
Submitted:
16 January 2025
Posted:
17 January 2025
You are already at the latest version
Abstract
The rapid identification of allele variants in target genes is crucial for accelerating marker-assisted selection (MAS) in plant breeding. Although current high-throughput genotyping methods are efficient in detecting known polymorphisms, they are limited when multiple variant sites are scat-tered along the gene. This study presents a target amplicon sequencing approach using Oxford Nanopore Technologies (ONT-TAS) to rapidly sequence full-length genes and identify allele variants in sunflower and wheat collections. This procedure combines multiplex PCR and a rapid sequencing kit, significantly reducing the time and cost compared to previous methods. The efficiency of the approach was demonstrated by sequencing four genes (Ahasl1, Ahasl2, Ahasl3, and FAD2) in 40 sunflower genotypes and three genes (Ppo, Wx, and Lox) in 30 wheat genotypes. Phylogenetic analysis based on SNPs identified five distinct groups in the sunflower collection and multiple alleles for each target wheat gene. The ONT-TAS approach enabled rapid characterization of the genetic diversity of the target genes in the germplasm collections, providing a complete picture of SNPs and InDels along the full-length genes. This method offers a high-throughput, cost-effective solution for genotyping and identifying novel allele variants in plant breeding pro-grams.
Keywords:
nanopore sequencing
; amplicons
; wheat
; sunflower
; SNPs
; InDels
1. Introduction
Rapid identification of allele variants of target genes in plant collections is essential for accelerating the plant breeding process using marker-assisted selection (MAS). Allele-specific DNA markers are typically used in MAS to detect certain polymorphic regions of a gene. This strategy is rapid and cost-effective owing to the availability of modern detection systems (for example, KASP, ASPCR and PCR-SSCP) that allow genotyping hundreds of individual plants [1,2,3,4,5]. These high-throughput genotype systems have been widely used in plant breeding for various crop species [6,7,8,9,10,11,12,13,14,15]. KASP and other systems for high-throughput genotyping, for example, allele-specific PCR (ASPCR) and single strand conformation polymorphism (SSCP), allow discrimination of polymorphisms with a high degree of specificity and sensitivity [4].
The low cost and high throughput of these genotyping approaches make them ‘a way to choose’ when the DNA variant sites linked to certain traits are known, and they are not scattered along the gene. However, the more diverse germplasm sources involved in breeding, the more DNA variant sites at different gene parts have been dis-covered. For example, glutenin genes have multiple single nucleotide polymorphism (SNP) variants that are distributed over the entire gene and can differ between distinct Glu alleles [16,17]. Genes that improve the quality of wheat flour, such as lipoxygenase (Lox), polyphenol oxidase 1 (Ppo), and the granule-bound starch synthase (Wx), are of particular interest for allele identification and marker development. Identifying any structural variations in genes associated with baking quality is crucial for the breeding process. Another example of the detrimental genes for important traits is the family of AHASL genes, which are common targets of herbicides, and crop resistance to different herbicides is associated with distinct SNPs in different gene parts [18,19,20]. PCR-based methods have shown the C-to-T mutation in codon 205 of the Ahasl1-1 gene, offering moderate resistance to imazamox (IMI) [21,22]. Analysis of Ahasl1-2 has revealed a C-to-T mutation in codon 197, rendering a substantial level of SU (sulfonyl-urea) tolerance [21]. Ahasl1-3 involves a G-to-A mutation in codon 122, which confers strong resistance to IMI [23]. Ahasl1-4 contains a G-T mutation in codon 574, granting broad resistance to herbicides targeting AHAS in four different families [24]. Nevertheless, herbicide resistance is not restricted to only four alleles [25].
The existence of multiple polymorphic sites in the gene sequence and different variants makes the design of marker systems more complex and expensive [26]. Therefore, another approach based on target gene sequencing (TGS) was used. Different TGS methods have been developed over the last few years, but they are still time-consuming and require a certain level of molecular biology skills. The TGS methods can be divided into two groups: target amplicon sequencing (TAS) and target DNA ‘fishing’ and sequencing (TAFS). While the former approaches are based on PCR amplification, the latter (e.g., nCATS [27] and hybridization-based capture sequencing [28]) can be used without an amplification step. The biggest disadvantage of TAFS methods is the requirement of a large amount of DNA and labor-intensive procedures to achieve target DNA enrichment. The use of these methods for high-throughput and rapid TGS is challenging. The TAS methods are more sensitive and easier to scale up. Both methods involve a DNA sequencing step, and next-generation sequencing (NGS) is usually used for this [29,30,31]. Short-read NGS is commonly applied and has numerous advantages for TGS, including high throughput, low cost per sample, and low error rate. Short-read NGS sequencing of amplicons has been widely used for MAS [17,32].
It is essential to build TGS systems for plants to make the entire TGS procedure more rapid, cheaper, and less labor available for plant breeders worldwide. Short-read TAS requires expensive equipment and a high initial investment; therefore, the application of this approach in crop fields is not possible. Oxford Nanopore Technologies (ONT) provides a tiny sequencer, MinION, which allows sequencing to run even in the field, as it only requires a USB connection to the laptop [33,34]. ONT sequencing has been used for TGS [35,36,37,38]. In our earlier study, we showed that we could quickly detect structural variations in promoters and coding regions, as well as new allele variants in large and complex plant genomes, by combining PCR amplification of individual genes and ligation-based barcoding [39]. However, when analyzing a large number of samples, this strategy can be costly and time-consuming because of the time re-quired to amplify each individual gene and the length of time required to assemble the library.
In this study, we significantly improved and simplified the ONT-TAS procedure by using multiplex PCR and a rapid sequencing kit. We showed the efficiency of this novel strategy by rapid sequencing of four genes (Ahasl1, Ahasl2, Ahasl3, FAD2) in forty sunflower (Helianthus annuus) plants and three genes (Ppo, Wx, Lox) in thirty wheat (Triticum aestivum) plants. The results revealed a high level of polymorphism scattered over the gene sequences. We also identified alleles of three 3 sunflower genes (Ahasl1, Ahasl3, and FAD2) and two wheat genes (Ppo-D1b and Lox1) that differed by InDels and verified the results by PCR and Sanger sequencing. Overall, we demonstrated that the described ONT-TAS procedure is relatively simple, takes only a few days, and is not laborious for sequencing and genotyping target genes in sunflower (Helianthus annuus) and wheat (Triticum aestivum).
2. Materials and Methods
2.1. Plant Materials
For this study, the sunflower seeds (Supplementary Table S1) obtained from V.S. Pustovoit All-Russian Research Institute of Oil Crops (Krasnodar, Russia) and spring bread wheat cultivars (Supplementary Table S2) of different geographical ori-gins were used. Seeds were germinated at room temperature on wet filter paper disks. High-molecular-weight DNA was isolated from 200 to 500 mg of the material, which was homogenized in liquid nitrogen. DNA isolation was done according to the pub-lished protocol (https://www.protocols.io/view/plant-dna-extraction-and-preparation-for389-ont-seque-bcvyiw7w, accessed on 4 September 2021). The concentration and quality of isolat-ed DNA were assessed using a NanoDrop One UV-Vis Spectrophotometer (Thermo Scientific, Waltham, MA, USA). For amplification, equal concentrations of DNA were used, according to the manufacturer’s instructions.
2.2. Multiplex-PCR
PCR amplification was conducted using specific primers (Supplementary Table S3), which were designed using Primer 3.0 software (https://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi, accessed on Sep-tember 1, 2022). In multiplex PCR, two or more primer sets designed for the amplifica-tion of different targets are included in the same PCR reaction. Thus, the primer sets must be amplified under the same conditions. The PCR conditions were optimized for primer pairs, and optimal conditions for multiplex PCR were achieved.
