
Submitted:
24 October 2024
Posted:
24 October 2024
You are already at the latest version
Abstract
Our ongoing research focuses on developing imipridone derivatives capable of complete and selective eradication of cancer cells after relatively short treatment. We have synthetized systematically designed novel hybrids and evaluated their antiproliferative activity on PANC-1 and Fadu cell lines. We have also conducted preliminary studies on the mechanism including colony formation as well as dose-response tests in HEK293T wild-type (WT) and HEK293T CLPP-/- cells. Following gradual structural fine-tuning based on high throughput screening we identified two imipridone hybrids as the most potent derivatives. Their unique substitution pattern includes N-methylated propargylamine and ferrocenyl/phenyltriazole moieties on the benzyl groups attached to opposite sides of the imipridone core. The compounds with IC50 values similar to those of ONC201 were found capable of complete eradication of cancer cells at ca. 4 M, while ONC201 treatment at even higher concentrations left 30-50% of viable cells behind. Both compounds were found to exert equal activity in WT and CLPP-/- HEK293T cells, indicating a ClpP-independent mechanism. Further development is needed to improve the tumor selectivity of the two potent imipridone derivatives in order to develop novel drug candidates that evade resistance and can be applied in a sufficiently broad therapeutic window.
Keywords:
imipridone
; propargylamine
; 1
; 2
; 3-triazole
; CuAAc reaction
; Sonogashira coupling
; PANC-1 and Fadu cells
; cell viability
; dose-response measurements
; colony formation
; complete eradication of cancer cells
; ClpP-independent mechanism
1. Introduction
Imipridones represent a novel class of small molecule anticancer agents, with high oncotherapeutic potential. ONC201 is the first-in-class lead compound of the imipridone family, which is currently engaged in multiple clinical trials for solid and hematological malignancies, like gliomas, neuroendocrine tumors, multiple myeloma, breast cancer, endometrial cancer (https://clinicaltrials.gov/) featuring wide therapeutic windows and an exceptional safety profile [1]. Accordingly, the FDA has granted ONC201 Fast Track Designation for the treatment of adult recurrent H3 K27M-mutant HGG, Rare Pediatric Disease Designation for treatment of H3 K27M-mutant glioma, and Orphan Drug Designations for the treatment of glioblastoma and malignant glioma. The anticancer activity of ONC201 was originally identified in a target-agnostic screen searching for activators of TNF-related apoptosis-inducing ligand (TRAIL) in tumor cells [2]. Second-generation imipridones, such as ONC206 and ONC212 (Figure 1) were also identified as highly potent anticancer agents [3]. Compared to ONC201, ONC206 demonstrated improved inhibition of cell migration and was transferred to phase I trials in treatment of diffuse midline gliomas, while ONC212 was tested in >1000 human cancer cell lines in vitro featuring the lowest efficient doses in this series, typically in nanomolar range, and evaluated for safety and antitumor efficacy in vivo. Exhibiting rapid pharmacokinetics, ONC212 selectively kills tumor cells and has broad-spectrum preclinical activity across both solid tumors and hematological malignancies, including pancreatic cancer and leukemias prioritized for clinical indications [4].
Molecular targets of ONC201 and other imipridones have been intensively investigated in the past few years in order to clarify their mechanisms of action. Besides inducing TRAIL, ONC201 also upregulates the cell surface TRAIL receptor, death receptor 5 (DR5) [5,6], acts as a selective antagonist of dopamine receptor D2 (DRD2) [3] and has been identified as an allosteric agonist of mitochondrial protease caseinolytic protease P (ClpP) [7,8,9]. It was also disclosed that ONC212 selectively activates protein-coupled receptor 132 (GPCR-GPR132) [10]. However, it was highlighted that, beyond the multiple effects on TRAIL, DRD2 and GPCR-GPR132 signaling pathways, it is the alteration in mitochondrial metabolism initiated by ClpP activation that is by far the most efficient and dominant pathway leading to apoptosis [11]. While normally ClpP is regulated by the chaperone ClpX, hyperactivation of ClpP by imipridones and related structures causes an unregulated increase in the degradation of mitochondrial respiratory complexes I and II, leading to structural and functional damage of mitochondria and impairing oxidative phosphorylation (OXPHOS). Degradation of mitochondrial respiratory proteins is accompanied by, among other consequences, lower oxygen consumption rates, ATP depletion, increased amounts of mitochondrial reactive oxygen species (ROS) and depletion of mitochondrial DNA. These effects collectively activate the integrated stress response (ISR), which directs the cell to arrest the cell cycle and induce apoptosis. Cancer cells are highly sensitive to chemical activation of ClpP, while similar treatments do not induce cell death in normal cells despite a detectable degradation of ClpP substrate proteins [12]. This tumor-specific cytotoxicity and the wide therapeutic window of anticancer activity of imipridones may be due in part to an apparently lower dependence of normal cells on OXPHOS.
Despite the promising anti-proliferative and pro-apoptotic effects of ClpP-activators, the resistance of cancer cells to the treatment seems to be a persistent problem. Accordingly, several studies [13,14,15,16,17,18,19,20,21] identified imipridones as effective cytotoxic agents in micro- or nanomolar concentrations, however the dose-response curves showed that approximately 10–50% of the cells survived the treatment, even at higher imipridone concentrations and after prolonged exposure times.
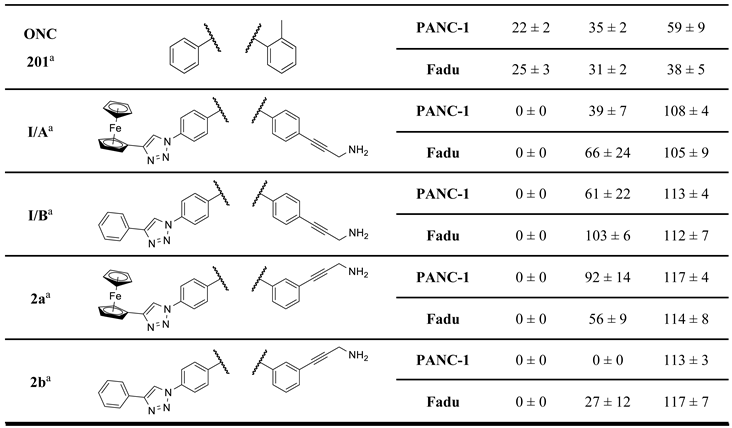
Our research group hypothesized that the tumor cytotoxicity of imipridone derivatives can be improved by boosting ROS formation in the mitochondria through supplementing the hybrid molecule with a ferrocene moiety that activates a Fenton-like pathway. This approach is supported by convincing preclinical evidence for the interplay between TRAIL and redox signaling pathways implicated in cancer [22]. In this context in a previous article [23], we have presented ferrocene-impridone derivatives that exhibit much more pronounced long-term cytotoxic effects on A2058 melanoma cell line than ONC201 and ONC212. These ferrocenylalkyl-substituted imipridones also displayed a marked efficacy against COLO-205 and EBC-1 cell lines, presumably due to the activation of ROS-implicating apoptotic pathways by the ferrocenyl moieties and their contribution to the antiproliferative effect. Thereafter, inspired by these results, we constructed a small library of organometallic imipridone hybrids, which contain ferrocene fragments tethered by triazole and/or alkyne linkers to different positions of the N4- and N5-benzyl groups pending on the imipridone core [24]. Complex evaluation of this set of hybrids identified propargylamines I/A and I/B as the most efficient anti-proliferative agents (Figure 2).
Contrary to ONC201, these two compounds were able to eliminate the resistance in PANC-1 pancreas ductal adenocarcinoma, Fadu pharyngeal squamous carcinoma, A2058 and EBC-1 cell lines, and no viable cells could be observed after treatments during colony formation assays at 10 µM concentrations. However, the IC50 values of these compounds were found to be 5-10 times higher than those of ONC201. On the other hand, we also demonstrated that in shorter treatment (24 hours exposure time instead of 72 hours) in PANC-1 and A2058 cells, I/A and I/B are far more efficient in apoptosis induction than ONC201, which caused no detectable apoptotic signal in the investigated cells after 24 hours of exposure time. Finally, in comparative cytotoxic studies on PANC-1 and A2058 cells and nontumorous primary fibroblasts, the organometallic derivative I/A emerged as the most potent anticancer agent in the presented research. This ferrocene derivative proved to be capable of complete and selective eradication of the tumor cells at 10 µM concentration without exerting an observable toxicity on the fibroblasts.
In our current work, we aim to expand the library of imipridone derivatives with members that not only have the potency to eradicate cancer cells completely and selectively, but also lower efficient doses and IC50 values compared to I/A and I/B.
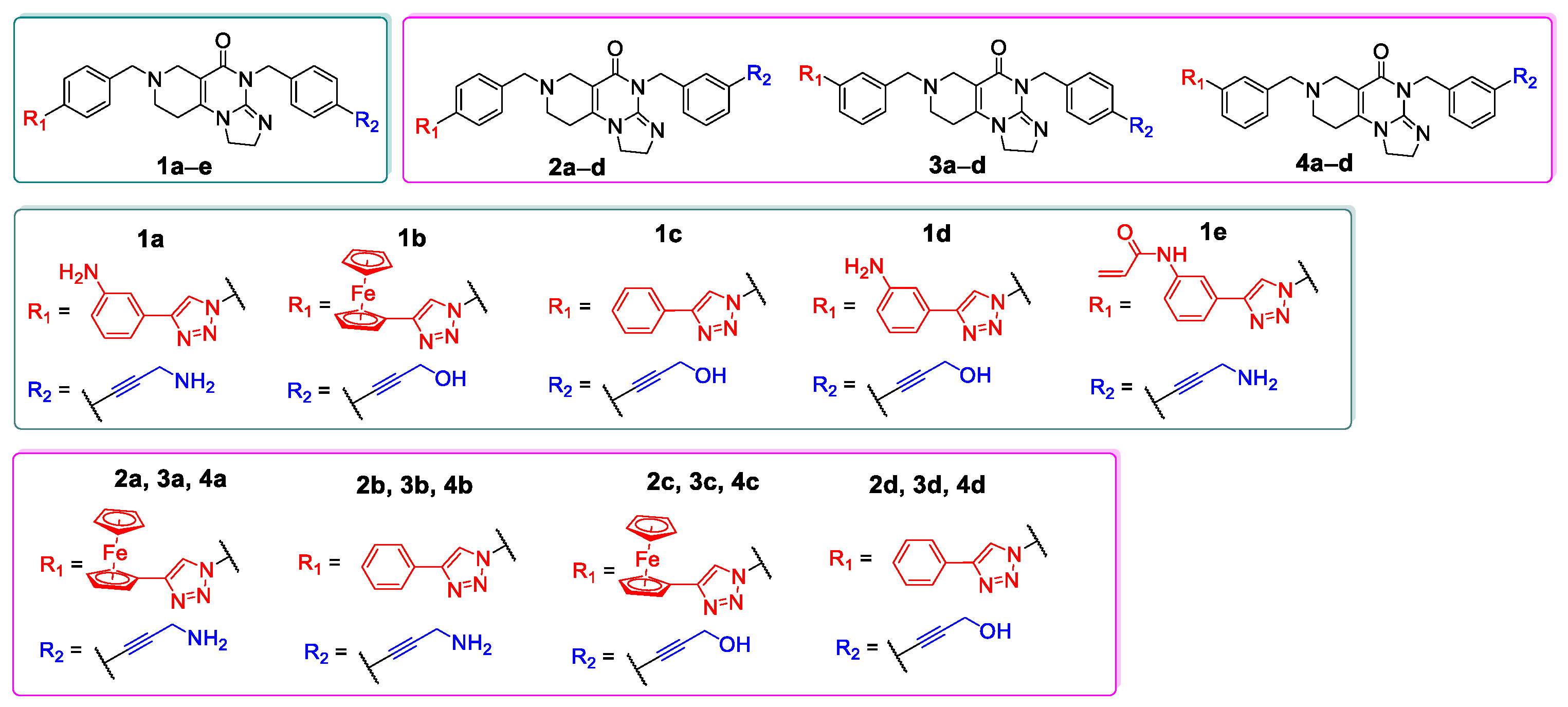
In the initial phase of our diversity-oriented research, we synthesized an array of imipridone hybrids structurally related to I/A and I/B carrying identical or systematically modified substituents in different positions of the terminal benzyl groups (1a–e, 2a–d, 3a–d and 4a–d: Figure 3). Compounds 1a–e can be regarded as the derivatives of I/A and I/B with para-substituted benzyl groups on the terminals of the imipridone scaffold ensuring maximal spatial separation of the arytriazole- and alkyne-containing functional groups, the two potential pharmacophoric warheads. Compounds 2a–4a and 2b–4b, containing at least one meta-substituted benzyl group, are regioisomers of I/A and I/B, respectively, with spatially less separated and differently oriented pharmacophoric warheads. As an additional variation, we introduced a 3-hydroxypropynyl group into hybrids 1b–d, 2c,d 3c,d and 4c,d replacing the 3-aminopropynyl moiety to investigate the role of basicity of this outward-protruding functional group. Moreover, for this comprehensive study, which also aimed at a preliminary investigation of the mechanism of action, we synthetized hybrid 1e, the acroylamino analogue of I/B, which might be capable to covalently bind to cysteine-containing cellular targets.
