Submitted:
01 May 2025
Posted:
02 May 2025
You are already at the latest version
Abstract
Cancer has a threatening impact on human health, and it is one of the primary causes of fatalities worldwide. Different conventional treatments have been employed to treat cancer, but their non-specific nature reduces their therapeutic efficacy. This study employs a C5N2-based targeted drug carrier to study the delivery mechanism of anticancer drugs, particularly cisplatin, carmustine, and mechlorethamine, using density functional theory (DFT). The geometries of the drugs, the C5N2 substrate, and the drug@C5N2 complexes were optimized at the PBE0-D3BJ/def2SVP level of theory. Interaction energy has been computed for the complexes which follow the trend, i.e., cisplatin@C5N2 > carmustine@C5N2 > mechlorethamine@C5N2. The NCI and QTAIM analyses confirmed the presence of van der Waals forces between the carmustine@C5N2 and mechlorethamine@C5N2 complexes, while weak hydrogen bonding has also been observed between the cisplatin@C5N2 complex. ELF analysis has been performed to analyze the degree of delocalization of electrons within the complexes, which manifests consistency with the findings of NCI and QTAIM analyses. The electronic properties of the analytes and the C5N2 substrate have been examined through FMO, CRDs, DOS, NBO, and EDD analyses. FMO, CRDs, and DOS analysis confirmed the enhanced reactivity of the complexes. NBO illustrated an electron density shift between the drugs and the C5N2 sheet, while EDD exhibited a substantial correlation with the NBO findings. Recovery time has been determined to assess the biocompatibility and the desorption behavior of the drugs. Moreover, negative solvation energies and increased dipole moments in a solvent phase manifested enhanced solubility and easy circulation of the drugs in biological media. Subsequently, this study illustrates that cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes can be utilized as efficient drug delivery systems.
Keywords:
density functional theory
; anticancer
; drug delivery
; interaction energy
; electronic properties
; therapeutic
; substrate
1. Introduction
Over the past few years, cancer has adversely affected human health and caused millions of deaths worldwide [1,2]. Various conventional approaches, including surgical treatment [3], radiation therapy [4], and chemotherapy [5] have been considered definitive solutions for cancer treatment. However, their non-specific nature and cytotoxicity further minimize their pharmacological effects [6,7,8]. The therapeutic efficiency of different anticancer drugs, such as doxorubicin (DOX) [9], anthracycline [10], and hydroxycamptothecin (HCPT)[11], has been evaluated for targeting cancer cells. Cisplatin (cis-diamminedichloroplatinum (II)) [12] is the most widely used anticancer drug to treat various types of malignancies related to the head, neck [13], ovaries [14], and bladder [15]. It forms an adduct with DNA, inhibiting its replication further and causing cell death [16]. Moreover, its therapeutic efficiency has been validated with various drug delivery systems, including selenium nanoparticles, to treat lung carcinoma [17]. It has also been delivered using polypeptide vesicles [18], magnetic iron oxide nanoparticles [19], and graphitic carbon nitride-based drug delivery systems [20]. Carmustine (BCNU), an alkylating agent commonly known as 1,3-bis(2-chloroethyl)-1-nitrosourea [21], has been popular in cancer treatment [22]. Its efficiency has been investigated for curing and suppressing brain tumors such as glioblastoma multiforme [23] using substrates such as polymeric nanoparticles [24] and polyamidoamine (PAMAM) dendrimers [25]. Also, its interaction with chitosan-coated nanoparticles was evaluated and approved to cure brain-associated tumors [26]. Recently, carmustine has been used for the treatment of ovarian cancer, employing a liposomal substrate [27]. Its therapeutic efficacy has also been reported with carbon [26] and boron nitride nanotubes [28] to treat malignant cancers. Consequently, mechlorethamine (mustine), an anticancer medication, has been employed to cure Hodgkin’s lymphoma [29] and cutaneous T-cell lymphomas [30,31]. Its anticancer efficacy has been investigated with nanocages, including silicon, carbon, and aluminium nitride [32]. However, severe health problems have been associated with the delivery of these anticancer drugs as they lack precision in differentiation between cancerous and healthy cells [33]. Moreover, anticancer drugs can cause serious health problems, mainly cell toxicity, due to their non-specific interaction, further minimizing their remedial efficacy [34,35]. Therefore, support systems have been evaluated for their controlled and direct release at the cancerous site to overcome associated health complications [36,37]. The drug carriers facilitate the medications to cross complex biological defense systems without causing widespread malignancies [38]. Theoretical [39] and experimental [40] evaluations of the drugs with different carriers have also been performed to enhance the cell targeting and bioimaging properties of anticancer drugs. Various two-dimensional (2D) [41,42], and three-dimensional (3D) [43,44] substrates have been examined including, hexagonal boron nitride (h-BN) [45], manganese dioxide [46], transition-metal di-chalcogenides (TMDs) [47], metallic nanoparticles [48], MXenes [49], and phosphorene [50] for the drug delivery. Additionally, nanocarriers comprising carbon have also been reported as potential drug carriers with low cytotoxicity, and good chemical stability i.e., chitosan [51], fullerene [52], and fluorinated graphene nanocarbon [53]. The reactivity of carbon nanotubes has been validated after encapsulation of an anticancer drug i.e., cisplatin [54]. In some studies, preparation, and cytotoxicity of dendrimers [55] and liposomal complexes [56] conjugated with doxorubicin and cisplatin have also been examined to improve the cellular uptake of the drugs. Comparatively, carbon nitride offers more benefits in the targeted delivery of anticancer drugs because of its distinctive physicochemical characteristics [57,58].
Different experimental and theoretical studies have been performed on the drug delivery efficiency of carbon nitrides comprising different ratios of carbon and nitrogen such as CN [59], C2N [60], and C3N4 [61,62] nanosheets. Recent studies on carbon nitrides manifest their suitable band gaps, which make them good photocatalysts for water splitting [63], CO oxidation [64], CO2 reduction [65], H2O2 production [66], and nitrogen reduction reaction [67,68]. Furthermore, graphitic carbon nitride g-C3N4 has been reported as an efficient substrate for delivering carboplatin [69], and melphalan [70] to treat malignant tumors. Significant stability [71], pH sensitivity [72], and photoluminescence [73] of carbon nitrides have been revealed which make them capable of delivering anticancer drugs such as lonidamine [74], flutamide [75], and cisplatin [20] at the cancerous site. Among carbon nitrides, C5N2 has emerged as a novel biocompatible substrate with a narrow band gap of 1.10 eV [76]. It possesses excellent carrier mobility and electrical conductivity further manifesting its considerable potential in biomedicine. The synthesis of C5N2 material is carried out through a condensation reaction between 1,2,4,5-benzene tetramine and hexaketocyclohexane [76]. Because of its stability and activity, it has been employed in photocatalysis to synthesize H2O2 [66]. In contrast to other substrates, it has a substantial drug-loading rate because of micropore abundance and large surface area [77].
In particular, no findings were reported in the literature about the drug-delivering capability of the C5N2 substrate. Hence, these inspiring properties and results encouraged us to explore and evaluate the theoretical efficiency of the C5N2 carrier to deliver anticancer drugs, mainly cisplatin, carmustine, and mechlorethamine, at the tumor site. In this work, DFT calculations have been performed to assess the interaction between the drugs and the substrate in both the gas and the solvent media. The intermolecular interactions between cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 have been evaluated using NCI, QTAIM, and ELF analyses. To ensure efficient and controlled drug release at the targeted site, evaluation of the interaction between the drugs and the C5N2 substrate has been carried out through FMO, DOS, NBO, and EDD analyses. Moreover, the reactivity, stability, and binding affinity of the complexes were analyzed through chemical reactivity descriptors.
2. Computational Methodology
Structures of the cisplatin, carmustine, mechlorethamine, C5N2 substrate, and their complexes were modelled and visualized utilizing the Gauss View program [78,79]. Computational calculations of different configurations of the drugs and the C5N2 carrier were performed using DFT (density functional theory) employing Gaussian 16 rev.C.01 software [80] at the PBE0-D3BJ/def2-SVP level of theory. PBE0 is a hybrid density functional preferred for the optimization of various structures such as analytes anchored two-dimensional supports [81]. Also, it provides a more accurate illustration of the molecular characteristics and electronic structures of various materials, including solids, organic molecules, and complexes of metals and non-metals [82]. It addresses the restrictions of pure density functional theory approximations and accurately predicts the structural and electronic properties of the molecules across the periodic table [83]. It provides significant estimations for the systems exhibiting non-covalent interactions, electronic excitations, and electron density shifts. Moreover, it is commonly employed in various fields, including drug delivery, to simulate optimizations and binding energy calculations [84,85]. Also, structural, optical, and vibrational properties of graphitic carbon nitrides [86] along with efficient hydrogen storage [87] were studied using PBE0 functional.
Different studies have highlighted its efficiency in predicting molecular phenomena, such as the photoisomerization of unsubstituted spirooxazine [88]. It has also been employed to deliver anticancer drugs, mainly hydroxyurea [89,90] and cisplatin [91,92], through borospherenes and metallacage drug delivery systems, respectively. It is scrutinized to study the charge transfer mechanism of phosphorus, oxygen, and sulfur-doped graphitic carbon nitride [93]. Interpretation of adsorption and separation phenomena of NO and CO2 over the surface of the C5N2 substrate has been computed at this hybrid functional. Adsorption of metformin [94], CO2, and CH4 [95] on functionalized C3N4 have also been validated using PBE0. It has been used to investigate the adsorption mechanism of H2, N2, CO, NH3, H2S, and SO2 [96], and modifications of electronic properties in platinum and silicon-doped C3N4 have also been investigated [97]. Evaluation of CO2 reduction on copper [98] and analysis of electronic properties on Zn/Ga doped C3N4 [99] have also been computed. PBE0-D3BJ/def2-SVP level of theory has been employed to facilitate the reaction kinetics of lithium-sulfur batteries [100], on carbon nitride substrate. Stefan Grimme’s empirical dispersion correction (D3 correction) with Becke Johnson damping (D3-BJ) [101] has been utilized to correct the energies for nonbonding interactions within the molecular system. The accuracy of this method in describing the structural and molecular variations of non-covalent systems has also been reported in the literature [102]. For solvent based calculations, SMD solvation model has been employed at the same level of theory using water as a solvent. CYLview software has been used to obtain and visualize detailed and precise portraits of the drug@C5N2 complexes [103]. The geometry optimization has been carried out using the simulated structure of the C5N2 sheet. A variety of possible orientations were considered to determine the stable arrangement of the drug on the C5N2 surface. Following optimization, vibrational frequency analysis was performed on the structures to confirm their minimal energy configurations on the potential energy surface. The significant parameter, interaction energy ( has been obtained to evaluate the interaction between the drug and the C5N2 surface within the complex using the following expression,
The interaction energy of the drug@C5N2 system is represented by , where , and represents the interaction energies of the complex, the drug, and the C5N2 sheet, respectively [104]. Non-covalent interactions (NCI) analysis also known as the reduced density gradient (RDG) method, has been performed to analyze the weak interactions between the drug and the C5N2 substrate using Multiwfn [105] and visual molecular dynamics (VMD) [106] programs. Electron density and reduced density gradient (s) are the two key derivatives used to analyze the type of interactions, such as stearic repulsions, van der Waals interactions, and hydrogen bonding between the complexes. The RDG is a dimensionless gradient norm function, and it is inversely related to the electron density Moreover, it identifies the regions of non-covalent interactions with relatively low electron densities. Mathematically, RDG (s) is expressed as [107],
The magnitude of electronic density plays a significant role in determining the strength of non-covalent interactions. According to the equation, the greater value of RDG further decreases the electronic density suggesting a minimal possibility of non-covalent interaction. Comparatively, the lower value of RDG enhances the potential of non-covalent interactions by increasing the electronic density at a particular site of interaction. The attractive and repulsive nature of interaction and spatial distribution of electron density is determined by the Laplacian of electronic density . Also, it provides necessary information about the electron-rich and electron-depleted regions. A negative value illustrates the region of concentrated electronic density confirming the formation of covalent bonds. Moreover, the positive value corresponds to the depletion of electronic density as found in van der Waals interactions. Mathematically, is expressed as a trace of the Hessian matrix of electronic density as shown below [108],
Here represents the eigenvalues of the Hessian matrix which assist in describing the behavior of interactions. Among these eigenvalues, plays a crucial role in identifying the nature of interactions i.e., ( represents strong and attractive interaction, ( corresponds to strain and stearic repulsion, and predicts the presence of weak interactions such as van der Waals forces. To visualize and evaluate the type, strength, and spatial pattern of non-covalent interactions, we generated a 2D RDG scatter plot and a 3D isosurface representing interactions using Multiwfn and VMD software. The 2D graph combines the sign with the electron density to explain the type of interaction ranging from 0.05 to 0.05). To augment the 2D scatter plot, the 3D isosurface is represented by the colors of the sign of such as blue color indicates the presence of strong and attractive interactions illustrated by the region where , red color represents repulsive stearic interactions indicated by and green corresponds to the weak van der Waals interactions as illustrated by on the x- axis of the 2D plot. The color representation facilitates a direct visualization of the interaction domains within the complex systems [109]. The nature of the interaction that exists during the complexation of the drug@C5N2 complex has also been visualized through the quantum theory of atoms in molecules analysis (QTAIM). It locates all the interactions among atoms by pairing up the densities of electrons spinning oppositely which further illustrate the non-covalent interactions within the complexes [110]. Topological analysis of the electronic density is the foundation of atoms in molecules (AIM) analysis [111]. Further, it interprets the interactions between the drug@substrate complexes i.e., cisplatin@C5N2, carmustine@C5N2, mechlorethamine@C5N2 through certain parameters including electronic density , the Laplacian of electronic density the total energy density (H), the potential energy density (V), and the kinetic energy density (G) computed at the bond critical points (BCPs) and the curvature of the bond. The strength of the non-covalent interaction has been specified by the values of electronic density . If ( 0.1), it indicates the presence of intermolecular attractive forces, while ( 0.1) suggests the presence of van der Waals interactions between the drug@C5N2 complex [112]. Overall energy distribution of the complex systems has been evaluated through the total energy density (H), which is the sum of the potential (V) and kinetic (G) energy densities, and it is expressed as [113],
The signs of the potential energy density (V) and the kinetic energy density (G) manifest coherency, with (V) being negative and (G) being positive. Moreover, the nature of the interactions between the drugs and the C5N2 sheet whether covalent or non-covalent, have also been validated by analyzing the values of the key indicators such as Laplacian of electron density , and energy density (H). Additionally, the greater value of and (H) manifests the presence of non-covalent interactions between the drug and the C5N2 substrate, i.e., (). In contrast, drug@C5N2 complexes exhibit strong attraction when these parameters manifest a relation, i.e., , (H 0) [113]. Electronic properties of the drug@C5N2 complexes have been computed through different analyses. Frontier molecular orbital (FMO) analysis was employed to analyze the electronic structure and charge transfer characteristics of the complexes mainly concentrating on the HOMO and LUMO molecular orbitals. For the C5N2 substrate, the drugs, and their complexes, calculations were performed to determine the energies of the HOMO and the LUMO orbitals. The energy of the highest occupied molecular orbital (HOMO) illustrates the electron donating capability (high value of ) while the energy of the lowest unoccupied molecular orbital (LUMO) predicts the electron-accepting ability (high value of ) of a molecule after the complex formation. represents the ionization potential (I) of the molecule and corresponds to the electron affinity (A) of the system. According to Koopman’s theorem, FMOs are correlated with the (I) and (A) values by a relation, [114].
