
Submitted:
22 December 2024
Posted:
23 December 2024
You are already at the latest version
Abstract
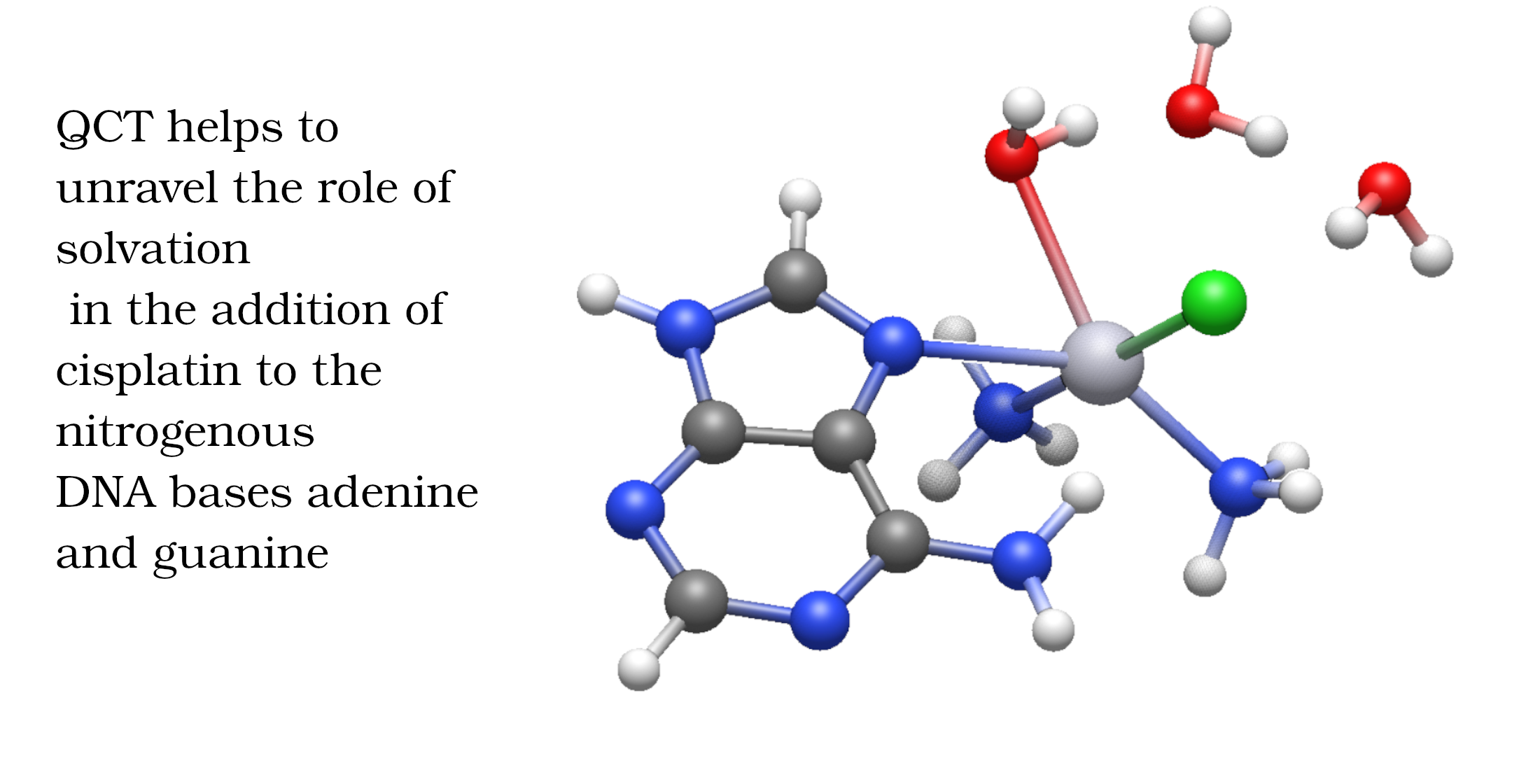
Cisplatin is still a first-line agent in cancer treatment due to its effectiveness. Despite the large body of research concerning this drug, the role of explicit water molecules in its mechanism remains uncertain. We addressed the addition of cisplatin with the nitrogenous DNA bases adenine and guanine, with an emphasis on the impact of explicit microsolvation on every step of the action pathway of this pharmaceutical. We used electronic structure calculations to explore the energetics of the key reactions of this mechanism. We also exploited state-of-the-art methods of wave function analyses, namely the Quantum Theory of Atoms in Molecules and the Interacting Quantum Atoms partition, to explore the chemical bonding throughout such steps. Our results reveal that microsolvation significantly affects electronic and Gibbs free activation energies differently as previously reported (F.P. Cossio et. al. ChemPhysChem, 17, 3932, 2016). The changes in activation energies are consistent with Hammond’s postulate in terms of the changes of the chemical bonding scenario between reactants and transition states. Overall, we provide an in-depth description of the importance of the surrounding water molecules of cisplatin, which aids in understanding the mechanism of pharmaceuticals in the pursuit of more effective cancer treatments.
