
Submitted:
20 April 2025
Posted:
21 April 2025
You are already at the latest version
Abstract
The integration of molecular markers has significantly advanced the accuracy and speed of genetic analysis in plants, thereby accelerating the adoption of molecular breeding in field crops and forestry species. Among these, single nucleotide polymorphisms (SNPs) have emerged as the most widely utilized markers due to their high abundance across genomes and compatibility with high-throughput genotyping platforms. These functional markers hold great promise for enhancing the efficiency of marker-assisted selection (MAS) in crop improvement programs. Next-generation sequencing (NGS) technologies continue to play a transformative role in SNP genotyping, particularly for large-scale discovery applications. A key factor in scaling up SNP genotyping lies in minimizing the number of biochemical reactions required per assay. Therefore, the development of robust, multiplexed genotyping methods capable of amplifying numerous genomic loci simultaneously has become essential. Despite these advancements, there remains a gap in the availability of ultra-high-throughput and cost-efficient genotyping platforms suitable for routine use in applied breeding. This review highlights and evaluates four contemporary SNP genotyping assays and platforms, illustrating their roles in achieving scalable and accurate genotyping. Additionally, the paper provides an overview of current progress in SNP marker discovery, validation, and application in quantitative trait locus (QTL) analysis and modern plant breeding strategies.
Keywords:
Single Nucleotide Polymorphisms (SNPs)
; Molecular Markers
; Genotyping Platforms
; Marker-Assisted Selection (MAS)
; Next-Generation Sequencing (NGS)
; Plant Breeding
1.0. Introduction
The advent of molecular markers has transformed plant genetic analysis, significantly enhanced both the pace and precision of research and enabled the widespread employment of molecular breeding in crops and trees. Over the past three decades, marker systems and their detection platforms have undergone remarkable evolution. Molecular markers facilitate the detection of allelic variation within the genome of a species, including differences due to simple sequence repeats (SSRs), single nucleotide polymorphisms (SNPs), and insertions or deletions (InDels) (Wang et al., 1998; Ferguson et al., 2011). These variations have been developed into reliable marker systems for detecting genetic polymorphisms (Benardo, 2008; Jiang, 2013).
Molecular markers are broadly categorized based on their detection method, throughput, and cost into: (1) low-throughput, hybridization-based markers (e.g., RFLPs); (2) medium-throughput, PCR-based markers (e.g., RAPDs, AFLPs, SSRs); and (3) high-throughput, sequence-based markers (e.g., SNPs) (Wang et al., 1998; Benardo, 2008; Ferri et al., 2010). Prior to the advent of PCR technology, RFLPs were widely used due to their reproducibility and co-dominant inheritance. However, their labor-intensive, time-consuming, and costly detection process limited automation and large-scale application.
The introduction of PCR revolutionized marker development, enabling the emergence of more efficient systems such as RAPDs, AFLPs, and SSRs. While RAPDs can detect multiple loci, their low reproducibility due to random primer binding has limited their use. In contrast, AFLPs offer higher reproducibility and sensitivity using longer selective primers and discriminatory nucleotides at the 3’ ends, making them suitable for organisms with limited genomic information. Nevertheless, the laborious detection steps and lack of automation hindered their widespread application in breeding programs (Zhang et al., 2011).
SSRs became the marker of choice in the 1990s due to their hyper-variability, co-dominant nature, reproducibility, locus specificity, and genome-wide distribution. SSRs offer additional advantages such as multiplexing capability, compatibility with high-throughput genotyping, and simple PCR-based detection via gel electrophoresis at relatively low cost (Jiang, 2013; Vieira, 2016). These features allowed SSRs to overcome many limitations of earlier marker technologies and dominate plant molecular genetics and breeding into the early 21st century (Powell et al., 1996; Mammadov et al., 2012a).
However, in the last two decades, SSRs have been largely supplanted by SNP markers. SNPs, first extensively characterized in the human genome, are now recognized as the most abundant and widely distributed form of genetic variation in plant genomes (Rasheed et al., 2017; Shahabzadeh et al., 2022). Despite being bi-allelic and therefore less polymorphic than SSRs, SNPs offer key advantages, including genome-wide abundance, uniform distribution, and suitability for high-throughput and ultra-high-throughput genotyping platforms (Yan et al., 2009; Allen et al., 2011). These advantages have made SNPs the preferred marker type in plant genetics and breeding (Karim et al., 2020; Korsa & Feyissa, 2022).
SNP discovery has increasingly relied on computational approaches fueled by the rapid growth of publicly available genomic data. However, the complexity of plant genomes poses challenges in identifying informative SNPs, necessitating alternative discovery strategies for certain crops. The cost of genotyping remains a key factor influencing marker adoption, particularly in breeding programs that require genotyping of large segregating populations (Hiremath et al., 2012; Raj et al., 2022).
The development of high-throughput genotyping platforms and chemistries has facilitated the generation of dense genetic linkage maps, enhancing the appeal of SNPs for marker-assisted breeding (Collard & Mackill, 2008; You et al., 2018). Advances in next-generation sequencing (NGS) have further improved the speed, accuracy, and affordability of SNP discovery and genotyping. NGS-based approaches have proven invaluable for identifying, validating, and deploying genome-wide SNP markers and for developing array-based genotyping systems in a wide range of plant species (Davey et al., 2011; Manivannan et al., 2021).
This review provides an overview of SNP discovery, validation, and application in crop improvement. It also highlights key developments in modern genotyping platforms and assays, emphasizing their roles in advancing genomic-assisted plant breeding in the 21st century.
2.0. Methodology
This review employed a comprehensive and systematic literature analysis to synthesize current knowledge on the discovery, validation, and application of single nucleotide polymorphisms (SNPs) in modern plant breeding. Relevant peer-reviewed journal articles, technical reports, and books published between 2000 and 2024 were retrieved using scientific databases such as PubMed, Scopus, Web of Science, Google Scholar, and ScienceDirect. The search employed keywords and Boolean operators such as “SNP markers AND plant breeding,” “genotyping platforms,” “marker-assisted selection,” “next-generation sequencing AND crops,” and “SNP validation AND QTL mapping.” The selection criteria for inclusion were based on relevance to SNP development, technological advancements in genotyping, their integration in breeding programs, and evidence of practical applications in crop improvement. Emphasis was placed on studies that addressed the scalability, cost-effectiveness, throughput, and flexibility of genotyping platforms. Articles focusing on major and underutilized crops, particularly those discussing high-resolution applications like genome-wide association studies (GWAS), marker-assisted backcrossing (MABC), and genomic selection, were prioritized.
Data extracted from the selected studies were thematically organized into five core areas: (1) historical overview and importance of molecular markers in breeding; (2) strategies for SNP discovery, especially in complex genomes; (3) SNP validation and genotyping chemistry; (4) comparative analysis of major SNP genotyping platforms; and (5) practical applications of SNPs in trait mapping, germplasm characterization, and modern breeding strategies.
Critical evaluation of genotyping technologies was informed by benchmarking throughput (samples × markers), cost per data point, platform flexibility, and suitability for polyploid crops. This approach enabled a holistic assessment of how SNP-based tools are transforming the landscape of precision breeding and crop improvement across diverse agricultural systems.
3.0. Strategies for SNP Discovery in Complex Plant Genomes
Uncovering SNPs in crops characterized by low genomic complexity, such as Arabidopsis thaliana and Nicotiana tabacum, is relatively straightforward. In contrast, identifying SNPs in crops with complex genomes presents significant challenges due to the high prevalence of repetitive sequences. The development of SNP markers for genome-wide screening typically begins with identifying a broad pool of candidate SNPs. This is commonly achieved through non-targeted methods like resequencing or high-resolution melting (HRM) analysis in a diverse set of genotypes. These approaches enhance the likelihood of discovering informative polymorphisms that are suitable for downstream applications in breeding programs.
Earlier efforts, such as the resequencing of unigene-derived amplicons via Sanger sequencing and mining SNPs from EST databases followed by PCR validation, enabled researchers to target gene-rich regions while avoiding repetitive sequences. Although these strategies yielded gene-based SNPs (Ganal et al., 2009), they were limited by low polymorphism rates in conserved coding sequences and were ineffective in capturing SNPs located in non-coding regulatory or intergenic regions. Moreover, these methods were labor-intensive and costly.
The advent of next-generation sequencing (NGS) technologies, including platforms like Roche 454, SOLiD, Ion Torrent, and Illumina HiSeq, revolutionized SNP discovery by enabling high-throughput sequencing of targeted exonic regions through sequence capture and enrichment strategies (Hodges et al., 2007; Mammadov et al., 2012a). However, the design of probe-based sequence capture requires prior reference genomes, limiting their use to well-characterized crops. While transcriptome (EST) and exon resequencing remain valuable for detecting SNPs in coding regions and causal mutations (Okou et al., 2007), they fail to capture regulatory SNPs often found in non-coding regions (Dean, 2006; Emberton et al., 2005).
To address genome complexity, several reduction techniques have been developed (Costa et al., 2017; Kim et al., 2024). Methods such as methylation filtering, High-Cot selection, and microarray-based genomic selection reduce repetitive content but cannot eliminate all duplicated sequences, leading to false positives (Van Orsouw et al., 2007; Akbari et al., 2021). Alternative strategies like DOP-PCR, combined with in silico analysis, have also been proposed to simplify genome-wide SNP genotyping (Jordan et al., 2002; Lopes et al., 2022; Panahi et al., 2024).
Genome-wide sampling sequencing (GWSS) has further advanced SNP discovery. Despite its promise, GWSS is often hampered by data inconsistency, missing reads, and variable site coverage (Jiang et al., 2016). Optimizing library preparation to ensure even genome coverage remains essential.
SNP detection in polyploid crops is particularly difficult (Clevenger et al., 2015). Homoeologous and paralogous sequences can introduce false-positive SNPs. Unlike diploids, where minor allele frequencies suffice, distinguishing true SNPs in polyploids may require incorporating haplotype information (Chutimanitsakun et al., 2011; Bus et al., 2012; Yu et al., 2008). Accurate SNP validation in allopolyploids hinges on differentiating these sequence variation classes (Yu et al., 2011). Computational pipelines and genome complexity reduction methods are thus indispensable for reliable SNP detection in polyploid species (Narechania et al., 2009; Mammadov et al., 2012b).
