
Submitted:
14 April 2025
Posted:
15 April 2025
You are already at the latest version
Abstract
Background/Objectives: Colon cancer remains a significant health challenge world-wide, with inflammatory pathways such as tumor necrosis factor-alpha (TNF-α) playing a central role in its progression. TNF-α, a key proinflammatory cytokine, is implicated in various stages of colon cancer development, including inflammation, tumor growth, and metastasis. This review provides a comprehensive overview of the molecular mechanisms through which TNF-α contributes to colon cancer progression, with a focus on its interaction with signaling pathways like NF-κB, oxidative stress, and Wnt/β-catenin.
Methods: A comprehensive review of the literature was conducted to elucidate the role of tumor necrosis factor-alpha in the pathogenesis of colon cancer. Peer-reviewed clin-ical and preclinical studies involving human subjects were identified through electronic databases, including PubMed, Scopus, and Web of Science. Selection criteria focused on studies examining the molecular mechanisms of TNF-α signaling, its interaction with key oncogenic pathways and therapeutic interventions targeting TNF-α. Data were synthesized to highlight both mechanistic insights and therapeutic applications.
Results: TNF-α’s involvement in promoting tumorigenesis and its complex role in the tumor microenvironment highlight its potential as both a therapeutic target and a challenge for effective treatment. The review also delves into the potential of an-ti-TNF-α therapies and the emerging role of combination strategies with immune checkpoint inhibitors. Despite promising preclinical findings, clinical application faces challenges due to the dual role of TNF-α in both promoting and inhibiting tumor pro-gression.
Conclusion: Future research should focus on overcoming resistance mechanisms, de-veloping personalized therapeutic strategies, and balancing the effects of TNF-α in cancer therapy.
Keywords:
Colon Cancer
; Tumor necrosis factor-alpha: Inflammatory pathway
; Tumor
; Oxidative stress
; DNA damage
; Therapeutic targeting
1. Introduction
Colon cancer is the third most frequent cancer globally, and it is more common in developed nations, especially in North America, Europe, and Australia. Rates are rising in Asia and Eastern Europe, particularly among older age groups, whereas they are stabilizing or declining in high-income regions [1]. Colon cancer is classified into four main stages based on the TNM (Tumor, Node, Metastasis) system. In Stage I, the cancer is confined to the inner lining or muscle layer of the colon wall. Stage II involves the spread of cancer to nearby tissues without lymph node involvement. In Stage III, the cancer affects regional lymph nodes but has not metastasized to distant sites. Finally, in Stage IV, the cancer has spread to distant organs. Early detection at stages 0, I, or II raises the survival rate to over 80%; however, the chance of survival drops to below 10 percent in late-stage cases (III or IV) when cancer has spread. [2].
Treatment options for colon cancer include radiation therapy, chemotherapy, targeted therapy, cryosurgery, and surgery. Surgery is the main treatment for colon cancer, and while it cures about 50% of patients, recurrence is a common problem. Chemotherapy is a crucial treatment approach, but it has severe side effects such hematologic issues gastrointestinal toxicity, and systemic toxicity from non-targeted drug distribution, which also leads to drug resistance. Although results are still not ideal, improvements in drug delivery techniques and molecular biomarker identification are meant to increase treatment selectivity and decrease side effects [3]. Rather than genetic mutations, environmental factors have been implicated in the majority of colon cancer cases. Certain intestinal pathogens and commensals, environmental and food-borne mutagens, and persistent intestinal inflammation—which occurs before tumor development are risk factors. An important factor in the onset and spread of colon cancer is inflammation. In diseases like inflammatory bowel disease (IBD), chronic intestinal inflammation encourages DNA damage, epigenetic modifications, and the generation of reactive oxygen and nitrogen species, all of which can result in tumor suppressor gene mutations. By secreting proinflammatory cytokines and chemokines, this inflammatory environment also promotes tumor growth, metastasis, and progression. One of the main proinflammatory cytokines in this process is tumor necrosis factor-alpha (TNF-α). In colon cancer, TNF-α stimulates transcription factors like Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and signal transducer and activator of transcription 3 (Stat3), which promote angiogenesis, cell proliferation, survival, and immune evasion. Both sporadic and colitis-associated colorectal cancers have been found to have elevated TNF-α level, which makes it an important target for therapeutic intervention [4].
The main objective of this study is to examine how TNF-α affects the pathophysiology of colon cancer, with a strong focus on how it regulates inflammation, tumor growth, and immune response. In order to improve treatment outcomes for individuals with colon cancer, the study intends to investigate TNF-α as a possible therapeutic target.
2. Role of Tumor Necrosis Factor-alpha in colon cancer pathophysiology
2.1. Overview of Tumor Necrosis Factor Alpha (TNF-α)
Tumor necrosis factor alpha (TNF-α), as the name suggests, is capable of causing necrosis of tumor. It is an inflammatory cytokine produced by activated macrophages, T lymphocytes, and various immune cells. TNF-α is responsible for initiating various signaling pathways such as inflammation, cell survival and proliferation, and apoptosis as well. These pathways are activated upon binding of TNF-α to two receptors TNF-α type 1 receptor (TNFR1) and TNF-α type 2 receptor (TNFR2). It is noted that TNF-α is also produced by tumor cells however, its effect largely depends on the type of tumor and is influenced by the tumor microenvironment [5].
2.1.1. Structure and Function of TNF-α
TNF-α is well known for the capacity to destroy tumors and was known as cachexin for this particular function; however, this cytokine has a complex role in the tumor microenvironment of colorectal carcinoma (CRC). The family of TNF proteins consists of membrane proteins and secreted cytokines that can bind to the cell surface receptors. As stated earlier, activated macrophages, T cells, various immune cells including natural killer cells and even tumor cells produce TNF-α [5]. At molecular level, the TNF-α is synthesized as a type 2 transmembrane precursor protein (tmTNF) weighing 26 kilodaltons (kDa). The TNF-α converting enzyme (TACE), a matrix metalloproteinase, then cleaves the tmTNF releasing the soluble molecule (sTNF) that weighs 17 kDa. The tmTNF and sTNF unify into groups of three forming a homotrimer to activate their cell function. The TNF gene located on chromosome 6, codes for the homotrimer TNF-α protein consisting of 157 amino acids. The two distinct cell surface receptors, TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2) mediate the biological actions and signaling pathways of TNF-α. Among these two receptors, TNFR1 is capable of initiating the majority of functions of TNF-α. It is heavily expressed on almost all cell types whereas TNFR2 is mainly expressed by immune cells [6]. The TNFR1 is widely studied for its double role; it helps in facilitating cell death or apoptotic signals due to the presence of the death domain (DD) and on contrary, it also has the ability to induce cell proliferation and survival signals. Death domain is the cytoplasmic domain of the type 1 receptor that aids in initiating signaling pathways as the TNFR lacks its own enzyme [7]. The primary signaling pathway in the case of TNFR1 is the anti-apoptotic pathway where nuclear factor kappa-B (NF-κB) is activated for cell survival. TNFR2 has a pro-survival function where the signaling pathway is activated via NF-κB [6].
2.2. Tumor Necrosis Factor Alpha (TNF-α) and Colorectal Carcinoma
One of the common malignancies of the gastrointestinal tract is the colorectal carcinoma and it has also become one of the leading causes of cancer-related deaths worldwide [8]. Tumor cells release inflammatory cytokines to induce inflammation that significantly contributes to the progression of malignancy. TNF-α has various biological actions like inflammation, apoptosis, cell proliferation and differentiation. However, in the TME of colorectal cancer, elevated levels of TNF-α have been recognized to play a role in tumor progression and metastasis. The tumor surrounding cells produce proinflammatory cytokines as well as various growth factors that accelerate the process of cell transition. The epithelial cells of a tumor acquire mesenchymal-like properties, and this process is called epithelial mesenchymal transition (EMT). These cells gain invasive and migratory properties and play an important role in the progression and metastasis of CRC. To evaluate the proliferation and migratory potential of cells treated with TNF-α, a wound healing assay was performed. In both cell lines TNF-α treatment rapidly closed the wound compared with the controls (p < 0.0001) and this result demonstrated the role of TNF-α induced EMT in colorectal cancer cases [9]. Another study conducted by Wang et al. revealed that TNF-α induced EMT in highly aggressive human colorectal carcinoma cell line HCT116 accelerated the cancer invasion and metastasis [10]. A study investigating the relation of serum levels of TNF-α with survival and progression of cancer in CRC patients had significant results. The study included 119 CRC cases and 177 controls recording highest levels of TNF-α in terminal stage CRC (42.7 ± 12.5 pg/mL). Additionally, patients with low serum levels of TNF-α had significantly higher median survival rate compared to those with high levels of TNF-α [11].
