
Submitted:
31 May 2024
Posted:
31 May 2024
You are already at the latest version
Abstract
In the frame of our diversity-oriented research on multitarget small molecule anticancer agents, utilizing convergent synthetic sequences terminated by Sonogashira coupling reactions, a preliminary selection of representative alkyne-tethered vindoline hybrids was synthesized. The novel hybrids with additional pharmacophoric fragments of well-documented anticancer agents, including FDA-approved tyrosine-kinase inhibitors (imatinib and erlotinib) or ferrocene or chalcone units, were evaluated for their antiproliferative activity on malignant cell lines MDA-MB-231 (triple negative breast cancer), A2780 (ovarian cancer), HeLa (human cervical cancer) and SH-SY5Y (neuroblastoma) as well as human embryonal lung fibroblast MRC-5 served as reference non-malignant cell line for the assessment of the therapeutic window of the tested hybrids. The biological assays identified a trimethoxyphenyl-containing chalcone-vindoline hybrid (36) as a promising lead compound exhibiting submicromolar activity on A2780 cells with a marked therapeutic window. These results suggest that ovarian cancer might be considered as a prioritized target of treatment with 36.
Keywords:
vindoline
; imatinib
; erlotinib
; chalcone
; hybrid molecules
; Sonogashira reaction
; antiproliferative effect
; selectivity
; ovarian cancer
1. Introduction
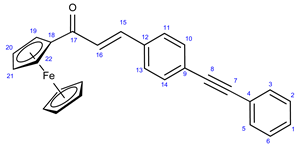
Cancer is one of the most severe health problems. Even though the survival rate has improved, different types of tumorous diseases are still among the leading causes of mortality with low survival rates [1,2]. Although in clinical practice, chemotherapy is generally considered one of the most widely used tools for cancer treatment, often in combination with other therapies, such as surgery, radiation, or hormone therapy, the efficacy of most anticancer chemotherapies is decreased by several therapy-limiting factors, including multidrug resistance (MDR) [3,4] and adverse effects. Consequently, developing more potent novel drugs possessing improved activity, selectivity, and enhanced potency to overcome MDR is continuously in the focus of interest. One of the most appealing new strategies for developing improved chemotherapy is the fragment-based design and synthesis of hybrid compounds by coupling a reasonable selection of pharmacophoric fragments [5,6,7]. Such hybrid drugs capable of interacting with more than one cellular molecular target can be considered highly potent anticancer agents with enhanced efficiency as they trigger cell death by multiple mechanisms, thus having a real potential to overcome typical disadvantages of single anticancer agents, including resistance and adverse effects. For an expansion of novel potent therapeutic agents, the implication of compounds of natural origin and their chemically modified versions also seems an attractive strategy. In this regard, several representatives from alkaloid families are of pronounced interest [8,9,10,11]. The emblematic tubulin inhibitor bis-indole alkaloid Vinblastine (1: Figure 1) can also be considered as a hybrid composed of coupled monomers catharanthine (1a) and vindoline (1b). This alkaloid is one of the most widely used therapeutic agents for treating, e.g., breast cancer and acute lymphocytic leukemia, either as a single agent or in combination with other drugs, exerting its activity by inhibiting tubulin polymerization [12].
Vinca alkaloids, including vinblastine, can be isolated from Catharanthus roseus. The monomers occur in much greater quantity in the plant than the dimers; however, these single fragments do not display perceptible anticancer effects [13,14]. To address this problem, various vinca alkaloids have been synthesized as a hybrid, such as amino acid-, steroid-containing hybrids, and with other pharmacophore moieties [15,16,17]. In this context, pursuing our diversity-oriented research on the development of novel small-molecule anticancer agents with enhanced anticancer potency, we aimed at constructing a preliminary selection of novel alkyne-tethered vindoline-based hybrids in which position 10 is coupled to fragments of well-documented pharmacophore residues replacing catharanthine (1a) present in vinblastine 1. Accordingly, we planned to introduce FDA-approved tyrosine-kinase inhibitor anticancer agents imatinib (2) [18] and erlotinib (3) [19] as well as ferrocene- and chalcone containing moieties 4 and 5 (Figure 2) in the targeted vindoline hybrids.
Besides its commercial availability and enhanced stability compared to catharanthine, the choice of vindoline as an essential fragment in our targeted hybrids can be justified by the results of recent years pointing to the possibility of such types of structural engineering that can produce real anticancer agents incorporating this alkaloid conjugated to suitable pharmacophores including amino acids [20,21], steroids [22] and N-heterocycles [21].
The introduction of ferrocene-containing fragments can be reasoned by the following findings. In 1984, Köpf-Maier et al. reported the anticancer properties of ferrocene salts [23]. Later, more studies reported ferrocene derivatives exhibiting antiproliferative effects on several cancer cell lines and low toxicity on non-transformed cells. It is of importance that due to its stability, super-aromaticity, elevated membrane-penetrating ability, and implication in substituent-dependent, thus fine-tuneable, ROS-generating single electron transfer (SET) events, organoferrocene fragments became the most widespread structural motifs in the emerging group of organometallics displaying anticancer activity triggered by versatile mechanisms of actions [24,25,26,27,28,29,30,31,32,33,34]. It has also been demonstrated that replacing an aromatic nucleus of certain organic compounds for a ferrocene unit can lead to products with antiproliferative activity that is absent or less manifested in the parent molecule [35,36,37,38].
Chalcones are also privileged scaffolds embedded in a plethora of highly potent anticancer drug candidates inducing cancer cell death by versatile mechanisms of actions, including cell cycle arrest in subG1, S, and G2/M phases, inhibition of tubulin polymerization, enzyme dynamics [39,40,41,42,43,44] and signal transductions initiated by nuclear factor κB [45]. On the other hand, it is also of pronounced importance that various chalcone-containing scaffolds feature marked potency even in overcoming drug resistance as they were found to exhibit in vitro and in vivo effects on both drug-susceptible and drug-resistant cancers by targeting aromatase enzyme (CIP19A1), breast cancer resistance protein (BCRP), vascular endothelial growth factor (VEGF) and ATP binding cassette subfamily G member 2 (ABCG2) [46,47]. Since indole is an integrated structural motif in vindoline, chalcone-indol hybrids are particularly worth to be highlighted as further examples with demonstrated antiproliferative activity against A549, MCF-7, HepG2, paclitaxel-resistant HCT-8/T, and vincristine-resistant HCT-8/V cell lines [48]. In this regard, Yan et al. reported low nanomolar activity for chalcone-indol hybrids detected again on A549, MCF-7, and HCT-8 cancer cells [49].
2. Results and Discussion
The targeted alkyne-tethered hybrids were accessed via two straightforward convergent synthetic pathways both terminated by Sonogashira reactions involving the readily accessible 10-iodovindoline [50] and the primarily prepared propargylated pharmacophoric moieties, or the 10-iodovindoline-derived silyl-protected 10-ethynylvindoline and iodinated chalcones as alternative coupling partners. Besides synthetic aspects, the introduction of acetylenic linker into anticancer drug candidates can also be beneficial in terms of bioactivity, as justified by characteristic literature examples reporting on alkyne derivatives identified as potent antitumor agents of natural and synthetic origin [51,52,53,54,55,56,57]. An emblematic alkyne-coupled pyrrolo [2,3-d]pyrimidine, BIIB028 displaying therapeutic activity with excellent drug-like properties and acceptable safety profile in treatment of breast cancer, melanoma, gastrointestinal cancer, lymphoma and myeloma [58,59,60] further supports the view about the benefits of building carbon-carbon triple bond as a rigid spacer in potential anticancer agents.
2.1. Multistep Synthesis of the Alkyne-Tethered Vindoline Hybrids
2.1.1. Synthesis of Propargylated Imatinib Fragments
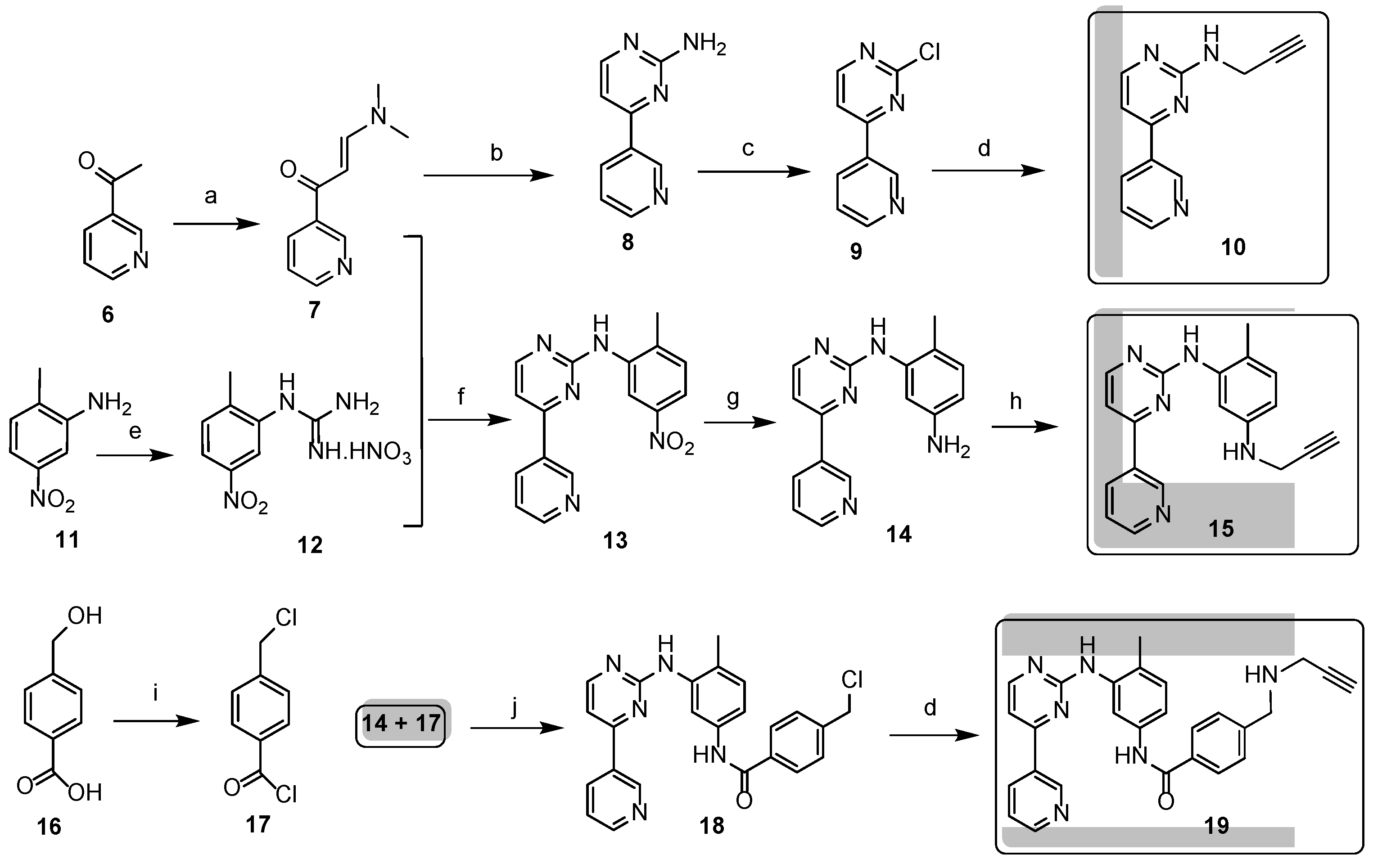
The propargylated imatinib fragments were synthesized utilizing the feasibility of introduction of propargyl group in the intermediates 9, 14 and 18 accessible by the reaction sequences developed by Liu et al. [61], as shown in Scheme 1. Accordingly, 3-acetylpyridine (6) was reacted with DMF-DMA to obtain the intermediate enaminone 7 which was cyclized with guanidine-nitrate to obtain aminopyrimidine 8 of which diazotization followed by chlorination afforded chloropyrimidine 9. This electrophilic intermediate was reacted with propargylamine to obtain compound 10, the alkyne-functionalized imatinib moiety suitable for Sonogashira coupling. Starting an other pathway the pyrimidine-forming ring closure of enaminone 7 was performed with N-(2-methyl-5-nitrophenyl)guanidine nitrate 12 previously generated from nitroaniline 11. The catalytic hydrogenation of the resulting nitroaryl derivative 13 gave aniline 14 as a nucleophilic key intermediate of which N-alkylation with propargyl bromide conducted under standard conditions led to the formation of the next imatinib-based terminal alkyne 15 featuring a more extended structural motif compared to the biaryl-type intermediate 10. Finally, the reaction pathway starting with double chlorination of 4-(hydroxymethyl)benzoic acid (16 → 17) followed byN-acylation (14 + 17 → 18) and sequential N-propargylation of the benzyl chloride type intermediate 18 constructed 19, the most complex propargylated fragment comprising the majority of the structural motifs of imatinib.