Genotyping-by-sequencing (GBS) has emerged as a cost-effective, scalable method for SNP discovery and genotyping (Pootakam, 2023). It enables the construction of high-density linkage maps, facilitating genome-wide association studies (GWAS) (Elshire et al., 2011; Nelson et al., 2011). GBS leverages restriction enzymes like ApeKI to reduce genome complexity and has been successfully used in species like maize and sorghum (Mammadov et al., 2009; Wang et al., 2020). Although GBS does not require a reference genome, its output contains significant missing data, necessitating imputation using tools such as BEAGLE or IMPUTE2 (Howie et al., 2009; Marchini & Howie, 2010; Mora-Márquez et al., 2024).
As sequencing technologies improve, GBS continues to gain popularity despite challenges such as PCR amplification bias, missing data, and the need for robust bioinformatics pipelines (Hindorff et al., 2009; Davey et al., 2013; Panahi et al., 2024). These innovations have driven the discovery of SNPs associated with economically important traits. For instance, GWAS has successfully identified markers for dry matter and carotenoid content in cassava (Esuma et al., 2016; Rabbi et al., 2017), and disease resistance loci in potato and cassava (Wolfe et al., 2016; Kansup et al., 2020; Okada et al., 2019). These advances underscore the growing impact of SNP-based technologies in modern plant breeding
4.0. SNP Validation and Modern Genotyping Platforms and Chemistries
SNP marker development for genome screening generally involves three key stages: detection, validation, and final selection (Figure 1). However, the identification of SNPs, even with the support of reference sequences and advanced software, does not guarantee their suitability as reliable genetic markers. To confirm the Mendelian nature of a discovered SNP, ongoing validation in different or independent populations is essential (Sun et al., 2010; Andelkovic et al., 2020).
Marker validation involves designing a genotyping assay based on the polymorphism and then testing it across a diverse panel of germplasm and segregating populations (Mammadov et al., 2012b). For effective genome-wide SNP assay development, it is crucial to ensure adequate genome coverage by the selected SNP set. Once SNPs are selected, screening can be performed using high-throughput techniques on segregating germplasm sets, tailored to the breeding goal or research objective (Myles et al., 2010).
Using segregating populations for marker validation is generally more informative than unrelated lines, as it enables the evaluation of segregation patterns and the marker’s discriminative power. This also helps distinguish true Mendelian loci from repetitive or duplicated sequences that may escape detection by bioinformatics pipelines (Velazco et al., 2019). Given the cost constraints of validating every discovered SNP, most programs validate a representative subset, using it to define parameter thresholds for selecting SNPs in final assays (Senthilvel et al., 2019).
Recent advances in multiplexing and automation have greatly improved SNP genotyping efficiency (Zhang et al., 2020). Various high-throughput platforms are now capable of genotyping from a single SNP to up to a million in parallel. Some of the widely adopted platforms include Illumina’s BeadArray-based technologies (GoldenGate® and Infinium®), Affymetrix arrays, Life Technologies’ TaqMan OpenArray® system, and KASP (Kompetitive Allele-Specific PCR) offered by KBiosciences through the SNP Line™ platform (Fan et al., 2003; Steemers et al., 2007; Mammadov et al., 2012b; Fernando et al., 2015; Lui et al., 2022).
These platforms vary in chemistry, throughput capacity, and cost. Selecting a platform depends on factors such as the total number of SNPs required, the SNP context sequence length, and available budget, as genotyping remains a cost-intensive aspect of molecular breeding (Kumpatla et al., 2012; Mammadov et al., 2012a). A more detailed comparison of these genotyping platforms will be discussed later in this review.
Although most platforms perform well across various crops, SNP analysis in polyploid species remains challenging due to complex allelic combinations.
In such species, SNPs can be categorized into simple SNPs, hemi-SNPs, and homoeo-SNPs. Mammadov et al. (2012a) classified these using examples from diploid and tetraploid cotton species. The genome of tetraploid cotton, Gossypium hirsutum (AD1) and G. barbadense (AD2)—includes two subgenomes: A (from progenitors such as G. herbaceum and G. arboreum) and D (from G. raimondii).
- Simple SNPs identify allelic differences at corresponding loci within a single subgenome, leading to distinct genotype groupings.
- Hemi-SNPs reveal genetic variations that appear as homozygous in one organism but heterozygous in another.
- Homoeo-SNPs, on the other hand, identify variation at homoeologous or paralogous loci across A and D subgenomes but are often monomorphic in tetraploid species (Mammadov et al., 2012a; Abdelraheem et al., 2017; Akter et al., 2019).
Simple and hemi-SNPs are considered valuable for genetic mapping and diversity analysis. Simple SNPs generally segregate like diploid markers and represent about 10–30% of polymorphic loci in polyploid crops, while hemi-SNPs can account for 30–60% and are particularly useful in F2, RIL, and DH populations. In contrast, homoeo-SNPs are less suitable for mapping due to their inconsistent genotype patterns resulting from polymorphism among homoeologous sequences or duplicated loci (Mammadov et al., 2012a; Kumpatla et al., 2012).
Numerous high-throughput genotyping platforms, both multiplex and single-plex, have been proposed for marker-assisted selection (MAS) in crops such as wheat, rice, maize, and legumes. However, large-scale adoption in practical breeding is limited by high costs, lower customization efficiency, and expensive equipment requirements (Aurélie et al., 2009; Masouleh et al., 2009; Varshney et al., 2016).
The development of ultra-high-throughput, cost-efficient genotyping systems tailored for applied breeding remains a crucial goal for enhancing genetic gain. This review explores such platforms currently utilized in SNP genotyping and discusses their implications for accelerating crop improvement. We anticipate that the perspectives provided here will foster further discourse on optimizing genotyping technologies for future breeding applications.
5.0. Comparative Analysis of Four Genotyping Assays and Platforms
5.1. The OpenArray Technology (TaqMan System)
OpenArray technology, developed by Life Technologies, utilizes endpoint TaqMan chemistry to perform high-throughput SNP genotyping (Brenan & Morrison, 2005). It employs a miniaturized OpenArray plate, approximately one-eighth the size of a standard 384-well plate, yet capable of holding up to 3,072 individual assays. This is achieved through a design comprising 48 subarrays, each containing 64 through-holes. These holes are coated with hydrophobic and hydrophilic surfaces, allowing reagents to remain suspended via capillary action (Schleinitz et al., 2011; Mammadov et al., 2012a).
OpenArray offers high flexibility, enabling varied SNP-to-sample combinations. The plates typically come preloaded with assay reagents, with users only needing to add DNA samples. To further increase throughput, Kumpatla et al. (2012) suggested integrating slide towers with PACR thermocyclers; using two such systems, up to 2,048 samples across 128 SNPs can be processed daily in a 128 x 24 format. While high-throughput systems like Affymetrix GeneChip and Illumina BeadChip are preferable for genome-wide projects with numerous SNPs and limited sample volumes, OpenArray is more suitable for scenarios involving fewer SNPs and larger sample populations (Thomson, 2014; Dagnall et al., 2018).
OpenArray is particularly beneficial in marker-assisted selection (MAS), where a limited number of SNPs are used to track specific QTLs across many samples. For such targeted applications, single-plex genotyping methods like KASP and TaqMan may also be ideal (Neelam et al., 2013; Fernando et al., 2015). For instance, Rienzo et al. (2018) employed TaqMan assays in tomato to detect resistance-breaking isolates of Tomato Spotted Wilt Virus with high sensitivity, outperforming high-resolution melting (HRM) techniques. The protocol developed was not only accurate but also streamlined, utilizing crude leaf RNA extractions (Patil et al., 2017; Rienzo et al., 2018).
5.2. Illumina’s BeadArray Platform
Illumina's BeadArray (microarray) technology uses silica beads embedded in microwells and coated with oligonucleotide probes specific to genomic loci (Adler et al., 2013). It supports ultra-high-throughput SNP genotyping and structural variation analysis. Technologies like GoldenGate (GG) and Infinium assays fall under this platform, which can genotype up to 3,072 and 1.1 million SNPs, respectively (Fan et al., 2003; Rasheed et al., 2017).
BeadArray analysis utilizes the iScan confocal scanner and GenomeStudio software for data acquisition and interpretation. GG assays use universal primers that prevent primer interactions, allowing the amplification of all loci in a single tube. In Infinium, the assays are immobilized on a chip, while GG uses a suspension format. Both use distinct bead types based on SNP structure: Infinium I (for A/G and T/C SNPs) require two bead types, while Infinium II requires one.
Processing throughput is constrained to three 96-well plates per day due to TECAN robot limitations. Despite this, the platform remains a preferred choice for validating numerous SNPs in fewer samples, with a minimum of 24 and a maximum of 288 samples processed across one to twelve BeadChips.
5.3. High-Resolution Melting (HRM)
HRM is a post-PCR genotyping technique that uses intercalating dyes to detect DNA melting transitions with high sensitivity and specificity (Reed et al., 2007). Fluorescence decreases as double-stranded DNA melts, and the unique melting curve profiles can distinguish even single-nucleotide differences (Er & Chang, 2012).
This method is especially valuable for identifying SNPs in exploratory studies or routine diagnostics (Krypuy et al., 2006; Singh et al., 2012; Song et al., 2015). Homozygous samples yield single-peak curves, while heterozygous ones show broader, dual-peak profiles due to imperfect duplex formation (Mader et al., 2008). HRM is economical and minimizes contamination risk through its closed-tube format (Ebili & Ilyas, 2015; Slomka et al., 2017).
Nonetheless, challenges remain. Pipetting inconsistencies and environmental variations can compromise accuracy (Tucker & Huynh, 2014). Additionally, certain SNP types (e.g., A>T or T>A transitions) may produce indistinct profiles on some instruments (Vossen et al., 2009; Slomka et al., 2017).
5.4. Kompetitive Allele-Specific PCR (KASP)
KASP is a single-plex SNP genotyping method renowned for its cost-effectiveness, high sensitivity, and adaptability across different plate formats (Semagn et al., 2014). It uses a PCR mix containing allele-specific primers and a common primer, along with a fluorescence-based reporting system (Alvarez-Fernandez et al., 2021).
The assay employs Förster resonance energy transfer (FRET) for detection, using quenched fluorescent oligonucleotides that emit a signal upon binding to their targets. KASP is compatible with standard lab equipment and has been used widely in crop breeding programs, such as in chickpeas, wheat (Yu et al., 2017), and cassava (Udoh et al., 2017).
KASP assays are also used by major research centers like CIMMYT for high-volume genotyping (Rabbi et al., 2014; Semagn et al., 2015). SNPs linked to traits like heat stress tolerance in maize have been validated through KASP using RNA-seq data (Jagpot et al., 2020; Schaarschmidt et al., 2020).