Early stage of CRC has analyzed low serum levels of TNFα whereas the TNFα serum levels remain elevated in advanced stages of CRC. High levels of TNF-α also influence the activation of pro-oncogenic signaling pathways like nuclear factor κB (NF-κB) and activator protein 1 (AP-1) that help in cell survival and proliferation in CRC. A recent study evaluating the effect of TNFα on HT-29 colorectal cancer cells, observed that TNFα secreted high levels of pro-tumorigenic cytokines IL-8 and IL-6 that aided tumor progression [8]. A downstream transcription factor of the cytokine IL-6 is STAT3, which has an indispensable role in the development of various cancers including CRC. TNFα plays an important function in increasing the phosphorylation and the expression of STAT3 and its target genes that are related to the cell survival and metastasis of colorectal cancer [12]. Tumor necrosis factor-α-induced protein 8 (TNFAIP8/TIPE) is a family of proteins whose expression is regulated by TNFα. This family has many types of proteins including TNFAIP8-like 1 (TIPE1), TNFAIP8-like 2 (TIPE2), and TNFAIP8-like 3 (TIPE3) proteins. It is to be noted that over-expression of TNFAIP8 is associated with pathogenesis of various cancers including CRC [13]. A 2014 study was conducted to better understand the mechanism of TIPE2 via caspase 8 in colon cancer patients and the study discovered that TIPE2 was dominantly expressed in colon cancer tissues and its expression was linked to lymph node distant metastasis and Dukes stage of CRC [14]. Multiple other studies have recorded the relationship between TNF-α, and colon cancer as seen in Table 1.
2.3. Tumor Necrosis Factor Alpha (TNF-α) and immune response
Upon recognition of tumor cells, the body's immune response fuels inflammatory changes that contribute to carcinogenic processes. This inflammation feeds tumor growth by releasing bioactive molecules like growth factors, cytokines and chemokines. A declined immune response aids in the progression of malignant neoplasm. In the case of CRC, tumor cells acquire characteristics that help the tumor in evading immunological surveillance [15].
2.3.1. Pro-Inflammatory Role and Immune Activation
In the early stages of CRC, TNF-α generates an inflammatory response in which the immune cells such as neutrophils and macrophages are recruited to the site of tumor. The prominent subtype of neutrophils known as N1 is initially released and gets transformed into NF2 as the cancer progresses [16]. One of the key signaling pathways that is responsible for transcription of pro-inflammatory genes is the NF-κB pathway that gets activated via the TNF-α receptors (TNFR1 and TNFR2). Natural killer (NK) cells are a primary defence against the tumor cells. These cells include both types of receptors, activating and inhibitory receptors. The laboratory investigations in CRC patients have shown a decrease in type 1 NK cells whilst there has been an increase in type 2 NK cells in the early stages of cancer. The member D of group 2 (NKG2D) acts as an activating receptor while the member A (NKG2A) is an inhibitory receptor. The interaction between the ligands of NKG2D (NKG2DL) and NKG2D itself influences the survival, expansion and cytotoxic activity of NK cells that activates a strong immune response against colorectal cancer cells [17].
TNF-α activates the T cells and helps in maturating the antigen presenting dendritic cells. Certain inflammatory cytokines studied earlier like IL-6 which are produced by TNF-α also amplify the immune response in TME in early stages [7].
2.3.2. Immune Evasion
Immune evasion is a strategic mechanism that allows cancerous cells to go unnoticed by the immune system. In CRC patients, this mechanism is mediated by the dual role of TNF-α. The regulatory T cells (Tregs) are expressed by the action of TNF-α and they undermine the function of the effector T cells. Tregs contribute by activating a complex network of signaling pathways leading to immunosuppression and tumor progression in CRC. Two most important pathways activated by the Tregs are PI3K/Akt/mTOR and the NF-κB pathway, both of which affect the effector T cells as those cells are the pathways activation site. The effector T cells are responsible for fighting off tumor cells however due to the activation of these pathways, the prime ability of the effector T cells, to produce inflammatory cytokines and eliminate the tumor cells, is greatly reduced. Moreover, the Tregs helps in activation of the STAT3 pathway by acting on tumor cells in order to promote immune evasion and tumor growth. Another factor that helps tumor cells to evade the immune system include the immune checkpoint inhibitors expressed by the tumor cells in response to TNF-α. The increased expression of immune checkpoint inhibitor programmed death-ligand 1 (PD-L1) downregulates the T-cell responses, helping the tumor cells to escape the destruction by the immune system [14].
3. Mechanistic Insights of Pathways
3.1. NF-κB Pathway Activation
The NF-κB signaling pathway plays a central role in inflammation, immune response, and cancer, particularly in the context of colon cancer. Dysregulation of this pathway is a key factor in the development and progression of chronic inflammatory diseases like inflammatory bowel disease (IBD) and colitis-associated cancer (CAC). The transcription factor NF-κB, a family of proteins, is essential for maintaining intestinal homeostasis, regulating epithelial integrity, and managing the interactions between the gut epithelium and the microbiota. In IBD, NF-κB is activated in macrophages and epithelial cells of the inflamed mucosa, where it contributes to the inflammatory response and epithelial barrier dysfunction [28,29,30,31].
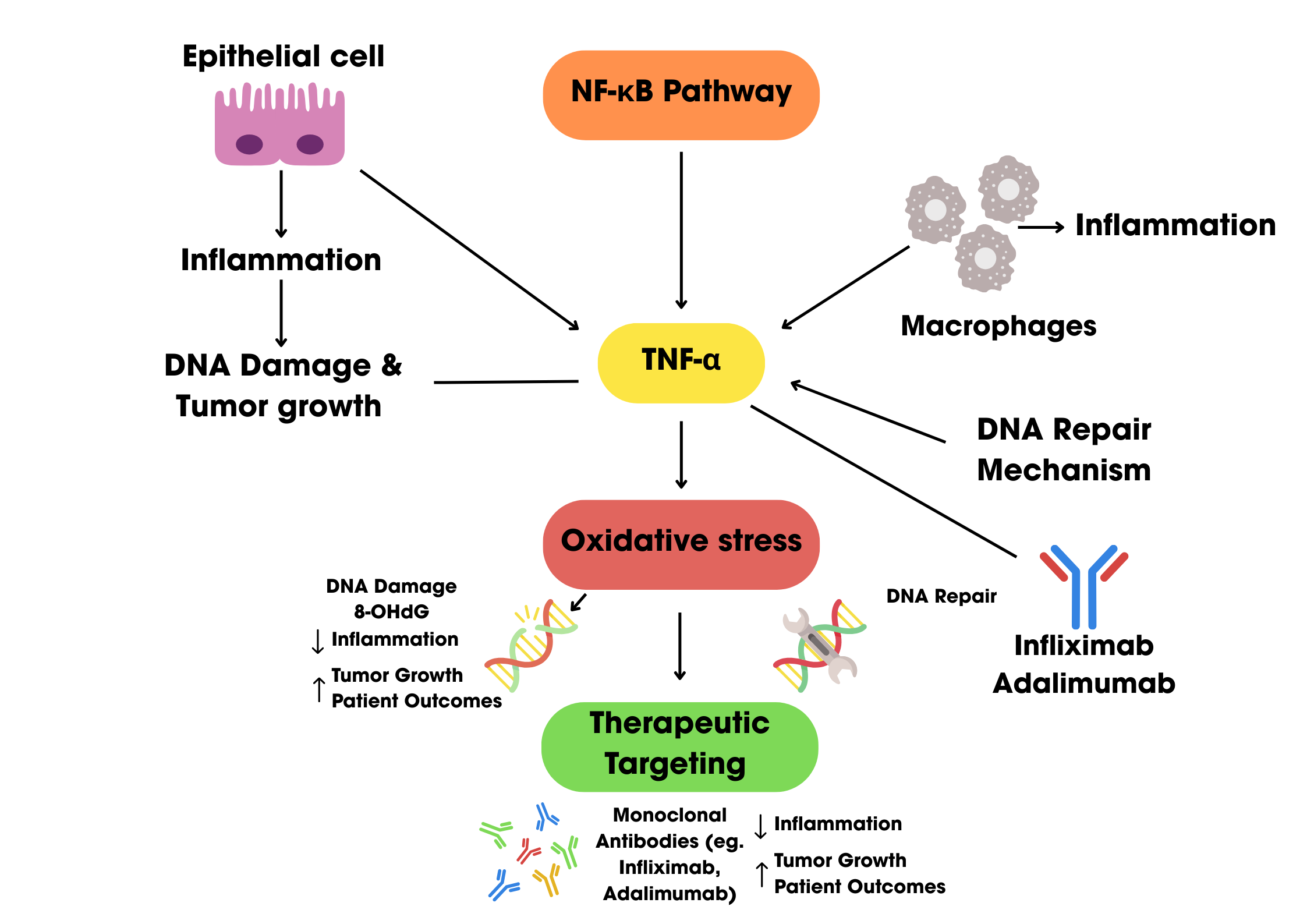
NF-κB is activated in response to various stimuli, including cytokines like TNF-α (Tumor Necrosis Factor alpha), which is a potent inducer of NF-κB. TNF-α binds to its receptor TNFR1, triggering a cascade of signaling events that lead to NF-κB activation. Upon activation, NF-κB translocates to the nucleus, where it regulates the expression of genes involved in inflammation, immune response, and cell survival. The interaction between TNF-α and NF-κB is critical in controlling both the immune response and the resolution of inflammation. TNF-α can also induce cell death mechanisms, including apoptosis and necroptosis, which are regulated by NF-κB. In these pathways, NF-κB activation can prevent apoptosis by inducing the expression of survival proteins like c-FLIP, whereas its absence may lead to cell death. The relationship between NF-κB and TNF-α is therefore essential for balancing immune responses, tissue repair, and cell fate decisions, making it a key player in both immune defence and disease processes such as cancer and inflammatory disorders [32].