Moreover, another parameter is the energy gap gap between the HOMO and the LUMO orbitals of the complex system which provides significant insights into the adsorption behavior, reactivity, and stability of the drug at the C5N2 sheet. It has been calculated using the expression, where is the lowest energy level of the unoccupied molecular orbital and represents the highest energy level of the occupied molecular orbital. The global reactivity descriptors including chemical potential (μ), electrophilicity index (ω), hardness (η), and softness (s), were computed from the energies of the HOMO and the LUMO at the same level of theory. The reactivity, stability, and the interaction behavior of the drugs at the C5N2 surface have been analyzed by employing expressions [115],
(μ) is related to the electronegativity of the molecule and its more negative value depicts the difficulty in the removal of an electron showing less reactivity with higher stability. (ω) provides insight into the stabilization energy acquired after fulfilling the electrons (gained from the surrounding environment) [116]. A lower value of (ω) corresponds to the nucleophilicity of a molecule (acts as a nucleophile) while its higher value depicts electrophilicity of a molecule (acts as an electrophile). The values of (η) and (S) significantly play their role in the evaluation of the polarizability of the chemical system such as a small energy gap corresponds to a soft molecule (high polarizability) while a large energy gap is related to a hard molecule (less polarizability) [117]. The density of states (DOS) justifies the FMO analysis by providing a comprehensive view of the energy gap, and the electronic properties of the complexes. Evaluation of the orbitals has been performed by the FMO analysis while the states of electron density near the HOMOLUMO gap (gap) were analyzed through DOS using the GaussSum program [118]. Moreover, electron localization function (ELF) analysis has also been carried out to understand the weak interactions including van der Waals forces by visualizing electron density changes after the formation of a complex [119]. ELF measures the extent of electron density localization i.e., (ELF ≈ 1) refers to high electron localized regions which indicates the presence of covalent bond, (ELF ≈ 0.5) refers to electron delocalized regions indicating the presence of non-covalent interactions, and (ELF ≈ 0) highlights the non-interacting regions with minimum electron density [120]. We generated color-coded ELF maps to analyze and distinguish between the localized (covalent) and delocalized (non-covalent) interactions between the drug@C5N2 complexes through Multiwfn v. 3.0.8 program. Natural bond orbital (NBO) analysis is used to analyze the electronic density shifts between the complexes [121]. Also, it helps in identifying the charge transfer mechanism between the donor and the acceptor sites within the drug@C5N2 complexes. Electron Density Distribution (EDD) analysis provides insights into the charge distribution between atoms within the molecular system [122]. We have visualized the flow of electrons by analyzing the electron density changes after the association of the drug@C5N2 system.
3. Results and Discussion
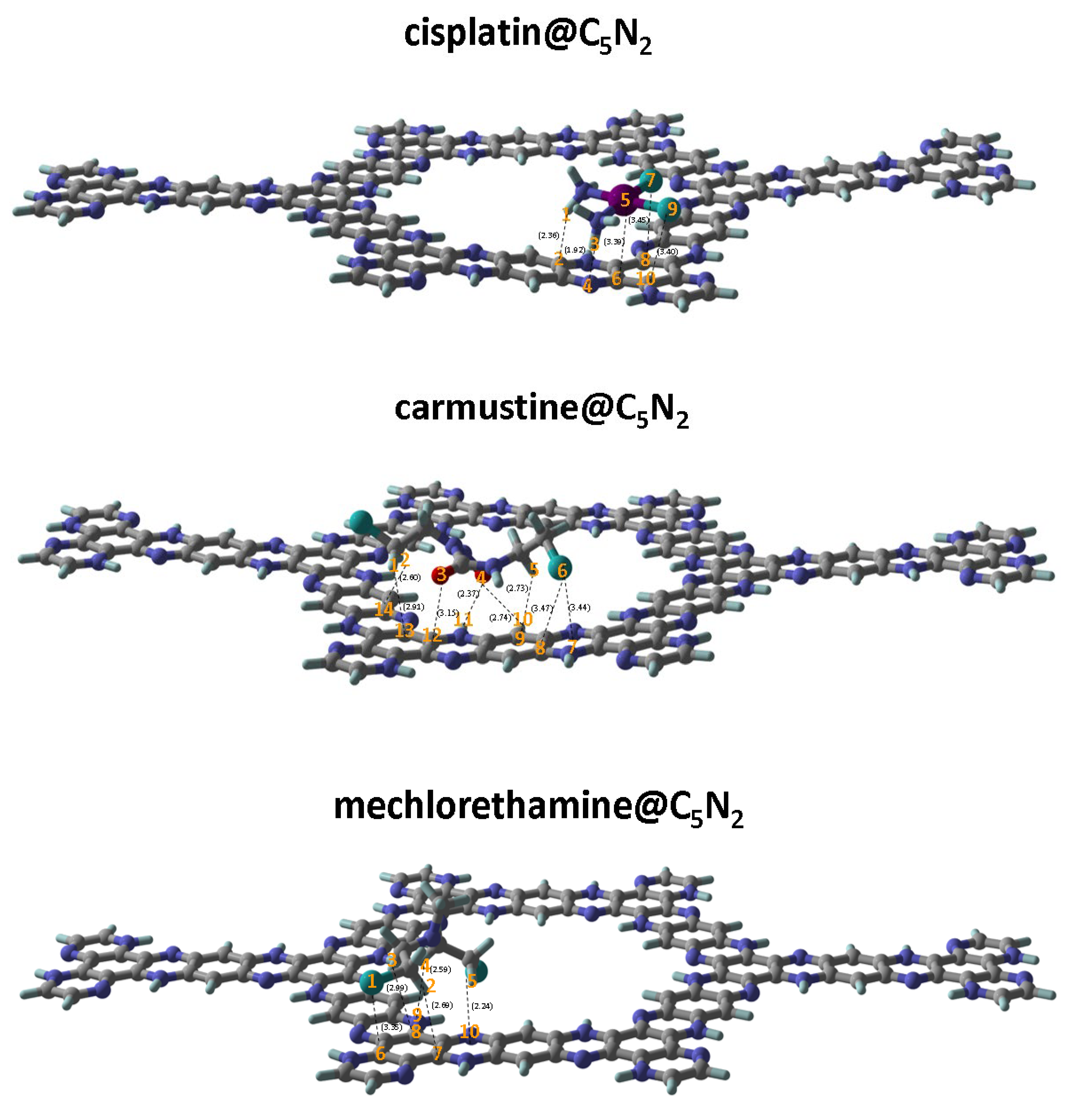
C5N2 is a porous crystalline structure with five fused benzene rings comprising two types of CC bonds having bond lengths i.e., 1.42 Å and 1.37 Å. Each benzene ring is bridged by a pyrazine ring which constitutes CN, and CC bond with bond lengths 1.36 Å, and 1.39 Å, respectively. Different orientations of the drugs (cisplatin, carmustine, and mechlorethamine) were considered at different locations on the C5N2 sheet to determine the most stable configuration of the complex. The spatial distinction of these binding sites can easily be evaluated through their designations such as (a) located in the central cavity of the sheet, (b) positioned directly above the benzene ring, (c) located above the pyrazine ring, (d) aligned just above the hydrogenated benzene ring, and (e) forming a pseudo-cycle with the nitrogen and hydrogen atoms present at the inside edges of the sheet. The optimized sheet of C5N2 representing possible active sites is shown in Figure 1.
After examining different orientations and positions (see Figure S1), the most stable geometry of the cisplatin is observed over the benzene (b) and pyrazine ring (c) of the C5N2 sheet with an interaction energy of 27.60 kcal . The two hydrogen atoms (H1 and H3) of the cisplatin drug interacted with the C2 and N4 of the C5N2 sheet while keeping a distance of 2.36 Å and 1.92 Å, respectively. The interaction between H1C2 suggests the presence of van der Waals interaction while H3N4 interacted with a relatively shorter distance which manifests the formation of a hydrogen bond (NHN) because it ranges between (1.5 Å2.0 Å) [123]. The platinum Pt5 is linked with the C6 of the C5N2 sheet with a bond distance of 3.39 Å and the two Cl atoms of the cisplatin (Cl7 and Cl9) show interaction with the C8 and C10 of the benzene ring of the C5N2 sheet with a highest bond length of 3.45 Å and 3.40 Å illustrating van der Waals interaction between the cisplatin@C5N2 complex as shown in Figure 2 and Table 1.
For the carmustine drug, among different orientations (see Figure S2) the most suitable position is oriented parallelly above the sites (b), (c), and (d) of the C5N2 sheet with an interaction energy of 19.69 kcal . The negative values of the interaction energy ensured the stability of the drug carriers. In addition, the hydrogen atoms i.e., H10 and H11 of the C5N2 sheet interacted with O4 of the carmustine drug while maintaining a bond distance of 2.74 Å, and 2.37 Å, respectively, which depicts the formation of a weak hydrogen bond between O4H11 of the complex. The bonding interactions with a distance of 2.73 Å, 2.91 Å, and 2.60 Å between H5C9, H1C13, and H2C14 atoms, sequentially, indicate non-covalent interactions between the carmustine and the C5N2 sheet as they appear outside the normal range of a covalent bond (1.09 Å) [124]. The oxygen O3 interacted with C12 of C5N2 with a bond distance of 3.15 Å. Moreover, the chlorine (Cl6) atom of the carmustine exhibit interaction with N7 and C8 of the C5N2 sheet with a bond length of 3.44 Å, and 3.47 Å, respectively, further suggesting van der Waals interaction as shown in Figure 2. For the mechlorethamine drug, among different feasible orientations (see Figure S 3), the most stable configuration is attained at the top of the sites (b), (c), and (e). However, a chlorine atom (Cl1) of the drug is moved slightly and oriented above the pyrazine ring of the C5N2 sheet.

Table 1.
Interaction energies (kcal) with their corresponding bond lengths (Å) between the associated atoms of the drugs and the C5N2 carrier after complex formation.
Table 1.
Interaction energies (kcal) with their corresponding bond lengths (Å) between the associated atoms of the drugs and the C5N2 carrier after complex formation.
| Drug@C5N2 complex | Drug | Bond Lengths (Å) | |
|---|---|---|---|
| cisplatin@C5N2 | C2H1 N4H3 C6Pt5 C8Cl7 C10Cl9 |
2.36 1.92 3.39 3.45 3.40 |
27.60 |
| carmustine@C5N2 | C13H1 C14H2 C12O3 H11O4 H10O4 C9H5 C8Cl6 N7Cl6 |
2.91 2.60 3.15 2.37 2.74 2.73 3.47 3.44 |
19.69 |
| mechlorethamine@C5N2 | C6Cl1 C7H2 C8H3 N9H4 N10H5 |
3.35 2.69 2.99 2.59 2.24 |
17.73 |
The suitable interaction energy of mechlorethamine complexed with the C5N2 is 17.73 kcal . Hydrogen atoms (H2, and H3) of the drug exhibit non-covalent interaction with the C7, and C8 of the C5N2 sheet by keeping a distance of 2.69 Å and 2.99 Å. The H4 atom of the mechlorethamine drug is interacted with the N9 of the C5N2 sheet by 2.59 Å. Besides this, a distance of 2.24 Å is observed between H5 and N10 of the drug and the substrate complex. These bond lengths are outside the normal range of a covalent bond (1.01 Å) [125] and can be considered weak hydrogen bonds between NH or van der Waals interactions (~ 2.0 Å to 3.5 Å) [126] between HC atoms. Moreover, chlorine (Cl1) is also linked with the carbon (C6) of the C5N2 sheet while maintaining a bond distance of 3.35 Å further confirming that ClC is connected through van der Waals interaction. Hence the negative values of the interaction energies and suitable bond distances manifested the potential of cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes to be evaluated as drug delivery systems.
The results of interaction energies manifested that the most stable geometry is identified for the cisplatin@C5N2 complex because of weak hydrogen bonding between the drug and the substrate. Additionally, the presence of two electron-rich oxygen atoms in the carmustine@C5N2 complex is responsible for its stronger adsorption on the surface of the C5N2 carrier as compared to mechlorethamine@C5N2 complex as shown in Table 1. The order of interaction energies based on the bond length and the nature of the interaction between the drugs and the substrate is cisplatin@C5N2 carmustine@C5N2 mechlorethamine@C5N2. The most suitable alignment and the position over the C5N2 sheet for cisplatin, carmustine, and mechlorethamine is represented in Figure 2. The resulting interaction energies of the complexes (drug@C5N2) with different orientations of the drugs over the substrate are presented in Table S1.
