
Submitted:
19 February 2025
Posted:
20 February 2025
You are already at the latest version
Abstract
Long noncoding RNAs (lncRNAs) have emerged as pivotal regulators in plant immune responses, yet their roles in rice resistance against Magnaporthe oryzae (M. oryzae) remain inadequately explored. In this study, we integrated translatome data with conventional genome annotations to construct an optimized protein-coding dataset. Subsequently, we developed a robust pipeline (“RiceLncRNA”) for the accurate identification of rice lncRNAs. Using strand-specific RNA sequencing (ssRNA-seq) data from the resistant (IR25) and susceptible (LTH) and Nipponbare (NPB) varieties under M. oryzae infection, we identified 9,003 high-confidence lncRNAs, significantly improving identification accuracy over traditional methods. Among the differentially expressed lncRNAs (DELs), those unique to IR25 were enriched in the biosynthetic pathways of phenylalanine, tyrosine, and tryptophan, which suggests that they enhance the production of salicylic acid (SA) and auxin (IAA) precursors to trigger defense responses. Conversely, DELs specific to LTH primarily clustered within carbon metabolism pathways, indicating a metabolic reprogramming mechanism. Notably, 21 DELs responded concurrently in both IR25 and LTH at 12 h and 24 h post-inoculation, indicating a synergistic regulation of jasmonic acid (JA) and ethylene (ET) signaling while partially suppressing IAA pathways. Weighted gene coexpression network analysis (WGCNA) and competing endogenous RNA (ceRNA) network analysis revealed that key lncRNAs (e.g., LncRNA.9497.1) function as miRNA “sponges,” thereby indirectly modulating the expression of receptor-like kinases (RLKs), resistance (R) proteins, and hormone signaling pathways. The reliability of these findings was confirmed through qRT-PCR and cloning experiments. In summary, our study provides an optimized rice lncRNA annotation framework and reveals the mechanism by which lncRNAs enhance rice blast resistance through the regulation of hormone signaling pathways. These findings offer an important molecular basis for rice disease-resistant breeding.
Keywords:
Long noncoding RNAs
; Magnaporthe oryzae
; Plant Immunity
; ceRNA
; WGCNA
; Hormone signaling
1. Introduction
Long noncoding RNAs (lncRNAs) are transcripts longer than 200 nucleotides with limited or no protein-coding potential [1]. Emerging evidence indicates that lncRNAs participate in a wide range of biological processes across various organisms, regulating gene expression through diverse mechanisms at the chromatin, transcriptional, posttranscriptional, translational, and posttranslational levels [2,3]. In plants, lncRNAs have been shown to regulate key physiological activities, such as flowering time [4,5], crop yield [6], fruit development [7], photomorphogenesis [8], gene silencing [9], and responses to biotic and abiotic stresses [10,11,12].
Despite these advances, the functional characterization of plant lncRNAs remains relatively limited, with only a few regulatory mechanisms fully elucidated. For example, the lncRNAs COLDWARP, COLDAIR, and COOLAIR mediate vernalization by silencing FLOWERING LOCUS C (FLC) [9,13,14]. The long noncoding RNA LRK Antisense Intergenic RNA (LAIR) interacts with OsMOF and OsWDR5 to promote the expression of leucine-rich repeat receptor kinase (LRK) gene clusters, significantly increasing rice yield [6]. Importantly, the crucial roles of lncRNAs in plant responses to biotic stress and immunity are gradually coming to light. In tomato, lncRNA16397 induces the expression of SlGRX, reducing reactive oxygen species (ROS) accumulation and thereby increasing resistance to Phytophthora infestans [11]. In Arabidopsis, the lncRNA ELENA1 interacts with the mediator complex subunit MED19a to regulate PR1 expression, bolstering immunity against pathogens [10]. In rice, the lncRNA ALEX1 enhances resistance to bacterial blight by modulating the jasmonic acid (JA) pathway [15], whereas the long noncoding RNA SABC1 helps balance plant immunity and growth by regulating salicylic acid (SA) synthesis [16]. Moreover, lncRNA23468 functions as a competing endogenous RNA (ceRNA) that suppresses miR482b accumulation, consequently elevating the expression of NBS-LRR genes and fortifying defense against P. infestans in tomato [17].
Rice (Oryza sativa L.), one of the world’s most important staple crops (particularly in Asia and Africa), faces a major threat from rice blast disease caused by the fungus M. oryzae. Globally, rice blast can lead to yield losses up to 30%, posing a severe challenge to food security and agricultural economies, with estimated annual losses of up to $66 billion [18]. Hence, understanding the molecular mechanisms of rice blast disease is of paramount importance for global food security [19,20].
Earlier research demonstrated that protein-coding genes play significant roles in the defense response of rice against M. oryzae [21,22]. However, the involvement of lncRNAs in this process has not been extensively explored. A recent rice telomere-to-telomere genome annotation revealed an additional 1,373 protein-coding genes, indicating that the genome annotation is still incomplete, especially in intergenic regions [23]. Such gaps might contribute to false positives in lncRNA identification and thus hamper downstream functional analyses.
To more accurately identify and characterize rice lncRNAs expressed during M. oryzae infection, we combined multiple software tools and methods suitable for lncRNA identification in rice and developed a pipeline named RiceLncRNA (https://github.com/njausxl/RiceLncRNA). This pipeline begins by integrating rice translatome data to construct a novel rice protein-coding gene annotation database (the CodingRNA dataset). By merging this dataset with Michigan State University Rice Genome Annotation Project version 7 (MSU v7) [24], we substantially improved annotation completeness (adding approximately 18.9% more gene loci) and accuracy in intergenic regions. With this optimized annotation, we analyzed strand-specific RNA-seq data from IR25 (a monogenic line harboring the blast resistance gene Pikm), LTH (a susceptible variety), and NPB (a conventional variety) at multiple time points postinoculation (0 h, 12 h, 24 h, 72 h). Through systematic analyses of these data, we identified 9,003 high-fidelity lncRNAs, with 605 differentially expressed under inoculation—including 415 that respond directly to stress. Notably, the resistant variety IR25 exhibited 293 specific DELs, whereas LTH showed only 70, suggesting that lncRNAs are substantially involved in the resistance signaling network. Functional enrichment analysis revealed that the target genes of these DELs are significantly enriched in multiple hormones signaling pathways, notably SA, JA, ET, and IAA. By constructing a ceRNA regulatory network and performing WGCNA, we discovered that several core regulatory lncRNAs (e.g., LncRNA.9497.1, LncRNA.9562.1) either directly target resistance (R) genes, receptor kinases, and disease-resistance proteins, or indirectly regulate them by competitively binding specific miRNAs. Through these complementary mechanisms, these lncRNAs synergistically modulate hormone signaling pathways to improve resistance. This study not only refines the rice genome annotation and establishes a reliable lncRNA identification pipeline but also, more importantly, elucidates the regulatory network of lncRNAs in rice–M. oryzae interactions, providing essential theoretical foundations and potential targets for advancing plant immune mechanism research and implementing novel disease-resistant breeding strategies.
2. Results
2.1. Analysis of the CodingRNA Dataset Assembly
On the basis of translatome data, we constructed a rice protein-coding gene annotation database, which we termed “CodingRNA.” Compared with the MSU v7 reference genome alone, this translatome-based annotation covered approximately 57% of the genome, and merging both annotations yielded an additional ~18.9% of gene loci (Table S2). As shown in Figure S1, several protein-coding genes were identified within regions previously annotated as intergenic in MSU v7. For example, in the region from 4,643,010 to 4,659,242 on chromosome 4 (Figure S1A), MSU v7 had originally classified the entire stretch as intergenic; however, our detailed analysis of the corresponding translatome data revealed a novel protein-coding gene with clear ribosome footprint signals (labeled “CD48328”). This discovery confirms that the locus encodes a functional protein rather than a non-coding transcript. In summary, by incorporating such findings into the CodingRNA dataset, we significantly enhance the accuracy and completeness of rice genome annotation by ensuring that protein-coding transcripts are not erroneously classified as lncRNAs.
2.2. Integrated Translatome and Transcriptome-Based Lncrna Identification Pipeline and Analysis
To systematically uncover rice lncRNAs, we analyzed strand-specific RNA-seq data from IR25 (carrying the blast resistance gene Pikm), LTH (susceptible), and NPB (a conventional variety), sampled at different postinoculation time points (0 h, 12 h, 24 h, 48 h, 72 h) under M. oryzae. Overall, 40 RNA-seq libraries that have been validated for strand-specificity were utilized (Table S3), though two (LTH-24 h-2, LTH-24 h-3) were discarded due to poor quality.