In comparison to KASP, the cost and design lead time for TaqMan, GG, and Infinium assays vary. TaqMan assays can be synthesized within two weeks, while GG and Infinium take six and nine weeks, respectively (Ahn et al., 2018; Ayalew et al., 2019). Additionally, platforms like Illumina enforce minimum sample requirements per batch (480 for GG and 1,152 for Infinium), which may limit their use for smaller validation studies (Kumpatla et al., 2012).
Selecting a genotyping platform involves considering several key factors, including, SNP number, sequence context length, project scale, budget, and urgency (Kwok, 2002; Bui et al., 2017; Ruff et al., 2020). For example, Illumina HiSeq reads (~70 nt) are compatible with GG and Infinium assays that require 50 nt on either side of the SNP (Benardo et al., 2015), but may not suit TaqMan designs, which require longer sequences (~100 nt) to accommodate probe and primer binding (Jatayev et al., 2017; Yang et al., 2020). Thus, no one platform is universally ideal; rather, the decision must align with the project’s specific needs.
6.0. Next-Generation Sequencing (NGS)
Next-generation sequencing (NGS) platforms have emerged as transformative tools for cost-effective genotyping, particularly in underutilized and orphan crops. These technologies enable simultaneous SNP discovery and genotyping with minimal bias across diverse genetic backgrounds (Davey et al., 2013). NGS techniques have proven invaluable for the discovery, validation, and assessment of genetic markers, thereby accelerating the development of genome-wide SNP arrays and high-throughput genotyping chemistries (Thomson, 2014; Rasheed et al., 2017).
To optimize SNP discovery in complex genomes, it is crucial to apply genome complexity reduction strategies in tandem with NGS. These strategies help avoid repetitive and duplicated DNA regions, improving the efficiency and specificity of sequencing efforts (Yuan et al., 2003; Benardo et al., 2015). Historically, Sanger sequencing dominated genomic studies; however, unlike Sanger’s dependence on capillary electrophoresis, NGS leverages massively parallel sequencing, advanced imaging technologies, and computational algorithms to decode sequence data at a much faster rate.
NGS platforms offer broad applications, including microRNA sequencing, transcriptome profiling, target sequencing, whole-genome resequencing, de novo assembly, and chromatin immunoprecipitation sequencing. Based on sequencing read length, NGS methods are generally classified into short-read and long-read technologies (Schilbert et al., 2020; Satam et al., 2023).
The term "genotyping-by-sequencing" (GBS) serves as an umbrella description for sequencing-based genotyping approaches (Rasheed et al., 2017). Scheben et al. (2016) identified thirteen distinct GBS methodologies used in crop plants, each offering unique features. These include classical GBS (Elshire et al., 2011; Poland et al., 2012; Kim & Tai, 2013), diversity arrays technology sequencing (DArT-seq) (Li et al., 2015), sequence-based genotyping (SBG) (Van Poecke et al., 2013), restriction fragment sequencing (REST-seq) (Stolle & Moritz, 2013), and restriction enzyme site comparative analysis (RESCAN) (Kim & Tai, 2013).
Despite its strengths, GBS is not without limitations. Common issues include genotyping errors due to low sequencing coverage, particularly misclassification of heterozygotes as homozygotes. These errors are especially pronounced in outcrossing species and early-generation mapping populations. The absence of a reference genome and the presence of polyploidy further complicate SNP identification, as paralogous sequences may be mistakenly interpreted as identical reads. Increasing sequencing depth, using rare-cutting restriction enzymes, and reducing the number of multiplexed samples during library preparation are effective strategies to mitigate these challenges (Davey et al., 2013).
Additional drawbacks include amplification bias favoring shorter fragments with higher GC content, labor-intensive library preparation protocols, high levels of missing data, limited bioinformatics tools for accurate imputation, and challenges in data storage and analysis (Hindorff et al., 2009). Nevertheless, the rapid advancements in sequencing chemistries, long-read platforms, and enhanced reference genomes have fueled the growing popularity of GBS in crop genetics and breeding.
NGS has dramatically accelerated genomic research across plants, animals, and microbes due to its high-throughput, cost efficiency, and precision. Table 1 presents a comparative summary of array-based and NGS-based genotyping platforms, detailing differences in cost, throughput, flexibility, and application. For more in-depth insights into NGS capabilities, readers are referred to reviews by Rasheed et al. (2017) and Scheben et al. (2016).
7.0. Application of SNPs in Crop Breeding
Molecular markers derived from modern genomics tools, as illustrated in Figure 1, possess high density, scalability, and cost-effectiveness. They have proven especially useful in genome-wide association studies (GWAS), quantitative trait loci (QTL) mapping, and gene discovery. The information extracted through these approaches enables the precise manipulation of trait variation for diverse breeding objectives, thereby improving crop productivity and resilience (Varshney et al., 2016; Rasheed et al., 2016).
Despite their potential, the integration of marker-assisted and genomic selection into plant breeding programs has not yet reached its full potential. This underutilization may stem from infrastructural limitations, lack of technical expertise, or insufficient investment in capacity-building. Yet, with the advent of commercial platforms, most genotyping tasks can now be outsourced at affordable rates, democratizing access to molecular tools.
Once molecular markers such as SNPs are validated in independent populations, they become reliable assets for a wide array of genetic applications, including gene and QTL mapping, linkage disequilibrium-based association mapping, map-based cloning, germplasm characterization, genetic diagnostics, event detection, and marker-assisted trait introgression (Berdugo-Cely et al., 2017; Xu et al., 2019). Such validation efforts are essential for ensuring robustness and reproducibility across genetically diverse backgrounds.
Constructing recombination-based genetic linkage maps is a prerequisite for understanding marker order along chromosomes. These maps are typically generated using segregating populations such as backcrosses, F2s, recombinant inbred lines (RILs), or doubled haploids (DHs). While major crops now possess dense SNP-based genetic maps, many minor or underutilized crops rely on mixed marker systems, combining SNPs with older markers like RFLPs or SSRs (Cheema & Dicks, 2009; Poland et al., 2012). Notably, SNP genotyping lends itself well to automation, facilitating high-throughput breeding applications (Jones et al., 2007) (Figure 2).
Rafalski (2002) highlighted that the availability of efficient SNP genotyping platforms enables the genetic dissection of economically important traits. Their widespread deployment accelerates both marker-assisted and genomic selection, increasing genetic gain per unit time. SNPs are ideal for fine-mapping QTLs due to their abundance and genome-wide distribution, often yielding the highest possible map resolution.Throughput refers to the total data output from a single sequencing run, typically described as ‘number of samples × number of markers’ (where K = 1000). Although all next-generation sequencing (NGS) technologies offer high-throughput capabilities, the actual throughput depends largely on how many samples can be multiplexed in a single run. Flexibility describes the ability to freely select the number of samples and markers based on research needs. NGS-based approaches are generally more adaptable in this regard, allowing greater customization in sample and marker selection. Applications can be broadly categorized into two tiers: Tier 1: Includes high-resolution genetic studies such as genome-wide association studies (GWAS), quantitative trait loci (QTL) mapping, genomic selection, and assessments of genetic diversity. Tier 2: Encompasses targeted applications like gene tagging, marker-assisted backcrossing (MABC), and background selection. Several inherent features make SNPs highly suitable for molecular breeding: their ubiquitous distribution across genomes, bi-allelic nature (which facilitates straightforward data analysis), frequent location within gene-rich regions, and relative stability due to low mutation rates (Gore et al., 2009; Tian et al., 2020). These properties render SNPs powerful tools not only for high-resolution mapping but also for genome-wide predictions in genomic selection frameworks.
In cassava, SNP markers have been instrumental in assessing genetic diversity and parental selection. Markers have been linked to traits such as cassava mosaic disease (CMD) resistance (Wolfe et al., 2016; Rabbi et al., 2020), provitamin A content, dry matter content (Esuma et al., 2016; Rabbi et al., 2017; Udoh et al., 2017), and starch composition (de Oliveira et al., 2014). Recent studies confirm the complementary value of combining agro-morphological and molecular data to effectively distinguish genotypes, thus maximizing selection accuracy in breeding pipelines (Zhang et al., 2018).
SNP development and validation have also been advanced in maize. The NSF-funded project on maize genomic diversity (Zhao et al., 2012) contributed significantly by identifying SNPs in over 3,000 genes and mapping over 1,100 SNPs in Nested Association Mapping (NAM) populations (Yu et al., 2008). Although validation can be hindered by unknown polymorphisms in uncharacterized germplasm (Jones et al., 2009), this is mitigated using high-quality sequence alignments and validated marker databases across diverse genetic backgrounds.
Given the demand for high-throughput marker technologies in maize improvement, multiple SNP arrays have been developed (Ganal et al., 2011; Unterseer et al., 2014; Xu et al., 2017). Buckler et al. (2009) used NAM populations to dissect flowering time in maize, revealing its polygenic nature with 29 QTLs identified through 1.6 million SNPs. Similarly, SNP markers have been linked to resistance genes in soybean, such as Rag2 for aphid resistance and markers linked to root-knot nematode resistance (Kim et al., 2009; Monteros et al., 2010; Kim et al., 2010).
Ultimately, the strategic identification of tightly linked SNPs enhances the precision of MAS, reduces breeding cycle time, and provides cost-effective tools for trait introgression and genome-wide selection across multiple crops.
8.0. Conclusions
Plant breeding plays a pivotal role in achieving global food security amid challenges such as land scarcity, a growing population, and climatic variability. The synergy between genotypic and phenotypic data is critical for unraveling the genetic basis of complex traits, enabling the implementation of precise and efficient breeding strategies.
The explosion of sequence data has catalyzed the development of single nucleotide polymorphism (SNP) markers across a wide spectrum of crop species. Once validated across diverse populations, these markers become invaluable tools for genetic mapping, trait introgression, and accelerated gene discovery. Their integration into high-throughput genotyping platforms has led to the construction of dense genetic linkage maps, laying the foundation for marker-assisted and genomic selection in both major and underutilized crops.
Unlike earlier marker systems, SNPs enable the generation of saturated genetic maps, facilitating genome-wide tracking, fine mapping of target loci, and rapid identification and cloning of key genes or QTLs. The development of robust, large-scale SNP genotyping protocols is a cornerstone of modern breeding pipelines, supporting scalable trait dissection and selection.
Nonetheless, several challenges persist. While SNP chips exist for many crops, their design is often based on limited reference panels, which can reduce their applicability to genetically distant populations. This limitation underscores the need for customizable and economically viable genotyping solutions that accommodate broader genetic diversity. In this context, next-generation sequencing (NGS)-based platforms offer a promising alternative, enabling concurrent SNP discovery and genotyping with less bias toward specific genetic backgrounds.