2.1.2. Sonogashira Coupling Reactions Terminating the Synthetic Pathways to the Targeted Alkyne-Tethered Vindoline Hybrids
The first group of the hybrids containing the fragments of FDA-approved anticancer agents 21–24 was synthesized by coupling 10-iodovindoline 20 with propargyalated imatinib fragments (10, 15, 19) and intact erlotinib (3) as outlined in Scheme 2. The reactions were conducted for 24 h at room temperature in DMF using CuI(20%)/PdCl2(PPh3)2(10%) and N,N-diisopropylethylamine (DIPEA) (3 eq.) as catalyst system and base, respectively (Scheme 4). Under the same conditions ferrocene-containing hybrids 28 and 29 were obtained when the commercially available ethynylferrocene 25 and N-propargyl ferrocene carboxamide 27, respectively, were used as alkyne components in the coupling reactions. The carboxamide 27 was obtained by the well-established acylation of propargylamine with N-ferrocenoylimidazole 26 [62].
In the first step of the synthetic route to representative chalcone-containing hybrids 10-iodovindoline 20 was coupled with trimethylsilylacetylene under the same Sonogashira conditions to afford protected alkyne 30. In a one-pot procedure, without isolation and purification, the unsTable 30 was subjected to TBAF-mediated desilylation followed by Sonogashira coupling of the resulting non-isolated 10-ethynylvindoline with iodinated chalcones 34 and 35 to obtain hybrids 36 and 37, respectively (Scheme 2). Iodochalcones 34 and 35 were previously prepared by Claisen-Schmidt condensation of 4-iodobenzaldehyde 31 with 3,4,5-trimethoxyacetophenone (32) and acetylferrocene (33), respectively (Scheme 2).
The reactions of 34 and 35 with phenylacetylene 38 led to the formation of chalcones 36a and 37a (Scheme 3), serving as reference models in the biological assays.
2.2. In Vitro Antiproliferative Evaluation of the Novel Vindoline Hybrids and Reference Compounds
The antiproliferative effect of the novel vindoline-containing hybrids was initially tested on MRC-5 cells (non-cancerous human embryonal lung fibroblasts) to obtain results concerning the cytotoxicity of the tested molecules. The anticancer properties of the substances were characterized utilizing MDA-MB-231 (breast adenocarcinoma), HeLa (human cervical cancer), A2780 (ovarian cancer), SH-SY5Y (neuroblastoma) cells. Two concentrations, 10 and 30 µM, were applied at the initial screening. In the case of the most potent analogs, i.e., when the cell growth inhibition was higher than 50% at 10 µM on any cancer cell lines, the assay was repeated with a set of dilutions to determine the IC50 values. The results of the grown inhibition screening and the IC50 values of the most potent compounds are listed in Table 1 and Table 2, respectively.
The percentage of cell growth inhibition caused by vindoline (1b) and the newly synthesized hybrids (21–24, 28, 29, 36, 37, 36a and 37a) are listed in Table 1. It can be seen that vindoline extended with pharmacophore units shows some cytotoxic effects. Of the hybrids containing imatinib fragments (21–23), component 22 showed cell division inhibition above 85% on 3 cell lines (MDA-MB-231, A2780, SH-SY5Y) at concentration of 30 µM. Ferrocene-containing compounds (28 and 29) also showed less substantial cell division inhibitory effects at a concentration of 30 µM.
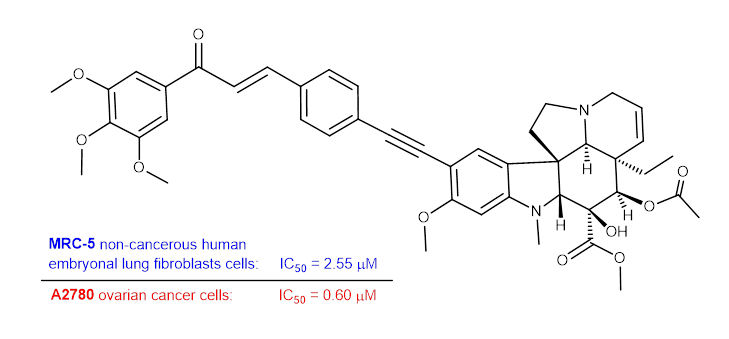
Trimethoxyphenyl derivative 36 was identified as the most potent antiproliferative agent (IC50 = 0.6–2.55 µM), especially against the A2780 cell line. Comparison of the effects of 36 and its simplified analog 36a led to the conclusion that the contribution of the vindoline residue to the antiproliferative effect on malignant cells is 3–5-fold more substantial than that of the phenyl group (Table 2). Though the IC50 values measured on the malignant and MRC-5 cells show comparable therapeutic windows for 36 and 36a, the latter seems less toxic against non-cancerous cells. However, it must be emphasized that based on the ratio of IC50 values, ovarian cancer (MRC-5/A2780 = 4.25 and 3.35 for 36 and 36a, respectively) might be considered the prioritized target of treatment with 36. The exact identification of the potential therapeutic target requires further investigations, including in vivo experiments. The effect was significantly reduced by replacing the trimethoxyphenyl group with ferrocene (37). However, the effect became more favorable by replacing the vindoline residue in 37 for the phenyl group (37a), but it was not as good as the others.
4. Materials and Methods
The Materials and Methods should be described with sufficient details to allow others to replicate and build on the published results. Please note that the publication of your manuscript implicates that you must make all materials, data, computer code, and protocols associated with the publication available to readers. Please disclose at the submission stage any restrictions on the availability of materials or information. New methods and protocols should be described in detail while well-established methods can be briefly described and appropriately cited.
All chemicals were obtained from commercially available sources (Merck, Budapest, Hungary; Fluorochem, Headfield, UK; Molar Chemicals, Halásztelek, Árpád street 1., 2314 Hungary; VWR, Debrecen, Hungary) and used without further purifications. Merck Kieselgel (230–400 mesh, 60 Å) was used for flash column chromatography. Melting points (uncorrected) were determined with a Büchi M-560. The 1H and 13C-NMR spectra were recorded in DMSO-d6 solution in 5 mm tubes at room temperature on a Bruker DRX-500 spectrometer (Bruker Biospin, Karlsruhe, Baden Württemberg, Germany) at 500 (1H) and 125 (13C) MHz, with the deuterium signal of the solvent as the lock and TMS as internal standard (1H and 13C). The 2D-HSQC-, HMBC- and NOESY spectra, which support the exact assignments of 1H- and 13C NMR signals, were registered by using the standard Bruker pulse programs. Exact mass measurements were performed on a high-resolution Waters ACQUITY RDa Detector (Waters Corp., Wilmslow, U.K.) equipped with electrospray ionization source using on-line UHPLC coupling. UHPLC separation was performed on a Waters ACQUITY UPLC H-Class PLUS system using a Waters Acquity UPLC BEH C18 column (2.1 x 150 mm, 1.7 µm). Samples were dissolved in MeOH/Water 5:95 V/V, 5-5 µL sample solutions were injected. Linear gradient elution (0 min 5% B, 1.0 min 5% B, 7.0 min 80% B, 7.1 min 100% B, 8.0 min 100% B, 8.1 min 5% B, 12.0 min 5% B) with eluent A (0.1% formic acid in water, V/V) and eluent B (0.1% formic acid in Methanol, V/V) was used at a flow rate of 0.200 mL/min at 45 °C column temperature. High-resolution mass spectra were acquired in the m/z 50-2000 range in positive ionization mode. Leucine enkephalin peptide was used for single Lock Mass calibration correction.
For each compound characterized in this session, the numbering of atoms used for the assignment of 1H- and 13C NMR signals do not correspond to IUPAC rules reflected from the given systematic names. Imatinib fragments (7–9, 12–14, 17 and 18) were synthesized by reported procedures [61]. N-Ferrocenoylimidazole (26) was prepared using the method reported by Imrie et al. [62].
3.1. General Procedure for the Synthesis of Propargylated Imatinib Fragments 10 and 19
2-Chloro-4-(pyridine-3-yl)pyrimidine (9) or 4-(chloromethyl)-N-(4-methyl-3-((4-(pyridine-3-yl)pyrimidine-2-yl)amino)phenyl)benzamide (18) (2 mmol.) was dissolved in MeCN (20 mL), then propargylamine (1.27 mL, 1.10 g, 20 mmol) was added dropwise to the solution. The obtained mixture was stirred at reflux temperature for 12 h and concentrated in vacuo. The residue was purified by column chromatography on silica using solvent mixture DCM:MeOH (15:1) as eluent, followed by crystallization with Et2O to obtain the pure product (10, 19).
3.1.1. N-(Prop-2-yn-1-yl)-4-(Pyridin-3-yl)Pyrimidin-2-Amine (10)
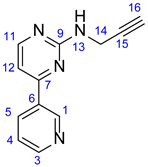
Light grey solid. Mp: 93–95 ◦C; Yield: 104 mg (26%). 1H-NMR (DMSO-d6): 9.30 (br s, 1H, H1); 8.65 (dd, J = 4.5 Hz and 1.6 Hz, 1H, H3); 8.46 (two coalesced d’s, J = 5.0 Hz, 2H, H5 and H11); 7.67 (t, J = 5.9 Hz, 1H, H13); 7.55 (dd, J = 7.8 Hz and 4.6 Hz, 1H, H4); 7.32 (d, J = 5.1 Hz, H12); 4.15 (dd, J = 5.9 Hz and 2.2 Hz, 1H, 2H, H14); 3.02 (t, J = 2.2 Hz, 1H, H17). 13C-NMR (DMSO-d6): 164.9 (C7); 162.3 (C9); 159.9 (C11); 151.9 (C3); 148.6 (C1); 134.8 (C5); 133.0 (C6); 124.3 (C4); 107.2 (C12); 82.7 (C15); 72.6 (C17); 30.8 (C14);
3.1.2. N-(4-Methyl-3-((4-(Pyridin-3-yl)Pyrimidin-2-yl)Amino)Phenyl)-4-((Prop-2-yn-1-Ylamino)-Methyl)- Benzamide (19)
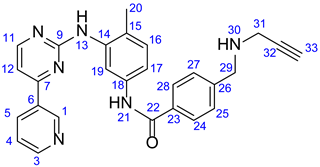
Light orange solid. Mp: 155–156 ◦C; Yield: 843 mg (94%). 1H-NMR (DMSO-d6): 10.17 (s, 1H, H21); 9.22 (d, J = 1.6 Hz, 1H, H1); 8.91 (s, 1H, H13); 8.65 (dd, J = 4.6 Hz and 1.6 Hz, 1H, H3); 8.48 (d, J = 5.1 Hz, 1H, H11); 8.44 (dt, J = 7.8 Hz and 1.6 Hz, 1H, H5); 7.92 (d, J = 8.5 Hz, 2H, H24 and H28); 7.50–7.46 (overlapping m’s, 4H, H4, H17, H25 and H27); 7.33 (d, J = 5.1 Hz, 1H, H12); 6.93 (d, J = 8.2 Hz, 1H, H16); 6.86 (d, J = 2.0 Hz, 1H, H19); 3.92 (s, 2H, H29); 3.44 (d, J = 2.0 Hz, 2H, H31); 3.24 (t, J = 2.0 Hz, 1H, H33); 2.04 (s, 3H, H20); 13C-NMR (DMSO-d6): 165.7 C22); 164.9 (C7); 162.3 (C7); 161.6 (C9); 159.2 (C11); 151.9 (C3); 148.6 (C1); 141.6 (C26); 138.3 (C14); 137.7 (C18); 135.1 (C5); 132.5 (C6); 130.8 (C16); 129.4 (C23); 129.0 (C25 and C27); 128.1 (C24 and C28); 126.4 (C15); 124.4 (C4); 117.9 (C19); 117.5 (C17); 107.8 (C12); 81.0 (C32); 76.0 (C33); 50.8 (C29); 36.9 (C31); 18.1 (C20).