After trimming low-quality reads and adapters, we retained approximately 10,070,404,673 high-quality reads with an average GC content of 48.89% (Table S4). The mean alignment rate to MSU v7 was 96.89%, with an 86.23% rate of correctly paired reads.
We eventually assembled 114,267 transcripts (≥200 nt, fragments per kilobase of transcript per million mapped reads (FPKM) ≥0.5). To mitigate false positives, an updated rice lncRNA identification pipeline (Figure 1A) was implemented. Its key innovation lies in employing the refined CodingRNA dataset to avoid misclassifying protein-coding transcripts as lncRNAs. Through GFFcompare, we extracted transcripts labeled with classcodes i, x, o, u, and p (totaling 12,836 candidates; see Figure S2A for an explanation of the classcodes). Subsequent filtering against protein families database (Pfam), RNA families database (Rfam), and NCBI nonredundant protein database (NR) removed additional protein-coding or known noncoding RNAs, giving 11,543 candidates (Figure 1B).
Moreover, we applied Coding Potential Calculator 2 (CPC2) (11,748 transcripts), Predictor of Long non-coding RNAs and mEssenger RNAs based on an improved K-mer scheme (PLEK) (11,133), and Coding-Non-Coding Index (CNCI) (10,098) to evaluate coding potential (Figure 1C). Only transcripts consistently deemed “noncoding” by all three tools were retained (9,350). Taking the intersection of these results, we ultimately identified 9,003 high-fidelity lncRNAs (Figure 1D, Table S5). Classification indicated 2,803 intergenic, 2,076 antisense, 419 bidirectional, 3,056 intronic, and 649 sense lncRNAs (Figure 1E, Table S6).
A more conventional pipeline that did not integrate translatome data identified 10,464 putative lncRNAs, which included the aforementioned 9,003 high-confidence lncRNAs plus an additional 1,461 transcripts (Figure S2B). Further analysis revealed that approximately 96.3% of these 1,461 transcripts exhibited protein-coding potential, mostly located in insufficiently annotated intergenic regions. When we compared these 1,461 transcripts against newly predicted genes in the T2T rice genome (Table S7), about 10.1% (147 transcripts) aligned with those newly predicted coding genes. These findings underscore that incorporating translatome data substantially reduces the false-positive rate in lncRNA identification.
The final set of 9,003 lncRNAs was unevenly distributed across chromosomes (Figure 2A, Figure S3A), with Chr1 bearing the most (1,177) and Chr10 the fewest (549). These lncRNAs featured a mean GC content of 41%, notably lower than the ~53% typical of protein-coding genes (Table S8). Approximately 64% spanned 200–400 bp (median 333 nt) (Figure 2B), much shorter than mRNAs (median 1,008 nt), and displayed lower expression levels (Figure 2C). Notably, ~84.75% contained only one exon (Figure 2D), and they had fewer isoforms than mRNAs (Figure 2E).
Further comparison to publicly available rice lncRNA databases (PlantNATdb, PNRD, RNAcentral, NONCODE, CANTATAdb, GreeNC) revealed that 3,778 (41.96%) of our identified lncRNAs matched known records (E value <1e-10, identity >80%, coverage >50%), of which 2,446 were shared across all four sample sets (Figure S3B).
In summary, the optimized identification pipeline that integrates translatome and transcriptome data not only significantly reduces the false-positive rate and ensures high accuracy of the results, but also provides a convenient workflow and reference standard for future rice lncRNA identification, thereby greatly expanding the rice lncRNA resource.
2.3. DELs and Their Differential Expression (DE) Target Genes Regulatory Networks Reveal the Mechanisms Underlying Rice Response to M. oryzae Infection
To investigate the transcriptional regulation differences in rice varieties under M. oryzae infection and to discover potential resistance- or susceptibility-related lncRNAs, we systematically analyzed the DE of both mRNAs and lncRNAs in the resistant variety IR25 and the susceptible variety LTH at 0 h, 12 h, and 24 h postinoculation. A comprehensive comparison of transcriptomic differences between inoculated and noninoculated conditions and across time points revealed 4,426 differentially expressed mRNAs (DEGs), including 3,821 DEGs and 605 differentially expressed lncRNAs (DELs). Of these, 415 DELs and 3,338 DEGs were stress-responsive (Table S9, Figure 3A).
When comparing conditions before and after inoculation, the resistant genotype IR25 displayed 293 lineage-specific DELs, whereas the susceptible genotype LTH presented only 70 (Figure 3B). Further analysis indicated that IR25 had the highest number of specific DELs (137) at 12 h postinoculation, suggesting a faster transcriptional response during the early stage of M. oryzae stress. By applying cis and trans target gene prediction (20 kb upstream and downstream, |r| > 0.5; RIblast combined with correlation analysis), we identified 61 cis and 341 trans target genes for the 293 IR25-specific DELs (Table S10, Table S11). GO enrichment analysis showed that these target genes are primarily involved in photosynthesis, “small molecule biosynthetic process” (GO: 0006508), and “organic acid metabolic process” (GO: 0016053)—all of which are closely tied to aromatic amino acid metabolism (Figure 3C). KEGG pathway analysis further revealed significant enrichment in the “phenylalanine, tyrosine, and tryptophan biosynthesis” pathway (osa00400) (Figure 3D), with notable enrichment of genes such as LOC_Os01g55870 (chorismate mutase 3, chloroplastic) and LOC_Os09g08130 (indole-3-glycerol phosphate synthase). These genes play critical roles in phenylalanine and tryptophan metabolism, respectively, serving as key precursor nodes for SA and IAA biosynthesis. These results suggest that the DELs specific to IR25 may enhance plant defense against M. oryzae by modulating aromatic amino acid metabolic pathways and expediting the synthesis of resistance-related hormones.
In contrast, the susceptible genotype LTH exhibited only 70 specific DELs, which were associated with 18 cis and 74 trans target genes (corresponding to 15 and 12 DELs, respectively). GO enrichment showed that these target genes were enriched in carbon metabolism-related terms such as “starch metabolic process” (GO: 0019250) and “sugar metabolic process” (GO: 0006006) (Figure 3E). Meanwhile, KEGG analysis indicated that “starch and sucrose metabolism” (osa00500) and “glyoxylate and dicarboxylate metabolism” (osa00630) pathways were significantly enriched (Figure 3F). Thus, under M. oryzae stress, LTH-specific DELs seem more inclined to govern carbon metabolism reprogramming rather than potent defense hormone pathways, suggesting that LTH predominantly undergoes a metabolic adjustment-based stress pattern at early stages, failing to trigger strong hormone-mediated defenses in a timely manner.
Furthermore, we detected 52 DELs shared by IR25 and LTH (Figure 3B), all of which were consistently up- or downregulated in both varieties. GO enrichment analysis on the cis and trans target genes of these 52 DELs uncovered terms related to “rhythmic process” (GO: 0048511) and sugar metabolism, such as “hexose metabolic process” (GO: 0019318) (Figure S4A). Their KEGG enrichment highlighted the “pentose phosphate pathway” (osa00030), “Calvin cycle carbon fixation” (osa00710), and “amino acid biosynthesis” (osa01230) (Figure S4B). These findings point to a conserved role for these commonly responsive genes in energy metabolism and fundamental physiological regulation, helping coordinate basic adaptive responses in both resistant and susceptible genotypes.
We further examined DELs in the NPB variety at 24, 48, and 72 h post-inoculation (Figure S5), observing only a few DELs—just two overlapped with the aforementioned set of 52 DELs—indicating significant differences in how LTH, IR25, and NPB respond at the lncRNA level. This highlights genotype-specific defense mechanisms.
A direct comparison of the susceptible and resistant genotypes (LTH vs. IR25) at 12 h and 24 h postinoculation revealed 21 lncRNAs co-responsive to M. oryzae infection (Figure S4C). Their cis/trans target enrichment suggested potential synergy between JA and ET biosynthesis, as well as IAA modulation. GO enrichment analysis indicated that phospholipase A1 (PLA1) and cysteine synthase genes are closely associated with these 21 lncRNAs, with PLA1 being crucial for JA biosynthesis during early infection stages and cysteine serving directly as a precursor for ET (Figure S4D) [25,26]. Furthermore, within the KEGG-enriched “plant hormone signal transduction” pathway (Figure S4E), two key genes—JAZ (LOC_Os04g32480) and SAUR (LOC_Os02g52990)—were identified (Figure S4F-G) [27,28]. The marked upregulation of JAZ suggests a potential negative feedback mechanism isn JA signaling, whereas the pronounced downregulation of SAUR may indicate suppression of IAA, thereby prioritizing the synthesis of defense-related hormones.