For sunflower, PCR was performed in a 20 µL mixture containing 2× BioMaster LR HS-PCR (Biolabmix, Novosibirsk, Russia) reaction mixture, 0.2 µL of working solution of each primer, and 20 ng of DNA. Amplification was performed under the following temperature conditions using a mixture of four primer pairs: 35 cycles of denaturation at 94°С for 20 s, primer annealing at 60°С for 30 s, and elongation at 68°С for 2 min.
For wheat, PCR was performed in a 20 µL mixture containing 2× BioMaster LR HS-PCR (Biolabmix, Novosibirsk, Russia) reaction mixture, 0.4 µL of working solution of each primer, 40 ng of DNA, and 5% DMSO. Amplification was performed under the following temperature conditions using a mixture of three primer pairs: 35 cycles of denaturation at 94°С for 20 s, primer annealing at 65°С for 30 s, and elongation at 68°С for 3 min.
The multiplex PCR results were visualized via gel electrophoresis using a 1% aga-rose gel with ethidium bromide staining.
Multiplex amplicons were purified using 1× Agencourt AMPure XP Beads (Beck-man Coulter, Pasadena, CA, USA) in accordance with the manufacturer’s instructions. The purified amplicon concentration and integrity were estimated using a NanoDrop One UV-Vis Spectrophotometer (Thermo Scientific, Waltham, MA, USA) and Qubit (Qubit dsDNA BR Assay Kits, Thermo Fisher Scientific, Waltham, MA, USA), respec-tively, and checked by gel electrophoresis. The PCR products were equalized in con-centration for Nanopore sequencing.
2.3. Library Preparation and Sequencing
For Native barcoding kit, we used phosphorylated primers for PCR amplification. For Nanopore sequencing, a library was prepared from pooled samples using the nanopore native barcoding genomic DNA SQK-NBD110-24 (Oxford Nanopore Technologies, Oxford, UK), with some modifications in the process of using the NEBNext Companion Module for Oxford Nanopore Technologies Ligation Sequencing (New England Biolabs, MA, USA). Briefly, ~100 ng of each pooling sample in 4.5 µL was mixed with 0.5 µL Native Barcode and 5 µL Blunt/TA Ligase Master Mix and incubated on a Hula mixer for 10 min at room temperature. Purification by Agencourt AMPure XP Beads was per-formed during phosphorylation. Each of the 12 barcoded samples was resuspended in 2.7 µL of nuclease-free water and transferred to a new LoBind tube for adapter ligation. Then, ~32.5 µL were pooled and barcoded amplicons were mixed with 10 µL NEBNext Quick Ligation Reaction Buffer (5X), 5 µL Quick T4 DNA Ligase, and 2.5 µL Adapter Mix II (AMII). Adapter Ligation Mix was incubated on a Hula mixer for 10 min at room temperature. Double washing was performed using 125 µL of Short Fragment Buffer (SFB). Incubation was performed in a water bath at 37 °C for 10 min, and then for 5 min at room temperature. Sequencing was carried out using MinION and a flow cell SQK-LSK109. Basecalling was performed by Guppy (Version 6.3.8).
For Rapid barcoding kit, each sample was mixed with 9μl of the multiplexing PCR product (50 ng) and 1 μl of one rapid barcode. The mixture was incubated at 30°C for 2 min followed by incubation at 80°C for 2 min. All barcoded DNA samples were pooled, and 800 μl of pooled DNA was mixed with an equal volume of AMPure XP Beads (Beckman Coulter, Pasadena, CA, USA). After 5 min of incubation at room temperature on a Hula mixer, the barcoded DNA was cleaned twice with 80% ethanol and eluted with 15 μl of Elution Buffer (EB). Incubation was performed at 37°С for 30 min.
An aliquot of the barcoded DNA was used to obtain a total volume of 11 μl with EB. One microliter of Rapid Adapter F (RAP F) was added to the barcoded DNA and the mixture was incubated at room temperature for 5 min. Then, 12 µL barcoded am-plicons were mixed with 37.5 µL Sequencing Buffer II (SBII), 25.5 µL Loading Beads II (LBII). Sequencing was performed using MinION and the flow cell SQK-LSK109. Basecalling was performed by Guppy (Version 6.3.8).
2.4. PCR Validation of InDels
To validate the InDels located in the sunflower and wheat genes, which were iden-tified by amplicon sequencing, the primers listed in Supplementary Table S4 were used. PCR was performed using Encyclo DNA polymerase (Evrogen, Moscow, Russia) ac-cording to the manufacturer’s instructions. PCR conditions were specific for each InDel (Supplementary Table S5). To confirm the presence of InDels, the PCR products were directly sequenced by Sanger sequencing.
2.5. SNP Calling
For variant calling, the ONT reads were mapped to the reference sequences using minimap2 [40]. The obtained BAM files were sorted and indexed using Samtools [41].
Primary SNP calling was carried out using Calir3 (https://github.com/HKU-BAL/Clair3, accessed on July 28, 2024) with r941_prom_hac_g360+g42 model. The obtained vcf files of different samples were fil-tered (--remove-filtered-all --minQ 10) and merged using vcftools [42]. Phylogenetic tree was built from the merged vcf files using VCF2PopTree [43]. SNPs were annotated using SNPeff [44].
3. Results
3.1. Three Approaches for Nanopore Amplicon Sequencing.
Three amplicon sequencing experiments were performed to determine the optimal library preparation method for sunflower and wheat collections (Table 1).
For sunflower, we choose three genes (Ahasl1, Ahasl2, Ahasl3) involved in herbicide – resistance [21,22,23,24] and one gene (FAD2) that is responsible for high oleic acid content of sunflower seeds [26]. The first ONT sequencing experiment was performed using the native barcoding kit. Primers (Figure 1A) were designed to amplify the four sunflower genes (Ahasl1, Ahasl2, Ahasl3, and FAD2) (Figure 1C). The PCR products were barcoded according to sunflower genotype using a Native Barcoding Kit and sequenced on a single MinION flow cell. The sequencing procedure took 2 h, resulted in 341,477 reads. The number of reads per gene per sample varied between 6,222 and 20,850, providing sufficient gene coverage for downstream analysis. Each primer pair was amplified separately for the sunflower genotypes. All PCR products were purified, pooled according to the sample number, and barcoded according to the manufacturer’s protocol. Owing to the limited number of barcodes, 12 barcodes were first loaded into the flow cell, and another part of the barcodes was loaded after washing. Analysis of allelic variants was carried out by comparing the obtained reads with the reference sequences Ahasl1 (NC_035441.2), Ahasl2 (NC_035438.2), Ahasl3 (AY541458.1), and FAD2 (FJ791046.1).
To make the ONT-TAS procedure more robust and routine, we performed two im-portant modifications in the next experiment. First, we optimized the multiplex ampli-fication procedure and achieved amplification of all four genes in a single tube during one PCR reaction (Figure 1D). Amplicons were successfully generated from the 40 sun-flower genotypes. Each amplicon contained the following PCR products: Ahasl1 (2,000 bp), Ahasl2 (2,010 bp), Ahasl3 (1,921 bp), and FAD2 (2,000 bp). Second, we used a Rapid Barcoding Kit instead of the native barcoding kit. The Rapid Barcoding Kit per-formed transpose-mediated barcoding and allowed the barcoding procedure to be completed within 10 minutes. These changes in the ONT-TAS significantly reduced the time required for library preparation and made it less laborious. Thus, 40 sunflower samples were amplified using a mixture of the four primer pairs, resulting in 40 amplicons at a time with four different products of each size. Mapping of the reads to the reference genes showed that all target genes were sequenced for all sunflower genotypes, implying the successful amplification of the genes by multiplex PCR. We evaluated the length of the obtained reads and the target gene coverage resulted from the application of the Native Barcoding Kit (Figure 1F) and the Rapid Barcoding Kit (Figure 1G). We found that the median coverage values for Ahasl1, Ahasl2, Ahasl3, and FAD2 was obtained using the Rapid Barcoding Kit varied between 13x-214x, 198x-575x, 360x-1097x and 14x-240x, respectively (Figure 1H). These values were lower than those of the Native Barcoding kit. However, the obtained coverage values for the Rapid Barcoding Kit were still sufficient for SNP calling and downstream analyses.