Genotyping-by-sequencing (GBS) offers a low-cost, high-resolution means of identifying SNPs across numerous samples. However, the successful application of GBS still hinges on adequate computational infrastructure, including reliable imputation algorithms and sufficient data processing capacity. The bioinformatics demands of these technologies remain a bottleneck for many breeding programs in low-resource settings.
Despite these hurdles, the combination of SNP markers, genomics tools, and NGS technologies presents a powerful avenue for accelerating genetic gains in crop breeding. Their application not only enhances selection efficiency but also supports the rapid deployment of improved varieties that are resilient, high-yielding, and tailored to future agricultural challenges.
As we look ahead, the continued development of flexible, scalable, and accessible SNP-based genotyping platforms, paired with investments in data analysis capabilities, will be essential for realizing the full promise of genomics-assisted plant breeding
Acknowledgements
The authors express their gratitude to Dr. Olasanmi Bunmi (PhD) for his invaluable supervision and guidance throughout this work. We also acknowledge the Pan African University for providing institutional support that made this study possible.
Conflict of Interest
All authors contributed equally to the preparation of this manuscript.
References
- Abdelraheem, A.; Fang, D.D.; Zhang, J. “Quantitative trait locus mapping of drought and salt tolerance in an introgressed recombinant inbred line population of upland cotton under the greenhouse and field conditions”. Euphytica 2017, 214, 8. [Google Scholar] [CrossRef]
- Adler, A.J.; Wiley, G.B.; Gaffney, P.M. “Infinium Assay for Large-scale SNP Genotyping Applications”. Journal of Visualized Experiments 2013, 81, e50683. [Google Scholar] [CrossRef]
- Ahn, Y.K.; Manivannan, A.; Karna, S.; Jun, T.H.; Yang, E.Y.; Choi, S.; Kim, J.H.; Kim, D.S.; Lee, E.S. “Whole Genome Resequencing of Capsicum baccatum and Capsicum annuum to Discover Single Nucleotide Polymorphism Related to Powdery Mildew Resistance”. Sci. Rep 2018, 8, 51–88. [Google Scholar] [CrossRef]
- Akter, T; Islam, A.K.M.A.; Rasul, M.G.; Kundu, S.; halequzzaman; Ahmed, J.U. “Evaluation of genetic diversity in short duration cotton (Gossypium hirsutum L.)”. Journal of Cotton Research 2019, 2, 1–10. [Google Scholar] [CrossRef]
- Allen, A.M.; Barker, G.L.A.; Berry, S.T.; Coghill, J.A.; Gwilliam, R.; Kirby, S.; Robinson, P.; et al. “Transcript-specific, single-nucleotide polymorphism discovery and linkage analysis in hexaploid bread wheat (Triticum aestivum L.)”. Plant Biotechnology Journal 2011, 9, 1086–1099. [Google Scholar] [CrossRef]
- Alvarez-Fernandez, A.; Bernal, M.J.; Fradejas, I. Alvarez-Fernandez, A., M.J. Bernal, I. Fradejas. 2021. “KASP: a genotyping method to rapid identification of resistance in Plasmodium falciparum”. Malaria Journal. 2021, 20, 1–16. [Google Scholar] [CrossRef]
- Andelkovic, V.; Cvejić, S.; Jocić, S.; Kondic-Spika, A.; Jeromela, A. Marjanović; Mikić, S.; Prodanović, S. “Use of plant genetic resources in crop improvement–example of Serbia”. Genetic Resources and Crop Evolution 2020, 67, 1935–1948. [Google Scholar] [CrossRef]
- Appleby, N.; Edwards, D.; Batley, J. “New technologies for ultra-high throughput genotyping in plants”. Plant Genomics 2009, 513, 19–39. [Google Scholar] [CrossRef]
- Aurélie B., M.C.L. Paslier, M. Dardevet, F. Exbrayat-Vinson, I. Bonnin, A. Cenci, A. Haudry, D. Brunel, and C. Ravel. 2009. “High-throughput single nucleotide polymorphism genotyping in wheat (Triticum spp.)”. Plant Biotechnology Journal (7): 364-374.
- Ayalew, H.; Tsang, P.W.; Chu, C.; Wang, J.; Liu, S.; Chen, C.; Ma, X.F. “Comparison of TaqMan, KASP, and rhAmp SNP genotyping platforms in hexaploid wheat”. PLoS One 2019, 14, e0217222. [Google Scholar] [CrossRef]
- Berdugo-Cely, J.; Valbuena, R.I.; Sánchez-Betancourt, E.; Barrero, L.S.; Yockteng, R. “Genetic diversity and association mapping in the Colombian Central Collection of Solanum tuberosum L. Andigenum group using SNPs markers”. PLoS ONE, 2017, 12, e0173039. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.; Wang, S.; Amand, P.S.; Bai, G.; Fang, D.D. “Using next-generation sequencing for multiplexed trait-linked markers in wheat”. PLoS One 2015, 10, e0143890. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, R. “Molecular markers and selection for complex traits in plants: learning from the last 20 years,” Crop Science 48: 1649–1664. 2008. [Google Scholar]
- Brenan, C.; Morrison, T. High throughput, nanoliter quantitative PCR. Drug Discovery Today Technologies 2005, 2, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Buckler, E.S.; Holland, J.B.; Bradbury, P.J.; Acharya, C.B.; Brown, P.J.; Browne, C.E.E.; Flint-Garcia, S.; et al. “The Genetic Architecture of Maize Flowering Time”. Science 2009, 325, 714–718. [Google Scholar] [CrossRef]
- Bui, T.G.T., N.T.L. Hoa, J. Yen, and R. Schafleitner. 2017. “PCR-based assays for validation of single nucleotide polymorphism markers in rice and mungbean”. Hereditas 154: 1-3. [CrossRef]
- Bus, A.; Hecht, J.; Huettel, B.; Reinhardt, R.; Stich, B. “High-throughput polymorphism detection and genotyping in Brassica napus using next-generation RAD sequencing”. BMC Genomics 2012, 13, 281. [Google Scholar] [CrossRef]
- Cheema, J.; Dicks, J. “Computational approaches and software tools for genetic linkage map estimation in plants”. Briefings in Bioinformatics 2009, 10, 595–608. [Google Scholar] [CrossRef]
- Chleinitz, D.; DiStefano, J.K.; Kovacs, P. “Disease gene identification: targeted SNP genotyping using the TaqMan assay”; Humana Press: New York, 2011; pp. 77–87. [Google Scholar]
- Chutimanitsakun, Y., R.W. Nipper, A.C.L. Cuesta-Marcos, A. Corey, T. Filichkina, P.M. Hayes. 2011. “Construction and application for QTL analysis of a Restriction Site Associated DNA (RAD) linkage map in barley”. BMC Genomics 12 (1): 1-4. [CrossRef]
- Clevenger, J., Chavarro, C., Pearl, S. A., Ozias-Akins, P., & Jackson, S. A. 2015. Single nucleotide polymorphism identification in polyploids: A review, example, and recommendations. Molecular Plant, 8:, 831–846. [CrossRef]
- Collard, B.C.; Mackill, D.J. “Marker-assisted selection: an approach for precision plant breeding in the twenty-first century”. Philosophical Transactions of the Royal Society of London, Series B, Biological Sciences 2008, 363, 557–72. [Google Scholar] [CrossRef]
- Costa, J. R., Bejcek, B. E., McGee, J. E., et al. 2017. Genome editing using engineered nucleases and their use in genomic screening. In S. Markossian, A. Grossman, M. Arkin, et al. (Eds.), Assay Guidance Manual. Eli Lilly & Company and the National Center for Advancing Translational Sciences. https://www.ncbi.nlm.nih.gov/books/NBK464635/.