We showed that the resistant variety IR25 exhibits a more robust transcriptomic response to M. oryzae infection than the susceptible LTH, largely through lncRNAs involved in aromatic amino acid metabolism and hormone signaling. Moreover, we identified a subset of lncRNAs that coordinate JA, ET, and IAA pathways, indicating that JA and ET signals play pivotal roles under M. oryzae stress while IAA signaling functions as an auxiliary route, collectively maintaining a dynamic balance between stress tolerance and immune responses.
2.4. Construction of a ceRNA Network Reveals the Role of lncRNAs in Rice Blast Resistance
This study constructed a ceRNA network containing 20 miRNAs, 17 lncRNAs, and 115 mRNAs (Figure 4), systematically analyzing how lncRNAs indirectly regulate resistance genes and hormone signals by “sponging” miRNAs during M. oryzae infection. Four lncRNAs—LncRNA.9497.1, LncRNA.9562.1, LncRNA.13491.1, and LncRNA.33800.3—emerged as major nodes, closely linked to hormone signaling pathways (e.g., JA; IAA; ABA; and gibberellin, GA) as well as the expression of various resistance-related genes.
LncRNA.9497.1 indirectly regulates several hormone-related genes by “sponging” osa-miR395a, osa-miR2864.1, and osa-miR5830. For instance, osa-miR395a targets OsSultr2;2 (a sulfate transporter), potentially bolstering sulfur metabolism to supply more substrates for defense [29]. In addition, osa-miR2864.1 targets a variety of receptor kinases, including OsLRK6 (leucine-rich repeat receptor kinase), OsMRLK16 (wheat germ agglutinin domain kinase), OsRLCK204 (receptor-like cytoplasmic kinase), and SDRLK-40 (receptor-like kinase). Among these, OsLRK6 is vital for immune signal transduction, whereas OsMRLK16, OsRLCK204, and SDRLK-40 facilitate microbe-associated molecular pattern (MAMP) perception and potentially abiotic stress cross-tolerance. Significantly higher expression of these receptor kinases in IR25 at 24 h post-inoculation suggests that certain lncRNAs may facilitate early-stage pathogen recognition through miRNA “sponging”.
Moreover, osa-miR5830 targets OsABA8ox3 and OsMETS2, which regulate ABA degradation and methionine metabolism, respectively. OsABA8ox3 modulates ABA levels, influencing stress adaptability [30]. In IR25 inoculated with M. oryzae, OsABA8ox3 was notably downregulated by 24 h, whereas OsMETS2 was upregulated, thus promoting ethylene biosynthesis and reinforcing disease resistance [31]. Thus, in the resistant IR25 line, LncRNA.9497.1 may indirectly downregulate ABA signaling and upregulate ethylene signaling via miRNA sponging, thereby enhancing the downstream immune response in rice.
LncRNA.33800.3 indirectly regulates OsWRKY70, OsCPS1, and OsGH3-2 by “sponging” osa-miR5075. OsWRKY70 is a key transcription factor in the JA signaling pathway that positively regulates JA biosynthesis while negatively affecting GA synthesis, thus prioritizing defense over growth [32]. OsCPS1 participates in GA biosynthesis [33], and OsGH3-2 is an IAA amino acid synthase implicated in broad-spectrum resistance by limiting auxin levels [34].
In LTH (susceptible) at 12 h postinoculation, OsGH3-2 was significantly downregulated, whereas OsWRKY70 was markedly upregulated, indicating that under early pathogen stress, lncRNAs such as LncRNA.33800.3 may simultaneously reduce auxin and raise JA signals to enhance disease resistance.
Furthermore, LncRNA.9562.1 and LncRNA.13491.1 regulate several resistance genes and hormone signals by targeting osa-miR529a, osa-miR5830, and osa-miR2090.
osa-miR2090 targets OsRLCK42 (receptor-like protein kinase) and Cht5 (chitinase). OsRLCK42 aids in early stress signal transduction, whereas Cht5 degrades fungal cell walls, a process pivotal to JA-mediated immunity [35]. In LTH, OsRLCK42 was strongly downregulated while Cht5 was upregulated, suggesting partial defense activation yet compromised early signaling in a susceptible background.
osa-miR529a targets OsABA8OX2 (ABA metabolic balance) and OsbHLH148 (a transcription factor regulating JA signaling), which contribute to both drought and disease stress responses [36] . In resistant IR25, OsABA8OX2 tended to be downregulated at 24 h postinfection, while OsbHLH148 was upregulated, reflecting a shift from ABA to JA-driven defenses [37,38].
osa-miR5830 targets OsABA8ox3 and interacts with OsMETS2, a methionine synthase supporting ethylene biosynthesis [31,39]. This further underscores intricate hormone crosstalk controlled by hub lncRNAs.
Collectively, these ceRNA interactions indicate that lncRNAs can coordinate hormone pathways (ABA, ET, JA, IAA) and receptor kinases to bolster rice blast resistance.
2.5. WGCNA Uncovers Key Hormone, R genes / Proteins, and Receptor Kinase Networks Under M. oryzae Stress
After removing batch effects, normalizing expression, and filtering outliers, a total of 2,583 lncRNAs and 15,701 genes were retained for WGCNA (Table S12). With a soft threshold power of 18 (scale-free topology index ~0.85; Figure 5A-B), 26 modules were identified (TOMType=Unsigned, deepSplit=2, minModuleSize=30, mergeCutHeight=0.2). Notably, around 85.23% of all genes/lncRNAs gathered into the top 10 modules (Figure 5C).
By correlating module eigengenes (MEs) with phenotypic traits (blast resistance vs. susceptibility) (Figure 5D), three notable correlations emerged. The Darkgreen Module showed strong positive correlation with resistance (r=0.96, p=3×10^-21), while the Lightyellow Module demonstrated negative correlation with resistance (r=–0.87, p=1×10^-12). The Gray60 Module exhibited negative correlation with susceptibility (r=–1.0, p=9×10^-40). Scatter plots of transcript significance (TS) vs. module membership (MM) (Figure 6E, Figure S6A-B) confirmed these relationships, prompting further network analysis of hub lncRNAs and their target genes.
In the darkgreen module (Figure 6A, Table S13), LncRNA.9497.1, LncRNA.21901.1, and LncRNA.35959.1 serve as core nodes, associating with disease/stress resistance genes such as wall-associated kinase 1 (WAK1), putative disease resistance protein (RGA4), OsWRKY125, and mitogen-activated protein kinase 17 (MAPK17). WAK1 mediates cell wall strengthening and defense signal transduction [40]. Meanwhile, RGA4 and RGA3 are NB-LRR-type resistance genes detecting pathogen effectors [41], and OsWRKY125 is a WRKY TF driving the expression of defense genes [42]. MAPK17 sits at a crucial position in MAPK signaling cascades under pathogen attack [43].
Within the lightyellow module (Figure 7C), LncRNA.10688.1 and LncRNA.36066.1 emerged as central regulatory points. LncRNA.10688.1 is directly linked to RGA5 (LOC_Os11g37740, LOC_Os12g37770) and Bph40 (LOC_Os11g39209), both contributing to pathogen-triggered immunity (PTI) and physical defense barriers [44,45]. It also relates to OsLP2 (LOC_Os12g08240), implying cross-disease and environmental stress crosstalk [46]. In LTH inoculated with M. oryzae, RGA5 and OsLP2 were notably upregulated, whereas Bph40 was slightly downregulated, suggesting partial immune enhancement yet weaker mechanical defenses in a susceptible background.
Piks-1 and Piks-2, recognized CC-NBS-LRR pairs defending against M. oryzae [47], and Pb1 (LOC_Os04g06280), a durable blast resistance gene from indica [48], also converged in this module’s coexpression network, implying multiple layers of early PTI and defense synergy. In terms of stress resistance, OsPUB69 (LOC_Os12g33180), an E3 ubiquitin ligase gene associated with LncRNA.21855.1, has been shown to play a crucial role in stress tolerance, particularly in protein degradation and cellular homeostasis regulation [49]. Similarly, OsGLP8-12 (LOC_Os12g28015), associated with LncRNA.36066.1, regulates reactive oxygen species (ROS) levels, which is essential for rice adaptation and disease resistance under oxidative stress conditions [50,51].