Previously, we used a Native Barcoding Kit to sequence the target wheat genes [39]. In this study we applied the Multiplex + Rapid Barcoding strategy to sequence three wheat genes (Ppo, Wx, and Lox) of 30 wheat lines. To amplify target genes, primers were designed for the coding and promoter regions specific to the three subgenomes (Figure 1B). Although the primers were designed for multiplex PCR, the PCR conditions for each primer pair were optimized to achieve optimal multiplex conditions. Subsequently, 30 amplicons containing three different-sized products, Ppo (2,200 bp), Wx (2,900 bp), and Lox (3,000 bp), were obtained (Figure 1E). The obtained reads were aligned to the reference sequences of wheat genes from NCBI: EF070148.1 (Ppo-A1b), GQ303713.1 (Ppo-B1a), EF070150.1(Ppo-D1b), GQ166692.1 (Lox1), HQ406780.1 (Lox-B1b), KC679302.1 (Lpx-D1), MT048401.1 (Wx-A1q), KF861808.1 (Wx-B1l), LC373576.1 (Wx-D1g). The sequencing yield >200,000 reads and resulting in up to 40× target gene coverage.
Thus, the results showed that TAS using a combination of Multiplex PCR and Rapid Barcoding Kit allowed rapid sequencing of target genes with high coverage values.
3.2. SNP Validation in Sunflower Varieties.
We then performed SNP calling using Clair3 [45] followed by variant annotation using SNPeff [46]. The resulting vcf files contained 246 SNPs among the target genes of the sunflower varieties. Ahasl1 (Figure 2A) had the highest number (189 SNPs), of which 72% (137 SNPs) belonged to synonymous SNPs, and 28% (52 SNPs) were nonsynonymous SNPs (nsSNPs). Ahasl3 (Figure 2B) showed 6 nsSNPs and 48 synonymous SNPs, representing 22% (54 SNPs) of all identified SNPs. Overall, according to SNP annotation, 23.6% (58 SNPs) of the identified SNPs were missense, while more than 76% (188 SNPs) were synonymous.
In terms of the number of SNPs, the most divergent sunflower sample was RHA450, which had 43 SNPs, including 16 synonymous and 6 non-synonymous SNPs for Ahasl1 and 17 synonymous and 4 nsSNPs for Ahasl3. The minimum number of SNPs was observed for Ahasl1 (one synonymous SNP) and FAD2 (two synonymous SNPs) in the VK464 and ZS samples, respectively.
Further the proportions of 12 mutation types were considered (Figure 2C). The most frequent mutations were C-to-T (34 SNPs), A-to-G (30 SNPs), A-to-C (30 SNPs), G-to-A (29 SNPs), G-to-T (25 SNPs), and G-to-C (19 SNPs). Only the G-to-T mutation was 60% represented by non-synonymous SNPs, while the rest (A-to-G, G-to-A, C-to-T) were 70% or more represented by synonymous mutations. Among the 12 possible substitutions, only 4 (A-to-C, G-to-C, A-to-T, T-to-A) were represented by fully synonymous mutations of all substitutions, of which the least represented substitutions were A-to-T and T-to-A, with one and two synonymous SNPs, respectively. It was found that those synonymous mutations that change the GC content represent for about 35% of all mutations and are represented by A-to-C (40%), A-to-G (40%), T-to-C (16%), T-to-G (4%), of which are not synonymous with A-to-G (8 SNPs), T-to-C (6 SNPs), T-to-G (2 SNPs).
Based on the analysis of four target genes in the sunflower collection, Ahasl1 (Fig-ure 2C) accounted for the largest number of variations (189 SNPs), most of which (137 SNPs) were synonymous. The remaining 52 SNPs were non-synonymous substitutions. The Ahasl3 gene (six SNPs) had almost nine times fewer non-synonymous substitutions than the Ahasl1 gene in the sunflower collection. The smallest number of SNPs was found in Ahasl2 (one SNP) and FAD2 (two SNPs). To further explore the genetic diversity of the sunflower collections using ONT-TAS data, we constructed a phylogenetic tree based on the detected SNPs. This analysis revealed five groups of sunflower genotypes (Supplementary Figure S1, Supplementary Table S6) that differed significantly by the SNPs in Ahasl1. Additionally, we identified 1, 3, 1, 1, and 34 sunflower genotypes that clustered into five groups according to Ahasl3 SNPs (Supplementary Table S6). The results showed significant diversity of Ahasl1 and Ahasl3 genes in the sunflower collection, unraveled by the ONT-TAS procedure.
Using a similar approach, we analyzed the diversity of the target wheat genes. We detected two, three, and three different alleles for the Ppo-A, Ppo-B, and Ppo-D genes, respectively. Six, one, and two alleles were identified for the Wx-A, Wx-B, and Wx-D genes, respectively. Additionally, one, six, and one allele were found for the Lox-A, Lox-B, and Lox-D genes, respectively. Thus, Wx-A and Lox-B genes had the highest number of alleles in our collections.
Taken together, the ONT-TAS approach allowed us to rapidly characterize the ge-netic diversity of the target genes in sunflower and wheat genes in the germplasm col-lections.
3.3. Analysis of Structural Variations
Structural variations were discovered during the analysis of the sunflower SNPs. In the sunflower genotypes analyzed, a deletion of approximately 10 bp was found within the tandem regions of FAD2 (Supplementary Figure S2). This deletion, which occurs in repetitive regions, highlights the complexity of assembling and analyzing long tandem repeats in these plants. Additionally, a deletion of approximately 10 bp was detected in Ahasl1. This deletion was observed in ten sunflower samples and was located in the coding region of the gene. Upon comparing the obtained Ahasl3 reads with the reference sequences, an insertion of approximately 15 bp was discovered (Figure 3A). This insertion was located within the coding region of the gene in RHA450 and ZC samples. Flanking primers were designed for all identified InDels for PCR analysis (Figure 3C) and Sanger sequencing (Figure 3D).
Structural variations were identified during the analysis of the wheat ONT amplicon sequencing data. InDels within the Ppo-D1 gene (~ 30 bp) were identified in a single sample, whereas the Lox1 gene (~ 300 bp) was identified in four samples (Figure 2B). Both InDels were confirmed by Sanger sequencing (Figure 3D).