Keywords:
cisplatin action pathway
; implicit solvation model
; surrounding water molecules
; electronic and Gibbs free activation energies
; quantum chemical topology
; quantum theory of atoms in molecules
; interacting quantum atoms
; delocalisation indices
1. Introduction
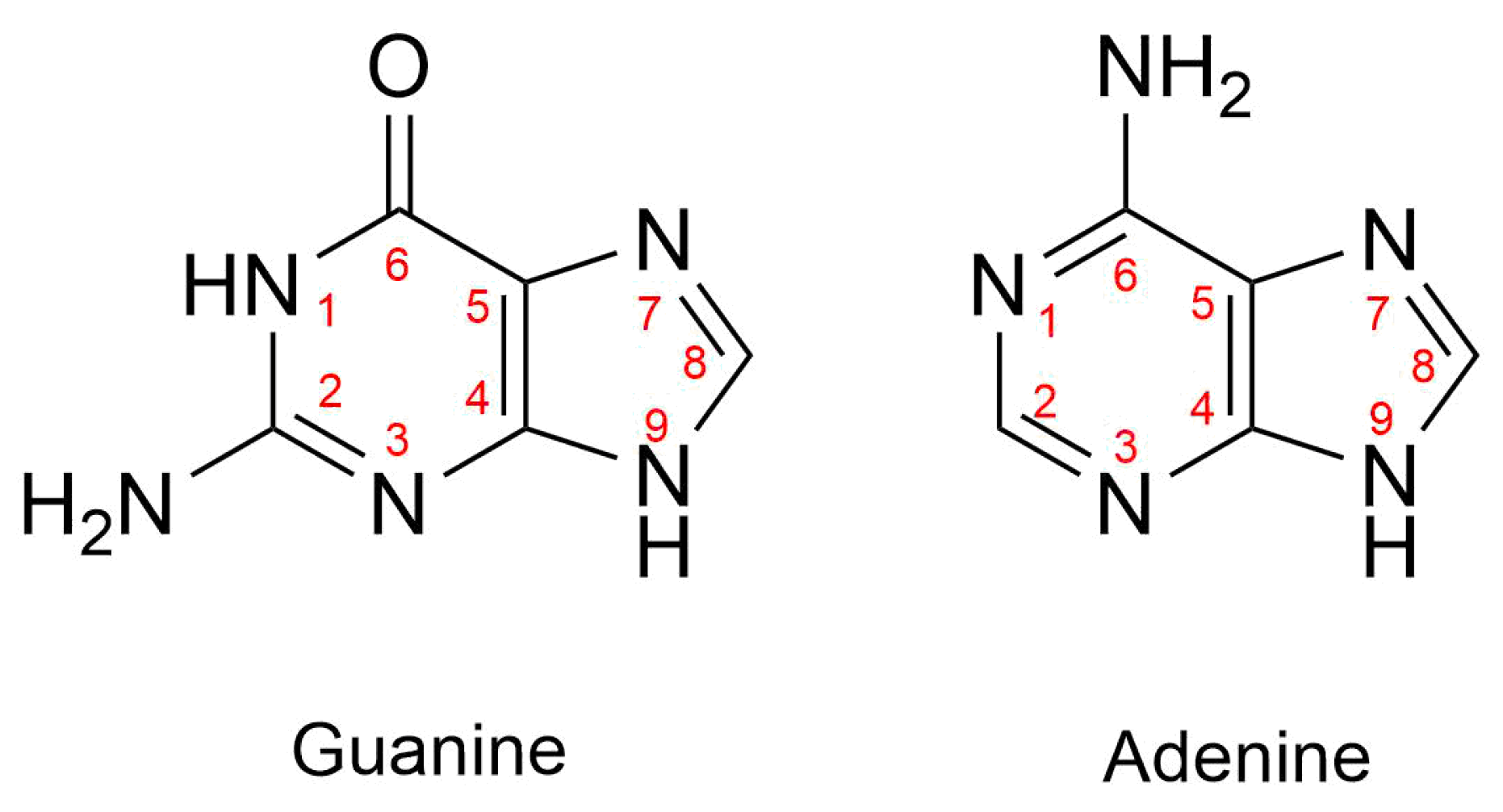
Cisplatin, cis-Pt(NH3)2Cl2 (1), is one of the quintessential drugs in cancer treatment [1]. Since its discovery in 1965 [2], it has been be widely used in the fight against various types of cancer such as testicular [3], ovarian [4], lung [5], prostate [6], among many others [7]. For almost six decades, the elucidation of its mechanism of action has been a topic of interest from experimental [1] and theoretical [8] perspectives. Cisplatin is a platinum (II) metal complex with a square planar geometry. This complex, however, does not react directly with DNA [9]. It must first undergo hydration reactions, in which it loses one or both chloride anions (Figure 1). These transformations occur via a characteristic substitution reaction mechanism of square planar complexes [10], wherein the transition state has a pentacoordinate trigonal bipyramid geometry [11]. It is still not clear which of the activated complexes, cis-diamminechloroaquaPt(II) (2) or cis-diamminediaquaPt(II) (3) binds directly to the DNA bases[12]. This addition is the rate-limiting step of the whole process. Previous in vitro [13] and computational [14] studies indicated the propensity of cisplatin to bind to the N7 of adenine and guanine nitrogenous bases (Figure 2).
Despite the large body of research about cisplatin, there are still unsolved issues concerning its reaction mechanism with DNA. For example, the role of the solvation water molecules in this mechanism is still unclear. Given the biological relevance of the action pathway of drugs, the availability of a hydration model capable of reliably reproducing experimental values is often pivotal. Due to this circumstance, there are studies that compare distinct solvation methods for biologically relevant reactions. For example, Lakbaibi et. al. evaluated the role of explicit solvation in the Darzens reaction, one of the most useful synthetic procedures to form epoxides, as well as natural and pharmacological products [15]. These authors found that explicit solvation significantly reduces the values of Gibbs free activation energies values. This result was attributed to the capacity of the explicit solvation model to account for crucial hydrogen-bonding interactions, which are not properly described by implicit models. In another study, Wang and Cao [16] investigated the hydration of CO2 with both explicit and implicit methods of microhydration. Their results show that explicit solvation is critical for a reasonable description of the proton relay mechanism which occurs in this system. Alberto et. al [17]. addressed the hydrolysis mechanism of a platinum derivative anticancer drug [18], Nedaplatin, with an implicit solvation model and the influence of an extra explicit water molecule. These authors concluded that the explicit water molecule incorporates the effects of hydrogen bonding and provides a more accurate description of the stabilisation of the leaving group. More specifically for the investigation reported herein, there are still unsolved issues regarding the role of microsolvation in the mechanism of cisplatin in spite of the considerable interest about this pharmaceutical. A related study on this matter [19] highlighted the significant influence of explicit water molecules for the underlying interactions governing the addition reaction mechanisms of cisplatin with DNA bases. In this investigation, Cossio et. al. concluded that the incorporation of explicit water molecules reduces the Gibbs free activation energy in every step of the action mechanism of cisplatin. The utilisation of explicit microhydration led to computational results which agree closely with experimental observations [19]. Unfortunately, the consideration of electronic energies without explicit H2O solvation molecules in this same study yields questionable negative activation energies for the second hydration of cisplatin [19]. Besides, the examination of interatomic distances of some reactants and the corresponding transition states indicates that the water molecules surrounding the reactive system interact more strongly with the reactants than with the transition state. Hence, one would expect that the activation energy of such steps of the action mechanism of cisplatin increases as a result of the consideration of explicit microhydration, as opposed to the conclusions reported by Cossio et al. [19]. Therefore, we decided to revisit the effect of explicit microhydration on the biological mechanism of cisplatin. For this purpose, we also exploited state-of-the-art methods of wave function analyses to get further insights about the changes in the chemical bonding scenario in these reactive systems due to the incorporation of explicit H2O solvation molecules. It is well known, indeed, that non-covalent interactions play a fundamental role in a drug mechanism of action [20]. Since the hydration of cisplatin is the key step for the activation of this drug, it is vital to understand the role of hydrogen bonding in this reaction. Indeed, we have previously investigated in detail the role of H-bonds in the catalysis of chemical reactions [21,22]. More specifically for the matter at hand, Robertazzi et al. [23], considered thermodynamical properties and effects of explicit solvation in the first hydration shell of cisplatin using electronic structure calculations and the Quantum Theory of Atoms in Molecules (QTAIM). As opposed to the conclusions of Cossio et. al. [19], these authors found that explicit solvation substantially distorts the cisplatin geometry and leads to higher activation energies while forming a solvation sphere with ten explicit water molecules.