The gray60 module features LncRNA.13491.1, LncRNA.9562.1, and LncRNA.9997.2 (Figure 7B), which coordinate multiple receptor kinases (OsRLCK366, OsRLCK5), resistance genes (RGA5-L1), and hormone regulators (OsPP2C19). For example, LncRNA.13491.1 intersects with RGA4, RGA5, RPM1, and Xa1, all tied to effector-triggered immunity (ETI) [52,53]. It also influences OsMADS56 and OsbHLH179, potentially modulating strigolactone (SL) and GA signals [54,55]. Similarly, LncRNA.9562.1 correlates with OsRLCK366 (a receptor kinase) and OsPP2C19 (an ABA regulator) [56,57], facilitating hormone interplay and stress responses [58,59,60]. LncRNA.9997.2 impacts the regulation of GA, ABA, and even cytoskeletal elements via OsRLCK5 and VLN4 [61]. Interestingly, based on the expression heatmap of lncRNA within the module (Figure S6C-E), LncRNA.13491.1 and LncRNA.9562.1 peaked in LTH at 12 h but remained low in IR25, indicating potential genotype-specific temporal regulation of immune-associated lncRNAs.
Collectively, these findings demonstrate that multiple lncRNAs within WGCNA modules coordinate hormone pathways (ABA, GA, SL), receptor kinases (OsRLCK5, OsRLCK366), and R genes/proteins (RGA4, RGA5, Pb1, Piks-1, Piks-2) to bolster both early PTI and downstream ETI, thereby establishing a robust multi-layered defense framework in rice.
2.6. Mechanistic Insights into the Functional Roles of Key lncRNAs in Rice Blast Resistance
To examine the mechanistic roles of the top lncRNAs identified by WGCNA—e.g., LncRNA.9497.1, LncRNA.9562.1, LncRNA.10688.1, and LncRNA.13491.1—we performed gene set enrichment analysis (GSEA) for single key lncRNAs, focusing on pathways implicated in secondary metabolism and hormone signals, and structural diagrams for these four lncRNAs were also generated (Figure S7).
LncRNA.9497.1, annotated as an antisense transcript (Figure 7A), was significantly associated with the phenylpropanoid biosynthesis pathway (osa00940) and broader plant secondary metabolite biosynthesis (osa00999) (Figure 7A). These metabolic processes contribute to lignin and flavonoid production, which reinforce cell walls and produce antimicrobial compounds. Thus, LncRNA.9497.1 likely boosts disease resistance through robust metabolic defense.
Genes linked to LncRNA.9562.1 were mainly enriched in plant hormone signal transduction (osa04075) (Figure 7B, Figure S8), encompassing SA, ET, brassinosteroid (BR), GA, and JA pathways. Critical components like NPR1 and PR1 and ET-related genes appear among its targets, indicating a capacity to orchestrate hormone crosstalk and secondary metabolism, thereby enabling a rapid immune response to M. oryzae.
LncRNA.10688.1 was significantly enriched in the phenylpropanoid biosynthesis pathway (osa00940) and the carotenoid biosynthesis pathway (osa00906) (Figure 7C). Phenylpropanoids foster structural defenses and are precursors to SA, while carotenoids enhance antioxidant capacity and can feed into ABA production, thus regulating plant stress adaptation.
LncRNA.13491.1 exhibited notable enrichment in pigment biosynthetic processes (GO: 0046148) and flavonoid biosynthesis (osa00941) (Figure 7D). Flavonoids help mitigate oxidative stress and may act as signaling molecules during pathogen attacks, suggesting that LncRNA.13491.1 aligns plant defense responses with redox equilibrium.
Altogether, these key lncRNAs enhance plant adaptation and defense under M. oryzae infection by integrating basic metabolic, secondary metabolic, and multiple hormones signaling pathways—particularly the phenylpropanoid, flavonoid, JA, SA, ET, and IAA pathways. Their multifaceted roles highlight potential targets for breeding disease-resistant rice varieties using gene editing or advanced selection techniques.
2.7. qRT–PCR Analysis and LncRNA Cloning Validation
To validate these bioinformatic results (Table S14), four important lncRNAs and seven of their target genes were chosen from key WGCNA modules for quantitative real-time PCR (qRT–PCR) (Figure 8A–C, M, G–I, N-Q). The expression patterns largely matched the RNA-seq data, reinforcing the reliability of differential expression analysis. Another three random DELs and their three target genes were likewise confirmed (Figure 8D–F, J–L), supporting the computational pipeline’s accuracy.
One high-priority lncRNA (lncRNA.9562.1) was subsequently cloned, and Sanger sequencing verified its length and sequence fidelity (Figure S9A-B). The cloned sequence fully matched the predictions, indicating the pipeline’s robust annotation. These validation results pave the way for potential functional analyses, such as overexpression or knockout experiments, to assess the phenotypic impacts of this lncRNA on rice disease resistance.
3. Discussion
3.1. Optimized lncRNA Identification Pipeline and Its Contribution to Rice Genome Annotation
In this study, we developed a more comprehensive rice protein-coding gene database (CodingRNA) by merging translatome data with existing genome annotations, thereby refining the lncRNA identification pipeline (Figure 1A). Compared with conventional annotation approaches, our pipeline demonstrates an enhanced ability to exclude transcripts with protein-coding potential, effectively reducing false-positive lncRNA calls. This improvement aligns with recent telomere-to-telomere genome assembly work [23] reporting the discovery of 1,571 additional coding genes beyond MSU v7, underscoring the importance of updated annotation strategies (Table S7).
We identified 9,003 high-fidelity lncRNAs (Figure 1D, Table S5)—exceeding previous tallies by Wang et al. (4,787) and Priyanka et al. (5,337) [62,63]. Such discrepancies likely stem from differences in rice varieties, stress conditions, sequencing strategies, and lncRNA pipelines. Notably, 40% of our lncRNAs matched those in public databases (Figure S3B), whereas 60% appeared novel, highlighting possible genotype-, tissue-, or stress-specific expression. Our pipeline incorporates translatome data integration for excluding potential coding transcripts, three-pronged coding potential prediction (CPC2, PLEK, CNCI) supplemented by Pfam/Rfam/NR filtering, rice-specific TAD features for lncRNA target gene identification, and RIblast for trans-target detection, replacing older methods.
Despite substantial progress, there remain limitations. The focus on certain genotypes and time points leaves open questions about broader spectrum resistance and lncRNA behavior under other M. oryzae races or environmental contexts. Additionally, although qRT–PCR verified expression changes (Figure 8), functional assays such as overexpression or knockout are still needed to confirm direct phenotypic influences. Lastly, this work primarily addressed leaf tissue, calling for future multi-tissue or single-cell omics analyses to reveal dynamic spatiotemporal patterns of lncRNAs. Nevertheless, by offering an integrated pipeline on GitHub, this study establishes a rigorous foundation for more accurate lncRNA discovery and functional research in rice.
3.2. LncRNAs Participate in Rice Blast Resistance by Regulating JA, ET, and IAA Signaling Pathways in a Coordinated Manner
We observed that during M. oryzae infection, the target genes of DELs — which are themselves DEGs — in IR25 (resistant) and LTH (susceptible) varieties were significantly enriched in the JA and ET signaling pathways. This aligns with prior evidence highlighting the importance of the JA pathway in rice blast resistance. [62]. Yet, our study also unveils the ET pathway’s role. For example, some lncRNA targets in both IR25 and LTH—such as PLA1 (involved in JA biosynthesis) and PCO (affecting ethylene-related transcription factors [25,26])—suggest overlapping ET–JA crosstalk (Figure S4D). This synergy aligns with known JA–ET cooperation in defense against necrotrophic pathogens [64,65].
In IR25-specific responses, many DEL targets clustered within aromatic amino acid metabolism, implying potential enhancement of phenylalanine- and tryptophan-derived SA and IAA. Notably, previous studies have demonstrated that IAA and SA exhibit antagonistic effects, and this balance helps conserve energy during pathogen infection, thereby optimizing plant defense responses, which is consistent with our findings [66]. Meanwhile, JAZ genes (negative regulators of the JA pathway) [27] and SAUR genes (auxin-responsive [28]) also appear among the common DEL targets (Figure S4E-G), implying dynamic hormone crosstalk. Overall, IR25’s greater number of specific DELs (293) versus LTH’s 70 indicates a more robust or earlier activation of these hormone-regulated defenses in the resistant genotype. Conversely, LTH-specific DELs were concentrated in carbon metabolism, illustrating a metabolic shift that might be less effective at early defense induction.
