Submitted:
09 April 2025
Posted:
10 April 2025
You are already at the latest version
Abstract
Neural cell membranes consist of phospholipids, sphingolipids, cholesterol and proteins. Phospholipids and sphingolipids contribute to the lipid asymmetry, whereas cholesterol and sphingolipids form lipid microdomains or lipid rafts. Lipid rafts float within the membrane, and certain groups of proteins unite within these rafts. A large number of signaling molecules are concentrated within rafts. In response to cell stimulation or injury, phospholipids, sphingolipids, and cholesterol generate lipid metabolites, which promote or block inflammation in the body. Thus, phospholipids release arachidonic acid or docosahexaenoic acid from the sn-2 position of glycerol moiety by the action of phospholipase A2. Arachidonic acid is a precursor for eicosanoids, a group of metabolites, which includes prostaglandins, leukotrienes, thromboxane, and lipoxins. These metabolites are not only involved in the cell proliferation, differentiation, blood clotting, and blood vessel permeability, but also promote the induction of inflammation, immune responses, and gene expression. In contrast, metabolism of docosahexaenoic acid generates, specialized pro-resolving lipid metabolites called resolvins, protectins, and maresins. These metabolites regulate immune function by producing resolution of inflammation, a cell protective mechanism. Sphingolipid derived-metabolites, particularly ceramide, ceramide-1-phosphate, sphingosine, and sphingosine-1-phosphate regulating a diverse range of cellular processes including inflammation. Among cholesterol-derived metabolites (24-hydroxy, 25-hydroxy, and 7-ketocholesterlcholesterol), unesterified 7-ketocholesterol disrupts fluidity and alters signaling pathways associated with inflammation, oxidative stress, and apoptosis leading to cellular damage and death. The cross-talk among phospholipid, sphingolipid, and cholesterol-derived lipid metabolites modulates the intensity of inflammation, a process that contributes to lipo-toxicity and cell death.
Keywords:
arachidonic acid
; docosahexaenoic acid
; eicosanoids
; ceramide
; ceramide1-phosphate
; sphingosine
; sphingosine 1-phosphate
1. Introduction
Lipids are fundamental organic molecules that constitute the backbone of neural cell membranes. They not only provide cell membranes with suitable environment, fluidity, and ion permeability, but also play crucial roles in signal transduction processes and regulation of energy metabolism [1]. Among various lipids, phospholipids and sphingolipids contribute to the lipid asymmetry, whereas cholesterol and sphingolipids form lipid microdomains or lipid rafts. Lipid rafts float within the membrane, and certain groups of proteins unite within these rafts. A large number of signaling molecules are concentrated within rafts, which function as signaling centers capable of facilitating efficient and specific signal transduction pathways [2]. Metabolism of lipids not only affects many cellular processes critical for maintaining normal neural cell function and homeostasis, but can also act as energy storage depot (triglycerides). Fatty acids are essential components of phospholipids and sphingolipids. They also serve as an important energy source through mitochondria-mediated beta-oxidation and tricarboxylic acid (TCA) [3]. In addition, they may also major determinants of physicochemical properties of membrane and modulators of cellular signaling pathways [3,4]. Collective evidence suggests that intricate interactions among phospholipids, sphingolipids, cholesterol and proteins provide neural membranes with delicate, dynamic, and stable shape responsible for numerous cell membrane activities. Low levels of phospholipid-, sphingolipid-, and cholesterol-derived lipid metabolites are necessary for normal cellular functions such as inflammation, gene expression, growth arrest, differentiation, and adhesion. However, high levels of phospholipid-, sphingolipid-, and cholesterol-derived lipid metabolites may cause induction of inflammation, oxidative stress, and apoptosis. Among these processes, inflammation is modulated not only by arachidonic acid (ARA, 20:4n-6) and docosahexaenoic acid (DHA, 22:6n-3)-derived metabolites [5,6], but also by cross-cross-talk among phospholipid, sphingolipid and cholesterol-derived metabolites. The purpose of this review article is to describe the proinflammatory and anti-inflammatory effects of lipid metabolites to a broader audience with the hope that this discussion would jumpstart more studies on cross-talk among lipid metabolites leading to modulation of inflammation and induction of lipo-toxicity.
2. Inflammation
Inflammation is a protective mechanism, which is characterized by the activation of immune (neutrophils, macrophages, and lymphocytes) and non-immune cells at the injury or infection site. Onset of inflammation results in restoration of homeostasis in body systems after injuries or infections [7,8]. The main generators of inflammation in the visceral tissues are macrophages and infiltrating immune cells. In the brain and spinal cord, inflammation is mediated by the mild and short-term activation of microglia and astrocytes. Mild and short-term activation of microglia and astrocytes usually produce neuroprotective effects and ameliorates early symptoms of neurodegeneration. Mild and short-term activation of microglia and astrocytes promote the release of low levels of cytokines, which help in maintaining synaptic plasticity, modulating neuronal excitability, and stimulating toll-like receptors (TLRs), a family of pattern recognition receptors that recognize microbial pathogens and respond by activating the innate immune system. These processes support neurogenesis and neurite outgrowth. In contrast, long-term activation of microglia and astroglia contribute to the overexpression of cytokines leading to neurodegeneration. Long-term activation of microglia and astroglia also results in marked alterations in the regulation of p53 protein and transcription factor NF-κB. These processes are crucial for shifting the regenerative effects into detrimental effects during inflammatory reactions. At the molecular level, the long-term inflammatory reaction is characterized by the release of prostaglandins (PGs), leukotrienes (LTs), thromboxane (TX), and nitric oxide (NO) by resident cells in the injured/infected tissue (i.e., tissue macrophages, dendritic cells, lymphocytes, endothelial cells, fibroblasts, and mast cells). This leads to a noticeable increase in cerebral blood flow and accumulation of circulating leukocytes [9] at the injury or infection site. Additionally, increased levels of proinflammatory cytokines (tumor necrosis factor-alpha (TNF-α), interleukin-1 (IL-1) and interleukin-6 (IL-6)) secreted from activated immune cells enhance the vascular permeability of leukocytes through raising the levels of leukocyte adhesion molecules on endothelial cells [10,11]. It is reported that prolonged presence of inflammation at the injury site may lead to serious problems, including impaired proteolysis, apoptosis, and epigenetic modifications [12,13]. The current knowledge of inflammatory signaling has been gained not only from studying members of the IL-1 and TNF receptor families, but also from the Toll-like microbial pattern recognition receptors (TLRs) [10,13] (Figure 1). TLRs are ubiquitously expressed in macrophages, neutrophils, dendritic cells (DCs), natural killer (NK) cells, mast cells, eosinophils, and basophils. These cells are instrumental in mounting innate immune responses against invading pathogens and tissue injury. In the presence of high levels of inflammatory metabolites (PGs, LTs, and TX) at the injury or infection site is associated with the onset of inflammation (redness, swelling, heat, and pain) and loss of cell function.