3.2. 4-Methyl-N1-(Prop-2-yn-1-yl)-N3-(4-(Pyridine-3-yl)Pyrimidine-2-yl)Benzene-1,3-Diamine (15)
6-Methyl-N-(4-(pyridine-3-yl)pyrimidine-2-yl)benzene-1,3-diamine (14) (1.109 g, 4 mmol) and NaHCO3 (336 mg, 4 mmol) were dissolved in DMSO (12 mL). To this solution propargylbromide (0.37 mL, 571 mg, 4.8 mmol) was added dropwise, and the reaction mixture was stirred for 2.5 h at 70 °C. The mixture was poured into water and extracted with DCM (3 x 25 mL). The combined organic phase was washed with brine and water, then dried over Na2SO4 and evaporated to dryness. The residue was subjected to column chromatography on silica using EtOAc:MeOH:TEA (10:1:0.03) as eluent, then crystallized from Et2O to obtain the pure product as a yellow powder.
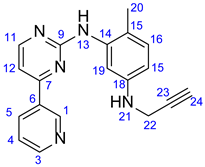
Light grey solid. Mp: 101–103 ◦C (dec.); Yield: 215 mg (17%). 1H-NMR (DMSO-d6): 9.22 (d, J = 1.6 Hz, 1H, H1); 8.68 (s, 1H, H13); 8.65 (dd, J = 4.6 Hz and 1.6 Hz, 1H, H3); 8.43 (d, J = 5.1 Hz, 1H, H11); 8.34 (dt, J = 7.8 Hz and 1.6 Hz, 1H, H5); 7.48 (dd, J = 7.8 Hz and 4.6 Hz, 1H, H4); 7.33 (d, J = 5.1 Hz, 1H, H12); 6.94 (d, J = 8.3 Hz, 1H, H16); 6.86 (d, J = 2.0 Hz, 1H, H19); 6.38 (dd, J = 8.3 Hz and 2.0 Hz, 1H, H15); 5.73(t, J = 6.1 Hz, 1H, H21); 3.78 (dd, J = 6.1 Hz and 2.1 Hz, 2H, H22); 2.96 (t, J = 2.1 Hz, 1H, H24); 2.05 (s, 3H, H20); 13C-NMR (DMSO-d6): 162.0 (C7); 161.2 (C9); 160.0 (C11); 151.9 (C3); 148.7 (C1); 146.7 (C18); 138.7 (C14); 134.8 (C5); 132.7 (C6); 130.8 (C16); 124.3 (C4); 120.9 (C15); 110.4 (C19); 110.0 (C15); 107.8 (C12); 83.1 (C23); 73.3 (C24); 33.0 (C22); 17.7 (C20).
3.3. Synthesis of N-Propargylferrocenecarboxamide (27)
Propargylamine (0.32 mL, 0.275 g, 5 mmol), ferrocenoylimidazolide 26 (1.704 g; 6 mmol, 1.2 eq.) and DMAP (0.184 g; 6 mmol, 1.2 eq.) were dissolved in freshly distilled pyridine (15 mL). This reaction mixture was purged with argon and stirred for 12 h at room temperature, then poured onto crushed ice. The resulting suspension was extracted with DCM (5 × 20 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2SO4 and evaporated to dryness on a rotary evaporator. The dark solid residue was purified by column chromatography on silica using solvent mixture DCM:MeOH (20:1) as eluent, followed by sequential crystallization with water and Et2O to obtain the pure product as a light orange solid. Yield: 1.00 g (75%).
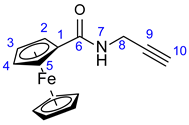
Light orange powder. Mp: 125–126 ◦C (dec.); 1H-NMR (DMSO-d6): 8.30 (t, J = 5.6 Hz, 1H, H7); 4.83 (t, J = 1.8 Hz, 2H, H2 and H5); 4.35 (t, J = 1.8 Hz, 2H, H3 and H4); 4.18 (s, 5H, η5-C5H5); 3.95 (dd, J = 5.6 Hz and 2.4 Hz, 2H, H8); 3.11 (t, J = 2.4 Hz, 1H, H10). 13C-NMR (DMSO-d6): 169.2 (C6); 82.7 (C9); 76.2 (C1); 69.8 (η5-C5H5); 69.6 (C4 and C5); 64.7 (C2 and C5); 28.4 (C8).
3.4. Synthesis of Iodochalcone Intermediates 34 and 35
The corresponding methyl ketone 32 or 33 (1 mmol) and 4-iodobenzaldehyde 31 (232 mg, 1 mmol) were dissolved in EtOH (3 mL). To this solution 2% NaOH/H2O (2 mL) was added, and the resulting mixture was stirred for 12 h at room temperature under argon atmosphere. The precipitated crystals were filtered out and first purified by column chromatography on silica using solvent mixture DCM:MeOH (40:1) as eluent and crystallized from EtOH to obtain the pure product.
3.4.1. (E)-3-(4-Iodophenyl)-1-(3,4,5-Trimethoxyphenyl)Prop-2-en-1-one (34)
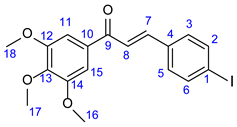
Pale yellow crystals. Mp: 182–184 ◦C (dec.); Yield: 390 mg (87%). Yield: 391 mg (87%). 1H-NMR (DMSO-d6): 7.98 (d, J = 15.6 Hz, 1H, H8); 7.84 (d, J = 8.2 Hz, 2H, H2 and H6); 7.71 (d, J = 8.2 Hz, 2H, H3 and H5); 7.68 (d, J = 15.6 Hz, 1H, H7); 7.43 (s, 2H, H11 and H15); 3.90 (s, 6H, H16 and H18); 3.77 (s, 3H, H17). 13C-NMR (DMSO-d6): 188.2 (C9); 153.4 (C12 and C14); 143.2 (C7); 142.6 (C13); 138.2 (C2 and C6); 134.7 (C4); 133.3 (C10); 131.3 (C3 and C5); 123.3 (C8); 106.7 (C11 and C15); 98.1 (C1); 60.7 (C17), 56.6 (C16 and C18).
3.4.2. (E)-3-(4-Iodophenyl)-1-Ferrocenylprop-2-en-1-one (35)
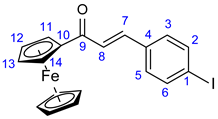
Deep red crystals. Mp: 133–135 ◦C (dec.); Yield: 314 mg (71%). 1H-NMR (DMSO-d6): 7.82 (d, J = 8.0 Hz, 2H, H2 and H6); 7.67 (d, J = 8.0 Hz, 2H, H3 and H5); 7.56 (d, J = 15.6 Hz, 1H, H7); 7.46 (d, J = 15.6 Hz, 1H, H8); 5.04 (br s, 2H, H11 and H14); 4.67 (br s, 2H, H12 and H13); 4.21 (s, 5H, η5-C5H5). 13C-NMR (DMSO-d6): 192.4 (C9); 139.1 (C7); 138.2 (C2 and C6); 135.0 (C4); 131.0 (C3 and C5); 124.8 (C8); 97.1 (C1); 81.1 (C10); 73.3 (C12 and C13); 70.2 (C11 and C14); 70.3 (η5-C5H5).
3.5. General Procedure for the Sonogashira Reactions Using 10-Iodovindoline (20) as Coupling Partner. Synthesis of Hybrids 21–24, 28, 29 and Silyl-Protected Intermediate 30
10-Iodovindoline (20) (1 mmol), the corresponding alkyne component (3, 10, 15, 19, 25, 27or trimethylsilylacetylene) (1 mmol), CuI (38 mg, 0.2 mmol,), PdCl2(PPh3)2 (70 mg, 0.1 mmol) and DIPEA (0.53 mL, 390 mg, 3 mmol) was dissolved in DMF (20 mL). The mixture was stirred for 24 h, at room temperature under argon atmosphere then poured into water. The precipitate was filtered off, washed with water (5 x 10 mL), dried, extracted with DCM (4 x 15 mL), and filtered off again. The organic solution was washed with water (1 x 30 mL) and brine, then dried over Na2SO4. After evaporation of the solvent, the solid residue was subjected to column chromatography on silica using solvent mixture DCM:MeOH (15:1) as eluent and crystallized from Et2O to obtain the pure product. Since in the course of chromatography 30 underwent decomposition, this silylated intermediate was used without purification for subsequent Sonogashira reactions with the iodochalcones.
3.5.1. Methyl (3aR,3a1R,4R,5S,5aR,10bR)-4-Acetoxy-3a-Ethyl-5-Hydroxy-8-Methoxy-6-Methyl-9-(3-((4- Methyl-3-((4-(Pyridin-3-yl)Pyrimidin-2-yl)Amino)Phenyl)Amino)Prop-1-yn-1-yl)-3a,3a1,4,5,5a,6,11,12-Octahydro-1H-Indolizino [8,1-cd]Carbazole-5-Carboxylate (21)
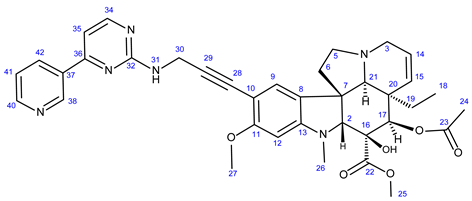
Light yellowih solid. Mp: 162–163 ◦C; Yield: 160 mg (24 %). 1H-NMR (DMSO-d6): 9.10 (d, J = 2.0 Hz, 1H, H38); 8.80 (s, 1H, OH on C16); 8.65 (dd, J = 4.6 Hz and 1.6 Hz, 1H, H40); 8.58 (d, J = 5.1 Hz, 1H, H42); 8.49 (d, J = 5.3 Hz, 1H, H34); 7.52 (dd, J = 7.8 Hz and 4.6 Hz, 1H, H41); 7.21 (s, 1H, H9); 7.13 (d, J = 5.3 Hz, 1H, H35); 6.26 (s, 1H, H12); 6.24 (t, J = 5.0 Hz, 1H, H31); 5.83 (ddd, J = 9.9 Hz, 4.8 Hz and 1.8 Hz, 1H, H14); 5.14 (s, 1H, H17); 5.11 (d, J = 9.9 Hz, 1H, H15); 4.03 (d, J = 5.0 Hz, 1H, H30); 3.77 (s, 3H, H27); 3.62 (s, 3H, H25); 3.58 (s, 1H, H2); 3.42 (m, 1H, H3/A); 3.28 (m, 1H, H5/A); 2.91 (br d, J = 16.2 Hz, 1H, H3/B); 2.72, (s, 1H, H21); 2.68 (m, 1H, H5/B); 2.59 (s, 3H, H26); 2.24 (m, 2H, H6A and H6B); 1.92 (s, 3H, H24); 1.49 (m, 1H, H19/A), 0.95 (m, 1H; H19/B); 0.46 (t, J = 6.9 Hz, 3H, H18). 13C-NMR (DMSO-d6): 172.6 (C22); 170.7 (C23); 163.1 (C36); 161.4 (C11); 157.2 (C34); 156.5 (C32); 153.4 (C13); 151.7 (C40); 131.2 (C37); 130.3 (C15); 127.3 (C9); 125.6 (C8); 124.8 (C14); 124.0 (C41); 103.9 (C35); 101.7 (C10); 93.4 (C12); 91.2 (C29); 80.8 (C2); 81.3 (C28); 76.3 (C17); 65.5 (C21); 60.4 (C5); 58.4 (C27); 53.0 (C7); 51.5 (C25); 50.5 (C3); 43.6 (C6); 42.5 (C20); 39.2 (C26); 30.4 (C19); 21.1 (C24); 8.1 (C18).