2. Results and Discussion
2.1. Multistep Synthesis of the First Group of the Targeted Imipridone Hybrids
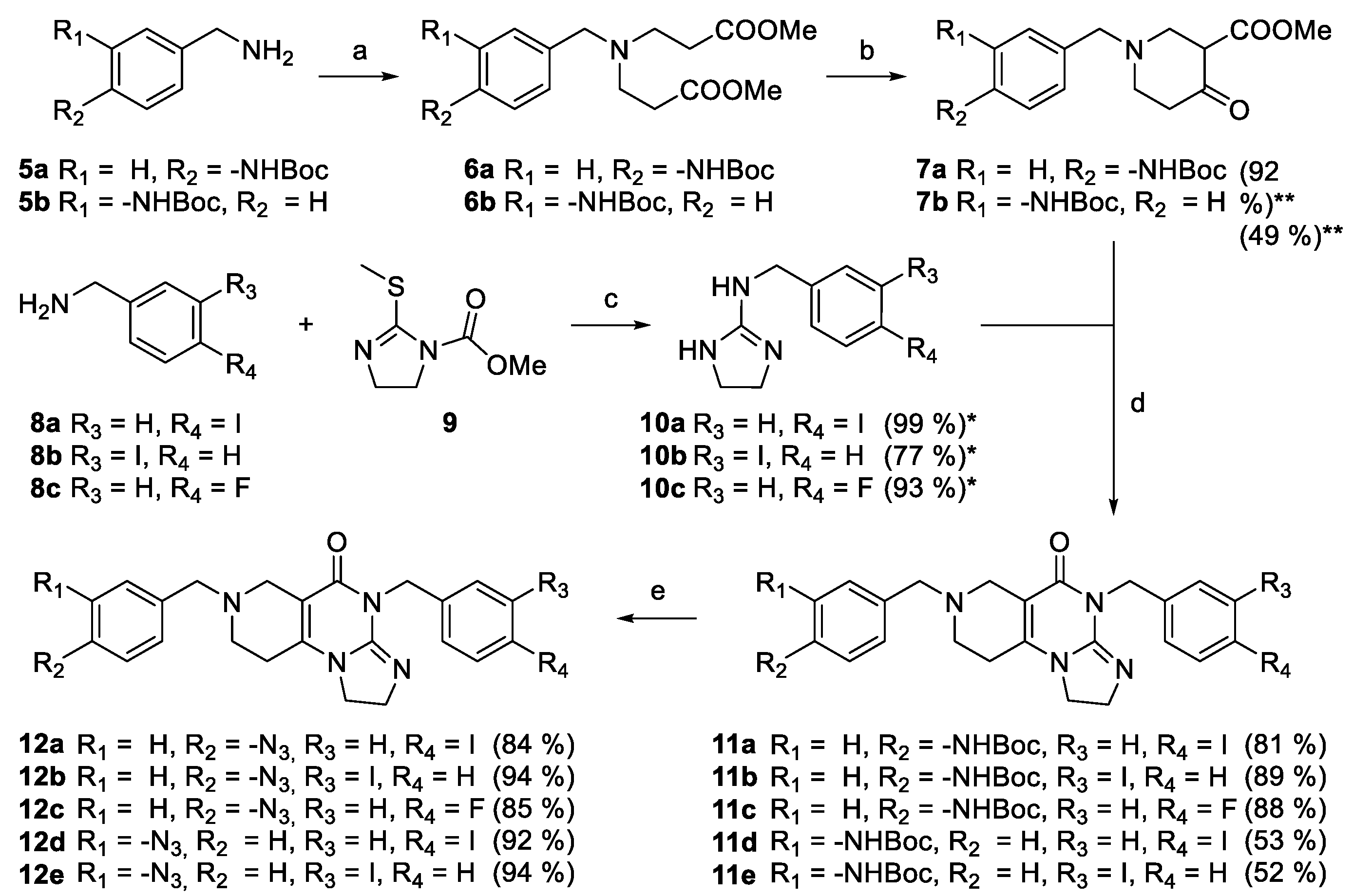
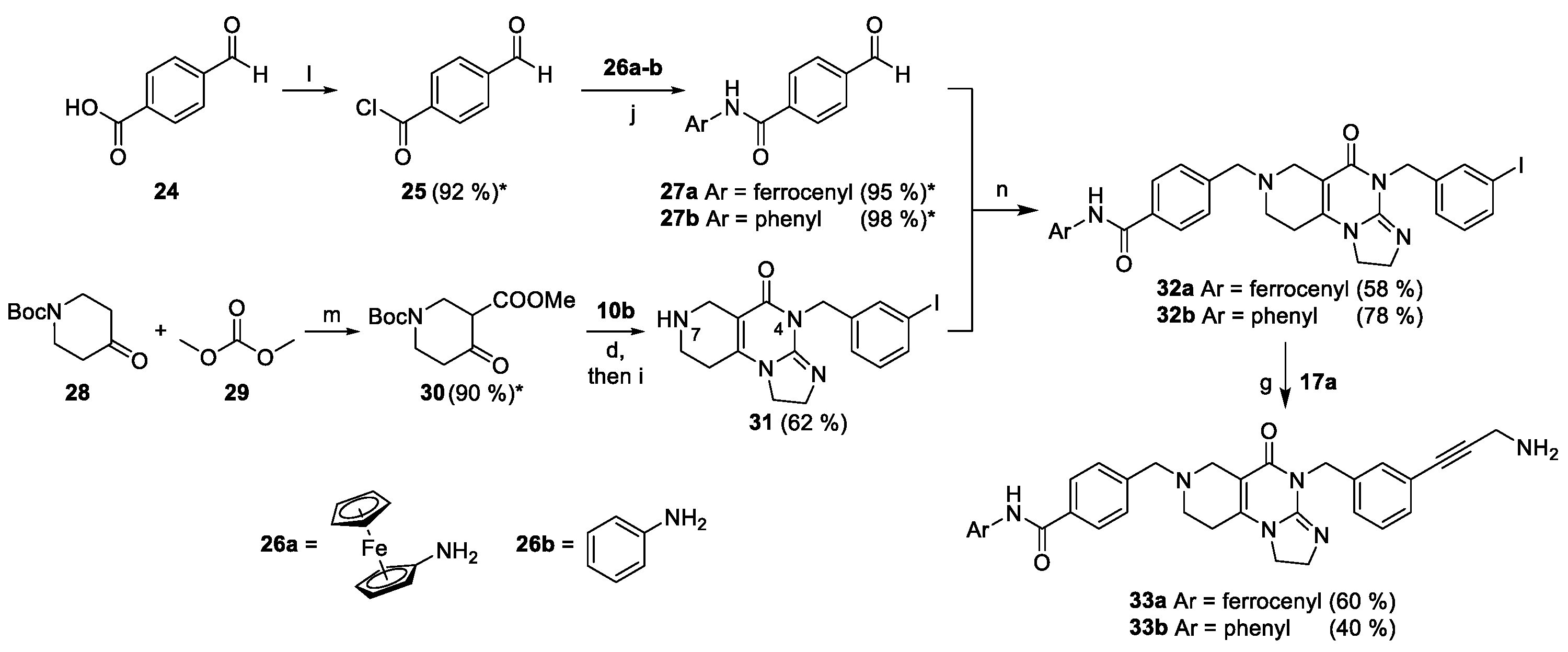
The synthesis of the designed derivates were performed following the multistep convergent synthetic pathway described in our previous publication [24]. Accordingly, by means of double acrylate addition followed by base-catalyzed cyclization mono-Boc-protected 3- and 4-aminobenzylamines 5a and 5b were converted into piperidones 7a and 7b, respectively (5 → 6 → 7: Scheme 1). On the other hand, 3- and 4-halobenzylamines 8a–c were coupled with activated methyltioimidazoline 9 affording cyclic guanidines 10a–c, which were then reacted without purification with 7a,b to construct angular tricyclic imipridones 11a–e carrying Boc-protected amino group (Scheme 1). By sequential N-deprotection, diazotation and treatment with an excess of NaN3, 11a–e were converted to regioisomeric halogenated azides 12a–e in good yields required for the preparation of the designed hybrids (Scheme 1).
We continued with copper-catalyzed azide-alkyne cycloadditions of the 12a–e using ethynylferrocene (13), phenylacetylene (14) and 3-aminophenylacetylene (15) as coupling partners resulting in triazoles 16a–j in mediocre-to-good yields (Scheme 2). Afterwards, switching to the conditions of Sonogashira reaction, iodinated 1,3-triazolyl compounds 16a–e and 16g–j were coupled with propargylamine 17a and propargyl alcohol 18 to obtain 1a–d, 2a–d, 3a–d and 4a–d, the first set of the designed compounds intended for in vitro viability screening (Scheme 2).
Starting from 16c, acrylamido derivative 1e was accessed employing primary Sonogashira coupling with N-Boc propargylamine 17b followed by standard acryloylation and sequence-terminating deprotection of the propargylamine moiety (Scheme 3).
2.2. In Vitro Cell Viability Screening of the First Set of the Novel Imipridone Hybrids
Cell viability screenings of the first set of compounds were performed on PANC-1 and Fadu human cancer cell lines at three concentrations (2.8, 8.3 and 25 μM: Table 1) to establish structure-activity relationships. The same cell lines were used in our previous work on antiproliferative assessment [24].
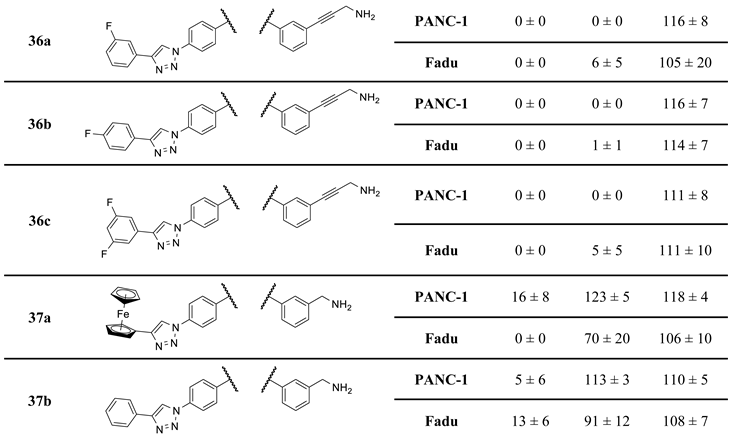
The experiments conducted at 25 µM imipridone concentration disclose the most characteristic SAR indicating that the presence of the protruding basic amino group, kept in a constant distance from the imipridone residue by the rigid linker, is indispensable to achieve the desired drop in viability to 0–1% in either of the cancer cell lines, as reflected from the viability data measured exclusively for aminopropynyl derivatives I/B, 2a,b, 3a,b and 4a,b, while the hydroxypropynyl compounds 1b–d, 2c,d, 3c,d and 4c,d as well as the fluorinated derivative 16f displayed substantially lower activity even at this high drug concentration. Moreover, the results unambiguously show that at 8.3 µM concentration only the ferrocenyltriazolyl derivative 2a and its phenyltriazolyl counterpart 2b can achieve practically complete eradication of both types of cancer cells outperforming their regioisomers I/A and I/B, respectively, as well as every other member of the first set of novel imipridone hybrids screened. It is worth pointing out that the substantial activity upboost reflected by the relative performance of isomeric pairs I/A-2a and I/B-2b is definitely associated with the change of orientation of the rigid basic side chain, which was achieved by moving this pharmacophoric warhead from para to meta position on the N4-benzyl group with retained para position of the ferrocenyl/phenyltriazolyl residue on the N7-benzyl group pending on the imipridone core. Typically, other isomers of 2a and 2b (3ab and 4ab) along with the further members of the first set of hybrids showed little or no improvement in efficacy compared to I/A and I/B. Accordingly, we selected 2a and 2b as the leads of group obviously worthy of further structural optimization.
Finally, it should also be highlighted that 4c and 4d hydroxypropynyl derivatives proved to be the best performing derivatives at 2.8 µM, even though they were not able to completely abolish cell viability at any concentration. In this regard, they were more similar to the original imipridone ONC201 than I/A or I/B in the character of the antiproliferative effect.
2.3. Design of the Second Group of Potential Drugs With Activity Expected to be Superior to that Displayed By the Members of the First Set of Hybrids
With the overarching goal to identify additional hybrids with further increased anticancer potency, we also devised strategies for reasonable fragment refinements in 2a,b (Figure 4), which proved to be prominently efficient in the primary viability screening. Since 1a–e, the bis-para-benzyl substituted analogues of I/A and I/B, hydroxypropynoyl derivatives 2c and 2d, as well as the regioisomers 3a–d and 4a–d displayed decreased activity in the primary screening, we did not consider their structural tune-up for subsequent molecular design. On the other hand, since 2a and 2b displayed superior activity compared to their isomers I/A and I/B, respectively, these models were selected for the following structural modifications in the terminal functional groups. The triazole ring was envisaged to be replaced for amide bond generally recognized as its bioisostere [25,26,27,28]. Additional envisaged modifications included the introduction of fluorine substituents in the phenyl ring of the 4-phenyl-1H-1,2,3-triazol-1-yl moiety, fine-tuning the basicity of the 3-aminopropynyl nitrogen and, by exclusion of the carbon-carbon triple bond, reducing the distance between the phenyl ring and the amino group, which might be critical for antiproliferative activity. These modifications were motivated by the potential of the redesigned functionalization to influence parameters such as polarity, basicity, conformation, membrane permeability, as well as the pharmacodynamic profile of the drug candidates.
2.4. Multistep synthesis of the first group of the targeted imipridone hybrids
We prepared two types of isomeric amides, carrying N-acylamino- or N-arylcarbamoyl moiety in the para position of the N7-benzyl group with 3-aminopropynylbenzyl group pending on the N4 position of the central imipridone core (N-acylamino derivatives 23a,b and N-arylcarbamoyl derivatives 33a,b presented in Scheme 4 and 5, respectively). N-Ferrocenyl- and benzoylamino derivatives 23a,b were accessed by a three-step procedure started with the acid-catalyzed deprotection of Boc-protected amine 11b (cf. Scheme 1) followed by sequential N-acylation of the deprotected iodoamine XIb with ferrocenoylchloride 20 and benzoic acid anhydride 21 and terminating propargylamine-mediated Sonogashira coupling of the resulting amides 22a,b conducted under standard conditions (Scheme 4).
The second type of amides 33a,b were accessed by multistep convergent synthetic pathways (Scheme 5). The first pathway led to N-arylamides of 4-formylbenzoic acid 27a and 27b obtained from 4-formylbenzoylchloride 25 by treatment with aminoferrocene 26a and aniline 26b, respectively. The second pathway started with carboxymethylation of N-Boc-oxopiperidine (28) effected by dimethyl-carbonate-mediated (29). The generated oxocarboxylate (30) was condensed with 3-iodobenzyl-substituted 2-amino-3,4-dihydroimidazole (10b: cf. Scheme 1) constructing N7-Boc protected imipridone scaffold of which treatment with concentrated hydrochloride acid afforded 4-(3-iodobenzyl)-substituted cyclic secondary amine 31. Finally, the convergent synthesis was continued by triacetoxyhydroborate-mediated reductive alkylation of 31 by 27a and 27b to obtain amides 32a and 32b, respectively, which were then subjected to Sonogashira-coupling with propargylamine 17 resulting in the formation of the targeted imipridone models 33a,b.
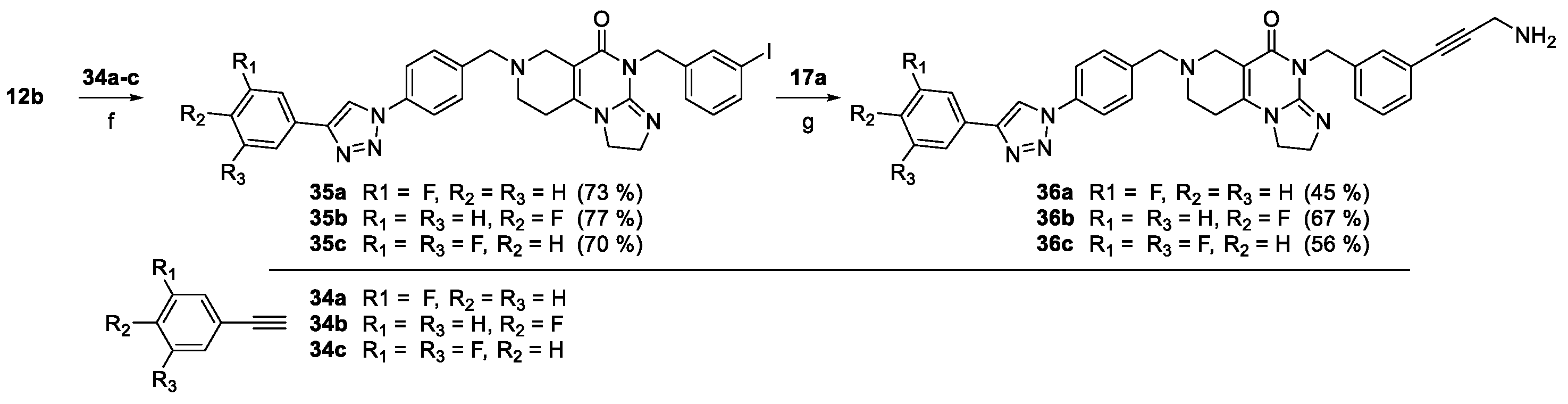
Compounds 36a–c, the fluorinated analogues of 2b were also synthesized by the sequence involving two orthogonal transition-metal-catalyzed procedures (Scheme 6). In the first step the CuAAc coupling of azido-imipridone 12b (cf. Scheme 1) and fluorinated phenylacetylenes 34a–c afforded iodinated fluorophenyltriazoles 35a–c which were then converted into 36a–c under the conditions of Sonogashira reaction using propargylamine 17a as alkyne component.