Detailed investigations have indicated that the inflammation is also supported by oligomeric protein complexes known as inflammasomes (Figure 1) [14]. Inflammasomes reside in cytoplasm and act as platforms for the induction of inflammatory signaling. Inflammasomes are made up of several proteins, including NLRP3, NLRC4, AIM2 and NLRP6. Activation of the inflammasome during infection or injury results in a neuroprotective or detrimental effects in the brain. Thus, interactions of NLRP3 inflammasome with mitochondria in the presence of Ca2+ facilitates the accumulation of reactive oxygen species (ROS) and reactive nitrogen species (RNS). It is not known whether interactions between NLRP3 and mitochondria are for surveying the mitochondrial integrity and sensing mitochondrial damage, or whether mitochondria simply serve as a physical platform for inflammasome assembly [15]. Recognition of injury and stress signals by inflammasomes do not regulate transcription of immune response genes, but activate caspase-1, a proteolytic enzyme that cleaves and activates the secreted pro-cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18), and interleukin-33 (IL-33) into IL-1β, IL-18, and IL-33 [10,15]. In addition, TLR-mediated increased expression of above cytokines, autophagy and epigenetic factors have also been reported to modulate intensity of inflammation [16] (Figure 1).
Two types of inflammations have been reported to occur in humans and animals: (a) acute inflammation develops rapidly with the experience of pain, whereas (b) chronic inflammation develops slowly without the experience of pain. The onset of acute inflammation occurs in neurotraumatic diseases such as stroke, traumatic brain and spinal cord injuries. In contrast, chronic inflammation develops slowly in neurodegenerative diseases (Alzheimer’s disease and Parkinson’s disease) (Table 1) [17]. Chronic inflammation differs from acute inflammation in the threshold of pain perception. As a result, the immune system continues to attack at the cellular level. Chronic inflammation lingers for years causing continued insult to the brain tissue, ultimately reaching the threshold of detection [18]. Chronic inflammation disrupts hormonal signaling networks in the brain and induces deficits in long-term potentiation (LTP), the major neuronal substrate for learning and memory. Induction of inflammation is closely interlinked with oxidative stress, a process that overwhelms the antioxidant defenses of the cells through the generation of reactive oxygen and nitrogen species (ROS and RNS) [19]. This may be either due to an overproduction of ROS and RNS or to a failure of cell buffering mechanisms. It is difficult to establish the temporal sequence of their relationship.
The onset of inflammation is followed by a process called resolution, a turning off mechanism by cells to limit tissue injury [20]. Resolution not only results in reduction of numbers of immune cells at the core of the injury site, clearance of apoptotic cells, and debris by increased phagocytic activity, but also in the induction of recently identified new molecules that activate tissue regeneration in the brain tissue. Levels of ARA and DHA-derived lipid metabolites in brain and visceral tissues are partly regulated by diet. At the molecular level, the resolution of inflammation is orchestrated by DHA-derived-bioactive lipid metabolites called resolvins (Rvs), neuroprotectins (NPDs), and maresins (MaR). NPD1 has been reported to play endogenous neuroprotective role by inhibiting apoptotic DNA damage, up-regulating anti-apoptotic proteins Bcl-2 and BclxL, and down-regulating pro-apoptotic Bax and Bad expression [20,21,22].
Enzymatic metabolites of DHA metabolism not only downregulate proinflammatory cytokines but also produce anti-oxidant, anti-inflammatory, anti-thrombotic, anti-arrhythmic, hypo-lipidemic, and vasodilatory effects [23,24]. Accumulating evidence supports the view that levels of ARA and DHA, and their lipid metabolites not only orchestrate and control the onset of inflammation and oxidative stress, but also cooperate in maintaining appropriate downstream actions and responses [17,23,24,25].
2.1. Phospholipid Metabolites in Inflammation
In response to cell stimulation or injury, phospholipids release polyunsaturated free fatty acids ARA or DHA from the sn-2 position of glycerol moiety through the action of phospholipase A2 (PLA2) [1,17,23]. Enzymatic mediators of ARA metabolism are called as eicosanoids. They include PGs, LTs, TXs, and LXs (Figure 2). These metabolites not only play important roles in internal and external communication, but also modulate inflammation through the upregulation of the expression of proinflammatory cytokines [17,24,25]. Eicosanoids also modulate other cellular responses such as the growth arrest, differentiation, adhesion, migration, and apoptosis [23]. Furthermore, eicosanoids also contribute to vascular function by regulating cerebral blood flow, angiogenesis, and pain [24,25]. Collective evidence suggests that inflammation involves several converging mechanisms responsible for sensing, transducing, amplifying, and turning off mechanisms that involve the participation of various eicosanoids [24,25]. Non-enzymically oxidation of ARA results in the production of 4-hydroxynonenal (4-HNE), isoprostanes (IsoP), isoketals (IsoK), and isofurans (IsoF). These metabolites produce pro-oxidant, pro-thrombotic, pro-aggregatory, and pro-inflammatory effects. (Figure 2).
Plasma membrane (PM); N-methyl-D-aspartate receptor (NMDA-R); glutamate (Glu); phosphatidylcholine (PtdCho); lyso-phosphatidylcholine (Lyso-PtdCho); cytosolic phospholipase A2 (cPLA2); plasmalogen-selective PLA2 (PlsEtn-PLA2); platelet activating factor (PAF); arachidonic acid (ARA); cyclooxygenase-2 (COX-2); 5-lipoxygenase (5-LOX); prostaglandins (PGs); leukotrienes (LTs); thromboxane (TXs); reactive oxygen species (ROS); 4-hydroxynonel (4-HNE); isoprostane (IsoP), acrolein (AC); malondialdehyde (MDA); nuclear factor kappaB (NF-κB); nuclear factor kappaB response element (NF-κB-RE); inhibitory subunit of NFκB (IκB); tumor necrosis factor-α (TNF-α); interleukin-1β (IL-1β); matrix metalloproteinases (MMPs); neuroprostane (NP); neurofuran (NF); neuroketal (NK); 4-hydroxyhexenal (4-HHE); Resolvin D.
The non-enzymatic lipid metabolites of DHA metabolism include 4-hydroxyhexanal (4-HHE), neuroprostanes (NPs), neuroketals (NKs), and neurofurans (NFs) (Figure 2). Enzymatic metabolites of DHA metabolism not only downregulate proinflammatory cytokines but also produce anti-oxidant, anti-inflammatory, anti-thrombotic, anti-arrhythmic, hypo-lipidemic, and vasodilatory effects [24,25]. Accumulating evidence suggests that ARA and DHA-derived lipid metabolites compete with each other and regulate induction and regulation of inflammation by controlling the duration and magnitude of acute inflammation, oxidative stress as well as the return of the injury site to homeostasis [17,24,25]. Level of ARA and DHA-derived lipid metabolites in brain and visceral tissues are partly regulated by diet. Collective evidence suggests that levels of ARA and DHA, and their lipid metabolites not only orchestrate and control the onset of inflammation and oxidative stress, but also regulate the action of lipid-dependent enzymes to execute appropriate downstream actions and responses [17,24,25].
Lyso-phosphatidylcholine, the other product of PLA2 catalyzed reaction either reacylated through acylation/ de-acylation cycle into native glycerophospholipids or converted into platelet activating factor (PAF; 1-O-alkyl-2-acetyl-sn-glycerophosphocholine), which is a proinflammatory lipid molecule [2]. It exerts its inflammatory effects by activating the PAF receptors that consequently activate leukocytes, stimulate platelet aggregation, and induce the release of cytokines and expression of cell adhesion molecules [2]. It is reported that PAF is also involved in allergic reactions, and circulatory system disturbances such as atherosclerosis [2].