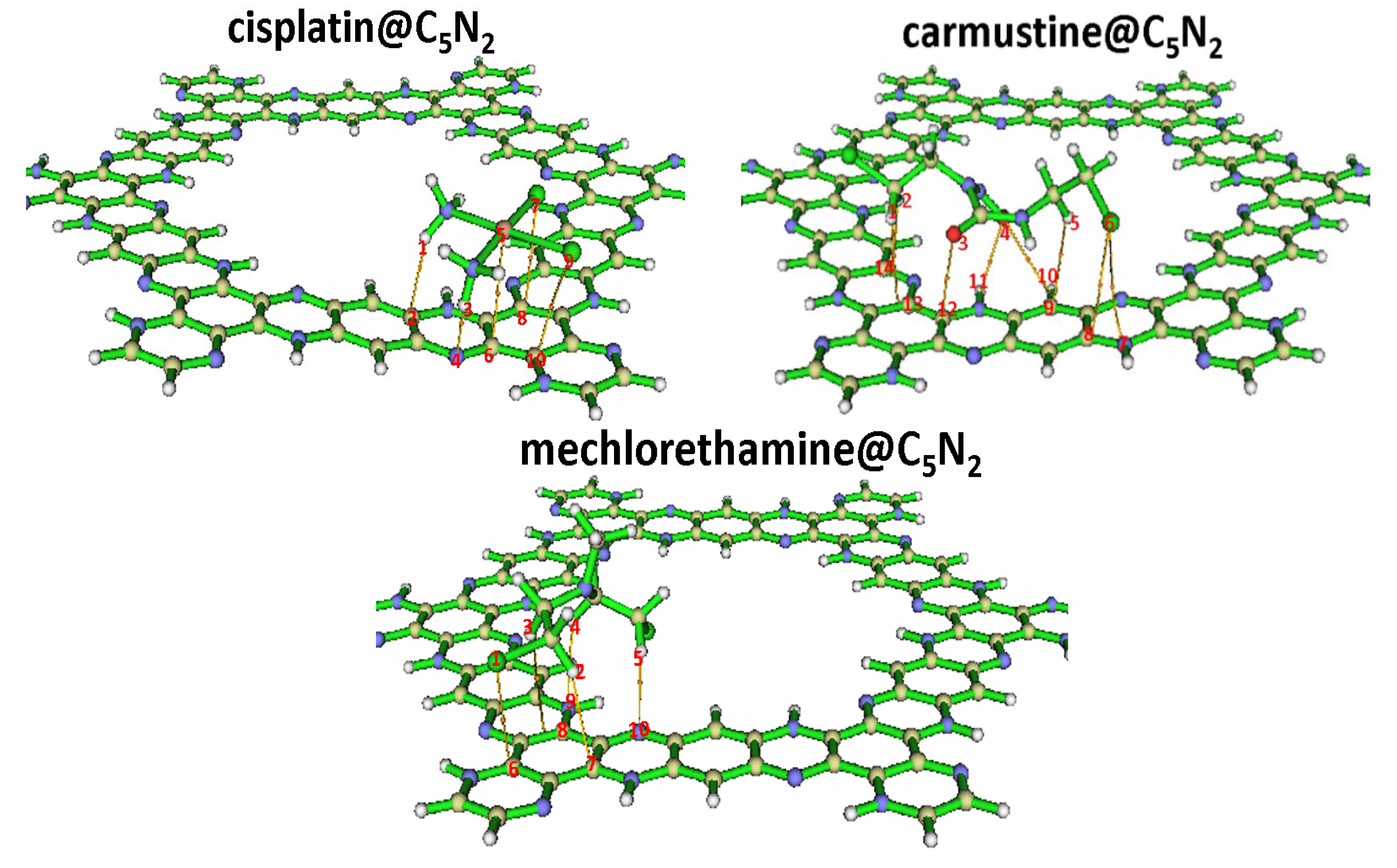
Figure 2.
Stable geometries of the cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes highlighting interacting atoms and their respective bond lengths (represented by spotted lines) between the drugs and the C5N2 sheet.
Figure 2.
Stable geometries of the cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes highlighting interacting atoms and their respective bond lengths (represented by spotted lines) between the drugs and the C5N2 sheet.
3.1. Topological Analysis
3.1.1. Non-Covalent Interaction (NCI) Analysis
NCI analysis has been performed on the most stable complexes of the cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 to visualize the non-covalent interactions. 2D RDG plots and 3D iso surfaces are displayed to visualize the attraction and repulsion between the drug@C5N2 complexes.
Three color codes have been employed to examine the nature of interactions. In the 3D isosurface, the red color reveals the stearic hindrance, and the green color represents van der Waals interactions. In contrast, the blue color demonstrates the presence of attractive forces between the drugs and the C5N2 substrate such as hydrogen bonding. Besides this, in a 2D plot, red, green, and blue spikes depict stearic repulsions, van der Waals interactions, and hydrogen bonding, respectively (as shown in Figure 3). The strength of the non-covalent interactions is directly related to the thickness of a particular patch. For instance, a thick patch leads to a stronger interaction as compared to a speckled streak which represents weak interactions between the complexes. Moreover, the electron density (on the x-axis) is directly related to the nature of interactions i.e., high electron density corresponds to stronger interactions between the complex systems. In 2D plots of cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes, the green color spikes manifest the presence of van der Waals interactions ranging from showing a significant correlation with the green color 3D isosurface aligned parallelly over pyrazine, benzene, and hydrogenated benzene rings of the C5N2 sheet. The red color spikes range from manifest the presence of repulsive interactions between the drugs and the substrate. These spikes show consistency with the red color 3D isosurface between the conjugated rings of the C5N2 substrate. The widespread blue color spots in a 2D plot range from represents weak hydrogen bonding between cisplatin@C5N2 complex. In contrast, the absence of the blue color spikes in 2D RDG plots of carmustine@C5N2 and mechlorethamine@C5N2 complexes manifest the lack of hydrogen bonding between the drugs and the surface as displayed with the value ranging from on the x-axis. Moreover, in the case of the carmustine@C5N2 complex, the green patches are more thicker showing its greater van der Waals interaction with the C5N2 as compared to the cisplatin@C5N2, and the mechorethamine@C5N2 complexes. However, the cisplatin@C5N2 complex exhibits stronger interaction with the substrate due to weak hydrogen bonding. Hence, NCI results revealed significant coherence with the computed results of the interaction energies, and the bond lengths further validating the presence of non-covalent interactions between the drug@C5N2 systems.
3.1.2. Quantum Theory of Atoms in Molecules (QTAIM) Analysis
Quantum theory of atoms in molecules is a primary analyzing tool that provides a comprehensive understanding of the strength and the nature of atomic interactions within the molecular system. It helps in evaluating the presence of covalent and non-covalent bonds at the quantum level. The bonding properties of cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 have been fundamentally validated through electronic density at the bond critical points (BCPs) representing the bonding region between the atoms of the drug and the C5N2 sheet. For the cisplatin@C5N2 complex, three BCPs were found between HC, NH, and PtC interacting atoms and two were observed between ClC atoms of the complex (as shown in Figure 4). The values of () range from (0.032 a.u. to 0.0008 a.u.) which reflects the presence of van der Waal interaction between the cisplatin and the C5N2 sheet. The highest value of electron density is (0.03 a.u.) between H3 of the cisplatin and N4 of the C5N2 sheet manifesting weak hydrogen bonding which substantially correlates with its high interaction energy and decreased bond length. Moreover, the positive values of energy density (H) and Laplacian of electron density range from (0.00008 0.0002 a.u.) and (0.02 0.08 a.u.), respectively, exhibiting weak interactions between the drug and the substrate. The V/G ratio is used to estimate the interatomic interactions. It suggests the presence of non-covalent interaction when V/G 1, while it reveals the formation of a covalent bond if V/G 2. These values vary from 0.83 a.u. to 0.98 a.u. illustrating non-covalent interactions between cisplatin@C5N2 system. Also, the values of Lagrangian of kinetic energy (G), and potential energy density (V) indicate non-bonding interactions (as presented in Table 2). In the case of the carmustine@C5N2 complex, eight BCPs were located in which three correspond to CH interactions, two were observed between OH bonds, and one is identified between OC, ClN, and ClC interacted atoms (Figure 4). The values of electron density ( vary from (0.003 a.u.) to (0.01 a.u.) indicating van der Waals interaction. Additionally, the values of the Laplacian of electron density , energy density (H,) and V/G ratio range from (0.0180.041 a.u.), (0.00050.0012 a.u.), and (0.73 a.u. to 0.87 a.u.), sequentially, manifesting non-covalent interactions between carmustine@C5N2 complex. Moreover, the values of other parameters including Lagrangian of kinetic energy (G), and potential energy density (V) also illustrate van der Waals interactions (shown in Table 2).
For mechlorethamine, five BCPs were located between the C5N2 and the mechlorethamine complex. Among them, four BCPs were found between each of the two atoms (HN and HC), while one BCP was observed between Cl1C6 interacted atoms (see Figure 4). The values of the electron density ( shift from (0.0040.01 a.u.) exhibiting non-bonding interactions. Furthermore, the positive values of Laplacian of electron density change from (0.0150.045 a.u.) illustrating long-range connections between the mechlorethamine@C5N2 complex. The values of V/G ratio and energy density (H) among individual atoms ranging from (0.001 to 0.9 a.u.) and (0.0009 to 0.0001), respectively, also manifested weak interactions between the mechlorethamine@C5N2 complex. Other topological parameters including (G), and (V) have exhibited van der Waals interaction between the complex as shown in Table 2. These QTAIM parameters revealed consistency in finding the nature and strength of bonding further manifesting van der Waals interactions between carmustine@C5N2, and mechlorethamine@C5N2 complexes in combination with weak hydrogen bonding between cisplatin@C5N2 complex. Moreover, these computed results are aligned well and illustrate a significant correlation with the values of interaction energies and NCI analysis.
Figure 4.
QTAIM analysis of studied drug@C5N2 complexes highlighting interacting atoms with corresponding bond critical points (BCPs).
Figure 4.
QTAIM analysis of studied drug@C5N2 complexes highlighting interacting atoms with corresponding bond critical points (BCPs).
3.1.3. Electron Localization Function (ELF) Analysis
ELF analysis has been used to analyze the electron density changes and the nature of interaction after the formation of the complexes. Moreover, it depicts the covalent nature of bonding (indicated by red color) when its value on the y-axis (representing the degree of electron localization) varies between 0.8001.00 (on a color scale). However, it suggests the delocalization of electrons and confirms the presence of non-covalent interactions when the degree of localization ranges from (0.1000.500) as represented by the blue color in ELF plots (Figure 5). For the cisplatin@C5N2 complex, the changes in the electron density have been occurred due to the interaction between the drug and the C5N2 sheet (highlighted in Figure 5). The blue color region reflects the presence of non-covalent interaction with a scale range from 0.1000.400 (on the bond axis). It also suggests shifting of electronic density between the interacting atoms of the complex. In the carmustine@C5N2 complex, the electron density changes are reflected clearly after the interaction of the drug and the C5N2 sheet. The blue color indicates the area of electron delocalization further depicting the presence of van der Waals interaction between the complex. Moreover, the red color scale of the C5N2 sheet is shifted from 0.900 (along the bond axis) to the blue color scale (0.200) manifesting the reduction of the localized electrons (covalent nature) after the formation of the mechlorethamine@C5N2 complex. These findings indicate the changes and shifting of electron densities and illustrate the presence of non-covalent interactions between the drug@C5N2 complexes further correlating with the results of NCI and QTAIM analyses.
3.2. Analysis of Electronic Properties
3.2.1. Frontier Molecular Orbital (FMOs) Analysis and Chemical Reactivity Descriptors
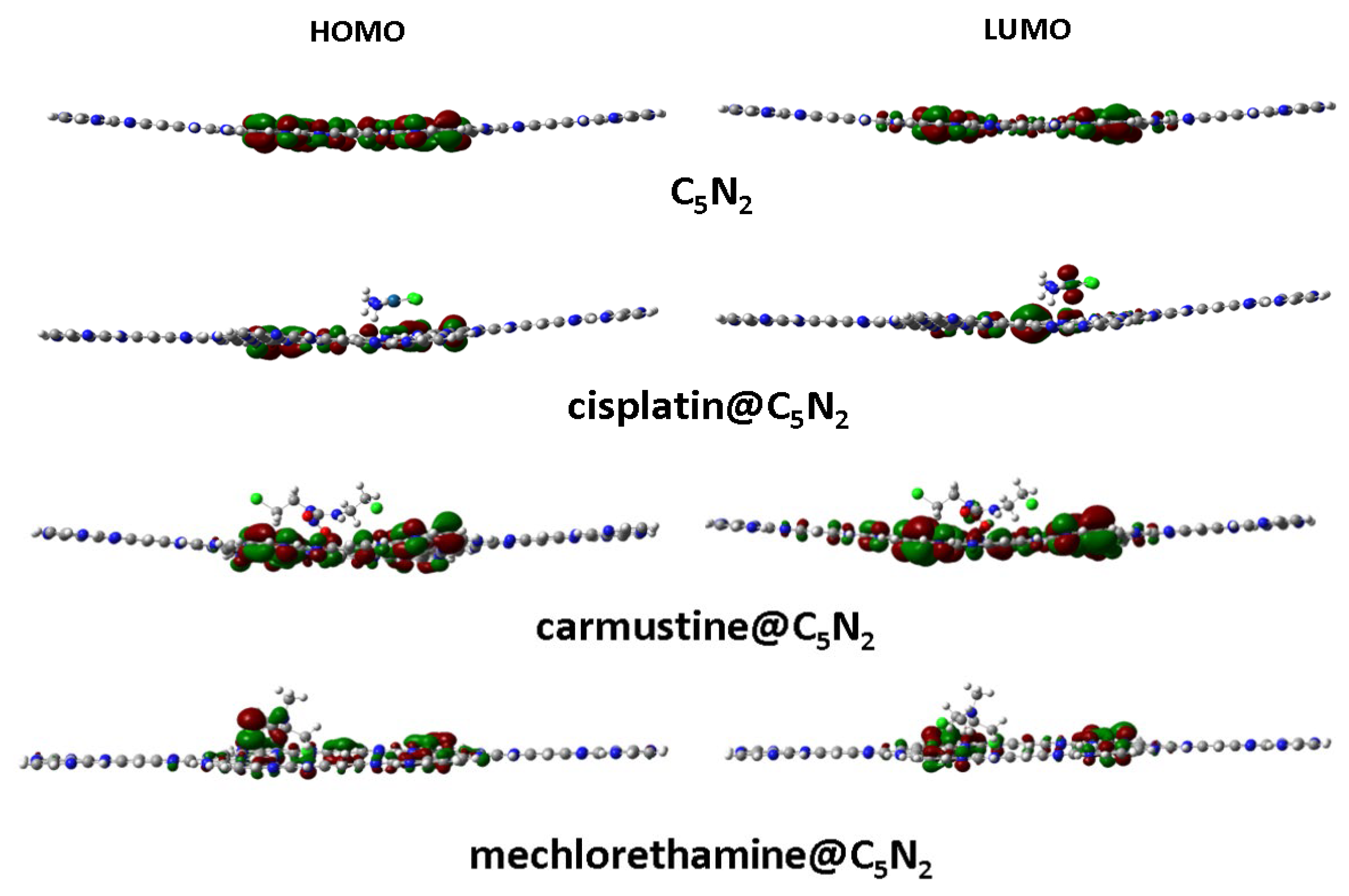
Electronic properties of cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes were computed to analyze the reactivity of the complexes using the FMO analysis. It is used to identify the active sites of the drugs to check their potential in biological systems. During complex formation, HOMO is responsible for the donation of electronic charges while LUMO corresponds to the acceptance of charges. Furthermore, energies of HOMOLUMO and spatial distribution of electronic charges have been evaluated and visualized for the drug@C5N2 complexes through FMO analysis. The high value of HOMO indicates its ability to electron donation while the high value of LUMO (less negative) illustrates its electron-accepting nature as shown in Table 3. The reactivity and chemical stability of the complexes i.e., cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 have been explained by a significant variable, HL gap (the gap between HOMO and LUMO orbitals) represented by . A small band gap reflects high reactivity and polarizability with low kinetic stability of the complexes and vice versa for a large band gap i.e., low reactivity and polarizability with high kinetic stability of the complexes. Figure 6 represents the HOMOLUMO of the C5N2 substrate, cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes, respectively.