In view of this background, we aim to elucidate the role of non-covalent interactions in the reactivity of cisplatin across its biological mechanism with the aid of wave function analyses. We also focused on the chemical bonds, which are formed and destroyed throughout the whole reaction mechanism of cisplatin, based on electronic structure theory and Quantum Chemical Topology (QCT) methods, namely QTAIM and the Interacting Quantum Atoms (IQA) energy partition. In short, we address in this study: (i) the steps of the biological mechanism of cisplatin and their corresponding activation energies and (ii) the influence of explicit water molecules on the chemical bonding of the reactants and transition states, for a better understanding of their effect on the activation energies along the biological mechanism of cisplatin. Electronic structure and QCT calculations indicate that the interaction of the explicit solvation water molecules stabilises to a greater extent (i) reactants or (ii) transition states, leading to an increase or decrease of the corresponding activation energy, respectively. Finally, we observe the changes in the chemical bonding scenario of leaving and entering groups throughout the biological mechanism of cisplatin via delocalisation indices as definided by QTAIM. Overall, this paper illustrates how the combination of suitable electronic structure calculations and QCT analyses provides (i) a well-suited assessment of the effects of explicit solvation water molecules on activation energies and (ii) valuable insights into such effects in terms of changes in the chemical bonding scenario within the actin pathway of cisplatin.
Figure 2.
Numbering of atoms in guanine and adenine. We emphasise that cisplatin binds N7 when it interacts with either of these nitrogenous bases.
Figure 2.
Numbering of atoms in guanine and adenine. We emphasise that cisplatin binds N7 when it interacts with either of these nitrogenous bases.
2. Theoretical Framework
In this section we briefly discuss the QCT methods used in this work, namely the QTAIM and IQA methods of wave function analyses. The QTAIM approach leads to a partition of the 3D-space into disjoints regions , , ...identified with the atoms of chemistry [24]. The QTAIM defines expectation values of quantum-mechanical observables within these atomic basins. These observables include energy and multipole moments. For example, the QTAIM atomic charge is defined as,
wherein is the atomic number of the nucleus inside , is the electronic charge distribution and is the expectation value of the number of electrons within . Topological analyses of the electronic density can be performed in terms of critical points of , i.e., points where ∇ vanishes. The QTAIM allows to compute the number of electrons shared between two atoms, denoted as Delocalization Index (DI),
in which is the covariance between the expectation value of the number of electrons in and . The value of yields a measure of the exchange of electrons between and , the distinctive feature of covalency.
On the other hand, the IQA energy partition can be used to divide the electronic energy of a system into net, , and interatomic, , contributions,
The quantity is the net energy of atom A, while the value of is the interaction energy between atoms and . In turn, we can split the IQA interaction energy into a classical, , and an exchange-correlation, component, i.e.,
wherein and are respectively related to ionic and covalent contributions to the chemical bond between basins and . These quantities can be approximated using QTAIM charges and DIs [25] as follows,
in which R is the distance between the nuclei within and .
3. Computational Details
We performed geometry optimisations of all Pt complexes involved in the cisplatin reaction mechanism using both the M06-2X [26] and PBE0 [27] exchange-correlation functionals as implemented in the Orca 5.0.3 program package [28,29,30]. For these electronic structure calculations, we used the def2-SVP basis set for all the main-group elements and def2-TZVP basis set for platinum (hereafter referred to as Basis Set 1), along with a Stuttgart-Dresden relativistic effective core potential for the Pt centre [31]. To identify the most suitable exchange-correlation functional to carry out the QCT computations, we benchmarked the first hydration step of cisplatin using the more extensive def2-QZVP basis set (Basis Set 2) developed by Ahlrichs [32]. The PBE0 functional was used together with the Grimme’s D3 dispersion correction [33,34], as this combination accurately accounts for non-covalent interactions such as hydrogen bonding between different molecular strands which are critical factors in the second addition of nitrogenous bases to cisplatin. Moreover, PBE0+D3 reliably captures the structure and energetics of heavy metal complexes [35], while M06-2X agrees very well with experimental results for the hydration steps of cisplatin [36,37]. Besides, the last mentioned exchange-correlation functional is particularly accurate to predict energy barriers across reaction mechanisms [38]. Notably, both functionals yielded similar trends throughout the reaction pathway involving (i) cisplatin and (ii) adenine or guanine. We emphasise that all calculations accounted for solvent effects via the conductor-like polarizable continuum model (C-PCM) [39]. We proceeded in this way, so that we could better assess the effect of the sorrunding water molecules on the reactive system.
In order to generate suitable electron densities for QTAIM analyses, we performed single-point calculations for every minimum and transition state using the Zero-Order Regular Approximation (ZORA) [40]. These electronic structure computations were carried out with relativistically contracted orbital and auxiliary basis sets [41]. Finally, QTAIM analyses [24] were performed using the Aimall program [42].
4. Results and Discussion
We discuss first the hydration of cisplatin as schematised in Figure 1. It is well known that the first hydration of cisplatin is the rate-limiting step for its activation in the cell [7]. Therefore, we set out to investigate under different approximations the effect of microhydration on the activation energy for this first hydration step. We considered two explicit water molecules along the entire reaction mechanism to have comparable results with previously reported activation energies. As Cossio et al. [19] wrote: “going from zero to two surrounding water molecules, the computed activation and reaction energies can vary substantially, particularly the Gibbs activation and reaction energies. However, our results also show that for a reliable representation of the system, only two molecules of water need to be considered ” [19]. The corresponding results are shown in Table 1, where the consideration of microhydration in the system increases the electronic and the Gibbs free activation energies in contrast with the results of reference [19]. We also observed that there are no large differences between and (>1.7 kcal/mol).The electronic activation energy computed with the M06-2X approximation along with Basis Set 1 with implicit solvation effects (20.57 kcal/mol) agrees with that experimentally determined by Repta and Long [37] (19–20 kcal/mol). However, by using the same approximate functional and basis set, but considering explicit microsolvation (22.04 kcal/mol), our results agree with the experimental findings of Brancroft et al. [36] (22.25 kcal/mol). Regardless of the consideration of microsolvation, the values of , i.e., the change in Gibbs free energy for the overall reaction, indicate that the reaction is endergonic. Nevertheless, microsolvation results in a higher value in as represented in Table 1. In summary, implicit and explicit solvation represent experimental results adequately. Therefore, we will consider in the following section both sets of results with and without the effect of explicit water molecules to understand the role of microsolvation in the reaction mechanism of cisplatin with the nitrogenous bases adenine and guanine.