2.2. Markers for Inflammation
Cytokines, chemokines, eicosanoids and platelet activating factor (PAF) are major biomarkers for inflammation. Levels of these biomarkers are increased in body tissues and fluids during inflammation [17,24]. These metabolites act on neural and non-neural cells directly as well as through eicosanoid receptors. Four types of eicosanoid receptors (EP1, EP2, EP3, and EP4) have been cloned [25]. These receptors are G protein-coupled receptors, which evoke cellular responses through distinct intracellular mechanisms. Some prostaglandins, PGE1, PGE2 and PGD2, are inflammatory [26], whereas others PGs are anti-inflammatory such as PGD2 and prostaglandin J2 [27]. High levels of eicosanoids contribute to the development of cytotoxicity, vasogenic edema, and cellular damage through the participation of NF-κB, isoforms of PLA2, PLC, PKC and cytokines [17,24].
2.3. Sphingolipid Metabolites in Inflammation
Sphingolipids form a broad class of amphiphatic lipids with diverse functions. Sphingolipids consist of sphingomyelin, ceramide, ceramide 1-phosphate (C 1-P), sphingosine, and sphingosine1-phosphate (S1P). They can be rapidly converted into each other. Among above mentioned metabolites sphingolipids, ceramide, C 1-P) and sphingosine-1-phosphate (S1P) have received the greatest attention (Figure 3). In addition to serving as a precursor to complex sphingolipids, ceramide also regulates vital cellular functions, such as cell growth, viability, differentiation, proliferation, migration and angiogenesis, apoptosis, inflammation, regulation of enzyme activities, neutrophil adhesion to the vessel wall, and vascular tone [28]. Several studies have indicated that ceramide, C 1-P, and S1P an important role in inflammation [29].
Sphingomyelin is hydrolyzed by enzyme called sphingomyelinases (SMases). These enzymes are abundantly present in the brain and visceral tissues. They catalyze the hydrolysis of sphingomyelin into ceramide and phosphorylcholine [30]. These enzymes are a major, rapid source of ceramide and S1P production not only in response to receptor stimulation, but also in pathological conditions such as oxidative stress and inflammation. Stimulation of TNF-α receptor results in activation of acid and neutral SMases and sphingosine kinases (SK). Activation of these enzymes contribute to the generation of ceramide and C1-P, S1P, and subsequent activation of the pro-inflammatory transcription factor, nuclear factor-κB (NF-κB) [31]. Under normal conditions, NF-κB/ Iκ-Bα complex (inhibitory form) is present in the cytoplasm. During oxidative stress and pathological conditions, NF-κB/ Iκ-Bα complex breaks down and free NF-κB migrates to the nucleus, where it interacts with NF-κB response element (NF-κB-RE). The binding of NF-κB to NF-κB-RE promotes the expression of proinflammatory cytokines (TNF-α, IL-1β, and IL-6) and chemokine [30] and free IκBα is degraded by the ubiquitin proteasome system. Cytokines and chemokines act via specific receptors, which are expressed on the neural and non-neural cell membranes of their target cells. Their action involves a complex network linked to feedback loops and cascade through the activation of protein kinases and PLA2. At low levels, cytokines and chemokines are required for cell metabolism and survival under normal, but at higher levels an imbalance among cytokines and chemokines is detrimental to cells. The activation of NF-κB is also coupled with the stimulation of PLA2, cyclooxygenases (COXs) and lipoxygenases (LOXs) and generation of PG, LTs, and TX [2].
Often inflammatory responses are temporary only occurring locally, and activated to protect from the invasion of pathogens or injury and to promote repair of tissue damage. However, when uncontrolled or inappropriate, acute inflammation can lead to persistent chronic inflammation, causing the development of autoimmune or circulatory system diseases, neurological disorders (Alzheimer’s disease and Parkinson’s disease) and metabolic diseases, including type 2 diabetes, obesity, cardiovascular disease, and even cancer; if the inflammatory response is left unchecked, many inflammatory mediators are released into the blood, causing sepsis, which can lead to death [24,32]. It is suggested that timely resolution of inflammation is crucial for preventing severe and chronic inflammation in neurodegenerative diseases [24,32].
2.4. Cholesterol Metabolites in Inflammation
Cholesterol is an essential component of the mammalian cell membranes [2,33]. It not only contributes to membrane fluidity and permeability, but also major component of lipid rafts, which are platforms for signal transduction processes [2]. In brain, about 70% of cholesterol is present in myelin sheath, where it is involved in synaptogenesis, maintenance, turnover, and stabilization of synapses [34]. Cholesterol has many other biological functions, such as the synthesis of vitamin D, steroid hormones (e.g., cortisol, aldosterone, and adrenal androgens), and sex hormones (e.g., testosterone, estrogens, and progesterone). Cholesterol cannot cross the blood–brain barrier, and thus the brain synthesizes and metabolizes its own pool of cholesterol. The primary metabolic enzymes for brain cholesterol are cholesterol hydroxylases (CH24H), which metabolize cholesterol into 24S-hydroxycholesterol (24HC), and 25-hydroxycholesterol (25HC) (Figure 4) [34,35]. Dysregulation of CH24H and 24HC can affect neuronal excitability through a range of mechanisms. 24HC is a positive allosteric modulator of N-methyl-D-aspartate (NMDA) receptors. These receptors are coupled with the increase in glutamate release and stimulation of PLA2 resulting in degradation of phospholipid in neural membranes [17,35,36]. Among cholesterol metabolites, 7-KC is a pro-inflammatory oxysterol usually associated with oxidized lipoprotein deposits present in aged retinas [37]. Increased levels of 7-KC are also found in the tissues, plasma, and/or cerebrospinal fluid of patients with cardiovascular diseases, eye diseases, and neurodegenerative diseases [32,38]. ARA also interacts with 7-KC in the presence of Acyl-CoA cholesterol acyltransferase (ACAT) to form the 7KC-ARA complex [39].
This complex is closely associated with various pathological processes, including cardiovascular diseases and age-related macular degeneration. It is proposed that 7-KC-mediated inflammatory responses in endothelial cells elevates the risk of cardiovascular diseases, Alzheimer’s, disease and age-related macular degeneration [39,40,41]. In addition, treatment of murine neuronal N2a cells with 7-KC not only results in overproduction of ROS, but alterations in plasma membrane, and drop of transmembrane mitochondrial potential (ΔΨm) leads to cell death. Unesterified 7-KC is also known to disrupt membrane fluidity promotes inflammation, oxidative stress, and apoptosis.
During oxidative stress, production of ROS contributes to inflammation in two ways: (a) interactions of ROS with NF-κB/IκB complex in the cytoplasm producing free NF-κB, which migrates to the nucleus, where it binds with NF-κB response element and promotes the expression of proinflammatory cytokines, chemokines, adhesion molecules [17,41] and (b) in macrophages 7-KC activates the immune sensor TLR4, which also promotes the translocation of free NF-κB to the nucleus promoting the expression of proinflammatory cytokines and chemokines [17,41]. Levels of oxysterols (including 7-KC) can be regulated through several mechanisms of cholesterol clearance. ATP-binding cassette (ABC) efflux pumps play important roles in exporting oxysterols from cells in various tissues, and the expression of these transporters is one mechanism by which intracellular oxysterol levels can be regulated [39].