Thus, multiple hormone signals—SA, ET, IAA, JA—likely collaborate under M. oryzae stress, and each genotype’s capacity to coordinate these pathways promptly could decide the strength and timeliness of immune responses.
3.3. lncRNAs Mediate Immune Responses by Regulating RLKs and R Genes/Proteins and by Participating in ceRNA Networks
Our results further indicate that, within the WGCNA modules correlated to blast resistance, RLKs and resistance proteins are markedly enriched. For instance, in the dark green module, LncRNA.13491.1 interacts with OsRLCK366 (a receptor-like kinase), OsRGA5-L1 (a resistance protein), and OsPP2C19 (a signaling regulator). This observation is consistent with findings in mulberry [67], where the lncRNA MuLRR-RLK-AS negatively modulates RLK expression to influence disease resistance.
Moreover, our ceRNA network showed that several lncRNAs—LncRNA.9497.1, LncRNA.9562.1, LncRNA.13491.1, and LncRNA.33800.3—competitively bind specific miRNAs (osa-miR395a, osa-miR2864.1, osa-miR5830), thereby influencing the expression of OsSultr2, OsWRKY70, and OsGH3-2, genes affecting sulfur metabolism, JA signaling, and auxin homeostasis, respectively. These discoveries validate the endogenous target mimic (eTM) hypothesis by Franco-Zorrilla et al. (2007) [68]. Similarly, studies in tomato [69] and sweet sorghum [70] suggest that lncRNAs strengthen resistance by regulating transcription factors and stress response genes via ceRNA-based interactions.
Furthermore, subcellular localization predictions (Table S15) provide further insights into how these lncRNAs may exert their regulatory functions. For instance, LncRNA.9562.1 exhibits high-confidence localization in extracellular, mitochondrial, and nuclear compartments, suggesting multifaceted roles in perceiving pathogen signals or orchestrating nuclear transcriptional events. Conversely, LncRNA.9497.1 is predicted to reside largely in the extracellular region, supporting a putative role in intercellular communication or apoplastic signaling. Such compartment-specific predictions enrich our understanding of how each lncRNA may spatially coordinate defense responses under M. oryzae infection.
In summary, lncRNAs in rice not only directly modulate RLKs and R genes/proteins but also indirectly shape hormone signaling pathways through ceRNA networks, forming an extensive regulatory web to determine the eventual disease resistance phenotype.
4. Materials and Methods
4.1. LncRNA Strand-Specific Library Data Sources
A total of 513 translatome (datasets were collected from various tissues of japonica rice under different experimental conditions [71,72,73,74,75,76], along with 40 strand-specific RNA-seq datasets derived from leaves of multiple japonica rice varieties [77,78] (Table S1, Table S3). These datasets served two major purposes: (i) constructing the rice coding gene dataset for improved annotation and subsequent lncRNA identification, and (ii) conducting differential expression analyses as well as coexpression network inference. Specifically, all 40 strand-specific datasets were used for lncRNA identification and WGCNA, whereas 16 of these datasets were devoted to differential expression analysis. We focused on the monogenic line IR25, harboring the blast resistance gene Pikm, and the susceptible line LTH (Lijiangxintuanhe) [79]. Among the 513 translatome datasets, various experimental replicates spanned stress conditions such as drought, temperature fluctuation, salinity, submergence, and heavy metals, in addition to multiple tissues (roots, stems, leaves, buds, panicles, and flowers). Detailed information on library construction is provided in Supplementary Materials 2.
4.2. LncRNA Identification and Classification
For the 40 strand-specific RNA-seq datasets, we first used FastQC (v0.11.9) to check read quality [80]. Strand specificity was determined via Rseqc (v5.0.1) [81], with any nonstrand-specific libraries excluded (Table S3). Adapter sequences and low-quality reads were removed using TrimGalore (v0.6.7) [82] (quality cutoff q30) and Fastp (v0.20.1) [83]. Bowtie2 (v2.4.2) was then employed to discard ribosomal RNA (rRNA) reads [84], after which HISAT2 (v2.2.1) [85] aligned the cleaned reads to the MSU v7 reference genome with strand-specific parameters (--rna-strandness FR or RF) [24]. Transcriptome assembly was carried out using Stringtie (v2.1.5) under the --rf or --fr options, and all resulting GTF files were merged using Stringtie merge. Transcripts below 200 nt in length or FPKM < 0.5 were discarded [86].
To remove protein-coding transcripts, we used GFFcompare (v0.9.8) to compare the assembled annotation against our integrated rice coding gene fusion annotation (hereafter referred to as the CodingRNA dataset) [87]. Candidate lncRNAs were those with class codes i, x, o, u, and p. Additional filtering steps were taken by comparing these candidates to the protein families database (Pfam) (v37.0) [88] and NCBI nonredundant protein database (NR) databases (e-value <1e-5) to eliminate any potential protein-coding transcripts [89], as well as against the RNA families database (Rfam) (v14.10) database (e-value <1e-5) to remove known small RNAs [90]. Next, three coding potential prediction tools—Coding Potential Calculator 2 (CPC2) (v1.0.1) [91], Predictor of Long non-coding RNAs and mEssenger RNAs based on an improved K-mer scheme (PLEK) (v1.2) [92], and Coding-Non-Coding Index (CNCI) (v2) [93]—were simultaneously applied, and only transcripts consistently predicted to be noncoding by all three tools were retained.
To further classify the final lncRNAs, we compared them once more with MSU v7 via GFFcompare. Class i was defined as an intronic lncRNA, class o as a sense lncRNA, class x as an antisense lncRNA, and classes p and u as general lncRNAs (Figure S2B). Additionally, bedtools closest (v2.30.0) identified bidirectional lncRNAs, while the rest were assigned as intergenic [94].
4.3. Identification of cis- and trans-Targets and Known lncRNAs
We extracted both differentially expressed genes (DEGs) and differentially expressed lncRNAs (DELs) from the expression analyses and used Pearson correlation analysis (the cor function in R) to evaluate DEL–DEG pairs with an absolute correlation >0.5 and p <0.05. For trans-target identification, we employed RIblast (interaction energy < –14 kcal/mol, interaction length ≥ 15 bp), alongside the same correlation threshold, to confirm putative lncRNA–gene associations [95].
Considering the median size of rice topologically associated domains (TADs) is ~35 kb [96], a ±20 kb window was chosen for cis-target searches. Any genes located within 20 kb upstream or downstream of the lncRNA, exhibiting |r| >0.5, were deemed cis-targets. We then downloaded six rice lncRNA databases—PlantNATdb (v1.4) [97], PNRD (v1.0) [98], RNAcentral (v22) [99], NONCODE (v6.0) [100], CANTATAdb (v2.0) [101], and GreeNC (v2.0) [102]—merging them via CD-hit (v4.8.1) [103]. Finally, we utilized Blastn (v2.9.0) [104] (E-value<1e-5, identity>95%) to compare our candidate lncRNAs with these known datasets, thus identifying novel lncRNAs versus those present in public repositories.
4.4. Differential Expression Analysis and Functional Enrichment
We performed differential expression analysis using the DESeq2 (v1.22.1) [105] package in R. Prior to analysis, batch effects were removed, and each group of lncRNAs and mRNAs was analyzed separately. Thresholds of |log2FC| ≥1.5 (for lncRNAs) or |log2FC| ≥2 (for mRNAs) and adjusted p ≤0.05 were used to designate significant differential expression. Principal component analysis (PCA) was then conducted via prcomp package, with visualization through ggplot2 (v3.5.1) [106] and pheatmap (v1.0.12).
Functional enrichment (Gene Ontology, GO; Kyoto Encyclopedia of Genes and Genomes, KEGG; Gene Set Enrichment Analysis, GSEA) was carried out using clusterProfiler (v4.10.1)[107], referencing the org.Osativa.eg.db (v0.01) rice database [108]. We applied a significance cutoff of p-value < 0.05 for all enrichment analyses.
4.5. Transcription Factor Identification and lncrna Localization
Transcription factors (TFs) are crucial in rice’s response to M. oryzae stress [20]. Hence, PlantTFDB (v5.0) [109] was utilized to predict TFs within the DEGs, the ceRNA network, and the WGCNA modules. LncRNA subcellular localization was assessed via RNALocate (v3.0) [110], providing an initial insight into nuclear- vs. cytoplasmic-located lncRNAs.