4. Discussion
In this study, we showed that the combination of multiplex PCR and Rapid Barcoding procedure provides a rapid and cost-effective approach to detect SNPs and SVs in full-length target genes across multiple samples of plant collections. We verified this approach to determine the genetic variation in four (Ahasl1, Ahasl2, Ahasl3, and FAD2) and three (Ppo, Wx, and Lox) wheat genes in collections comprising 30 and 40 samples, respectively. The obtained data explicitly showed the distribution of SNPs and InDels along full-length genes, providing a complete picture of the target gene di-versity in the germplasm collection. Previously, we [39] and others [47] used Native Barcoding Kit for amplicon sequencing. We performed a comparative evaluation of Native Barcoding Kit and Rapid Barcoding Kit + Multiplex. These approaches differ in terms of price and time required. For the native barcoding approach, the amplicons of each wheat sample (92 amplicons in our case) were individually amplified, purified, and pooled to obtain the final samples for barcoding. The phosphorylated PCR amplicons from each sample were barcoded, combined in one tube, and sequenced. Alt-hough this approach resulted in longer and intact reads, it was more expensive because it requires third party reagent (Ligase Master Mix) and more PCR reactions. Ad-ditionally, the library preparation for the Native Barcoding Kit is more time-consuming and takes nearly 3 hours compared to 1.5 hours required for Rapid Barcoding Kit. The estimated price of sequencing a single sample using this approach was approximately 16.15$, or 4$ per gene (Table 2). Application of multiplex PCR together with the Rapid Barcoding Kit further reduced the cost of ONT-TAS sequencing to 13.61$, or 3.4$ per gene. This price was calculated considering that up to six sequencing runs could be carried out for a single MinION flow cell. This price is even lower than that of Sanger sequencing (for example, 5.25$ per sample proposed by https://eurofinsgenomics.com/en/products/pricing/). However, in contrast to Sanger sequencing, the ONT-TAS procedure can easily be used for long PCR products (up to 3Kb in this study).
There are still some obstacles that limit the broad application of ONT-TAS in full-length gene sequencing. First, there is no user-friendly software for easy down-stream analysis of ONT-TAS data, including variant calling, phylogeny analysis, and novel and known allele identification. Second, ONT reads possess a relatively high error rate compared to traditional short-read sequencing methods, such as Illumina [48]. Although single nucleotide sequencing errors can be easily discriminated as they occur randomly along the reads, homopolymeric regions are particularly problematic. Errors in these areas often manifest as deletions, making it difficult to accurately determine the length of homopolymers [48]. The error rate in the ONT data is directly related to the version of the library preparation chemistry and flow cell type. In this study, we used V10 chemistry and R9 nanopore flow cells, which are prone to produce reads with high error rates. However, a combination of the R10 flow cell (for example, R10.4.1) and the latest Q20+ ONT chemistry results in significantly lower error rate and can further improve ONT-TAS data in future [49,50]. Overcoming these challenges is necessary to make ONT-TAS routinely used for marker-assisted selection, where hundreds of individuals are usually genotyped within a limited time.
ONT amplicon sequencing has been widely used for 16S ribosomal RNA (rRNA) sequencing in metagenomic studies. This method offers significant advantages over traditional short-read sequencing techniques, allowing for comprehensive analysis of complex microbial communities [51]. There are also several reports on the application of ONT-TAS for sequencing eukaryotic protein-coding genes [37,38,39,47,52,53]. These and our current studies highlight the key advantages of ONT-TAS, which make it useful for broader applications for plant genotyping and marker-assisted selection. First, ONT-TAS allows the accurate identification of SNPs across multiple samples simultaneously. Second, ONT-TAS can be used in combination with multiplex PCR to reduce price and turnaround time. Third, in addition to SNPs, ONT-TAS can also characterize structural variants owing to its long-read capabilities. Fourth, the real-time sequencing capability of nanopore technology allows immediate data analysis and interpretation of results. Finally, the portable nature of nanopore sequencers enables their use in field settings, which makes them suitable for rapid genotyping in diverse environments. These features offer several distinct benefits for the application of ONT-TAS in other areas including CRISPR validation. ONT-TAS allows for comprehensive coverage of target regions and adjacent sequences and helps to accurately identify structural variants, confirming the integrity of the modified genomic context, which is often missed by short-read technologies. This is especially important when several sgRNAs are ap-plied, and different structural modifications can occur [54,55,56].
5. Conclusions
This study demonstrates the potential of the ONT-TAS method for high-throughput genotyping of full-length target genes in plant collections. The combination of multiplex PCR and Rapid Sequencing Kit for the ONT-TAS approach provides sufficient in-formation for SNP calling and InDel detection in the target genes across multiple genotypes and is useful for the rapid identification of known and novel alleles of desired genes.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Supplementary Figure S1. Phylogenetic tree of Ahasl1 genes of sunflower genotypes; Supplemen-tary Figure S2. Sequence analysis of FAD2 gene helps to match the reference sequence with the obtained reads for sunflower for deletion identifying; Supplementary table S1. 40 cultivars of sun-flower used in this study; Supplementary table S2. 30 cultivars of spring bread wheat used in this study; Supplementary table S3. Primers used for PCR; Supplementary table S4. Primers used for SV identifications; Supplementary table S5. PCR conditions for SV validation; Supplementary table S6. Ahasl and Ahasl3 alleles identified in this study.
Author Contributions