- Dagnall, C.L.; Morton, L.M.; Hicks, B.D.; et al. “Successful use of whole genome amplified DNA from multiple source types for high-density Illumina SNP microarrays”. BMC Genomics 2018, 19, 182. [Google Scholar] [CrossRef]
- Davey, J.W.; Cezard, T.; Fuentes-Utrilla, P.; Eland, C.; Gharbi, K.; Blaxter, M.L. “Special features of RAD sequencing data: implications for genotyping”. Molecular Ecology 2013, 22, 3151–3164. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. “Genome-wide genetic marker discovery and genotyping using next-generation sequencing”. Nature Review Genetics 2011, 12, 499–510. [Google Scholar] [CrossRef]
- De Oliveira, E.J.; Ferreira, C.F.; Santos, V. da Silva; Jesus, O.N.E.; Oliveira, G.A.; Silva, M.S. da. “Potential of SNP markers for the characterization of Brazilian cassava germplasm”. Theroritical and applied genetics 2014, 127, 1423–1440. [Google Scholar] [CrossRef] [PubMed]
- Dean, A. “On a chromosome far, far away: LCRs and gene expression”. Trends in Genetics, 2006, 22, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Ebili, H.O.; Ilyas, M. “Cancer mutation screening: Comparison of high-resolution melt analysis between two platforms”. ecancermedicalscience 2015, 9, 522. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Poland, J.A.; Kawamoto, K.; Buckler, E.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 2011, 6, e19379. [Google Scholar] [CrossRef]
- Emberton, J.; Ma, J.; Yuan, Y.; SanMiguel, P.; Bennetzen, J.L. “Gene enrichment in maize with hypomethylated partial restriction (HMPR) libraries”. Genome Research 2005, 15, 1441–1446. [Google Scholar] [CrossRef]
- Er, T.K.; Chang, J.G. “High-resolution melting: Applications in genetic disorders”. Clinica Chimica Acta 2012, 414, 197–201. [Google Scholar] [CrossRef]
- Esuma, W.; Herselman, L.; Labuschagne, M.T.; Ramu, P.; Lu, F.; Baguma, Y.; Buckler, E.S.; Kawuki, R.S. “Genome-wide association mapping of provitamin A carotenoid content in cassava”. Euphytica. 2016. [Google Scholar] [CrossRef]
- Fan, J. B.; Oliphant, A.; Shen, R.; Kermani, B.G.; Garcia, F.; Gunderson, K.L.; Hansen, M.; et al. “Highly parallel SNP genotyping”. Cold Spring Harbor Symposia on Quantitative Biology 2003, 68, 69–78. [Google Scholar] [CrossRef]
- Ferguson, M.E.; Hearne, S.J.; Close, T.J.; Wanamaker, S.; Moskal, W.A.; Town, C.D.; et al. “Identification, validation, and high-throughput genotyping of transcribed gene SNPs in cassava”. Theoretical and Applied Genetics 2011, 124, 685–695. [Google Scholar] [CrossRef]
- Fernando, H.K.D.H.; Kajenthini, T.J.C.; Rebeira, S.P.; Bamunuarachchige, T.C.; Wickramasinghe, H.A.M. Validation of Molecular Markers for the Analysis of Genetic Diversity of Amylase Content and Gel Consistency among Representative Rice Varieties in Sri Lanka. Tropical Agricultural Research 2015, 26, 317–328. [Google Scholar] [CrossRef]
- Ferri, L., E. Perrin, S. Campana, S. Tabacchioni, G. Taccetti, P. Cocchi, N. Ravenni, C. Dalmastri..(2010). “Application of multiplex single nucleotide primer extension (mSNuPE) to the identification of bacteria: the Burkholderia cepacia complex case”. Journal of Microbiological Methods 80: 251–256. [CrossRef]
- Ganal, M.W.; Altmann, T.; Roder, M.S. “SNP identification in crop plants”. Current Opinion in Plant Biology 2009, 12, 211–217. [Google Scholar] [CrossRef]
- Ganal, M.W.; Durstewitz, G.; Polley, A.; Bérard, A.; Buckler, E.S.; Charcosset, A. “Large maize (Zea mays L.) SNP genotyping array: development and germplasm genotyping and genetic mapping to compare with the B73 reference genome”. PLoS One 2011, 6, 28334. [Google Scholar] [CrossRef] [PubMed]
- Gore, M.A.; Chia, J.M.; Elshire, R.J.; Sun, Q.; Ersoz, E.S.; Hurwitz, B.L.; et al. “A First-Generation Haplotype Map of Maize”. Science 2009, 326, 1115–1117. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Holme, J.; nd; Anthony, J. “SNP genotyping: the KASP assay”. Methods Molecular Biology. 2014, 1145, 75–86. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. “Potential etiologic and functional implications of genome-wide association loci for human diseases and traits”. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef]
- Hiremath, P.J.; Kumar, A.; Penmetsa, R.V.; Farmer, A.; Schlueter, J.A; Chamarthi, S.K.; et al. “Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes”. Plant Biotechnology Journal 2012, 10, 716–732. [Google Scholar] [CrossRef]
- Hiremath, P.J.; Kumar, H.A.; Penmetsa, R.V.; Farmer, A.; Schlueter, J.A.; Chamarthi, K.S; et al. “Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes”. Plant Biotechnology Journal 2012, 10, 716–732. [Google Scholar] [CrossRef]
- Hodges, E.; Xuan, Z.; Balija, V. “Genome-wide in situ exon capture for selective resequencing”. Nature Genetics 2007, 39, 1522–1527. [Google Scholar] [CrossRef]
- Howie, B.N.; Donnelly, P.; Marchini, J. “A flexible and accurate genotype imputation method for the next generation of genome-wide association studies”. PLoS Genetics 2009, 5, e1000529. [Google Scholar] [CrossRef]
- Huang, X.; Han, B. “Natural variations and genome-wide association studies in crop plants”. Annual review of plant biology 2014, 65, 85–551. [Google Scholar] [CrossRef]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C. “Genome-wide association studies of 14 agronomic traits in rice landraces”. Nature Genetics 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Jagtap, A.B.; Vikal, Y.; Johal, G.S. “Genome-Wide Development and Validation of Cost-Effective KASP Marker Assays for Genetic Dissection of Heat Stress Tolerance in Maize”. International Journal of Molecular Sciences 2020, 21, 7386. [Google Scholar] [CrossRef] [PubMed]
- Jatayev, S., A. Kurishbayev, L. Zotova, G. Khasanova, D. Serikbay, A. Zhubatkanov, et al. 2017. “Advantages of Amplifluor-like SNP markers over KASP in plant genotyping”. BMC Plant Biol 17: 254. [CrossRef]
- Jiang, G.L. “Molecular Markers and Marker-Assisted Breeding in Plants”. Plant Breeding from Laboratories to Fields. 2013. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, H.; Michal, J.J.; Zhou, X.; Liu, B.; Woods, L.C.S.; Fuchs, R.A. “Genome-Wide Sampling Sequencing for SNP Genotyping: Methods, Challenges, and Future Development”. International Journal of Biological Sciences 2016, 12, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Jones, H., J. Kawauchi, P. Braglia. 2007. “RNA polymerase I in yeast transcribes dynamic nucleosomal rDNA”. Nature Structural and Molecular Biology 14: 123–130. [CrossRef]
- Jones, M.A.; Gargano, J.W.; Rhodenizer, D.; Martin, I.; Bhandari, P.; Grotewiel, M. “A forward genetic screen in Drosophila implicates insulin signaling in age-related locomotor impairment”. Experimental Gerontology 2009, 44, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.; Charest, A.; Dowd, J.F.; Blumenstiel, J.P.; Yeh, R.F.; Osman, A.; Housman, D.E.; Landers, J.E. “Genome complexity reduction for SNP genotyping analysis”. Proceedings of the National Academy of Sciences 2002, 99, 2942–2947. [Google Scholar] [CrossRef]
- Kansup, J.; Amawan, S.; Wongtiem, P.; Sawwa, A.; Ngorian, S.; Narkprasert, D.; Hansethasuk, J. “Marker-assisted selection for resistance to cassava mosaic disease in Manihot esculenta Crantz". Thai Agricultural Research Journal 2020, 38, 68–79. [Google Scholar] [CrossRef]
- Karim, K.Y.; Ifie, B.; Dzidzienyo, D.; Danquah, E.Y.; Blay, E.T.; Whyte, J.B.A.; et al. “Genetic characterization of cassava (Manihot esculenta Crantz) genotypes using agro-morphological and single nucleotide polymorphism markers”; 317–330: Physiol Mol Biol Plants 26, 2020. [Google Scholar] [CrossRef]
- Kassa, M.T.; M, You F.; W, Hiebert C.; J, Pozniak C.; R, Fobert P.; G, Sharpe A.; et al. “Highly predictive SNP markers for efficient selection of the wheat leaf rust resistance gene Lr16”. BMC Plant Biology 2017, 17, 45. [Google Scholar] [CrossRef]
- Kim, K.S.; Bellendir, S.; Hudso, K.A.; Hill, C.B.; Hartman, G.L.; Hyten, D.L.; et al. “Fine mapping the soybean aphid resistance gene Rag1 in soybean”. Theoretical and Applied Genetics, 2009, 120, 1063–1071. [Google Scholar] [CrossRef]
- Kim, K.S.; Hill, C.B.; Hartman, G.L.; Hyten, D.L.; Hudson, M.E.; Diers, B.W. “Fine mapping of the soybean aphid-resistance gene Rag2 in soybean PI 200538”. Theoretical and Applied Genetics 2010, 121, 599–610. [Google Scholar] [CrossRef]
- Kim, S.I.; Tai, T.H. “Identification of SNPs in closely related Temperate Japonica rice cultivars using restriction enzyme-phased sequencing”. PLoS One 2013, 8, 60176. [Google Scholar] [CrossRef]
- Kim, K.D.; Kang, K.; Kim, C. “Application of Genomic Big Data in Plant Breeding: Past, Present, and Future”. Plants 2020, 9, 1454. [Google Scholar] [CrossRef]
- Kim, K., Choe, D., Cho, S., Palsson, B., and Cho, B.-K. Reduction-to-synthesis: The dominant approach to genome-scale synthetic biology. Trends in Biotechnology 2024, 42, 1048–1063. [CrossRef] [PubMed]
- Komar, A.A. 2009. “[Methods in Molecular Biology] Single Nucleotide Polymorphisms Volume 578 The TaqMan Method for SNP Genotyping”., 10.1007/978-1-60327-411-1(Chapter 19), 293–306. [CrossRef]
- Korsa, F., & Feyissa, T. 2022. Effects of functional single nucleotide polymorphisms on plant phenotypes. Archives of Crop Science, 5: 185–192. [CrossRef]