3. Cross-Talk Among Phospholipid, Sphingolipid, Cholesterol Metabolites in Inflammation
In visceral tissues and brain, the metabolism of phospholipid, sphingolipid, and cholesterol is interrelated and interconnected. Many cellular stimuli (neurotransmitters, cytokines, and growth factors) modulate more than one enzyme of phospholipid, sphingolipid, and cholesterol metabolism at the same time. Thus, sphingolipid metabolism metabolites (ceramide, C 1-P, and S1P) stimulate cPLA2 activity and metabolites of phospholipid metabolism (arachidonate and ROS) stimulate SMases suggesting a cross-talk between metabolites of phospholipid and sphingolipid metabolism [42,43] (Figure 5). The significance of cross-talk between phospholipid and sphingolipid metabolites and normal cellular processes lies in their levels, exposure time, and crucial roles of lipid metabolites not only in maintaining overall cellular health, but also influencing the onset of metabolic and neurological disorders. Thus, cross-talk between different lipid families commonly occurs during phospholipid and sphingolipid metabolism and lipid metabolites act as signaling molecules, influencing cellular functions and impacting processes like energy production, inflammatory cell signaling, and even progression of metabolic and neurological disorders [17,40]. In addition, 7-KC produces mitochondrial dysfunction not only by triggering oxidative stress and inflammation, but also impairing fatty acid oxidation, which, in turn, can contribute to fatty acid oxidation, lipid accumulation, and lipo-toxicity [44]. Furthermore, 7-KC has been reported to produce an excessive mitophagy (intracellular degradation of damaged mitochondria) by means of autophagy [45]. It is reported that mitochondrial dysfunction is closely associated with the onset and progression of many chronic diseases involving inflammatory response through NF-κB and inflammasome activation. Damaged mitochondria can also play an important role in inflammasome activation by releasing danger-associated molecular patterns (DAMPs) such as ROS, mtDNA, and ATP [46]. These DAMPs can trigger pro-inflammatory signaling pathways, such as the activation of the NLRP3 inflammasome and the release of pro-inflammatory cytokines like IL-1β and IL-18 [47,48]. Impaired mitochondrial function can disrupt cellular metabolism and bioenergetics, further enhancing the inflammatory responses. Collective evidence suggests that dysregulation of the balance between sphingolipids, phospholipids, and cholesterol metabolites may result in “lipo-toxicity”, a term coined by Unger 17 years ago to describe the toxic effects of excessive free fatty acids on pancreatic beta cell survival [49]. The term has never been formally defined but has been used variably to describe cellular injury and death caused by free fatty acids, their metabolites, increase in ceramide/sphingosine pools, and 7-KC mediated mitochondrial dysfunction in non-neural tissues [50,51,52,53]. The molecular mechanisms underlying lipo-toxicity may include high levels of lipid metabolites, oxidative stress, mitochondrial dysfunction, impaired autophagy, and onset of inflammation [50]. The induction of these processes may result in cellular dysfunction and death in diverse tissues including brain, kidney, and heart [51,52,53].
Plasma membrane (PM); Plasma membrane (PM); N-methyl-D-aspartate receptor (NMDA-R); glutamate (Glu); phosphatidylcholine (PtdCho); cytosolic phospholipase A2 (cPLA2); arachidonic acid (ARA); sphingomyelin (SM); cyclooxygenase-2 (COX-2); prostaglandin E2 (PGE2); ceramide 1-phosphate (C 1-P); sphingosine 1 phosphate (S 1 P); reactive oxygen species (ROS); tumor necrosis factor-alpha (TNF-α); interleukin-1beta (IL-1β); ceramide and C 1-P stimulates cPLA2; ARA metabolites and ROS stimulate sphingomyelinases (SMases); Hydroxycholesterols and 7-ketocholesterol stimulate caspase cascade and promote mitochondrial; 1. ceramide kinase; 2. ceramidase; 3. sphingosine kinase; and (+) indicate stimulation.
Under physiological conditions, homeostasis between phospholipid sphingolipid, and cholesterol-derived metabolites is based not only signaling network, but also optimal levels of lipid metabolites. However, under pathological conditions, marked increases in levels of phospholipid sphingolipid, and cholesterol-derived metabolites may disturb the signaling networks, and result in loss of communication among lipid metabolites. This process not only threatens the integrity of cellular lipid homeostasis, but also facilitates cell death [25,44]. The severity of the lipo-toxic insult can be modulated by the specific cellular genetic vulnerability and toxicity induced by lipid metabolites.
4. Conclusion
The generation of phospholipid, sphingolipid, and cholesterol-derived lipid metabolites is necessary for normal cellular function. ARA-derived lipid metabolites (PGs, LTs, TXs, and LXs) produce inflammation. In contrast, DHA-derived lipid metabolites block inflammation not only by promoting potent anti-inflammatory and pro-resolution effects, but also by enhancing clearance of apoptotic cells and debris from inflamed tissues. Sphingolipid-derived lipid metabolites include ceramide, C 1-P, and S 1 P. These mediators are involved in the modulation of signal transduction processes associated with apoptosis, inflammation, oxidative stress, and cell growth. The cross-talk among phospholipid, sphingolipid, and cholesterol-derived lipid metabolites modulates the intensity of inflammation and oxidative stress. These processes may contribute to lipo-toxicity and cell death.
Author Contributions
Conceptualization, writing and ending -AAF & TF. Both authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Farooqui AA, Horrocks LA, Farooqui T. Glycerophospholipids in brain: their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem. Phys. Lipids 2000; 106:1-29.
- Farooqui, A.A. Hot Topics in Neural Membrane Lipidology; 2009, Springer, New York.
- Raghu P. Functional diversity in a lipidome. Proc. Natl. Acad. Sci. U. S. A, 2020; 117: 11191–11193.
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296; Correction in Nat. Rev. Mol. Cell Biol. 2019, 20, 715. [CrossRef]
- Dennis WD, Norris PC. Eicosanoid Storm in Infection and Inflammation. Nat Rev Immunol. 2015; 15: 511-523.
- Calder PC (2017) Omega-3 fatty acids and inflammatory processes: from molecules to man. Biochem Soc Trans 2017; 45: 1105–1115.
- Medzhitov, R. Inflammation 2010: New Adventures of an Old Flame. Cell 2010, 140, 771–776. [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [CrossRef]
- Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012;4: a006049.
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [CrossRef]
- Cignarella A, Bolego C. Innate Immunity in Inflammation. In: Riccardi C, Levi-Schaffer F, Tiligada E, editors. Immunopharmacology and Inflammation. Cham: Springer International Publishing p. 179–90, 2018.
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking Aging to Chronic Disease. Cell 2014, 159, 709–713. [CrossRef]
- Chen, Y.-H.; Wu, K.-H.; Wu, H.-P. Unraveling the Complexities of Toll-like Receptors: From Molecular Mechanisms to Clinical Applications. Int. J. Mol. Sci. 2024, 25, 5037. [CrossRef]
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010; 140:821–832.
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [CrossRef]
- Netea-Maier, R.T.; Plantinga, T.S.; van de Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [CrossRef]
- Farooqui, A.A. Inflammation and Oxidative Stress in Neurological Disorders; Springer Nature: Dordrecht, GX, Netherlands, 2014; ISBN: .
- Wood PL. Neuroinflammation: Mechanisms and Management, Humana Press, Totowa, New Jersey, 1998.