4.6. Weighted Gene Coexpression Network Analysis (WGCNA)
Weighted gene coexpression network analysis (WGCNA) was undertaken to explore interaction relationships among lncRNAs and mRNAs [111], excluding those already used for differential expression analysis. Following expression normalization (via DESeq2) and batch effect removal, low-expression data and outliers were discarded, retaining genes with higher variance (top 75% by median absolute deviation). A soft threshold power of 18 was used, constructing an adjacency matrix via the adjacency function. A topological overlap measure (TOM) was then calculated to define the similarity matrix of lncRNA–mRNA expression.
Hierarchical clustering was performed, and modules were defined or merged using the dynamic tree cut method (deepSplit=2, minModuleSize=30, mergeCutHeight=0.25). Each module’s eigengene was correlated with specific phenotypes (e.g., rice blast resistance vs. susceptibility), generating correlation matrices. Modules showing an absolute correlation coefficient |r|≥0.8 and p<0.05 were considered significantly associated with the trait. The core hub genes or lncRNAs within those modules were visualized using Cytoscape (v3.10.2) [112].
4.7. Single Key lncRNA Analysis
A normalized, batch-corrected expression dataset of both lncRNAs and mRNAs was used for Spearman correlation analyses. For each chosen lncRNA, all rice genes were sorted by the absolute value of their correlation (descending order), creating an ordered gene list.
Next, gene set enrichment analysis (GSEA) was performed via clusterProfiler (v4.14.4) on this ranked list to determine the biological processes or functional categories most associated with the lncRNA in question [107]. The ggplot, ridgeplot, and gseaplot functions in ggplot2 were utilized to visualize results from GSEA (v1.68.0) [113].
4.8. Competing Endogenous RNA (ceRNA) Network Construction
A total of 713 rice miRNAs were sourced from miRBase, forming the foundation for our ceRNA prediction library. psRNATarget (v2) [114], with parameter settings adapted from Zhang et al. [115], was used to predict lncRNA–miRNA and mRNA–miRNA interactions. The integrated mRNA–miRNA–lncRNA coexpression network was finally visualized in Cytoscape (v3.10.2).
4.9. qRT-PCR Method and lncRNA Cloning
To validate the reliability of the sequencing results, we randomly selected three long noncoding RNAs (lncRNAs), four key lncRNAs, and ten of their target genes for quantitative real-time polymerase chain reaction (qRT-PCR) analysis. Primers for qRT-PCR were designed using Primer3Plus software [116] and verified for specificity using PrimerBlast [117]. All primers were synthesized by Genscript Biotech (primer sequences are provided in Table S16).
qRT-PCR experiments were performed using the Applied Biosystems 7500 Real-Time PCR System, with three biological replicates for each sample. The reaction program was as follows: pre-denaturation at 98°C for 2 minutes, followed by 40 cycles of denaturation at 98°C for 2 seconds, annealing and extension at 59°C for 10 seconds. A melt curve analysis was performed after each run to confirm the specificity of the amplification products. The 18S rRNA gene was used as the reference gene. The relative expression levels of the target genes were calculated using the 2^(-ΔΔCt) method.
Specific primers (Table S16) for amplifying the full-length sequences of target lncRNAs were designed based on the RNA-seq data. Total RNA was reverse-transcribed into cDNA using SuperScript III Reverse Transcriptase (Invitrogen). PCR amplification was performed using PrimeSTAR GXL DNA Polymerase (TaKaRa), with the following reaction conditions: 98°C for 5 minutes; (98°C for 10 seconds, 60°C for 15 seconds, 72°C for 1 minute per kb) × 30 cycles; and 72°C for 10 minutes.
PCR products were separated by agarose gel electrophoresis, purified, ligated into the pNC-Cam1304-35S vector, and then transformed into DH5α competent cells. Positive clones were confirmed by Sanger sequencing (Tsingke). Sequencing results were aligned with the reference genome to verify the accuracy of the cloned sequences.
5. Conclusions
This study provides significant advancements in understanding the roles of lncRNAs in rice’s defense against M. oryzae. By creating an optimized lncRNA identification pipeline—incorporating translatome data and existing annotations—we identified 9,003 high-confidence rice lncRNAs with improved accuracy. In-depth analyses of differential expression, WGCNA, and ceRNA network construction revealed their critical involvement in multiple hormones signaling pathways (JA, SA, ET, IAA), as well as in regulating receptor-like kinases and resistance proteins. Key lncRNAs (such as LncRNA.9497.1 and LncRNA.9562.1) emerged as central regulators enhancing rice adaptability and immune response.
These findings offer a comprehensive framework for delineating lncRNA functions in plant immunity and propose molecular targets for breeding resistant rice varieties. Future efforts should emphasize the functional validation of top candidate lncRNAs through overexpression or knockout lines, exploring their applicability across diverse genotypes and M. oryzae strains.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Utilization of the CodingRNA dataset improves the accuracy of lncRNA identification. Figure S2. The impact of the CodingRNA dataset on lncRNA identification and classification code annotation categories. Figure S3 Distribution and comparison of lncRNAs across rice varieties and reference databases. Figure S4 Functional analysis of DE target genes predicted for co-expressed DELs in IR25 and LTH in response to rice blast stress. Figure S5 Venn diagram of 52 commonly responding lncRNAs in NPB and LTH/IR25 under rice blast stress. Figure S6 Correlation and expression analysis of key WGCNA modules. Figure S7. Structural analysis of key lncRNAs in rice. Figure S8 GSEA KEGG enrichment pathway of lncRNA.9562.1-related genes in plant hormone signal transduction (osa04075). Figure S9 Full-length amplification and sequencing results of lncRNA.9562.1 cloning. Table S1: Translatome Data Information. Table S2: Comparison of Translatome Assembly Data and the Merged Dataset with MSU v7 Rice Annotation. Table S3: Validation Information for Strand-Specific RNA-seq Data. Table S4: Statistical Summary of Strand-Specific Transcriptome Sequencing Data. Table S5: Complete GTF Annotation Information of Long Non-Coding RNAs (lncRNAs). Table S6: Classification of Long Non-Coding RNAs (lncRNAs). Table S7 Distinguishing False Positive lncRNAs from Novel Protein-Coding Genes in the Telomere-to-Telomere (T2T) Genome Assembly. Table S8 Comparative Statistics of lncRNA and Gene Features. Table S9 Statistics of Differential Expression Analysis for lncRNAs and mRNAs. Table S10 Statistics of Cis-Target Genes for Differentially Expressed lncRNAs Across Groups. Table S11 Statistics of Trans-Target Genes for Differentially Expressed lncRNAs Across Groups. Table S12 Expression Statistics of Filtered and Normalized Genes and lncRNAs for WGCNA Analysis. Table S13 Functional Annotations of Genes in Grey60, Darkgreen, and Lightyellow Modules Identified by WGCNA. Table S14 Transcriptome-Predicted Expression Results of Experimentally Validated lncRNAs and mRNAs. Table S15 Multiple subcellular localization prediction and scoring for four key lncRNAs. Table S16 Primer Information for qRT-PCR Validation Experiments.
Author Contributions
X.S., L.P., S.X. and H.Z. wrote the manuscript and interpreted the data. X.S. and S.X. contributed to the analysis and interpretation of data. X.S. and H.Z. designed the work, H.Z. L.P. and A.B. revised the manuscript. X.S., T.C. and S.X. conducted the experiments. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (grant number 32372556) and the Yangtze River Delta Science and Technology Innovation Consortium Key Research Project (grant number 24CSJ140200) to HZ.
Data Availability Statement
The data sets are included within the article and its Additional files. The raw sequencing data have been deposited in the NCBI Sequence Read Archive under accession number PRJNA545418 and PRJNA1058262, and in the BIG Data Center at the Beijing Institute of Genomics under accession number CRA003133. In addition, the RiceLncRNA pipeline developed for this study is publicly available on GitHub (https://github.com/njausxl/RiceLncRNA). All other data and material analyzed in the current study are included in the manuscript and the Supplementary Information.
Acknowledgments
We thank Jing Fan for providing the LTH and IR25 rice seeds. We also appreciate the valuable discussions and guidance on sequencing data analysis from Jing Fan and Lianguang Shang. Additionally, we are grateful to Zhilin Yuan and Hengfu Yin for their insightful suggestions and revisions of the manuscript. We are grateful for the support by the high-performance computing platform of Bioinformatics Center, Nanjing Agricultural University.