For the bare C5N2 surface, the energy values of the HOMO and LUMO are identified as 3.42 eV, and 2.82 eV, respectively, while their is 0.60 eV. After the formation of the cisplatin@C5N2, and carmustine@C5N2 complexes, the energy of the HOMO and LUMO has been elevated to 3.41 eV and 2.80 eV, sequentially, with the decrease in the energy gap (0.57 eV). The decrease in the values of the energy gap for cisplatin@C5N2 and carmustine@C5N2 complexes is directly associated with the increase of energy of HOMOLUMO (indicating their presence at slightly higher energy levels). Moreover, in the case of the mechlorethamine@C5N2 system, the energy gap is reduced to 0.58 eV resulting from the elevation of the energy of HOMO (3.39 eV), and LUMO (2.81 eV). However, the decrease in the energy gap of the complexes corresponds to the enhancement of the chemical reactivity of the C5N2 substrate (as compared to the pristine C5N2 sheet) as shown in Table 3.
Among these complexes, the energy of HOMO of mechlorethamine@C5N2 complex (3.39 eV) indicates its tendency of electron donation as it is shifted on a higher energy level and the energy of LUMO of cisplatin and carmustine (2.80 eV) reflects its ability of electron acceptance within the complex. The reduction in the energy gap manifests a feasible interaction owing to an effective charge transfer between the drugs and the C5N2 sheet resulting in a stabilized complex. The orbital spatial distribution of the cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes is represented in Figure 6. In the case of cisplatin@C5N2, and carmustine@C5N2 complexes, the isosurface of the HOMO is completely distributed over the C5N2 surface while LUMO is spread slightly over their respective drugs. In contrast, in the mechlorethamine@C5N2 complex, the HOMO isosurface is widely distributed over the mechlorethamine drug and the substrate while the LUMO is entirely dispersed over the surface of the C5N2 sheet. These electron cloud patterns reflect electron density shifts between the drugs and the C5N2 carrier after complex formation and exhibit a major correlation with the findings of the interaction energy and the topology analyses. The reactivity and stability of the drugs, the C5N2 substrate, and the drugs@C5N2 complexes are explained by the computed values of the chemical reactivity descriptors. The more negative value of (μ) in the mechlorethamine@C5N2 complex (3.10 eV) indicates its least reactivity and higher stability as compared to the cisplatin@C5N2 and carmustine@C5N2 complexes with relatively less negative (μ) value (3.09 eV). Moreover, a lower value of (ω) in the mechlorethamine@C5N2 complex (16.51 eV) manifests its nucleophilic nature towards the C5N2 sheet while a high (ω) value in the cisplatin@C5N2, and carmustine@C5N2 complexes (17.68 eV) indicate their electrophilic nature towards the C5N2 substrate. Between the drug@C5N2 complexes, the highest hardness value is (0.29 eV) in the mechlorethamine@C5N2 complex manifesting its less polarizability and a harder nature having a large energy gap as compared to the cisplatin@C5N2 and carmustine@C5N2 complexes as presented in Table 3. These findings demonstrate a better interaction of the drugs with the C5N2 sheet and illustrate enhanced stability in (mechlorethamine@C5N2 complex) and higher reactivity in cisplatin@C5N2, and carmustine@C5N2 complexes.
Figure 6.
Spatial distribution of the HOMOLUMO orbitals of the bare C5N2 sheet along with the drug@C5N2 complexes.
Figure 6.
Spatial distribution of the HOMOLUMO orbitals of the bare C5N2 sheet along with the drug@C5N2 complexes.
3.2.2. Density of States (DOS) Analysis
The electronic structure of the complexes i.e., cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 has been evaluated through the DOS analysis to visualize the energy levels and their relative positions. The contributions of the HOMOLUMO orbitals and the gap within the complexes have also been verified through the DOS plots as shown in Figure 7. The DOS spectra of the cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes manifest a slight shift in the HOMOLUMO peaks as compared to the peaks of the pristine C5N2 sheet (3.42 eV and 2.82 eV, respectively). The HOMO of the cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes is shifted to a less negative position (3.41 eV and 3.39 eV) while LUMO emerged at (2.80 eV and 2.81 eV) suggesting enhanced chemical reactivity and a charge transfer between the interacting atoms of the complexes. The peak intensities reflect the contribution of the orbitals and the availability of the electronic states at a specific energy level. Hence, the shifting of the peaks and the enhancement of the orbitals of HOMOLUMO result in the reduction of the energy gap which further reinforces the findings of the FMO analysis.
3.2.3. NBO and EDD Analyses
Natural bond orbital analysis plays a significant role in illustrating the transfer of charges between the drug@C5N2 complexes. After the formation of the cisplatin@C5N2 complex, the value of NBO charge on the cisplatin drug is increased to 0.039 which shows the electronic density is shifted from the C5N2 sheet to the cisplatin drug. The negative charge of nitrogen and chlorine atoms bonded with H1, H3, and Pt5 has also been elevated to 0.98, 0.99, and 0.39 in sequence from 0.20 and 0.33 after complex formation. Moreover, the charge on H1, H3, and Pt5 atoms of the cisplatin drug has also been increased from 0.180, 0.169, 0.04 to 0.419, 0.430, 0.24, respectively after the interaction with C2, N4, and C6 making H1, H3, and Pt5 atoms of the drug electron deficient. However, the amount of charge attracted from the sheet is greater than the charge donated by the drug, manifesting that the major electron density is shifted from the C5N2 sheet to the cisplatin drug after interaction. In the carmustine@C5N2 complex, the negative value of the NBO charge 0.031 illustrates the transfer of electronic density from the C5N2 sheet to the carmustine drug. The oxygen atoms of the carmustine attract electrons towards themselves making the C5N2 sheet electron-deficient. The negative charges on O3 and O4 of the carmustine have been increased from 0.24 and 0.25 to 0.62 and 0.39, respectively after complexation with C12, H10, and H11 of the C5N2 substrate. Whereas the value of negative charge on the nitrogen of carmustine has also been elevated from 0.32 to 0.34. Moreover, the positive values of the electronic charges in the alkyl chain of carmustine intensified up to 0.40,0.24,0.25, and 0.38 after complex formation. These clear escalations in the charge values of cisplatin and carmustine manifested their electron-accepting nature while presenting C5N2 as a donor substrate in the cisplatin@C5N2, and carmustine@C5N2 complexes. In contrast, the positive value of electronic charge (0.479) over mechlorethamine indicates the transfer of charge from the drug to the C5N2 sheet. The nitrogen (N10) is attracting electron density from the H5 atom of the mechlorethamine and its charge value is increased from 0.51 to 0.56 in a complex. The charge on Cl1 is decreased from 0.21 to 0.10 after interacting with C6 of the C5N2 sheet. These findings manifested that the charge had been transferred from the mechlorethamine drug to the C5N2 sheet in a complex system. Hence in the cisplatin@C5N2 complex, the highest amount of charge i.e., (0.039) is shifted from the C5N2 surface to the drug molecule, while in mechlorethamine@C5N2, 0.479 of charge is captivated from the drug molecule to the pyrazine and benzene rings of the C5N2 substrate after association as shown in Table 3.
To validate and comprehend the NBO results, EDD has been performed to elucidate the electron density shifts between the drug and the C5N2 sheet in the drug@C5N2 complexes. Two color codes have been used to analyze the charge accumulation and depletion i.e., red and blue, sequentially, after the association of the drug and the C5N2 carrier. The EDD results have been computed by subtracting the sum of the individual electron densities of drugs and the C5N2 sheet from the electron density of drug@C5N2 complexes as represented by [122],
The negative values of are represented by red (charge accumulation) while the positive value of is indicated by blue color (charge depletion) as represented by EDD portrayals in Figure 8. For the cisplatin@C5N2 complex, the electron density is shifted from the benzene ring of the C5N2 carrier (as represented by the blue color isosurface) to the electronegative chlorine and nitrogen atoms of the drug (depicted by the red color isosurface over the drug molecule). Moreover, the nitrogen and carbon atoms of the pyrazine ring of the C5N2 sheet accepted electron density (depicted by the red isosurface) from the hydrogen atoms of the cisplatin (illustrated by the blue color isosurface over H1, and H3 atoms) after interaction as shown in Figure 8. In the carmustine@C5N2 complex, the electron density is shifted from the conjugated pyrazine and benzene rings of the C5N2 surface (as illustrated by the blue color isosurface) to the electronegative atoms of the drug (indicated by the red color) after their interaction within a complex. The red color isosurface is also distributed over the edged aromatic pyrazine ring of the C5N2 sheet because it exhibit the least interaction with the drug molecule. In the mechlorethamine@C5N2 complex, the blue color isosurface of the drug (mainly on H atoms) represents electron deficiency after interaction with the substrate atoms. Moreover, the red isosurface over pyrazine and benzene rings of the substrate depicts the withdrawal of electron density from the drug (H5 and C7) to the C5N2 sheet after complex formation as indicated by Figure 8. Consequently, the isosurface and findings of EDD correlate significantly with the results of FMO, and NBO analyses.
4. Recovery Time
The desorption of the anticancer drugs plays a crucial part in targeted drug delivery which has been evaluated by analyzing the recovery times of the drugs. The theoretical calculation of the recovery time (τ) [127] for the desorption of cisplatin, carmustine, and mechlorethamine from the surface of the C5N2 sheet has been performed using the expression,
Here, T is the temperature, ʋ is the attempt frequency (ʋ = [128] and K is the Boltzmann’s constant. At 473 K, the recovery time of the cisplatin@C5N2 and the carmustine@C5N2 complexes is s, and s, respectively. Moreover, for the mechlorethamine@C5N2 complex, the recovery time is s. These findings manifest that mechlorethamine and carmustine will desorb faster having shorter recovery times while cisplatin will desorb slowly following the order i.e., cisplatin@C5N2 carmustine@C5N2 mechlorethamine@C5N2 further correlating with the results of interaction energies. Moreover, the recovery time of the drugs is exponentially decreased with the increase in the temperature as shown in Table S2.
5. Solvent Effect
To gain insight into the interaction and stability of the drug@C5N2 complexes in a realistic media, SMD (solvation model based on density) is employed at the same level of theory. This model provides a precise illustration of both the electrostatic and non-electrostatic interactions of the system and is parameterized for a wider range of solvents [129,130]. Moreover, it can calculate the solvation energies that are crucial for bonding and interaction strengths of large and complex geometries with different solvents [131,132]. It has also been used to evaluate the hydration-free energies in drug design [133]. Since water is a key component of the human biochemical system, it has been used as a solvent in this study. The influence of the solvent on interactions between the drug molecules and the C5N2 substrate has been investigated using the expression [134],
∆E corresponds to the solvation energy of the complex, where the total energy of the complexes i.e., drugs@C5N2 in solvent and gas phases is represented by , and , respectively. The solvation energy of the cisplatin@C5N2, and carmustine@C5N2 complexes is increased to 65.13 kcal and 51.11 kcal, respectively, which confirms their stability in the aqueous phase. Additionally, for the mechlorethamine@C5N2 complex, the solvation energy is increased to 46.82 kcal, manifesting its significant adsorption in water media. These negative values revealed stable interaction of the drugs with the C5N2 substrate in the solvent phase. The dipole moment is another parameter that is used to investigate the solubility and polarity of a complex in an aqueous phase. The calculated value of a dipole moment for the C5N2 substrate, cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 are observed as 0.02 D, 6.10 D, 2.69 D, 1.83 D in gaseous and 0.30 D, 7.43 D, 5.33 D, 2.43 D in solvent phases, respectively. Compared to the C5N2 sheet, the high dipole moment of the complexes indicates better solubility, localization, and directional interaction in polar solvents. This manifests that the hydrophilicity of the cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes has also been increased after complexation that facilitates easy circulation of the drugs within the biological systems. The values of the solvation energy of cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes with their respective dipole moments in gaseous and solvent phases are presented in Table S3. These results depict better stability and improved interaction of the drug@C5N2 complexes with the cellular targets which is further beneficial in drug delivery.
6. Conclusion
Computational studies have been performed to analyze the interaction behavior of anticancer drugs including cisplatin, carmustine, and mechlorethamine over the surface of the C5N2 sheet using DFT calculations. Geometry optimization of the drugs, C5N2 sheet, and their complexes (cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2) has been performed at the PBE0-D3BJ/def2SVP level of theory. Different positions of the drugs over the substrate have been validated and the most suitable configuration has been used to analyze the nature of interaction between the complexes. The highest interaction energy has been obtained for cisplatin@C5N2 (27.60 kcal ) following carmustine@C5N2 (19.69 kcal) and mechlorethamine@C5N2 (17.79 kcal complexes. Topology analysis of the complexes has been performed to analyze the strength and the type of bonding between the drug@C5N2 systems. The QTAIM and NCI analyses of the complexes manifested the presence of non-covalent interactions between the drug and the C5N2 sheet. In the case of carmustine@C5N2 and mechlorethamine@C5N2 complexes, van der Waals interactions have been observed as green color isosurface and patches in 3D and 2D RDG plots. Conversely, weak hydrogen bonding has also been observed in the cisplatin@C5N2 complex as depicted by the blue color spots in a 2D NCI plot. The higher values of electron density (ρ) and Laplacian of electron density (∇2ρ) manifested non-covalent interactions correlating with the NCI results. ELF analysis has been carried out to estimate the degree of electron delocalization between the complexes. Reactivity and interaction of the complexes have been enhanced with the reduction of the HOMOLUMO gap (. The chemical potential and electrophilicity index of the cisplatin@C5N2, and carmustine@C5N2 complexes indicate their higher reactivity while mechlorethamine@C5N2 is the least reactive among them. DOS analysis exhibited a significant correlation with the findings of the FMO analysis further confirming the decrease in the energy gap due to the shifting of the peaks and the enhancement of the orbitals. Electron density shifts have been computed and analyzed through NBO analysis and EDD portrayals. Recovery time has been calculated to analyze the desorption behavior of the drugs from the C5N2 surface. The mechlorethamine drug recovered faster while carmustine and cisplatin drugs manifested slow recovery from the surface of the C5N2 sheet exhibiting a substantial correlation with the results of the interaction energy. SMD solvation model has been employed to evaluate the solvation energy of the complexes and the calculations have been performed at the same level of theory by employing water as a solvent. The interaction of the complexes has been enhanced in solvent media. The higher values of the dipole moment indicated better solubility, localization, and directional interaction of the complexes in a solvent phase. Conclusively, the findings manifested that the reported systems including cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 can be utilized as efficient targeted drug delivery systems for the treatment of cancer.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1S3: Optimized geometries of the cisplatin@C5N2, carmustine@C5N2 and mechlorethamine@C5N2 complexes with different orientations of the drugs over the C5N2 sheet; Table T1T3: Interaction energies (kcal, recovery time (τ) at different temperatures, solvation energies (kcal, and dipole moment (D) in gas and solvent media of the drug@C5N2 complexes.