- Ramos-González EJ, Bitzer-Quintero OK, Ortiz G, Hernández-Cruz JJ, Ramírez-Jirano LJ. Relationship between inflammation and oxidative stress and its effect on multiple sclerosis. Neurologia (Engl Ed). 2024 ;39: 292-301.
- Serhan, C.N.; Savill, J. Resolution of inflammation: the beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [CrossRef]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [CrossRef]
- Vidar Hansen T, Serhan CN. Protectins: Their biosynthesis, metabolism, and structure-function. Biochem Pharmacol. 2022; 206:115330.
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 1–32. [CrossRef]
- Farooqui AA, Farooqui T. Phospholipids, Sphingolipids, and Cholesterol-Derived Lipid Mediators and Their Role in Neurological Disorders. Int J Mol Sci 2024; 25: 10672.
- Phillis, J.W.; Horrocks, L.A.; Farooqui, A.A. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: Their role and involvement in neurological disorders. Brain Res. Rev. 2006, 52, 201–243. [CrossRef]
- Lautier J-L, Wautier M-P. Pro- and Anti-Inflammatory Prostaglandins and Cytokines in Humans: A Mini Review. Int J Mol Sci. 2023; 24:9647.
- Ricciotti E, FitzGerald GA. Prostaglandins and Inflammation. Arterioscler Thromb Vasc Biol. 2011;1: 986–1000.
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell. Signal. 2008, 20, 1010–1018. [CrossRef]
- Nixon, G.F. Sphingolipids in inflammation: pathological implications and potential therapeutic targets. Br. J. Pharmacol. 2009, 158, 982–993. [CrossRef]
- Ong WY, Herr DR, Farooqui T, Ling EA, Farooqui AA. Role of sphingomyelinases in neurological disorders Expert Opin Ther Targets. 2015; 19:1725-1742.
- Schütze S, Potthoff K, Machleidt T, Berkovic D, Wiegmann K, Krönke M. TNF activates NF-κB by phosphatidylcholine-specific phospholipase C-induced ‘acidic’ sphingomyelin breakdown. Cell. 1992;71: 765–776.
- Boyd, R.J.; Avramopoulos, D.; Jantzie, L.L.; McCallion, A.S. Neuroinflammation represents a common theme amongst genetic and environmental risk factors for Alzheimer and Parkinson diseases. J. Neuroinflammation 2022, 19, 1–20. [CrossRef]
- Ikonen, E.; Zhou, X. Cholesterol transport between cellular membranes: A balancing act between interconnected lipid fluxes. Dev. Cell 2021, 56, 1430–1436. [CrossRef]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [CrossRef]
- Wheless JW, Rho JM. Role of cholesterol metabolism and homeostasis in the brain hyperexcitability. Epilepsia 2025; 66; 33-46.
- Lawrence, T. The Nuclear Factor NF-kappa B Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [CrossRef]
- Pariente, A.; Peláez, R.; Pérez-Sala, A.; Larráyoz, I.M. Inflammatory and cell death mechanisms induced by 7-ketocholesterol in the retina. Implications for age-related macular degeneration. Exp. Eye Res. 2019, 187, 107746. [CrossRef]
- Andersen A, Campo A, Fulton E, Corwin A, Jerome III WG, O’Connor MS, 7-Ketocholesterol in disease and aging. Redox Biol. 2019; 29:101380.
- Freeman, N.E.; Rusinol, A.E.; Linton, M.; Hachey, D.L.; Fazio, S.; Sinensky, M.S.; Thewke, D. Acyl-coenzyme A:cholesterol acyltransferase promotes oxidized LDL/oxysterol-induced apoptosis in macrophages. J. Lipid Res. 2005, 46, 1933–1943. [CrossRef]
- Weigel, T.K.; Kulas, J.A.; Ferris, H.A. Oxidized cholesterol species as signaling molecules in the brain: diabetes and Alzheimer’s disease. Neuronal Signal. 2019, 3, NS20190068. [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [CrossRef]
- Kihara, A.; Igarashi, Y. Cross Talk between Sphingolipids and Glycerophospholipids in the Establishment of Plasma Membrane Asymmetry. Mol. Biol. Cell 2004, 15, 4949–4959. [CrossRef]
- Kihara, A. Sphingosine 1-phosphate is a key metabolite linking sphingolipids to glycerophospholipids. Biochim. et Biophys. Acta (BBA) - Mol. Cell Biol. Lipids 2014, 1841, 766–772. [CrossRef]
- Sukhorukov, V.N.; Orekhov, A.N. Molecular Aspects of Inflammation and Lipid Metabolism in Health and Disease: The Role of the Mitochondria. Int. J. Mol. Sci. 2024, 25, 6299. [CrossRef]
- Wajner, M.; Amaral, A.U. Mitochondrial dysfunction in fatty acid oxidation disorders: insights from human and animal studies. Biosci. Rep. 2016, 36, e00281. [CrossRef]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer's disease. Ageing Res. Rev. 2020, 64, 1–101191. [CrossRef]
- Vringer, E.; Tait, S.W.G. Mitochondria and cell death-associated inflammation. Cell Death Differ. 2022, 30, 304–312. [CrossRef]
- Hamzeh, O.; Rabiei, F.; Shakeri, M.; Parsian, H.; Saadat, P.; Rostami-Mansoor, S. Mitochondrial dysfunction and inflammasome activation in neurodegenerative diseases: Mechanisms and therapeutic implications. Mitochondrion 2023, 73, 72–83. [CrossRef]
- Lee, Y.; Hirose, H.; Ohneda, M.; Johnson, J.H.; McGarry, J.D.; Unger, R.H. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc. Natl. Acad. Sci. 1994, 91, 10878–10882. [CrossRef]
- Malhi, H.; Gores, G.J. Molecular Mechanisms of Lipotoxicity in Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2008, 28, 360–369. [CrossRef]
- Yoon, H.; Shaw, J.L.; Haigis, M.C.; Greka, A. Lipid metabolism in sickness and in health: Emerging regulators of lipotoxicity. Mol. Cell 2021, 81, 3708–3730. [CrossRef]
- Lytrivi, M.; Castell, A.-L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [CrossRef]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid Metabolism and Toxicity in the Heart. Cell Metab. 2012, 15, 805–812. [CrossRef]
Figure 1.
Molecular mechanisms of neuroinflammation along with priming of NLRP3 inflammasomes activation. Plasma membrane (PM); phosphatidylcholine (PtdCho); cytosolic phospholipase A2 (cPLA2); arachidonic acid (ARA); Toll-like receptors (TLRs); proinflammatory endotoxin lipopolysaccharide (LPS); adaptor protein (MyD88); reactive oxygen species (ROS); tumor necrosis factor receptor-associated factor adaptor protein 6 (TRAF6); NF-κB-inducing kinase (NIK); IκB kinase (IKK); NF-kappaB (NF-kB); NF-kappaB response element (NF-kB-RE); inhibitory subunit of NF-κB (I-κB); tumor necrosis factor-alpha (TNF-α); interleukin-1beta (IL-1β); interleukine-6 (IL-6); pro-interleukin-1β (pro-IL-1β); NLR family, pyrin domain containing 3 (NLRP3) and apoptosis-associated speck-like protein containing a CARD (ASC), collectively known as the NLRP3 inflammasome.