- Krypuy, M.; Newnham, G.M.; Thomas, D.M.; Conron, M.; Dobrovic, A. “High resolution melting analysis for the rapid and sensitive detection of mutations in clinical samples: KRAS codon 12 and 13 mutations in non-small cell lung cancer”. BMC Cancer 2006, 6, 295. [Google Scholar] [CrossRef] [PubMed]
- Kumpatla, S.P.; Buyyarapu, R.; Abdurakhmonov, I.Y.; Mammadov, J.A. “Genomics-assisted plant breeding in the 21st century: technological advances and progress in Plant Breeding”. I. Y. Abdurakhmonov, Ed., pp. 131–184. 2012. [Google Scholar]
- Kwok, P.Y. “Single Nucleotide Polymorphisms”. Current Issues in Molecular Biology 2002, 5, 43–60. [Google Scholar]
- Li, H.; Vikram, P.; Singh, R.P.; Kilian, A.; Carling, J.; Song, J.; Burgueno-Ferreira, J.A.; et al. “A high-density GBS map of bread wheat and its application for dissecting complex disease resistance traits”. BMC Genomics 2015, 16, 216. [Google Scholar] [CrossRef]
- Liu, P., Lv, J., Ma, C., Zhang, T., Huang, X., Yang, Z., Zhang, L., Hu, J., Wang, S., and Bao, Z. 2022. Targeted genotyping of a whole-gene repertoire by an ultrahigh-multiplex and flexible HD-Marker approach. Engineering, 13: 186–196. [CrossRef]
- Lopes, U. V.; ires; L, J.; ramacho; P, K.; rattapaglia. Genome-wide SNP genotyping as a simple and practical tool to accelerate the development of inbred lines in outbred tree species: An example in cacao (Theobroma cacao L.). PLOS ONE, 2022, 17, e0270437. [Google Scholar] [CrossRef]
- Mader, E.; Lukas, B.; Novak, J. “A strategy to setup co-dominant microsatellite analysis for high-resolution-melting-curve-analysis (HRM)”. BMC Genetics 2008, 9, 69. [Google Scholar] [CrossRef]
- Mammadov, J.A.; Chen, W.; Mingus, J.; Thompson, S.; Kumpatla, S. “Development of versatile gene-based SNP assays in maize (Zea mays L. )”. Molecular Breeding 2012, 29, 779–790. [Google Scholar] [CrossRef]
- Mammadov, J.A.; Chen, W.; Ren, R. “Development of highly polymorphic SNP markers from the complexity reduced portion of maize (Zea mays L.) genome for use in marker-assisted breeding”. Theoretical and Applied Genetics. 2009, 121, 577–588. [Google Scholar] [CrossRef]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. “SNP Markers and Their Impact on Plant Breeding”. International Journal of Plant Genomics 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Manivannan, A., Choi, S., Jun, T. H., Yang, E. Y., Kim, J. H., Lee, E. S., Lee, H. E., Kim, D. S., and Ahn, Y. K. (2021). Genotyping by sequencing-based discovery of SNP markers and construction of linkage maps from F5 population of pepper with contrasting powdery mildew resistance trait. BioMed Research International 2021: 6673010. [CrossRef]
- Marchini, J.; Howie, B. “Genotype imputation for genome-wide association studies”. Nature Reviews Genetics 2010, 11, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Masouleh, A.K.; Waters, D.L.; Reinke, R.F.; Henry, R.J. “A high1016 throughput assay for the rapid and simultaneous analysis of perfect markers for 1017 important quality and agronomic traits in rice using multiplexed MALDI-TOF 1018 mass spectrometry”. Plant Biotechnology Journal 2009, 7, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Monteros, M.J., B.K. Ha, D.V. Phillips, et al. 2010. “SNP assay to detect the ‘Hyuuga’ red-brown lesion resistance gene for Asian soybean rust”. Theoretical and Applied Genetic 121: 1023–1032. [CrossRef]
- Mora-Márquez, F., Nuño, J. C., Soto, Á., and López de Heredia, U. Missing genotype imputation in non-model species using self-organizing maps. Molecular Ecology Resources, 2025, 25, e13992. [CrossRef] [PubMed]
- Morishige, D.T.; Klein, P.E.; Hilley, J.L.; et al. “Digital genotyping of sorghum – a diverse plant species with a large repeat-rich genome”. BMC Genomics, 2013. [Google Scholar] [CrossRef]
- Myles, S.; Chia, J.M.; Hurwitz, B.; Simon, C.; Zhong, G.Y.; et al. “Rapid genomic characterization of the genus Vitis”. PloS One 2010, 5, e8219. [Google Scholar] [CrossRef]
- Narechania, A.; Gore, M.A.; Buckler, E.S.; et al. “Large-scale discovery of gene-enriched SNPs”. The Plant Genome 2009, 2, 121–133. [Google Scholar]
- Neelam, K; Brown-Guedira, G.; Huang, L. “Development and validation of a breeder-friendly KASPar marker for wheat leaf rust resistance locus Lr21”. Molecular Breeding 2013, 31, 233–237. [Google Scholar] [CrossRef]
- Nelson, J.C.; Wang, S.; Wu, Y.; et al. “Single-nucleotide polymorphism discovery by high-throughput sequencing in sorghum,” BMC Genomics 12: 352. 2011. [Google Scholar]
- Okou, T.; Steinberg, K.M.; Middle, C.; Cutler, D.J.; Albert, T.J.; Zwick, M.E. “Microarray-based genomic selection for high-throughput resequencing”. Nature Methods 2007, 411, 907–909. [Google Scholar] [CrossRef]
- Panahi, B., Mohammadzadeh Jalaly, H., and Hamid, R. 2024. Using next-generation sequencing approach for discovery and characterization of plant molecular markers. Current Plant Biology 40: 100412. [CrossRef]
- Patil, G.; Chaudhary, J.; Vuong, T.D.; Jenkins, B.; Qiu, D.; Kadam, S.; Shannon, G.J.; Nguyen, H.T. “Development of SNP Genotyping Assays for Seed Composition Traits in Soybean”. International Journal of Plant Genomics 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Poland, J. A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L.; Yin, T. “Development of High-Density Genetic Maps for Barley and Wheat Using a Novel Two-Enzyme Genotyping-by-Sequencing Approach”. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef]
- Pootakham, W. 2023. Genotyping by sequencing (GBS) for genome-wide SNP identification in plants. In Plant Genotyping: Methods and Protocols (pp. 1–14). Springer. [CrossRef]
- Powel, W.; Machray, G.C.; Proven, J. “Polymorphism revealed by simple sequence repeats,” Trends in Plant Science 1: 215–222. 1996. [Google Scholar]
- Rabbi, I.; Hamblin, M.; Gedil, M.; et al. “Genetic mapping using genotyping-by-sequencing in the clonally propagated cassava”. Crop Science 2014, 54, 1384–1396. [Google Scholar] [CrossRef]
- Rabbi, I.; Udoh, L.I.; Wolfe, M.; Parkes, E.Y.; Gedil, M.; Dixon, A.; Ramu, P.; Jannink, J.; Kulakow, P. “Genome-Wide Association Mapping of Correlated Traits in Cassava: Dry Matter and Total Carotenoid Content”. The Plant Genome 2017, 10, 0. [Google Scholar] [CrossRef] [PubMed]
- Rabbi, I.Y.; Kayondo, S.I.; Bauchet, G.; et al. “Genome-wide association analysis reveals new insights into the genetic architecture of defensive, agro-morphological, and quality-related traits in cassava”. Plant Mol Biol, 2020. [Google Scholar] [CrossRef]
- Rafalski, A. “Applications of single nucleotide polymorphisms in crop genetics”. Current Opinion in Plant Biotechnology 2002, 5, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Raj, R. N., Qureshi, N., & Pourkheirandish, M. 2022. Genotyping by sequencing advancements in barley. Frontiers in Plant Science 13: 931423. [CrossRef]
- Rasheed, A.; Hao, Y.; Xia, X.; Khan, A.; Xu, Y.; Varshney, R.K.; He, Z. “Crop Breeding Chips and Genotyping Platforms: Progress, Challenges, and Perspectives”. Molecular Plant. 2017, 10, 1047–1064. [Google Scholar] [CrossRef]
- Rasheed, A.; Wen, W.; Gao, F.; Zhai, S.; Jin, H.; Lui, J.; Guo, Q.; et al. “Development and validation of KASP assays for functional genes underpinning key economic traits in wheat”. Theoretical and Applied Genetics 2016, 129, 1843–1860. [Google Scholar] [CrossRef]
- Reed, G.H.; Kent, J.O.; Wittwer, C.T. “High-resolution DNA melting analysis for simple and efficient molecular diagnostics”. Pharmacogenomics 2007, 8, 597–608. [Google Scholar] [CrossRef]
- Rienzo, D.V.; Bubici, G.; Montemurro, C.; Cillo, F.; Shu-Biao, W. “Rapid identification of tomato Sw-5 resistance-breaking isolates of Tomato spotted wilt virus using high resolution melting and TaqMan SNP Genotyping assays as allelic discrimination techniques”. PLOS ONE 2018, 13, e0196738. [Google Scholar] [CrossRef]
- Ruff, T.M.; Marston, E.J.; Eagle, J.D.; Sthapit, S.R.; Hooker, M.A.; Skinner, D.Z.; et al. “Genotyping by multiplexed sequencing (GMS): A customizable platform for genomic selection”. PLoS ONE 2020, 15, e0229207. [Google Scholar] [CrossRef]
- Satam, B., Maheshwari, S., and Dangi, C. B. Next-generation sequencing technology: Current trends and advancements. Biology 2023, 12, 997. [CrossRef]
- Schaarschmidt, S.; Fischer, A.; Zuther, E.; Hincha, D.K. “Evaluation of Seven Different RNA-Seq Alignment Tools Based on Experimental Data from the Model Plant Arabidopsis thaliana”. Int. J. Mol. Sci 2020, 21, 1720. [Google Scholar] [CrossRef]
- Scheben, A.; Batley, J.; Edwards, D. “Genotyping-by-sequencing approaches to characterize crop genomes: choosing the right tool for the right application”. Plant Biotechnology Journal 2016, 15, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Schilbert, H.M.; Rempel, A.; Pucker, B. “Comparison of Read Mapping and Variant Calling Tools for the Analysis of Plant NGS Data”. Plants 2020, 9, 439. [Google Scholar] [CrossRef] [PubMed]
- Schleinitz, D, J.K. DiStefano, and P. Kovacs. 2011. “Disease gene identification: targeted SNP genotyping using the TaqMan assay”. New York: Humana Press; p. 77–87.
