
Submitted:
13 April 2025
Posted:
14 April 2025
You are already at the latest version
Abstract
In recent years, many studies have pointed out the excellent properties of materials composed of collagen and chitosan for wound-healing applications. Combining both polymers in a wound dressing together with a bioactive compound could complement and potentiate these materials as new-generation wound-healing products. Hydroxytyrosol (HT), an antioxidant from olive oil, may contribute to wound healing due to its already reported anti-inflammatory, antimicrobial, and angiogenesis-stimulating properties. It could be a beneficial addition to collagen-chitosan dressings, improving their therapeutic effects. This study screens the potential of collagen-chitosan composites with HT for wound-healing applications and assesses the influence of the compound's incorporation on the materials' properties. The material production involved incorporating chitosan and HT into a marine collagen extract. The resulting collagen-chitosan-HT material was obtained by freeze-drying. Prototype dressing characterization included morphology by scanning electron microscopy, solid and hydrated state by textural and rheologic studies, and in vitro HT release studies. The materials’ cytocompatibility screening was assessed using a mouse fibroblast cell line, and the antibacterial activity was evaluated against microorganisms commonly implicated in wound infections. Results indicate that chitosan contributed to the material's mechanical robustness by maintaining a high viscosity and preserving the material gel structure. The in vitro release studies suggest an HT-controlled release profile with a maximum release (70%) achieved after 10 h. Biological experiments proved the materials' cytocompatibility with skin cells and very promising antibacterial efficacy against S. aureus and P. aeruginosa. In conclusion, HT was successfully incorporated into a collagen-chitosan matrix, enhancing the therapeutic prospect of the resultant material. The collagen-chitosan-HT composite presents a promising potential as an advanced wound-healing material.
Keywords:
Collagen
; Chitosan
; Hydroxytyrosol
; Biomaterials characterization
; Advanced wound dressings
; Biomedical applications
1. Introduction
Wound management remains a critical challenge in clinical practice, with various types of wounds requiring tailored approaches for effective healing. Acute wounds, such as surgical incisions and traumatic injuries, typically follow a predictable healing course. In contrast, chronic wounds, including diabetic ulcers, pressure sores, and venous leg ulcers, often exhibit prolonged inflammation and impaired healing, posing significant burdens on patients and healthcare systems [1,2]. The wound-healing process is a complex and dynamic sequence of events that involves four overlapping phases: hemostasis, inflammation, proliferation, and remodeling [3]. Traditional wound dressings, such as gauze and bandages, primarily serve as protective barriers. However, they often fail to address the complex biological processes of wound healing. Understanding wound healing has led to developing a new generation of bioactive dressings. These dressings are not just physical barriers but active participants in the healing process. They deliver therapeutic agents, maintain a moist wound environment, and promote cellular activity essential for tissue repair [1,4].
Furthermore, these dressings may present several forms, including hydrated-state hydrogels, films, and hydrocolloid dressings or solid-state fibers, acrylics, and foams, each tailored to specific wound types and healing stages [4]. Solid wound dressings offer several advantages over hydrogels and other hydrated materials in wound management. They are typically easier to sterilize and handle and do not require special storage conditions. Due to the low moisture content, they can absorb exudate efficiently, maintaining an optimal wound moisture balance in highly exudative wounds [5]. Solid-state materials also provide a longer shelf life, are less prone to microbial contamination, and prevent the degradation of unstable bioactive compounds.
Emergent new-generation solid dressings comprise various synthetic and natural polymeric matrices. Synthetic polymers include poly(lactic-co-glycolic acid), polyurethane, and polycaprolactone. On the other hand, natural polymers such as alginate, chitosan, and collagen are also frequently explored as wound dressing components [4,5]. Some natural polymers possess inherent bioactive properties and offer several advantages over synthetic compounds as they are highly biocompatible and biodegradable, reducing the risk of adverse reactions. Moreover, several natural polymers, such as chitosan and collagen, can be derived from undervalued food industry by-products, potentially making the wound dressing production process more sustainable and eco-friendlier [6,7]. Collagen, a primary structural protein in the human extracellular matrix, provides a scaffold that supports cell adhesion, migration, and proliferation, thereby facilitating tissue regeneration [8]. Several works have mainly presented the potential of marine collagen due to its excellent wound-healing properties and abundant sources [7,9,10]. On the other hand, chitosan has also emerged as a promising polymer for wound dressings, presenting biocompatibility, biodegradability, and antimicrobial activity properties [6]. Additionally, chitosan could significantly enhance the mechanical robustness of wound dressing materials, ensuring resistance to physical stresses without tearing or losing integrity.
Incorporating bioactive compounds in novel polymeric dressings is crucial for enhancing wound healing outcomes. These bioactive agents, such as antimicrobial peptides, growth factors, and anti-inflammatory molecules, actively participate in the healing process by promoting cell proliferation, reducing infection risks, and modulating the inflammatory response [4,5,11]. Hydroxytyrosol (HT), a phenolic compound usually present in olive oil products and processing sub-products, has garnered significant attention for its potential in wound healing applications. Its distinctive antioxidative properties help to mitigate oxidative stress at the wound site, thereby protecting cells from damage and promoting tissue repair [12]. HT also exhibits anti-inflammatory and antimicrobial activities, crucial for reducing inflammation and preventing infections during wound healing [12,13]. Several studies have also shown that HT can enhance angiogenesis and collagen synthesis, further accelerating wound closure and improving overall healing outcomes [13,14]. These multifaceted benefits make HT a valuable bioactive compound that can be incorporated into advanced wound care products.
This work explores collagen-chitosan composites’ production and wound-healing potential enhanced with HT for topical applications. The authors intend to combine marine collagen’s structural and regenerative properties with chitosan’s antimicrobial and mechanical robustness, together with the incorporation of HT, to further enhance the therapeutic efficacy of the composites. This study evaluates the morphology, the solid and hydrated-state mechanical properties, and the HT release profile. Bioassays were also conducted to determine the toxicity screening of the skin cells’ contact and assess the compound’s synergic antimicrobial activity against microorganisms recognized as skin commensals and nosocomial pathogens.
2. Materials and Methods
2.1. Materials
HT (≥ 98%) was supplied by Greenway Biotech Co., Ltd (China). Low molecular weight (50–190 kDa, 75–85% deacetylated) chitosan, Eagle’s minimum essential medium (EMEM), phosphate-buffered saline (PBS), pH = 7.4, dimethyl sulfoxide (DMSO) and hydrochloric acid (HCl, ≥37%) were purchased from Sigma (USA). Nonessential amino acids (NEAA), fetal bovine serum (FBS), and 0.25% (w/v) Trypsin-EDTA were purchased from Gibco (Life Technologies, United States). CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assay (MTS) reagent assay was obtained from Promega (Madison, USA). Tryptic soy broth (TSB) was purchased from Scharlau (Spain), and tryptone soya agar (TSA) and nutrient agar (NA) were purchased from Oxoid (UK). Cation-adjusted Mueller Hinton broth (CAMHB) was acquired from BD Difco (USA). Antibiotic test discs and SnakeSkin™ dialysis tubing (10 kDa) were purchased from Fisher Scientific (EU). All solvents used for the HT quantification studies were of HPLC grade. Staphylococcus aureus ATCC 6538 (WDCM 00193) and Pseudomonas aeruginosa ATCC 27853 (WDCM 00025) were the strains selected to represent gram-positive and gram-negative bacteria, respectively. These species are recognized as skin commensals as well as nosocomial pathogens [15,16].