The activation of NF-κB in colon cancer is primarily through the canonical and non-canonical pathways. The canonical NF- κB activation pathway, primarily triggered by TNF-α, begins with its binding to the TNF-α receptor (TNFR), leading to the recruitment of the IκB kinase (IKK) complex, which includes IKKα, IKKβ, and NEMO, through adaptor proteins like TRAF2 and RIP1. This interaction induces K63 ubiquitination of RIP1, facilitated by E3 ligases cIAP-1 and cIAP-2, providing a platform for IKK activation. IKK is subsequently activated through phosphorylation by upstream kinases, such as MEKK3 and TAK1, leading to the phosphorylation of IκB proteins, marking them for polyubiquitination and proteasomal degradation as shown in Figure 1. This degradation releases NF-κB dimers (e.g., p65/p50), allowing their translocation to the nucleus where they initiate the transcription of genes involved in inflammation, immune responses, and cell survival.
In the context of CAC, studies have shown that the deletion of IKKβ in intestinal cells can reduce tumor incidence, although it does not significantly affect tumor size, suggesting that NF-κB activation in non-epithelial cells, like macrophages, may be more critical for tumor progression. In contrast, deletion of IKKβ in myeloid cells leads to reduced tumor size, highlighting the importance of cell-specific NF-κB activation in CAC development. Moreover, inhibition of NF-κB in intestinal epithelial cells through conditional ablation of the NF-κB essential modulator (NEMO) results in severe colitis and epithelial barrier compromise, further emphasizing the importance of NF-κB in maintaining intestinal epithelial integrity and controlling inflammation.
In addition to canonical NF-κB signaling, the non-canonical pathway also plays a role in colon cancer, particularly in immune regulation and cell survival. This pathway is activated by TNF receptors such as BAFFR, CD40, and LTβR and involves the stabilization of NIK (NF-κB-inducing kinase), leading to the processing of the precursor protein p100 into p52. The p52/RelB dimer then translocate to the nucleus to regulate gene expression as shown in figure 1. Although slower and more persistent, this pathway is crucial for long-term immune responses and tissue remodelling in the colon.
Furthermore, redox signaling significantly influences NF-κB activation. Reactive oxygen species (ROS), produced by various stimuli such as TNF-α and IL-1, can modify NF-κB subunits, particularly by oxidizing cysteine residues, which enhances DNA binding and transcriptional activation. Lipid signaling molecules like ceramide, derived from diacylglycerol (DAG), also contribute to NF-κB activation. The generation of ceramide through sphingomyelinase activation further supports NF-κB signaling by influencing key kinases and phosphatases.
Given the complex role of NF-κB in regulating both inflammation and tumorigenesis, targeting this pathway offers a promising therapeutic strategy for treating colon cancer. Preclinical studies have demonstrated the involvement of TNF-NF-κB signaling in both tumor initiation and progression as well as in maintaining intestinal homeostasis. As such, targeting specific components of the NF-κB pathway, including IKKβ, NEMO, or TNF receptors, could potentially serve as an effective therapeutic approach for treating CAC and other related inflammatory disorders [28,29,30,31].
3.2. Induction of Oxidative Stress and DNA Damage
Oxidative stress plays a major role in causing DNA damage, potentially resulting in mutations and genomic instability, which is a characteristic of numerous diseases, such as cancer. DNA is highly vulnerable to oxidative harm from reactive oxygen species (ROS), including hydroxyl radicals, which can lead to base alterations, single-strand breaks (SSBs), and double-strand breaks (DSBs). Oxidative stress can indeed lead to around 10,000 DNA changes per cell each day, with a significant share of this damage resulting from ROS-induced lesions. A widely researched and prevalent oxidative DNA damage is 8-hydroxydeoxyguanosine (8-OHdG), which is formed through the oxidation of guanine, a base especially susceptible to changes because of its low oxidation potential. If left unrepaired, 8-OHdG can cause mutations, especially G:C to T:A transversions, commonly observed in mutated oncogenes and tumor suppressor genes.
To mend oxidative DNA damage, cells depend on various DNA repair mechanisms, such as base excision repair (BER) and nucleotide excision repair (NER). In BER, DNA glycosylases, including 8-oxoguanine DNA glycosylase (OGG1), identify and eliminate damaged bases such as 8-OHdG, resulting in an abasic site. An AP endonuclease subsequently processes the site, kicking off the repair mechanism and returning the DNA sequence to its original state. NER, conversely, tackles a broader spectrum of DNA lesions, including those caused by ROS, by excising the damaged oligonucleotide and resynthesizing the strand utilizing the complementary DNA template. NER functions via two sub-pathways: Global Genome NER (GG-NER), which examines the complete genome for helix-distorting damage, and Transcription-Coupled NER (TC-NER), which addresses damage that hinders transcription. The XPC protein and its accessory subunits are essential for recognizing DNA damage in GG-NER. Shortcomings in NER components, such as XPC, can lead to greater susceptibility to UV radiation and an elevated risk of skin cancer.
Additionally, ROS can trigger DNA repair genes via the influence of transcription factors such as AP-1 and CREB/c-Jun. These elements attach to DNA and trigger the transcription of repair genes, including apex1 and neil1, which play a role in BER. This illustrates how the repair system acts in a coordinated way to address oxidative stress and preserve genomic integrity. Nevertheless, the exact processes through which ROS directly influence the expression of DNA repair genes remain unclear.
The effects of oxidative DNA damage are considerable, as it can result in replication mistakes and genomic instability. The 8-OHdG lesion, for instance, may mispair with adenine or cytosine while DNA is replicated, thereby enhancing mutation. Moreover, ROS is capable of altering nucleotides within the nucleotide pool, generating compounds such as OH8dG, which may be integrated into DNA during replication, possibly leading to mutations. Increased amounts of oxidative DNA damage indicators like 8-OHdG and 8-nitroguanine are often linked to multiple cancers and other illnesses as displayed in Figure 2. These biomarkers act as crucial instruments for assessing oxidative stress and comprehending its function in disease progression [33,34].
3.3. Crosstalk with Wnt/β-catenin and STAT3 signaling Pathways
Colorectal cancer (CRC) is often characterized by the dysregulation of the Wnt/β-catenin signaling pathway, a crucial cascade responsible for maintaining intestinal homeostasis. In normal conditions, the Wnt signaling pathway is tightly regulated to ensure appropriate cell proliferation, differentiation, and apoptosis. However, in CRC, epigenetic alterations frequently disrupt this regulation. DNA methylation of key Wnt antagonists such as the SFRP family (Secreted Frizzled-Related Proteins), WIF1 (Wnt Inhibitory Factor 1), and DKK3 (Dickkopf-related protein 3) has been well documented. These DNA methylation events lead to the transcriptional silencing of these critical Wnt inhibitors, removing their ability to restrain the pathway. As a result, the Wnt/β-catenin signaling cascade becomes abnormally activated, contributing to uncontrolled cell proliferation, survival, and tumor progression, hallmark features of CRC.
The epigenetic silencing of Wnt antagonists results in the stabilization and nuclear translocation of β-catenin, a central player in the Wnt pathway. In the absence of Wnt inhibitors, β-catenin accumulates in the cytoplasm, where it escapes degradation and translocates into the nucleus. Once in the nucleus, β-catenin binds to the T-cell factor/lymphoid enhancer-binding factor (TCF/LEF) family of transcription factors, forming a transcriptional complex that drives the expression of genes that promote cellular proliferation, survival, and the prevention of apoptosis. This dysregulated activation of the Wnt/β-catenin pathway plays a central role in CRC tumorigenesis, highlighting the potential of targeting epigenetic modifications to restore normal Wnt signaling and suppress tumor growth.