Author Contributions
Validation, analysis, investigation, data curation, writing—original draft preparation, writing—review and editing, visualization, S.H.M.Z.; Conceptualization, methodology, computational resources, supervision, M.A.H.; validation, computational resources, A.L; writing—review and editing, M.A., M.A.H, and A.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
Data is contained within the article and supplementary file.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA: a cancer journal for clinicians 2023, 73, 17-48. [CrossRef]
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA: a cancer journal for clinicians 2023, 73, 233-254. [CrossRef]
- Cao, S.; Wei, Y.; Huang, J.; Yue, Y.; Deng, A.; Zeng, H.; Wei, W. A bibliometric worldview of breast-conserving surgery for breast cancer from 2013 to 2023. Frontiers in Oncology 2024, 14, 1405351. [CrossRef]
- Srinivasan, D.; Subbarayan, R.; Srivastava, N.; Radhakrishnan, A.; Adtani, P.N.; Chauhan, A.; Krishnamoorthy, L. A comprehensive overview of radiation therapy impacts of various cancer treatments and pivotal role in the immune system. Cell Biochemistry and Function 2024, 42, e4103. [CrossRef]
- Sharma, A.; Jasrotia, S.; Kumar, A. Effects of chemotherapy on the immune system: implications for cancer treatment and patient outcomes. Naunyn-Schmiedeberg’s Archives of Pharmacology 2024, 397, 2551-2566. [CrossRef]
- Liu, B.; Zhou, H.; Tan, L.; Siu, K.T.H.; Guan, X.-Y. Exploring treatment options in cancer: tumor treatment strategies. Signal transduction and targeted therapy 2024, 9, 175. [CrossRef]
- Kar, A.; Agarwal, D.G.; Agarwal, D.S. A Review On Nanostructure Drug Carriers for Treatment and Management of Neuroendocrine Cancer.(2023). Int J Pharm Sci 2023, 14, b1-9.
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes & Diseases 2023, 10, 1367-1401. [CrossRef]
- Gökşen Tosun, N. Enhancing therapeutic efficacy in breast cancer: a study on the combined cytotoxic effects of doxorubicin and MPC-3100. Naunyn-Schmiedeberg’s Archives of Pharmacology 2024, 397, 3249-3259. [CrossRef]
- Wei, Y.; Li, Q.; Mo, H.; Qi, Y.; Ge, H.; Sun, X.; Fan, Y.; Zhang, P.; Wang, J.; Luo, Y. Comparative efficacy of anthracycline-free and anthracycline-containing neoadjuvant chemoimmunotherapy regimens for triple-negative breast cancer. Translational Oncology 2025, 51, 102171. [CrossRef]
- Liu, Z.; Yuan, Y.; Wang, N.; Yu, P.; Teng, Y. Drug combinations of camptothecin derivatives promote the antitumor properties. European Journal of Medicinal Chemistry 2024, 116872. [CrossRef]
- Romani, A.M. Cisplatin in cancer treatment. Biochemical pharmacology 2022, 206, 115323. [CrossRef]
- Strojan, P.; Vermorken, J.B.; Beitler, J.J.; Saba, N.F.; Haigentz Jr, M.; Bossi, P.; Worden, F.P.; Langendijk, J.A.; Eisbruch, A.; Mendenhall, W.M. Cumulative cisplatin dose in concurrent chemoradiotherapy for head and neck cancer: A systematic review. Head & neck 2016, 38, E2151-E2158.
- Song, M.; Cui, M.; Liu, K. Therapeutic strategies to overcome cisplatin resistance in ovarian cancer. European journal of medicinal chemistry 2022, 232, 114205. [CrossRef]
- Li, F.; Zheng, Z.; Chen, W.; Li, D.; Zhang, H.; Zhu, Y.; Mo, Q.; Zhao, X.; Fan, Q.; Deng, F. Regulation of cisplatin resistance in bladder cancer by epigenetic mechanisms. Drug Resistance Updates 2023, 68, 100938. [CrossRef]
- Sala, L.; Perecko, T.; Mestek, O.; Pinkas, D.; Homola, T.; Kocisek, J. Cisplatin-cross-linked DNA origami nanostructures for drug delivery applications. ACS Applied Nano Materials 2022, 5, 13267-13275. [CrossRef]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.-W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer 2015, 1856, 189-210.
- Shirbin, S.J.; Ladewig, K.; Fu, Q.; Klimak, M.; Zhang, X.; Duan, W.; Qiao, G.G. Cisplatin-induced formation of biocompatible and biodegradable polypeptide-based vesicles for targeted anticancer drug delivery. Biomacromolecules 2015, 16, 2463-2474. [CrossRef]
- Wagstaff, A.J.; Brown, S.D.; Holden, M.R.; Craig, G.E.; Plumb, J.A.; Brown, R.E.; Schreiter, N.; Chrzanowski, W.; Wheate, N.J. Cisplatin drug delivery using gold-coated iron oxide nanoparticles for enhanced tumour targeting with external magnetic fields. Inorganica Chimica Acta 2012, 393, 328-333. [CrossRef]
- Perveen, M.; Nazir, S.; Arshad, A.W.; Khan, M.I.; Shamim, M.; Ayub, K.; Khan, M.A.; Iqbal, J. Therapeutic potential of graphitic carbon nitride as a drug delivery system for cisplatin (anticancer drug): A DFT approach. Biophysical chemistry 2020, 267, 106461. [CrossRef]
- Weiss, R.B.; Issell, B.F. The nitrosoureas: carmustine (BCNU) and lomustine (CCNU). Cancer treatment reviews 1982, 9, 313-330. [CrossRef]
- Khanizadeh, A.; Ghaemi, A.; Pourmadadi, M.; Javadi, S.; Rahdar, A.; Yazdian, F.; Ghazy, E.; Pandey, S. Advancing cancer therapy: unveiling the cutting-edge potential of carmustine nano carriers for targeted treatment. Journal of Drug Delivery Science and Technology 2024, 105943. [CrossRef]
- Ahmad, S.; Khan, I.; Pandit, J.; Emad, N.A.; Bano, S.; Dar, K.I.; Rizvi, M.M.A.; Ansari, M.D.; Aqil, M.; Sultana, Y. Brain targeted delivery of carmustine using chitosan coated nanoparticles via nasal route for glioblastoma treatment. International Journal of Biological Macromolecules 2022, 221, 435-445. [CrossRef]
- Chen, S.; Qiu, Q.; Wang, D.; She, D.; Yin, B.; Chai, M.; He, H.; Heo, D.N.; Wang, J. Long acting carmustine loaded natural extracellular matrix hydrogel for inhibition of glioblastoma recurrence after tumor resection. Frontiers of Chemical Science and Engineering 2022, 16, 536-545. [CrossRef]
- Bayat, M.; Taherpour, A.A.; Elahi, S.M.; Fellowes, T. Separation of anticancer medicines carmustine, lomustine, semustine and melphalan by PAMAM dendrimer: a theoretical study. Journal of the Iranian Chemical Society 2018, 15, 1223-1234. [CrossRef]
- Kamel, M.; Mohammadi, M.; Mohammadifard, K.; Mahmood, E.A.; Heravi, M.R.P.; JM, A.H.; Hossaini, Z. Comprehensive theoretical prediction of the stability and electronic properties of hydroxyurea and carmustine drugs on pristine and Chitosan-functionalized graphitic carbon nitride in vacuum and aqueous environment. Vacuum 2023, 207, 111565. [CrossRef]
- Rani, V.; Venkatesan, J.; Prabhu, A. Carmustine-Loaded Liposomal Delivery Effectively Targets Malignant Glioma Cells and Seizes Endothelial Sprouting In vitro. Journal of Cluster Science 2024, 1-11. [CrossRef]
- Mortazavifar, A.; Raissi, H.; Akbari, A. DFT and MD investigations on the functionalized boron nitride nanotube as an effective drug delivery carrier for Carmustine anticancer drug. Journal of Molecular Liquids 2019, 276, 577-587. [CrossRef]
- Solomon, J.; Jacobs, E.M.; Bateman, J.R.; Lukes, R.J.; Weiner, J.M.; Donohue, D.M. Chemotherapy of lymphoma with mechlorethamine and vinblastine. Archives of Internal Medicine 1973, 131, 407-417. [CrossRef]
- Jacobs, E.M.; Peters, F.C.; Luce, J.K.; Zippin, C.; Wood, D.A. Mechlorethamine HCl and cyclophosphamide in the treatment of Hodgkin’s disease and the lymphomas. Jama 1968, 203, 392-398. [CrossRef]
- Vonderheid, E.C.; Tan, E.T.; Kantor, A.F.; Shrager, L.; Micaily, B.; Van Scott, E.J. Long-term efficacy, curative potential, and carcinogenicity of topical mechlorethamine chemotherapy in cutaneous T cell lymphoma. Journal of the American Academy of Dermatology 1989, 20, 416-428. [CrossRef]
- Mekkey, S.M.; Al-dolaimy, F.; Hussein, U.A.-R.; Younis, S.M.D.; Kadhim, A.J.; Kareem, M.W.A.; Abed, N.K.; Asiri, M.; Alkhayyat, S.; Alsalamy, A.H. Investigation of Drug Delivery of Mechlorethamine (Anticancer Drug) by Si76, C76, Al38N38 Nanocages. Silicon 2024, 16, 585-592. [CrossRef]
- de Vries Schultink, A.; Suleiman, A.; Schellens, J.; Beijnen, J.; Huitema, A. Pharmacodynamic modeling of adverse effects of anti-cancer drug treatment. European journal of clinical pharmacology 2016, 72, 645-653. [CrossRef]
- Zhong, Q.; Zee, K.; Rasmussen, K.; McKinley, B.J.; Linger, R.M.; Ray, S.D. Side effects of anti-cancer medications. In Side Effects of Drugs Annual; Elsevier: 2022; Volume 44, pp. 431-445.
- Torino, F.; Barnabei, A.; Paragliola, R.M.; Marchetti, P.; Salvatori, R.; Corsello, S.M. Endocrine side-effects of anti-cancer drugs: mAbs and pituitary dysfunction: clinical evidence and pathogenic hypotheses. European journal of endocrinology 2013, 169, R153-R164. [CrossRef]
- Kashkooli, F.M.; Soltani, M.; Souri, M. Controlled anti-cancer drug release through advanced nano-drug delivery systems: Static and dynamic targeting strategies. Journal of controlled release 2020, 327, 316-349. [CrossRef]
- Qin, S.-Y.; Zhang, A.-Q.; Cheng, S.-X.; Rong, L.; Zhang, X.-Z. Drug self-delivery systems for cancer therapy. Biomaterials 2017, 112, 234-247. [CrossRef]
- Rashidi, N.; Davidson, M.; Apostolopoulos, V.; Kelley, M.R.; Nurgali, K. Targeted combination nano-drug delivery system to enhance anti-cancer efficacy and reduce side effects. Cancer Research 2024, 84, 478-478. [CrossRef]
- Xu, H.; Wang, Q.; Fan, G.; Chu, X. Theoretical study of boron nitride nanotubes as drug delivery vehicles of some anticancer drugs. Theoretical Chemistry Accounts 2018, 137, 1-15. [CrossRef]
- Li, Y.; Xiao, K.; Luo, J.; Lee, J.; Pan, S.; Lam, K.S. A novel size-tunable nanocarrier system for targeted anticancer drug delivery. Journal of controlled release 2010, 144, 314-323. [CrossRef]
- Zhang, H.; Fan, T.; Chen, W.; Li, Y.; Wang, B. Recent advances of two-dimensional materials in smart drug delivery nano-systems. Bioactive Materials 2020, 5, 1071-1086. [CrossRef]
- Molaei, M.J. Two-dimensional (2D) materials beyond graphene in cancer drug delivery, photothermal and photodynamic therapy, recent advances and challenges ahead: A review. Journal of Drug Delivery Science and Technology 2021, 61, 101830. [CrossRef]
- Gangrade, A.; Mandal, B.B. Drug delivery of anticancer drugs from injectable 3D porous silk scaffold for prevention of gastric cancer growth and recurrence. ACS Biomaterials Science & Engineering 2020, 6, 6195-6206. [CrossRef]
- Shi, K.; Aviles-Espinosa, R.; Rendon-Morales, E.; Woodbine, L.; Maniruzzaman, M.; Nokhodchi, A. Novel 3D printed device with integrated macroscale magnetic field triggerable anti-cancer drug delivery system. Colloids and Surfaces B: Biointerfaces 2020, 192, 111068. [CrossRef]
- Weng, Q.; Wang, B.; Wang, X.; Hanagata, N.; Li, X.; Liu, D.; Wang, X.; Jiang, X.; Bando, Y.; Golberg, D. Highly water-soluble, porous, and biocompatible boron nitrides for anticancer drug delivery. ACS nano 2014, 8, 6123-6130. [CrossRef]
- Zhang, Z.; Yan, W.; Ji, Y. A novel manganese dioxide-based drug delivery strategy via in situ coating γ-polyglutamic acid/cisplatin for intelligent anticancer therapy. Journal of Materials Chemistry B 2023, 11, 667-674. [CrossRef]
- Chen, H.; Liu, T.; Su, Z.; Shang, L.; Wei, G. 2D transition metal dichalcogenide nanosheets for photo/thermo-based tumor imaging and therapy. Nanoscale horizons 2018, 3, 74-89. [CrossRef]
- Chandrakala, V.; Aruna, V.; Angajala, G. Review on metal nanoparticles as nanocarriers: Current challenges and perspectives in drug delivery systems. Emergent Materials 2022, 5, 1593-1615. [CrossRef]
- Wu, M.; Yang, J.; Ye, T.; Wang, B.; Tang, Y.; Ying, X. Efficient Drug Delivery of Ti3C2T x MXenes for Synergistic Treatment of Human Hypopharyngeal Squamous Cell Carcinoma. ACS Applied Materials & Interfaces 2023, 15, 29939-29947.