3.5.2. Methyl (3aR,3a1R,4R,5S,5aR,10bR)-4-Acetoxy-3a-Ethyl-5-Hydroxy-8-Methoxy-6-Methyl-9-(3-((4- Methyl-3-((4-(Pyridin-3-yl)Pyrimidin-2-yl)Amino)Phenyl)Amino)Prop-1-yn-1-yl)-3a,3a1,4,5,5a,6,11,12-Octahydro-1H-Indolizino [8,1-cd]Carbazole-5-Carboxylate (22)
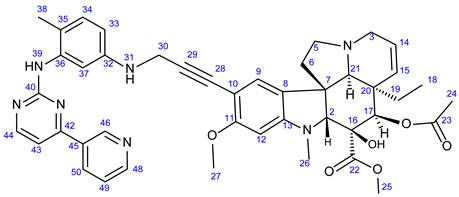
Light yellowish solid. Mp: 157–158 ◦C; Yield:193 mg (25 %). 1H-NMR (DMSO-d6): 9.22 (d, J = 1.8 Hz, 1H, H55); 8.73 (s, 1H, OH on C16); 8.69 (s, 1H, H39); 8.65 (dd, J = 4.6 Hz and 1.6 Hz, 1H, H48); 8.65 (dd, J = 4.6 Hz and 1.8 Hz, 1H, H57); 8.43 (d, J = 5.1 Hz, 1H, H44); 8.34 (dt, J = 7.8 Hz and 1.8 Hz, 1H, H50); 7.48 (dd, J = 7.8 Hz and 4.8 Hz, 1H, H49); 7.33 (d, J = 5.1 Hz, 1H, H43); 7.12 (s, 1H, H9); 6.93 (d, J = 8.2 Hz, 1H, H34); 6.86 (d, J = 2.0 Hz, 1H, H37); 6.38 (dd, J = 8.2 Hz and 2.0 Hz, 1H, H33); 6.26 (s, 1H, H12); 5.84 (ddd, J = 9.9 Hz, 4.8 Hz and 1.8 Hz, 1H, H14); 5.82 (t, J = 6.1 Hz, 1H, H31); 5.13 (s, 1H, H17); 5.11 (d, J = 9.9 Hz, 1H, H15); 4.05 (d, J = 6.1 Hz, 1H, H30); 3.76 (s, 3H, H27); 3.64 (s, 3H, H25); 3.57 (s, 1H, H2); 3.44 (m, 1H, H3/A); 3.31 (m, 1H, H5/A); 2.93 (br d, J = 16.2 Hz, 1H, H3/B); 2.72, (s, 1H, H21); 2.63 (m, 1H, H5/B); 2.59 (s, 3H, H26); 2.26 (m, 2H, H6A and H6B); 2.05 (s, 3H, H38); 1.92 (s, 3H, H24); 1.52 (m, 1H, H19/A), 0.95 (m, 1H; H19/B); 0.46 (t, J = 6.9 Hz, 3H, H18). 13C-NMR (DMSO-d6): 172.5 (C22); 170.6 (C23); 162.0 (C42); 161.2 (C40); 161.5 (C11); 160.0 (C44); 153.4 (C13); 151.9 (C48); 148.7 (two coalesced lines, C32 and C46); 138.7 (C36); 134.8 (C50); 132.7 (C45); 130.8 (C34); 130.3 (C15); 125.3 (C8); 124.3 (C49); 124.8 (C14); 159.2 (C53); 151.9 (C57); 141.6 (C33); 138.3 (C45); 137.7 (C41); 135.1 (C59); 132.5 (C54); 130.8 (C43); 129.4 (C36); 129.0 (C34 and C38); 128.1 (C35 and C37); 127.4 (C9); 126.4 (C44); 124.4 (two coalesced lines, C8 and C58); 117.9 (C46); 117.5 (C42); 107.8 (C52); 101.7 (C10); 93.2 (C12); 89.9 (C29); 83.5 (C2); 81.3 (C28); 76.3 (C17); 65.5 (C21); 60.4 (C5); 58.4 (C27); 53.0 (C7); 51.5 (two coalesced lines, C25 and C32); 50.5 (C3); 43.6 (C6); 42.5 (C20); 39.2 (C26); 38.2 (C30); 30.4 (C19); 21.1 (C24); 18.3 (C47); 8.1 (C18). Elemental composition: C44H47N7O6, HRMS: calculated m/z: 770.3661, measured m/z: 770.36549, mass error: 0.8 ppm (M+H+).
3.5.3. Methyl (3aR,3a1R,4R,5S,5aR,10bR)-4-Acetoxy-3a-Ethyl-5-Hydroxy-8-Methoxy-6-Methyl-9-(3-((4- ((4-Methyl-3-((4-(Pyridin-3-yl)Pyrimidin-2-yl)Amino)Phenyl)Carbamoyl)Benzyl)Amino)Prop-1-yn-1-yl)-3a,3a1,4,5,5a,6,11,12-Octahydro-1H-Indolizino [8,1-cd]Carbazole-5-Carboxylate (23)
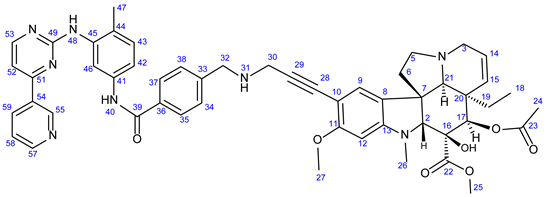
Light orange solid. Mp: 138–140 ◦C; Yield: 181 mg (20 %) 1H-NMR (DMSO-d6):10.17 (s, 1H, H40); 9.20 (d, J = 1.8 Hz, 1H, H55); 8.90 (s, 1H, H48); 8.74 (s, 1H, OH on C16); 8.65 (dd, J = 4.6 Hz and 1.8 Hz, 1H, H57); 8.48 (d, J = 5.1 Hz, 1H, H53); 8.44 (dt, J = 7.8 Hz and 1.8 Hz, 1H, H59); 8.02 (d, J = 2.0 Hz, 1H, H46); 7.94 (d, J = 8.5 Hz, 2H, H35 and H37); 7.52 (dd, J = 7.8 Hz and 4.8 Hz, 1H, H58); 7.49 (d, J = 8.5 Hz, 2H, H34 and H38); 7.44 (dd, J = 8.2 Hz and 2.0 Hz, 1H, H42); 7.33 (d, J = 5.1 Hz, 1H, H52); 7.17 (s, 1H, H9); 6.93 (d, J = 8.2 Hz, 1H, H43); 6.26 (s, 1H, H12); 5.82 (ddd, J = 9.9 Hz, 4.8 Hz and 1.8 Hz, 1H, H14); 5.15 (s, 1H, H17); 5.12 (d, J = 9.9 Hz, 1H, H15); 3.89 (s, 2H, H32); 3.51 (s, 2H, H30); 3.76 (s, 3H, H27); 3.62 (s, 3H, H25); 3.57 (s, 1H, H2); 3.42 (m, 1H, H3/A); 3.32 (m, 1H, H5/A); 2.91 (br d, J = 16.2 Hz, 1H, H3/B); 2.72, (s, 1H, H21); 2.63 (m, 1H, H5/B); 2.57 (s, 3H, H26); 2.26 (m, 2H, H6A and H6B); 2.05 (s, 3H, H47); 1.92 (s, 3H, H24); 1.49 (m, 1H, H19/A), 0.95 m, 1H; H19/B); 0.46 (t, J = 6.9 Hz, 3H, H18). 13C-NMR (DMSO-d6): 172.5 (C22); 170.6 (C23); 165.7 (C39); 162.3 (C51); 161.6 (C49); 161.5 (C11); 159.2 (C53); 153.4 (C13); 151.9 (C57); 148.6 (C55); 141.6 (C33); 138.3 (C45); 137.7 (C41); 135.1 (C59); 132.5 (C54); 130.8 (C43); 130.3 (C15); 129.4 (C36); 129.0 (C34 and C38); 128.1 (C35 and C37); 127.4 (C9); 126.4 (C44); 125.4 (C8); 124.8 (C14); 124.4 (two coalesced lines, C8 and C58); 117.9 (C46); 117.5 (C42); 107.8 (C52); 101.7 (C10); 93.2 (C12); 89.9 (C29); 83.5 (C2); 81.3 (C28); 76.3 (C17); 65.5 (C21); 60.4 (C5); 58.4 (C27); 53.0 (C7); 51.5 (two coalesced lines, C25 and C32); 50.5 (C3); 43.6 (C6); 42.5 (C20); 39.2 (C26); 38.2 (C30); 30.4 (C19); 21.1 (C24); 18.3 (C47); 8.1 (C18). Elemental composition: C52H54N8O7, HRMS: calculated m/z: 903.4188, measured m/z: 903.41853, mass error: 0.3 ppm (M+H+).
3.5.4. Methyl (3aR,3a1R,4R,5S,5aR,10bR)-4-Acetoxy-9-((3-((6,7-bis(2-Methoxyethoxy)Quinazolin-4-yl)- Amino)Phenyl)Ethynyl)-3a-Ethyl-5-Hydroxy-8-Methoxy-6-Methyl-3a,3a1,4,5,5a,6,11,12-Octahydro-1H-Indolizino [8,1-cd]Carbazole-5-Carboxylate (24)
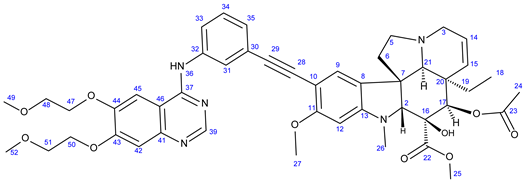
Light orange solid. Mp: 158–159 ◦C (dec.); Yield: 228 mg (27 %) 1H-NMR (DMSO-d6): 9.46 (s, 1H, H36); 8.73 (s, 1H, OH on C16); 8.52 (s, 1H, H39); 8.23 (t, J = 5.8 Hz, 1H, H31); 7.93 (t, J = 2.0 Hz, 1H, H31); 7.87 (dt, J = 7.6 Hz and 2.0 Hz, 1H, H35); 7.40 (t, J = 7.6 Hz, 1H, H34); 7.23 (s, 2H, H42 and H45); 7.17 (s, 1H, H9); 7.13 (dt, J = 7.6 Hz and 2.0 Hz, 1H, H33); 6.26 (s, 1H, H12); 5.82 (ddd, J = 9.9 Hz, 4.8 Hz and 1.8 Hz, 1H, H14); 5.15 (s, 1H, H17); 5.12 (d, J = 9.9 Hz, 1H, H15); 4.26 (m, 4H, H47 and H50); 3.78 (m, 2H, H48); 3.76 (s, 3H, H27); 3.71 (m, 2H, H51); 3.62 (s, 3H, H25); 3.57 (s, 1H, H2); 3.42 (m, 1H, H3/A); 3.34 (s, 6H, H49 and H52); 3.32 (m, 1H, H5/A); 2.91 (br d, J = 16.2 Hz, 1H, H3/B); 2.72, (s, 1H, H21); 2.63 (m, 1H, H5/B); 2.57 (s, 3H, H26); 2.26 (m, 2H, H6A and H6B); 1.92 (s, 3H, H24); 1.49 (m, 1H, H19/A), 0.95 m, 1H; H19/B); 0.46 (t, J = 6.9 Hz, 3H, H18). 13C-NMR (DMSO-d6): 172.5 (C22); 170.8 (C23); 161.5 (C11); 156.7 (C37); 154.3 (C43); 153.4 (two coalesced lines, C13 and C39); 148.8 (C44); 147.5 (C41); 140.2 (C32); 130.3 (C15); 129.3 (C34); 127.4 (C9); 126.0 (C8); 125.0 (C33); 124.8 (C14); 124.2 (C31); 124.1 (C30); 109.5 (C46); 108.8 (C42); 104.0 (C45); 101.7 (C10); 93.4 (C12); 91.3 (C29); 88.4 (C28); 83.5 (C2); 79.4 (C16); 76.1 (C17); 70.6 and 70.5 (C47 and C50, interhageable assignements); 68.9 and 68.6 (C48 and C51, interchangeable assignements); 65.5 (C21); 60.4 (C5); 58.9 (two coalesced lines, C49 and C52); 58.4 (C27); 53.0 (C7); 51.5 (C25); 50.5 (C3); 43.6 (C6); 42.5 (C20); 39.2 (C26); 30.4 (C19); 21.1 (C24); 8.1 (C18). Elemental composition: C47H53N5O10, HRMS: calculated m/z: 848.3865, measured m/z: 848.38561, mass error: 1.1 ppm (M+H+).
3.5.5. Methyl (3aR,3a1R,4R,5S,5aR,10bR)-4-Acetoxy-3a-Ethyl-5-Hydroxy-8-Methoxy-6-Methyl-9-(Ferro- Cenylethynyl)-3a,3a1,4,5,5a,6,11,12-Octahydro-1H-Indolizino [8,1-cd]Carbazole-5-Carboxylate (28)
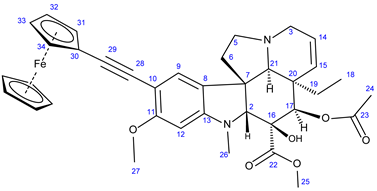
Light orange solid. Mp: 218–219 ◦C (dec.); Yield: 160 mg (24 %) 1H-NMR (DMSO-d6): 8.73 (s, 1H, OH on C16); 7.20 (s, 1H, H9); 6.29 (s, 1H, H12); 5.84 (ddd, J = 9.9 Hz, 4.8 Hz and 2.0 Hz, 1H, H14); 5.14 (d, J = 9.9 Hz, 1H, H15); 5.13 (s, 1H, H17); 4.42 (br s, 2H, H31 and H34); 4.24 (br s, 2H, H32 and H34); 4.20 (s, 5H, η5-C5H5); 3.76 (s, 3H, H27); 3.64 (s, 3H, H25); 3.57 (s, 1H, H2); 3.40 (m, 1H, H3/A); 3.28 (m, 1H, H5/A); 2.91 (br d, J = 16.2 Hz, 1H, H3/B); 2.69, (s, 1H, H21); 2.60 (m, 1H, H5B); 2.59 (s, 3H, H26); 2.28 (m, 2H, H6/A and H6/B); 1.92 (s, 3H, H24); 1.49 (m, 1H, H19/A), 0.95 (m, 1H; H19/B); 0.46 (t, J = 6.9 Hz, 3H, H18). 13C-NMR (DMSO-d6): 172.5 (C22); 170.6 (C23); 160.2 (C11); 152.9 (C13); 130.3 (C15); 127.4 (C9); 124.8 (C14); 124.4 (C8); 100.1 (C10); 93.4 (C12); 92.1 (C29); 83.0 (C2); 79.1 (C16); 76.3 (C17); 76.2 (C28); 71.5 (C31 and C34); 70.2 (η5-C5H5); 68.9 (C32 and C34); 65.6 (C21); 65.4 (C30); 60.1 (C5); 58.4 (C27); 52.9 (C7); 51.4 (C25); 50.5 (C3); 43.4 (C6); 42.5 (C20); 39.0 (C26); 30.4 (C19); 21.1 (C24); 8.1 (C18); Elemental composition: C37H40FeN2O6, HRMS: calculated m/z: 664.2230, measured m/z: 664.22229, mass error: 1.1 ppm (M+., Fe(II) oxidized to Fe(III)).