Figure 1.
Molecular mechanisms of neuroinflammation along with priming of NLRP3 inflammasomes activation. Plasma membrane (PM); phosphatidylcholine (PtdCho); cytosolic phospholipase A2 (cPLA2); arachidonic acid (ARA); Toll-like receptors (TLRs); proinflammatory endotoxin lipopolysaccharide (LPS); adaptor protein (MyD88); reactive oxygen species (ROS); tumor necrosis factor receptor-associated factor adaptor protein 6 (TRAF6); NF-κB-inducing kinase (NIK); IκB kinase (IKK); NF-kappaB (NF-kB); NF-kappaB response element (NF-kB-RE); inhibitory subunit of NF-κB (I-κB); tumor necrosis factor-alpha (TNF-α); interleukin-1beta (IL-1β); interleukine-6 (IL-6); pro-interleukin-1β (pro-IL-1β); NLR family, pyrin domain containing 3 (NLRP3) and apoptosis-associated speck-like protein containing a CARD (ASC), collectively known as the NLRP3 inflammasome.
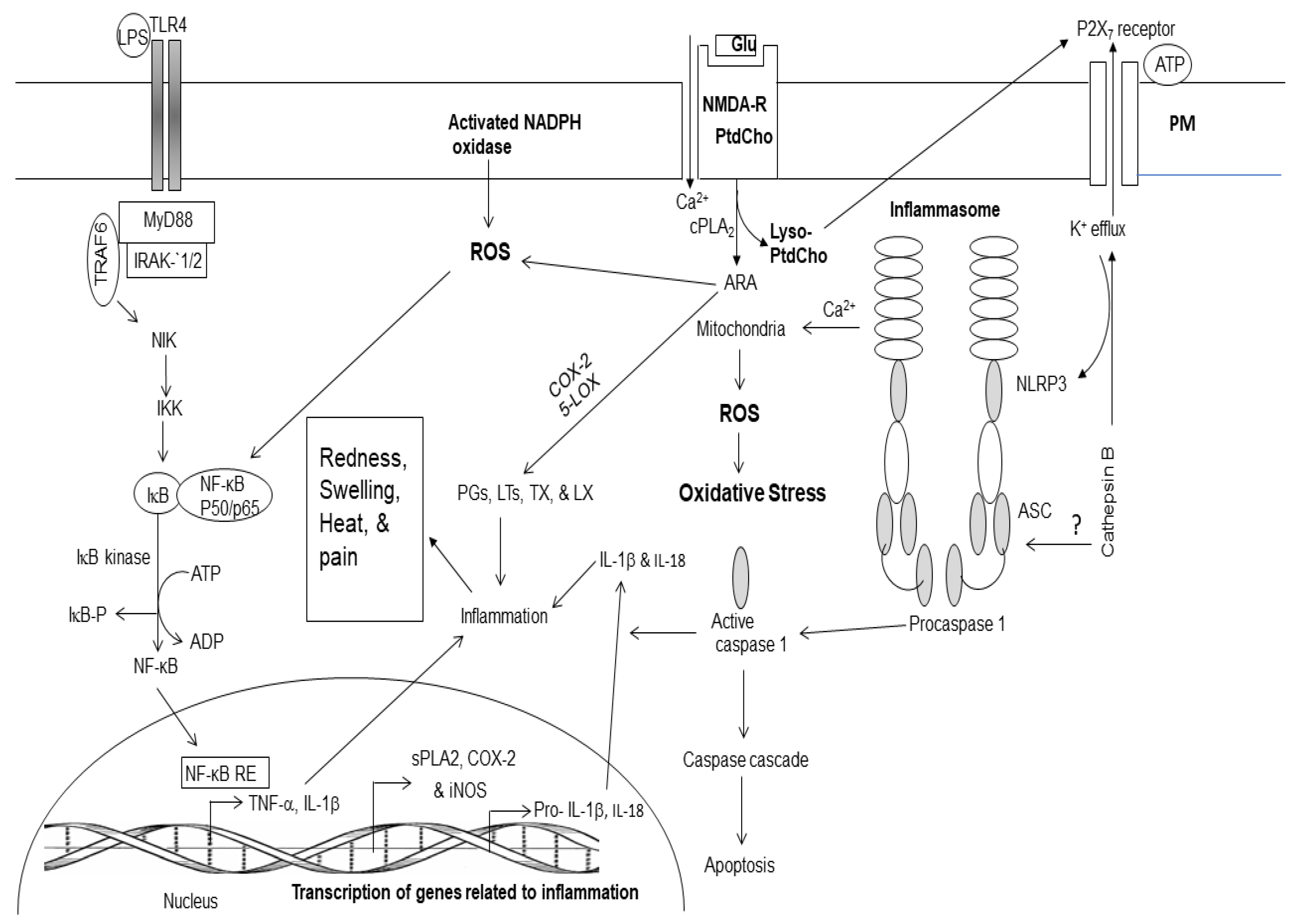
Figure 2.
Generation of enzymatic and non-enzymatic mediators of arachidonic and docosahexaenoic acid metabolism.
Figure 2.
Generation of enzymatic and non-enzymatic mediators of arachidonic and docosahexaenoic acid metabolism.
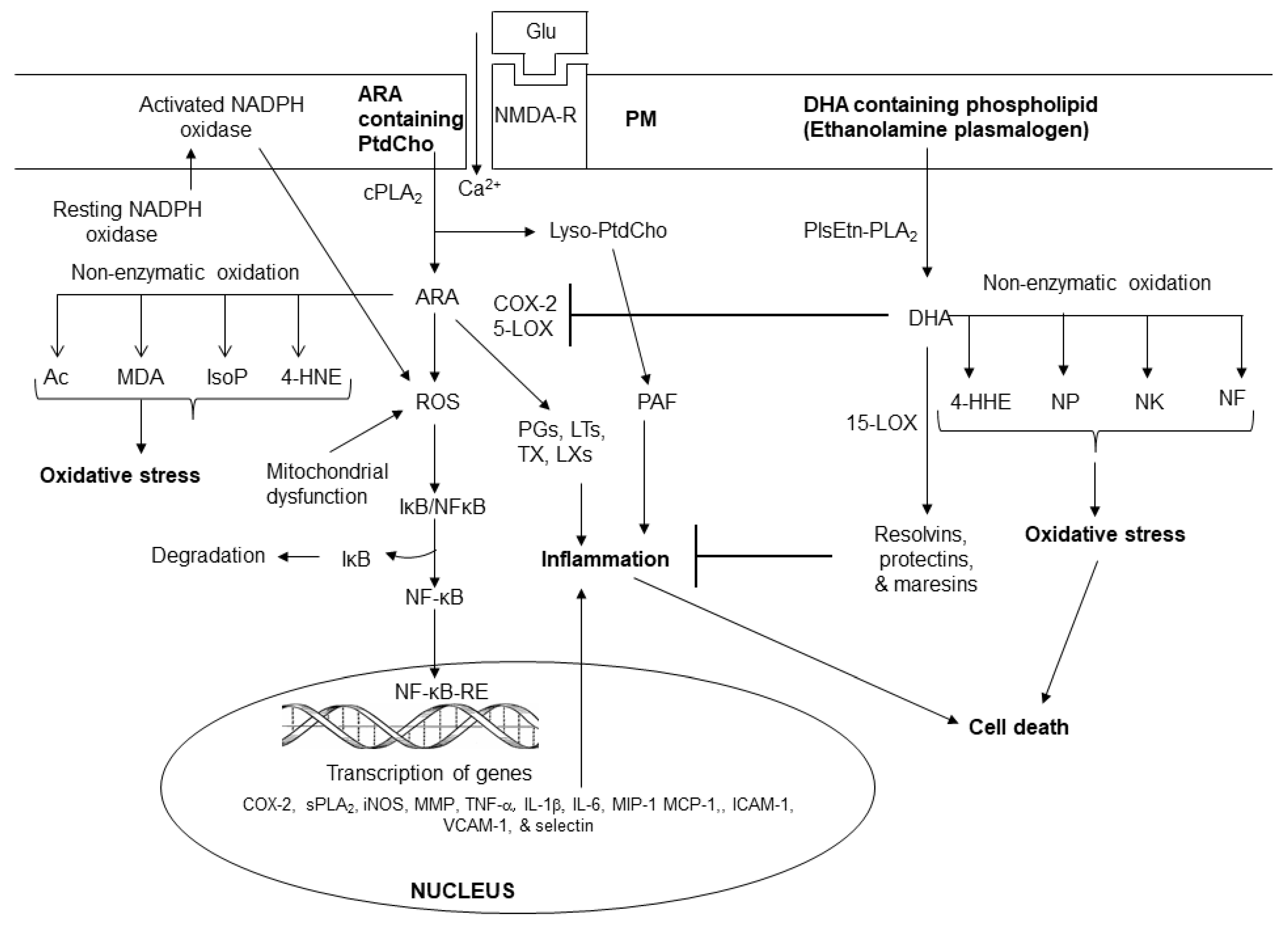
Figure 3.