Conceptualization, I.K.; methodology, E.P. and I.K.; software, I.K.; validation, E.P., E.M., P.M., K.D., I.G. and M.D.; formal analysis, E.P.; investigation, E.P., E.M., P.M. and K.D.; resources, A.S and Y.D.; data curation, I.K.; writing—original draft preparation, E.P.; writing—review and editing, I.K.; visualization, E.P.; supervision, I.K.; project administration, A.S.; funding acquisition, A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation (grant no. 22-64-00076, method development and its application for the sunflower gene sequencing) and the Ministry of Education and Science of the Russian Federation (goszadanie No. FGUM-2022-0005) (wheat gene sequencing).
Data Availability Statement
The nanopore data produced for this study are available in the Sequence Read Archive (SRA) NCBI under the Bioproject Accession PRJNAXXXX.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Xiong, H.; Li, Y.; Guo, H.; Xie, Y.; Zhao, L.; Gu, J.; Zhao, S.; Ding, Y.; Liu, L. Genetic Mapping by Integration of 55K SNP Array and KASP Markers Reveals Candidate Genes for Important Agronomic Traits in Hexaploid Wheat. Front. Plant Sci. 2021, 12, 628478. [CrossRef]
- Kaur, B.; Mavi, G.S.; Gill, M.S.; Saini, D.K. Utilization of KASP Technology for Wheat Improvement. Céréal. Res. Commun. 2020, 48, 409–421. [CrossRef]
- Hou, Y.; Luo, Q.; Chen, C.; Zhou, M. Application of Cycleave PCR to the Detection of a Point Mutation (F167Y) in the Β2-tubulin Gene of Fusarium Graminearum. Pest Manag. Sci. 2011, 67, 1124–1128. [CrossRef]
- Bettinaglio, P.; Galbusera, A.; Caprioli, J.; Orisio, S.; Perna, A.; Arnoldi, F.; Bucchioni, S.; Noris, M.; Group, on be-half of the B.S. Single Strand Conformation Polymorphism (SSCP) as a Quick and Reliable Method to Genotype M235T Polymorphism of Angiotensinogen Gene. Clin. Biochem. 2002, 35, 363–368. [CrossRef]
- Kovalchuk, S.N.; Arkhipova, A.L. Development of TaqMan PCR Assay for Genotyping SNP Rs211250281 of the Bovine Agpat6 Gene. Anim. Biotechnol. 2023, 34, 3250–3255. [CrossRef]
- Kang, D.-Y.; Cheon, K.-S.; Oh, J.; Oh, H.; Kim, S.L.; Kim, N.; Lee, E.; Choi, I.; Baek, J.; Kim, K.-H.; et al. Rice Genome Resequencing Reveals a Major Quantitative Trait Locus for Resistance to Bakanae Disease Caused by Fusarium Fu-jikuroi. Int. J. Mol. Sci. 2019, 20, 2598. [CrossRef]
- Ayalew, H.; Tsang, P.W.; Chu, C.; Wang, J.; Liu, S.; Chen, C.; Ma, X.-F. Comparison of TaqMan, KASP and RhAmp SNP Genotyping Platforms in Hexaploid Wheat. PLoS ONE 2019, 14, e0217222. [CrossRef]
- Salgotra, R.K.; Stewart, C.N. Functional Markers for Precision Plant Breeding. Int. J. Mol. Sci. 2020, 21, 4792. [CrossRef]
- Devran, Z.; Kahveci, E. Development and Validation of a User-Friendly KASP Marker for the Sw-5 Locus in Tomato. Australas. Plant Pathol. 2019, 48, 503–507. [CrossRef]
- Mangal, V.; Sood, S.; Bhardwaj, V.; Kumar, V.; Kumar, A.; Singh, B.; Dipta, B.; Dalamu, D.; Sharma, S.; Thakur, A.K.; et al. Diagnostic PCR-Based Markers for Biotic Stress Resistance Breeding in Potatoes (Solanum Tuberosum L.). Australas. Plant Pathol. 2023, 52, 227–240. [CrossRef]
- Jagtap, A.B.; Vikal, Y.; Johal, G.S. Genome-Wide Development and Validation of Cost-Effective KASP Marker Assays for Genetic Dissection of Heat Stress Tolerance in Maize. Int. J. Mol. Sci. 2020, 21, 7386. [CrossRef]
- Nair, S.K.; Babu, R.; Magorokosho, C.; Mahuku, G.; Semagn, K.; Beyene, Y.; Das, B.; Makumbi, D.; Kumar, P.L.; Olsen, M.; et al. Fine Mapping of Msv1, a Major QTL for Resistance to Maize Streak Virus Leads to Development of Production Markers for Breeding Pipelines. Theor. Appl. Genet. 2015, 128, 1839–1854. [CrossRef]
- Sieber, A.-N.; Longin, C.F.H.; Leiser, W.L.; Würschum, T. Copy Number Variation of CBF-A14 at the Fr-A2 Locus Determines Frost Tolerance in Winter Durum Wheat. Theor. Appl. Genet. 2016, 129, 1087–1097. [CrossRef]
- Yang, F.; Liu, Q.; Wang, Q.; Yang, N.; Li, J.; Wan, H.; Liu, Z.; Yang, S.; Wang, Y.; Zhang, J.; et al. Characterization of the Durum Wheat-Aegilops Tauschii 4D(4B) Disomic Substitution Line YL-443 With Superior Characteristics of High Yielding and Stripe Rust Resistance. Front. Plant Sci. 2021, 12, 745290. [CrossRef]
- Dipta, B.; Sood, S.; Mangal, V.; Bhardwaj, V.; Thakur, A.K.; Kumar, V.; Singh, B. KASP: A High-Throughput Gen-otyping System and Its Applications in Major Crop Plants for Biotic and Abiotic Stress Tolerance. Mol. Biol. Rep. 2024, 51, 508. [CrossRef]
- Nucia, A.; Okoń, S.; Tomczyńska-Mleko, M. Characterization of HMW Glutenin Subunits in European Spring Common Wheat (Triticum Aestivum L.). Genet. Resour. Crop Evol. 2019, 66, 579–588. [CrossRef]
- Bernardo, A.; Wang, S.; Amand, P.St.; Bai, G. Using Next Generation Sequencing for Multiplexed Trait-Linked Markers in Wheat. PLoS ONE 2015, 10, e0143890. [CrossRef]
- Schuppert, G.F.; Tang, S.; Slabaugh, M.B.; Knapp, S.J. The Sunflower High-Oleic Mutant Ol Carries Variable Tandem Repeats of FAD2-1, a Seed-Specific Oleoyl-Phosphatidyl Choline Desaturase. Mol. Breed. 2006, 17, 241–256. [CrossRef]
- Lacombe, S.; Souyris, I.; Bervillé, A.J. An Insertion of Oleate Desaturase Homologous Sequence Silences via SiRNA the Functional Gene Leading to High Oleic Acid Content in Sunflower Seed Oil. Mol. Genet. Genom. 2009, 281, 43–54. [CrossRef]
- Dimitrijevic; Imerovski; Miladinović; Jocković; Cvejić; Jocić; Sakač Screening of the Presence of Ol Gene in NS Sunflower Collection. Proceedings, 19th International Sunflower Conference 2003, С. 660-666.
- Kolkman, J.M.; Slabaugh, M.B.; Bruniard, J.M.; Berry, S.; Bushman, B.S.; Olungu, C.; Maes, N.; Abratti, G.; Zam-belli, A.; Miller, J.F.; et al. Acetohydroxyacid Synthase Mutations Conferring Resistance to Imidazolinone or Sul-fonylurea Herbicides in Sunflower. Theor. Appl. Genet. 2004, 109, 1147–1159. [CrossRef]
- Bruniard, J.M.; Miller, J.F. Inheritance of imidazolinone-herbicide resistance in sunflower/herencia de la resistencia a imidazolinonas en girasol/hérédité de la résistance à l’herbicide imidazolinone chez le tournesol. HELIA 2001, 24, 11–16. [CrossRef]
- Sala, C.A.; Bulos, M.; Echarte, M.; Whitt, S.R.; Ascenzi, R. Molecular and Biochemical Characterization of an Induced Mutation Conferring Imidazolinone Resistance in Sunflower. Theor. Appl. Genet. 2008, 118, 105. [CrossRef]
- Sala, C.A.; Bulos, M. Inheritance and Molecular Characterization of Broad Range Tolerance to Herbicides Targeting Acetohydroxyacid Synthase in Sunflower. Theor. Appl. Genet. 2012, 124, 355–364. [CrossRef]
- Sala, C.A.; Bulos, M.; Altieri, E.; Ramos, M.L. Genetics and Breeding of Herbicide Tolerance in Sunflower. Helia 2012, 35, 57–69. [CrossRef]
- Dimitrijević, A.; Imerovski, I.; Miladinović, D.; Cvejić, S.; Jocić, S.; Zeremski, T.; Sakač, Z. Oleic Acid Variation and Marker-Assisted Detection of Pervenets Mutation in High- and Low-Oleic Sunflower Cross. Crop Breed. Appl. Biotechnol. 2017, 17, 235–241. [CrossRef]