3.5.6. Methyl (3aR,3a1R,4R,5S,5aR,10bR)-4-Acetoxy-9-(3-Ferroceneamidoprop-1-yn-1-yl)-3a-Ethyl-5- Hydroxy-8-Methoxy-6-Methyl-3a,3a1,4,5,5a,6,11,12-Octahydro-1H-Indolizino [8,1-cd]Carbazole-5-Carboxylate (29)
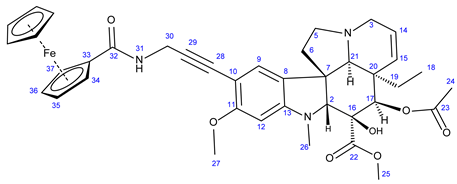
Light orange solid. Mp: 202–204 ◦C (dec.); Yield: 195 mg (27 %) 1H-NMR (DMSO-d6): 8.73 (s, 1H, OH on C16); 8.23 (t, J = 5.8 Hz, 1H, H31); 7.12 (s, 1H, H9); 6.28 (s, 1H, H12); 5.83 (ddd, J = 9.9 Hz, 4.8 Hz and 1.8 Hz, 1H, H14); 5.13 (s, 1H, H17); 5.11 (d, J = 9.9 Hz, 1H, H15); 4.52 (br s, 2H, H34 and H37); 4.26 (br s, 2H, H35 and H36); 4.16 (d, J = 5.8 Hz, 2H, H30); 4.01 (s, 5H, η5-C5H5); 3.76 (s, 3H, H27); 3.64 (s, 3H, H25); 3.57 (s, 1H, H2); 3.42 (m, 1H, H3/A); 3.30 (m, 1H, H5/A); 2.91 (br d, J = 16.2 Hz, 1H, H3/B); 2.68, (s, 1H, H21); 2.60 (m, 1H, H5/B); 2.26 (m, 2H, H6A and H6B); 2.59 (s, 3H, H26); 1.92 (s, 3H, H24); 1.50 (m, 1H, H19/A), 0.96 m, 1H; H19/B); 0.47 (t, J = 6.9 Hz, 3H, H18). 13C-NMR (DMSO-d6): 172.5 (C22); 170.6 (C23); 169.3 (C32); 161.8 (C11); 153.7 (C13); 130.3 (C15); 127.4 (C9); 126.1 (C8); 124.9 (C14); 124.4 (C8); 101.8 (C10); 93.6 (C29); 93.2 (C12); 83.3 (C2); 79.2 (C28); 79.0 (two coalesced lines, C33 and C16); 76.3 (C17); 69.6 (C35 and C36); 69.9 (η5-C5H5); 65.7 (C21); 64.7 (C32 and C34); 60.1 (C5); 58.4 (C27); 52.9 (C7); 51.4 (C25); 50.5 (C3); 43.4 (C6); 42.5 (C20); 39.0 (C26); 30.4 (C19); 29.6 (C30); 21.1 (C24); 8.1 (C18). Elemental composition: C39H43FeN3O7, HRMS: calculated m/z: 722.2523, measured m/z: 722.2508, mass error: 2.1 ppm (M+H+).
3.6. Synthesis of Chalcone-Containing Hybrids (36 and 37) by Sonogashira Coupling
The mixture of 600 mg of crude silyl-protected acetylene 30 (in pure form 1 mmol is 550 mg), TBAF (523 mg, 2 mmol), the appropriate chalcone (34, 424 mg, 1 mmol) or 35 (442 mg, 1 mmol), CuI (38 mg, 0.2 mmol,), PdCl2(PPh3)2 (70 mg, 0.1 mmol) and DIPEA (0.53 mL, 390 mg, 3 mmol) was dissolved in DMF (20 mL). The mixture was stirred for 24 h, at room temperature, under an argon atmosphere, and then poured into water. The precipitate was filtered off, washed with water (5 x 10 mL), dried, extracted with DCM (4 x 15 mL), and filtered off again. The organic solution was washed with water (1 x 30 mL) and brine, then dried over Na2SO4. After evaporation of the solvent, the solid residue was subjected to column chromatography on silica using solvent mixture DCM:MeOH (15:1) as eluent and crystallized from Et2O to obtain the pure product.
3.6.1. Methyl (3aR,3a1R,4R,5S,5aR,10bR)-4-Acetoxy-3a-Ethyl-5-Hydroxy-8-Methoxy-6-Methyl-9-((4-((E)- 3-oxo-3-(3,4,5-Trimethoxyphenyl)Prop-1-en-1-yl)Phenyl)Ethynyl)-3a,3a1,4,5,5a,6,11,12-Octahydro-1H-Indolizino [8,1-cd]Carbazole-5-Carboxylate (36)
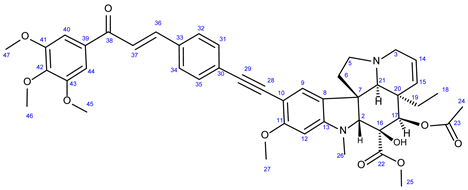
Light yellow powder. Mp: 149–150 ◦C Yield: 185 mg (24 %) 1H-NMR (DMSO-d6): 8.87 (s, 1H, OH on C16); 7.98 (d, J = 16.2 Hz, 1H, H37); 7.94 (d, J = 8.2 Hz, 2H, H32 and H34); 7.76 (d, J = 16.2 Hz, 1H, H36); 7.51 (d, J = 8.2 Hz, 2H, H31 and H35); 7.45 (s, 2H, H40 and H44); 7.38 (s, 1H, H9); 6.26 (s, 1H, H12); 5.84 (ddd, J = 9.8 Hz, 4.8 Hz and 2.0 Hz, 1H, H14); 5.13 (s, 1H, H17); 5.11 (d, J = 9.8 Hz, 1H, H15); 3.91 (s, 6H, H45 and H47); 3.79 (s, 3H, H46); 3.76 (s, 3H, H27); 3.64 (s, 3H, H25); 3.57 (s, 1H, H2); 3.42 (m, 1H, H3/A); 3.30 (m, 1H, H5/A); 2.91 (br d, J = 16.2 Hz, 1H, H3/B); 2.71 (s, 1H, H21); 2.60 (m, 1H, H5/B); 2.26 (m, 2H, H6A and H6B); 2.59 (s, 3H, H26); 1.92 (s, 3H, H24); 1.48 (m, 1H, H19/A), 0.95 m, 1H; H19/B); 0.47 (t, J = 6.9 Hz, 3H, H18). 13C-NMR (DMSO-d6): 188.2 (C38); 172.6 (C22); 170.6 (C23); 162.3 (C11); 153.7 (C13); 153.4 (C41 and C43); 143.5 (C36); 142.5 (C42); 133.5 (two coalesced lines, C33 and C39); 131.6 (C31 and C35); 130.3 (C15); 129.8 (C32 and C34); 127.4 (C9); 126.1 (C8); 124.9 (C14); 124.4 (C8); 122.6 (C37); 106.2 (C40 and C44); 101.8 (C10); 93.4 (C12); 91.5 (C29); 91.3 (C28); 83.5 (C2); 79.0 (C16); 76.1 (C17); 65.4 (C21); 60.8 (C46); 60.1 (C5); 58.4 (C27); 56.8 (C45 and C47); 52.9 (C7); 51.4 (C25); 50.5 (C3); 43.4 (C6); 42.5 (C20); 39.1 (C26); 30.5 (C19); 21.1 (C24); 8.1 (C18). Elemental composition: C45H48N2O10, HRMS: calculated m/z: 777.3382, measured m/z: 777.34096, mass error: -3.6 ppm (M+H+).
3.6.2. Methyl (3aR,3a1R,4R,5S,5aR,10bR)-4-Acetoxy-3a-Ethyl-5-Hydroxy-8-Methoxy-6-Methyl-9-((4- ((E)-3-oxo-3-Ferrocenylprop-1-en-1-yl)Phenyl)Ethynyl)-3a,3a1,4,5,5a,6,11,12-Octahydro-1H-Indolizino [8,1-cd]Carbazole-5-Carboxylate (37)
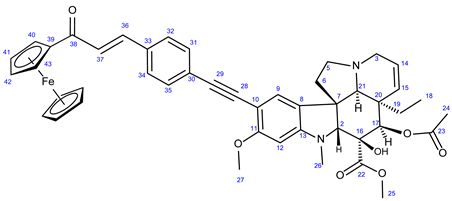
Orange powder. Mp: 163–165 ◦C (dec.); Yield: 207 mg (26 %) 1H-NMR (DMSO-d6): 8.85 (s, 1H, OH on C16); 7.89 (d, J = 8.2 Hz, 2H, H32 and H34); 7.63 (d, J = 16.2 Hz, 1H, H36); 7.50 (d, J = 8.2 Hz, 2H, H31 and H35); 7.47 (d, J = 16.2 Hz, 1H, H37); 7.32 (s, 1H, H9); 6.26 (s, 1H, H12); 5.85 (ddd, J = 9.8 Hz, 4.8 Hz and 2.0 Hz, 1H, H14); 5.12 (s, 1H, H17); 5.11 (d, J = 9.8 Hz, 1H, H15); 5.07 (t, J = 1.8 Hz, 2H, H40 and H43); 4.68 (t, J = 1.8 Hz, 2H, H41 and H42); 4.23 (s, 5H, η5C5H5); 3.76 (s, 3H, H27); 3.65 (s, 3H, H25); 3.57 (s, 1H, H2); 3.43 (m, 1H, H3/A); 3.30 (m, 1H, H5/A); 2.91 (br d, J = 16.2 Hz, 1H, H3/B); 2.71 (s, 1H, H21); 2.60 (m, 1H, H5/B); 2.26 (m, 2H, H6A and H6B); 2.59 (s, 3H, H26); 1.92 (s, 3H, H24); 1.49 (m, 1H, H19/A), 0.94 m, 1H; H19/B); 0.48 (t, J = 6.9 Hz, 3H, H18). 13C-NMR (DMSO-d6): 192.4 (C38); 172.7 (C22); 170.6 (C23); 162.3 (C11); 153.7 (C13); 153.4 (C41 and C43); 143.5 (C36); 142.5 (C42); 139.4 (C36); 134.6 (C33); 131.6 (C31 and C35); 130.3 (C15); 129.4 (C32 and C34); 127.4 (C9); 126.4 (C8); 125.6 (C30); 124.9 (C14); 124.6 (C37); 124.4 (C8); 101.8 (C10); 93.4 (C12); 91.5 (C29); 91.3 (C28); 83.5 (C2); 81.2 (C39); 79.0 (C16); 76.2 (C17); 73.3 (C41 and C42); 70.3 (η5C5H5); 65.4 (C21); 60.8 ((C46); 60.1 (C5); 58.4 (C27); 56.8 (C45 and C47); 52.9 (C7); 51.4 (C25); 50.5 (C3); 43.4 (C6); 42.4 (C20); 39.0 (C26); 30.6 (C19); 21.1 (C24); 8.2 (C18). Elemental composition: C46H46FeN2O7, HRMS: calculated m/z: 795.2727, measured m/z: 795.27252, mass error: 0.2 ppm (M+H+).