4.1. Monofunctional and Bifunctional Addition of DNA Bases to Cisplatin
Another significant step in the action pathway of cisplatin is the addition of nitrogenous bases. Table 2 presents the results concerning activation enthalpies for the first and second functionalisations of cisplatin with guanine as schematised in Figure 3. Relevant effects occur with the inclusion of explicit water molecules for each of the additions of the nitrogenous bases. We found for the monofunctionalisation of cisplatin that explicit microsolvation decreases the activation energy, while for the second functionalisation, it presents the opposite effect. The M06-2X functional closely aligns with the experimental activation enthalpy for the first functionalisation of cisplatin (18 ± 1 kcal/mol) [36] for both implicit and explicit solvation models. The experimental activation enthalpy for the subsequent addition of guanine is 21 ± 2 kcal/mol, a value which is accurately reproduced by the PBE0 functional with the Grimme’s correction for dispersion and explicit solvation water molecules. Still, the M06-2X functional with explicit solvation remains competitive in modelling the bifunctionalisation reaction of guanine failing just by 1 kcal/mol in the superior limit of the experimental data and it yields significantly better results regarding the first functionalisation of cisplatin. Thus, the remainder of the discussion will focus exclusively on the results obtained using the M06-2X functional.
4.2. Energetics for the Reaction of Cisplatin with Bases Adenine and Guanine
We will examine in this section the biological addition of adenine and guanine to cisplatin (Figure 4) with implicit and explicit solvation effects. More specifically, we will analyse in detail each of the reaction steps for the addition of these nitrogenous bases to cisplatin along with the corresponding changes in their electronic activation energies due to explicit microhydration. As shown in Figure 4, the mechanisms with guanine and adenine share certain steps, viz., the hydration of cisplatin (TS1, TS2). The mechanism involving the addition of adenine consists of transition states TS3A–TS7A. Ditto for the addition of guanine and the transition states TS3G–TS7G. The profiles corresponding to the Gibbs free activation energies exhibit the same trends as the activation electronic energies (see Figures S1–S24 in the Supporting Information). Given the lack of a direct relationship between (i) the respective quality and nucleophilicity of the leaving and entering groups and (ii) changes in the activation energies, we ruled out that the entering or leaving group determine the variations in activation energies. As mentioned above, a preliminary examination of interatomic distances led us to realise that in cases where the activation energy increases due to the contemplation of explicit water molecules (see e.g TS4A and TS6G in Table 3), the apparent role of the surrounding water molecules is a greater stabilisation in the reactants than in the corresponding transition states. The consideration of the differences in electronic activation energies,
in Table 3 shows that the explicit consideration of solvation water molecules increases overall the activation energy of the reaction steps considered in Figure 4 with a few exceptions. These results are in direct opposition with the conclusions offered by Cossio et. al. [19]. The lower and upper limits of are −4.58 and 15.07 kcal/mol. We considered for further wave function analyses the reactants and transition states of the steps for which 3 kcal/mol, i.e, those elementary reactions for which the incorporation of explicit water solvation molecules is most conspicuous.
4.3. QTAIM and IQA Wave Function Analyses
In order to better understand the effect of the surrounding water molecules on the activation energies of the above-mentioned reaction steps, we determined the IQA interaction energy of the atoms with which the encompassing water molecules form a bond path with the reactive system, as defined in equations (4)−(6) of the Theoretical Framework section. Table 4 reports these differences for these IQA interaction energies for the transition state and the corresponding reactants,
When , then the surrounding water molecules stabilise the reactants more strongly than the transition state and therefore we would expect that the encompassing H2O monomers increase the activation energy of the reaction under consideration, i.e., . Correspondingly, when , then the encircling water molecules stabilise the transition state more substantially than the reactants, and hence the solvation H2O monomers conduce to a reduction of the activation energy, viz., . There is indeed a correspondence between the signs of and for all the systems shown in Table 4 apart from TS1, i.e., the transition state with the smallest value of for which we performed the QTAIM and IQA analyses.
It is noteworthy that the interaction energies () differ significantly between the TS3A and TS3G steps, despite their expected similarity. In TS3A, the interaction is highly attractive, partly due to a charge transfer of 0.03 a.u. from the water molecules to cisplatin. In contrast, TS3G shows strongly repulsive interactions, as there is no such charge transfer. Additionally, TS3A forms a closed coordination cycle, whereas in TS3G, one of the water molecules primarily acts as a spectator, accumulating a charge of . This leaves the other water molecule to engage in predominantly electrostatically repulsive interactions with the cisplatin molecule.
Finally, to get further insights into the adidtion of nitrogenous bases to cisplatin, we used QTAIM delocalisation indices as chemical bond descriptors. We proceeded in this way to describe the changes in the bond formation from reactants to transition states. There is a displacement of bonded species with the arrival of a nucleophilic ligand in substitution reactions of square planar complexes. The substitution takes place via an associative mechanism in which the nucleophilic ligand first binds to the metal centre, forming a trigonal bipyramidal structure in the transition state. The leaving group is subsequently replaced by the entering nucleophile. More concretely, the entering and leaving species are in equatorial positions throughout the geometric rearrangement of the transition state and the spectator ligands do not affect the direct substitution of the reaction. In the case of the reaction of cisplatin with nitrogenous bases of DNA, the entering group will always be the nitrogenous base and the leaving group will be either chloride, water, or a second nitrogenous base depending on the reaction step being analysed. Table 5 shows the variations in the values of DIs for the leaving and entering groups involved in the formation and breaking of chemical bonds of each transition state with explicit and implicit solvation models for the structures shown in Figure 5. The corresponding changes in delocalisation indices are,
wherein X is an entering or leaving group as shown in Figure 5. The greater the absolute value of , the greater the change in the chemical bonding of the Pt–X interaction. As expected, for the entering and leaving groups are positive and negative respectively (Table 5). The addition of , for the leaving and the entering groups,
represents a measure of the effects of the rupture and the formation of chemical bonds from the reactants to the transition state. Table 6 reports the values of for the implicit and the explicit solvation models addressed herein. Because
- the breaking of chemical bonds involves an energy cost, while the formation of these interactions release energy to the surroundings and,
- the energy of the transition states is higher than those of the reactants in every case (all values of in Table 3),
we expect the effects of the rupture of chemical bonds to be more important than those of the formation of these contacts. In other words, we anticipate that the value of since is positive for the entering group and negative for the leaving group, being the latter influence more relevant as mentioned above. Table 6 shows that this is indeed the case. The difference,
indicates whether the effects of rupture of chemical bonds are more important for the explicit () or the implicit () solvation models. Apart from TS1, which is the transition state with the smallest value of for which we carried out QCT analyses, we note that for those transition states wherein , it holds that . Indeed, the inclusion of explicit solvation water molecules makes the effect of rupture of chemical bonding from reactants to transition state more stringent and, hence, a higher cost in activation energy is observed. We note the opposite behaviour when . These observations are consistent with Hammond’s postulate “structures close in energy that transform directly into each other are also similar in structure” [43]. In other words, when the inclusion of explicit solvation water molecules results in a more/less drastic change in the chemical bonding scenario, we expect a rise/diminution in activation energy with respect to implicit solvation models.