- Kirov, I.; Polkhovskaya, E.; Dudnikov, M.; Merkulov, P.; Vlasova, A.; Karlov, G.; Soloviev, A. Searching for a Needle in a Haystack: Cas9-Targeted Nanopore Sequencing and DNA Methylation Profiling of Full-Length Glutenin Genes in a Big Cereal Genome. Plants 2021, 11, 5. [CrossRef]
- Ceballos-Garzon, A.; Comtet-Marre, S.; Peyret, P. Applying Targeted Gene Hybridization Capture to Viruses with a Focus to SARS-CoV-2. Virus Res. 2024, 340, 199293. [CrossRef]
- Taheri, S.; Abdullah, T.L.; Yusop, M.R.; Hanafi, M.M.; Sahebi, M.; Azizi, P.; Shamshiri, R.R. Mining and Devel-opment of Novel SSR Markers Using Next Generation Sequencing (NGS) Data in Plants. Molecules 2018, 23, 399. [CrossRef]
- Michael, T.P.; Bryant, D.; Gutierrez, R.; Borisjuk, N.; Chu, P.; Zhang, H.; Xia, J.; Zhou, J.; Peng, H.; Baidouri, M.E.; et al. Comprehensive Definition of Genome Features in Spirodela Polyrhiza by High-depth Physical Mapping and Short-read DNA Sequencing Strategies. Plant J. 2017, 89, 617–635. [CrossRef]
- Onda, Y.; Takahagi, K.; Shimizu, M.; Inoue, K.; Mochida, K. Multiplex PCR Targeted Amplicon Sequencing (MTA-Seq): Simple, Flexible, and Versatile SNP Genotyping by Highly Multiplexed PCR Amplicon Sequencing. Front. Plant Sci. 2018, 9, 201. [CrossRef]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.-X.; Liu, H.; Li, Z. Genotyping-by-Sequencing (GBS), an Ultimate Marker-Assisted Selection (MAS) Tool to Accelerate Plant Breeding. Front. Plant Sci. 2014, 5, 484. [CrossRef]
- Liou, C.-H.; Wu, H.-C.; Liao, Y.-C.; Lauderdale, T.-L.Y.; Huang, I.-W.; Chen, F.-J. NanoMLST: Accurate Multilocus Sequence Typing Using Oxford Nanopore Technologies MinION with a Dual-Barcode Approach to Multiplex Large Numbers of Samples. Microb. Genom. 2020, 6, e000336. [CrossRef]
- Sheka, D.; Alabi, N.; Gordon, P.M.K. Oxford Nanopore Sequencing in Clinical Microbiology and Infection Diagnostics. Brief. Bioinform. 2021, 22, bbaa403. [CrossRef]
- Latz, M.A.C.; Grujcic, V.; Brugel, S.; Lycken, J.; John, U.; Karlson, B.; Andersson, A.; Andersson, A.F. Short- and Long-read Metabarcoding of the Eukaryotic RRNA Operon: Evaluation of Primers and Comparison to Shotgun Metagenomics Sequencing. Mol. Ecol. Resour. 2022, 22, 2304–2318. [CrossRef]
- Karst, S.M.; Ziels, R.M.; Kirkegaard, R.H.; Sørensen, E.A.; McDonald, D.; Zhu, Q.; Knight, R.; Albertsen, M. High-Accuracy Long-Read Amplicon Sequences Using Unique Molecular Identifiers with Nanopore or PacBio Sequencing. Nat. Methods 2021, 18, 165–169. [CrossRef]
- Marcolungo, L.; Passera, A.; Maestri, S.; Segala, E.; Alfano, M.; Gaffuri, F.; Marturano, G.; Casati, P.; Bianco, P.A.; Delledonne, M. Real-Time On-Site Diagnosis of Quarantine Pathogens in Plant Tissues by Nanopore-Based Sequencing. Pathogens 2022, 11, 199. [CrossRef]
- Mimosa, M.L.; Alameri, W.; Simpson, J.T.; Nakhla, M.; Boissinot, K.; Munoz, D.G.; Das, S.; Feilotter, H.; Fattouh, R.; Saleeb, R.M. A Novel Approach to Detect IDH Point Mutations in Gliomas Using Nanopore Sequencing Test Validation for the Clinical Laboratory. J. Mol. Diagn. 2023, 25, 133–142. [CrossRef]
- Polkhovskaya, E.; Gruzdev, I.; Moskalev, E.; Merkulov, P.; Bolotina, A.; Soloviev, A.; Kirov, I. Nanopore Amplicon Sequencing Allows Rapid Identification of Glutenin Allelic Variants in a Wheat Collection. Agronomy 2023, 14, 13. [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, 1000 Genome Project Data Processing The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [CrossRef]
- Subramanian, S.; Ramasamy, U.; Chen, D. VCF2PopTree: A Client-Side Software to Construct Population Phylogeny from Genome-Wide SNPs. PeerJ 2019, 7, e8213. [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff. Fly 2012, 6, 80–92. [CrossRef]
- Su, J.; Zheng, Z.; Ahmed, S.S.; Lam, T.-W.; Luo, R. Clair3-Trio: High-Performance Nanopore Long-Read Variant Calling in Family Trios with Trio-to-Trio Deep Neural Networks. Brief. Bioinform. 2022, 23, bbac301. [CrossRef]
- Cingolani, P. Variant Calling, Methods and Protocols. Methods Mol. Biol. 2012, 2493, 289–314. [CrossRef]
- Whitford, W.; Hawkins, V.; Moodley, K.S.; Grant, M.J.; Lehnert, K.; Snell, R.G.; Jacobsen, J.C. Proof of Concept for Multiplex Amplicon Sequencing for Mutation Identification Using the MinION Nanopore Sequencer. Sci. Rep. 2022, 12, 8572. [CrossRef]
- Delahaye, C.; Nicolas, J. Sequencing DNA with nanopores: Troubles and biases. PLoS One 2021, 16, e0257521. [CrossRef]
- Koren, S.; Bao, Z.; Guarracino, A.; Ou, S.; Goodwin, S.; Jenike, K.M.; Lucas, J.; McNulty, B.; Park, J.; Rautiainen, M.; et al. Gapless assembly of complete human and plant chromosomes using only nanopore sequencing. Genome Res 2024, 34, 1919-1930. [CrossRef]
- Chen, Y.; Nie, F.; Xie, S.Q.; Zheng, Y.F.; Dai, Q.; Bray, T.; Wang, Y.X.; Xing, J.F.; Huang, Z.J.; Wang, D.P.; et al. Efficient assembly of nanopore reads via highly accurate and intact error correction. Nat Commun 2021, 12, 60. [CrossRef]
- Matsuo, Y.; Komiya, S.; Yasumizu, Y.; Yasuoka, Y.; Mizushima, K.; Takagi, T.; Kryukov, K.; Fukuda, A.; Morimoto, Y.; Naito, Y.; et al. Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION nanopore sequencing confers species-level resolution. BMC Microbiol 2021, 21, 35. [CrossRef]
- Eaton, K.M.; Bernal, M.A.; Backenstose, N.J.C.; Yule, D.L.; Krabbenhoft, T.J. Nanopore Amplicon Sequencing Reveals Molecular Convergence and Local Adaptation of Rhodopsin in Great Lakes Salmonids. Genome Biol Evol 2021, 13. [CrossRef]
- Dyshlovoy, S.A.; Paigin, S.; Afflerbach, A.K.; Lobermeyer, A.; Werner, S.; Schuller, U.; Bokemeyer, C.; Schuh, A.H.; Bergmann, L.; von Amsberg, G.; et al. Applications of Nanopore sequencing in precision cancer medicine. Int J Cancer 2024, 155, 2129-2140. [CrossRef]
- Fagan, K.J.; Chillon, G.; Carrell, E.M.; Waxman, E.A.; Davidson, B.L. Cas9 editing of ATXN1 in a spinocerebellar ataxia type 1 mice and human iPSC-derived neurons. Mol Ther Nucleic Acids 2024, 35, 102317. [CrossRef]
- Sato, R.; Nanasato, Y.; Takata, N.; Nagano, S.; Fukatsu, E.; Fujino, T.; Yamaguchi, K.; Moriguchi, Y.; Shigenobu, S.; Suzuki, Y.; et al. Efficient selection of a biallelic and nonchimeric gene-edited tree using Oxford Nanopore Technologies sequencing. Tree Physiol 2024, 44. [CrossRef]
- Chey, Y.C.J.; Corbett, M.A.; Arudkumar, J.; Piltz, S.G.; Thomas, P.Q.; Adikusuma, F. CRISPR-mediated megabase-scale transgene de-duplication to generate a functional single-copy full-length humanized DMD mouse model. BMC Biol 2024, 22, 214. [CrossRef]
Figure 1.