3.7. Synthesis of Reference Chalcone-Containing Hybrids (36a and 37a) by Sonogashira Coupling
The mixture of phenylacetylene (102 mg, 1mmol), the appropriate iodochalcone (34, 424 mg, 1 mmol) or 35 (442 mg, 1 mmol), CuI (38 mg, 0.2 mmol,), PdCl2(PPh3)2 (70 mg, 0.1 mmol) and DIPEA (0.53 mL, 390 mg, 3 mmol) was dissolved in DMF (20 mL). The mixture was stirred for 24 h at room temperature under an argon atmosphere then poured into water. The precipitate was filtered off, washed with water (5 x 10 mL), dried, extracted with DCM (4 x 15 mL) and filtered off again. The organic solution was washed with water (1 x 30 mL) and brine, then dried over Na2SO4. After evaporation of the solvent the solid residue was subjected to column chromatography on silica using solvent mixture DCM:MeOH (15:1) as eluent and crystallized from Et2O to obtain the pure product.
3.7.1. (E)-3-(4-(Phenylethynyl)Phenyl)-1-(3,4,5-Trimethoxyphenyl)Prop-2-en-1-One (36a)
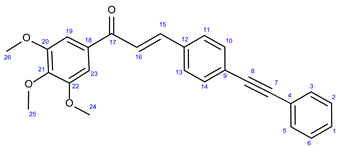
Light yellowish solid. Mp: 138–140 ◦C; Yield: 316 mg (79 %) 1H-NMR (DMSO-d6): 7.82 (d, J = 16.2 Hz, 1H, H15); 7.65 (d, J = 8.2 Hz, 2H, H11 and H13); 7.59 (d, J = 8.2 Hz, 2H, H10 and H14); 7.57 (m, 2H, H3 and H5); 7.51 (d, J = 16.2 Hz, 1H, H16); 7.43-7.36 (m, 3H, H1, H2 and H6); 7.31 (s, 2H, H19 and H23); 3.98 (s, 6H, H24 and H26); 3.97 (s, 3H, H25). 13C-NMR (DMSO-d6): 189.0 (C17); 153.2 (C20 and C22); 143.8 (C15); 142.6 (C21); 134.7 (C12); 133.4 (C18); 131.7 (C2 and C6); 132.1 (C10 and C14); 128.6 (C1); 128.44 (C3 and C5); 128.40 (C11 and C13); 125.5 (C9); 131.7 (C2 and C6); 122.9 (C4); 122.2 (C16); 106.2 (C19 and C23); 91.8 (C8); 89.1 (C7); 61.0 (C25); 56.5 (C24 and C26). Elemental composition: C26H22O4, HRMS: calculated m/z: 399.1591, measured m/z: 399.15923, mass error: - 0.3 ppm (M+H+).
3.7.2. (E)-1-(Ferrocenyl)-3-(4-(Phenylethynyl)Phenyl)Prop-2-en-1-One (37a)
Light orange solid. Mp: 203–204 ◦C (dec.); Yield: 308 mg (74 %) 1H-NMR (DMSO-d6): 7.80 (d, J = 16.2 Hz, 1H, H15); 7.66 (d, J = 8.2 Hz, 2H, H11 and H13); 7.61 (d, J = 8.2 Hz, 2H, H10 and H14); 7.58 (m, 2H, H3 and H5); 7.43-7.36 (m, 3H, H1, H2 and H6); 7.16 (d, J = 16.2 Hz, 1H, H16); 4.95 (t, J = 1.8 Hz, 2H, H19 and H24); 4.63 (t, J = 1.8 Hz, 2H, H20 and H21); 4.26 (s, 5H, η5C5H5). 13C-NMR (DMSO-d6): 192.8 (C17); 140.0 (C15); 135.0 (C12); 132.1 (C10 and C14); 131.7 (C2 and C6); 128.6 (C1); 128.4 (C2 and C5); 128.2 (C11 and C13); 125.0 (C9); 123.5 (C16); 123.0 (C4); 91.8 (C8); 89.1 (C7); 80.6 (C18); 72.9 (C20 and C21); 70.2 (η5C5H5); 69.8 (C19 and C22). Elemental composition: C27H20FeO HRMS: calculated m/z: 416.0858, measured m/z: 416.08557, mass error: 0.6 ppm (M+., Fe(II) oxidized to Fe(III)).
3.8. Determination of Antiproliferative Activities
The antiproliferative properties of a selected set of the prepared vindoline analogs were determined by the standard MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay using MDA-MB-231, HeLa, A2780, and SH-SY5Y cell lines isolated from human breast, cervical, ovarian cancer, and neuroblastoma, respectively [63]. Non-cancerous human fibroblasts MRC-5 cells were additionally used to characterize the cancer selectivity of the tested agents. Cells were obtained from the European Collection of Cell Cultures (Salisbury, UK) and maintained in Eagle’s minimal essential medium (EMEM) supplemented with 10% fetal calf serum, 1% non-essential amino acids, and 1% antibiotic–antimycotic at 37 °C in a humidified atmosphere with 5% CO2. All media and supplements were purchased from Capricorn Scientific Ltd. (Ebsdorfergrund, Germany). Cells were seeded into 96-well plates (5000/well), and after overnight incubation, the tested substances were added at two concentrations (10 or 30 µM). After incubation for 72 h under cell-culturing conditions, the MTT solution was added (20 µL of 5 mg/mL per well), and the medium was removed after 4 hours. The generated formazan crystals were solubilized in 100 µM dimethylsulfoxide, and the absorbance was measured at 545 nm using a microplate reader (BMG Labtech, Ortenberg, Germany). In the case of the test substances exhibiting higher than 50% growth inhibition at 10 µM on any cancer cell lines, the assays were repeated using a set of dilutions. Two independent experiments were performed with five parallel wells. The IC50 values were calculated by fitting sigmoid concentration-response curves using GraphPad Prism 10.0 software (GraphPad Software, San Diego, CA, USA).
5. Conclusions
This contribution presents feasible synthetic pathways to the first representatives of alkyne-tethered vindoline-based hybrids as potential anticancer agents. The antiproliferative assays identified a trimethoxyphenyl-containing chalcone-vindoline hybrid (36) as a highly efficient and selective lead compound featuring a wide therapeutic window determined by its submicromolar activity on A2780 cells and a substantially decreased activity on MRC-5 fibroblast cells. Consequently, ovarian cancer might be considered a prioritized target of treatment with 36, which merits more extended investigation to disclose its cellular targets and mechanism of action, paving the way for developing rationally designed follow-up molecules with enhanced potency in clinical applications.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Copies of the NMR spectra of the novel propargylated pharmacophore fragments S2–S5; Copies of the NMR spectra of the novel iodinated chalcones S6–S7; Copies of the NMR spectra of the novel alkyne-tethered vindoline hybrids S8–S16; Copies of the HRMS spectra of the novel alkyne-tethered vindoline hybrids S17–S20.
Author Contributions
“Conceptualization, A.C. and L.H.; methodology, E.F. and P.K.; validation, G.S. and I.Z.; formal analysis, G.S.; investigation, E.F.; B.A.T.; R.M.; D.P. and A.C.; resources, L.H.; I.Z. and A.C.; data curation, E.F.; writing—original draft preparation, E.F.; writing—review and editing, I.Z. and A.C.; visualization, A.C.; supervision, I.Z.; project administration, E.F.; funding acquisition, L.H. I.Z. and A.C.
Funding
“This work was funded by the Hungarian Scientific Research Fund [OTKA K_129037], ELTE Thematic Excellence Programme Synth+, supported by the Hungarian Ministry for Innovation and Technology and by the Ministry of Innovation and Technology of Hungary from the Na-tional Research, Development and Innovation Fund [TKP2021-EGA-32]. This work has also been connected to the project no. RRF-2.3.1-21-2022-00015 with the support provided by the European Union (Széchenyi Plan Plus, National Laboratory Program, PharmaLab).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data generated and analyzed during our research are not available in any public database or repository but will be shared by the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Garcia-Mayea, Y.; Mir, C.; Masson, F.; Paciucci, R.; Leonart, M.E. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin Cancer Biol. 2020, 60, 166–180. [Google Scholar] [CrossRef]
- Kucuksayan, E.; Ozben, T. Hybrid Compounds as Multitarget Directed Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 907–918. [Google Scholar] [CrossRef]
- Fortin, S.; Bérubé, G. Advances in the development of hybrid anticancer drugs. Expert Op. Drug Disc. 2013, 8, 1029–1047. [Google Scholar] [CrossRef]
- Zheng, W.; Zhao, Y.; Luo, Q.; Zhang, Y.; Wu, K.; Wang, F. Multi-Targeted Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 3084–3098. [Google Scholar] [CrossRef]
- Ferguson, P.J.; Phillips, J.R.; Seiner, M.; Cass, C.E. Differential Activity of Vincristine and Vinblastine against Cultured Cells. Cancer Res. 1984, 44, 3307–3312. [Google Scholar]
- Isah, T. Anticancer Alkaloids from Trees: Development into Drugs. Pharmacogn. Rev. 2016, 10, 90–99. [Google Scholar] [CrossRef]
- Noble, C.O.; Guo, Z.; Hayes, M.E.; Marks, J.D.; Park, J.W.; Benz, C.C.; Kirpotin, D.B.; Drummond, D.C. Characterization of Highly Stable Liposomal and Immunoliposomal Formulations of Vincristine and Vinblastine. Cancer Chemother. Pharmacol. 2009, 64, 741–751. [Google Scholar] [CrossRef]
- Binet, S.; Chaineau, E.; Fellous, A.; Lataste, H.; Krikorian, A.; Couzinier, J.P.; Meininger, V. Immunofluorescence study of the action of navelbine, vincristine and vinblastine on mitotic and axonal microtubules. Int. J. Cancer 1990, 46, 262–266. [Google Scholar] [CrossRef]
- Owellen, R.J.; Owens, A.H.; Donigian, D.W. The binding of vincristine, vinblastine and colchicine to tubulin, Biochem. and Biophys. Res. Comm. 1972, 47, 685–691. [Google Scholar] [CrossRef]
- Moudi, M.; Go, R.; Yien, C.Y.; Nazre, M. Vinca Alkaloids. Int. J. Prev. Med. 2013, 4, 1231–1235, PMID: 24404355; PMCID: PMC3883245. [Google Scholar]
- Martino, E.; Casamassiva, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca alkaloids and analogues as anticancer agents: Looking back, peering ahead. Bioorg & Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef] [PubMed]
- Keglevich, A.; Dányi, L.; Rieder, A.; et al. ; Synhesis and Cytotoxic Activity of New Vindoline Derivatives Coupled to Natural and Synthetic Pharmacophores. Molecules. 2020, 25, 1010. [Google Scholar] [CrossRef]
- Keglevich, A.; Zsiros, V.; Keglevich, P.; Szigetvári, Á.; et al. ; Synthesis and In Vitro Antitumor Effect of New Vindoline-steroid Hybrids. Curr. Org. Chem. 2019, 23, 959–967. [Google Scholar] [CrossRef]
- Mayer, S.; Keglevich, P.; Hazai, L. Vinca Hybrids with Antiproliferative Effect. Med. Res. Arch. 2022, 10, 3. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: a breakthrough of targeted therapy in cancer. Chemotherapy research and practice 2014, 357027. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Kong, J.; Wang, J.; Wang, C.; Shen, H. Severe megaloblastic anemia in a patient with advanced lung adenocarcinoma during treatment with erlotinib: a case report and literature review. BMC Pulm. Med. 2024, 24, 121. [Google Scholar] [CrossRef]
- Keglevich, P.; Hazai, L.; Gorka-Kereskényi, Á.; Péter, L.; Gyenese, J.; Lengyel, Z.; Kalaus, G.; Dubrovay, Z.; Dékány, M.; Orbán, E.; Bánóczi, Z.; Szántay, Cs., Jr.; Szántay, C. Synthesis and in vitro antitumor effect of new vindoline derivatives coupled with amino acid esters. Heterocycles 2013, 87, 2299–2317. [Google Scholar] [CrossRef]
- Keglevich, A.; Dányi, L.; Rieder, A.; Horváth, D.; Szigetvári, Á.; Dékány, M.; Szántay, C., Jr.; Latif, A.D.; Hunyadi, A.; Zupkó, I.; Keglevich, P.; Hazai, L. Synthesis and Cytotoxic Activity of New Vindoline Derivatives Coupled to Natural and Synthetic Pharmacophores. Molecules 2020, 25, 1010. [Google Scholar] [CrossRef] [PubMed]