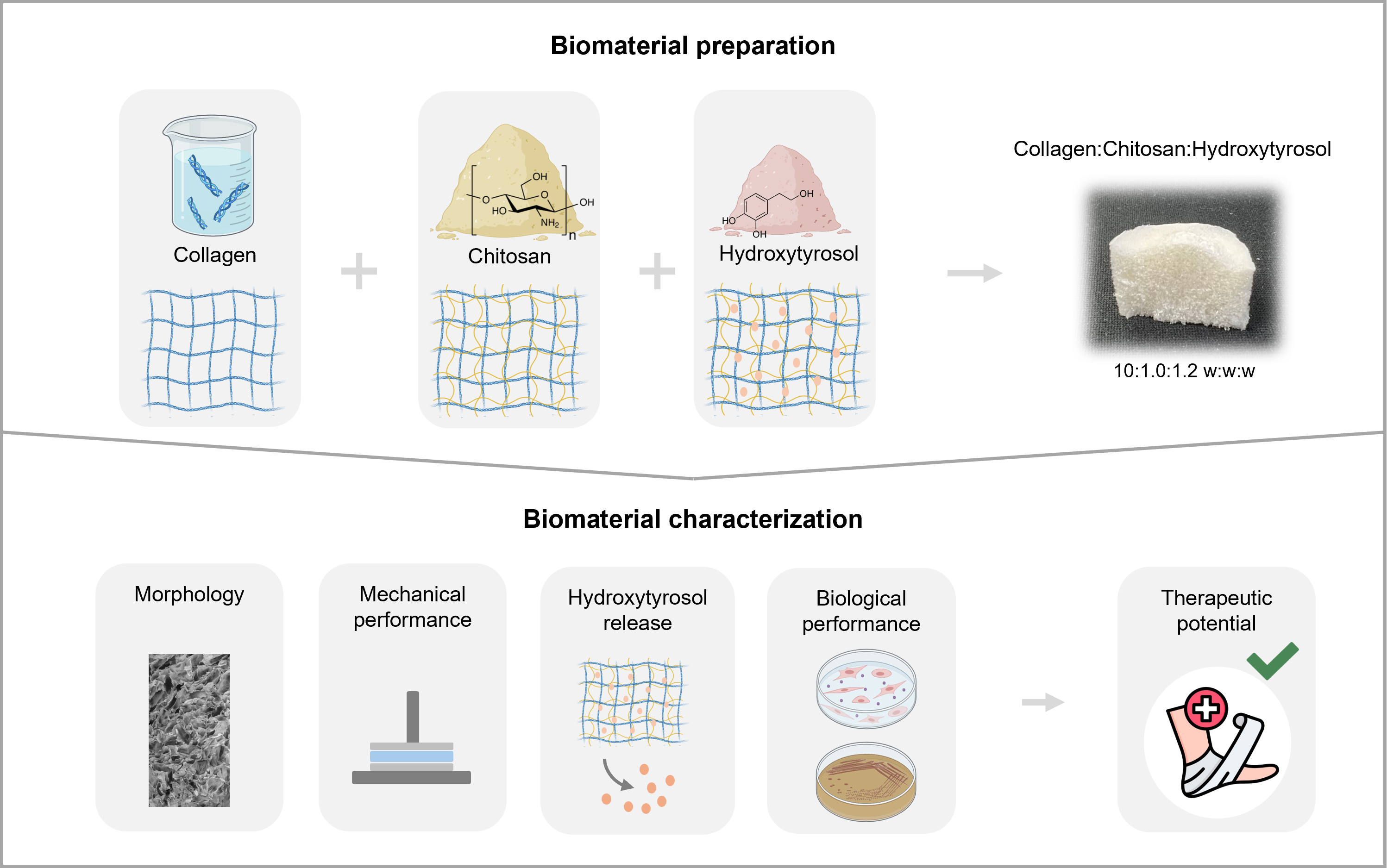
2.2. Biomaterials Preparation
The collagen extract used to produce the solid biomaterials in this study was obtained by extraction according to our previous work [7]. The extract is composed of collagen (0.4 wt.%) and a natural deep eutectic solvent (NADES) consisting of citric acid, xylitol, and water (1.0:1.0:10 mol ratio). Chitosan was added to the collagen extract in a 10:1.0 w:w (collagen:chitosan) ratio, taking advantage of the acidic nature of the solution required for the chitosan solubilization. Chitosan was dissolved under magnetic stirring at room temperature for 2 h. Then, the collagen-NADES extract and chitosan solution were dialyzed against distilled water to remove the NADES. Dialysis was performed for 5 days at 4 °C, with the solutions changed every 12 h. After dialysis, HT was dissolved in the dialyzed collagen-chitosan solution for 15 min in a concentration of 1.6 mg/mL. Finally, the collagen-chitosan-HT solution was transferred into molds (12-well and 24-well plates) and gelled under 4 °C for 18 h. The hydrogels were frozen (-20 °C) and freeze-dried for 48 h. All freeze-dried samples were prepared in triplicates. The collagen (Coll)-chitosan (Chit)-hydroxytyrosol (HT) biomaterial production process is summarized in Figure 1.
2.3. Morphology
The morphology of the samples was analyzed using field emission gun scanning electron microscopy (SEM), using a Phenom ProX G6 Desktop SEM (Thermo Fisher Scientific) at an accelerating voltage of 10 kV and working distances of 2–6 mm. The samples were imaged at several magnifications.
2.4. Mechanical Properties
2.4.1. Textural Analysis
Before textural analysis, the samples were pressed into discs to normalize the thickness of each material. Thickness normalization was done using a compressed air press, with the materials being pressed three times for 5 s at 5 bar. Textural analysis measured the burst strength (BS) and distance to burst (DB) calculated from the puncture test. This test determines the material’s strength to puncture a cylindrical probe at a constant speed and the distance at the breaking point. This analysis was performed using a texture analyzer (TA.XT plus, Stable Microsystems, Godalming Surrey, UK). Samples were fixed to a film support rig (HDP/FSR) and compressed with a probe adaptor (AD/100) at 1 mm/s until rupture. A load cell of 30 kg was used, and the puncture test was carried out in triplicate.
2.4.2. Rheology Studies
The rheological studies were conducted in a controlled stress Kinexus Lab + Rheometer (NETZSCH Analyzing & Testing, Selb, Germany), using the program rSpace for Kinexus (Version 1.76.2398.0, 2019, Selb, Germany) at 32°C. Solid samples were hydrated with PBS (pH = 7.4), mimicking wound fluid exudate absorption, and characterized regarding viscosity, oscillation, tackiness, and adhesion. All methodologies were assessed using a smooth plate-plate geometry with an upper diameter of 20 mm (PU20). For viscosity measurements, the shear rate range varied from 0.1 to 10 s−1. Regarding the oscillatory method, first, an amplitude sweep test was performed to define the linear viscoelasticity range (shear strain was 1% for all samples). The oscillation frequency of samples was determined using frequencies ranging from 0.1 to 10 Hz. For tackiness and adhesion measurements, a pull-away test was performed with 0.5 mm/s for gaping speed, 20 mm for the final gap, and 0.5 mm for the working gap. Testing was performed in triplicates for viscosity and oscillation. Tackiness and adhesion were carried out with n=5 replicates.
2.4.3. In Vitro HT Release Studies
The release of HT from the Coll:Chit:HT biomaterial prepared was studied using Franz diffusion cells (4 mL receptor volume; 1 cm2 permeation area), provided with hydrophilic polysulfone membrane filters (Supor® 450 PES Membrane Disc Filters, 0.45 µm, Pall Corporation, New York, USA) using PBS as the receptor phase. The membranes were washed and equilibrated with the receptor phase and then set between the donor and receiver compartments of the Franz diffusion cells. The system was maintained at 32 °C ± 1 °C for 30 min before the experiment started. The donor phase consisted of 62 mg of each biomaterial, with a total HT loaded content of 7.8 mg, quantified by HPLC-DAD. Each biomaterial replicate was evenly applied on the surface of the membrane in the donor compartment and immediately sealed with Parafilm® to prevent water evaporation. Samples of 200 µL were collected from the receptor phase at 1 h, 2 h, 4 h, 6 h, 10 h, and 24 h, and the collected volume was immediately replaced with a fresh receptor phase kept at the same temperature. Studies were performed using four Franz cells, and the amount of permeated HT was determined using the HPLC method described below. The percentage of HT released into the medium was calculated using the following equation:
Where Mt is the cumulative amount of HT released at each sampling time point t, and M0 is the initial weight of the HT in the materials.
The data obtained from in vitro release studies were computed using DDsolver [17], which is an Excel plugin module, and the resultant data were fitted to different kinetic models:
2) Zero order kinetics
Where K0 is the zero-order release constant
3) First order kinetics
Where K1 is the first-order release constant.
4) Higuchi model
Where KH is the Higuchi release constant.
5) Korsmeyer-Peppas model
Where KKP is the release constant incorporating the structural and geometric characteristics of the drug-dosage form, and n is the diffusional exponent indicating the drug-release mechanism.
In all models, F is the fraction (%) of the released drug in time, t. The adjusted coefficient of determination (R2 adjusted) was estimated for each model, fitted, and used as a measure of the model’s ability to describe a given dataset. The R2 adjusted values and the Akaike minimum information theoretical criterion (AIC) were used as a measure of fit to compare the different models. The AIC technique relies on fitting the model to the existing data and assessing its potential for predicting future values. When comparing several competing models, the best-fitting model gives the minimum AIC value [18,19].
2.4.4. HT Identification and Quantification by HPLC
Identification and quantification of HT were performed by an HPLC-DAD Vanquish (Thermo Fisher Scientific, USA) equipped with a quaternary pump, a solvent degasser, an autosampler (10 μL injection volume at 12 °C), and a column oven at 35 °C. The equipment was also coupled to a Photodiode Array Detector Waters 996 PDA (Waters, Ireland, UK), scanning wavelength absorption from 190 nm to 680 nm. The column used was a Luna 5 µm C18(2) 100 Å (250 x 4 mm). A gradient method was applied with two eluents: eluent A (Milli-Q water with 0.5% formic acid) and eluent B (90% acetonitrile, 9.5% Milli-Q water, and 0.5% formic acid). A flow rate of 0.6 mL/min was set, and the following elution program was applied: 0−15 min 94.4% of A and 5.6% of B; 15−22 min 80% of A and 20% of B; 22−45 min 60% of A and 40% B; 45−55 min 100% of B, and finally returning to the initial conditions for 10 min.
2.5. Bioassays
For the biological assays, materials’ sterilization was performed and validated using ultraviolet (UV) irradiation and sterility confirmation tests following the methodology referenced in the literature [20].
2.5.1. Cytocompatibility
To mimic the contact of the materials with skin cells, cytocompatibility was evaluated following the direct contact methodology described in ISO 10993–5, a sensitive test used to determine medical devices’ toxicity [20,21]. Mouse fibroblasts NCTC clone 929 (ECACC 88102702) cells, purchased from the European Collection of Authenticated Cell Cultures (ECACC, Public Health England, Salisbury, UK), were routinely grown in the standard EMEM medium supplemented with 1% (v/v) NEAA and 10% (v/v) heat-inactivated FBS. Stock cells were maintained as monolayers in 75 cm2 culture flasks, subcultured weekly (seeding 30,000 cells/cm), and incubated at 37 °C in a 5% CO2 humidified atmosphere. For cell passage, the cells were detached when confluence reached about 80% using 0.25% (v/v) trypsin/EDTA at 37 °C. The cells were collected, and viability was determined using the standard trypan blue staining procedure. Cell counting was performed using an automated cell counter (Invitrogen Countess™ 3, Thermo Fisher Scientific). All cellular assays were performed with cells between passages 10 and 20.