5. Conclusions
We examined the reaction mechanism of cisplatin with guanine and adenine, emphasising the impact of microsolvation on activation energies, which are shown to closely match reported experimental values. Our findings highlight that the presence of explicit water molecules can significantly influence activation energies by preferentially stabilising either the reactants or the transition state. State-of-the-art methods of wave function analyses, namely, the QTAIM and IQA approaches, have been instrumental in elucidating the role of microsolvation by providing detailed insights into the interaction energies and changes in the chemical bonding landscape of reactants and transition states. Overall, this research enhances our understanding of how explicit solvation shapes the biological mechanism of cisplatin, demonstrating that microsolvation can modulate activation barriers through a complex interplay of structural and energetic factors, consistent with Hammond’s postulate.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
J. Iván Salazar-Barrientos: Formal analysis; Investigation; Writing – original draft. José Manuel Guevara-Vela: Methodology; Software; Writing – review & editing. Marco A. García-Revilla: Data curation; Writing – review & editing. Evelio Francisco: Resources; Writing – review & editing. Miguel Gallegos: Supervision; Writing – review & editing. Tomás Rocha-Rinza: Supervision; Conceptualization; Writing – review & editing. Ángel Martín Pendás: Supervision; Funding acquisition; Writing – review & editing. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
We gratefully acknowledge DGTIC/UNAM for computer time (project LANCAD-UNAM-DGTIC 250). AMP thanks Grant PID2021-122763NB-I00 funded by MICIU 10.13039/501100011033 and by ERDF "A way of making Europe” of the European Union..
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Romani, A.M. Cisplatin in cancer treatment. Biochem. Pharmacol. 2022, 206, 115323. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of Cell Division in Escherichia coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef]
- de Vries, G.; Rosas-Plaza, X.; van Vugt, M.A.; Gietema, J.A.; de Jong, S. Testicular cancer: Determinants of cisplatin sensitivity and novel therapeutic opportunities. Cancer Treatment Reviews 2020, 88, 102054. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Sun, G.; Zhong, M.; Yu, Y.; Brewer, M. Anticancer efficacy of cisplatin and trichostatin A or 5-aza-2′-deoxycytidine on ovarian cancer. Br. J. Cancer 2013, 108, 579–586. [Google Scholar] [CrossRef]
- Go, R.S.; Adjei, A.A. Review of the Comparative Pharmacology and Clinical Activity of Cisplatin and Carboplatin. J. Clin. Oncol. 1999, 17, 409–409. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, X.; Wang, X.; Wei, X.; Wang, D.; Liu, X.; Xu, L.; Batu, W.; Li, Y.; Guo, B.; others. RSL3 enhances the antitumor effect of cisplatin on prostate cancer cells via causing glycolysis dysfunction. Biochem. Pharmacol. 2021, 192, 114741. [Google Scholar] [CrossRef] [PubMed]
- Makovec, T. Cisplatin and beyond: molecular mechanisms of action and drug resistance development in cancer chemotherapy. J. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef]
- Mantri, Y.; Baik, M. Computational Studies: Cisplatin, 2005. [CrossRef]
- Goodsell, D.S. The Molecular Perspective: Cisplatin. Stem Cells 2006, 24, 514–515, [https://academic.oup.com/stmcls/article-pdf/24/3/514/41878088/stmcls_24_3_514.pdf]. [Google Scholar] [CrossRef]
- Martin Jr, D.S. Anomalies in ligand exchange reactions for platinum (II) complexes. Inorg. Chim. Acta Rev. 1967, 1, 87–97. [Google Scholar] [CrossRef]
- Atkins, P. Shriver and Atkins’ inorganic chemistry; Oxford University Press, USA, 2010.
- Tullius, T.D.; Ushay, H.M.; Merkel, C.M.; Cardonna, J.P.; Lippard, S.J., Structural Chemistry of Platinum—DNA Adducts. In Platinum, Gold, and Other Metal Chemotherapeutic Agents; chapter 3, pp. 51–74. [https://pubs.acs.org/doi/pdf/10.1021/bk-1983-0209.ch003]. [CrossRef]
- Fichtinger-Schepman, A.M.J.; Van der Veer, J.L.; Den Hartog, J.H.J.; Lohman, P.H.M.; Reedijk, J. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation. Biochemistry 1985, 24, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Baik, M.H.; Friesner, R.A.; Lippard, S.J. Theoretical study of cisplatin binding to purine bases: why does cisplatin prefer guanine over adenine? J. Am. Chem. Soc. 2003, 125, 14082–14092. [Google Scholar] [CrossRef] [PubMed]
- Lakbaibi, Z.; Jaafar, A.; EL Aatiaoui, A.; Tabyaoui, M. Effect of the explicit solvation of 2-propanol on the Darzens reaction mechanism: A computational study. Comput. Theor. Chem. 2022, 1209, 113628. [Google Scholar] [CrossRef]
- Wang, B.; Cao, Z. How water molecules modulate the hydration of CO2 in water solution: Insight from the cluster-continuum model calculations. J. Comput. Chem. 2012, 34, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Alberto, M.E.; Lucas, M.F.A.; Pavelka, M.; Russo, N. The Second-Generation Anticancer Drug Nedaplatin: A Theoretical Investigation on the Hydrolysis Mechanism. J. Phys. Chem. B 2009, 113, 14473–14479. [Google Scholar] [CrossRef]
- Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: an historical perspective and an update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- de Cózar, A.; Larrañaga, O.; Bickelhaupt, F.M.; San Sebastián, E.; Ortega-Carrasco, E.; Maréchal, J.D.; Lledós, A.; Cossío, F.P. New insights into the reactivity of cisplatin with free and restrained nucleophiles: microsolvation effects and base selectivity in cisplatin–DNA interactions. ChemPhysChem 2016, 17, 3932–3947. [Google Scholar] [CrossRef] [PubMed]
- Huque, F.T.; Platts, J.A. The effect of intramolecular interactions on hydrogen bond acidity. Org. Biomol. Chem. 2003, 1, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Romero-Montalvo, E.; Guevara-Vela, J.M.; Narváez, W.E.V.; Costales, A.; Pendás, Á.M.; Hernández-Rodríguez, M.; Rocha-Rinza, T. The bifunctional catalytic role of water clusters in the formation of acid rain. Chem. Commun. 2017, 53, 3516–3519. [Google Scholar] [CrossRef] [PubMed]
- Sauza-de la Vega, A.; Salazar-Lozas, H.; Narváez, W.E.V.; Hernández-Rodríguez, M.; Rocha-Rinza, T. Water clusters as bifunctional catalysts in organic chemistry: the hydrolysis of oxirane and its methyl derivatives. Org. Biomol. Chem. 2021, 19, 6776–6780. [Google Scholar] [CrossRef]
- Robertazzi, A.; Platts, J.A. Hydrogen bonding, solvation, and hydrolysis of cisplatin: A theoretical study. J. Comput. Chem. 2004, 25, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Martín Pendás, A.; Francisco, E. Real space bond orders are energetic descriptors. Phys. Chem. Chem. Phys. 2018, 20, 16231–16237. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: the ORCA program system, version 4.0. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152. [Google Scholar] [CrossRef]
- Figgen, D.; Peterson, K.A.; Dolg, M.; Stoll, H. Energy-consistent pseudopotentials and correlation consistent basis sets for the 5d elements Hf–Pt. J. Chem. Phys. 2009, 130. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Vetere, V.; Adamo, C.; Maldivi, P. Performance of the `parameter free’ PBE0 functional for the modeling of molecular properties of heavy metals. Chem. Phys. Lett. 2000, 325, 99–105. [Google Scholar] [CrossRef]
- Bancroft, D.P.; Lepre, C.A.; Lippard, S.J. Platinum-195 NMR kinetic and mechanistic studies of cis-and trans-diamminedichloroplatinum (II) binding to DNA. J. Am. Chem. Soc. 1990, 112, 6860–6871. [Google Scholar] [CrossRef]
- Repta, A.J.; Long, D.F. Reactions of cisplatin with human plasma and plasma fractions. In Cisplatin; Elsevier, 1980; pp. 285–304.