One therapeutic approach for reversing the methylation-driven silencing of Wnt antagonists involves the inhibition of DNA methyltransferases (DNMTs), the enzymes responsible for adding methyl groups to DNA. In CRC, DNMT inhibition has been shown to reduce the stem-like properties of cancer cells. This effect is mediated by the demethylation of key genes that normally inhibit Wnt signaling, such as SFRP1, leading to a reduction in the aberrant activation of the Wnt/β-catenin pathway. Specifically, SFRP1 methylation is critical for maintaining the stem cell population within colorectal tumors, suggesting that reactivating the expression of SFRP1 through DNMT inhibition could diminish the self-renewal potential of CRC stem cells, thereby limiting tumor progression and metastasis.
The relationship between Wnt/β-catenin signaling and TNF-α (Tumor Necrosis Factor-alpha) is complex, involving intricate interactions between these pathways, particularly in inflammation and immune regulation. Wnt signaling, especially through the canonical β-catenin-dependent pathway, regulates critical cellular processes such as proliferation, differentiation, and apoptosis. Under normal conditions, Wnt signaling stabilizes β-catenin, allowing it to translocate to the nucleus and activate transcription of genes involved in these processes. Conversely, TNF-α, a key pro-inflammatory cytokine, typically activates the NF-κB signaling pathway, driving inflammation and immune responses. Interestingly, β-catenin has been shown to interact with NF-κB, with studies indicating that the stabilization of β-catenin can modulate the activity of NF-κB, either enhancing or suppressing its transcriptional activity depending on the cellular context. In inflammatory diseases, such as inflammatory bowel disease (IBD), dysregulated Wnt/β-catenin signaling often exacerbates inflammation, while β-catenin’s role in immune cells, particularly dendritic cells (DCs), helps maintain a balance between pro-inflammatory Th1/Th17 and anti-inflammatory Treg cells. This cross-talk between Wnt/β-catenin and TNF-α/NF-κB pathways highlights a potential regulatory mechanism in controlling immune responses and inflammation, with β-catenin acting as a rheostat for immune tolerance and inflammation. Understanding this relationship could offer insights into therapeutic strategies for inflammatory and autoimmune diseases [35].
The canonical Wnt/β-catenin signaling pathway is initiated when Wnt ligands, such as Wnt1, Wnt3a, or Wnt5a, bind to receptors on the cell membrane, including the Frizzled family of receptors and the co-receptor LRP5/6 (Low-Density Lipoprotein Receptor-Related Protein 5/6). This ligand-receptor interaction triggers a cascade of intracellular events that result in the stabilization of β-catenin. Under normal conditions, β-catenin is tightly regulated by a destruction complex that includes proteins such as APC (Adenomatous Polyposis Coli), GSK3β (Glycogen Synthase Kinase 3 beta), and CK1 (Casein Kinase 1), which promote its phosphorylation and subsequent degradation. However, when Wnt ligands are present, this destruction complex is inhibited, leading to the accumulation of β-catenin in the cytoplasm. Stabilized β-catenin then translocate to the nucleus, where it interacts with TCF/LEF transcription factors to activate the transcription of genes involved in cell cycle progression, anti-apoptotic signaling, and cellular differentiation.
In the context of CRC, this canonical Wnt/β-catenin signaling not only drives tumorigenesis but also enhances the ability of cancer cells to survive under stressful conditions, such as oxidative stress, which is often present in the tumor microenvironment. Under oxidative stress, reactive oxygen species (ROS) can damage cellular components, leading to apoptosis. However, Wnt signaling can counteract this damage by activating pro-survival pathways. One such pathway involves the transcription factor STAT3 (Signal Transducer and Activator of Transcription 3). When Wnt signaling is active, it triggers the phosphorylation of STAT3 at tyrosine 705 (Tyr705), a modification that activates STAT3 and allows it to translocate into the nucleus. In the nucleus, STAT3 promotes the expression of genes involved in cell survival, including anti-apoptotic proteins such as Bcl-2 and Mcl-1. The activation of the Wnt-STAT3 signaling axis is particularly important in CRC cells, where it helps the tumor cells resist oxidative stress and survive in the face of harmful environmental conditions.
The importance of STAT3 in this protective mechanism is underscored by studies showing that depletion of STAT3 diminishes the pro-survival effects of Wnt signaling. This suggests that STAT3 is a critical mediator of Wnt-driven cellular defence in CRC. In fact, the Wnt-STAT3 pathway acts as a key pro-survival mechanism that enables CRC cells to endure stressful conditions and maintain tumorigenic potential, further supporting the notion that targeting components of this pathway could provide therapeutic benefit in CRC treatment.
In summary, the Wnt/β-catenin signaling pathway is central to the development and progression of CRC, with epigenetic silencing of Wnt antagonists driving its aberrant activation. This dysregulation enhances cellular survival, particularly under conditions of oxidative stress, through the activation of STAT3. Understanding the molecular mechanisms underlying Wnt/β-catenin and Wnt-STAT3 signaling in CRC provides critical insights into potential therapeutic strategies aimed at restoring normal signaling and inhibiting tumor growth. Targeting the epigenetic modifications that silence Wnt antagonists, as well as the Wnt-STAT3 pathway, may offer promising avenues for CRC treatment and prevention [34,36,37,38,39].
4. TNF-α as a therapeutic Target in Colon cancer
TNF-α plays a critical role in the progression of colon cancer, with its high expression associated with poor prognosis [25,40]. TNF-α promotes tumor growth by stimulating macrophages to produce pro-tumorigenic factors such as colony-stimulating factor-1 (CSF-1), vascular endothelial growth factor-A (VEGF-A), and matrix metalloproteinase-2 (MMP-2) [41]. It also enhances the epithelial-to-mesenchymal transition, increasing tumor invasiveness [42].
Targeting TNF-α with monoclonal antibodies like infliximab has shown promise in colon cancer treatment. This therapy induces antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity, promoting apoptosis and enhancing cell death [40]. When combined with chemotherapy agents such as oxaliplatin or 5-fluorouracil, anti-TNF-α treatment demonstrates synergistic effects, leading to tumor regression in preclinical models [25,40].
Moreover, TNF-α inhibitors, Infliximab, Adalimumab, and Etanercept, are used to modulate the inflammatory response by blocking TNF-α activity. These inhibitors have shown potential in reducing inflammation and tumor growth in colorectal cancer models [43]. For instance, the blockade of TNF-α signaling has been shown to suppress colorectal cancer progression and enhance the efficacy of immunotherapies by reducing regulatory T cell infiltration in the tumor microenvironment [27]. The therapeutic use of TNF-α inhibitors in colon cancer is promising, particularly in cases associated with chronic inflammation, such as ulcerative colitis. However, the use of TNF-α inhibitors is not without challenges. They can have side effects, including the potential for malignancy development, and their long-term safety requires careful monitoring [43]. Novel approaches, such as the use of selective inhibitors like Takinib, which sensitize cells to TNF-α-induced cell death, are being investigated for their potential to enhance cancer treatment efficacy [44]. Additionally, the identification of small-molecule inhibitors that disrupt TNF-α activity offers another avenue for therapeutic development as seen in Table 2 [45].
5. Limitations and future direction
This review focuses mainly on TNF-α's involvement in colon cancer, possibly ignoring the contributions of other cytokines, signaling pathways, and molecular mechanisms that might also be important in the development of the illness. Furthermore, some of the information presented originates from preclinical research, which might not accurately reflect clinical outcomes in humans. The therapeutic potential of treating TNF-α in patients with colon cancer requires more clinical data, especially from randomized controlled trials. Additionally, although TNF-α is examined as a therapeutic target in the review, the mechanisms of resistance that may emerge during anti-TNF-α therapy are not thoroughly covered, despite the fact that these processes are essential for creating more potent and long-lasting treatment plans.
Targeted molecular therapies and personalized immunotherapy are two examples of upcoming precision medicine developments that may assist overcome resistance and enhance results. The effectiveness of treatment may be improved, for instance, by targeting different immune checkpoints (such as Siglec-15) or combining immune checkpoint inhibitors with substances like CD40 agonists. Furthermore, more individualized and successful approaches may be made possible by genetic profiling and advanced methods like circulating tumor cell (CTC) detection, especially for high-risk individuals [46,47,48].
Targeting TNF-α therapeutically is made more difficult by its dual function as a tumor suppressor and promoter, as well as its capacity to encourage metastasis through mechanisms such microRNA-21 activation and TROP-2 overexpression. One of the challenges in creating TNF-α-based treatments is balancing these consequences [9,49,50].