Sphingolipid-derived metabolites and their role in metabolism. Ceramide 1-phosphate (C 1-P); sphingosine 1 phosphate (S 1 P); cytosolic phospholipase A2 (cPLA2); phospholipase C (PLC); phospholipase D (PLD); protein kinase (PKC); and steroidogenic factor 1 (SF-1).
Figure 3.
Sphingolipid-derived metabolites and their role in metabolism. Ceramide 1-phosphate (C 1-P); sphingosine 1 phosphate (S 1 P); cytosolic phospholipase A2 (cPLA2); phospholipase C (PLC); phospholipase D (PLD); protein kinase (PKC); and steroidogenic factor 1 (SF-1).
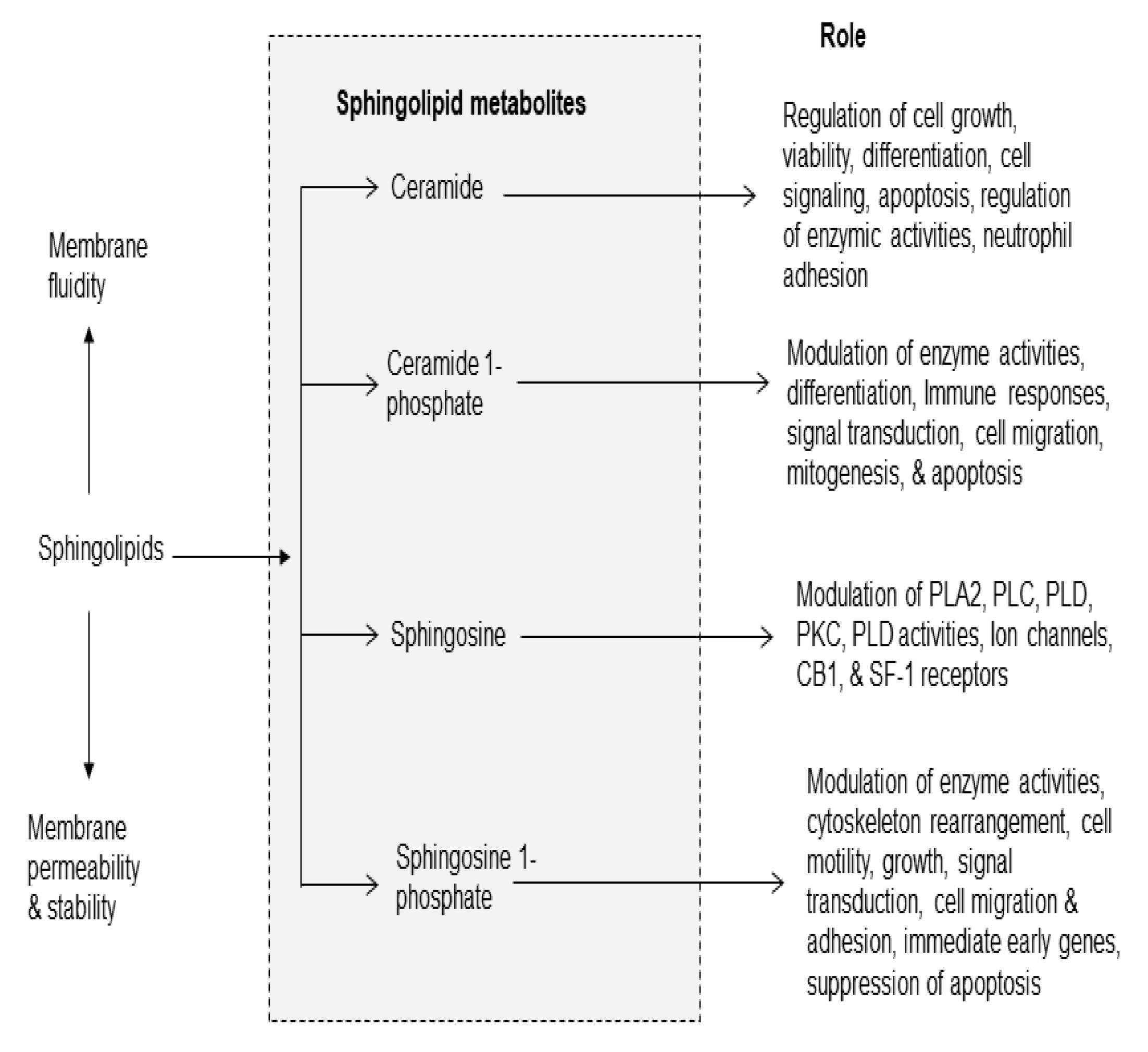
Figure 4.
Cholesterol-derived metabolites and their role in metabolism.