- Liang, W.; Luo, X. Theoretical studies of MoS2 and phosphorene drug delivery for antituberculosis drugs. The Journal of Physical Chemistry C 2020, 124, 8279-8287. [CrossRef]
- Wang, G.; Li, R.; Parseh, B.; Du, G. Prospects and challenges of anticancer agents’ delivery via chitosan-based drug carriers to combat breast cancer: A review. Carbohydrate polymers 2021, 268, 118192. [CrossRef]
- Vuong, B.X.; Hajali, N.; Asadi, A.; Baqer, A.A.; Hachim, S.K.; Canli, G. Drug delivery assessment of an iron-doped fullerene cage towards thiotepa anticancer drug. Inorganic Chemistry Communications 2022, 141, 109558. [CrossRef]
- Gong, P.; Du, J.; Wang, D.; Cao, B.; Tian, M.; Wang, Y.; Sun, L.; Ji, S.; Liu, Z. Fluorinated graphene as an anticancer nanocarrier: an experimental and DFT study. Journal of Materials Chemistry B 2018, 6, 2769-2777. [CrossRef]
- Guven, A.; Villares, G.J.; Hilsenbeck, S.G.; Lewis, A.; Landua, J.D.; Dobrolecki, L.E.; Wilson, L.J.; Lewis, M.T. Carbon nanotube capsules enhance the in vivo efficacy of cisplatin. Acta biomaterialia 2017, 58, 466-478. [CrossRef]
- Guo, X.-L.; Kang, X.-X.; Wang, Y.-Q.; Zhang, X.-J.; Li, C.-J.; Liu, Y.; Du, L.-B. Co-delivery of cisplatin and doxorubicin by covalently conjugating with polyamidoamine dendrimer for enhanced synergistic cancer therapy. Acta biomaterialia 2019, 84, 367-377. [CrossRef]
- Farooq, M.A.; Aquib, M.; Farooq, A.; Haleem Khan, D.; Joelle Maviah, M.B.; Sied Filli, M.; Kesse, S.; Boakye-Yiadom, K.O.; Mavlyanova, R.; Parveen, A. Recent progress in nanotechnology-based novel drug delivery systems in designing of cisplatin for cancer therapy: an overview. Artificial cells, nanomedicine, and biotechnology 2019, 47, 1674-1692. [CrossRef]
- Zhang, Y.; Sun, S.; Wu, Y.; Chen, F. Emerging Roles of Graphitic Carbon Nitride-based Materials in Biomedical Applications. ACS Biomaterials Science & Engineering 2024, 10, 4645-4661. [CrossRef]
- Ning, X.-F.; Zhu, Y.-Q.; Sun, H.; Yang, Y.; Liu, M.-X. The Latest Applications of Carbon-Nitride-Based Materials for Combination Treatment of Cancer. ACS Applied Materials & Interfaces 2024. [CrossRef]
- Zaboli, A.; Raissi, H.; Farzad, F.; Hashemzadeh, H. Assessment of adsorption behavior of 5-fluorouracil and pyrazinamide on carbon nitride and folic acid-conjugated carbon nitride nanosheets for targeting drug delivery. Journal of Molecular Liquids 2020, 301, 112435. [CrossRef]
- Ahsan, F.; Yar, M.; Gulzar, A.; Ayub, K. Therapeutic potential of C2N as targeted drug delivery system for fluorouracil and nitrosourea to treat cancer: a theoretical study. Journal of Nanostructure in Chemistry 2023, 13, 89-102. [CrossRef]
- Pourmadadi, M.; Rahmani, E.; Eshaghi, M.M.; Shamsabadipour, A.; Ghotekar, S.; Rahdar, A.; Ferreira, L.F.R. Graphitic carbon nitride (g-C3N4) synthesis methods, surface functionalization, and drug delivery applications: a review. Journal of Drug Delivery Science and Technology 2023, 79, 104001.
- Wang, X.; Liu, S.; Wang, J.; Liu, Y.; Guan, S.; Zhang, T. Spherical g-C3N4@ PDA nanocarrier for synergistic chemo-photothermal tumor therapy. Journal of Photochemistry and Photobiology A: Chemistry 2024, 454, 115736. [CrossRef]
- Asghar, S.; Roudgar-Amoli, M.; Alizadeh, A.; Shariatinia, Z. Water purification through adsorption of organic pollutant onto novel and effective phosphorus-containing g-C3N4/FeMo0. 5O3 nanocomposites. Water, Air, & Soil Pollution 2023, 234, 43.
- Singh, J.A.; Overbury, S.H.; Dudney, N.J.; Li, M.; Veith, G.M. Gold nanoparticles supported on carbon nitride: influence of surface hydroxyls on low temperature carbon monoxide oxidation. Acs Catalysis 2012, 2, 1138-1146. [CrossRef]
- Ou, H.; Ning, S.; Zhu, P.; Chen, S.; Han, A.; Kang, Q.; Hu, Z.; Ye, J.; Wang, D.; Li, Y. Carbon nitride photocatalysts with integrated oxidation and reduction atomic active centers for improved CO2 conversion. Angewandte Chemie International Edition 2022, 61, e202206579. [CrossRef]
- Liang, H.; Wang, A.; Cheng, R.; Tian, X.; Jing, S.; Tsiakaras, P. Efficient photocatalytic H2O2 production ability of a novel graphitic carbon nitride/carbon composites under visible light. Small 2023, 19, 2303813. [CrossRef]
- Peng, G.; Wu, J.; Wang, M.; Niklas, J.; Zhou, H.; Liu, C. Nitrogen-defective polymeric carbon nitride nanolayer enabled efficient electrocatalytic nitrogen reduction with high faradaic efficiency. Nano Letters 2020, 20, 2879-2885. [CrossRef]
- Tan, L.; Nie, C.; Ao, Z.; Sun, H.; An, T.; Wang, S. Novel two-dimensional crystalline carbon nitrides beyond gC 3 N 4: structure and applications. Journal of Materials Chemistry A 2021, 9, 17-33.
- Shamim, M.; Perveen, M.; Nazir, S.; Hussnain, M.; Mehmood, R.; Khan, M.I.; Iqbal, J. DFT study of therapeutic potential of graphitic carbon nitride (g-C3N4) as a new drug delivery system for carboplatin to treat cancer. Journal of Molecular Liquids 2021, 331, 115607. [CrossRef]
- Perveen, M.; Aslam, F.; Nazir, S.; Khan, M.I.; Zahra, G.; Iqbal, J. DFT study of therapeutic potential of graphitic carbon nitride as a carrier for controlled release of melphalan: an anticancer drug. Journal of Molecular Modeling 2022, 28, 359. [CrossRef]
- Xu, X.; Zhang, X.; He, H.; Dai, L.; Hu, J.; Si, C. Graphitic carbon nitride enters the scene: A promising versatile tool for biomedical applications. Langmuir 2024, 40, 15389-15406. [CrossRef]
- Bian, C.; Wang, Y.; Yi, Y.; Shao, S.; Sun, P.; Xiao, Y.; Wang, W.; Dong, X. Enhanced photocatalytic activity of S-doped graphitic carbon nitride hollow microspheres: Synergistic effect, high-concentration antibiotic elimination and antibacterial behavior. Journal of Colloid and Interface Science 2023, 643, 256-266. [CrossRef]
- Dong, J.; Zhao, Y.; Wang, K.; Chen, H.; Liu, L.; Sun, B.; Yang, M.; Sun, L.; Wang, Y.; Yu, X. Fabrication of graphitic carbon nitride quantum dots and their application for simultaneous fluorescence imaging and pH-responsive drug release. ChemistrySelect 2018, 3, 12696-12703. [CrossRef]
- Asif, K.; Perveen, M.; Khera, R.A.; Nazir, S.; Ayub, A.R.; Asif, T.; Shabbir, M.; Iqbal, J. Computational and theoretical study of graphitic carbon nitride (g-C3N4) as a drug delivery carrier for lonidamine drug to treat cancer. Computational and Theoretical Chemistry 2021, 1206, 113459. [CrossRef]
- Li, X. Aluminium nitride as an efficient catalyst in the synthesis of some chromeno [4, 3-b] chromenes and potential nanocarrier for delivery of flutamide. Chemical Papers 2024, 78, 1157-1166.
- Chen, W.; Liu, J.; Wang, Y.; Jiang, C.; Yu, B.; Sun, Z.; Lu, L. A C5N2 nanoparticle based direct nucleus delivery platform for synergistic cancer therapy. Angewandte Chemie 2019, 131, 6356-6360.
- Chuchev, K.; BelBruno, J. Electronic structure of C5N2, C6N2, and isoelectronic molecules. The Journal of Physical Chemistry A 2003, 107, 1887-1890.
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, version 6.0. 16. Semichem Inc Shawnee Mission KS 2016.
- Yaghoubi, A.; Ramazani, A. Using Gaussian and GaussView software for effective teaching of chemistry by drawing molecules. Research in Chemistry Education 2024, 6, 69-90.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01, Wallingford, CT, 2016.
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. Journal of Chemical Physics 1999, 110, 6158-6170. [CrossRef]
- Marsman, M.; Paier, J.; Stroppa, A.; Kresse, G. Hybrid functionals applied to extended systems. Journal of Physics: Condensed Matter 2008, 20, 064201. [CrossRef]
- Adamo, C.; Cossi, M.; Scalmani, G.; Barone, V. Accurate static polarizabilities by density functional theory: assessment of the PBE0 model. Chemical Physics Letters 1999, 307, 265-271. [CrossRef]
- Melissen, S.; Le Bahers, T.; Steinmann, S.N.; Sautet, P. Relationship between Carbon Nitride Structure and Exciton Binding Energies: A DFT Perspective. The Journal of Physical Chemistry C 2015, 119, 25188-25196. [CrossRef]
- Tuma, C.; Boese, A.D.; Handy, N.C. Predicting the binding energies of H-bonded complexes: A comparative DFT study. Physical Chemistry Chemical Physics 1999, 1, 3939-3947. [CrossRef]
- Lin, T.-J.; Chiu, C.-c. Influence of nonmetal dopants on charge separation of graphitic carbon nitride by time-dependent density functional theory. Physical Chemistry Chemical Physics 2020, 22, 647-657. [CrossRef]
- Veccham, S.P.; Head-Gordon, M. Density functionals for hydrogen storage: Defining the H2Bind275 test set with ab initio benchmarks and assessment of 55 functionals. Journal of chemical theory and computation 2020, 16, 4963-4982. [CrossRef]
- Galimov, E.R.; Kostjukov, V.V. Computational analysis of photoisomerization of unsubstituted spirooxazine by TD-DFT: solvent effect and functional choice. Theoretical Chemistry Accounts 2023, 143, 2. [CrossRef]
- Chong, D.P. Theoretical Study of Structures and Spectra of Small Anticancer Drugs: Fluorouracil, Hydroxyurea, and Tirapazamine. Journal of the Chinese Chemical Society 2016, 63, 109-120. [CrossRef]
- Zheng, Y.; Shan, K.; Zhang, Y.; Gu, W. Amino acid-functionalized borospherenes as drug delivery systems. Biophysical Chemistry 2020, 263, 106407. [CrossRef]
- de Almeida, C.A.; Pinto, L.P.; Dos Santos, H.F.; Paschoal, D.F. Vibrational frequencies and intramolecular force constants for cisplatin: assessing the role of the platinum basis set and relativistic effects. Journal of Molecular Modeling 2021, 27, 1-13. [CrossRef]
- Ugurlu, S. Investigation of metallacages for cisplatin encapsulation using Density Functional Theory (DFT); 2024.
- Chu, Y.-C.; Lin, T.-J.; Lin, Y.-R.; Chiu, W.-L.; Nguyen, B.-S.; Hu, C. Influence of P,S,O-Doping on g-C3N4 for hydrogel formation and photocatalysis: An experimental and theoretical study. Carbon 2020, 169, 338-348. [CrossRef]
- Ibarra-Rodríguez, M.; Sánchez, M. Adsorption of metformin on graphitic carbon nitride functionalized with metals of group 1–3 (Li, Na, K, Be, Mg, Ca, B, Al, and Ga), DFT calculations. Computational and Theoretical Chemistry 2022, 1207, 113532.
- Ibarra-Rodríguez, M.; Sánchez, M. Graphitic carbon nitride functionalized with four boron atoms for adsorption and separation of CO2/CH4: DFT calculations. Adsorption 2020, 26, 597-605.
- Ibarra-Rodríguez, M.; Sánchez, M. Adsorption of H2, N2, CO, H2S, NH3, SO2 and CH4 on Li-functionalized graphitic carbon nitride investigated by density functional theory. Bulletin of Materials Science 2020, 43, 144. [CrossRef]
- Gorai, D.K.; Kundu, T.K. Platinum-silicon doped graphitic carbon nitride: A first principle calculation. Physica B: Condensed Matter 2022, 627, 413547. [CrossRef]
- Pauly, M.; White, E.; Deegbey, M.; Fosu, E.A.; Keller, L.; McGuigan, S.; Dianat, G.; Gabilondo, E.; Wong, J.C.; Murphey, C.G.E.; et al. Coordination of copper within a crystalline carbon nitride and its catalytic reduction of CO2. Dalton Transactions 2024, 53, 6779-6790. [CrossRef]
- Ali, B.; Siddique, S.A.; Ahmed Siddique, M.B.; Ullah, S.; Ali, M.A.; Rauf, A.; Kamran, M.A.; Arshad, M. Insight on the structural, electronic and optical properties of Zn, Ga-doped/dual-doped graphitic carbon nitride for visible-light applications. Journal of Molecular Graphics and Modelling 2023, 125, 108603. [CrossRef]
- Liu, W.-W.; Niu, S.-T.; Xu, Z.-Q.; Zou, R.; Cui, C.-Y.; Lei, Y.-X.; Zhang, X.-B.; Ran, F. Highly-dispersed nickel on 2D graphitic carbon nitrides (g-C3N4) for facilitating reaction kinetics of lithium-sulfur batteries. Applied Surface Science 2023, 609, 155327. [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. Journal of computational chemistry 2011, 32, 1456-1465. [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. The Journal of chemical physics 2010, 132.
- Legault, C. CYLview, 1.0 b, Université de Sherbrooke. Sherbrooke, QC: http://www. cylview. org 2009.
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: a library for package-independent computational chemistry algorithms. J Comput Chem 2008, 29, 839-845. [CrossRef]
- Lu, T.; Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 2012, 33, 580-592. [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: visual molecular dynamics. Journal of molecular graphics 1996, 14, 33-38. [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. Journal of Chemical Theory and Computation 2011, 7, 625-632. [CrossRef]
- Vargas, R.; Garza, J.; Martínez, A.; Ibarra, I.A. Computational tools to study non-covalent interactions and confinement effects in chemical systems. Chemical Communications 2024, 60, 3008-3018. [CrossRef]
- Hajji, M.; Abad, N.; Habib, M.A.; Elmgirhi, S.M.H.; Guerfel, T. Computational chemistry methods for modelling non-covalent interactions and chemical reactivity— An overview. Journal of the Indian Chemical Society 2021, 98, 100208. [CrossRef]
- Popelier, P.L.A. The QTAIM Perspective of Chemical Bonding. In The Chemical Bond; 2014; pp. 271-308.
- Kumar, P.S.V.; Raghavendra, V.; Subramanian, V. Bader’s Theory of Atoms in Molecules (AIM) and its Applications to Chemical Bonding. Journal of Chemical Sciences 2016, 128, 1527-1536. [CrossRef]
- Foroutan-Nejad, C.; Shahbazian, S.; Marek, R. Toward a Consistent Interpretation of the QTAIM: Tortuous Link between Chemical Bonds, Interactions, and Bond/Line Paths. Chemistry - A European Journal 2014, 20, 10140–10152. [CrossRef]
- Wick, C.R.; Clark, T. On bond-critical points in QTAIM and weak interactions. Journal of Molecular Modeling 2018, 24, 142. [CrossRef]
- Phillips, J.C. Generalized Koopmans’ Theorem. Physical Review 1961, 123, 420-424.