- Semagn, K.; Babu, R.; Hearne, S.; Olsen, M. “Single nucleotide polymorphism genotyping using Kompetitive Allele-Specific PCR (KASP): an overview of the technology and its application in crop improvement”. Molecular breeding 2014, 33, 1–14. [Google Scholar] [CrossRef]
- Semagn, K.; Beyene, Y.; Babu, R.; Nair, S.; Gowda, M.; Das, B. “Quantitative Trait Loci Mapping and Molecular Breeding for Developing Stress Resilient Maize for Sub-Saharan Africa”. Crop Science 2015, 55, 1449. [Google Scholar] [CrossRef]
- Senthilvel, S.; Ghosh, A.; Shaik, M.; et al. “Development and validation of an SNP genotyping array and construction of a high-density linkage map in castor”. Sci Rep. 2019, 9, 3003. [Google Scholar] [CrossRef]
- Shahabzadeh, Z., Darvishzadeh, R., Mohammadi, R., Jafari, M., & Alipour, H. High-throughput single nucleotide polymorphism genotyping reveals population structure and genetic diversity of tall fescue (Festuca arundinacea) populations. Crop and Pasture Science 2022, 73, 1070–1084. [CrossRef]
- Shai, Z.; Song, W.; Xing, J.; et al. “Molecular mapping of quantitative trait loci for three kernel-related traits in maize using a doubled haploid population”. Molecular Breeding 2017, 37, 108. [Google Scholar] [CrossRef]
- Shen, G.Q.; Abdullah, K.G.; Wang, Q.K. “The TaqMan method of SNP genotyping. In: Komar A. (eds) Single Nucleotide Polymorphisms”. Methods in Molecular Biology™ (Methods and Protocols), Humana Press, Totowa, NJ 2009, 578, 293–306. [Google Scholar] [CrossRef]
- Singh, R.R.; Bains, A.; Patel, K.P.; Rahimi, H.; Barkoh, B.A.; Paladugu, A.; et al. “Detection of high-frequency and novel DNMT3A mutations in acute myeloid leukemia by high-resolution melting curve analysis”. J Mol Diagn. 2012, 14, 336–45. [Google Scholar] [CrossRef]
- Slomka, M.; Sobalska-Kwapis, M.; Wachulec, M.; Bartosz, G.; Strapagiel, D. “High-Resolution Melting (HRM) for High-Throughput Genotyping—Limitations and Caveats in Practical Case Studies”. International Journal of Molecular Sciences 2017, 18, 2316. [Google Scholar] [CrossRef]
- Song, C.; Castellanos-Rizaldos, E.; Bejar, R.; Ebert, B.L.; Makrigiorgos, G.M. “DMSO Increases Mutation Scanning Detection Sensitivity of High-Resolution Melting in Clinical Samples”. Clin Chem. 2015, 61, 1354–62. [Google Scholar] [CrossRef] [PubMed]
- Steele, K.A.; Quinton-Tulloch, M.J.; Amgai, R.B.; Dhakal, R.; Khatiwada, S.P.; Vyas, D.; et al. “Accelerating public sector rice breeding with high-density KASP markers derived from whole-genome sequencing of indica rice”. Molecular Breeding 2018, 38, 38. [Google Scholar] [CrossRef] [PubMed]
- Steemers, F.J.; Gunderson, K.L. “Whole-genome genotyping technologies on the BeadArray platform”. Biotechnology Journal 2007, 2, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Stolle, E.; Moritz, R.F. “RESTseq-efficient benchtop population genomics with restriction Fragment sequencing”. PLoS One 2013, 8, 63960. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, J.; Crouch, J.H.; Xu, Y. “The efficiency of selective genotyping for genetic analysis of complex traits and potential applications in crop improvement”. Molecular Breeding 2010, 26, 493–511. [Google Scholar] [CrossRef]
- Suo, W.; Shi, X.; Xu, S.; Li, X.; Lin, Y. “Towards low cost, multiplex clinical genotyping: 4-fluorescent Kompetitive Allele-Specific PCR and its application on pharmacogenetics”. PLoS ONE 2020, 15, e0230445. [Google Scholar] [CrossRef]
- Thomson, M.J. “High-throughput SNP genotyping to accelerate crop improvement”. Plant Breeding and Biotechnology 2014, 2, 195–212. [Google Scholar] [CrossRef]
- Thottathil, G.P.; Jayasekaran, K.; Othman, A.S. “Sequencing crop genomes: a gateway to improve tropical agriculture”. Tropical life science research 2016, 27, 93–114. [Google Scholar]
- Tian, H., Y. Yang, H. Yi, L. Xu, H. He, Y. Fan, and J. Zhao. 2020. “New resources for genetic studies in maize (Zea mays L.): a genome-wide Maize6H-60K SNP array and its application”. The Plant Journal. [CrossRef]
- Tucker, E.J.; Huynh, B.L. “Genotyping by high-resolution melting analysis”. Methods Mol. Biol. 2014, 1145, 59–66. [Google Scholar] [CrossRef]
- Udoh, L.I.; Melaku, G.; Parkes, Y.E.; Kulakow, P.; Adesoye, A.; Nwuba, C.; Rabbi, Y.I. “Candidate gene sequencing and validation of SNP markers linked to carotenoid content in cassava (Manihot esculenta Crantz)”. Molecular breeding. 2017, 37, 123. [Google Scholar] [CrossRef]
- Unterseer, S.; Bauer, E.; Haberer, G.; Seidel, M.; Knaak, C.; Ouzunova, M.; Meitinger, T. “A powerful tool for genome analysis in maize, development, and evaluation of the high-density 600k SNP genotyping array”. BMC Genomics 2014, 15, 823. [Google Scholar] [CrossRef] [PubMed]
- Van, O.N.J.; Hogers, R.C. J.; Janssen, A.; Yalcin, F.; Snoeijers, S.; Verstege, E. “Complexity reduction of polymorphic sequences (CRoPS): a novel approach for large-scale polymorphism discovery in complex genomes”. PLoS ONE 2007, 2, 1172. [Google Scholar] [CrossRef]
- Van, P.R.M.; Maccaferri, M.; Tang, J.; T.Truong; Janssen, A.; van-Orsouw, N.J.; Salvi, S.; et al. “Sequence-based SNP genotyping in durum wheat”. Plant Biotechnology Journal 2013, 11, 809–817. [Google Scholar] [CrossRef]
- Varshney, R.K.; Singh, V.K.; Hickey, J.M.; Xun, X.; Marshall, D.F.; Wang, J.; Ribaut, J.M. “Analytical and Decision Support Tools for Genomics-Assisted Breeding”. Trends in Plant Science 2016, 21, 354–363. [Google Scholar] [CrossRef]
- Varshney, R.K.; Hiremath, P.J.; Kashiwagi, P. Lekha. J.; Balaji, J.; Deokar, A.A.; Vadez, V.; Xiao, Y. “A comprehensive resource of drought- and salinity- responsive ESTs for gene discovery and marker development in chickpea (Cicer arietinum L.)”. BMC Genomics 2009, 10, 523. [Google Scholar] [CrossRef]
- Velazco, J.G., M. Malosetti, C.H. Hunt. “Combining pedigree and genomic information to improve prediction quality: an example in sorghum”. Theoritical and Applied Genetics 2019, 132, 2055–2067. [CrossRef]
- Vieira, M.L.C.; Santini, L.; Diniz, A.L.; Munhoz, C.D.F. “Microsatellite markers: what they mean and why they are so useful”. Genetics and Molecular Biology 2016, 39, 312–328. [Google Scholar] [CrossRef]
- Vossen, R.H.; Aten, E.; Roos, A.; Dunnen, J.T. “High-resolution melting analysis (HRMA): More than just sequence variant screening”. Hum. Mutat. 2009, 30, 860–866. [Google Scholar] [CrossRef]
- Wang, D.G.; Fan, J.B.; Siao, C.J.; Berno, A.; Young, P.; Sapolsky, R.; Ghandour, G.; Perkins, N.; et al. “Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome”. Science 1998, 280, 1077–1082. [Google Scholar] [CrossRef]
- Wang, N., Yuan, Y., Wang, H., Yu, D., Liu, Y., Zhang, A., Gowda, M., Nair, S. K., Hao, Z., Lu, Y., San Vicente, F., Prasanna, B. M., Li, X., & Zhang, X. Applications of genotyping-by-sequencing (GBS) in maize genetics and breeding. Scientific Reports 2020, 10, 16308. [CrossRef]
- Williams, J.G.K.; Kubelik, A.R.; Livak, K.J.; Rafalski, J.A.; Tingey, S.V. “DNA polymorphisms amplified by arbitrary primers are useful as genetic markers”. Nucleic Acids Research 1990, 18, 6531–6535. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.J., I.Y. Rabbi, C. Egesi, M. Hamblin, R. Kawuki, P. Kulakow, and J.L. Janniink. 2016. “Genome-wide association and prediction reveals genetic architecture of cassava mosaic disease resistance and prospects for rapid genetic improvement”. The plant genome 9(2). [CrossRef]
- Wosula, E.N., W. Chen, M. Amour, Z. Fei, and J.P. Legg. 2020. “KASP Genotyping as a Molecular Tool for Diagnosis of Cassava-Colonizing”. Bemisia tabaci. Insects 11: 305. [CrossRef]
- Wright, S. I., I.V. Bi, S.C. Schroeder, M. Yamasaki, J.F. Doebley, M.D McMullen, and B.S. Gaut..2005. “Evolution: the effects of artificial selection on the maize genome”. Science 308: 1310–1314. [CrossRef]
- Xu, Y., X. Liu, J. Fu, H. Wang, J. Wang, C. Huang, B.M. Prasanna, M.S. Olsen, G. Wang, and A. Zhang. 2019. “Enhancing genetic gain through genomic selection: from livestock to plants”. Plant Communications. 100005–. [CrossRef]
- Xu, C., Y. Ren, Y. Jian, Z. Guo, Y. Zhang, C. Xie, J. Fu, H. Wang, G. Wang, Y. Xu, and P. Li. 2017. “Development of a maize 55K SNP array with improved genome coverage for molecular breeding”. Molecular Breeding, 37 (3): 20. [CrossRef]
- Yan, J., X. Yang, T. Shah, H. Sanchez-Villeda, J. Li, M. Warburton, Y. Zhou, J.H. Crouch, and Y. Xu. 2009. “High-throughput SNP genotyping with the GoldenGate assay in maize”. Mol. Breed. 15: 441– 451.
- Yang, S., W. Yu, X. Wei, Z. Wang, Y. Zhao, X. Zhao, B. Tian, Y. Yuan, and X. Zhang.2020. “An extended KASP-SNP resource for molecular breeding in Chinese cabbage (Brassica rapa L. ssp. pekinensis)”. PLoS ONE 15: e0240042. [CrossRef]
- You, Q., Yang, X., Peng, Z., Xu, L., and Wang, J. 2018. Development and applications of a high-throughput genotyping tool for polyploid crops: Single nucleotide polymorphism (SNP) array. Frontiers in Plant Science, 9, Article 104. [CrossRef]
- Yu, H.; Xie, W.; Wang, J.; Xing, Y.; Xu, C.; Li, X.; Xiao, J.; Zhang, O. “Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers”. PLoS ONE 2011, 6, 10–13. [Google Scholar] [CrossRef]
- Yu, J.; Holland, J.B.; McMullen, M.D.; Buckler, E.S. “Genetic design and statistical power of nested association mapping in maize”. Genetics 2008, 178, 539. [Google Scholar] [CrossRef]
- Yuan, Y., SanMiguel, P.J. and Bennetzen, J.L. “High-Cot sequence analysis of the maize genome”. The Plant Journal 2003, 34, 249–255. [CrossRef]
- Zhang Z., X. Guo; Liu, B.; Tang, L.; Chen, F. “Genetic diversity and genetic relationship of Jatropha curcas between China and Southeast Asia revealed by amplified fragment length polymorphisms”. African Journal of Biotechnology 2011, 10, 2825–2832. [Google Scholar] [CrossRef]
- Zhang, S., Ye J., Lu K., Zou M., Chen X. and Zhou X. Genome-wide association studies of 11 Agronomical Traits in Cassava (Manihot esculenta Crantz). Frontier in plant science 2018, 9, 503. [CrossRef]
- Zhang, J., Yang, J., Zhang, L., Chen, L., Luo, J., Zhao, H., and Zhao, J. (2020). A new SNP genotyping technology Target SNP-seq and its application in genetic analysis of cucumber varieties. Scientific Reports 10: 5623. [CrossRef]
- Zhao Y., M. Gowda, W. Liu, T. Würschum, H.P. Maurer, F.H. Longin, N. Ranc, and J.C. Reif.2012. “Accuracy of genomic selection in European maize elite breeding populations”. Theoretical and Applied Genetics 124: 769–776. [CrossRef]
Figure 1.
A workflow system to develop practical Breeding Chips with Possible Genetic information from Linkage Mapping, GWAS, and Functional Genes.
Figure 1.
A workflow system to develop practical Breeding Chips with Possible Genetic information from Linkage Mapping, GWAS, and Functional Genes.