- Keglevich, A.; Zsiros, V.; Keglevich, P.; Szigetvári, Á.; Dékány, M.; Szántay, C., Jr.; Mernyák, E.; Wölfling, J.; Hazai, L. Synthesis and In Vitro Antitumor Effect of New Vindoline-steroid Hybrids. Current Organic Chemistry 2019, 23, 959–967. [Google Scholar] [CrossRef]
- Köpf-Maier, P.; Köpf, H.; Neuse, E.W. Ferricenium complexes: A new type of water-soluble antitumor agent. J. Cancer Res. Clin. Oncol. 1984, 108, 336–340. [Google Scholar] [CrossRef]
- Skoupilova, H.; Bartosik, M.; Sommerova, L.; Pinkas, J.; Vaculovic, T.; Kanicky, V.; Karban, J.; Hrstka, R. Ferrocenes as new anticancer drug candidates: Determination of the mechanism of action. Eur. J. Pharm. 2020, 867, 172825. [Google Scholar] [CrossRef]
- Věžník, J.; Konhefr, M.; Fohlerová, Z.; Lacina, K. ; Redox-dependent cytotoxicity of ferrocene derivatives and ROS-activated prodrugs based on ferrocenyliminoboronates. J. Inorg. Biochem. 2020, 224, 111561. [Google Scholar] [CrossRef]
- Jadhav, J.; Das, R.; Kamble, S.; Chowdhury, M.G.; Kapoor, S.; Gupta, A.; Vyas, H.; Shard, A. Ferrocene-based modulators of cancer-associated tumor pyruvate kinase M2. J. Organomet. Chem 2022, 968–969, 122338. [Google Scholar] [CrossRef]
- Yan, J.; Yue, K.; Fan, X.; Xu, X.; Wang, J.; Qin, M.; Zhang, Q.; Hou, X.; Li, X.; Wang, Y. Synthesis and bioactivity evaluation of ferrocene-based hydroxamic acids as selective histone deacetylase 6 inhibitors. Eur. J. Med. Chem. 2023, 246, 115004. [Google Scholar] [CrossRef]
- Wang, R.; Chen, H.; Weitao, W.; Zheng, M.; Zhang, T.; Zhang, Y. Ferrocene-containing hybrids as potential anticancer agents: Current developments, mechanisms of action and structure-activity relationships. Eur. J. Med. Chem. 2020, 190, 112109. [Google Scholar] [CrossRef]
- Resnier, P.; Galopin, N.; Yann Sibiril, Y.; Clavreul, A.; Cayon, J.; Briganti, A.; Legras, P.; Vessières, A.; Montier, T.; Jaouen, G.; Benoit, J.-P.; Passirani, C. Efficient ferrocifen anticancer drug and Bcl-2 gene therapy using lipid nanocapsules on human melanoma xenograft in mouse. Pharmacological Res. 2017, 126, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Ornelas, C. Application of ferrocene and its derivatives in cancer research. New J. Chem. 2011, 35, 1973–1985. [Google Scholar] [CrossRef]
- Braga, S.S.; Silva, A.M.S. A New Age for Iron: Antitumoral Ferrocenes. Organometallics 2013, 32, 5626–5639. [Google Scholar] [CrossRef]
- Quirante, J.; Dubar, F.; González, A.; Lopez, C.; Cascante, M.; Cortés, R.; Forfar, I.; Pradines, B.; Biot, C. Ferrocene-indole hybrids for cancer and malaria therapy. J. Organomet. Chem. 2011, 696, 1011–1017. [Google Scholar] [CrossRef]
- Ramirez-Vick, J.; Acevedo, C.; Melendez, E.; Singh, S. Cytotoxicity and Reactive Oxygen Species Generated by Ferrocenium and Ferrocene on MCF7 and MCF10A Cell Lines. J. Cancer Sci. Ther. 2012, 4, 271–275. [Google Scholar]
- Renschler, M.F. The emerging role of reactive oxygen species in cancer therapy. European Journal of Cancer. 2004, 40, 1934–1940. [Google Scholar] [CrossRef] [PubMed]
- Top, S.; Vessières, A.; Leclercq, G.; Quivy, J.; Tang, J.; Vaissermann, J.; Huché, M.; Jaouen, G. Synthesis, biochemical properties and molecular modelling studies of organometallic specific estrogen receptor modulators (SERMs), the ferrocifens and hydroxyferrocifens: evidence for an antiproliferative effect of hydroxyferrocifens on both hormone-dependent and hormone-independent breast cancer cell lines. Chemistry 2003, 9, 5223–5236. [Google Scholar] [CrossRef] [PubMed]
- Csókás, D.; Károlyi, B.I.; Bősze, S.; Szabó, I.; Báti, G.; Drahos, L.; Csámpai, A. 2,3-Dihydroimidazo [1,2-b]ferroceno[d]pyridazines and a 3,4-dihydro-2H-pyrimido [1,2-b]ferroceno[d]pyridazine: Synthesis, structure and in vitro antiproliferation activity on selected human cancer cell lines. J. Organomet. Chem. 2013, 750, 41–48. [Google Scholar] [CrossRef]
- Jernei, T.; Bősze, S.; Szabó, R.; Hudecz, F.; Majrik, K.; Csámpai, A. N-ferrocenylpyridazinones and new organic analogues: Synthesis, cyclic voltammetry, DFT analysis and in vitro antiproliferative activity associated with ROS-generation. Tetrahedron 2017, 73, 6181–6192. [Google Scholar] [CrossRef]
- Alaoui, N.-E.E.; Boulhaoua, M.; Hutai, D.; Oláh-Szabó, R.; Bősze, S.; Hudecz, F.; Csámpai, A. Synthetic and DFT Modeling Studies on Suzuki–Miyaura Reactions of 4,5-Dibromo-2-methylpyridazin-3(2H)-one with Ferrocene Boronates, Accompanied by Hydrodebromination and a Novel Bridge-Forming Annulation In Vitro Cytotoxic Activity of the Ferrocenyl–Pyridazinone Products. Catalysts 2022, 12, 578. [Google Scholar] [CrossRef]
- Sharma, V.; Kumar, V.; Kumar, P. Heterocyclic chalcone analogues as potential anticancer agents. Anticancer Agents Med. Chem. 2013, 13, 422–432. [Google Scholar]
- Karthikeyan, C.; Moorthy, N.S.; Ramasamy, S.; Vanam, U.; Manivannan, E.; Karunagaran, D.; Trivedi, P. Advances in chalcones with anticancer activities. Recent Pat. Anticancer Drug Discov 2015, 10, 97–115. [Google Scholar] [CrossRef]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Anti-cancer chalcones: Structural and molecular target perspectives. Eur J Med Chem 2015, 98, 69–114. [Google Scholar] [CrossRef]
- Gao, F.; Huang, G.; Xiao, J. Chalcone hybrids as potential anticancer agents: Current development, mechanism of action, and structure-activity relationship. Med Res Rev 2020, 40, 2049–2084. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Sood, A.K.; Goyal, K.; Singh, A.; Sharma, V.; Guliya, N.; Gulati, S.; Kumar, S. Chalcone Scaffolds as Anticancer Drugs: A Review on Molecular Insight in Action of Mechanisms and Anticancer Properties. Anticancer Agents Med. Chem. 2021, 21, 1650–1670. [Google Scholar] [CrossRef] [PubMed]
- Jernei, T.; Duró, C.; Dembo, A.; Lajkó, E.; Takács, A.; Kőhidai, L.; Schlosser, G.; Csámpai, A. Synthesis, Structure and In Vitro Cytotoxic Activity of Novel Cinchona-Chalcone Hybrids with 1,4-Disubstituted- and 1,5-Disubstituted 1,2,3-Triazole Linkers. Molecules 2019, 24. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B.; Johnson, T.E.; Lad, R.; Xing, C. Structure-activity relationship studies of chalcone leading to 3-hydroxy-4,3’,4’,5’-tetramethoxychalcone and its analogues as potent nuclear factor kappaB inhibitors and their anticancer activities. J. Med. Chem. 2009, 52, 7228–7235. [Google Scholar] [CrossRef]
- Riaz, S.; Iqbal, M.; Ullah, R.; Zahra, R.; Chotana, G.A.; Faisal, A.; Saleem, R.S.Z. Synthesis and evaluation of novel α-substituted chalcones with potent anticancer activities and ability to overcome multidrug resistance. Bioorg. Chem. 2019, 87, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Gao, M.; Diao, Q.; Gao, F. Chalcone Derivatives and their Activities against Drug-resistant Cancers: An Overview. Curr. Top. Med. Chem. 2021, 21, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, C.; He, L.; Lei, K.; Wang, F.; Pu, Y.; Yang, Z.; Cao, D.; Ma, L.; Chen, J.; et al. Design, synthesis and biological evaluation of a series of pyrano chalcone derivatives containing indole moiety as novel anti-tubulin agents. Bioorg. Med. Chem. 2014, 22, 2060–2079. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Chen, J.; Zhang, S.; Hu, J.; Huang, L.; Li, X. Synthesis, Evaluation, and Mechanism Study of Novel Indole-Chalcone Derivatives Exerting Effective Antitumor Activity Through Microtubule Destabilization in Vitro and in Vivo. J. Med. Chem 2016, 59, 5264–5283. [Google Scholar] [CrossRef]
- Johnson, P.D.; Sohn, J.-H.; Rawal, V.H. Synthesis of C-15 Vindoline Analogues by Palladium-Catalyzed Cross-Coupling Reactions. J. Org. Chem. 2006, 71, 7899–7902. [Google Scholar] [CrossRef]
- Siddiq, A.; Dembitsky, V. Acetylenic anticancer agents. Anticancer Agents Med. Chem. 2008, 8, 132–170. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, E.J. Cytotoxic anticancer candidates from natural resources. Curr. Med. Chem. Anticancer Agents 2002, 2, 485–537. [Google Scholar] [CrossRef]
- Christensen, L.P. Bioactive C(17) and C(18) Acetylenic Oxylipins from Terrestrial Plants as Potential Lead Compounds for Anticancer Drug Development. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Wang, S.; Liu, L.; Guo, X.; Li, G.; Wang, X.; Dong, H.; Li, Y.; Zhao, W. Synthesis of novel natural product-like diaryl acetylenes as hypoxia inducible factor-1 inhibitors and antiproliferative agents. RSC Adv. 2019, 9, 13878–13886. [Google Scholar] [CrossRef]
- Yang, C.; Shao, Y.; Li, K.; Xia, W. Bioactive selaginellins from Selaginella tamariscina (Beauv.) Spring. Beilstein J. Org. Chem. 2012, 8, 1884–1889. [Google Scholar] [CrossRef]
- Zhang, G.G.; Jing, Y.; Zhang, H.M.; Ma, E.L.; Guan, J.; Xue, F.N.; Liu, H.X.; Sun, X.Y. Isolation and cytotoxic activity of selaginellin derivatives and biflavonoids from Selaginella tamariscina. Planta Med. 2012, 78, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Carter-Cooper, B.; Du, Y.; Zhou, J.; Saeed, M.A.; Liu, J.; Guo, M.; Roembke, B.; Mikek, C.; Lewis, E.A.; et al. Alkyne-substituted diminazene as G-quadruplex binders with anticancer activities. Eur. J. Med. Chem. 2016, 118, 266–275. [Google Scholar] [CrossRef]
- Hong, D.; Said, R.; Falchook, G.; Naing, A.; Moulder, S.; Tsimberidou, A.-M.; Galluppi, G.; Dakappagari, N.; Storgard, C.; Kurzrock, R.; et al. Phase I Study of BIIB028, a Selective Heat Shock Protein 90 Inhibitor, in Patients with Refractory Metastatic or Locally Advanced Solid Tumors. Clin. Cancer Res. 2013, 19, 4824–4831. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, C.; Song, C. Pan- and isoform-specific inhibition of Hsp90: Design strategy and recent advances. Eur. J. Med. Chem. 2022, 238, 114516. [Google Scholar] [CrossRef]
- Liu, Y.-F.; Wang, C.-L.; Bai, Y.-J.; Han, N.; Jiao, J.-P.; Qi, X.-L. A Facile Total Synthesis of Imatinib Base and Its Analogues. Organic Process Research & Development. 2008, 12, 490–495. [Google Scholar]
- Imrie, C.; Cook, L.; Levendis, D.C. An investigation of the chemistry of ferrocenoyl derivatives. The synthesis and reactions of ferrocenoyl imidazolide and its derivatives. J. Organomet. Chem. 2001, 637–639, 266–275. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structure of vinblastine (1) as composed of the coupled units catharanthine (1a) and vindoline (1b).
Figure 1.
Structure of vinblastine (1) as composed of the coupled units catharanthine (1a) and vindoline (1b).