- Hackett, J.C. Chemical Reactivity Theory: A Density Functional View. Journal of the American Chemical Society 2010, 132, 7558-7558. [CrossRef]
- Parr, R.G.; Szentpály, L.v.; Liu, S. Electrophilicity index. Journal of the American Chemical Society 1999, 121, 1922-1924.
- Bendjeddou, A.; Abbaz, T.; Gouasmia, A.; Villemin, D. Quantum chemical studies on molecular structure and reactivity descriptors of some P-nitrophenyl tetrathiafulvalenes by density functional theory (DFT). Acta Chim. Pharm. Indica 2016, 6, 32-44.
- de Miranda, D.B.; Quintal, S.; Ferreira, G.B. Electronic analysis of n-propyl xanthate complexes with group 12 metals: a theoretical–experimental study. Journal of Molecular Modeling 2024, 30, 1-13. [CrossRef]
- Shilpa, D.; Sadasivam, K.; Thirumoorthy, M. Topology analysis of six phytochemicals through ELF and LOL basins–A DFT study. Indian Journal of Chemistry (IJC) 2023, 62, 1171-1177. [CrossRef]
- Jumabaev, A.; Koyambo-Konzapa, S.-J.; Hushvaktov, H.; Absanov, A.; Khudaykulov, B.; Holikulov, U.; Ernazarov, Z.; Issaoui, N.; Al-Dossary, O.M.; Nsangou, M. Intermolecular interactions in water and ethanol solution of ethyl acetate: Raman, DFT, MEP, FMO, AIM, NCI-RDG, ELF, and LOL analyses. Journal of Molecular Modeling 2024, 30, 349. [CrossRef]
- Weinhold, F. Natural bond orbital analysis: A critical overview of relationships to alternative bonding perspectives. Journal of Computational Chemistry 2012, 33, 2363-2379. [CrossRef]
- Koch, D.; Pavanello, M.; Shao, X.; Ihara, M.; Ayers, P.W.; Matta, C.F.; Jenkins, S.; Manzhos, S. The Analysis of Electron Densities: From Basics to Emergent Applications. Chemical Reviews 2024, 124, 12661-12737. [CrossRef]
- Leppert, J.r.; Urbinati, C.R.; Häfner, S.; Ohlenschläger, O.; Swanson, M.S.; Görlach, M.; Ramachandran, R. Identification of NH...N hydrogen bonds by magic angle spinning solid state NMR in a double-stranded RNA associated with myotonic dystrophy. Nucleic Acids Research 2004, 32, 1177-1183. [CrossRef]
- Pakiari, A.; Eskandari, K. The chemical nature of very strong hydrogen bonds in some categories of compounds. Journal of Molecular Structure: THEOCHEM 2006, 759, 51-60. [CrossRef]
- Grabowski, S.J. Hydrogen Bond – Definitions, Criteria of Existence and Various Types. In Understanding Hydrogen Bonds; The Royal Society of Chemistry: 2020; p. 0.
- Schiemenz, G.P. The sum of van der Waals radii–A pitfall in the search for bonding. Zeitschrift für Naturforschung B 2007, 62, 235-243. [CrossRef]
- Kaviani, S.; Shahab, S.; Sheikhi, M.; Potkin, V.; Zhou, H. A DFT study of Se-decorated B12N12 nanocluster as a possible drug delivery system for ciclopirox. Computational and Theoretical Chemistry 2021, 1201, 113246. [CrossRef]
- Nayini, M.M.R.; Sayadian, H.; Razavipour, N.; Rezazade, M. Chemical-sensing of Amphetamine drug by inorganic AlN nano-cage: A DFT/TDDFT study. Inorganic Chemistry Communications 2020, 121, 108237. [CrossRef]
- Kalhor, S.; Fattahi, A. Combinatorial MD/QM studies to develop novel ionic liquid-based anticancer drug delivery systems with aminium derived from carbohydrates as cationic components. Scientific Reports 2024, 14, 28980. [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. The Journal of Physical Chemistry B 2009, 113, 6378-6396. [CrossRef]
- Oğuz, I.C.; Vassetti, D.; Labat, F. Assessing the performances of different continuum solvation models for the calculation of hydration energies of molecules, polymers and surfaces: a comparison between the SMD, VASPsol and FDPB models. Theoretical Chemistry Accounts 2021, 140, 99. [CrossRef]
- Gwee, E.S.; Seeger, Z.L.; Appadoo, D.R.; Wood, B.R.; Izgorodina, E.I. Influence of DFT Functionals and Solvation Models on the Prediction of Far-Infrared Spectra of Pt-Based Anticancer Drugs: Why Do Different Complexes Require Different Levels of Theory? ACS omega 2019, 4, 5254-5269. [CrossRef]
- Zafar, A.; Reynisson, J. Hydration free energy as a molecular descriptor in drug design: a feasibility study. Molecular Informatics 2016, 35, 207-214. [CrossRef]
- Bibi, S.; Ur-Rehman, S.; Khalid, L.; Bhatti, I.A.; Bhatti, H.N.; Iqbal, J.; Bai, F.Q.; Zhang, H.-X. Investigation of the adsorption properties of gemcitabine anticancer drug with metal-doped boron nitride fullerenes as a drug-delivery carrier: a DFT study. RSC advances 2022, 12, 2873-2887. [CrossRef]
Figure 1.
Optimized structure of the C5N2 sheet at PBE0-D3BJ/def2-SVP level of theory. The central cavity (a), benzene ring (b), pyrazine ring (c), hydrogenated benzene ring (d), and a pseudo-cyclic triazine (e) are highlighted as possible active sites of the C5N2 sheet. Grey, blue, and bluish white colors represent carbon (C), nitrogen(N), and hydrogen (H) atoms, respectively.
Figure 1.
Optimized structure of the C5N2 sheet at PBE0-D3BJ/def2-SVP level of theory. The central cavity (a), benzene ring (b), pyrazine ring (c), hydrogenated benzene ring (d), and a pseudo-cyclic triazine (e) are highlighted as possible active sites of the C5N2 sheet. Grey, blue, and bluish white colors represent carbon (C), nitrogen(N), and hydrogen (H) atoms, respectively.
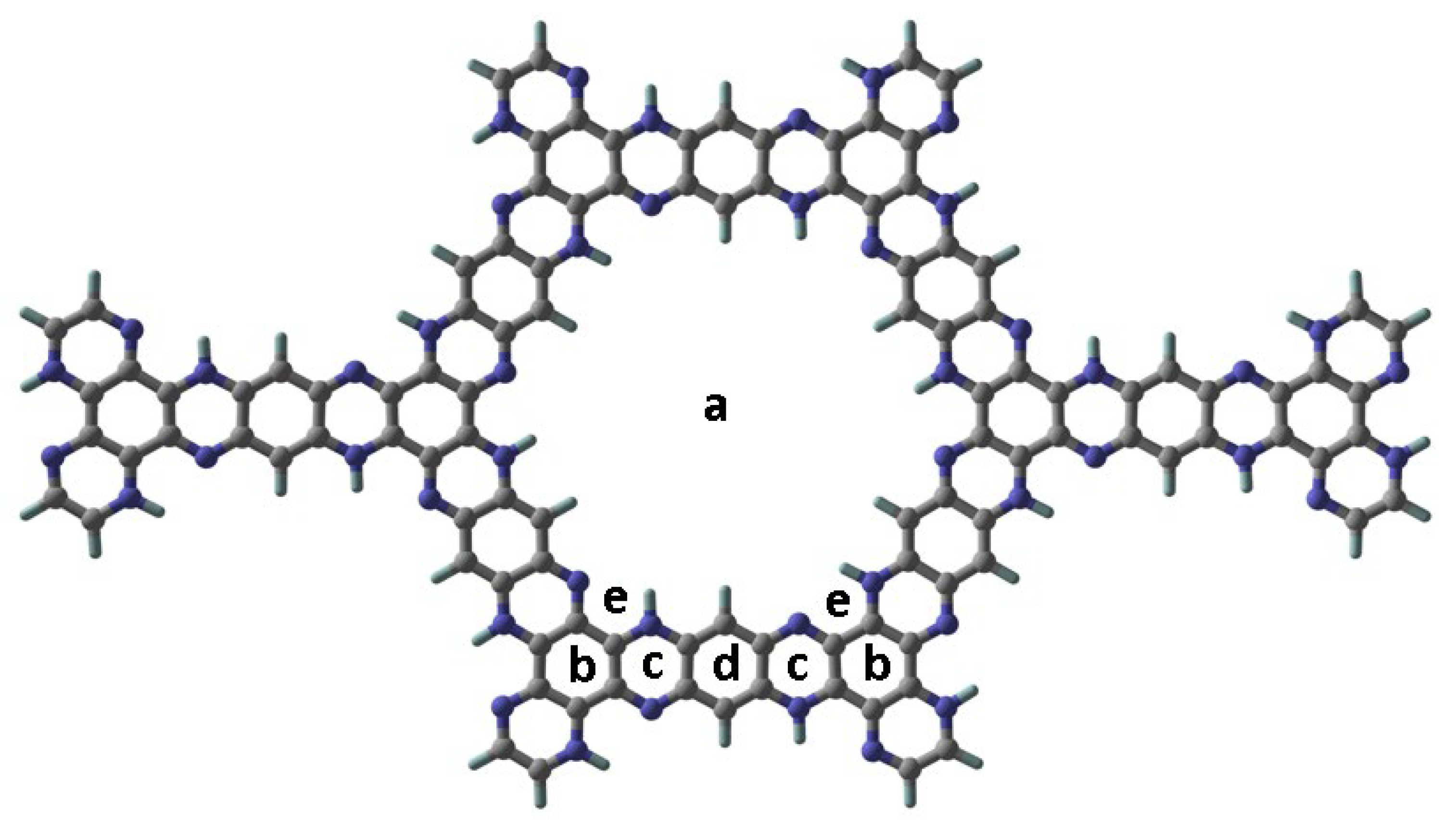
Figure 3.
3D isosurface and 2D RDG plot highlighting non-covalent interactions between the drugs and the C5N2 substrate.
Figure 3.
3D isosurface and 2D RDG plot highlighting non-covalent interactions between the drugs and the C5N2 substrate.
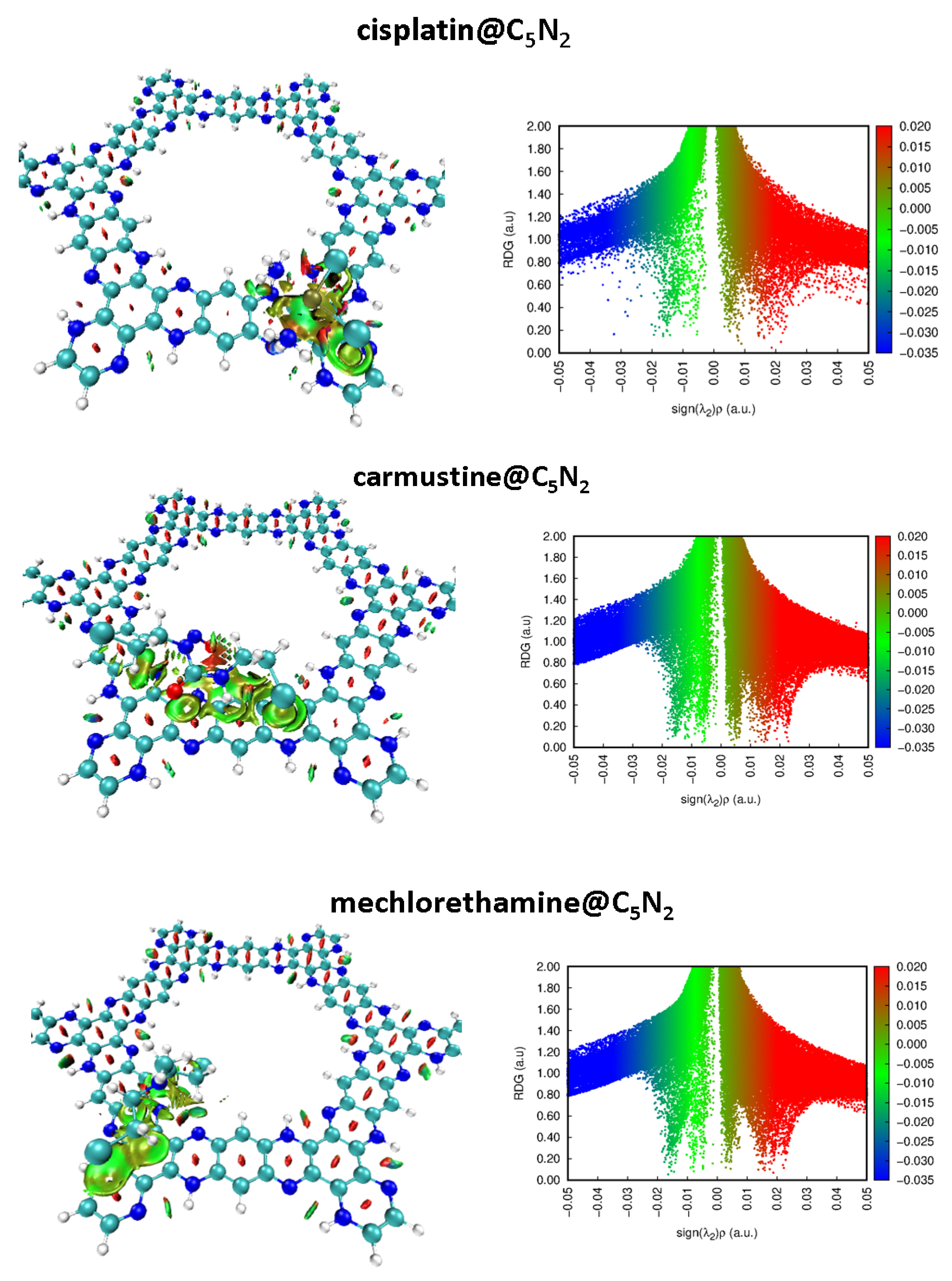
Figure 5.
Visual depiction of ELF plots of the bare C5N2 sheet, and the drug@C5N2 complexes.