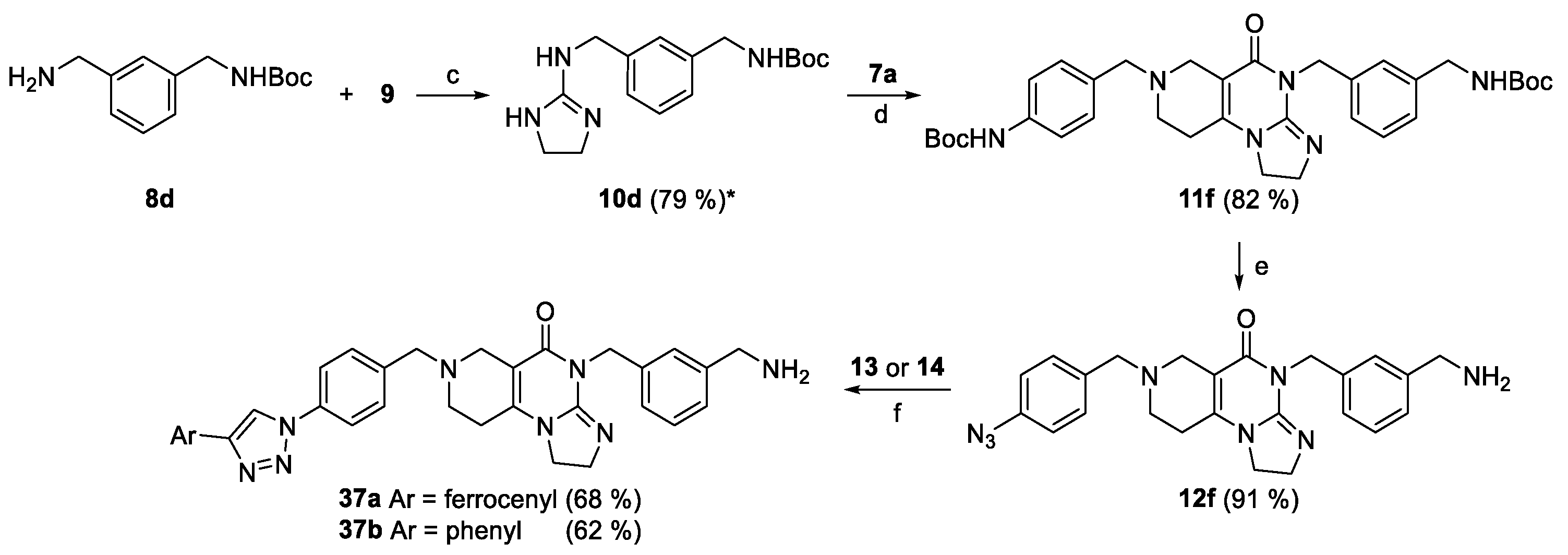
To achieve the envisaged distance shortening between the protruding basic amino group and the aryltriazolyl-substituted imipridone scaffold (see: Section 2.3), the aminopropynyl group was replaced for aminomethyl group by the reaction sequence outlined in Scheme 7. Accordingly, N-Boc-3-(aminomethyl)benzylamine 8d was incorporated in doubly protected imipridone-based diamine 11f by guanidine-formation (8d + 9 → 10d) and sequential annulation of intermediate 10d with methoxycarboxylated pipridone 7a. The standard deprotection of 11f was followed by selective diazotation-azidation sequence exploring the marked difference between the basicity of the aniline- and benzylamine moieties to obtain azide 12f, which was finally reacted with alkynes 13 and 14 under the conditions of CuAAc to obtain the targeted imipridone hybrids 37a and 37b, respectively.
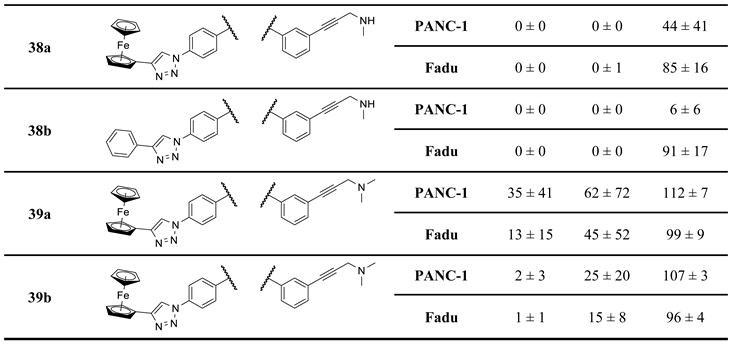
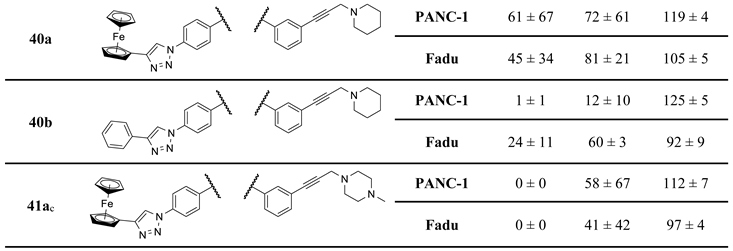
Finally, as the results of the first screen suggest that a basic aminopropynyl group is more beneficial in reducing cell viability than a hydroxypropynyl group (compare the activity of 2a,b and 2c,d: Section 2.2 and Table 1), we set out to investigate the effect of fine-tuning the basicity and size on antiproliferative activity. Thus, we introduced methylamino-, dimethylamino-, piperidinyl-, N-methylpiperazinyl- and morpholinyl groups in the aryltriazolyl-substituted imipridone scaffold using Sonogashira coupling of iodinated precursors 16a–d with N-propargylated amines 17c–g constructing a further subset of alkyne-tethered hybrids (38a,b, 39a,b, 40a,b, 41a,b and 42a,b: Scheme 8), the evaluation of which might lead to the recognition of further structure-activity relationships.
2.5. In vitro cell viability screening of the second set of the novel imipridone hybrids
Cell viability screenings of the second set of compounds were performed by the same protocols applied to evaluating the first set of models on PANC-1 and Fadu human cancer cell lines at three concentrations (2.8, 8.3 and 25 μM). The results of the viability screening are summarized in Table 2.
The cell viability values measured for 23a,b, 33a,b and 37a,b indicate that neither the replacement of triazole for any of the two types of amides, nor the reduction of the length of the basic side chain are beneficial for the antiproliferative activity.
On the other hand, mono- and difluorination at different sites of the phenyl group attached to the triazole ring led to a slight increase in efficacy at 8.3 µM on Fadu cells, but did not have appreciable influence on the activity against PANC-1 cells under the screening conditions, as reflected by the comparison of the data obtained for parent compound 2b and its fluorinated analogues 36a–c (Table 2). It is of pronounced importance that N-methylated compounds 38a,b were identified as the most potent models displaying the highest efficacy in the lowest concentration used in the screening, markedly outperforming primary amines 2a,b and 36a–c, particularly on PANC-1 cells. On the other hand, the viability data obtained with 39a,b, 40a,b, 41a,b and 42a,b indicate that further N-methylation or attachment of a six-membered cyclic amine to the propargyl residue substantially decreases the efficacy of the resulting tertiary amines, however, the basicity apparently slightly modulates their activity as reflected by the data measured for N-methylpiperazinyl and morpholinyl derivatives, the latter being almost completely ineffective on PANC-1 cells even at 25 µM concentration. This spectacular drop in efficacy can probably be ascribed to the increased bulkiness and/or the notably decreased basicity of the amine warhead.
Finally, based on the screening results, we hypothesized that fluorination of the triazolyl-attached phenyl group along with the monomethylation of the propargylamine moiety could further enhance the efficacy in an additive or even a synergistic manner leading to the development of the most potent representatives of the impiridone hybrids with aminopropynyl- and aryltriazolyl warheads. Accordingly, employing Sonogashira coupling of fluorinated iodoimipridones 35a–c with N-methylpropargylaine 17c, we prepared 43a–c as the last subset of our targeted hybrids (Scheme 9).
2.6. Dose–Response Study on Selected Compounds
The most efficacious compounds from the second set (38a,b) and 43a–c, the fluorinated analogues of 38b, were selected for systematic dose-response studies and determination of IC50 values. The measurements were conducted on PANC-1 and Fadu cells and non-tumorous primary fibroblast cells to assess the cytotoxicity profile and therapeutic window of the investigated compounds. For the sake of consistency, the dose-response curves were also detected for ONC201, I/A and I/B that served as relevant reference standards. The resulting dose-response curves are depicted in Figure 6, and the corresponding IC50 values are listed in Table 3.
Based on the results of viability screenings and the concepts of their rational design, we expected compounds 43a–c to display efficacy superior to that of 38a,b. However, the graphs and IC50 values indicate that 38a,b and 43a–c all exert highly similar activity on PANC-1 and Fadu cells, but compared to 38a,b fluorinated analogues 43a–c proved to be more cytotoxic towards the non-tumorous primary fibroblast cells featuring minimal if any therapeutic window as indicated by the selectivity indexes (SIs) falling in the range 1.0–1.4 (Table 3. columns 4 and 5). Nevertheless, pointing to a characteristic structure-activity relationship, compounds 38a,b and 43a–c with 1,3-disubstitutited N4-benzyl group produced 3–4 times lower IC50 in the two investigated cancer cell lines, than I/A and I/B, the previously published most potent imipridone derivatives carrying 1,4-disubstitutited benzyl group on N4. More importantly, besides the complete eradication of both types of cancer cells, 38a,b and 43a–c slightly outperform ONC201 in PANC-1 cells in terms of the efficient dose characterized by IC50 values. Nevertheless, the first-in-class imipridone features the lowest IC50 along with the highest SI in Fadu cells.
Taken together, 38a and 38b proved to be the most promising molecules among the novel imipridone hybrids presented in this study. However, their SI does not reach the acceptable level, and the concentration range (3–4 µM), in which these halogen-free drug candidates can almost completely eradicate both PANC-1 cells and Fadu cells, while displaying little to no cytotoxic effect towards non-tumorous fibroblast cells, is very narrow.
2.7. Colony forming assay
Based on the dose-response studies, the halogen-free molecules I/A,B and 38a–b were selected for a comparative colony forming assay in PANC-1 cells at 4 µM concentration, using ONC201 as positive control (Figure 6).
Compounds 38a,b substantially reduced the cancer cell population and left only traces of viable cells even at shorter (24 h) exposure. Following a longer (72 h) exposure with these hybrids, no viable cells were discernible in contrast to ONC201 and I/A,B. The results further support the outstanding efficacy of the newly designed 38a,b, in comparison with ONC201 and the parent compounds I/A,B.
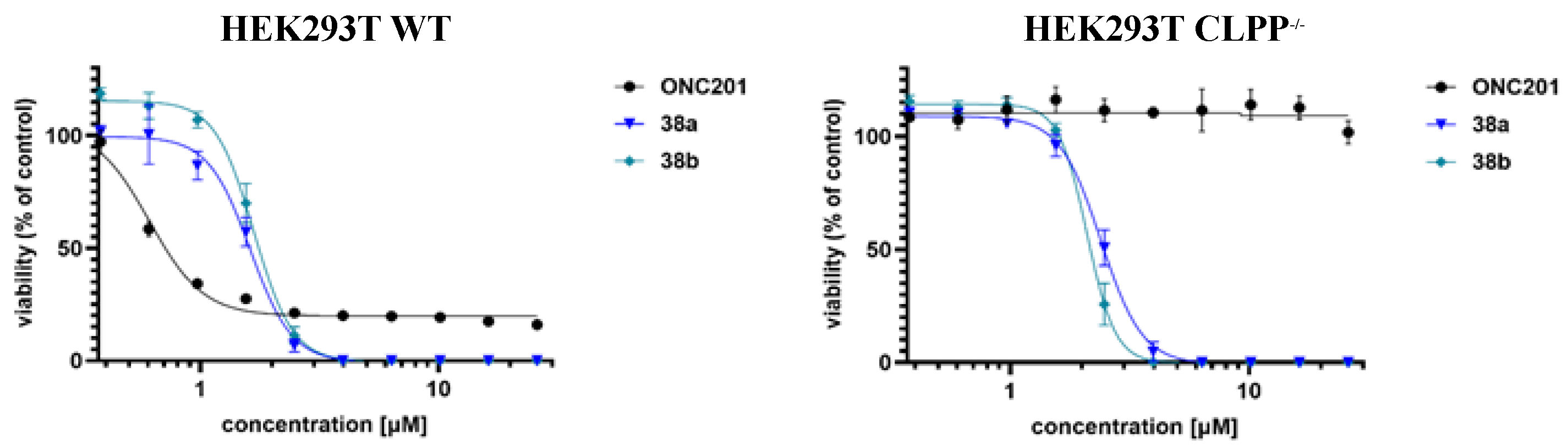
2.8. ClpP-Independent cytotoxicity of the selected imipridone hybrids
The role of mitochondrial ClpP activation in the cytotoxic activity of the most potent imipridone hybrids 38a,b was tested by comparing the dose-response curves from viability assays in HEK293T and HEK293T CLPP-/- cells. This approach is a well-established strategy for assessing the involvement of ClpP in the action of antiproliferative agents [29,30]. As it was expected, the reference imipridone ONC201 had no effect on the viability of HEK293T CLPP-/- cells in the lack of targetable ClpP, while the effect of 38a,b was maintained in the ClpP deficient cells (Figure 6), as also reflected by the IC50 values (Table 4). The results clearly indicated that the studied hybrid compounds reduce cell viability in a ClpP-independent manner under the applied experimental conditions. By means of in-depth x-ray diffraction studies, Houry et al. disclosed that the intercalation of the two terminal benzyl groups into the hydrophobic binding pockets of the ClpP-heptamer is critical for the formation of the activated peptidase complex [9], which is probably prevented by the two bulky and polar warheads of 38a,b. The activity of these compounds, on the other hand, might be attributed to the optimal separation and relative orientation of these warheads ensured by the imipridone skeleton. In addition, the tricyclic heterocycle comprising H-bond acceptor sites may also contribute to antiproliferative efficiency by binding to so far unidentified cellular target(s).