The study clearly outlines the crucial role that TNF-α plays in the pathophysiology of colon cancer, specifically its role in inflammation, tumor growth, and immune response. It also provides useful insights into the processes by which TNF-α contributes to the advancement of colon cancer by integrating existing preclinical and clinical findings. It also emphasizes the significance of additional research to improve therapy methods. Despite the drawbacks associated, TNF-α remains a promising therapeutic target in colon cancer.
6. Conclusion
TNF-α plays a multifaceted role in the pathophysiology of colon cancer, acting as both a promoter and a suppressor of tumor progression. The cytokine’s involvement in inflammation, immune modulation, and the tumor microenvironment makes it a crucial factor in cancer development. While preclinical studies support the therapeutic targeting of TNF-α, clinical applications have shown mixed results, highlighting the complexity of balancing TNF-α’s dual roles. Anti-TNF-α therapies, particularly monoclonal antibodies, have demonstrated promising effects in reducing inflammation and tumor progression, but the emergence of resistance mechanisms underscores the need for improved therapeutic strategies. Ultimately, a deeper understanding of TNF-α signaling and its interactions with other oncogenic pathways, such as NF-κB, Wnt/β-catenin, and STAT3, will pave the way for more effective and personalized treatments for colon cancer.
Author Contributions
Conceptualization, S.K. and M.A.V.M.; methodology, S.K.; software, Y.A.; validation, S,K., Y.A. and A.H.S.; formal analysis, S.K., Y.A., A.H.S., and A.Z.; investigation, A.H.S, U.N., H.M.A.; resources, H.M.A. and M.A.V.M.; data curation, S.K., A.Z. and U.N.; writing—original draft preparation, S.K., Y.A., A.H.S., A.Z., and U.N.; writing—review and editing, S.K., H.M.A.,M.A.V.M.; visualization, S.K., Y.A., M.A.V.M.; supervision, M.A.V.M., H.M.A.; project administration, S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare that this research was conducted without commercial or financial relationships that could create conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| TNF-α | Tumor necrosis factor alpha |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| Stat3 | Signal transducer and activator of transcription 3 |
| CRC | Colorectal Cancer |
| DNA | Deoxyribonucleic acid |
| ROS | Reactive oxygen species |
| Wnt | Wingless-Type |
| EMT | Epithelial mesenchymal transition |
| TNFR1 | TNF-α type 1 receptor |
| TNFR2 | TNF-α type 2 receptor |
| HT-29 | Human colorectal adenocarcinoma cell line |
| IBD | Inflammatory bowel disease |
| IKK | IκB kinase |
| TRAF2 | Tumor necrosis factor receptor-associated factor 2 |
| RIP1 | Kinase receptor interacting protein 1 |
| NEMO | Nuclear factor-kappa B Essential Modulator |
| 8-OHdG | 8-hydroxydeoxyguanosine |
| GG-NER | Global Genome NER |
| DNMTs | DNA methyltransferases |
| TROP-2 | Tumor-associated calcium signal transducer 2 |
References
- Farinha, P.; Pinho, J.O.; Matias, M.; Gaspar, M.M. Nanomedicines in the treatment of colon cancer: a focus on metallodrugs. Drug Deliv. Transl. Res. 2022, 12, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Alrushaid, N.; Alam Khan, F.; Al-Suhaimi, E.; Elaissari, A. Progress and Perspectives in Colon Cancer Pathology, Diagnosis, and Treatments. Diseases 2023, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Pathak, S.; Subramanium, V.D.; Dharanivasan, G.; Murugesan, R.; Verma, R.S. Strategies for targeted drug delivery in treatment of colon cancer: current trends and future perspectives. Drug Discov. Today 2017, 22, 1224–1232. [Google Scholar] [CrossRef]
- Cheng, E.; Shi, Q.; Shields, A.F.; Nixon, A.B.; Shergill, A.P.; Ma, C.; Guthrie, K.A.; Couture, F.; Kuebler, P.; Kumar, P.; et al. Association of Inflammatory Biomarkers With Survival Among Patients With Stage III Colon Cancer. JAMA Oncol. 2023, 9, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Jang, D.-I.; Lee, A.-H.; Shin, H.-Y.; Song, H.-R.; Park, J.-H.; Kang, T.-B.; Lee, S.-R.; Yang, S.-H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef] [PubMed]
- You, K.; Gu, H.; Yuan, Z.; Xu, X. Tumor Necrosis Factor Alpha Signaling and Organogenesis. Front. Cell Dev. Biol. 2021, 9, 727075. [Google Scholar] [CrossRef]
- Laha, D.; Grant, R.; Mishra, P.; Nilubol, N. The Role of Tumor Necrosis Factor in Manipulating the Immunological Response of Tumor Microenvironment. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Pakdemirli, A.; Kocal, G.C. TNF-alpha Induces Pro-Inflammatory Factors in Colorectal Cancer Microenvironment. Med Sci. Discov. 2020, 7, 466–469. [Google Scholar] [CrossRef]
- Alotaibi, A.G.; Li, J.V.; Gooderham, N.J. Tumour Necrosis Factor-Alpha (TNF-α)-Induced Metastatic Phenotype in Colorectal Cancer Epithelial Cells: Mechanistic Support for the Role of MicroRNA-21. Cancers 2023, 15, 627. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.-S.; Zhou, B.-H.; Li, C.-L.; Zhang, F.; Wang, X.-F.; Zhang, G.; Bu, X.-Z.; Cai, S.-H.; Du, J. Epithelial–Mesenchymal Transition (EMT) Induced by TNF-α Requires AKT/GSK-3β-Mediated Stabilization of Snail in Colorectal Cancer. PLOS ONE 2013, 8, e56664. [Google Scholar] [CrossRef]
- Stanilov, N.; Miteva, L.; Dobreva, Z.; Stanilova, S. Colorectal cancer severity and survival in correlation with tumour necrosis factor-alpha. Biotechnol. Biotechnol. Equip. 2014, 28, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Wang, J.; Huang, P.; Gou, S.; Yu, D.; Zong, L. Tumor necrosis factor-α induces proliferation and reduces apoptosis of colorectal cancer cells through STAT3 activation. Immunogenetics 2023, 75, 161–169. [Google Scholar] [CrossRef]
- Niture, S.; Dong, X.; Arthur, E.; Chimeh, U.; Niture, S.S.; Zheng, W.; Kumar, D. Oncogenic Role of Tumor Necrosis Factor α-Induced Protein 8 (TNFAIP8). Cells 2018, 8, 9. [Google Scholar] [CrossRef]
- Li, X.-M.; Su, J.-R.; Yan, S.-P.; Cheng, Z.-L.; Yang, T.-T.; Zhu, Q. A novel inflammatory regulator TIPE2 inhibits TLR4-mediated development of colon cancer via caspase-8. Cancer Biomarkers 2014, 14, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Al Obeed, O.A.; Alkhayal, K.A.; Al Sheikh, A.; Zubaidi, A.M.; Vaali-Mohammed, M.-A.; Boushey, R.; McKerrow, J.H.; Abdulla, M.-H. Increased expression of tumor necrosis factor-α is associated with advanced colorectal cancer stages. World J. Gastroenterol. 2014, 20, 18390–18396. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Molina, A.M.; Santana, T.T.; Redondo, M.; Romero, M.J.B. The Crucial Role of Inflammation and the Immune System in Colorectal Cancer Carcinogenesis: A Comprehensive Perspective. Int. J. Mol. Sci. 2024, 25, 6188. [Google Scholar] [CrossRef] [PubMed]
- Meckawy, G.R.; Mohamed, A.M.; Zaki, W.K.; Khattab, M.A.; Amin, M.M.; ElDeeb, M.A.; El-Najjar, M.R.; Safwat, N.A. Natural killer NKG2A and NKG2D in patients with colorectal cancer. J. Gastrointest. Oncol. 2019, 10, 218–225. [Google Scholar] [CrossRef]
- Li, M.; You, Q.; Wang, X. Association Between Polymorphism of the Tumor Necrosis Factor Alpha-308 Gene Promoter and Colon Cancer in the Chinese Population. Genet. Test. Mol. Biomarkers 2011, 15, 743–747. [Google Scholar] [CrossRef]