Cell viability was quantified using the MTS cytotoxicity test, following the methodology described in the literature with slight modifications [20,21]. NCTC clone 929 cells were seeded into 24-well plates (volume of 0.6 mL) with a density of 3.0 × 104 cells/cm2 and maintained in culture for 24 h (~1 doubling period) to form a semi-confluent monolayer. After 24 h, new EMEM supplemented with 0.5% FBS was replaced, and following the direct contact method, cells were incubated for 24 h with test samples. The materials used in this assay were previously prepared in 24-well plates, corresponding to samples of 32 mg/well. Following ISO 10993–5, these sterilized samples were directly incorporated into seeded wells, reaching a final testing concentration of 53.3 mg/mL. Lastly, the cytocompatibility testing samples were removed, and cells were rinsed three times with PBS and incubated for 3 h with 0.6 mL of MTS reagent assay, diluted according to the manufacturer’s information. The absorbance was recorded at 490 nm using a microplate spectrophotometer (EPOCH, 219 Bio-Tek, USA). Experiments were performed in triplicate in three independent assays. The positive control of cytotoxicity was done with a treatment of 10% (v/v) DMSO solution diluted in EMEM. Results were expressed as a percentage of cellular viability (Viab.%) relative to the negative control (untreated cells). A cytotoxic effect was considered for viability percentages below 70%, according to ISO 10993–5.
2.5.2. Antimicrobial Susceptibility Testing (AST)
Antimicrobial susceptibility testing (AST) assays were performed according to the broth microdilution method of CLSI M07-A10 guidelines [22]. Compound stock solutions were dispensed in a 96-well round bottom microtiter plate and twofold serially diluted in CAMHB to obtain a concentration range of solutions. A standardized inoculum was diluted in CAMHB to ensure that, after inoculation, each well contained approximately 5×104 CFU. Each inoculated microtiter plate was incubated under aerobic conditions at 37 °C for 24 h. Minimal inhibitory concentration (MIC) values were read as the lowest compound concentration for which visible growth was inhibited after 24 h of incubation. For each compound stock solution assayed, a growth control (CAMHB and diluted inoculum), a medium sterility control (CAMHB), and a stock solution sterility control (CAMHB and compound stock solution) were also tested. All compound stock solutions were previously filter-sterilized (0.45 µm) using a solvent-compatible filter, and the results of this experiment are expressed as median values of three biological replicates performed. The MIC values for the dissolving agent used (HCl) were determined to ensure that it did not inhibit bacterial growth in the assay conditions. All relevant compounds, dissolving agents, and respective highest tested concentrations are summarized in Table 1.
2.5.3. Antimicrobial Activity Determination of Biomaterials
The antibacterial performance of the biomaterials was measured using the absorption method and the quantification by plate count method from ISO 20743:2021, with some modifications [23]. All samples (biomaterials and the control fabric cotton disks) were prepared by weighing 20 mg and sterilized, as mentioned before. All sterile vials containing the samples were inoculated with 10 μL of a normalized bacterial suspension (1–3 × 105 CFU/mL), which was allowed to be fully absorbed. After inoculation and incubation steps (0 h and 24 h), the bacterial recovery was performed by adding 1 mL of shake-out physiological saline solution. This antimicrobial activity assay was performed for both selected target bacteria, and the results are expressed as mean values for three biological replicates. The antibacterial activity value was calculated according to ISO 20743:2013. Efficacy is defined as strong (for antibacterial activity value ≥ 3), significant (for 2 ≤ antibacterial activity value < 3), or negligible (for < 2 antibacterial activity value).
2.6. Statistical Analysis
The statistical analysis of the data was performed using GraphPad Prism 10 (GraphPad Software, Inc., CA). All values were tested for normal distribution and equal variance. When homogeneous variances were confirmed, data were analyzed by one-way analysis of variance (one-way ANOVA) coupled with Tukey’s post hoc analysis to identify means with significant differences. All registered statistical differences are mentioned when at least the p < 0.05 condition was verified.
3. Results and Discussion
Previously published works describe the production of collagen materials with chitosan and bioactive compounds for biomedical applications [10,24,25,26,27,28,29,30,31,32]. This article builds on that work and presents a new solid collagen material that leverages the NADES solubility properties of collagen, obtained according to our previous work [7], together with chitosan, while additionally incorporating a potent antioxidant (HT) barely studied as a biomaterial ingredient for potential biomedical applications. Three different materials composed of collagen (Coll), collagen and chitosan (Coll:Chit), and collagen with chitosan and HT (Coll:Chit:HT) were prepared to understand the influence of incorporating chitosan and HT in the collagen material. Chitosan was incorporated into the collagen extract in a 10:1.0 Coll:Chit w:w ratio, according to several solubilization tests indicating this maximum achievable chitosan incorporation. HT was incorporated according to the MIC obtained in the antimicrobial susceptibility testing performed against S. aureus and P. aeruginosa (Table 2). This test was also completed for chitosan to verify that both compounds used and incorporated in the collagen matrix were active antimicrobials against well-recognized skin commensals and nosocomial pathogens [15,16].
As expected, chitosan and HT displayed their antibacterial activity against both bacteria. This result agrees with the literature since, unlike collagen, chitosan and HT have a well-established bioactivity against these microorganisms [6,12,13,33,34,35]. HT is known to be unstable in aqueous media. It can degrade over time, especially when exposed to light and oxygen [36,37]. Nevertheless, preliminary testing revealed that aged HT solutions (24 h at 37 °C and 60 days at 4 ºC) retained their antimicrobial activities since the MIC remained, at least, the same as a freshly prepared HT solution (Table SM 2).
Based on chitosan solubility tests and the highest HT MIC value recorded for both bacteria (1.6 mg/mL), a material composed of collagen, chitosan, and HT was obtained in a 10:1.0:1.2 Coll:Chit:HT w:w:w ratio. The Coll and Coll:Chit materials were obtained by performing the Coll:Chit:HT production process described in the materials and methods section without the HT (Coll:Chit) and the chitosan and HT incorporation steps (Coll). The resulting materials’ macroscale photographs and the corresponding scanning electron microscopy (SEM) micrographs are shown to evaluate the morphology (Figure 2).
The resulting freeze-dried gels showed a similar monolithic macrostructure with a sponge-like appearance. SEM micrographs revealed that all the materials presented a similar microstructure consisting of large macropores (pore diameter in the range of several μm) with dense areas between pores. Several studies reported that this structure is typical of freeze-dried collagen gel materials [9,38]. The similar structure between all produced samples suggests that chitosan and HT did not influence the different collagen materials’ morphology.
Materials for topical application should exhibit acceptable mechanical characteristics and must be easy to apply [39]. So, mechanical robustness and malleability are important attributes of topical solid dressings for biomedical applications. These features ensure that dressings provide consistent protection against wounds’ external contaminants and maintain their structural integrity during handling [40,41]. In this sense, several techniques were applied to determine relevant features that characterize the samples. First, a puncture test was performed to determine the material’s strength to be punctured by a cylindrical probe at a constant speed and the distance at the breaking point. This distance is measured as the length from when the probe contacts the material until it breaks [42]. The results of the textural analysis of the solid materials are presented in Table 3.
The analyzed materials showed values of burst strength in the range of 13.9 N to 19.7 N. The solid sample only composed of collagen presented a lower burst strength value, whereas samples with chitosan ruptured at higher force values. Statistical analysis revealed that burst strength value differences are significant between the Coll sample and materials with chitosan. On the other hand, the difference between Coll:Chit and Coll:Chit:HT is not significant. These results suggest incorporating chitosan in the collagen material improved the sample’s mechanical resistance. The material with HT recorded a lower burst strength value than the Coll:Chit sample, which could suggest a plasticizing effect of this compound. However, this difference is not significant, indicating that HT’s presence showed no evident mechanical impact. These results agree with the literature, where several published works demonstrated the positive effect of chitosan in improving the mechanical robustness of collagen materials [43,44]. Nonetheless, a straightforward comparison is not easy as the tests implemented are not standardized, and a direct comparison cannot be established. Regarding the distance at burst, a larger distance to achieve the material burst suggests a higher elastic material, whereas smaller distances suggest a more brittle behavior [45]. However, the texturometer analysis showed similar results between samples with no significant differences, suggesting that chitosan and HT did not contribute to a different elastic behavior of the materials. Overall, the results revealed a higher robustness of materials prepared with chitosan, with the same elastic behavior as the sample only composed of collagen.
Evaluating the mechanical properties of solid-state materials for topical applications provides important information, particularly for handling dry material. Nonetheless, topical applications, particularly in wound management, may lead to exudate absorption, dramatically changing the material’s mechanical behavior from dry to hydrated. So, the viscosity, viscoelastic behavior, and adhesiveness of the hydrated materials were performed. The materials were characterized in two different mass:solvent proportions (1:1 w:w and 1:4 w:w) to mimic different fluid absorptions in two different wound fluid-draining conditions, from moderate (1:1 w:w) to copious (1:4 w:w) wound exudate volumes, after contact with the wounded skin area [46,47].
Figure 3a presents the viscosity results of the three materials in the two hydrated states studied. As expected, all the samples revealed a higher viscosity in the 1:1 w:w condition due to the lower liquid content of the materials. Viscosity behavior as a function of shear rate showed similar profiles for all samples in a 1:1 w:w ratio, with viscosity increasing from 0.1 s-1 to 1 s-1 and remaining constant between 1 s-1 and 10 s-1. The Coll:Chit sample presented higher viscosities at a lower shear rate than Coll and Coll:Chit:HT samples but tended to the same values at a higher shear rate (204 - 504 Pa·s at 10 s-1). On the other hand, the results of the tested samples under a 1:4 w:w ratio showed different viscosity profiles between samples with and without chitosan during all the tested shear rates. Samples containing chitosan exhibited viscosities at least two orders of magnitude higher than those composed solely of collagen. These results suggest that chitosan influenced the internal structure of the collagen materials, unlike HT, which did not change the materials’ viscosity in any of the tested conditions.
To further evaluate the mechanical properties of the prepared materials, the viscoelastic properties of the different hydrated materials were studied and are presented in Figure 3b.