Schematic depicting the primer positions for the four sunflower genes (А) and three wheat genes specific to subgenomes A, B, and D (B). Sunflower primers were used in two experiments: (C) PCR products were amplified separately, pooled, barcoded, and sequenced using the native barcoding kit; (D) PCR included four primer pairs, and the multiplex products were sequenced using the rapid barcoding kit. Read alignment to the reference sequence of the Ahasl1 gene after using the Native Barcoding Kit (F), the Rapid Barcoding Kit (G), and the sequence of three Wx genes (A, B, D, subgenomes) (H).
Figure 1.
Schematic depicting the primer positions for the four sunflower genes (А) and three wheat genes specific to subgenomes A, B, and D (B). Sunflower primers were used in two experiments: (C) PCR products were amplified separately, pooled, barcoded, and sequenced using the native barcoding kit; (D) PCR included four primer pairs, and the multiplex products were sequenced using the rapid barcoding kit. Read alignment to the reference sequence of the Ahasl1 gene after using the Native Barcoding Kit (F), the Rapid Barcoding Kit (G), and the sequence of three Wx genes (A, B, D, subgenomes) (H).
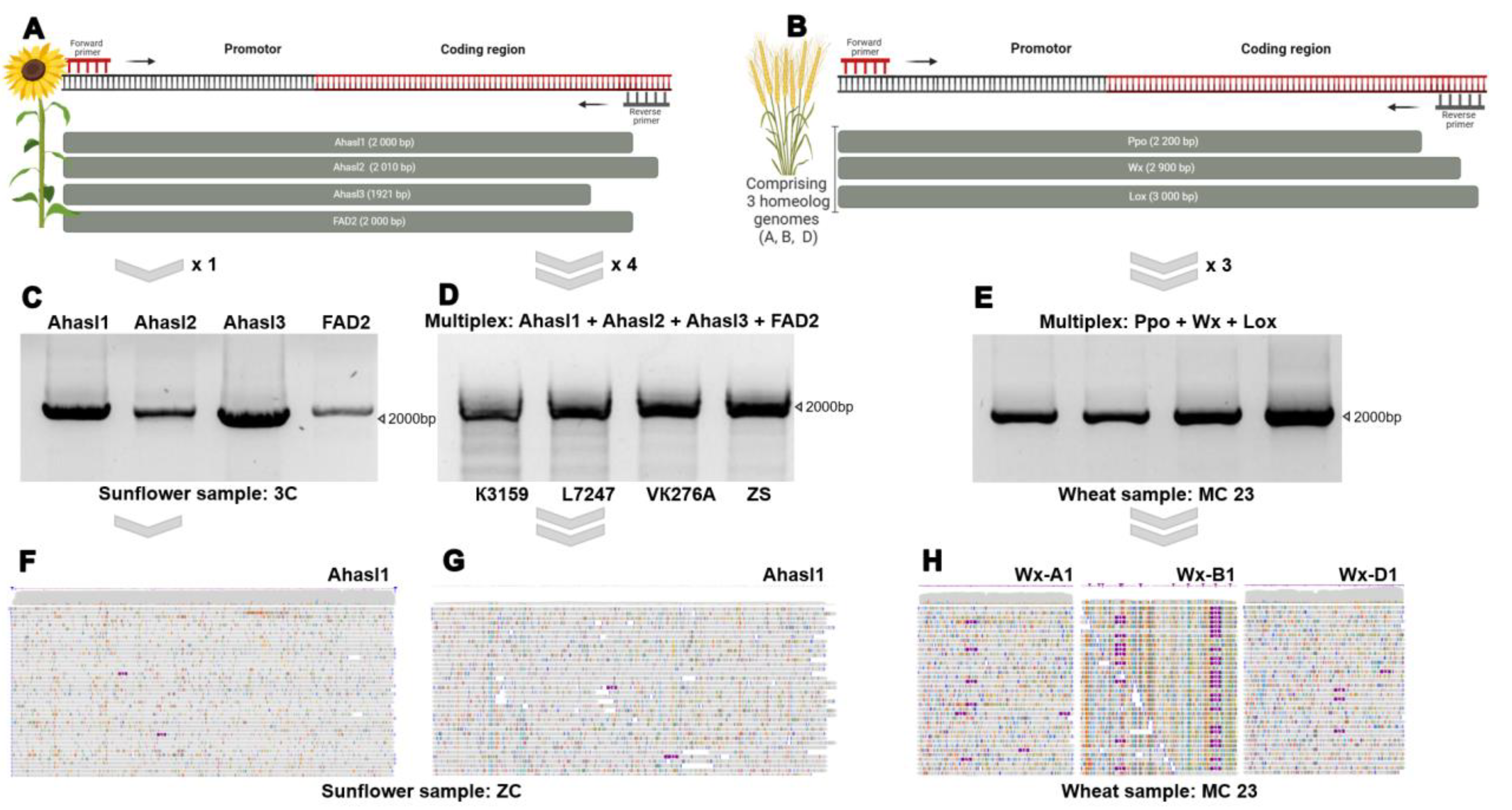
Figure 2.
Sequence analysis of AHASL genes helps match the reference sequence with the obtained reads for Ahasl1 (A) and Ahasl3 (B) for SNP identification. (C) Histogram illustrating the count of synonymous (orange) and non-synonymous (blue) SNPs detected in the four sunflower genes.
Figure 2.
Sequence analysis of AHASL genes helps match the reference sequence with the obtained reads for Ahasl1 (A) and Ahasl3 (B) for SNP identification. (C) Histogram illustrating the count of synonymous (orange) and non-synonymous (blue) SNPs detected in the four sunflower genes.
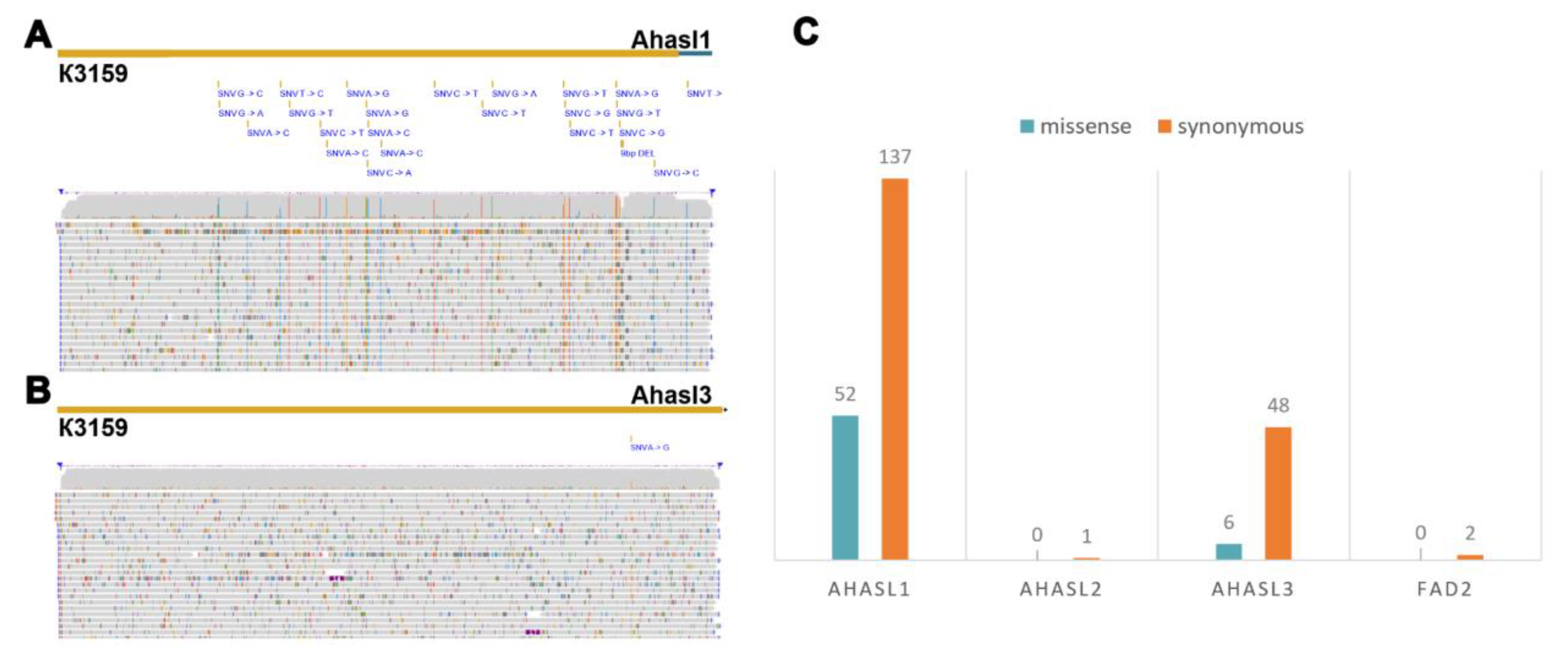
Figure 3.