- Kaminska, J.; Nowacki, M.; Kowalska, M.; Rysinska, A.; Chwalinski, M.; Fuksiewicz, M.; Michalski, W.; Chechlinska, M. Clinical Significance of Serum Cytokine Measurements in Untreated Colorectal Cancer Patients: Soluble Tumor Necrosis Factor Receptor Type I – An Independent Prognostic Factor. Tumor Biol. 2005, 26, 186–194. [Google Scholar] [CrossRef]
- Kemik, O.; Sumer, A.; Kemik, A.S.; Hasirci, I.; Purisa, S.; Dulger, A.C.; Demiriz, B.; Tuzun, S. The relationship among acute-phase response proteins, cytokines and hormones in cachectic patients with colon cancer. World J. Surg. Oncol. 2010, 8, 85–85. [Google Scholar] [CrossRef]
- Babic, A.; Shah, S.M.; Song, M.; Wu, K.; A Meyerhardt, J.; Ogino, S.; Yuan, C.; Giovannucci, E.L.; Chan, A.T.; Stampfer, M.J.; et al. Soluble tumour necrosis factor receptor type II and survival in colorectal cancer. Br. J. Cancer 2016, 114, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.T.; Ogino, S.; Giovannucci, E.L.; Fuchs, C.S. Inflammatory Markers Are Associated With Risk of Colorectal Cancer and Chemopreventive Response to Anti-Inflammatory Drugs. Gastroenterology 2011, 140, 799–808.e2. [Google Scholar] [CrossRef] [PubMed]
- Kapitanović, S.; Čačev, T.; Ivković, T.C.; Lončar, B.; Aralica, G. TNFα gene/protein in tumorigenesis of sporadic colon adenocarcinoma. Exp. Mol. Pathol. 2014, 97, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, Z. TNF-α promotes colon cancer cell migration and invasion by upregulating TROP-2. Oncol. Lett. 2018, 15, 3820–3827. [Google Scholar] [CrossRef]
- Li, W.; Xu, J.; Zhao, J.; Zhang, R. Oxaliplatin and Infliximab Combination Synergizes in Inducing Colon Cancer Regression. Med Sci. Monit. 2017, 23, 780–789. [Google Scholar] [CrossRef]
- Grimm, M.; Lazariotou, M.; Kircher, S.; Höfelmayr, A.; Germer, C.T.; von Rahden, B.H.A.; Waaga-Gasser, A.M.; Gasser, M. Tumor necrosis factor-α is associated with positive lymph node status in patients with recurrence of colorectal cancer—indications for anti-TNF-α agents in cancer treatment. Cell. Oncol. 2011, 34, 315–326. [Google Scholar] [CrossRef]
- Guo, Y.; Xie, F.; Liu, X.; Ke, S.; Chen, J.; Zhao, Y.; Li, N.; Wang, Z.; Yi, G.; Shen, Y.; et al. Blockade of TNF-α/TNFR2 signalling suppresses colorectal cancer and enhances the efficacy of anti-PD1 immunotherapy by decreasing CCR8+T regulatory cells. J. Mol. Cell Biol. 2023, 16. [Google Scholar] [CrossRef]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 2010, 14, 45–55. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef]
- A Baeuerle, P.; Henkel, T. Function and Activation of NF-kappaB in the Immune System. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef]
- Bhat, A.A.; Nisar, S.; Singh, M.; Ashraf, B.; Masoodi, T.; Prasad, C.P.; Sharma, A.; Maacha, S.; Karedath, T.; Hashem, S.; et al. Cytokine- and chemokine-induced inflammatory colorectal tumor microenvironment: Emerging avenue for targeted therapy. Cancer Commun. 2022, 42, 689–715. [Google Scholar] [CrossRef] [PubMed]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis. 2020, 8, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants 2024, 13, 312. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E.; Kamendulis, L.M.; Hocevar, B.A. Oxidative Stress and Oxidative Damage in Carcinogenesis. Toxicol. Pathol. 2009, 38, 96–109. [Google Scholar] [CrossRef]
- Jridi, I.; Canté-Barrett, K.; Pike-Overzet, K.; Staal, F.J.T. Inflammation and Wnt Signaling: Target for Immunomodulatory Therapy? Front. Cell Dev. Biol. 2021, 8. [Google Scholar] [CrossRef]
- Fragoso, M.A.; Patel, A.K.; Nakamura, R.E.I.; Yi, H.; Surapaneni, K.; Hackam, A.S. The Wnt/β-Catenin Pathway Cross-Talks with STAT3 Signaling to Regulate Survival of Retinal Pigment Epithelium Cells. PLOS ONE 2012, 7, e46892. [Google Scholar] [CrossRef]
- Kim, S.-J.; Kang, H.-G.; Kim, K.; Kim, H.; Zetterberg, F.; Park, Y.S.; Cho, H.-S.; Hewitt, S.M.; Chung, J.-Y.; Nilsson, U.J.; et al. Crosstalk between WNT and STAT3 is mediated by galectin-3 in tumor progression. Gastric Cancer 2021, 24, 1050–1062. [Google Scholar] [CrossRef]
- Kawada, M.; Seno, H.; Uenoyama, Y.; Sawabu, T.; Kanda, N.; Fukui, H.; Shimahara, Y.; Chiba, T. Signal Transducers and Activators of Transcription 3 Activation Is Involved in Nuclear Accumulation of β-Catenin in Colorectal Cancer. Cancer Res. 2006, 66, 2913–2917. [Google Scholar] [CrossRef]
- Li, Q.; Geng, S.; Luo, H.; Wang, W.; Mo, Y.-Q.; Luo, Q.; Wang, L.; Song, G.-B.; Sheng, J.-P.; Xu, B. Signaling pathways involved in colorectal cancer: pathogenesis and targeted therapy. Signal Transduct. Target. Ther. 2024, 9, 1–48. [Google Scholar] [CrossRef]
- Liu, F.; Ai, F.; Tian, L.; Liu, S.; Zhao, L.; Wang, X. Infliximab enhances the therapeutic effects of 5-fluorouracil resulting in tumor regression in colon cancer. OncoTargets Ther. 2016, ume 9, 5999–6008. [Google Scholar] [CrossRef]
- Zins, K.; Abraham, D.; Sioud, M.; Aharinejad, S. Colon Cancer Cell–Derived Tumor Necrosis Factor-α Mediates the Tumor Growth–Promoting Response in Macrophages by Up-regulating the Colony-Stimulating Factor-1 Pathway. Cancer Res. 2007, 67, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Bates, R.C.; Mercurio, A.M. Tumor Necrosis Factor-α Stimulates the Epithelial-to-Mesenchymal Transition of Human Colonic Organoids. Mol. Biol. Cell 2003, 14, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Zidi, I.; Mestiri, S.; Bartegi, A.; Ben Amor, N. TNF-α and its inhibitors in cancer. Med Oncol. 2009, 27, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Totzke, J.; Gurbani, D.; Raphemot, R.; Hughes, P.F.; Bodoor, K.; Carlson, D.A.; Loiselle, D.R.; Bera, A.K.; Eibschutz, L.S.; Perkins, M.M.; et al. Takinib, a Selective TAK1 Inhibitor, Broadens the Therapeutic Efficacy of TNF-α Inhibition for Cancer and Autoimmune Disease. Cell Chem. Biol. 2017, 24, 1029–1039.e7. [Google Scholar] [CrossRef]
- He, M.M.; Smith, A.S.; Oslob, J.D.; Flanagan, W.M.; Braisted, A.C.; Whitty, A.; Cancilla, M.T.; Wang, J.; Lugovskoy, A.A.; Yoburn, J.C.; et al. Small-Molecule Inhibition of TNF-. Science 2005, 310, 1022–1025. [Google Scholar] [CrossRef]
- Cornista, A.M.; Giolito, M.V.; Baker, K.; Hazime, H.; Dufait, I.; Datta, J.; Khumukcham, S.S.; De Ridder, M.; Roper, J.; Abreu, M.T.; et al. Colorectal Cancer Immunotherapy: State of the Art and Future Directions. Gastro Hep Adv. 2023, 2, 1103–1119. [Google Scholar] [CrossRef]
- Johnson, D.; Chee, C.E.; Wong, W.; Lam, R.C.; Tan, I.B.H.; Ma, B.B. Current advances in targeted therapy for metastatic colorectal cancer – Clinical translation and future directions. Cancer Treat. Rev. 2024, 125, 102700. [Google Scholar] [CrossRef]
- Tsai, K.-Y.; Huang, P.-S.; Chu, P.-Y.; Nguyen, T.N.A.; Hung, H.-Y.; Hsieh, C.-H.; Wu, M.-H. Current Applications and Future Directions of Circulating Tumor Cells in Colorectal Cancer Recurrence. Cancers 2024, 16, 2316. [Google Scholar] [CrossRef]
- Ben-Baruch, A. Tumor Necrosis Factor α: Taking a Personalized Road in Cancer Therapy. Front. Immunol. 2022, 13, 903679. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, Z. TNF-α promotes colon cancer cell migration and invasion by upregulating TROP-2. Oncol. Lett. 2018, 15, 3820–3827. [Google Scholar] [CrossRef]
Figure 1.