3. Conclusions
Supported by the results of high-throughput screens serving as guideline for iterative structural modifications, we synthesized a library of systematically designed novel imipridone hybrids with optimally positioned alkyne- and triazole-linked warheads and evaluated their antiproliferative activity on PANC-1 and Fadu cells. Preliminary mechanistic studies demonstrated that 38a,b, identified as the most potent targeted compounds exert their effect in a ClpP-independent manner. Their promising activity is obviously associated with special cooperative interactions of the so far undisclosed cellular target(s) with the ferrocenyl/phenyltriazolyl- and N-methylaminopropargyl groups optimally positioned on the central imipridone core. This distinctive feature sets them apart from the conventional ClpP agonist imipridone-based drugs. In terms of efficient doses, these drug candidates are comparable to ONC201. In terms of overcoming treatment-resistance, reflected by the ability to completely eradicate the treated cancer cells in an applicable concentration range around 4 μM, they have proved superior to the emblematic first-in-class imipridone. Nevertheless, they showed a limited tumor-selectivity with a small therapeutic window, which problem needs to be addressed during the ongoing development. The promising results and well-defined novel structure-activity relationships revealed in this study indicate that additional investigations are warranted identifying the cellular molecular target(s) of these drug candidates to elucidate their exact mechanism of action. The revealed mechanistic details can be explored in a reasonable design of further structurally related fine-tuned anticancer compounds. The aim is to decrease the toxicity on non-cancerous cells and thus widen the therapeutic window while retaining the potential in drastic eradication of cancer cells at lowered efficient doses. The desired enhancement in selectivity is planned to be achieved by conjugation of the drugs to peptide and nanocarriers leading to constructions that allow targeted drug release ensuring improved clinical potential in treatment of tumors resistant to traditional therapies.
4. Materials and Methods
All chemicals were obtained from commercially available sources (Merck, Budapest, Hungary; Fluorochem, Headfield, UK; Molar Chemicals, Halásztelek, Hungary; VWR, Debrecen, Hungary) and used without further purifications. Merck Kieselgel (230–400 mesh, 60 Å) was used for flash column chromatography. Melting points (uncorrected) were determined with a Büchi M-560. The 1H and 13C-NMR spectra were recorded in CDCl3 or in DMSO-d6 solution in 5 mm tubes at room temperature on a Bruker DRX-500 spectrometer (Bruker Biospin, Karlsruhe, Baden Württemberg, Germany) at 500 (1H) and 125 (13C) MHz, with the deuterium signal of the solvent as the lock and TMS as internal standard (1H and 13C). The 2D-HSQC and HMBC spectra, which support the exact assignments of 1H- and 13C NMR signals, were registered by using the standard Bruker pulse programs. Exact mass measurements were performed on a high-resolution Waters ACQUITY RDa Detector (Waters Corp., Wilmslow, UK) equipped with electrospray ionization source using on-line UHPLC coupling. UHPLC separation was performed on a Waters ACQUITY UPLC H-Class PLUS system using a Waters Acquity UPLC BEH C18 column (2.1 x 150 mm, 1.7 µm). Samples were dissolved in MeOH/Water 5:95 V/V, 5-5 µL sample solutions were injected. Linear gradient elution (0 min 5% B, 1.0 min 5% B, 7.0 min 80% B, 7.1 min 100% B, 8.0 min 100% B, 8.1 min 5% B, 12.0 min 5% B) with eluent A (0.1% formic acid in water, V/V) and eluent B (0.1% formic acid in Methanol, V/V) was used at a flow rate of 0.200 mL/min at 45 °C column temperature. High-resolution mass spectra were acquired in the m/z 50-2000 range in positive ionization mode. Leucine enkephalin peptide was used for single Lock Mass calibration correction.
A detailed spectral characterization with the 1H-, 13C-NMR and HRMS data of the targeted hybrid compounds screened in the biological assays (Table 1 and 2) are listed in the Supplementary Material (S1). The structures of other intermediates can be regarded as chemically evidenced through the spectroscopically confirmed structures of the derived imipridone hybrids.
For each spectroscopically characterized compound the numbering of atoms used for the assignment of 1H- and 13C NMR signals do not correspond to IUPAC rules reflected from the given systematic names (Supplementary Material).
Assignments of the NMR data, copies of the NMR spectra of the novel screened imipridone hybrids (S2–S13), the copies of the 1H- and 13C-NMR spectra (S14–S50) and the HRMS spectra (S51–S87) are included in the Supplementary Materials
4.1. Synthetic Procedures
The yields of the compounds of which preparation is presented in general procedures are listed in Scheme 1, Scheme 2, Scheme 3, Scheme 4 and Scheme 5.
4.1.1. Methods (a) and (b) – General procedure for the synthesis of the methyl 1-(tert-butoxycarbonylaminobenzyl)-4-oxopiperidine-3-carboxylates 7a,b (Methods a. and b.)
Methyl acrylate (15 mmol, 5 eq.) was added to a solution of a 3-(Boc-amino)benzylamine (5a) or 4-(Boc-amino)benzylamine (5b) (3 mmol, 1 eq.) in methanol (5 mL), and the mixtures were stirred at room temperature for 48 h. The solutions were concentrated in vacuo to obtain the crude diester products (6a,b). Without further purification, they were dissolved in dry THF (8 mL), and NaH (15 mmol, 5 eq.) was added to the solutions gradually at 0 °C while stirring the mixtures. The suspensions were cooled down to room temperature. After stirring for an additional 2 h, the suspensions were poured over crashed ice (100 g), and the pH of the resulting slurry was set to 7 with acetic acid. The aqueous phases were extracted with ethyl acetate (3 × 40 mL). The combined organic phases were dried over Na2SO4 and evaporated to dryness in vacuo to obtain the 7a,b, which were used without further purification in the subsequent cyclization step.
4.1.2. Synthesis of 2-(Methylthio)-4,5-dihydro-1H-imidazole-1-carboxylate (9)
2-Methylthio-4,5-dihydroimidazolium iodide (50 g, 0.205 mol, 1 eq.) and triethylamine (70 mL, 50.8 g, 0.502 mol, 2.45 eq.) were dissolved in DCM (250 mL). The solution was cooled down to 0 °C. Methylchloroformate (22 mL, 27 g, 0.28 mol, 1.4 eq.) was added dropwise to this cold solution which was then allowed to warm up to room temperature stirred overnight and concentrated in vacuo. After addition of EtOAc (400 mL) the suspension was stirred for 30 min. The precipitated ammonium salts were filtered off and washed with EtOAc (2 × 50 mL). The combined solution was evaporated to dryness. The solid residue was triturated with water (200 mL), filtered off and dried under vacuo to obtain 9 as a white solid (27.2 g, 0.156 mol, 76%) [23].
4.1.3. General Procedure for the Synthesis of Cyclic Guanidines 10a–d (Method c)
Primary amines (8a–d) (10 mmol, 1 eq.) were dissolved in a mixture of methanol (24 mL) and acetic acid (6 mL). To the solutions, 2-(methylthio)-4,5-dihydro-1H-imidazole-1-carboxylate (9) (2.18 g, 12.5 mmol, 1.25 eq.) was added, and the resulting mixtures were stirred at reflux for 18 h. The solutions were concentrated in vacuo, and the oily residues were dissolved in DCM (100 mL) and were washed with 3 M NaOH solution (2 × 10 mL), and brine (1 × 10 mL). The organic phases were dried over Na2SO4 and concentrated in vacuo. The products were used without further purification in the subsequent step [23].
4.1.4. Method (d) – General Procedure for the Synthesis of Imipridone Scaffold 11a–f (Method d).
The appropriate N-substituted methylcarboxylate piperidone 7a,b (2 mmol, 1 eq.) and cyclic guanidine (10a–d) (2 mmol, 1 eq.) were dissolved in methanol (10 mL). To this mixture a 3.6 M methanolic solution of NaOMe was added (0.69 mL, 2.5 mmol, 1.25 eq.) and stirred at reflux temperature for 12 h. In case of reactions with 10a, the mixture was cooled after 12 h, and the precipitated white crystalline product (11a, 11d) was filtered off, washed twice with cold methanol (2 × 1 mL) and dried in vacuo. In other cases, the mixtures were cooled, concentrated in vacuo, dissolved in DCM (100 mL), washed with water (2 × 10 mL) and brine (10 mL), dried over Na2SO4, and concentrated in vacuo. The crude yellow oily products were purified with column chromatography on silica using DCM:MeOH:NH3 = 15:1:0.1) as eluent. The collected band was crystallized from Et2O to obtain 11b–c and 11e,f, as yellowish white powder.
4.1.5. General Procedure for the Synthesis of Azidoimipridones 12a–f (Method e)
The appropriate N-Boc protected compound 11a–f (1 mmol, 1 eq.) was dissolved in cc. HCl (5 mL). After stirring for 30 min at 0 °C, using a syringe aqueous NaNO2 solution (2 mmol, 2 eq. in 1.5 mL water) was added dropwise to the solution which was then stirred for 45 min at 0 °C. To the resulting cold mixture solid NaN3 (10 mmol, 10 eq.) was added in portions over 15 min, stirred for further 30 min at 0 °C, then allowed to warm up to room temperature and stirred for an additional 1 h. The acidic mixture was neutralized with Na2CO3 and extracted with DCM (3 × 40 mL). The combined organic phases were washed with water (2 × 10 mL) and brine (1 ×10 mL), dried over Na2SO4, and concentrated in vacuo. The products were crystallized from Et2O to isolate 12a–f as white solids.
4.1.6. General Procedure for Copper(I) Catalyzed 1,4-Azide-alkyne Cycloadditions (Method f)
The appropriate azidoimipridone (12a–f) (0.5 mmol, 1 eq.), alkyne component (13–15, 34a–c) (0.5 mmol, 1 eq.) and CuI (9.5 mg, 0.05 mmol, 0.1 eq.) were dissolved in DMSO (2 mL) and stirred for 12 hours at room temperature in a closed vial. After 12 hours, the mixture was poured in water (20 mL) and stirred for 20 min. The precipitate was filtered off and washed with water (5 × 10 mL). The filter was washed with DCM (4 × 10 mL). The combined organic filtrates were dried over Na2SO4 and concentrated in vacuo. The crude product was purified using column chromatography (silicagel, DCM:MeOH:NH3 = 15:1:0.1) and crystallized from Et2O to yield amber/orange (in case of ferrocene-containing compounds – 16a, 16d, 16g, 16i, 37a) or white powder (16b–c, 16e–f, 16h, 16j, 35a–c, 37b).
4.1.7. General Procedure for the Sonogashira coupling reactions (Method g)
The appropriate iodoimipridone (16a–e, 16g–j, 22a,b, 32a,b or 35a–c) (0.2 mmol, 1 eq.), propargyl derivative (17a–g or 18) (0.2 mmol, 1 eq.), CuI (7.6 mg, 0.04 mmol, 0.2 eq.), Pd(PPh3)2Cl2 (14.0 mg, 0.02 mmol, 0.1 eq.) and DIPEA (129 mg, 1 mmol, 5 eq.) were dissolved in DMF (2 mL) and stirred for 24 h. After 24 h, the mixture was poured in water (30 mL) and stirred for 20 min. The precipitate was filtered off and washed with water (5 × 10 mL). The filter was washed with DCM (4 × 20 mL). The combined organic filtrate was dried over Na2SO4 and concentrated in vacuo. The crude product was purified using column chromatography (silicagel, DCM:MeOH:NH3 = 15:1:0.1), and crystallized from Et2O to yield amber/orange (in case of ferrocene-containing compounds – 1b, 2a, 2c, 3a, 3c, 4a, 4c, 23a, 33a, 38a, 39a, 40a, 41a, 42a) or white powder (1a, 1c, 2b, 2d, 3b, 3d, 4b, 4d, 19, 23b, 33b, 36a–c, 38b, 39b, 40b, 41b, 42b, 43a–c).
4.1.8. Synthesis of acrylamido imipridone 1e (Sequential application of Methods h. and i)
Compound 1e was accessed by sequential acylation and deprotection procedures.
Method h.): Aniline derivative 19 (68.3 mg, 0.1 mmol, 1 eq.) and DIPEA (26 mg, 0.2 mmol, 2 eq.) were dissolved in DCM (10 mL). To this mixture a solution of acryloyl chloride (12 mg, 0.13 mmol, 1.3 eq.) in DCM (5 mL) was added dropwise at room temperature under N2 flow. The resulting solution was stirred for 4 h at room temperature, then concentrated in vacuo.
Method i.): To the residue concentrated HCl (2 mL) was added, and the solution was stirred for 20 minutes at room temperature, then diluted with 8 mL of water. The pH was set to 10 with K2CO3 and the mixture was extracted with DCM (3 × 15 mL). The combined organic phase was washed with water (2 × 5 mL) and brine (5 mL), dried over Na2SO4, concentrated in vacuo. The residue was purified by column chromatography on silica using DCM:MeOH:NH3 (10:1:0.1) as eluent. The collected band was evaporated, and the oily residue was crystallized from Et2O to obtain 1e as a white powder.
4.1.9. Deprotection of N-Boc protected iodoimipridone 11b to produce iodinated free amine XIb (Method i)
Compound 11b (500 mg, 0.82 mmol) and concentrated HCl acid (5 mL) were added to a flask and stirred at room temperature. After 20 minutes the solution was diluted with water (45 mL) and the pH was set to 10 with K2CO3. The mixture was extracted with DCM (3 × 30 mL). The combined organic phase was washed with water (2 × 10 mL) and brine (10 mL), dried over Na2SO4, and concentrated in vacuo yielding a white solid (XIb) (414 mg, 0.81 mmol, 99%), which was used in the subsequent acylation reactions without further purification.
4.1.10. Synthesis of amides 22a and 27a,b (Method j)
Acyl chlorides [ferrocenoyl chloride (20) or 4-formylbenzoyl chloride (25)] (0.8 mmol, 2 eq.) were dissolved in DCM (10 mL) and a solution of the appropriate amino compound [XIb, aminoferrocene (26a) or aniline (26b)] (0.4 mmol, 1 eq.) and DIPEA (155 mg, 1.2 mmol, 3 eq.) in DCM (10 mL) was added dropwise at 0 °C under N2 flow. The mixtures were stirred at 0°C for 30 minutes, then allowed to warm up to room temperature and stirred for an additional 1 h. In the course of the synthesis of imipridone compound 22a the reaction mixture was washed with saturated Na2CO3 solution (2 × 5 mL), water (1 × 5 mL) and brine (1 × 5 mL). In the course of the synthesis of compounds 27a,b the reaction mixtures were subsequently washed with 1M HCl (2 × 5 mL) and saturated Na2CO3 solution. The combined organic phases were dried over Na2SO4, concentrated in vacuo to obtain an orange oil (22a) (280 mg), brown-red solid (27a) (127 mg, 0.38 mmol, 95%) or white powder (27b) (88 mg, 0.39 mmol, 98%). Compounds 27a,b were used in the subsequent reductive alkylation step without further purification, while 22a was purified by column chromatography on silicagel using DCM:MeOH:NH3 = 30:1:0.1 as eluent to yield an orange solid foam (61 mg, 0.084 mmol, 21%).