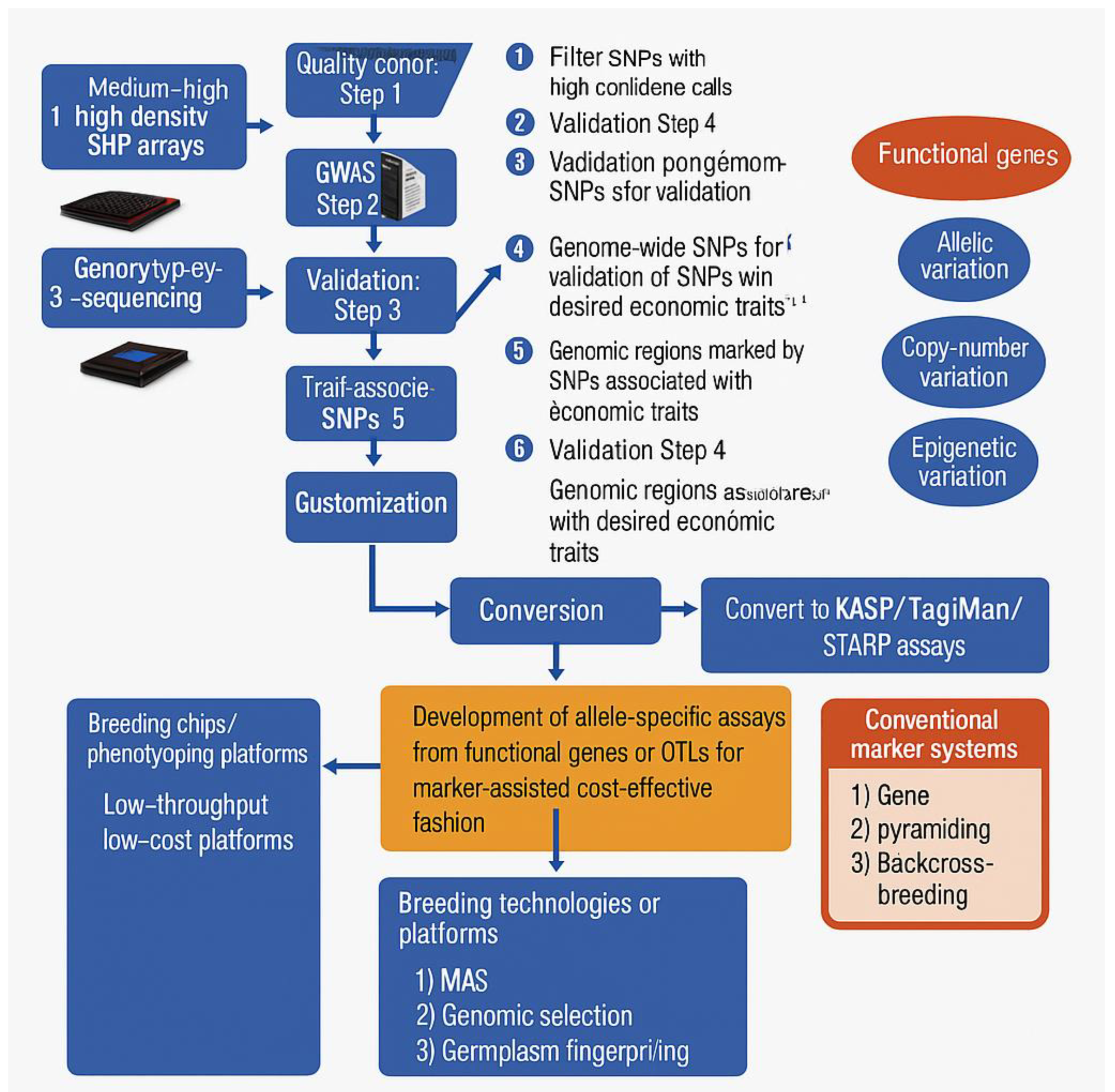
Figure 2.
Schematic representation of the application of SNP markers in modern plant breeding. The diagram outlines the process from marker discovery and validation to their use in gene/QTL mapping, genomic selection, germplasm characterization, and marker-assisted selection (MAS), highlighting the integration of SNPs into high-throughput and precision breeding pipelines.
Figure 2.
Schematic representation of the application of SNP markers in modern plant breeding. The diagram outlines the process from marker discovery and validation to their use in gene/QTL mapping, genomic selection, germplasm characterization, and marker-assisted selection (MAS), highlighting the integration of SNPs into high-throughput and precision breeding pipelines.
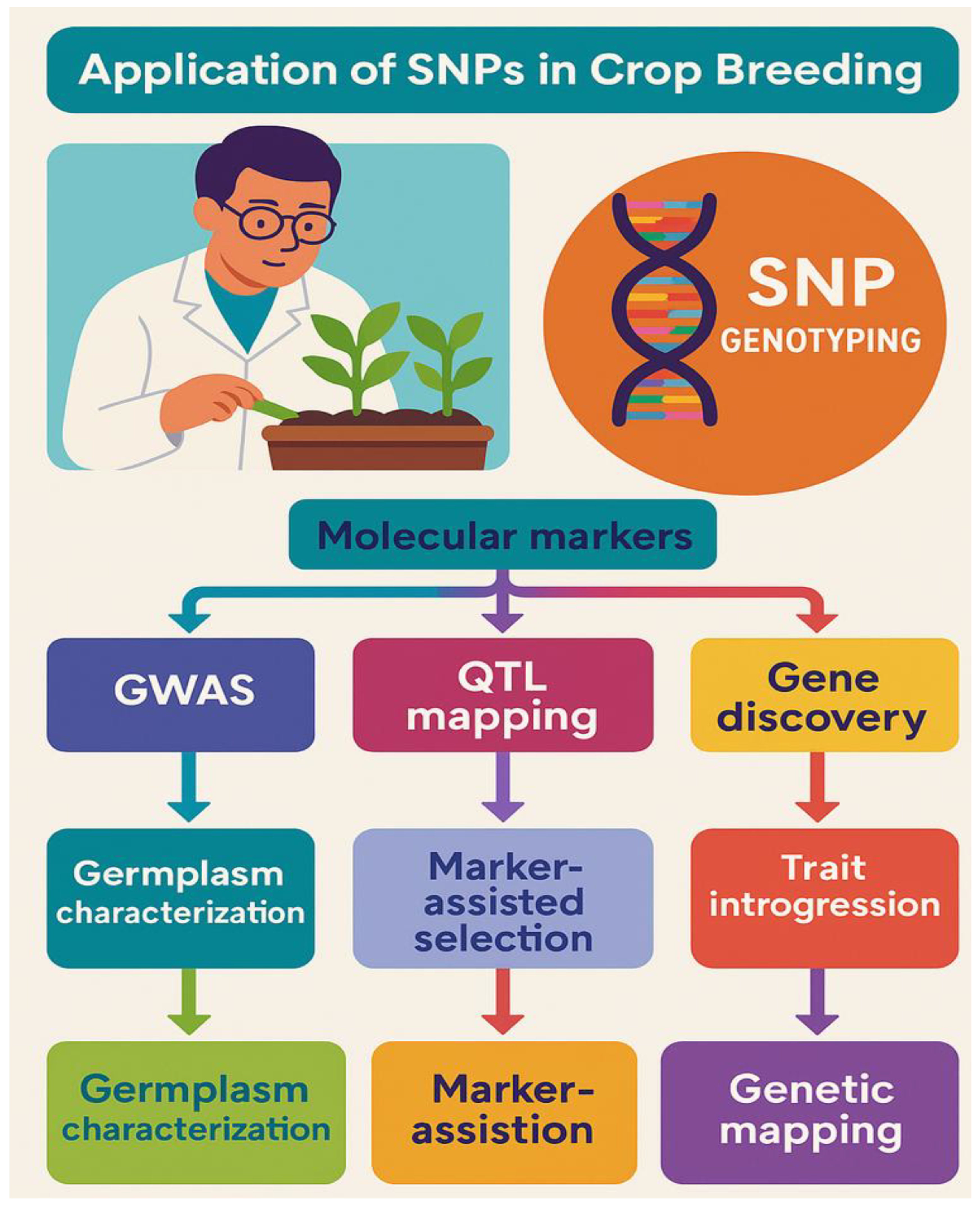

Table 1.
Features of Modern Genotyping assays and platforms available for gene Discovery and Modern Molecular Breeding in Crop Species.
Table 1.
Features of Modern Genotyping assays and platforms available for gene Discovery and Modern Molecular Breeding in Crop Species.
| Platform | Technology | Provider | Cost Per Sample | Cost Per Data Point | Analysis of Complexity | Prior Genomic Knowledge | Throughput | Flexibility | Application |
|---|---|---|---|---|---|---|---|---|---|
| Array-based | GoldenGate | Illumina | High | Moderate | Moderate | Yes | 172 × 1.5K | No | Tier 1 |
| Infinium XT | Illumina | Moderate | Low | Moderate | Yes | 96 × 50K | No | Tier 1 | |
| Infinium HD/HTS | Illumina | High | Low | Moderate | Yes | 24 × 90K/ 24 × 700K | No | Tier 1 | |
| Axiom | Affymetrix | Moderate to high | low | Moderate | Yes | 96 × 1000K Or 384 ×55K | No | Tier 1 | |
| NGS based | GBS | Non-commercial | Moderate | Low | Difficult | No | depend on sample multiples | Low | Tier 1 |
| RAD-seq | Non-commercial | Moderate | Low | Difficult | No | -do- | Low | Tier 1 | |
| SLAF-seq | Biomarker Tech | High | Low | Difficult | No | -do- | Low | Tier 1 | |
| Exome capture | Agilent/Nim-bleGen | High | Low | Difficult | Yes | -do- | Low to moderate | Tier 1 | |
| DArT-seq | DiveraityArray | Moderate | Low | Commercial support available | No | 96 × 50-100K | Low | ||
| rAmseq | Non-commercial | Very low | Low | Difficult | Yes | Multiplex | Low | Tier 1 | |
| Targeted GBS/low density array | fluidgm | fluidgm | Moderate | moderate | moderate | yes | 96 × 96/ 24 × 192/ 48 × 48 | moderate | Tier 2 |
| Sequenom MassARRAY | Agena Bioscience | Moderate | Moderate | Moderate | yes | 96 × 48 | Low | Tier 2 | |
| Eureka | Affymetrix | Moderate | Moderate | Moderate | Yes | At least 5K × 3K | Low | Tier 2 | |
| AmpliSeq | Thermo Fisher | Moderate | Moderate | Moderate | Yes | customizable | Moderate | Tier 2 | |
| Single Markers | KASP | LGC Group | Depend on reaction volume and assay number | High | Easy | Yes | Single-plex (up to ~150K data points/day) | Scalable | Tier 2 |
| TaqMan | Roche Molecular System | -do- | High | Easy | Yes | -do- | -do- | Tier 2 | |
| STARP | Non-commercial | -do- | Moderate | Easy | yes | -do- | -do- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