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- York, D.M.; Karplus, M. A smooth solvation potential based on the conductor-like screening model. J. Phys. Chem. A 1999, 103, 11060–11079. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Chen, X.Y.; Landis, C.R.; Neese, F. All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A. AIMAll (Version 19.10. 12). TK Gristmill Software: Overland Park, KS, USA 2019, p. 23.
- Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry; OUP Oxford, 2012.
Figure 1.
Hydration of cisplatin.
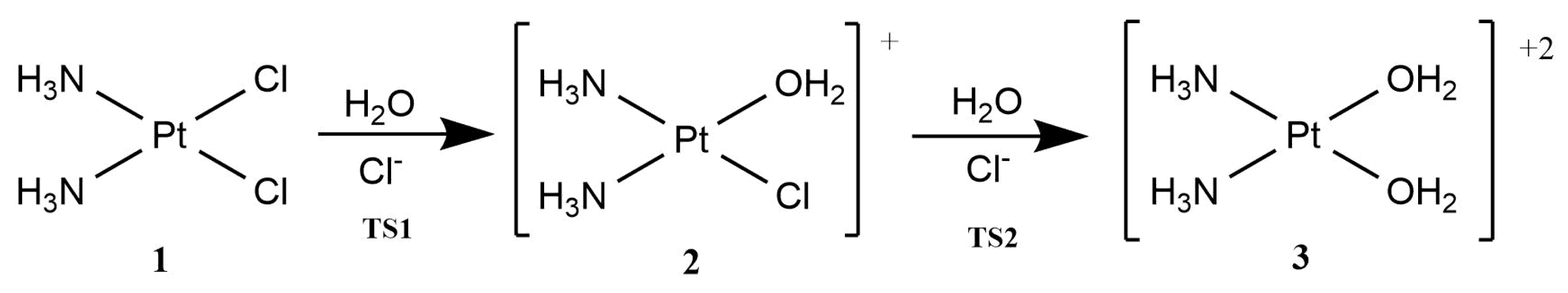
Figure 3.
Mono and bifunctionalisation schemes of cisplatin with guanine.
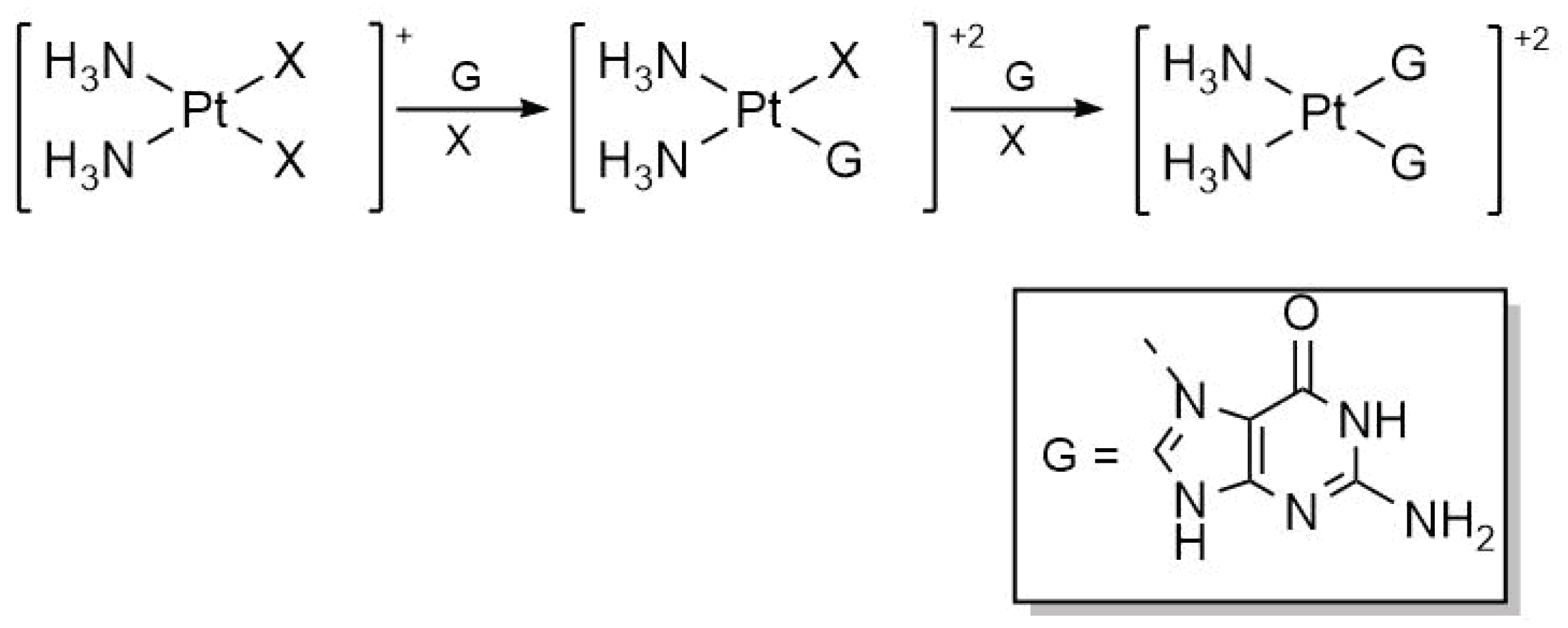
Figure 4.
Reaction mechanism of cisplatin hydration (with transition states TS1, TS2) as well as adenine (TS3A–TS7A) and (TS3G–TS7G) additions for which B=A and B=G respectively.
Figure 4.
Reaction mechanism of cisplatin hydration (with transition states TS1, TS2) as well as adenine (TS3A–TS7A) and (TS3G–TS7G) additions for which B=A and B=G respectively.