4.1.11. Synthesis of benzamide 22b (Methods k)
Compound XIb (205 mg, 0.4 mmol, 1 eq.), benzoic anhydride (21) (181 mg, 0.8 mmol, 2 eq.) and DIPEA (155 mg, 1.2 mmol, 3 eq.) were dissolved in DCM (20 mL) and the solution was stirred at reflux for 24 h, then cooled down, washed with 10% NaOH solution (2 × 5 mL), water (1× 5 mL) and brine (1× 5 mL). The organic phase was dried over Na2SO4, concentrated in vacuo, and crystallized from Et2O to obtain 22b (195 mg, 0.316 mmol, 79%) as a white powder.
4.1.12. Synthesis of ferrocenoyl chloride 20 and 4-formylbenzoyl chloride 25 (Method l)
The appropriate carboxylic acid [ferrocene carboxylic acid (460 mg, 2 mmol, 1 eq.) or 4-formylbenzoic acid (24) (300 mg, 2 mmol, 1 eq.)] was added to DCM (10 mL). To this mixture oxalyl chloride (1.02 g, 8 mmol, 4 eq.) dissolved in DCM (5 mL) then 2 drops of DMF were added sequentially at room temperature. The resulting mixture was stirred for 1 h, then concentrated in vacuo yielding an orange-brown oil (20) or a white solid (25), which was then triturated with hexane (10 mL). The insoluble impurities were filtered off and the filtrates were concentrated in vacuo to obtain ferrocenoyl chloride (20) (484 mg, 1.95 mmol, 97%) as an orange red oil or 4-formylbenzoyl chloride (25) (311 mg, 1.84 mmol, 92%) as a white powder.
4.1.13. Synthesis of the methyl N-Boc-4-oxopiperidine-3-carboxylate (30) (Method m)
N-Boc-4-oxopiperidin (28) (3.0 g, 15 mmol, 1 eq.) and dimethyl-carbonate (29) (6.8 g, 75 mmol, 5 eq.) were dissolved in dry toluene (22 mL) under Ar atmosphere. To this solution, NaH (1.8 g, 75 mmol, 5 eq.) was added in small portions and the resulting mixture was stirred at room temperature for 3 h and refluxed for an additional 2 h under Ar. The mixture was poured over ice (100 g) and the pH was set to 7 with acetic acid. The neutral slurry was extracted with EtOAc (3 × 100 mL). The combined organic phase was dried over Na2SO4 and concentrated in vacuo to yield 30 as a yellow oil (3.48 g, 13.5 mmol, 90%) which was used as cyclisation partner in the subsequent step without further purification.
4.1.14. Synthesis of 4-(3-iodobenzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]- pyrimidin-5(1H)-one (31) (sequential application of Methods d. and i.)
Compound 31 was accessed by the sequential procedures used for the construction of the imipridone scaffold.
Method d): Methyl N-Boc-4-oxopiperidine-3-carboxylate (30) (180 mg, 0.7 mmol, 1 eq.) and the appropriate cyclic guanidine (10b) (211 mg, 0.7 mmol, 1 eq.) were dissolved in methanol (10 mL) and 3.6 M NaOMe solution in methanol (0.24 mL, 0.825 mmol, 1.25 eq.) was added. The mixture was stirred at reflux for 12 h, then cooled, concentrated in vacuo, dissolved in DCM (40 mL), washed with water (2 × 7 mL) and brine (7 mL), dried over Na2SO4, and concentrated in vacuo. The crude yellow oily product was purified with column chromatography (silicagel, DCM:MeOH:NH3 = 20:1:0.1) yielding a light-yellow solid foam (234 mg, 0.46 mmol, 66%).
Method i.): The product of the previous step was dissolved in concentrated HCl (3 mL) and stirred at room temperature. After 20 minutes the solution was diluted with water (30 mL) and the pH was set to 10 with K2CO3. The mixture was extracted with DCM (3 × 20 mL). The combined organic phase was washed with water (2 × 5 mL) and brine (5 mL), dried over Na2SO4, and concentrated in vacuo yielding a light-yellow solid foam (31) (178 mg, 0.436 mmol, 95% for the deprotection, 62% cumulative yield), which was used without further purification.
4.1.15. Synthesis of compounds 32a–b by reductive alkylation (Method n.)
The appropriate aldehyde (27a–b, 0.25 mmol, 1 eq.) was dissolved in DCM (10 mL). To this solution 31 dissolved in DCM (10 mL) was added. The mixtures were stirred at room temperature for 20 minutes. Then NaHB(OAc)3 (74 mg, 0.35 mmol, 1.4 eq.) was added in small portions over the course of 15 minutes, and the resulting mixtures were stirred for an additional 2.5 hours at room temperature. The reaction was monitored by TLC and upon completion a saturated solution of K2CO3 (10 mL) was added and the mixtures were stirred vigorously for 15 minutes. The organic phases were separated, and the aqueous phases were extracted with DCM (3 × 5 mL). The combined organic phases were washed with water (2 × 5 mL) and brine (1 × 5 mL), dried over Na2SO4, and concentrated in vacuo. The crude product was purified using column chromatography (silicagel, DCM:MeOH:NH3 = 15:1:0.1) and crystallized from Et2O to obtain 32a (105 mg, 0.145 mmol, 58%) as an orange and 32b (121 mg, 0.196 mmol, 78%) as a white powder.
4.1.16. Synthesis of propargylamine derivatives 17e–g
The appropriate amine (piperidine, N-methylpiperazine or morpholine) (5 mmol, 1 eq.) was dissolved in acetonitrile (20 mL) and K2CO3 (1.4 g, 10 mmol, 2 eq.), then propargyl bromide (80% in toluene) (820 mg, 5.5 mmol, 1.1 eq.) was added while stirring at room temperature. The resulting suspensions were stirred overnight at room temperature in a closed vial under argon. The mixtures were concentrated in vacuo and triturated with Et2O (30 mL). The insoluble salts were filtered off and the filtrates were concentrated in vacuo and purified by column chromatography on silica using DCM:MeOH:NH3 (15:1:0.1) as eluent to obtain 17e (123 mg, 1 mmol, 20%), 17f (343 mg, 2.5 mmol, 50%) and 17g (463 mg, 3.7 mmol, 74%) as orange-yellow oils.
4.2. Biological Assays
4.2.1. Cell Culturing
Pharynx squamous cell carcinoma cell line Fadu (HTB-43™), pancreatic ductal adenocarcinoma cell line PANC-1 (CRL-1469™), primary human fibroblasts, and HEK293T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco/Thermo Fisher Scientific) supplemented with 10% (V/V) fetal bovine serum (FBS, Gibco/Thermo Fisher Scientific) in a humidified atmosphere at 37°C and 5% CO2. The Fadu and PANC-1 cells were obtained from the American Type Culture Collection (ATCC), and the authentication of these cell lines was validated by STR DNA analysis (Eurofins Scientific, Luxembourg, Luxembourg). All cell lines were routinely screened for the absence of mycoplasma infection (DAPI staining).
Primary fibroblast cultures were prepared from human skin biopsies and maintained in DMEM, supplemented with 10% FBS, 1% penicillin–streptomycin, and 1% MEM Non-Essential Amino Acids Solution (Thermo Fisher Scientific). We received HEK293T WT and HEK293T CLPP-/- cells as a gift from Aleksandra Trifunovic for the validation of the CLPP-independent mechanism of action.
4.2.2. CellTiter-Glo Cell Viability Assay Including the Assessment of ClpP-Coupled Action
Effects of the synthesized compounds on cell viability were measured by CellTiter-Glo® luminescent cell viability assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Briefly, cells were seeded onto a flat-bottomed, white 96 well plate (BRANDplates, cat. no.: 781965)), with the following density: PANC-1: 750 cells/well, Fadu: 2000 cells/well, Primary Fibroblast: 2000 cells/well, HEK293T WT: 1000 cells/well, HEK293T CLPP-/-: 2500 cells/well. After seeding, cells were incubated further for 48 h before the treatment. Cells were treated with the compounds for 72h. Three concentrations (25.0 µM; 8.3 µM; 2.8 µM) were applied at the initial cell viability screening, while at the determination of dose response curves 1.5-fold serial diluted compound concentrations were applied (range: 26 µM – 0.7 µM). After the treatment, the luminescence signal was recorded using a microplate reader (BioTek Synergy 2 Multi-Mode Reader, BioTek, Winooski, VT, USA). Dose–response curves (non-linear regression model) were generated and IC50 values were determined using Graph Pad Prism 8 software (GraphPad Software, San Diego, CA, USA). The selectivity index (SI) of each molecule tested was calculated as the IC50 on primary fibroblasts / IC50 on cancer cells.
4.2.3. Colony formation assay
Long-term cell survival after treatments was determined by clonogenic assay. PANC-1 cells were seeded in a transparent 6-well cell culture plate (VWR) (density: 1000 cells/well). After seeding, cells were incubated for 72 h before the treatment. Cells were treated with the compounds at 4 µM for 24 h or 72 h. Then, the medium was removed, and the cells were washed with phosphate-buffered saline (PBS). The cells formerly treated for 24 or 72 h were further incubated in cell culture medium for 5 or 3 days, respectively. Thereafter, the medium was removed, and the cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min. After fixation, the cells were washed with PBS, and 0.5% w/v crystal violet (CV) solution was added. After 1 h, the CV solution was removed, and the cells were washed thoroughly with water. Images were created by Corel Photo-Paint 2019.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Copies of the NMR spectra.
Author Contributions
Conceptualization, A.C., T.C and J.M.; methodology, T.C., J.M., I.M., B.G., A.V., D.P., and G.S.; validation, B.G, A.V., J.M. I.M. and G.S. formal analysis, B.G, A.V., J.M. I.M. D.P. and G.S; investigation, T.C., J.M., I.M., B.G., A.V. and A.C.; resources, A.C., M.C. and G.S.; data curation, J.M., I.M., B.G., A.V. and G.S.; writing—original draft preparation, T.C., J.M. and A.C.; writing—review and editing, J.M., I.M., B.G., A.V., M.C. and A.C.; visualization, A.C. T.C. and I.M.; supervision, A.C. J.M. and G.S; project administration, T.C.; funding acquisition, A.C., M.C., and G.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Hungarian Scientific Research Fund [OTKA K_129037], ELTE Thematic Excellence Programme Synth+, supported by the Hungarian Ministry for Innovation and Technology and the Lendület (Momentum) Programme of the Hungarian Academy of Sciences. Project no. TKP2021-EGA-24 has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA funding scheme.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data generated and analyzed during our research are not available in any public database or repository but will be shared by the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Crowder, R.N.; El-Deiry, W.S. First-In-Class Small Molecule ONC201 Induces DR5 and Cell Death in Tumor but Not Normal Cells to Provide a Wide Therapeutic Index as an Anti-Cancer Agent. PloS One 2015, 10, e0143082. [Google Scholar] [CrossRef]