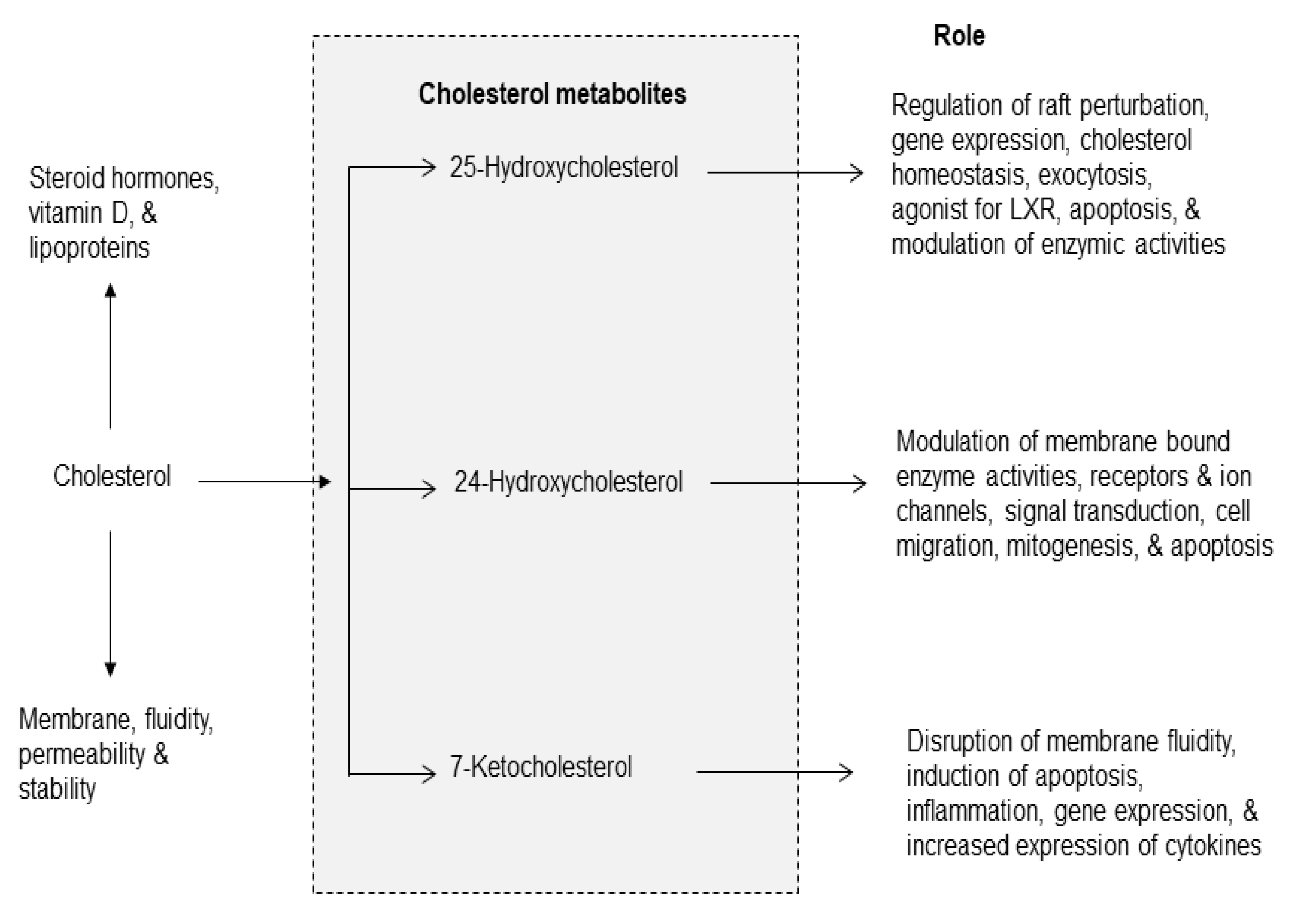
Figure 5.
Cross-talk among metabolites of phospholipid, sphingolipid, and cholesterol metabolism.
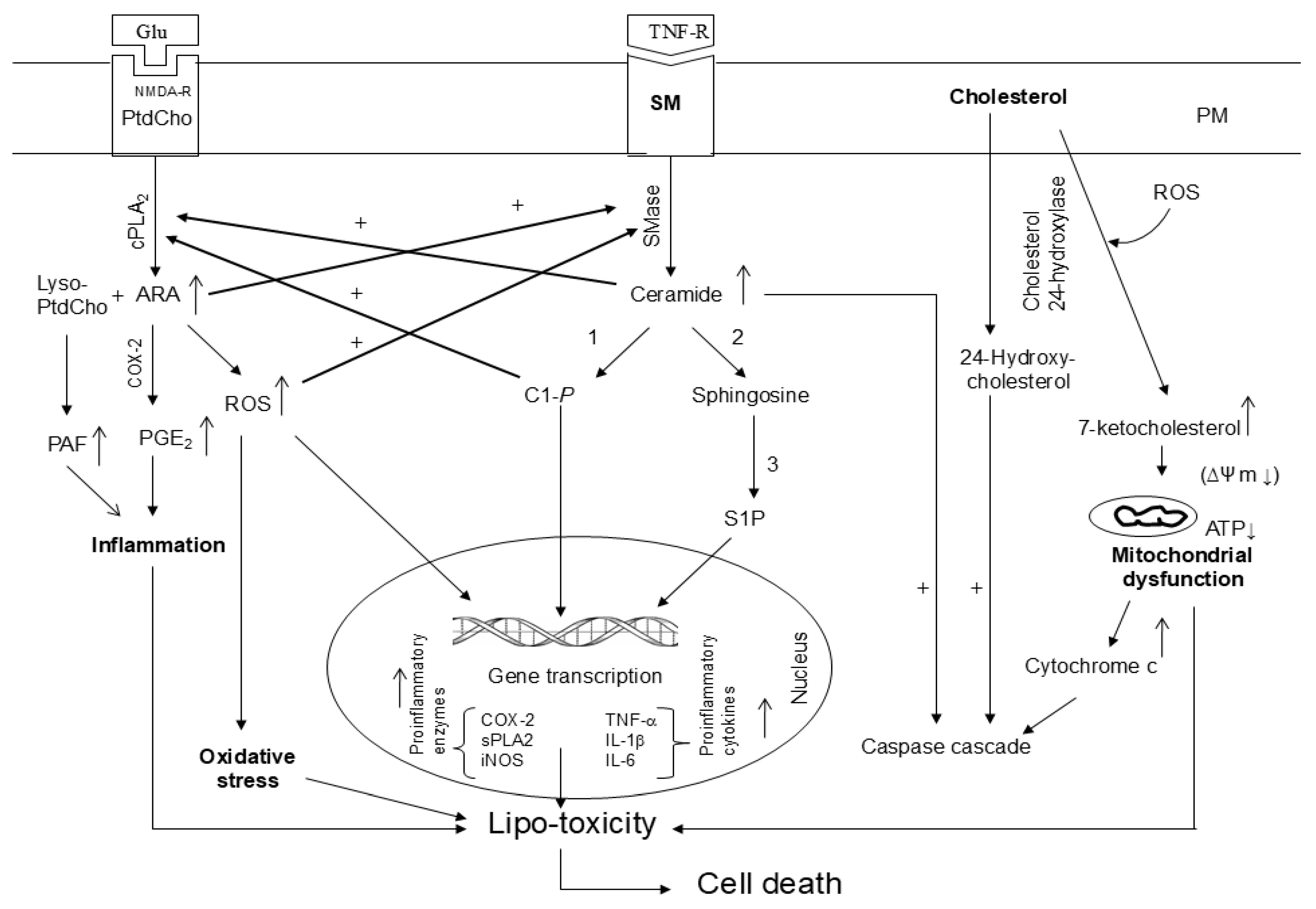

Table 1.
Biomarkers for acute and chronic inflammation.
| Parameters | Acute inflammation | Chronic inflammation |
|---|---|---|
| Color | Redness | No color |
| Rate of development | Rapid | Slow |
| Perception | Pain | No pain |
| Levels of cytokines | High | Low |
| Levels of chemokines | High | Low |
| Levels of eicosanoids | High | Low |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