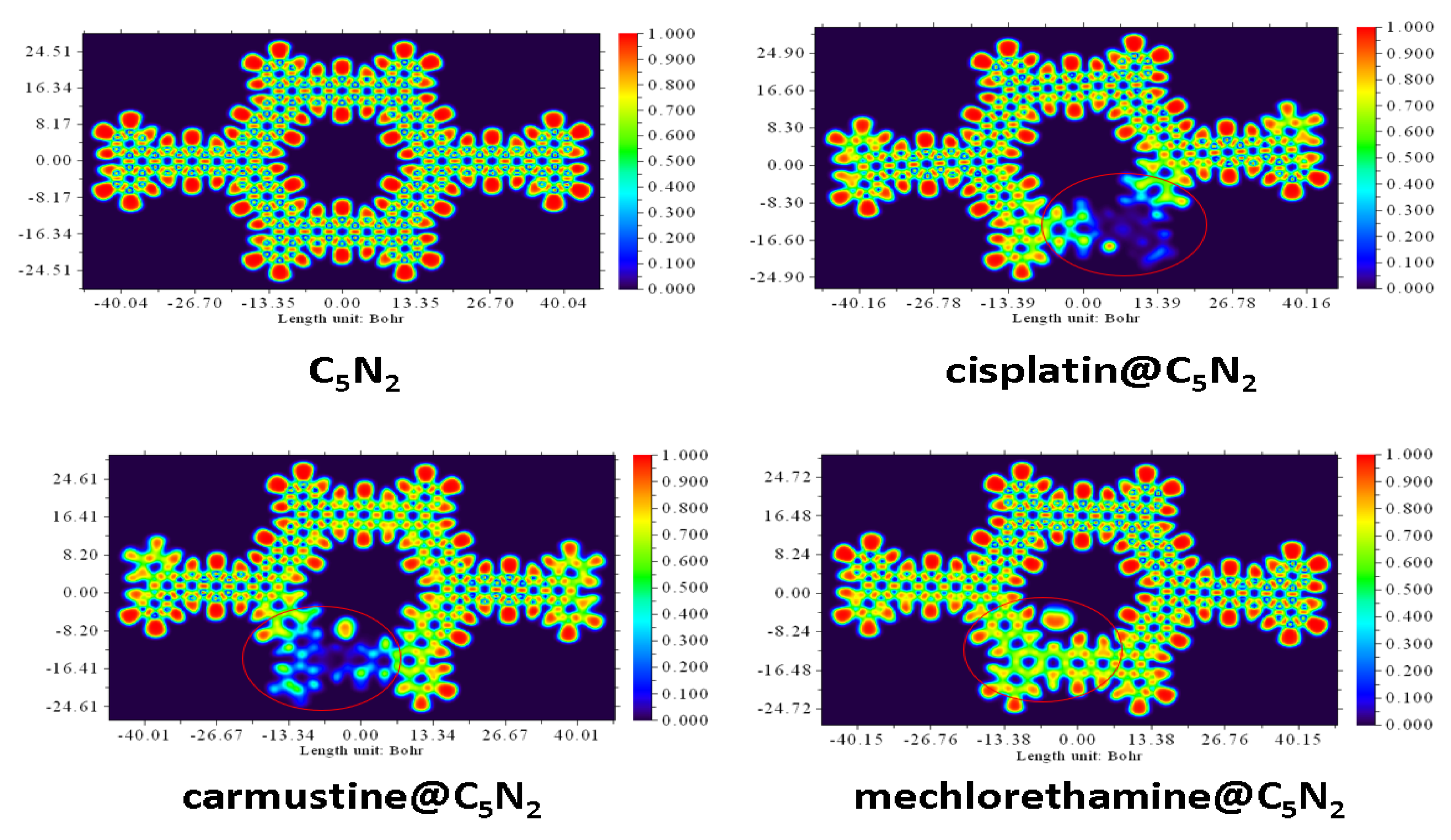
Figure 7.
Graphical illustration of DOS spectra highlighting the HOMOLUMO energy gap of the bare C5N2 sheet and drug@C5N2 complexes.
Figure 7.
Graphical illustration of DOS spectra highlighting the HOMOLUMO energy gap of the bare C5N2 sheet and drug@C5N2 complexes.
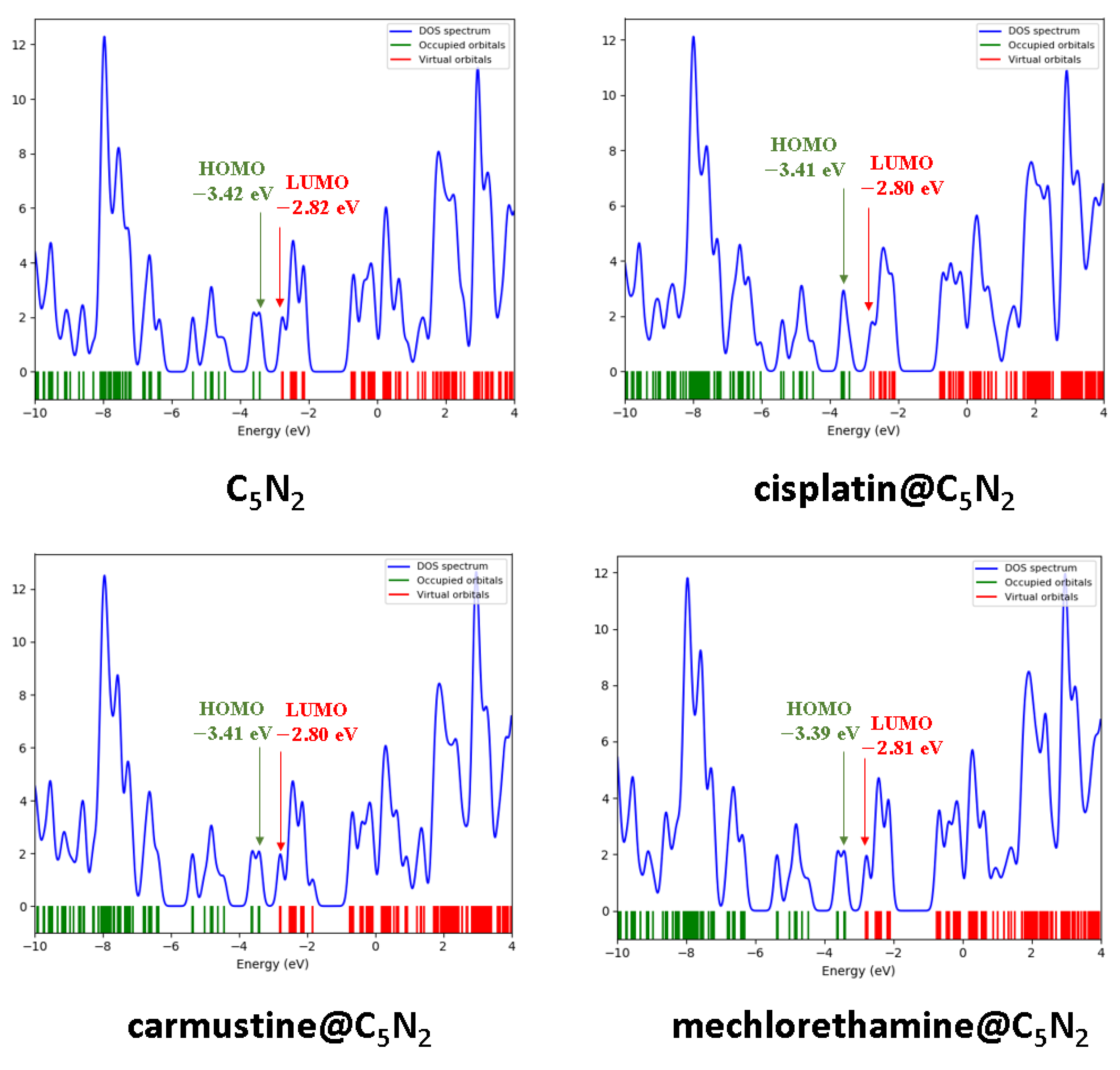
Figure 8.
The portrayal of the electron density distribution of cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes. The red color represents the electron density accumulated region while the blue color is related to the electron density depleted region.
Figure 8.
The portrayal of the electron density distribution of cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes. The red color represents the electron density accumulated region while the blue color is related to the electron density depleted region.
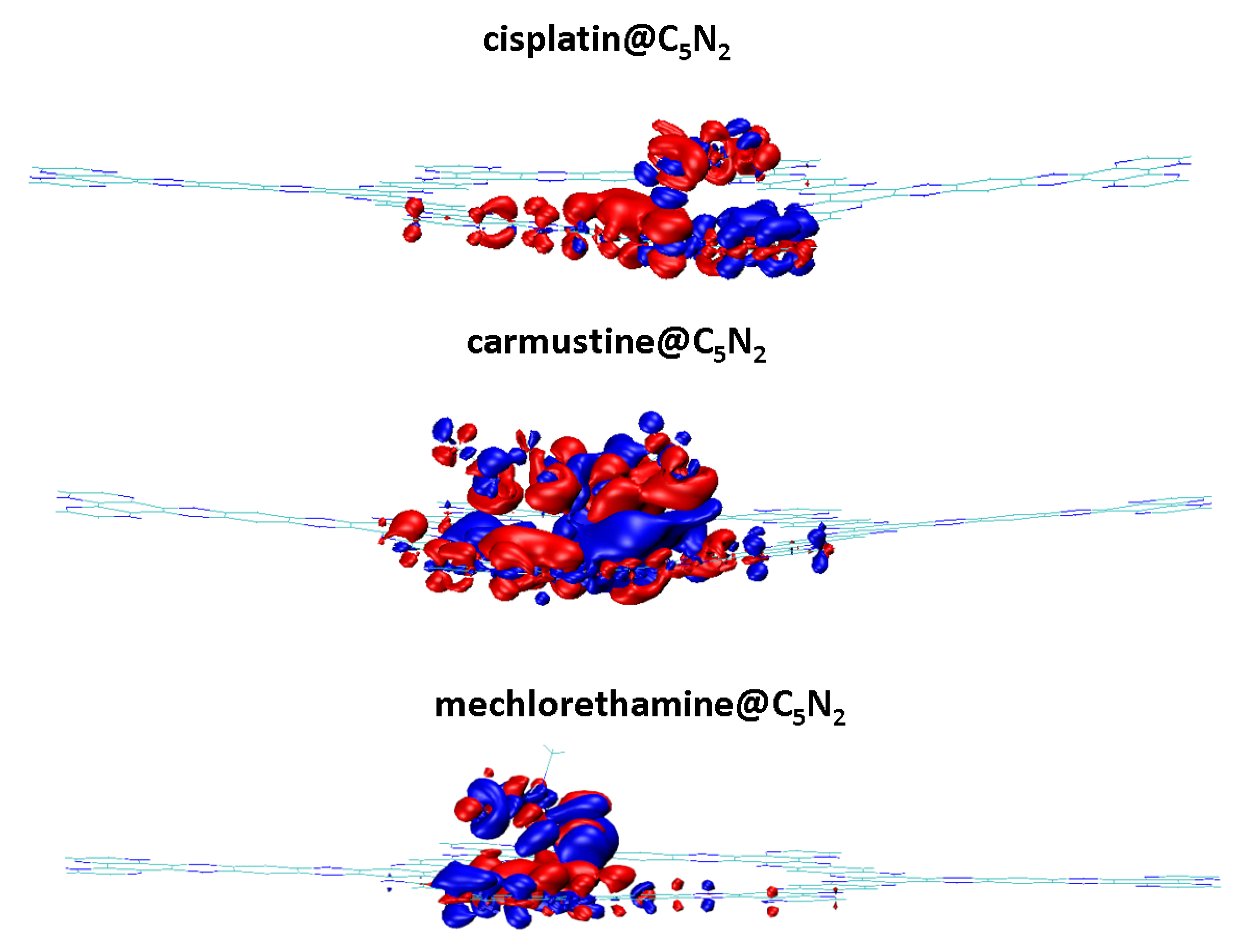
Table 2.
QTAIM topological parameters including electron density (), the Laplacian of electron density , the kinetic energy density (G), the potential energy density (V), the total energy density (H) and theV/G ratio of the cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes.
Table 2.
QTAIM topological parameters including electron density (), the Laplacian of electron density , the kinetic energy density (G), the potential energy density (V), the total energy density (H) and theV/G ratio of the cisplatin@C5N2, carmustine@C5N2, and mechlorethamine@C5N2 complexes.
| Drugs@C5N2 | drug | ρ(a.u.) |
(a.u.) |
G (a.u.) |
V (a.u.) |
H (a.u.) |
V/G |
|---|---|---|---|---|---|---|---|
| cisplatin@C5N2 | C2H1 N4H3 C6Pt5 C8Cl7 C10Cl9 |
0.0137 0.0329 0.0123 0.0075 0.0082 |
0.037 0.087 0.034 0.022 0.025 |
0.008 0.021 0.007 0.004 0.005 |
0.008 0.021 0.007 0.004 0.004 |
0.0004 0.0002 0.0007 0.0007 0.0008 |
0.95 0.98 0.90 0.83 0.85 |
| carmustine@C5N2 | C13H1 C14H2 C12O3 H11O4 H10O4 C9H5 C8Cl6 N7Cl6 |
0.0058 0.0030 0.0078 0.0100 0.0050 0.0070 0.0063 0.0064 |
0.018 0.016 0.026 0.041 0.025 0.023 0.022 0.021 |
0.003 0.004 0.005 0.009 0.004 0.004 0.004 0.004 |
0.002 0.003 0.004 0.008 0.003 0.003 0.003 0.004 |
0.0008 0.0006 0.0008 0.0009 0.0012 0.0009 0.0008 0.0005 |
0.75 0.79 0.85 0.89 0.73 0.79 0.80 0.87 |
| mechlorethamine@C5N2 | C6Cl1 C7H2 C8H3 N9H4 N10H5 |
0.0081 0.0082 0.0048 0.0091 0.0170 |
0.026 0.022 0.015 0.028 0.045 |
0.005 0.005 0.003 0.006 0.011 |
0.004 0.004 0.002 0.005 0.011 |
0.0009 0.0005 0.0007 0.0005 0.0001 |
0.84 0.90 0.77 0.90 0.01 |
Table 3.
Energies of the HOMO and LUMO of the drug@C5N2 complexes with their energy gap (eV), and corresponding chemical reactivity descriptors including chemical potential (μ), electrophilicity index (ω), hardness (η), softness (Ѕ) and their respective NBO charges.
Table 3.
Energies of the HOMO and LUMO of the drug@C5N2 complexes with their energy gap (eV), and corresponding chemical reactivity descriptors including chemical potential (μ), electrophilicity index (ω), hardness (η), softness (Ѕ) and their respective NBO charges.
| Complexes | EHOMO (eV) |
ELUMO (eV) |
(eV) |
μ (eV) |
ω (eV) |
η (eV) |
Ѕ (eV) |
) |
|---|---|---|---|---|---|---|---|---|
| C5N2 | 3.42 | 2.82 | 0.60 | 3.12 | 16.23 | 0.30 | 1.67 | |
| cisplatin@C5N2 | 3.41 | 2.80 | 0.57 | 3.09 | 17.68 | 0.27 | 1.65 | 0.039 |
| carmustine@C5N2 | 3.41 | 2.80 | 0.57 | 3.09 | 17.68 | 0.27 | 1.65 | 0.031 |
| mechlorethamine@C5N2 | 3.39 | 2.81 | 0.58 | 3.10 | 16.51 | 0.29 | 1.72 | 0.479 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