- Allen, J.E.; Kriegsfield, G.; Mayes, P.A.; Patel, L.; Dicker, D.T.; Patel, A.S. , Dolloff, N.G.; Messaris, E.; Scata, K.A., Wang, W.; Zhou, J-Y.; Wu, G.S.; El-Deiry, W.S.. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Science Transl. Med, 2013; 5, 171ra17. [Google Scholar] [CrossRef]
- Prabhu, V.V.; Morrow, S.; Rahman Kawakibi, A.; Zhou, L.; Ralff, M.; Ray, J.; Jhaveri, A.; Ferrarini, I.; Lee, Y.; Parker, C.; Zhang, Y.; Borsuk, R.; Chang, W.-Y.; Honeyman, J.N.; Tavora, F.; Carneiro, B.; Raufi, A.; Huntington, K.; Carlsen, L.; Louie, A.; El-Deiry, W.S. ONC201 and Imipridones: Anti-Cancer Compounds with Clinical Efficacy. Neoplasia 2020, 22, 725–744. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Kline, C.L.; Ralff, M. D, Lev, A.; Lulla. A.; Zhou, L., Olson, G.L.; Nallaganchu, B.R.; Benes, C.H.; Allen, J.E.; Prabhu, V.V; Stogniew, M.; Oster, W.; El-Deiry, W.S. Preclinical evaluation of the imipridone family, analogs of clinical stage anti-cancer small molecule ONC201, reveals potent anti-cancer effects of ONC212. Cell Cycle, 2017, 16, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, J.; Kojima, K.; Chachad, D.; Ruvolo, P.; Ruvolo, V.; Jacamo, R.O.; Borthakur, G.; Mu, H.; Zeng, Z.; Tabe, Y.; et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci. Signal 2016, 9, ra17. [Google Scholar] [CrossRef]
- Kline, C.L.; Van den Heuvel, A.P.; Allen, J.E.; Prabhu, V.V.; Dicker, D.T.; El-Deiry, W.S. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2α kinases. Sci. Signal 2016, 9, ra18. [Google Scholar] [CrossRef]
- Wedam, R.; Greer, Y. E.; Wisniewski, D. J.; Weltz, S.; Kundu, M.; Voeller, D.; Lipkowitz, S. Targeting mitochondria with CLPP agonists as a novel therapeutic opportunity in breast cancer. Cancers, 2023, 15, 1936. [Google Scholar] [CrossRef]
- Luo, B.; Ma, Y.; Zhou, Y.; Zhang, N.; Luo, Y. Human Clpp protease, a promising therapy target for diseases of mitochondrial dysfunction. Drug Discovery Today 2021, 26, 968–981. [Google Scholar] [CrossRef]
- Mabanglo, M.F.; Wong, K.-S.; Barghash, M.M.; Leung, E.; Chuang, S.H.W.; Ardalan, A.; Majaesic, E.M.; Wong, C.J.; Zhang, S.; Lang, H.; S. Karanewsky, D.S.; Iwanowicz, A.A.; Graves, L.M.; Iwanowicz, E.J.; Gingras, A.-C.; Houry, W.A. Potent ClpP agonists with anticancer properties bind with improved structural complementarity and alter the mitochondrial N-terminome. Structure 2023, 31, 185–200. [Google Scholar] [CrossRef]
- Nii, T.; Prabhu, V.V.; Ruvolo, V.; Madhukar, N.; Zhao, R.; Mu, H.; Heese, L.; Nishida, Y.; Kojima, K.; Garnett, M.J.; McDermott, U.; Benes, C.H.; Charter, N.; Deacon, S.; Elemento, O.; Allen, J.E.; Oster, W.; Stogniew, M.; Ishizawa, J.; Andreeff, M. Imipridone ONC212 activates orphan G protein-coupled receptor GPR132 and integrated stress response in acute myeloid leukemia. Leukemia 2019, 33, 2805–2816. [Google Scholar] [CrossRef]
- Bonner, E.R.; Waszak, S.M.; Grotzer, M.A.; Mueller, S.; Nazarian, J. Mechanisms of Imipridones in Targeting Mitochondrial Metabolism in Cancer Cells. Neuro Oncol. 2021, 23, 542–556. [Google Scholar] [CrossRef]
- 12. Ishizawa, J.; Zarabi, S,F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; Ruvolo, Samir, V.R.; Barghout, H.; Nishida, Y.; Hurren, R.; Ma, W.; Gronda, M.; Link, T.; Wong, K.; Mabanglo, M.; Kojima, K.; Borthakur, G.; MacLean, N.; Ma, M.C.J.; Leber, A.B.; Minden, M.D., Houry, W.; Kantarjian, H.; Stogniew, M.; Raught, B.; Pai, E.F.; Schimmer, A.D.; Andreeff, M. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737. [CrossRef] [PubMed]
- Lev, A.; et al. Anti-Pancreatic Cancer Activity of ONC212 Involves the Unfolded Protein Response (UPR) and Is Reduced by IGF1-R and GRP78/BIP. Oncotarget 2017, 8, 81776–81793. [Google Scholar] [CrossRef] [PubMed]
- Ferrarini, I.; Louie, A.; Zhou, L.; El-Deiry, W. S. ONC212 Is a Novel Mitocan Acting Synergistically with Glycolysis Inhibition in Pancreatic Cancer. Mol. Cancer Ther. 0962. [Google Scholar] [CrossRef]
- Wagner, J.; et al. Preclinical Evaluation of the Imipridone Family, Analogs of Clinical Stage Anti-Cancer Small Molecule ONC201, Reveals Potent Anti-Cancer Effects of ONC212. Cell Cycle 2017, 16, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Jacques, S.; et al. Imipridone Anticancer Compounds Ectopically Activate the ClpP Protease and Represent a New Scaffold for Antibiotic Development. Genetics 2020, 214, 1103–1120. [Google Scholar] [CrossRef]
- Nii, T.; et al. Imipridone ONC212 Activates Orphan G Protein-Coupled Receptor GPR132 and Integrated Stress Response in Acute Myeloid Leukemia. Leukemia 2019, 33, 2805–2816. [Google Scholar] [CrossRef]
- Ishizawa, J.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737. [Google Scholar] [CrossRef]
- Graves, P. R.; et al. Mitochondrial Protease ClpP Is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chem. Biol. 2019, 14, 1020–1029. [Google Scholar] [CrossRef]
- Prabhu, V. V.; et al. Single Agent and Synergistic Combinatorial Efficacy of First-in-Class Small Molecule Imipridone ONC201 in Hematological Malignancies. Cell Cycle 2018, 17, 468–478. [Google Scholar] [CrossRef]
- Tu, Y.; et al. The Imipridone ONC201 Induces Apoptosis and Overcomes Chemotherapy Resistance by Up-Regulation of Bim in Multiple Myeloma. Neoplasia 2017, 19, 772–780. [Google Scholar] [CrossRef]
- Voltan, R.; Secchiero, P.; Casciano, F.; Milani, D.; Zauli, G.; Tisato, V. Redox signalling and oxidative stress: Cross talk with TNF-related apoptosis inducing ligand activity. Int. J. Biochem. Cell Biol. 2016, 81, 364–374. [Google Scholar] [CrossRef]
- Bárány, P.; Oláh, R.S.; Kovács, I.; Czuczi, T.; Szabó, C.L.; Takács, A.; Lajkó, E.; Láng, O.; Kőhidai, L.; Schlosser, G.; et al. Ferrocene-Containing Impiridone (ONC201) Hybrids: Synthesis, DFT Modelling, In Vitro Evaluation, and Structure–Activity Relationships. Molecules 2018, 23, 2248. [Google Scholar] [CrossRef] [PubMed]
- Czuczi, T.; Murányi, J.; Bárány, P.; Móra, I.; Borbély, A.; Csala, M.; Csámpai, A. Synthesis and Antiproliferative Activity of Novel Imipridone–Ferrocene Hybrids with Triazole and Alkyne Linkers. Pharmaceuticals 2022, 15, 468. [Google Scholar] [CrossRef] [PubMed]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. ; The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discovery Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Carmona, A.V.; Tiwari, A.K.; Trippier, P.C. Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. J. Med. Chem. 2020, 63, 12290–12358. [Google Scholar] [CrossRef]
- Rečnik, L.-M.; Kandioller, W.; Mindt, T.L. 1,4-Disubstituted 1,2,3-Triazoles as Amide Bond Surrogates for the Stabilisation of Linear Peptides with Biological Activity. Molecules 2020, 25, 3576. [Google Scholar] [CrossRef]
- Huestis, M.P.; Terrett, J.A. Simple strategy towards amide bioisosteres. Nat. Chem. 2022, 14, 120–121. [Google Scholar] [CrossRef]
- Seiferling, D.; Szczepanowska, K.; Becker, C.; Senft, K.; Hermans, S.; Maiti, P.; Trifunovic, A. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. Scientific Reports 2016, 17, 953–964. [Google Scholar] [CrossRef]
- Wong, K.S.; Mabanglo, M.F.; Seraphim, T.V; ∙Mollica, A.; Mao, Y.-Q.; Rizzolo, K.; Leung, E.; Moutaoufik, M.T.; Hoell, L.; Phanse, S.; Goodreid, J.; Barbosa, L.R.S.; Ramos, C.H.I.; Babu, M.; Mennella, V.; ∙Robert, A.; Batey, R.A.; Schimmer, A.D.; Houry, W. A. Acyldepsipeptide Analogs Dysregulate Human Mitochondrial ClpP Protease Activity and Cause Apoptotic Cell Death. Cell Chem. Biol. 2018, 28, 1017–1030. [Google Scholar] [CrossRef]
Figure 1.
Emblematic imipridones under clinical (ONC201 and ONC206) or preclinical (ONC212) studies.
Figure 1.
Emblematic imipridones under clinical (ONC201 and ONC206) or preclinical (ONC212) studies.
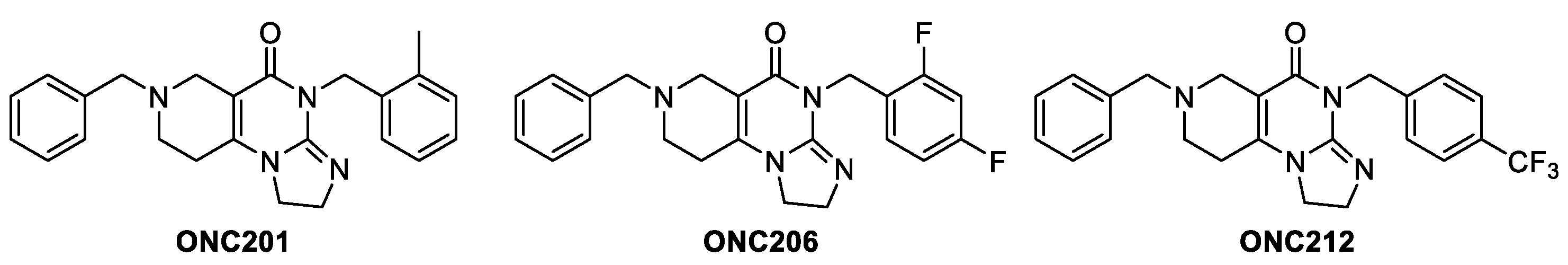
Figure 2.
The most promising compounds identified in our previous work [24].
Figure 2.
The most promising compounds identified in our previous work [24].
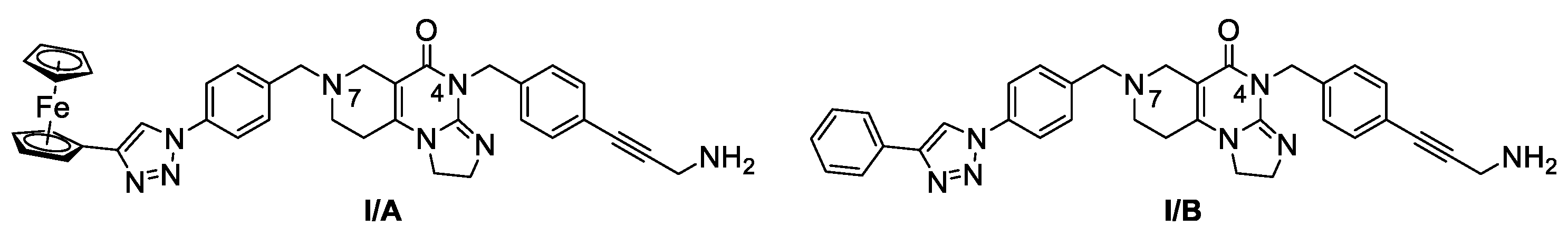
Figure 3.
Various regioisomers and structurally related derivatives of I/A and I/B intended for synthesis and in vitro cell viability screening as members of the first set of compounds.
Figure 3.
Various regioisomers and structurally related derivatives of I/A and I/B intended for synthesis and in vitro cell viability screening as members of the first set of compounds.
Scheme 1.
Convergent synthetic pathway leading to halogenated azidoimipridones. Reaction conditions: (a) methyl acrylate (5 eq.), MeOH, r.t., 48 h; (b) NaH (5 eq.), THF, 0 °C, then r.t., 2 h; (c) MeOH/AcOH = 4/1, reflux, 18 h; (d) 3.6 M NaOMe/MeOH (1.25 eq.), MeOH, reflux, 12 h; (e) 1) cc. HCl, r.t., 30 min. 2) NaNO2 (2 eq.), 0 °C, 45 min; 3) NaN3 (10 eq.), 0 °C, 45 min, then r.t., 1 h; (* = crude product; ** = cumulative yield, crude product).
Scheme 1.
Convergent synthetic pathway leading to halogenated azidoimipridones. Reaction conditions: (a) methyl acrylate (5 eq.), MeOH, r.t., 48 h; (b) NaH (5 eq.), THF, 0 °C, then r.t., 2 h; (c) MeOH/AcOH = 4/1, reflux, 18 h; (d) 3.6 M NaOMe/MeOH (1.25 eq.), MeOH, reflux, 12 h; (e) 1) cc. HCl, r.t., 30 min. 2) NaNO2 (2 eq.), 0 °C, 45 min; 3) NaN3 (10 eq.), 0 °C, 45 min, then r.t., 1 h; (* = crude product; ** = cumulative yield, crude product).
Scheme 2.
Sequential functionalization reactions on the terminal benzyl groups of the imipridone scaffold affording the first group of the targeted hybrids. Reaction conditions: (f) CuI (0.1 eq.), DMSO, r.t., 24 h; (g) CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h.
Scheme 2.
Sequential functionalization reactions on the terminal benzyl groups of the imipridone scaffold affording the first group of the targeted hybrids. Reaction conditions: (f) CuI (0.1 eq.), DMSO, r.t., 24 h; (g) CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h.
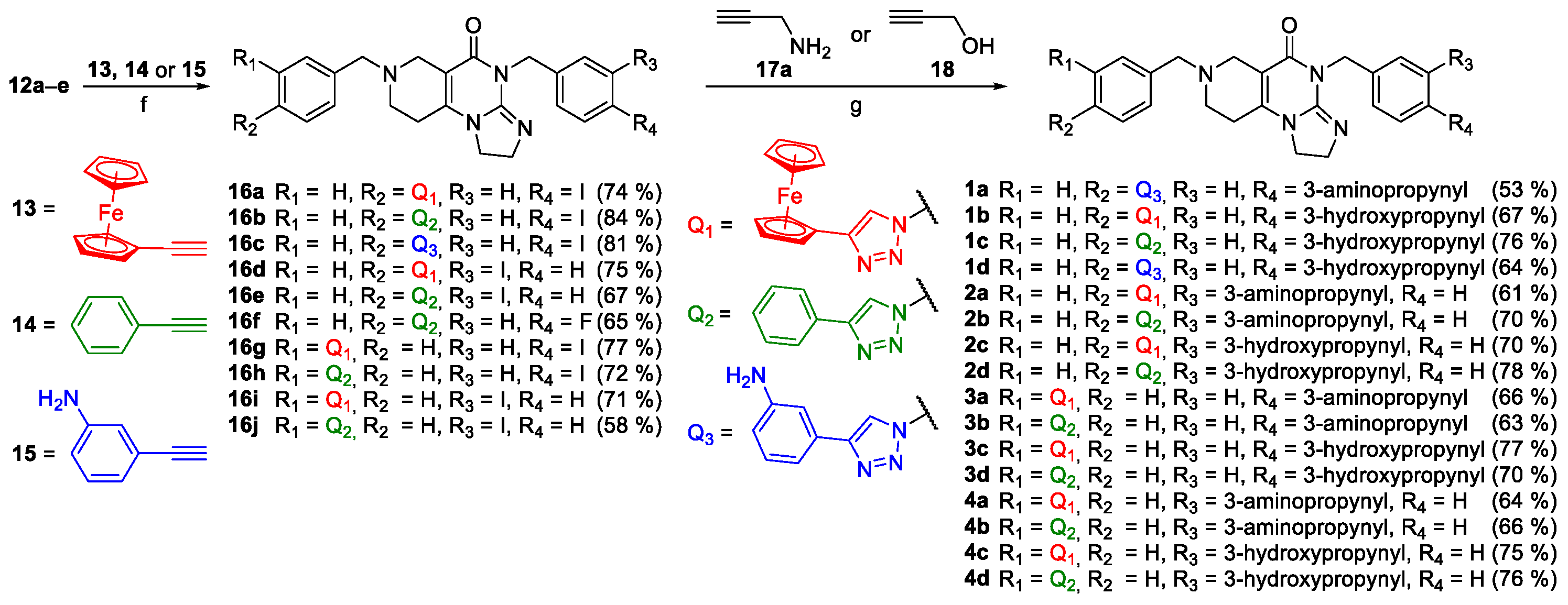
Scheme 3.
Synthesis of acrylamido derivative 1e employing a three-step procedure supported by selective N-protection strategy: Reaction conditions: (g) CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h; (h) acryloyl chloride (1.3 eq.), DIPEA (2 eq.), DCM, r.t, 4 h.; (i) cc. HCl, r.t, 20 min.
Scheme 3.
Synthesis of acrylamido derivative 1e employing a three-step procedure supported by selective N-protection strategy: Reaction conditions: (g) CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h; (h) acryloyl chloride (1.3 eq.), DIPEA (2 eq.), DCM, r.t, 4 h.; (i) cc. HCl, r.t, 20 min.
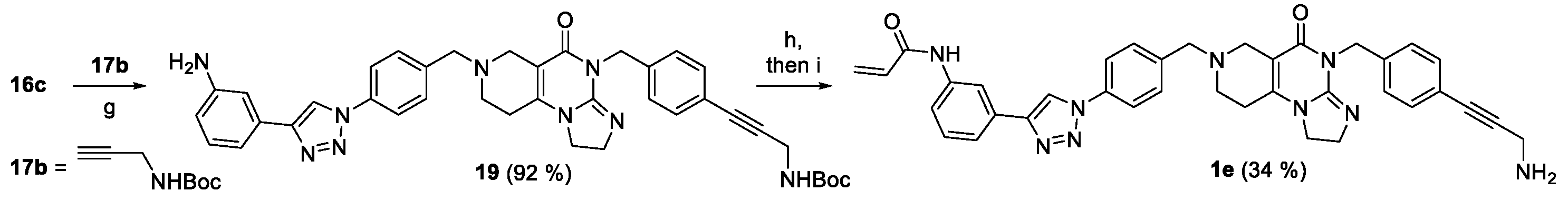
Figure 4.