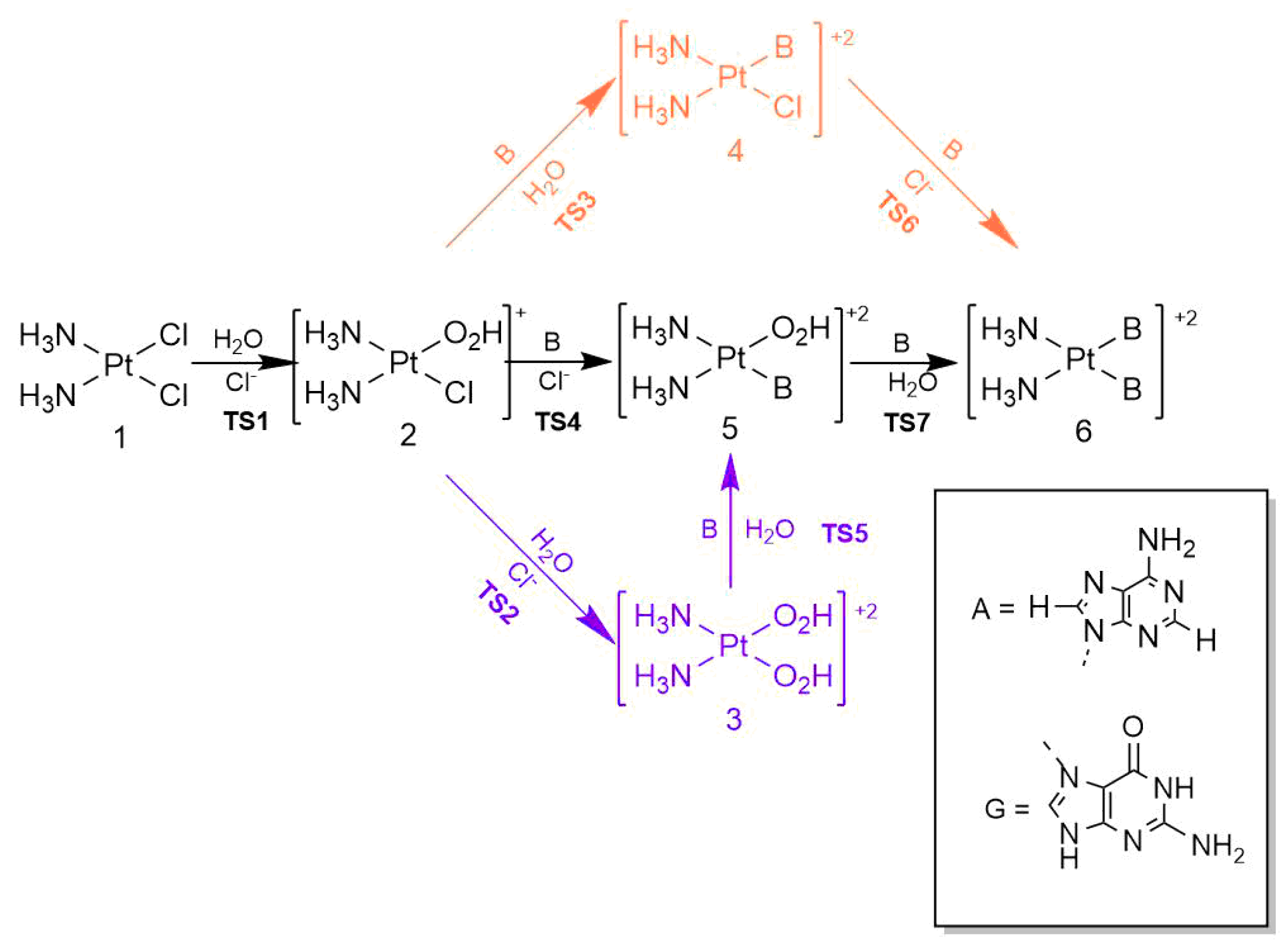
Figure 5.
Representation of three different skeletons displaying the entering and leaving groups of the investigated reaction in the ligands at equatorial positions in the geometric arrangement at every transition state. Ligands drawn above the Pt centre are the entering groups whereas those groups drawn below Pt atom are the leaving groups.
Figure 5.
Representation of three different skeletons displaying the entering and leaving groups of the investigated reaction in the ligands at equatorial positions in the geometric arrangement at every transition state. Ligands drawn above the Pt centre are the entering groups whereas those groups drawn below Pt atom are the leaving groups.
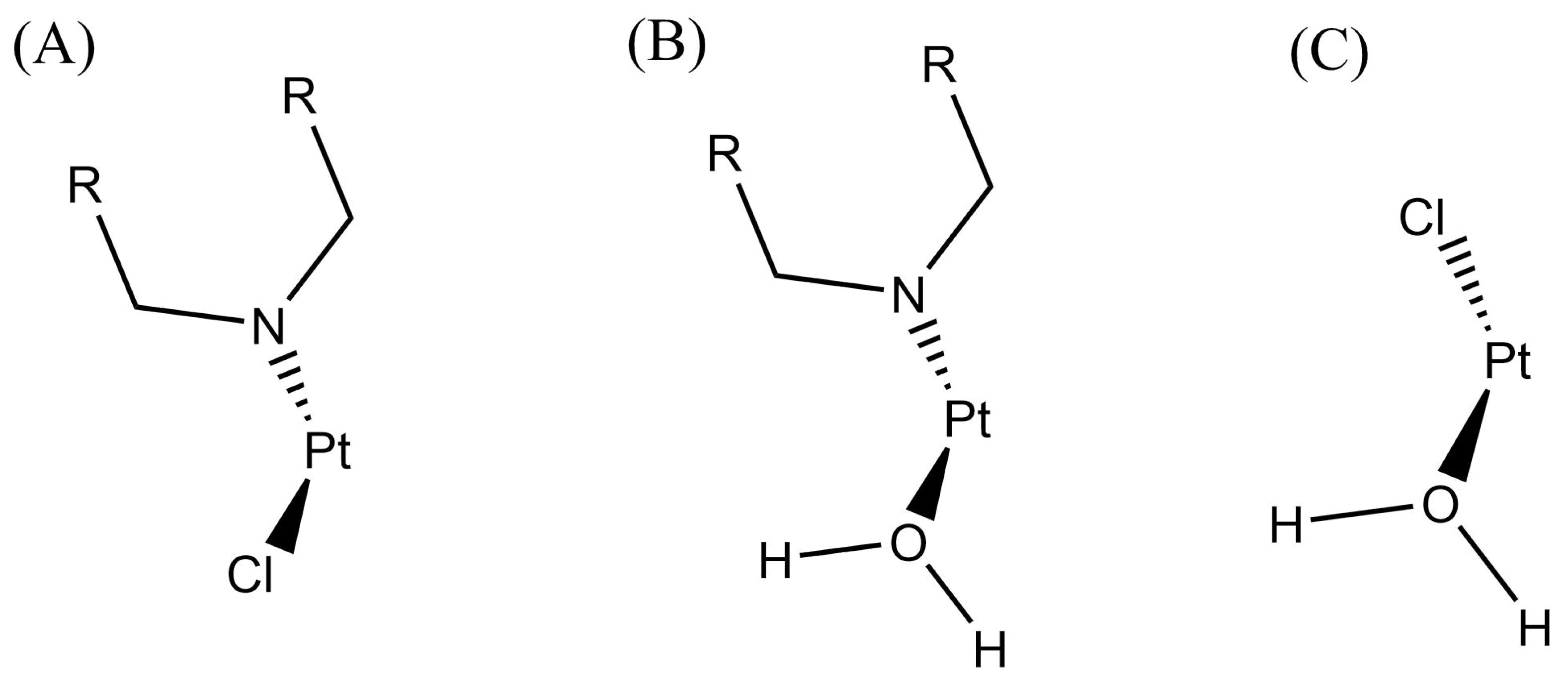

Table 1.
