.6 | |
| 10.0 | 0.49 | 0.87 | 1.28 | -2.44 | -1.83 | 3.86 | 0.46 | 1.33 | 4.94 | -2.09 | 3.92 | 114 | ||
Table 5.
In vitro transporters and permeability assay of SPH5030.
| Cell | Compounds | Papp*(10-6 cm/s) | Papp Ratio | |
|---|---|---|---|---|
| A→B | B→A | |||
| Caco-2 | 2 µM SPH5030 | 1.45 | 7.31 | 4.91 |
| 10 µM SPH5030 | 0.53 | 1.26 | 12.37 | |
| 50 µM SPH5030 | 0.43 | 0.99 | 2.34 | |
| 10µM SPH5030+GF120918 | 2.01 | 1.23 | 0.66 | |
| MDCK-MDR1 | 10 µM SPH5030 | 0.21 | 1.43 | 7 |
| 10 µM SPH5030+10 µM GF120918 | 0.25 | 0.13 | 0.51 | |
| MDCK-BCRP | 10 µM SPH5030 | 0.61 | 1.67 | 2.72 |
| 10 µM SPH5030+10 µM Ko143 | 0.72 | 0.51 | 0.70 | |
| MDCK-MDR1 | 10 µM Digoxin | 1.94 | 291 | 40.3 |
| 10 µM Digoxin + 10 µM SPH5030 | 5.07 | 324 | 17.2 | |
| 10 µM Digoxin + 50 µM SPH5030 | 8.31 | 224 | 7.25 | |
| MDCK-BCRP | 300 nM Imatinib | 0.49 | 6.67 | 3.64 |
| 300 nM Imatinib + 10 µM SPH5030 | 1.33 | 12.2 | 2.47 | |
| 300 nM Imatinib + 50 µM SPH5030 | 1.76 | 4.92 | 0.75 | |
* Papp: apparent permeability coefficient.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