The canonical and non-canonical NF-κB signaling pathways. The canonical pathway, triggered by TNF-α, leads to p65/p50 translocation into the nucleus. The non-canonical pathway, activated by CD40L, results in p52/RelB nuclear translocation.
Figure 1.
The canonical and non-canonical NF-κB signaling pathways. The canonical pathway, triggered by TNF-α, leads to p65/p50 translocation into the nucleus. The non-canonical pathway, activated by CD40L, results in p52/RelB nuclear translocation.
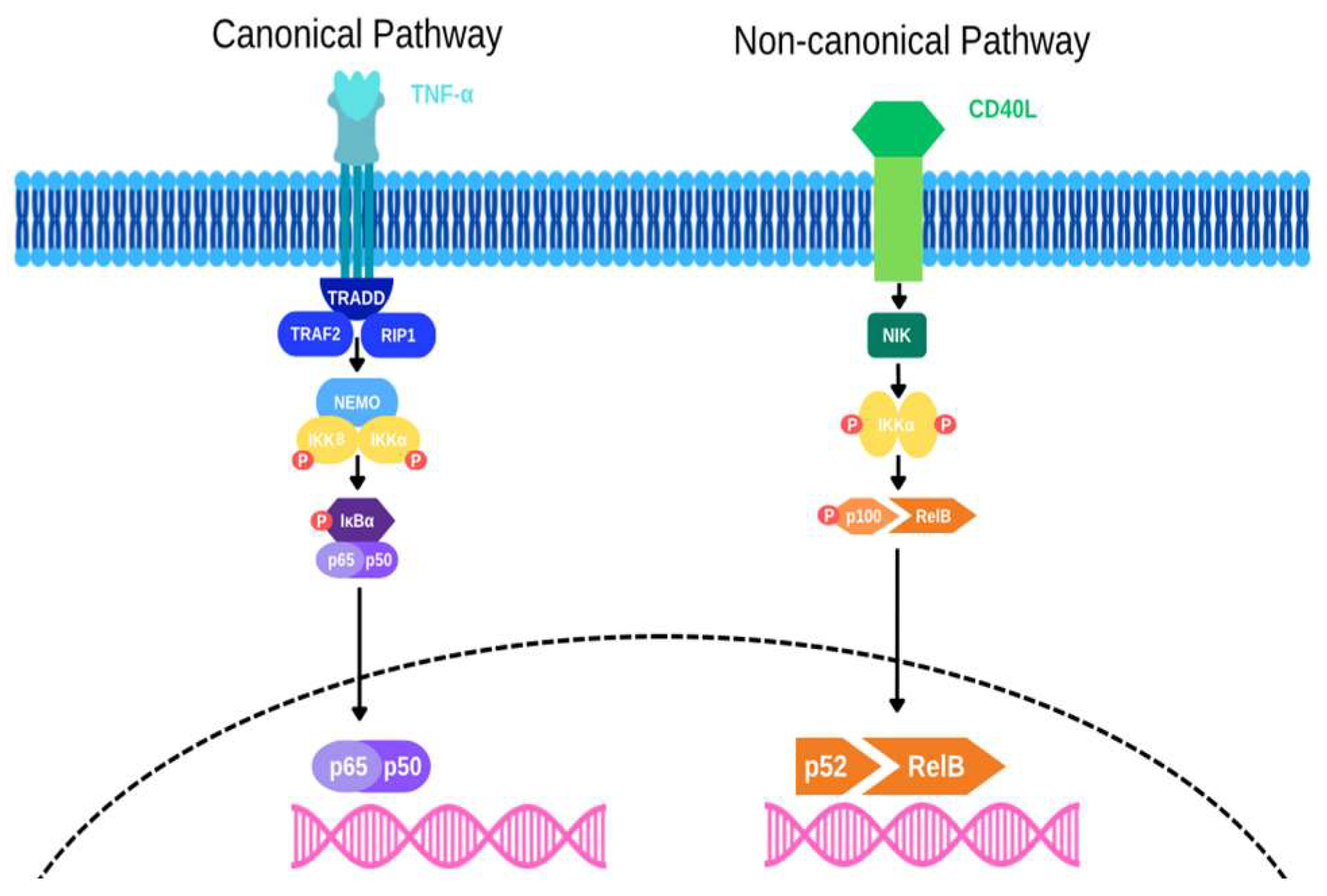
Figure 2.
Oxidative Stress and DNA Repair Mechanism. Oxidative stress induces DNA damage, leading to 8-OHdG formation and DNA repair responses. Successful repair via base or nucleotide excision pathways maintains genomic stability, while failure leads to mutations, genomic instability, and increased disease risk.
Figure 2.
Oxidative Stress and DNA Repair Mechanism. Oxidative stress induces DNA damage, leading to 8-OHdG formation and DNA repair responses. Successful repair via base or nucleotide excision pathways maintains genomic stability, while failure leads to mutations, genomic instability, and increased disease risk.
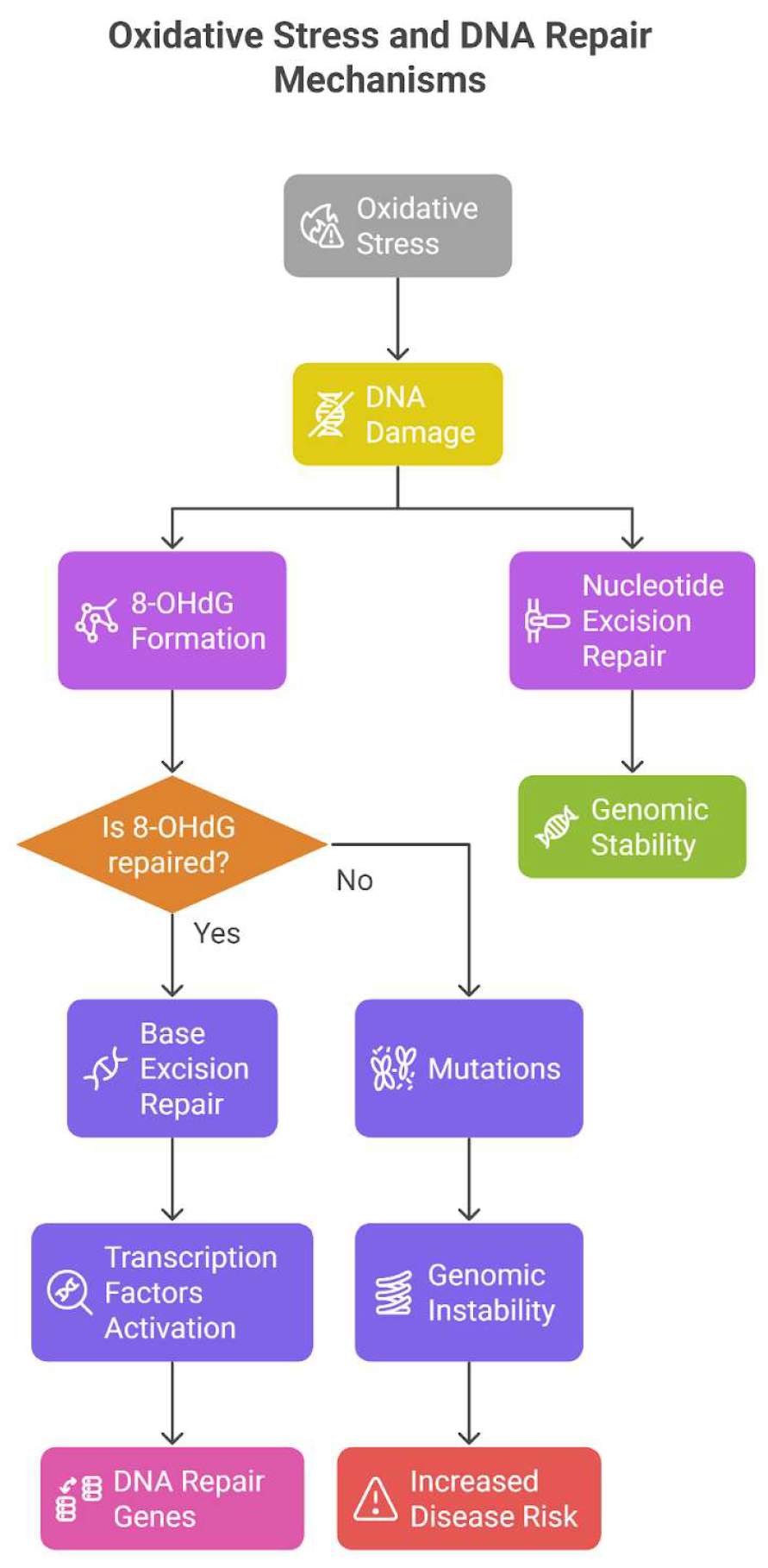

Table 1.