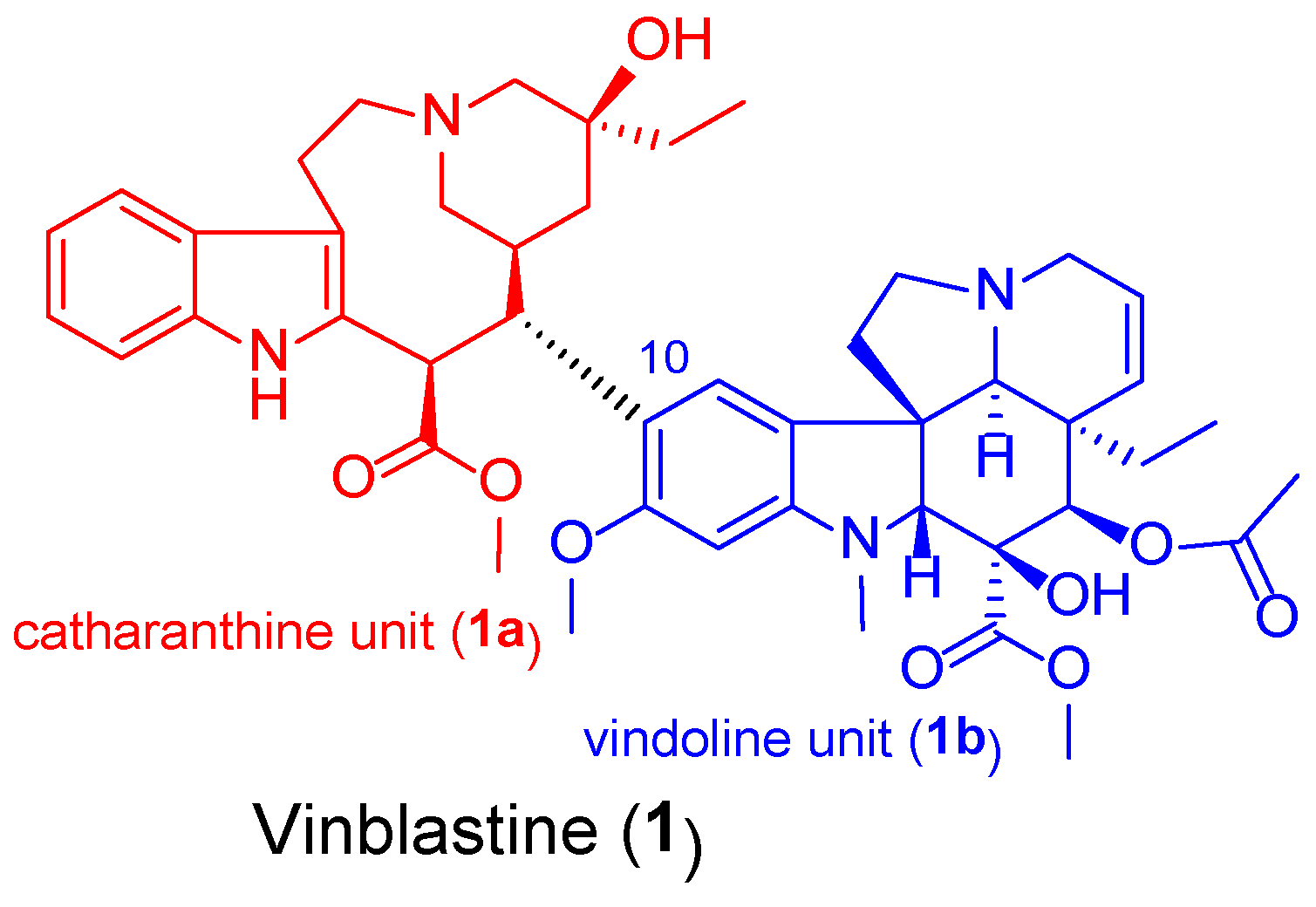
Figure 2.
Representative pharmacophores which, or their fragments were selected for introduction in the targeted vindoline hybrids: FDA-approved kinase inhibitors (imatinib 2 and erlotinib 3), ferrocene (4) and chalcone (5).
Figure 2.
Representative pharmacophores which, or their fragments were selected for introduction in the targeted vindoline hybrids: FDA-approved kinase inhibitors (imatinib 2 and erlotinib 3), ferrocene (4) and chalcone (5).
Scheme 1.
Synthesis of propargylated imatinib fragments suitable for Sonogashira reactions with 10-iodovindoline.
Scheme 1.
Synthesis of propargylated imatinib fragments suitable for Sonogashira reactions with 10-iodovindoline.
Scheme 2.
Synthetic pathways to the targeted hybrids starting from 10-iodovindoline (20) as common precursor.
Scheme 2.
Synthetic pathways to the targeted hybrids starting from 10-iodovindoline (20) as common precursor.
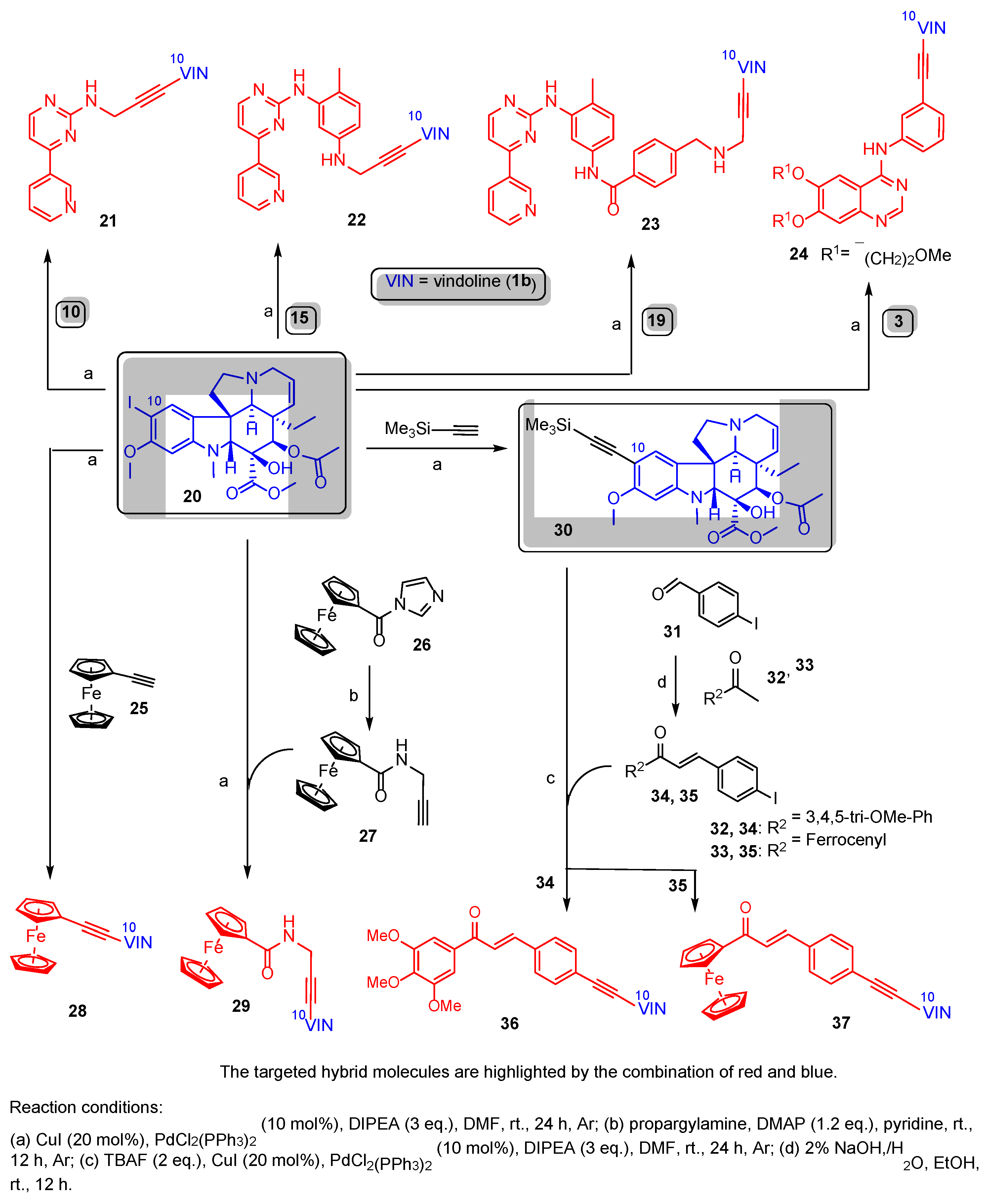
Scheme 3.
Synthesis of reference chalcones containing phenyl group in place of vindoline moiety.

Table 1.
Initial screening of the antiproliferative effect of the alkyne-tethered vindoline hybrids and reference compounds, including vindoline 1b, measured on the investigated cells.
Table 1.
Initial screening of the antiproliferative effect of the alkyne-tethered vindoline hybrids and reference compounds, including vindoline 1b, measured on the investigated cells.
| Compound | Conc. (µM) | Mean Growth Inhibition (%) ± SEM | ||||
|---|---|---|---|---|---|---|
| MRC-5 | MDA-MB-231 | HeLa | A2780 | SH-SY5Y | ||
| 21 | 10 | Not tested | < 20 | < 20 | < 20 | < 20 < 20 |
| 30 | < 20 | 21.41 ± 1.22 | 29.46 ± 1.03 | |||
| 22 | 10 | Not tested | < 20 | < 20 | 24.69 ± 2.03 | < 20 94.91 ± 1.85 |
| 30 | 89.92 ± 0.65 | 43.95 ± 1.74 | 97.13 ± 0.40 | |||
| 23 | 10 | Not tested | < 20 | < 20 | < 20 | < 20 < 20 |
| 30 | < 20 | < 20 | < 20 | |||
| 24 | 10 | < 20 | 47.84 ± 2.98 | < 20 | 75.74 ± 2.55 | 53.41 ± 0.89 |
| 30 | < 20 | 80.37 ± 1.83 | < 20 | 88.05 ± 1.08 | 82.74 ± 2.85 | |
| 28 | 10 | Not tested | < 20 | < 20 | < 20 | < 20 |
| 30 | < 20 | < 20 | 49.19 ± 1.98 | < 20 | ||
| 29 | 10 | Not tested | < 20 | < 20 | < 20 | < 20 91.64 ± 1.10 |
| 30 | 23.99 ± 0.81 | < 20 | 68.02 ± 1.37 | |||
| 36 | 10 | 87.09 ± 2.73 | 86.22 ± 0.79 | 89.62 ± 0.75 | 95.09 ± 0.50 | 89.00 ± 2.61 |
| 30 | 88.90 ± 2.80 | 85.01 ± 0.87 | 89.98 ± 0.46 | 94.50 ± 0.35 | 89.10 ± 2.74 | |
| 36a | 10 | 46.14 ± 2.17 | 86.37 ± 0.71 | 90.55 ± 0.72 | 95.53 ± 0.34 | 90.01 ± 2.84 |
| 30 | 89.88 ± 3.24 | 86.28 ± 0.47 | 90.29 ± 0.28 | 95.07 ± 0.27 | 91.67 ± 3.36 | |
| 37 | 10 | Not tested | < 20 | < 20 | < 20 | < 20 < 20 |
| 30 | < 20 | < 20 | 31.96 ± 2.40 | |||
| 37a | 10 | 27.48 ± 2.20 | 70.70 ±1.61 | 51.26 ± 0.79 | 57.17 ± 1.48 | < 20 |
| 30 | 52.25 ± 2.01 | 80.99 ± 1.49 | 71.19 ± 2.86 | 81.63 ± 2.56 | 51.77 ± 3.06 | |
| 1b | 10 | < 20 | < 20 | < 20 | < 20 | < 20 |
| 30 | < 20 | < 20 | < 20 | < 20 | < 20 | |
Table 2.
IC50 values obtained for the most active compounds identified in the initial viability tests.
Table 2.
IC50 values obtained for the most active compounds identified in the initial viability tests.
| Compound | IC50 (µM) | ||||
|---|---|---|---|---|---|
| MDA-MB-231 | HeLa | A2780 | SH-SY5Y | MRC-5 | |
| 29 | 11.78 | n.d. | 5.62 | 10.33 | n.d. |
| 36 | 1.15 (2.22a) | 1.57 (1.62a) | 0.60 (4.25a) | 1.26 (2.02a) | 2.55 |
| 36a | 4.22 (2.59a) | 4.54 (2.40a) | 3.26 (3.35a) | 5.60 (1.95a) | 10.92 |
| 37a | 7.79 (3.52a) | 11.47 (2.39a) | 8.84 (3.10a) | 29.16 (0.94a) | 27.42 |
a Selectivity indice (IC50 on MRC-5/IC50 on the cancer cell).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