Electronic () and Gibbs free () activation energies for the first hydration of cisplatin using different exchange-correlation functionals along with the Basis Set 1 defined in the Computational Details section. The quantity denotes the computed value for the change of Gibbs free energy for the overall reaction. The asterisk indicates that we performed these calculations using the Basis Set 2 [32].
Table 1.
Electronic () and Gibbs free () activation energies for the first hydration of cisplatin using different exchange-correlation functionals along with the Basis Set 1 defined in the Computational Details section. The quantity denotes the computed value for the change of Gibbs free energy for the overall reaction. The asterisk indicates that we performed these calculations using the Basis Set 2 [32].
| Level of Theory | (kcal/mol) | (kcal/mol) | (kcal/mol) |
|---|---|---|---|
| PBE0-D3BJ implicit solv. | 24.20 | 24.25 | 6.95 |
| PBE0-D3BJ explicit solv. | 26.02 | 26.77 | 5.12 |
| M06-2X implicit solv. | 20.57 | 19.81 | 5.98 |
| M06-2X explicit solv. | 22.04 | 23.28 | 3.51 |
| M06-2X implicit solv.* | 21.56 | 19.85 | 20.28 |
Table 2.
Activation enthalpies () for the mono and bifunctionalisation of cisplatin with guanine as schematised in Figure 3 computed with different approximations.
Table 2.
Activation enthalpies () for the mono and bifunctionalisation of cisplatin with guanine as schematised in Figure 3 computed with different approximations.
| Monofunctionalisation of cisplatin with guanine | |||
|---|---|---|---|
| Level of Theory | Solvation Type | (kcal/mol) | (kcal/mol) |
| PBE0-D3BJ | Implicit Solv. | 24.17 | 18 ± 1 |
| PBE0-D3BJ | Explicit Solv. | 23.91 | 18 ± 1 |
| M06-2X | Implicit Solv. | 19.17 | 18 ± 1 |
| M06-2X | Explicit Solv. | 18.07 | 18 ± 1 |
| Bifunctionalisation of cisplatin with guanine | |||
| Level of Theory | Solvation Type | (kcal/mol) | (kcal/mol) |
| PBE0-D3BJ | Implicit Solv. | 16.97 | 21 ± 2 |
| PBE0-D3BJ | Explicit Solv. | 21.01 | 21 ± 2 |
| M06-2X | Implicit Solv. | 10.20 | 21 ± 2 |
| M06-2X | Explicit Solv. | 24.30 | 21 ± 2 |
1 Experimental results from Branfort et.al. [36].
Table 3.
Electronic activation energies along with the values of as defined in Equation (7) for each step of the reaction of cisplatin with adenine and guanine with implicit and explicit hydration effects. The identity of each transition state is indicated in Figure 4.
| Transition State | Implicit (kcal/mol) | Explicit (kcal/mol) | (kcal/mol) |
|---|---|---|---|
| TS1 | 19.81 | 23.28 | 3.46 |
| TS2 | 18.89 | 18.79 | −0.10 |
| TS3A | 16.70 | 12.12 | −4.58 |
| TS4A | 20.66 | 32.24 | 11.58 |
| TS5A | 17.28 | 16.51 | −0.77 |
| TS6A | 16.11 | 17.86 | 1.75 |
| TS7A | 16.21 | 16.40 | 0.20 |
| TS3G | 14.95 | 21.08 | 6.13 |
| TS4G | 20.00 | 18.40 | −1.60 |
| TS5G | 14.55 | 12.17 | −2.38 |
| TS6G | 10.10 | 25.17 | 15.07 |
| TS7G | 16.71 | 18.89 | 2.18 |
Table 4.
IQA interaction energy changes () as defined in Equation (8) during the reaction steps for which 3 kcal/mol in atomic units and their corresponding values in kcal/mol.
Table 4.
IQA interaction energy changes () as defined in Equation (8) during the reaction steps for which 3 kcal/mol in atomic units and their corresponding values in kcal/mol.
| Reaction Step | (au) | (kcal/mol) |
|---|---|---|
| TS1 | −0.0283 | −17.76 |
| TS3A | −0.0811 | −50.87 |
| TS3G | 0.0740 | 46.40 |
| TS4A | 0.6122 | 384.13 |
| TS6G | 0.2314 | 145.16 |
Table 5.
Changes in the QTAIM delocalisation indices ( as defined in Equation (9)) for the entering () and leaving () groups of the atoms involved in the formation and rupture of chemical bonds under explicit ( (expl)) and implicit ( (impl)) solvation models. The structures referred by labels (A), (B) and (C) as well as the entering and leaving groups are shown in Figure 5. Atomic units are used throughout.
Table 5.
Changes in the QTAIM delocalisation indices ( as defined in Equation (9)) for the entering () and leaving () groups of the atoms involved in the formation and rupture of chemical bonds under explicit ( (expl)) and implicit ( (impl)) solvation models. The structures referred by labels (A), (B) and (C) as well as the entering and leaving groups are shown in Figure 5. Atomic units are used throughout.
| (A) | (B) | (C) | ||||||
|---|---|---|---|---|---|---|---|---|
| TS | (expl) | (impl) | TS | (expl) | (impl) | TS | (expl) | (impl) |
| 0.27 | 0.31 | 0.27 | 0.27 | 0.26 | 0.24 | |||
| −0.31 | −0.31 | −0.32 | −0.30 | −0.45 | −0.46 | |||
| 0.33 | 0.29 | 0.29 | 0.27 | |||||
| −0.45 | −0.38 | −0.33 | −0.32 | |||||
Table 6.
Values of as defined in equations (9) and (10) for the explicit and implicit solvation models considered herein. We report the quantity defined in formula (11) as well. Atomic units are used throughout.
Table 6.
Values of as defined in equations (9) and (10) for the explicit and implicit solvation models considered herein. We report the quantity defined in formula (11) as well. Atomic units are used throughout.
| Reaction Step | |||
|---|---|---|---|
| TS1 | −0.19 | −0.22 | 0.03 |
| TS3A | −0.04 | −0.05 | 0.01 |
| TS3G | −0.05 | −0.03 | −0.02 |
| TS4A | −0.12 | −0.09 | −0.03 |
| TS6G | −0.04 | 0.00 | −0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