Concerning the oscillation frequency test, all the samples exhibited predominantly elastic behavior at a higher mass:solvent ratio, evident from the greater magnitude of the elastic module (G′) compared to that of the viscous module (G″). This means that the structure of the gels remained intact through the entire range of frequencies, confirming that all the hydrated materials present a strong network and a solid-like behavior (G′ > G″) [43]. For the characterization under a lower mass:solvent ratio, all samples presented lower values of the complex shear modulus. However, the samples containing chitosan (Coll: Chit and Coll:Chit:HT) presented a solid-like behavior. In contrast, a transition to liquid-like behavior is observed in the collagen sample since G″ became higher than G′ at higher hydrated treatment. These results agree with the purpose of chitosan incorporation in conferring higher robustness to collagen materials. Viscosity and viscoelastic assessment revealed that the presence of this polymer is required to maintain higher viscosity and preserve a gel structure even in increased water absorption conditions. These rheology test results also suggested an approximated shear-thickening behavior of all materials since the materials’ viscosity increased with the applied stress [48]. This behavior indicates that the collagen materials could maintain the structural integrity in a wound bed under some stress and provide consistent coverage and protection, as previously described in the literature [49].
Another important aspect of screening analysis of the material’s potential for wound healing applications concerns its adhesiveness. Materials must be self-supported on the wound, otherwise, there is a need for an additional support dressing. On the other hand, dressings must also be easy to remove, avoiding traumatic and painful dressing removal for the patient, compromising wound healing [4,5]. Therefore, a tack and pull-away test was carried out, and the results are shown in Table 4. This assessment enables the evaluation of a sample’s tackiness and adhesive strength by measuring the peak normal force and area under the force-time curve (with a larger area indicating a stronger adhesive) as two parallel plates are pulled apart [50]. Tackiness in the context of material behavior is associated with stickiness and may result from adhesive forces between two materials in contact [51,52].
As expected, the results revealed that samples with lower water content display more prominent stickiness and adhesive strength with values of area under the force-time curve and absolute normal peak force up to 30 N·s and 15 N, respectively. However, the significant decrease in the sample’s stickiness in the 1:4 w:w treatment could be an interesting achievement. These results indicate that a reduction of material adhesiveness occurs at higher water absorption content, which could facilitate its removal painlessly for the patient. On the other hand, each treatment revealed similar results between samples, with slight differences that were not significant. These results suggest incorporating this chitosan and HT content did not significantly influence the collagen material’s stickiness.
In this study, HT was incorporated into the collagen-chitosan matrix, expecting that the HT release into the wound environment would boost the bioactive properties of the dressing material. In vitro release studies evaluated the HT release from the collagen-chitosan material over 24 hours. The study aimed to understand the kinetic release profile of HT, and the results are presented in Figure 4.
Results of HT release revealed an increasing release profile over time and a maximum release percentage (72.7%) at 24 h. However, the slight differences between the 24 h and 10 h time points are not statistically significant. So, the maximum HT release percentage would have already been at 10 h. The HT in vitro release was curve-fitted to zero-order (Eq. 2), first-order (Eq. 3), Higuchi (Eq. 4), and Korsmeyer-Peppas model (Eq. 5) to understand the release kinetics [18,53]. Controlled-release dosage forms are crucial for effective wound healing treatment, requiring an understanding of specific mass transport mechanisms for predicting quantitative kinetics and in vivo behavior. The release of polymer matrix content can occur by various mechanisms such as diffusion, erosion, and dissolution [53]. Table SM 1 presents the results of all tested models. The first-order model revealed an R2 adjusted of 0.92 ± 0.02 and the lowest AIC value of 45.5 ± 1.95. Thus, the release kinetics results suggest that the HT dissolution profile from the hydrated collagen-chitosan matrix is concentration-dependent [53].
The present work aimed to produce an innovative material for potential biomedical applications involving direct contact with skin cells. The cytotoxicity of the produced samples on a mouse fibroblast cell line was assessed to screen the biocompatible safety of the produced materials for potential wound healing applications. Figure 5 presents the ISO 10993-5 direct contact methodology results, which involved placing the material in direct contact with cultured cells. This method simulates real-life interactions between the material and skin cells, allowing for the identification of potential toxic effects from the material or its leachables and predicting biocompatibility.
All tested materials present similar cell viability percentages. However, statistical analysis revealed significant differences in cell viability percentages between the negative control (non-treated cells) and the Coll:Chit:HT sample. This is the only sample containing HT, suggesting that the release of this compound could contribute to the significant decrease in cell viability. However, none of the tested samples, including the Coll:Chit:HT sample, exhibited cell viability percentages below the potential cytotoxic effect threshold defined by ISO standard (70%). In this work, the HT concentration incorporated in the material was based on the MIC value of this compound against both bacterial targets selected. However, future works could directly investigate HT cytotoxicity by measuring the half-effective and inhibition concentrations (EC50 and IC50, respectively) if direct information regarding the cytotoxicity of HT is required.
After the materials were found to be non-cytotoxic, their antimicrobial activity was evaluated against S. aureus and P. aeruginosa. To determine the produced materials’ antimicrobial activity and assess the impact of incorporating chitosan and HT in the collagen matrix, the absorption method outlined in ISO 20743:2013 was performed. This method evaluates the microorganism’s reduction on the material’s surface by measuring the number of viable microorganisms after a specified contact period. It is particularly suitable for assessing the antimicrobial activity of materials intended for wound healing applications by simulating the material’s direct contact with the wound, potentially exposed to various pathogens, and due to its comprehensive and standardized approach. ISO 20743:2013 specifies K. pneumoniae as the surrogate gram-negative bacterial target to be used in the assay instead of P. aeruginosa. The scope of this standard is to assess textile antimicrobial efficacy, and the authors agree that P. aeruginosa is more relevant and adequate representative of gram-negative skin commensals and nosocomial pathogens. Table 5 shows the antimicrobial activity values according to the absorption method of ISO 20743:2013 and its corresponding efficacy.
Regarding S. aureus, the determination of antibacterial activity showed the efficacy of all tested materials. Even the sample composed only of collagen presented significant activity. On the other hand, Coll:Chit and Coll:Chit:HT presented strong antibacterial activity against S. aureus. Regarding P. aeruginosa, the Coll sample activity value was insufficient to be considered effective. In addition, although both samples containing chitosan revealed efficacy against P. aeruginosa, only Coll:Chit:HT showed a strong efficacy. These results indicate that the antibacterial activity of collagen materials was improved with chitosan incorporation. Also, the higher efficacy against P. aeruginosa demonstrates that not only chitosan but also incorporating HT enhances the antibacterial activity of collagen materials. These results agree with the literature’s reported works and the MIC values presented in Table 2, where these compounds showed antibacterial activity against S. aureus and P. aeruginosa [33,34,35]. As previously discussed, some HT degradation can occur in the wound-healing fluid environment. However, the antimicrobial evaluation of aged HT solutions (up to 60 days; Table SM 2) suggests that materials incorporated with this compound might maintain their activity against representative skin commensals and nosocomial pathogens and remain effective over time.
4. Conclusions
Collagen-chitosan composites with HT were successfully prepared. The solid materials, obtained by freeze-drying a Coll:Chit:HT gel mixture, displayed a sponge-like structure with a macroporous network, a feature shared by all the prepared materials. Textural solid-state analysis revealed a positive influence of chitosan on the material’s mechanical resistance performance. In addition, rheological studies suggested that chitosan incorporation preserved the material’s elastic behavior under a higher hydrated state. This study also indicated that a concentration-dependent mechanism released HT from the collagen-chitosan matrix with a controlled-release profile. Finally, bioassays proved the materials’ cytocompatibility with skin cells and strong antibacterial activity against S. aureus and P. aeruginosa. Incorporating chitosan and HT enhanced the collagen material’s antibacterial efficacy. In vivo studies are still necessary to assess the material’s wound healing performance and validate the compound’s biocompatibility and bioactivity in biological models closer to their final application (e.g. pH and temperature variations for certain distinct phases). In conclusion, in this study, HT was successfully incorporated in collagen-chitosan composites, enhancing the promising therapeutic effect of the resultant material. Coll:Chit:HT presents a very promising potential as an advanced wound-healing material for various topical diseases and pathologies.
Acknowledgments
The authors acknowledge the financial support received from the iNOVA4Health – UIDB/ 04462/2020 and UIDP/04462/2020, iMed.ULisboa – UIDB/04138/2020 and UIDP/04138/2020, J. Marto – CEECINST/00145/2018, programs financially supported by Fundação para a Ciência e Tecnologia/Ministério da Ciência, Tecnologia e Ensino Superior, through national funds are acknowledged. Financial support received from The Associate Laboratory LS4FUTURE supported by Fundação para a Ciência e a Tecnologia (FCT, Portugal) through the funding LA/ P/0087/2020 (DOI 10.54499/LA/P/0087/2020) are acknowledged. This work also received funding from the ERC-2016-CoG 725034 and was supported by the Associate Laboratory for Green Chemistry (LAQV), financed by national funds from FCT/ MCTES (UIDB/50006/2020). Miguel P. Batista acknowledges FCT for the financial support through 2020.05895.BD grant.
Declaration of competing interest
The authors declare that they have no competing financial interests or personal relationships that could have influenced the work reported in this paper.
CRediT authorship contribution statement
Miguel P. Batista: Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization, Funding acquisition. Margarida Pimenta: Methodology, Formal analysis, Data curation. Naiara Fernández: Writing – review & editing, Resources. Ana Rita C. Duarte: Writing – review & editing, Project administration, Funding acquisition, Supervision. Maria do Rosário Bronze: Writing – review & editing, Project administration, Funding acquisition, Resources, Supervision. Joana Marto: Writing – review & editing, Validation, Methodology, Formal analysis, Data curation, Conceptualization, Resources. Frédéric B. Gaspar: Writing – review & editing, Validation, Methodology, Formal analysis, Data curation, Conceptualization, Resources, Supervision, Funding acquisition.