Envisaged fragment refinements in 2a,b, the members of the first set of the targeted hybrids, which proved to be prominently efficient in the primary viability screening.
Figure 4.
Envisaged fragment refinements in 2a,b, the members of the first set of the targeted hybrids, which proved to be prominently efficient in the primary viability screening.

Scheme 4.
Synthetic route to the first type of amido-imipridone derivatives. Reaction conditions: (g) propargylamine (2 eq.), CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h; (i) cc. HCl, r.t., 20 min; (j) DIPEA (3 eq.), DCM, r.t., 1,5 h; (k) DIPEA (3 eq.), DCM, reflux, 24 h; * = crude product.
Scheme 4.
Synthetic route to the first type of amido-imipridone derivatives. Reaction conditions: (g) propargylamine (2 eq.), CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h; (i) cc. HCl, r.t., 20 min; (j) DIPEA (3 eq.), DCM, r.t., 1,5 h; (k) DIPEA (3 eq.), DCM, reflux, 24 h; * = crude product.
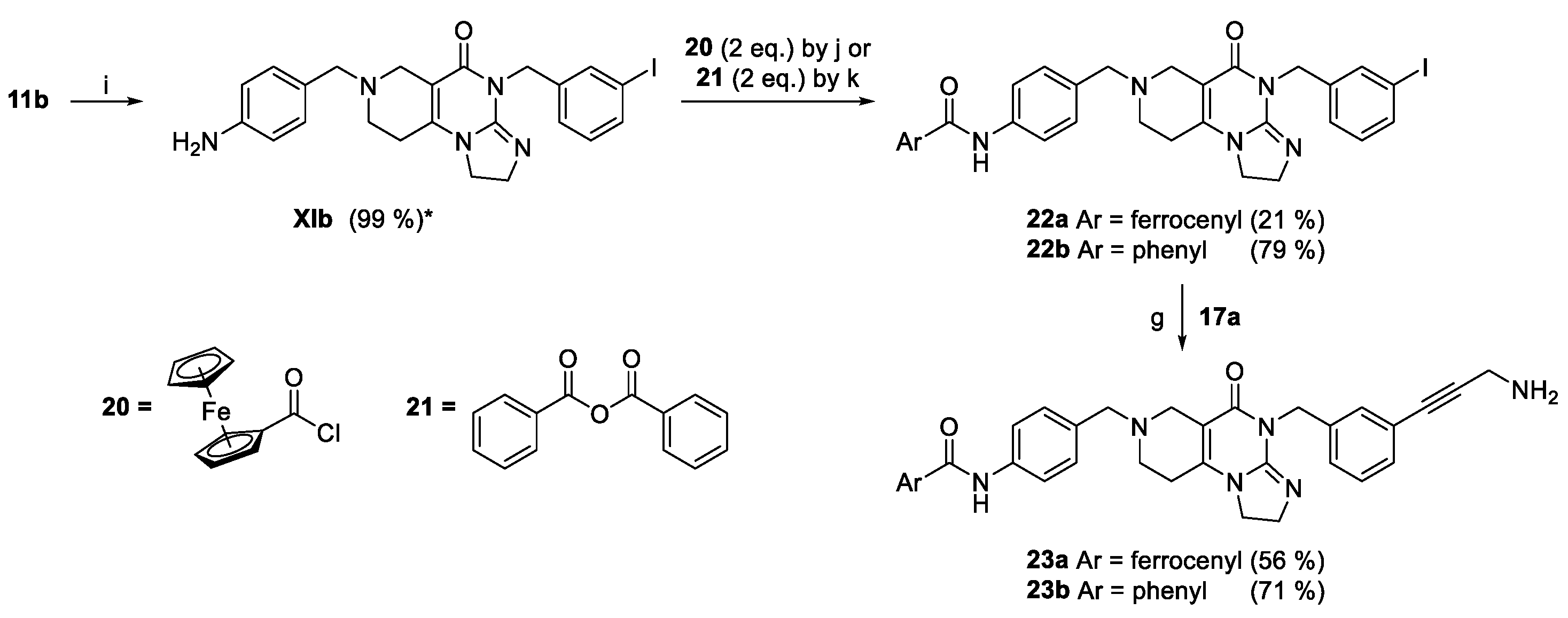
Scheme 5.
Synthetic route to the second type of amido-imipridone derivatives. Reaction conditions: (c) MeOH/AcOH = 4/1, reflux, 18 h; (d) 3.6 M NaOMe/MeOH (1.25 eq.), MeOH, reflux, 12 h; (g) propargylamine (2 eq.), CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h; (i) cc. HCl, r.t., 20 min; (j) DIPEA (3 eq.), DCM, 0°C, 30 min., then r.t., 1 h; (l) oxalyl chloride (2 eq.), DMF (cat.), DCM, r.t., 20 min; (m) NaH (5 eq.), toluene, r.t., 3 h, then reflux 2 h; (n) 1) DCM, r.t, 20 min., 2) NaHB(OAc)3 (1.4 eq.), r.t., 2.5 h.; * = crude product.
Scheme 5.
Synthetic route to the second type of amido-imipridone derivatives. Reaction conditions: (c) MeOH/AcOH = 4/1, reflux, 18 h; (d) 3.6 M NaOMe/MeOH (1.25 eq.), MeOH, reflux, 12 h; (g) propargylamine (2 eq.), CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h; (i) cc. HCl, r.t., 20 min; (j) DIPEA (3 eq.), DCM, 0°C, 30 min., then r.t., 1 h; (l) oxalyl chloride (2 eq.), DMF (cat.), DCM, r.t., 20 min; (m) NaH (5 eq.), toluene, r.t., 3 h, then reflux 2 h; (n) 1) DCM, r.t, 20 min., 2) NaHB(OAc)3 (1.4 eq.), r.t., 2.5 h.; * = crude product.
Scheme 6.
Synthetic route to 36a–c, the fluorinated derivatives of 2b. Reaction conditions: (f) CuI (0.1 eq.), DMSO, r.t., 24 h; (g) propargylamine (2 eq.), CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h.
Scheme 6.
Synthetic route to 36a–c, the fluorinated derivatives of 2b. Reaction conditions: (f) CuI (0.1 eq.), DMSO, r.t., 24 h; (g) propargylamine (2 eq.), CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h.
Scheme 7.
Synthetic route to the aminomethyl-imipridone derivatives. Reaction conditions: (c) MeOH/AcOH = 4/1, reflux, 18 h; (d) 3.6 M NaOMe/MeOH (1.25 eq.), MeOH, reflux, 12 h; (e) 1) cc. HCl, r.t., 30 min 2) NaNO2 (2 eq.), 0 °C, 45 min 3) NaN3 (10 eq.), 0 °C, 45 min, then r.t., 1 h; (f) CuI (0.1 eq.), DMSO, r.t., 24 h; * = crude product.
Scheme 7.
Synthetic route to the aminomethyl-imipridone derivatives. Reaction conditions: (c) MeOH/AcOH = 4/1, reflux, 18 h; (d) 3.6 M NaOMe/MeOH (1.25 eq.), MeOH, reflux, 12 h; (e) 1) cc. HCl, r.t., 30 min 2) NaNO2 (2 eq.), 0 °C, 45 min 3) NaN3 (10 eq.), 0 °C, 45 min, then r.t., 1 h; (f) CuI (0.1 eq.), DMSO, r.t., 24 h; * = crude product.
Scheme 8.
Synthesis of N-alkylated derivatives of 2a–b. Reaction conditions: (g) CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h.
Scheme 8.
Synthesis of N-alkylated derivatives of 2a–b. Reaction conditions: (g) CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h.
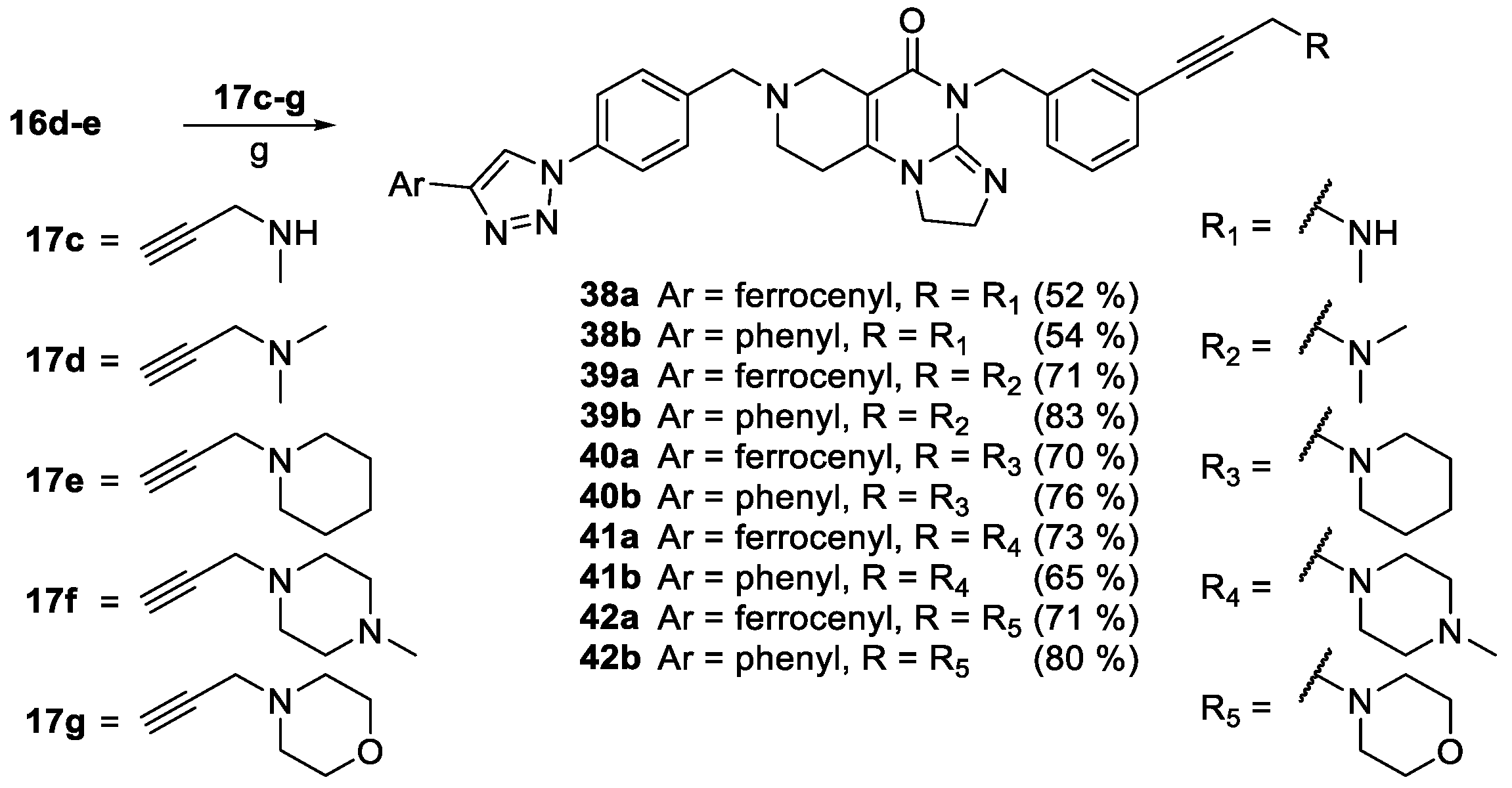
Figure 5.
Dose–response curves of selected imipridone hybrids I/A, I/B, 38a,b, 43a–c and ONC201 on PANC-1 and Fadu cells and non-tumorous primary fibroblast cells measured by CellTiter-Glo cell viability assay. Curves were fitted by GraphPad Prism 9.5.0 software using nonlinear regression (variable slope; 95% CI; n = 4). The concentration scale is logarithmic.
Figure 5.
Dose–response curves of selected imipridone hybrids I/A, I/B, 38a,b, 43a–c and ONC201 on PANC-1 and Fadu cells and non-tumorous primary fibroblast cells measured by CellTiter-Glo cell viability assay. Curves were fitted by GraphPad Prism 9.5.0 software using nonlinear regression (variable slope; 95% CI; n = 4). The concentration scale is logarithmic.

Figure 6.
Colony formation assay of selected imipridone derivatives on PANC-1 cell line at 4 µM concentration: (a) 24 h treatment followed by 5 days postincubation; (b) 72 h treatment followed by 3 days postincubation. 1: DMSO (control), 2: ONC201, 3: I/A, 4: I/B, 5: 38a and 6: 38b.
Figure 6.
Colony formation assay of selected imipridone derivatives on PANC-1 cell line at 4 µM concentration: (a) 24 h treatment followed by 5 days postincubation; (b) 72 h treatment followed by 3 days postincubation. 1: DMSO (control), 2: ONC201, 3: I/A, 4: I/B, 5: 38a and 6: 38b.
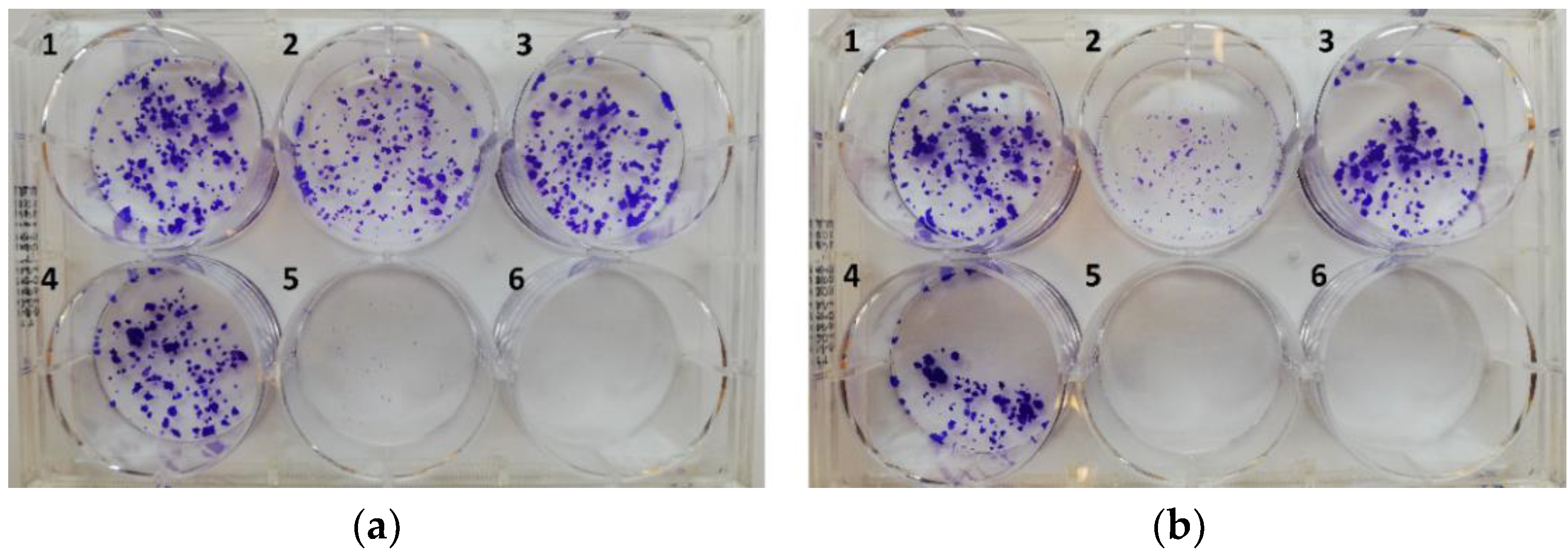
Figure 6.