Sequence analysis of AHASL genes helps match the reference sequence with the ob-tained reads for sunflower (A) and wheat (B) for identifying InDels. (C) PCR results with specific primer pairs flanking the InDels for sunflower genes (Ahasl1 and Ahasl3) and wheat genes (Ppo-D1 and Lox1). (D) Example of a chromatogram obtained by Sanger sequencing of amplicons with indels.
Figure 3.
Sequence analysis of AHASL genes helps match the reference sequence with the ob-tained reads for sunflower (A) and wheat (B) for identifying InDels. (C) PCR results with specific primer pairs flanking the InDels for sunflower genes (Ahasl1 and Ahasl3) and wheat genes (Ppo-D1 and Lox1). (D) Example of a chromatogram obtained by Sanger sequencing of amplicons with indels.
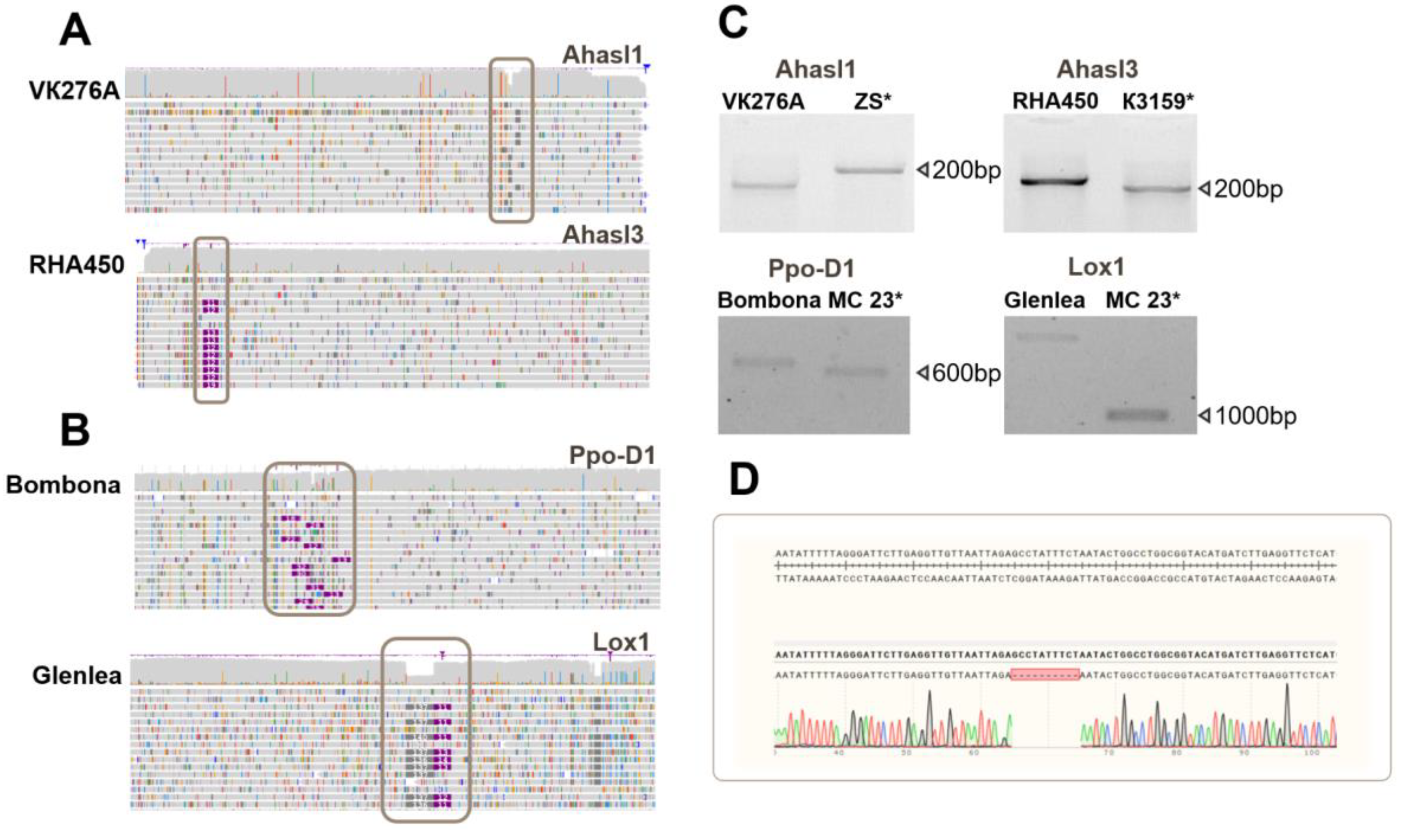
Figure 4.
Comparison of the timelines of the two amplicon sequencing approaches. The top line (green) shows amplicon sequencing using a Native Barcoding Kit and single PCR. The bottom line (red) shows the amplicon preparation using the Rapid Barcoding Kit and multiplex PCR.
Figure 4.
Comparison of the timelines of the two amplicon sequencing approaches. The top line (green) shows amplicon sequencing using a Native Barcoding Kit and single PCR. The bottom line (red) shows the amplicon preparation using the Rapid Barcoding Kit and multiplex PCR.

Table 1.
Summary of nanopore sequencing experiments.
| Experiments | Organism | Number of samples | Multiplex | Number of genes | Library preparation |
Number of reads per sample |
|---|---|---|---|---|---|---|
| Experiment 1 | Sunflower | 23 | no | 4 | Native barcoding kit |
>6,000 |
| Experiment 2 | Sunflower | 40 | yes | 4 | Rapid barcoding kit |
> 2,000 |
| Experiment 3 | Wheat | 30 | yes | 3 | Rapid barcoding kit |
> 2,000 |
Table 2.
The estimated price for sequencing four genes for a single sample us-ing the Native Barcoding kit and Rapid Barcoding kit.
Table 2.
The estimated price for sequencing four genes for a single sample us-ing the Native Barcoding kit and Rapid Barcoding kit.
| Native barcoding kit, cost/sample US$ | Rapid barcoding kit and mul-tiplex PCR for 4 genes, cost/sample US$ | |
|---|---|---|
| DNA extraction | 1.77 | 1.77 |
| Amplification and purification | 2.72 | 0.68 |
| Barcoding and library preparation | 8.3 (Native Barcoding Kit 96 V14) |
10.3 (Rapid Barcoding Kit 96 V14) |
| Third party consumables for library preparation kit | 2.5 (Ligation Mix) |
0 |
| Flow cell price (up to 6 individual runs) | 0.86 | 0.86 |
| Total cost | 16.15 | 13.61 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