References
- R.G. Frykberg, J. Banks, Challenges in the Treatment of Chronic Wounds, Adv Wound Care (New Rochelle) 4 (2015) 560–582.
- G. Han, R. Ceilley, Chronic Wound Healing: A Review of Current Management and Treatments, Adv Ther 34 (2017) 599–610.
- S.A. Eming, P. Martin, M. Tomic-Canic, Wound repair and regeneration: Mechanisms, signaling, and translation, Sci Transl Med 6 (2014). [CrossRef]
- L.I.F. Moura, A.M.A. Dias, E. Carvalho, H.C. De Sousa, Recent advances on the development of wound dressings for diabetic foot ulcer treatment--a review, Acta Biomater 9 (2013) 7093–7114. [CrossRef]
- L.J. Borda, F.E. Macquhae, R.S. Kirsner, Wound Dressings: A Comprehensive Review, Curr Dermatol Rep 5 (2016) 287–297.
- R.A.A. Muzzarelli, Chitins and chitosans for the repair of wounded skin, nerve, cartilage and bone, Carbohydr Polym 76 (2009) 167–182. [CrossRef]
- M.P. Batista, N. Fernández, F.B. Gaspar, M. do R. Bronze, A.R.C. Duarte, Extraction of Biocompatible Collagen From Blue Shark Skins Through the Conventional Extraction Process Intensification Using Natural Deep Eutectic Solvents, Front Chem 10 (2022). [CrossRef]
- S.S. Mathew-Steiner, S. Roy, C.K. Sen, Collagen in Wound Healing, Bioengineering 2021, Vol. 8, Page 63 8 (2021) 63. [CrossRef]
- D.N. Carvalho, R. Lopez-Cebral, R.O. Sousa, A.L. Alves, L.L. Reys, S.S. Silva, J.M. Oliveira, R.L. Reis, T.H. Silva, Marine collagen-chitosan-fucoidan cryogels as cell-laden biocomposites envisaging tissue engineering, Biomedical Materials 15 (2020) 055030. [CrossRef]
- P. Ramasamy, A. Shanmugam, Characterization and wound healing property of collagen–chitosan film from Sepia kobiensis (Hoyle, 1885), Int J Biol Macromol 74 (2015) 93–102. [CrossRef]
- V. Vivcharenko, A. Przekora, Modifications of Wound Dressings with Bioactive Agents to Achieve Improved Pro-Healing Properties, Applied Sciences 2021, Vol. 11, Page 4114 11 (2021) 4114. [CrossRef]
- N.D. Utami, A. Nordin, H. Katas, R.B.H. Idrus, M.B. Fauzi, Molecular Action of Hydroxytyrosol in Wound Healing: An In Vitro Evidence-Based Review, Biomolecules 2020, Vol. 10, Page 1397 10 (2020) 1397. [CrossRef]
- W.A. Batarfi, M.H. Mohd Yunus, A.A. Hamid, The Effect of Hydroxytyrosol in Type II Epithelial-Mesenchymal Transition in Human Skin Wound Healing, Molecules 2023, Vol. 28, Page 2652 28 (2023) 2652. [CrossRef]
- M.S. Duarte, V.M. Fuhro, J. de Souza Nogueira, B. Romana-Souza, Polyphenol hydroxytyrosol present olive oil improves skin wound healing of diabetic mice, Wound Repair and Regeneration (2024). [CrossRef]
- P.M. Alves, E. Al-Badi, C. Withycombe, P.M. Jones, K.J. Purdy, S.E. Maddocks, Interaction between Staphylococcus aureus and Pseudomonas aeruginosa is beneficial for colonisation and pathogenicity in a mixed biofilm, Pathog Dis 76 (2018) 3. [CrossRef]
- M.S. Mulani, E.E. Kamble, S.N. Kumkar, M.S. Tawre, K.R. Pardesi, Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: A review, Front Microbiol 10 (2019) 403107.
- Y. Zhang, M. Huo, J. Zhou, A. Zou, W. Li, C. Yao, S. Xie, DDSolver: An add-in program for modeling and comparison of drug dissolution profiles, AAPS Journal 12 (2010) 263–271. [CrossRef]
- S. Bom, C. Santos, R. Barros, A.M. Martins, P. Paradiso, R. Cláudio, P.C. Pinto, H.M. Ribeiro, J. Marto, Effects of starch incorporation on the physicochemical properties and release kinetics of alginate-based 3D hydrogel patches for topical delivery, Pharmaceutics 12 (2020) 1–20. [CrossRef]
- H. Akaike, A new look at the statistical model identification, IEEE Trans Automat Contr 19 (1974) 716–723. [CrossRef]
- M.P. Batista, V.S.S. Gonçalves, F.B. Gaspar, I.D. Nogueira, A.A. Matias, P. Gurikov, Novel alginate-chitosan aerogel fibres for potential wound healing applications, Int J Biol Macromol 156 (2020) 773–782. [CrossRef]
- ISO 10993-5:2009 - Biological evaluation of medical devices — Part 5: Tests for in vitro cytotoxicity, (n.d.). Available online: https://www.iso.org/standard/36406.html (accessed on 2 September 2024).
- M07 Ed12 | Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 12th Edition, (n.d.). Available online: https://clsi.org/standards/products/microbiology/documents/m07/ (accessed on 2 September 2024).
- ISO 20743:2021 - Textiles — Determination of antibacterial activity of textile products, (n.d.). Available online: https://www.iso.org/standard/79819.html (accessed on 2 September 2024).
- C.C. Wang, C.H. Su, C.C. Chen, Water absorbing and antibacterial properties of N-isopropyl acrylamide grafted and collagen/chitosan immobilized polypropylene nonwoven fabric and its application on wound healing enhancement, J Biomed Mater Res A 84A (2008) 1006–1017. [CrossRef]
- A. Deng, Y. Yang, S. Du, X. Yang, S. Pang, X. Wang, S. Yang, Preparation of a recombinant collagen-peptide (RHC)-conjugated chitosan thermosensitive hydrogel for wound healing, Materials Science and Engineering: C 119 (2021) 111555. [CrossRef]
- H. Xie, X. Chen, X. Shen, Y. He, W. Chen, Q. Luo, W. Ge, W. Yuan, X. Tang, D. Hou, D. Jiang, Q. Wang, Y. Liu, Q. Liu, K. Li, Preparation of chitosan-collagen-alginate composite dressing and its promoting effects on wound healing, Int J Biol Macromol 107 (2018) 93–104. [CrossRef]
- K.G. Shankar, S.U. Kumar, S. Sowndarya, J. Sridevi, S.S. Angel, C. Rose, Rumen tissue derived decellularized submucosa collagen or its chitosan-treated film as a cutaneous wound healant and 1H NMR-metabolite profiling of plasma, RSC Adv 6 (2016) 107403–107415. [CrossRef]
- W. Wang, S. Lin, Y. Xiao, Y. Huang, Y. Tan, L. Cai, X. Li, Acceleration of diabetic wound healing with chitosan-crosslinked collagen sponge containing recombinant human acidic fibroblast growth factor in healing-impaired STZ diabetic rats, Life Sci 82 (2008) 190–204. [CrossRef]
- M. Rezaii, S. Oryan, A. Javeri, Curcumin nanoparticles incorporated collagen-chitosan scaffold promotes cutaneous wound healing through regulation of TGF-β1/Smad7 gene expression, Materials Science and Engineering: C 98 (2019) 347–357. [CrossRef]
- K. Dutta, K. Sarkar, S. Karmakar, B. Gangopadhyay, A. Basu, S. Bank, S. De, B. Das, M. Das, D. Chattopadhyay, Asymmetric fabrication and in vivo evaluation of the wound healing potency of electrospun biomimetic nanofibrous scaffolds based on collagen crosslinked modified-chitosan and graphene oxide quantum dot nanocomposites, J Mater Chem B 11 (2023) 9478–9495. [CrossRef]
- M. Li, M. Han, Y. Sun, Y. Hua, G. Chen, L. Zhang, Oligoarginine mediated collagen/chitosan gel composite for cutaneous wound healing, Int J Biol Macromol 122 (2019) 1120–1127. [CrossRef]
- A.A. Mahmoud, A.H. Salama, Norfloxacin-loaded collagen/chitosan scaffolds for skin reconstruction: Preparation, evaluation and in-vivo wound healing assessment, European Journal of Pharmaceutical Sciences 83 (2016) 155–165. [CrossRef]
- M. Másson, Antimicrobial Properties of Chitosan and Its Derivatives, Advances in Polymer Science 287 (2021) 131–168. [CrossRef]
- C.L. Ke, F.S. Deng, C.Y. Chuang, C.H. Lin, Antimicrobial Actions and Applications of Chitosan, Polymers 2021, Vol. 13, Page 904 13 (2021) 904. [CrossRef]
- G. Bisignano, A. Tomaino, R. Lo Cascio, G. Crisafi, N. Uccella, A. Saija, On the In-vitro Antimicrobial Activity of Oleuropein and Hydroxytyrosol, Journal of Pharmacy and Pharmacology 51 (2010) 971–974. [CrossRef]
- J. Britton, R. Davis, K.E. O’Connor, Chemical, physical and biotechnological approaches to the production of the potent antioxidant hydroxytyrosol, Appl Microbiol Biotechnol 103 (2019) 5957–5974.
- C. Vilaplana-Pérez, D. Auñón, L.A. García-Flores, A. Gil-Izquierdo, Hydroxytyrosol and Potential Uses in Cardiovascular Diseases, Cancer, and AIDS, Front Nutr 1 (2014) 110584.