Dose–response curves of imipridone hybrids 38a,b and ONC201. Viability measured on HEK293T and HEK293T-/- cell lines based on CellTiter-Glo cell viability assay. Curves were fitted by GraphPad Prism 9.5.0 software using nonlinear regression (variable slope; 95% CI; n = 3). The concentration scale is logarithmic.
Figure 6.
Dose–response curves of imipridone hybrids 38a,b and ONC201. Viability measured on HEK293T and HEK293T-/- cell lines based on CellTiter-Glo cell viability assay. Curves were fitted by GraphPad Prism 9.5.0 software using nonlinear regression (variable slope; 95% CI; n = 3). The concentration scale is logarithmic.
Scheme 9.
Synthesis of N-methylated derivatives 43a–c carrying fluorinated phenyl groups on the triazolyl substituent. Reaction conditions: (g) CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h.
Scheme 9.
Synthesis of N-methylated derivatives 43a–c carrying fluorinated phenyl groups on the triazolyl substituent. Reaction conditions: (g) CuI (0.2 eq.), PdCl2(PPh3)2 (0.1 eq.), DIPEA (3 eq.), DMF, r.t., 24 h.
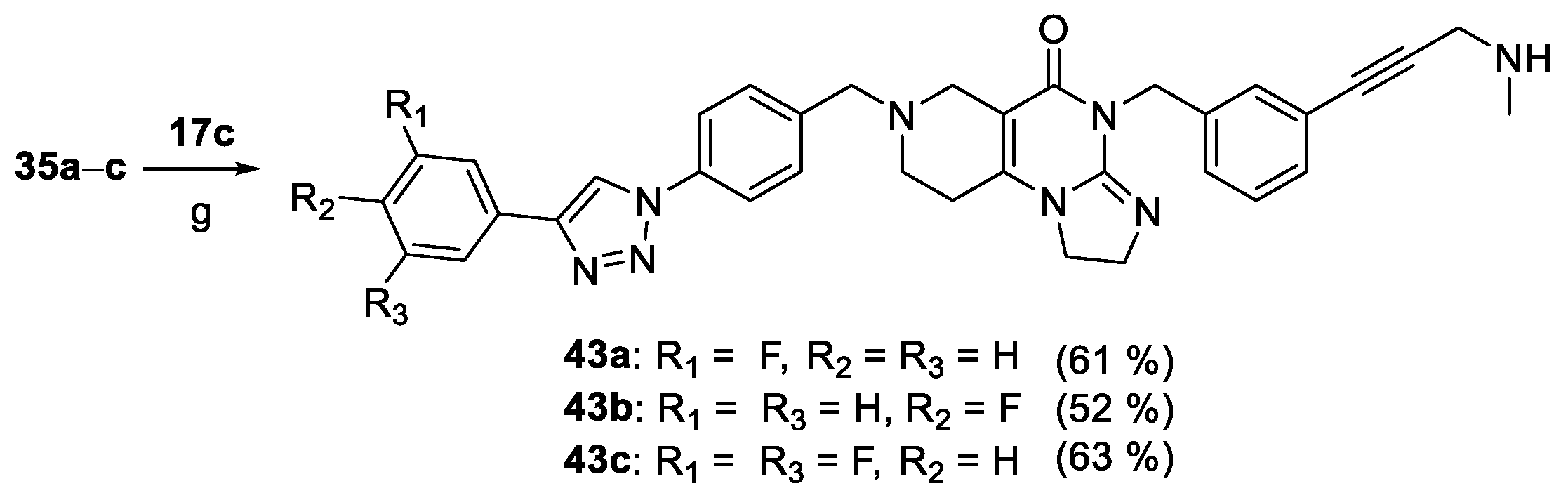

Table 1.
Results of the initial viability screening of the first set of novel imipridone derivatives on PANC-1 and Fadu cancer cell lines.
Table 1.
Results of the initial viability screening of the first set of novel imipridone derivatives on PANC-1 and Fadu cancer cell lines.
| Comp. |  |
Cell viability (% of control) ± SD (n = 2) | ||||
|---|---|---|---|---|---|---|
| R1 | R2 | Cell line | 25 µM | 8.3 µM | 2.8 µM | |
| ONC201 |  |
PANC-1 | 53 ± 2 | 58 ± 1 | 69 ± 1 | |
| Fadu | 27 ± 1 | 29 ± 1 | 31 ± 1 | |||
| I/A | 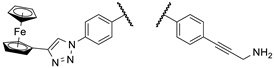 |
PANC-1 | 0 ± 0 | 39 ± 7 | 108 ± 4 | |
| Fadu | 0 ± 0 | 66 ± 24 | 105 ± 9 | |||
| I/B |  |
PANC-1 | 0 ± 0 | 61 ± 22 | 113 ± 4 | |
| Fadu | 0 ± 0 | 103 ± 6 | 112 ± 7 | |||
| 16f | 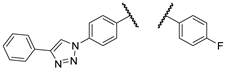 |
PANC-1 | 60 ± 1 | 68 ± 4 | 81 ± 3 | |
| Fadu | 77 ± 4 | 94 ± 1 | 100 ± 1 | |||
| 1a |  |
PANC-1 | 3 ± 2 | 93 ± 7 | 100 ± 5 | |
| Fadu | 10 ± 0 | 99 ± 2 | 106 ± 2 | |||
| 1b | 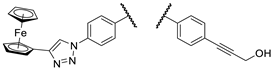 |
PANC-1 | 95 ± 2 | 93 ± 3 | 92 ± 0 | |
| Fadu | 68 ± 3 | 83 ± 0 | 88 ± 4 | |||
| 1c | 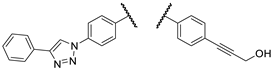 |
PANC-1 | 88 ± 6 | 87 ± 2 | 95 ± 3 | |
| Fadu | 64 ± 2 | 71 ± 0 | 79 ± 0 | |||
| 1d | 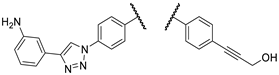 |
PANC-1 | 100 ± 5 | 101 ± 3 | 99 ± 3 | |
| Fadu | 84 ± 4 | 96 ± 7 | 99 ± 9 | |||
| 1e | 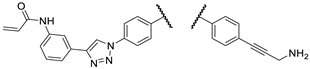 |
PANC-1 | 102 ± 0 | 91 ± 1 | 101 ± 7 | |
| Fadu | 69 ± 2 | 59 ± 4 | 96 ± 6 | |||
| 2a | 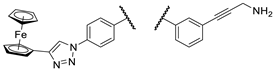 |
PANC-1 | 0 ± 0 | 0 ± 0 | 92 ± 2 | |
| Fadu | 0 ± 0 | 3 ± 0 | 107 ± 5 | |||
| 2b | 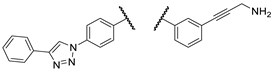 |
PANC-1 | 0 ± 0 | 0 ± 0 | 107 ± 1 | |
| Fadu | 0 ± 0 | 0 ± 0 | 102 ± 8 | |||
| 2c | 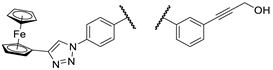 |
PANC-1 | 97 ± 2 | 95 ± 2 | 94 ± 2 | |
| Fadu | 65 ± 0 | 76 ± 4 | 79 ± 2 | |||
| 2d | 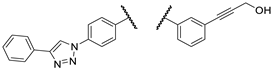 |
PANC-1 | 81 ± 0 | 87 ± 1 | 100 ± 8 | |
| Fadu | 78 ± 6 | 90 ± 10 | 99 ± 7 | |||
| 3a |  |
PANC-1 | 0 ± 0 | 88 ± 0 | 101 ± 7 | |
| Fadu | 0 ± 0 | 100 ± 6 | 104 ± 5 | |||
| 3b | 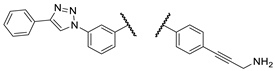 |
PANC-1 | 0 ± 0 | 84 ± 2 | 103 ± 8 | |
| Fadu | 22 ± 4 | 95 ± 3 | 103 ± 2 | |||
| 3c | 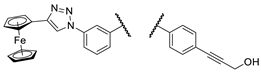 |
PANC-1 | 98 ± 1 | 98 ± 1 | 100 ± 3 | |
| Fadu | 85 ± 5 | 99 ± 2 | 103 ± 9 | |||
| 3d | 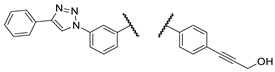 |
PANC-1 | 46 ± 7 | 55 ± 6 | 85 ± 1 | |
| Fadu | 23 ± 3 | 29 ± 2 | 87 ± 1 | |||
| 4a | 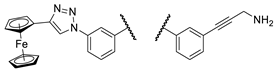 |
PANC-1 | 0 ± 0 | 73 ± 9 | 91 ± 4 | |
| Fadu | 0 ± 0 | 77 ± 3 | 102 ± 1 | |||
| 4b |  |
PANC-1 | 0 ± 0 | 70 ± 0 | 87 ± 5 | |
| Fadu | 0 ± 0 | 34 ± 3 | 76 ± 1 | |||
| 4cc |  |
PANC-1 | 47 ± 5 | 51 ± 5 | 67 ± 2 | |
| Fadu | 54 ± 4 | 68 ± 7 | 75 ± 1 | |||
| 4d |  |
PANC-1 | 53 ± 8 | 60 ± 2 | 74 ± 3 | |
| Fadu | 23 ± 3 | 27 ± 3 | 37 ± 1 | |||
Table 2.
Results of the initial viability screening of the second set of novel imipridone derivatives on PANC-1 and Fadu cancer cell lines.
Table 2.
Results of the initial viability screening of the second set of novel imipridone derivatives on PANC-1 and Fadu cancer cell lines.

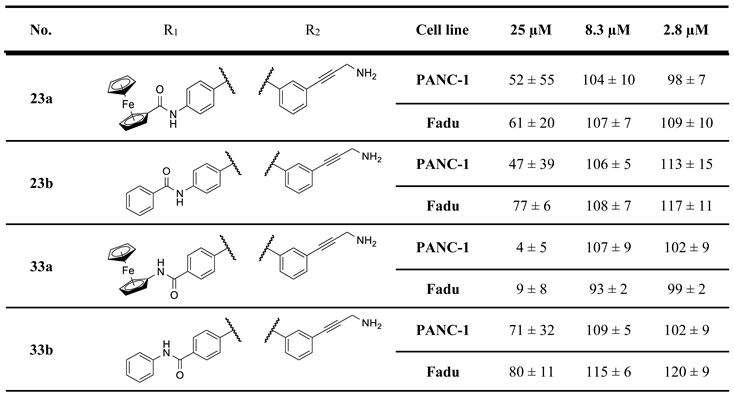
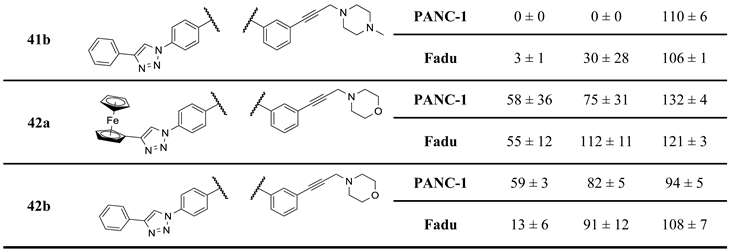
a For more reliable comparison and circumventing batch-effect ONC201, I/A, I/B, 2a and 2b, the latter two emerged as the most potent compounds in the screen of the first set, were repeatedly screened as references along with the samples of the second set of hybrid molecules.
Table 3.
IC50 values (µM) of the selected imipridone derivatives and reference compounds on PANC-1 and Fadu cell lines obtained by CellTiter-Glo cell viability assay after 72 h treatment using GraphPad Prism 9.5.0 software (nonlinear regression; variable slope; best-fit values; n = 4).
Table 3.
IC50 values (µM) of the selected imipridone derivatives and reference compounds on PANC-1 and Fadu cell lines obtained by CellTiter-Glo cell viability assay after 72 h treatment using GraphPad Prism 9.5.0 software (nonlinear regression; variable slope; best-fit values; n = 4).
| IC50 value | Selectivity index (SI)a | ||||
|---|---|---|---|---|---|
| PANC-1 | Fadu | Primary fibroblast |
Fibroblast / PANC-1 | Fibroblast / Fadu | |
| ONC201 | 3.1 | 1.3 | 26 < b | 1.6 < | 3.9 < |
| I/A | 10.7 | 10.0 | 14.9 | 1.4 | 1.5 |
| I/B | 8.2 | 10.3 | 11.0 | 1.3 | 1.1 |
| 38a | 2.7 | 2.9 | 4.7 | 1.7 | 1.6 |
| 38b | 2.5 | 3.3 | 4.4 | 1.8 | 1.3 |
| 43a | 2.5 | 3.2 | 3.2 | 1.3 | 1.0 |
| 43b | 3.2 | 3.2 | 3.5 | 1.1 | 1.1 |
| 43c | 2.6 | 3.6 | 3.7 | 1.4 | 1.0 |
a The Selectivity index (SI) is calculated as IC50 on primary fibroblasts / IC50 on cancer cells. b The IC50 is above the highest concentration applied in the assay, as viability remained above 50%.
Table 4.
IC50 values (µM) of 38a,b and ONC201 on HEK293T WT and CLPP-/- cells measured by CellTiter-Glo cell viability assay after 72 h treatment. IC50 values were determined by GraphPad Prism 9.5.0 software (nonlinear regression; variable slope; best-fit values; n = 4).
Table 4.
IC50 values (µM) of 38a,b and ONC201 on HEK293T WT and CLPP-/- cells measured by CellTiter-Glo cell viability assay after 72 h treatment. IC50 values were determined by GraphPad Prism 9.5.0 software (nonlinear regression; variable slope; best-fit values; n = 4).
| IC50 (µM) | ||
| HEK293T WT | HEK293T CLPP-/- | |
| ONC201 | 0.6 | n. d. |
| 38a | 1.6 | 2.4 |
| 38b | 1.7 | 2.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