Conflicts of Interest
The authors declare that they have no known competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.
References
- Mattick, J.S.; Rinn, J.L. Discovery and Annotation of Long Noncoding RNAs. Nat Struct Mol Biol 2015, 22, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Koch, L. Screening for lncRNA Function. Nat Rev Genet 2017, 18, 70–70. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I. Evolution to the Rescue: Using Comparative Genomics to Understand Long Non-Coding RNAs. Nat Rev Genet 2016, 17, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Ivanov, M.; Dittrich, A.N.; Nelson, A.D.; Marquardt, S. LncRNA FLAIL Affects Alternative Splicing and Represses Flowering in Arabidopsis. The EMBO Journal 2023, 42, e110921. [Google Scholar] [CrossRef]
- X, Z.; J, L.; B, L.; H, G.; Y, L.; Y, Q. Global Identification of Arabidopsis lncRNAs Reveals the Regulation of MAF4 by a Natural Antisense RNA. Nature communications 2018, 9. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, X.; Sun, F.; Hu, J.; Zha, X.; Su, W.; Yang, J. Overexpressing lncRNA LAIR Increases Grain Yield and Regulates Neighbouring Gene Cluster Expression in Rice. Nat Commun 2018, 9, 3516. [Google Scholar] [CrossRef]
- Tian, J.; Zhang, F.; Zhang, G.; Li, X.; Wen, C.; Li, H. A Long Noncoding RNA Functions in Pumpkin Fruit Development through S-Adenosyl-L-Methionine Synthetase. Plant Physiology 2024, 195, 940–957. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, X.; Lin, F.; He, G.; Terzaghi, W.; Zhu, D.; Deng, X.W. Arabidopsis Noncoding RNA Mediates Control of Photomorphogenesis by Red Light. Proceedings of the National Academy of Sciences 2014, 111, 10359–10364. [Google Scholar] [CrossRef]
- Marquardt, S.; Raitskin, O.; Wu, Z.; Liu, F.; Sun, Q.; Dean, C. Functional Consequences of Splicing of the Antisense Transcript COOLAIR on FLC Transcription. Mol Cell 2014, 54, 156–165. [Google Scholar] [CrossRef]
- Seo, J.S.; Sun, H.-X.; Park, B.S.; Huang, C.-H.; Yeh, S.-D.; Jung, C.; Chua, N.-H. ELF18-INDUCED LONG-NONCODING RNA Associates with Mediator to Enhance Expression of Innate Immune Response Genes in Arabidopsis. The Plant Cell 2017, 29, 1024–1038. [Google Scholar] [CrossRef]
- Cui, J.; Luan, Y.; Jiang, N.; Bao, H.; Meng, J. Comparative Transcriptome Analysis between Resistant and Susceptible Tomato Allows the Identification of lncRNA16397 Conferring Resistance to Phytophthora Infestans by Co-Expressing Glutaredoxin. The Plant Journal 2017, 89, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Long Non-Coding RNAs and Their Biological Roles in Plants. Genomics, Proteomics & Bioinformatics 2015, 13, 137–147. [CrossRef]
- Heo, J.B.; Sung, S. Vernalization-Mediated Epigenetic Silencing by a Long Intronic Noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Sung, S. Vernalization-Triggered Intragenic Chromatin Loop Formation by Long Noncoding RNAs. Developmental Cell 2017, 40, 302–312.e4. [Google Scholar] [CrossRef]
- Yu, Y.; Zhou, Y.-F.; Feng, Y.-Z.; He, H.; Lian, J.-P.; Yang, Y.-W.; Lei, M.-Q.; Zhang, Y.-C.; Chen, Y.-Q. Transcriptional Landscape of Pathogen-Responsive lncRNAs in Rice Unveils the Role of ALEX1 in Jasmonate Pathway and Disease Resistance. Plant Biotechnology Journal 2020, 18, 679–690. [Google Scholar] [CrossRef]
- Liu, N.; Xu, Y.; Li, Q.; Cao, Y.; Yang, D.; Liu, S.; Wang, X.; Mi, Y.; Liu, Y.; Ding, C.; et al. A lncRNA Fine-Tunes Salicylic Acid Biosynthesis to Balance Plant Immunity and Growth. Cell Host & Microbe 2022, 30, 1124–1138.e8. [Google Scholar] [CrossRef]
- Jiang, N.; Cui, J.; Shi, Y.; Yang, G.; Zhou, X.; Hou, X.; Meng, J.; Luan, Y. Tomato lncRNA23468 Functions as a Competing Endogenous RNA to Modulate NBS-LRR Genes by Decoying miR482b in the Tomato-Phytophthora Infestans Interaction. Hortic Res 2019, 6. [Google Scholar] [CrossRef]
- Wilson, R.A.; Talbot, N.J. Under Pressure: Investigating the Biology of Plant Infection by Magnaporthe Oryzae. Nat Rev Microbiol 2009, 7, 185–195. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Mitchell, T.; Hu, Y.; Liu, X.; Dai, L.; Wang, G.-L. Recent Progress and Understanding of the Molecular Mechanisms of the Rice–Magnaporthe Oryzae Interaction. Molecular Plant Pathology 2010, 11, 419–427. [Google Scholar] [CrossRef]
- Sakulkoo, W.; Osés-Ruiz, M.; Oliveira Garcia, E.; Soanes, D.M.; Littlejohn, G.R.; Hacker, C.; Correia, A.; Valent, B.; Talbot, N.J. A Single Fungal MAP Kinase Controls Plant Cell-to-Cell Invasion by the Rice Blast Fungus. Science 2018, 359, 1399–1403. [Google Scholar] [CrossRef]
- Li, W.; Chern, M.; Yin, J.; Wang, J.; Chen, X. Recent Advances in Broad-Spectrum Resistance to the Rice Blast Disease. Current Opinion in Plant Biology 2019, 50, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, Z.; Chern, M.; Yin, J.; Yang, C.; Ran, L.; Cheng, M.; He, M.; Wang, K.; Wang, J.; et al. A Natural Allele of a Transcription Factor in Rice Confers Broad-Spectrum Blast Resistance. Cell 2017, 170, 114–126.e15. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; He, W.; Wang, T.; Yang, Y.; Xu, Q.; Zhao, X.; Yang, L.; Zhang, H.; Li, X.; Lv, Y.; et al. A Complete Assembly of the Rice Nipponbare Reference Genome. Molecular Plant 2023, 16, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza Sativa Nipponbare Reference Genome Using next Generation Sequence and Optical Map Data. Rice 2013, 6, 4. [Google Scholar] [CrossRef]
- Xiong, Q.; Ma, B.; Lu, X.; Huang, Y.-H.; He, S.-J.; Yang, C.; Yin, C.; Zhao, H.; Zhou, Y.; Zhang, W.-K.; et al. Ethylene-Inhibited Jasmonic Acid Biosynthesis Promotes Mesocotyl/Coleoptile Elongation of Etiolated Rice Seedlings[OPEN]. Plant Cell 2017, 29, 1053–1072. [Google Scholar] [CrossRef]
- White, M.; Carbonare, L.D.; Puerta, M.L.; Iacopino, S. ; Martin; Edwards; Dunne, K.; Pires, E.; Levy, C.; McDonough, M.; et al. Structures of the Arabidopsis Thaliana Oxygen-Sensing Plant Cysteine Oxidases PCO4 and PCO5 Enable Targeted Manipulation of Their Activity. 2020.