- W. Lin, C. Mu, F. Liu, Q. Cheng, H. Li, B. Wu, G. Zhang, Collagen Cryogel Cross-Linked by Dialdehyde Starch, Macromol Mater Eng 295 (2010) 100–107. [CrossRef]
- L.C. Cefali, J.A. Ataide, A.R. Fernandes, I.M. de O. Sousa, F.C. da S. Gonçalves, S. Eberlin, J.L. Dávila, A.F. Jozala, M.V. Chaud, E. Sanchez-lopez, J. Marto, M.A. D’ávila, H.M. Ribeiro, M.A. Foglio, E.B. Souto, P.G. Mazzola, Flavonoid-Enriched Plant-Extract-Loaded Emulsion: A Novel Phytocosmetic Sunscreen Formulation with Antioxidant Properties, Antioxidants 2019, Vol. 8, Page 443 8 (2019) 443. [CrossRef]
- H.M. Nguyen, T.T. Ngoc Le, A.T. Nguyen, H.N. Thien Le, T.T. Pham, Biomedical materials for wound dressing: recent advances and applications, RSC Adv 13 (2023) 5509–5528. [CrossRef]
- J.G. Hodge, D.S. Zamierowski, J.L. Robinson, A.J. Mellott, Evaluating polymeric biomaterials to improve next generation wound dressing design, Biomaterials Research 2022 26:1 26 (2022) 1–39. [CrossRef]
- M.L. Spotti, J.P. Cecchini, M.J. Spotti, C.R. Carrara, Brea Gum (from Cercidium praecox) as a structural support for emulsion-based edible films, LWT 68 (2016) 127–134. [CrossRef]
- J. Becerra, M. Rodriguez, D. Leal, K. Noris-Suarez, G. Gonzalez, Chitosan-collagen-hydroxyapatite membranes for tissue engineering, J Mater Sci Mater Med 33 (2022) 1–16.
- K. Thongchai, P. Chuysinuan, T. Thanyacharoen, S. Techasakul, S. Ummartyotin, Integration of collagen into chitosan blend film composites: physicochemical property aspects for pharmaceutical materials, SN Appl Sci 2 (2020) 1–7.
- S. Suwas, R.K. Ray, Texture and Properties, (2014) 207–223. [CrossRef]
- R. White, K.F. Cutting, Modern exudate management: a review of wound treatments, World Wide Wounds 1 (2006).
- J. Tickle, Wound exudate: a survey of current understanding and clinical competency, British Journal of Nursing 25 (2016) 102–109. [CrossRef]
- M.M. Denn, J.F. Morris, D. Bonn, Shear thickening in concentrated suspensions of smooth spheres in Newtonian suspending fluids, Soft Matter 14 (2018) 170–184. [CrossRef]
- M. Zarei, J. Aalaie, Application of shear thickening fluids in material development, Journal of Materials Research and Technology 9 (2020) 10411–10433. [CrossRef]
- E.R. Johnston, Y. Miyagi, J.A. Chuah, K. Numata, M.A. Serban, Interplay between Silk Fibroin’s Structure and Its Adhesive Properties, ACS Biomater Sci Eng 4 (2018) 2815–2824.
- A. M. Grillet, N. B. Wyatt, L. M. Gloe, Polymer Gel Rheology and Adhesion, Rheology (2012) 338. Available online: https://books.google.com/books/about/Rheology.html?hl=pt-PT&id=LOOZDwAAQBAJ (accessed on 6 September 2024).
- Gonçalves, S. Raposo, H.M. Ribeiro, J. Marto, Useful In Vitro Techniques to Evaluate the Mucoadhesive Properties of Hyaluronic Acid-Based Ocular Delivery Systems, Pharmaceutics 2018, Vol. 10, Page 110 10 (2018) 110. [CrossRef]
- Mathematical models of drug release, Strategies to Modify the Drug Release from Pharmaceutical Systems (2015) 63–86. [CrossRef]
Figure 1.
Schematic representation of the collagen (Coll)-chitosan (Chit)-hydroxytyrosol (HT) biomaterials production process.
Figure 1.
Schematic representation of the collagen (Coll)-chitosan (Chit)-hydroxytyrosol (HT) biomaterials production process.
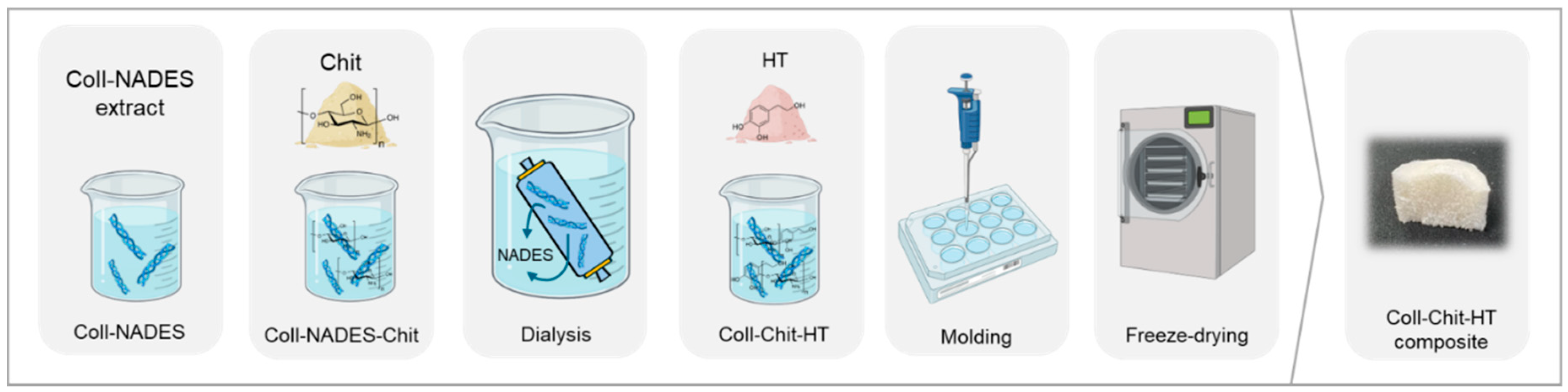
Figure 2.
Cross-sectional photographs (A, C, E) and SEM micrographs (B, D, F) at 100x magnification of the different materials produced with collagen (Coll), chitosan (Chit), and hydroxytyrosol (HT): Coll (A, B), Coll:Chit (C, D), and Coll:Chit:HT (E, F).
Figure 2.
Cross-sectional photographs (A, C, E) and SEM micrographs (B, D, F) at 100x magnification of the different materials produced with collagen (Coll), chitosan (Chit), and hydroxytyrosol (HT): Coll (A, B), Coll:Chit (C, D), and Coll:Chit:HT (E, F).
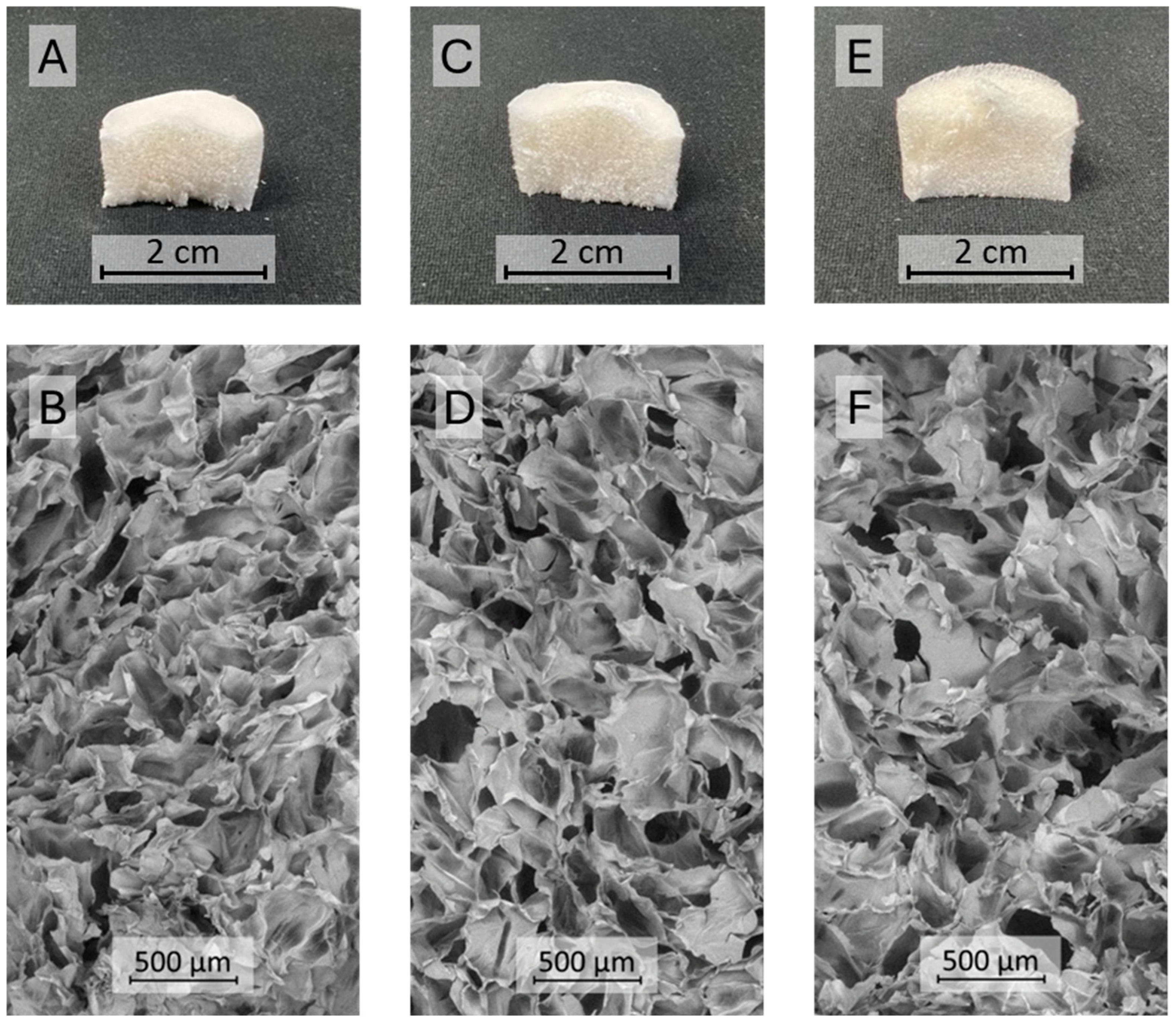
Figure 3.