Overview of studies investigating TNF-α role in colon cancer.
| Study | Study design | Sample size | Objective | Findings and results | Conclusion |
|---|---|---|---|---|---|
| Al Obeed et al. 2014 [15] | Retrospective cohort study |
30 |
To detect TNF-α expression in CRC cells among Saudi patients and correlate it with cancer stages. | TNF-α mRNA expression was higher in colorectal cancer than in normal tissue. High expression correlated with Stage III/IV neoplasms (P = 0.004). 83% of patients showed strong TNF-α staining, while 10% showed weak and 7% were negative. | High TNF-α expression may serve as a diagnostic marker and prognostic tool for advanced CRC stages. |
| Li et al,2011 [18] | Case-sontrol study | 180 colon cancer patients and 180 control subjects | To investigate association between TNF alpha 308G/A gene polymorphism and the risk of colon cancer. | Individuals with TNF-alpha-308AA genotype had a higher risk of developing colon cancer to those with other genotypes. | The TNF-alpha-308AA genotype is associated with an increased risk of colon cancer. |
| Kaminska et al.,2005 [19] | Observational study | 157 untreated colorectal cancer patients and 50 healthy volunteers | To evaluate clinical utility of measuring cytokines and their receptors in colorectal cancer patients and assess their correlation with clinicopathological features and prognosis. | Certain cytokines and their receptors were elevated in CRC patients compared to healthy controls.specifically, soluble tumor necrosis factor type 1 levels were identified as independent prognostic factors. | Measuring serum levels of specific cytokines,particularly sTNF RI, can provide valuable prognostic information in untreated colorectal cancer patients. |
| Kemik et al.,2010 [20] | Observational study | 126 colon cancer patients and 36 controls | To analyze the association between TNF-alpha and colon cancer related cachexia other inflammatory markers, cytokines, and hormones. | TNF-alpha levels were higher in patients with colon cancer patients compared to controls | In colon cancer-related cachexia, TNF-α is an essential cytokine that promotes weight loss, systemic inflammation, and the spread of the malignancy. |
| Babic et al.,2016 [21] | Prospective cohort study | 544 CRC patients (225 males from the Health Professionals Follow-Up Study and 319 women from the Nurses' Health Study). | To investigate into the association between colorectal cancer (CRC) patients' mortality and their levels of soluble tumour necrosis factor receptor type II (sTNF-RII). | Higher sTNF-RII levels were linked to higher overall and CRC-specific death rates. The study observed that sTNF-RII, a marker of inflammation, may contribute to the development of colorectal cancer. | TNF-alpha signaling may contribute to disease progression and resistance to treatment, as elevated sTNF-RII levels are associated with poorer survival outcomes in CRC patients. |
| Chan et al.,2011 [22] | Case-control study | from a group of 32,826 women, 280 cases of colorectal cancer and 555 matched controls. | This study looks at the association between colorectal cancer (CRC) risk and plasma inflammatory markers (CRP, IL-6, and sTNFR-2) and whether using aspirin or NSAIDs has a distinct impact on CRC risk depending on the levels of inflammatory markers. | An elevated risk of colorectal cancer was linked to higher plasma levels of sTNFR-2. Following blood collection, women with elevated sTNFR-2 levels who began taking aspirin or NSAIDs had a markedly decreased risk of colorectal cancer (CRC). Aspirin/NSAID use did not significantly lower the risk of colorectal cancer in women with low sTNFR-2 levels. |
CRC risk is linked to plasma sTNFR-2 levels (but not CRP or IL-6). Women with high sTNFR-2 levels are less likely to develop colorectal cancer (CRC) when taking aspirin or NSAIDs, but not those with low levels. This implies that anti-inflammatory medications may be more beneficial for preventing colorectal cancer in specific subgroups based on inflammation indicators. |
| Kapitanovic,2014 [23] | Case control study | 91 patients with sporadic colon adenocarcinoma and 100 healthy controls | Four TNF-α promoter SNPs were examined for allelic frequencies in patients with sporadic colon adenocarcinoma in order to look into their potential significance in sporadic colon cancer susceptibility. Furthermore, to evaluate the impact of these TNF-α SNPs on TNF-α mRNA and protein expression in colon tumours as well as their possible involvement in the initiation and spread of tumours. | TNF-α mRNA expression and tumor histological grade were shown to be significantly correlated, with grade 2 and grade 3 tumors exhibiting greater expression. A strong correlation was found between the TNF-α-857 C/T genotype and elevated TNF-α mRNA expression in tumor tissue. TNF-α protein expression was detected in 78.26% of tumors; however, neither TNF-α genotypes nor clinicopathological features were associated with this expression. Although it was not statistically significant, patients with malignancies that did not have TNF-α protein lived longer. |
Although TNF-α polymorphisms are not substantially linked to an increased risk of developing sporadic colon cancer, they may contribute to the disease's progression. Tumor behavior may be impacted by the TNF-α-857 C/T genotype's influence on TNF-α mRNA expression. |
| Zhao and Zhang, 2018 [24] | Experimental study Cell Culture: HCT-116 human colon cancer cells were used. |
Involved HCT-116 colon cancer cells treated with different TNF-α concentrations. | To investigate how TNF-α regulates TROP-2 expression and its impact on colon cancer cell migration and invasion in vitro. | -TROP-2 Expression: • Increased at low TNF-α concentrations |
Low concentrations of TNF-α promote colon cancer cell migration and invasion by upregulating TROP-2. |
| Li et al,2017 [25] | Tissue Sample Analysis Study | 108 human colon cancer tissue samples. |
To evaluate the expression and prognostic sig-nificance of TNF-α in colon cancer. | High TNF-α Expression: -Observed in colon cancer tissues and cell lines. Prognostic Significance: -High TNF-α levels were associated with poorer survival and identified as an independent adverse prognosticator in colon cancer. |
TNF-α is a poor prognostic marker in colon cancer. Blocking TNF-α may improve chemotherapy efficacy, potentially benefiting colon cancer patients. |
| M. Grimm et al., 2011 [26] | In Vitro Study | 104 patients | Association between TNF-α and lymph node status in CRC patients | 94% of the patients with CRC expressed TNF-α. High TNF-α expression was significantly associated with positive lymph node stage and recurrence of the tumor. | TNF-α expression by tumor cells may be an efficient immunological escape mechanism by inflammation enhanced metastases and probably by induction of apoptosis in tumor-infiltrating CD8+ immune cells resulting in a down reg-ulation of the tumoral immune re-sponse. |
| Guo et al., 2023 [27] | In vitro study | A total of 54 CRC and 60 gastric cancer samples were analysed with immunofluores-cence staining. | Association be-tween TNF-α sig-naling suppression and enhanced an-ti-PD1 immuno-therapy | Blocking TNF-α /TNFR2 signaling in colorectal cancer cells reduces CCR8+T regulatory cells, improving the efficacy of anti-PD1 immunotherapy and potentially improving prognosis in patients with CRC and gastric cancer. | Study findings provide insights into the regulatory mecha-nisms of CCR8+ Tregs, and we propose TNFR2 as a promising therapeutic target for the treatment of CRC. |
Table 2.
Therapeutic Targeting of TNF-α in Colon Cancer: Mechanisms, Monoclonal Antibody Therapy, and Clinical Implications.
Table 2.
Therapeutic Targeting of TNF-α in Colon Cancer: Mechanisms, Monoclonal Antibody Therapy, and Clinical Implications.
| Aspect | Details |
|---|---|
| Biological Role of TNF-α | Plays a critical role in colon cancer progression. High expression of TNF-α correlates with poor prognosis [25,40]. |
| Mechanisms of Action | Stimulates macrophages to produce pro-tumorigenic factors such as CSF-1, VEGF-A, and MMP-2 [41]. Enhances epithelial-to-mesenchymal transition (EMT), increasing invasiveness [42]. |
| Therapeutic Targeting | TNF-α is a key target in colon cancer due to its involvement in tumor-stromal interactions and inflammatory pathways. |
| Monoclonal Antibody Therapy | Infliximab (anti-TNF-α monoclonal antibody) induces antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC), promoting tumor cell apoptosis [40]. |
| Combination Therapy | Synergistic effects when combined with chemotherapy agents like oxaliplatin or 5-fluorouracil, resulting in tumor regression in preclinical models [25,40]. |
| Clinical Implications | TNF-α antagonists are currently being tested in clinical trials as a part of novel treatment strategies for colon cancer. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