Viscosity vs shear (a) and frequency sweep test (b) for all materials prepared with collagen (Coll), chitosan (Chit), and hydroxytyrosol (HT) in two different mass:solvent (w:w) hydrated states: 1:1 w:w and 1:4 w:w.
Figure 3.
Viscosity vs shear (a) and frequency sweep test (b) for all materials prepared with collagen (Coll), chitosan (Chit), and hydroxytyrosol (HT) in two different mass:solvent (w:w) hydrated states: 1:1 w:w and 1:4 w:w.
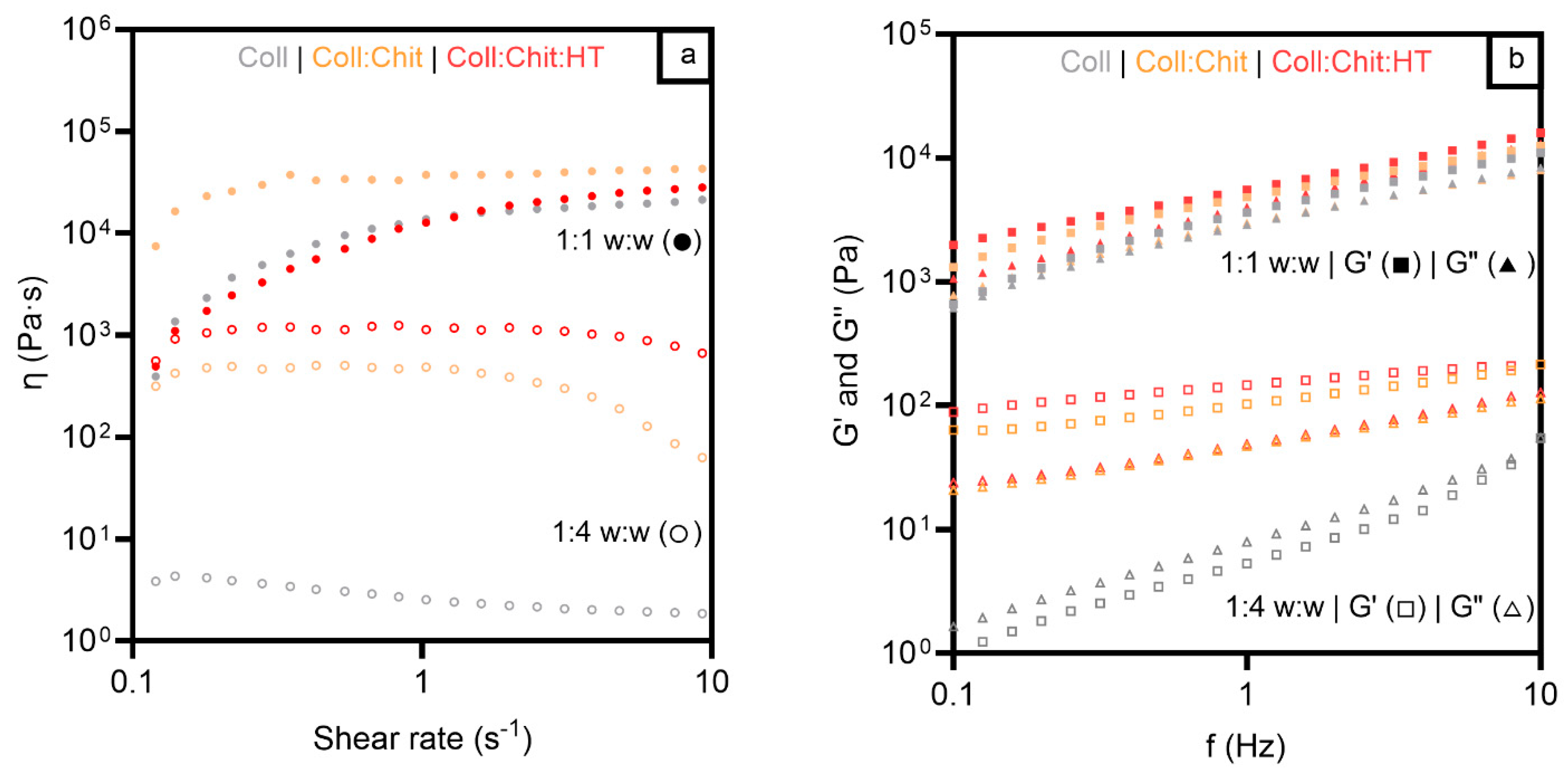
Figure 4.
Release profile and corresponding first-order model of hydroxytyrosol (HT) from the collagen (Coll)-chitosan (Chit)-HT biomaterial prepared. Statistically significant differences comparing all time points are indicated by different letters. (Mean ± SD, n=4).
Figure 4.
Release profile and corresponding first-order model of hydroxytyrosol (HT) from the collagen (Coll)-chitosan (Chit)-HT biomaterial prepared. Statistically significant differences comparing all time points are indicated by different letters. (Mean ± SD, n=4).
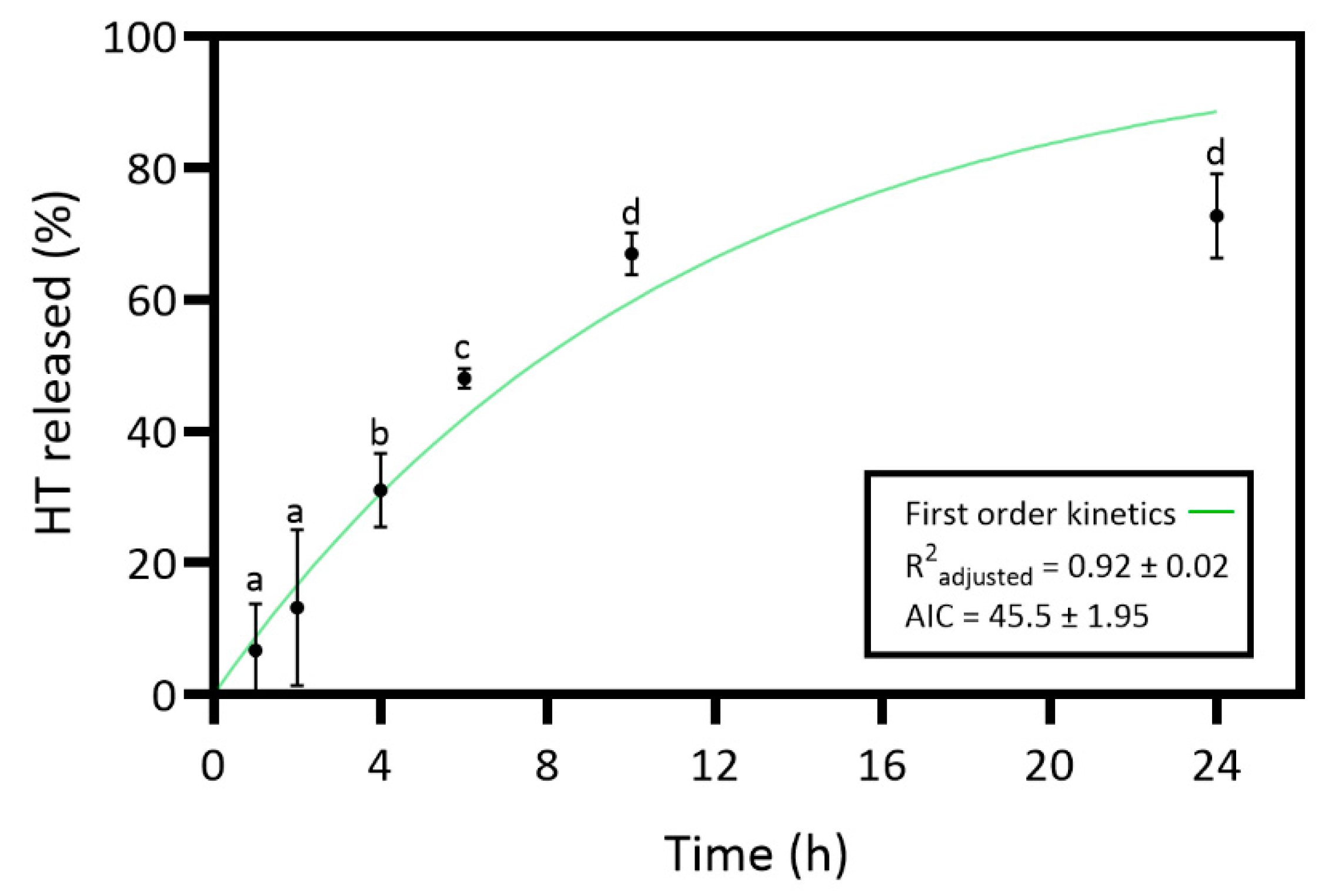
Figure 5.
Cytotoxicity assay using the MTS reagent: materials prepared with collagen (Coll), chitosan (Chit), and hydroxytyrosol (HT) were incubated with NCTC clone 929 cell line for 24 h at 37 °C and 5% CO2 humidified atmosphere (mean ± SD, n = 3). A 10% (v/v) solution of DMSO in cell culture media was used as a positive cytotoxic control. Samples have a cytotoxic potential if viability is reduced to < 70% of the negative control (untreated cells). Statistically significant differences comparing all conditions are indicated by different letters.
Figure 5.
Cytotoxicity assay using the MTS reagent: materials prepared with collagen (Coll), chitosan (Chit), and hydroxytyrosol (HT) were incubated with NCTC clone 929 cell line for 24 h at 37 °C and 5% CO2 humidified atmosphere (mean ± SD, n = 3). A 10% (v/v) solution of DMSO in cell culture media was used as a positive cytotoxic control. Samples have a cytotoxic potential if viability is reduced to < 70% of the negative control (untreated cells). Statistically significant differences comparing all conditions are indicated by different letters.
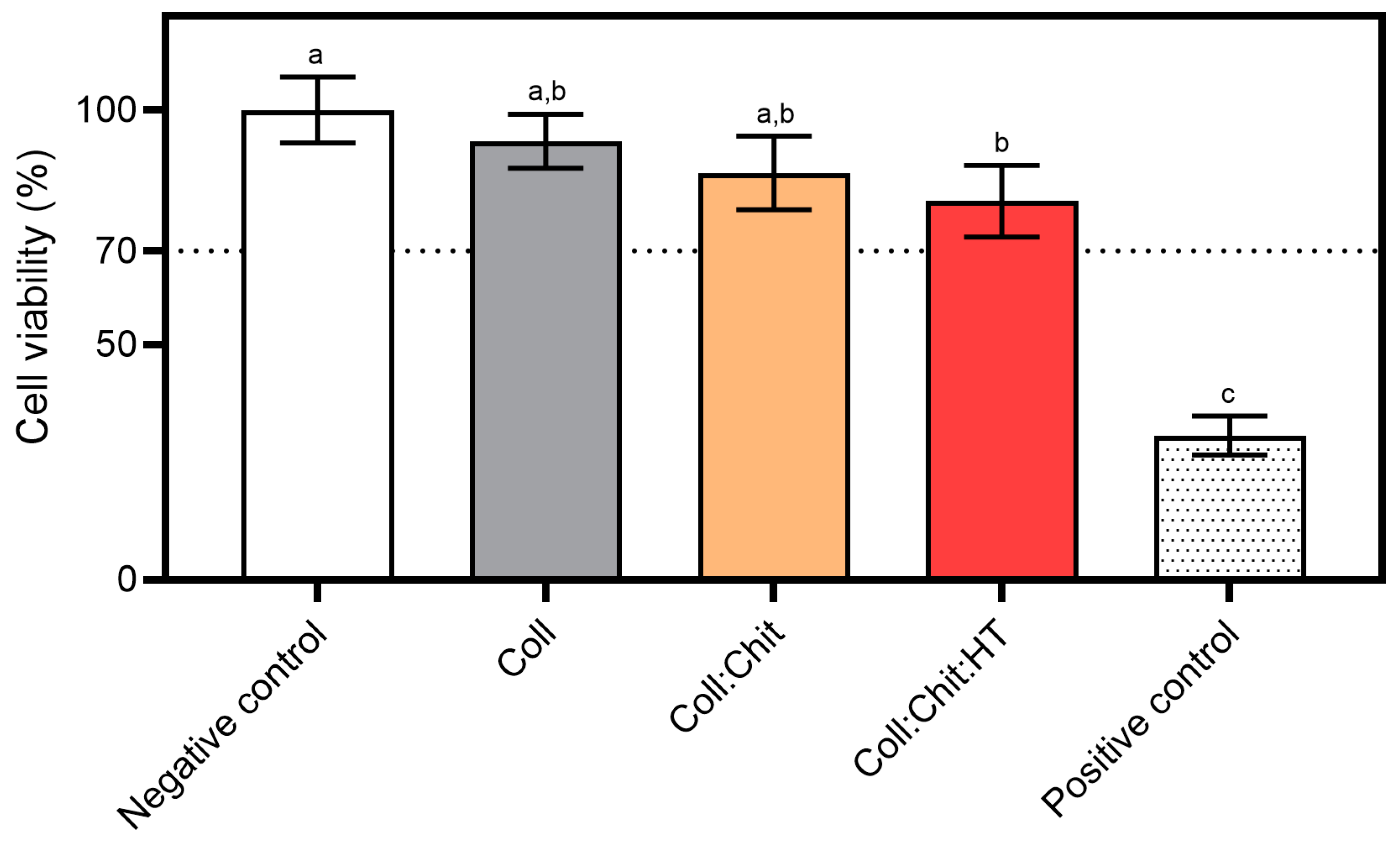

Table 1.
Compounds, dissolving agents, and respective highest tested concentrations in the antimicrobial susceptibility testing assays. ddH2O: sterilized double distilled water.
Table 1.
Compounds, dissolving agents, and respective highest tested concentrations in the antimicrobial susceptibility testing assays. ddH2O: sterilized double distilled water.
| Compounds | Dissolving Agent | Highest Tested Concentration |
|---|---|---|
| Chitosan | HCl 0.1 M | 10 mg/mL |
| Hydroxytyrosol | ddH2O | 20 mg/mL |
| HCl | ddH2O | 50 mM |
| Xylitol | ddH2O | 300 mg/mL |
| Citric acid | ddH2O | 100 mg/mL |
Table 2.
Antimicrobial susceptibility testing assays against Staphylococcus aureus ATCC 6538 (WDCM 00193) and Pseudomonas aeruginosa ATCC 27853 (WDCM 00025) of chitosan (Chit), and hydroxytyrosol (HT). Median values of MICs (mg/mL) are presented in this table, while replicate values are available in supplementary materials (Table SM 2).
Table 2.
Antimicrobial susceptibility testing assays against Staphylococcus aureus ATCC 6538 (WDCM 00193) and Pseudomonas aeruginosa ATCC 27853 (WDCM 00025) of chitosan (Chit), and hydroxytyrosol (HT). Median values of MICs (mg/mL) are presented in this table, while replicate values are available in supplementary materials (Table SM 2).
| Tested Compounds | MICMedian (mg/mL) | |
|---|---|---|
| S. aureus ATCC 6538 | P. aeruginosa ATCC 27853 | |
| Chit | 0.16 | 0.31 |
| HT | 0.39 | 1.56 |
Table 3.
Textural analysis of the solid materials prepared with collagen (Coll), chitosan (Chit), and hydroxytyrosol (HT). Statistically significant differences comparing all samples in each parameter are indicated by different letters.
Table 3.
Textural analysis of the solid materials prepared with collagen (Coll), chitosan (Chit), and hydroxytyrosol (HT). Statistically significant differences comparing all samples in each parameter are indicated by different letters.
| Samples | Burst Strength (N) | Distance at Burst (mm) |
|---|---|---|
| Coll | 13.9 ± 1.82a | 53.0 ± 0.40a |
| Coll:Chit | 19.7 ± 3.57b | 53.5 ± 0.20a |
| Coll:Chit:HT | 17.1 ± 0.89b | 53.5 ± 0.18a |
Table 4.
Adhesive properties for all materials prepared with collagen (Coll), chitosan (Chit), and hydroxytyrosol (HT) in two different mass:solvent hydrated states: 1:1 w:w and 1:4 w:w. Within each evaluated parameter (area under the force-time curve and peak of normal force), statistically significant differences comparing all samples and hydrated states are indicated by different letters.
Table 4.
Adhesive properties for all materials prepared with collagen (Coll), chitosan (Chit), and hydroxytyrosol (HT) in two different mass:solvent hydrated states: 1:1 w:w and 1:4 w:w. Within each evaluated parameter (area under the force-time curve and peak of normal force), statistically significant differences comparing all samples and hydrated states are indicated by different letters.
| Area Under Force-Time Curve (N·s) | Peak of Normal Force (N) | |||
|---|---|---|---|---|
| 1:1 w:w | 1:4 w:w | 1:1 w:w | 1:4 w:w | |
| Coll | 27.9 ± 2.26a | 1.44 ± 0.54b | -15.4 ± 1.30a | -0.12 ± 0.07b |
| Coll:Chit | 24.4 ± 1.76a | 1.18 ± 0.54b | -12.4 ± 2.01a | -0.27 ± 0.13b |
| Coll:Chit:HT | 25.1 ± 1.53a | 1.24 ± 0.50b | -12.5 ± 1.87a | -0.22 ± 0.08b |
Table 5.
Antimicrobial activity values and corresponding efficacy of the tested materials against Staphylococcus aureus ATCC 6538 (WDCM 00193) and Pseudomonas aeruginosa ATCC 27853 (WDCM 00025) from contact with samples at a 2 g/mL concentration during 24 h at 37 °C. Efficacy of antibacterial activity: strong (for antibacterial activity value ≥ 3); significant (for 2 ≤ antibacterial activity value < 3); negligible (for < 2 antibacterial activity value).
Table 5.
Antimicrobial activity values and corresponding efficacy of the tested materials against Staphylococcus aureus ATCC 6538 (WDCM 00193) and Pseudomonas aeruginosa ATCC 27853 (WDCM 00025) from contact with samples at a 2 g/mL concentration during 24 h at 37 °C. Efficacy of antibacterial activity: strong (for antibacterial activity value ≥ 3); significant (for 2 ≤ antibacterial activity value < 3); negligible (for < 2 antibacterial activity value).
| Strain | Sample | Antibacterial Activity Value | Efficacy of Antibacterial Activity |
|---|---|---|---|
| S. aureus ATCC 6538 | Coll | 2.1 ± 0.3 | Significant |
| Coll:Chit | 6.9 ± 0.1 | Strong | |
| Coll:Chit:HT | 7.1 ± 0.1 | Strong | |
| P. aeruginosa ATCC 27853 | Coll | 1.7 ± 0.2 | Negligible |
| Coll:Chit | 2.6 ± 0.2 | Significant | |
| Coll:Chit:HT | 5.8 ± 0.5 | Strong |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