- Chini, A.; Boter, M.; Solano, R. Plant Oxylipins: COI1/JAZs/MYC2 as the Core Jasmonic Acid-signalling Module. The FEBS Journal 2009, 276. [Google Scholar] [CrossRef]
- Bao, D.; Chang, S.; Li, X.; Qi, Y. Advances in the Study of Auxin Early Response Genes: Aux/IAA, GH3, and SAUR. The Crop Journal 2024, 12, 964–978. [Google Scholar] [CrossRef]
- Yang, Z.; Hui, S.; Lv, Y.; Zhang, M.; Chen, D.; Tian, J.; Zhang, H.; Liu, H.; Cao, J.; Xie, W.; et al. miR395-Regulated Sulfate Metabolism Exploits Pathogen Sensitivity to Sulfate to Boost Immunity in Rice. Molecular Plant 2022, 15, 671–688. [Google Scholar] [CrossRef]
- Zhu, G.; Ye, N.; Zhang, J. Glucose-Induced Delay of Seed Germination in Rice Is Mediated by the Suppression of ABA Catabolism Rather Than an Enhancement of ABA Biosynthesis. Plant and Cell Physiology 2009, 50, 644–651. [Google Scholar] [CrossRef]
- Zhai, K.; Liang, D.; Li, H.; Jiao, F.; Yan, B.; Liu, J.; Lei, Z.; Huang, L.; Gong, X.; Wang, X.; et al. NLRs Guard Metabolism to Coordinate Pattern- and Effector-Triggered Immunity. Nature 2022, 601, 245–251. [Google Scholar] [CrossRef]
- Li, R.; Zhang, J.; Li, J.; Zhou, G.; Wang, Q.; Bian, W.; Erb, M.; Lou, Y. Prioritizing Plant Defence over Growth through WRKY Regulation Facilitates Infestation by Non-Target Herbivores. eLife 2015, 4, e04805. [Google Scholar] [CrossRef]
- Otomo, K.; Kenmoku, H.; Oikawa, H.; König, W.A.; Toshima, H.; Mitsuhashi, W.; Yamane, H.; Sassa, T.; Toyomasu, T. Biological Functions of Ent - and Syn -copalyl Diphosphate Synthases in Rice: Key Enzymes for the Branch Point of Gibberellin and Phytoalexin Biosynthesis. The Plant Journal 2004, 39, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Yu, H.; Li, X.; Xiao, J.; Wang, S. Rice GH3 Gene Family: Regulators of Growth and Development. Plant Signaling & Behavior 2011, 6, 570–574. [Google Scholar] [CrossRef]
- Miyamoto, K.; Shimizu, T.; Lin, F.; Sainsbury, F.; Thuenemann, E.; Lomonossoff, G.; Nojiri, H.; Yamane, H.; Okada, K. Identification of an E-Box Motif Responsible for the Expression of Jasmonic Acid-Induced Chitinase Gene OsChia4a in Rice. J Plant Physiol 2012, 169, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Luo, Y.; Zhang, L.; Yao, Y.; Han, L.; Chen, Z.; Wang, L.; Li, Y. OsABA8ox2, an ABA Catabolic Gene, Suppresses Root Elongation of Rice Seedlings and Contributes to Drought Response. The Crop Journal 2020, 8, 480–491. [Google Scholar] [CrossRef]
- Seo, J.; Joo, J.; Kim, M.; Kim, Y.; Nahm, B.H.; Song, S.I.; Cheong, J.; Lee, J.S.; Kim, J.; Choi, Y.D. OsbHLH148, a Basic Helix-loop-helix Protein, Interacts with OsJAZ Proteins in a Jasmonate Signaling Pathway Leading to Drought Tolerance in Rice. The Plant Journal 2011, 65, 907–921. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, R.; Huang, K.; Huang, S.; Wang, H.; Wei, Z.; Li, Z.; Bian, M.; Jiang, W.; Wu, T.; et al. The OsWRKY63 – OsWRKY76 – OsDREB1B Module Regulates Chilling Tolerance in Rice. The Plant Journal 2022, 112, 383–398. [Google Scholar] [CrossRef]
- Saika, H.; Okamoto, M.; Miyoshi, K.; Kushiro, T.; Shinoda, S.; Jikumaru, Y.; Fujimoto, M.; Arikawa, T.; Takahashi, H.; Ando, M.; et al. Ethylene Promotes Submergence-Induced Expression of OsABA8ox1, a Gene That Encodes ABA 8’-Hydroxylase in Rice. Plant Cell Physiol 2007, 48, 287–298. [Google Scholar] [CrossRef]
- Li, H.; Zhou, S.-Y.; Zhao, W.-S.; Su, S.-C.; Peng, Y.-L. A Novel Wall-Associated Receptor-like Protein Kinase Gene, OsWAK1, Plays Important Roles in Rice Blast Disease Resistance. Plant Mol Biol 2009, 69, 337–346. [Google Scholar] [CrossRef]
- Okuyama, Y.; Kanzaki, H.; Abe, A.; Yoshida, K.; Tamiru, M.; Saitoh, H.; Fujibe, T.; Matsumura, H.; Shenton, M.; Galam, D.C.; et al. A Multifaceted Genomics Approach Allows the Isolation of the Rice Pia -blast Resistance Gene Consisting of Two Adjacent NBS-LRR Protein Genes. The Plant Journal 2011, 66, 467–479. [Google Scholar] [CrossRef]
- Yang, W.; Zhao, J.; Zhang, S.; Chen, L.; Yang, T.; Dong, J.; Fu, H.; Ma, Y.; Zhou, L.; Wang, J.; et al. Genome-Wide Association Mapping and Gene Expression Analysis Reveal the Negative Role of OsMYB21 in Regulating Bacterial Blight Resistance in Rice. Rice 2021, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Rohila, J.S.; Yang, Y. Rice Mitogen-Activated Protein Kinase Gene Family and Its Role in Biotic and Abiotic Stress Response. Journal of Integrative Plant Biology 2007, 49, 751–759. [Google Scholar] [CrossRef]
- Cesari, S.; Thilliez, G.; Ribot, C.; Chalvon, V.; Michel, C.; Jauneau, A.; Rivas, S.; Alaux, L.; Kanzaki, H.; Okuyama, Y.; et al. The Rice Resistance Protein Pair RGA4/RGA5 Recognizes the Magnaporthe Oryzae Effectors AVR-Pia and AVR1-CO39 by Direct Binding[W][OA]. Plant Cell 2013, 25, 1463–1481. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Wang, H.; Nie, L.; Tan, D.; Zhou, C.; Zhang, Q.; Li, Y.; Du, B.; Guo, J.; Huang, J.; et al. Bph30 Confers Resistance to Brown Planthopper by Fortifying Sclerenchyma in Rice Leaf Sheaths. Molecular Plant 2021, 14, 1714–1732. [Google Scholar] [CrossRef]
- Wu, F.; Sheng, P.; Tan, J.; Chen, X.; Lu, G.; Ma, W.; Heng, Y.; Lin, Q.; Zhu, S.; Wang, J.; et al. Plasma Membrane Receptor-like Kinase Leaf Panicle 2 Acts Downstream of the DROUGHT AND SALT TOLERANCE Transcription Factor to Regulate Drought Sensitivity in Rice. Journal of Experimental Botany 2015, 66, 271–281. [Google Scholar] [CrossRef]
- Xiao, G.; Wang, W.; Liu, M.; Li, Y.; Liu, J.; Franceschetti, M.; Yi, Z.; Zhu, X.; Zhang, Z.; Lu, G.; et al. The Piks Allele of the NLR Immune Receptor Pik Breaks the Recognition of AvrPik Effectors of the Rice Blast Fungus. Journal of integrative plant biology 2022. [Google Scholar] [CrossRef]
- Hayashi, N.; Inoue, H.; Kato, T.; Funao, T.; Shirota, M.; Shimizu, T.; Kanamori, H.; Yamane, H.; Hayano-Saito, Y.; Matsumoto, T.; et al. Durable Panicle Blast-Resistance Gene Pb1 Encodes an Atypical CC-NBS-LRR Protein and Was Generated by Acquiring a Promoter through Local Genome Duplication. The Plant Journal 2010, 64, 498–510. [Google Scholar] [CrossRef]
- Kim, M.-S.; Kang, K.-K.; Cho, Y.-G. Molecular and Functional Analysis of U-Box E3 Ubiquitin Ligase Gene Family in Rice (Oryza Sativa). International Journal of Molecular Sciences 2021, 22, 12088. [Google Scholar] [CrossRef]
- Li, L.; Xu, X.; Chen, C.; Shen, Z. Genome-Wide Characterization and Expression Analysis of the Germin-Like Protein Family in Rice and Arabidopsis. International Journal of Molecular Sciences 2016, 17. [Google Scholar] [CrossRef]
- Durrani, I.S.; Jan, A.; Shah, S.; Iqbal, A.; Ahmad, D.; Khan, H.; Naqvi, S.M.S. Bioinformatics Studies of OSGLP8-12 Gene from Oryza Sativa (Japonica) Reveal Its Role in Conferring Resistance against Disease and Stresses. Pakistan Journal of Botany 2020. [Google Scholar] [CrossRef]
- Ji, Z.; Ji, C.; Liu, B.; Zou, L.; Chen, G.; Yang, B. Interfering TAL Effectors of Xanthomonas Oryzae Neutralize R-Gene-Mediated Plant Disease Resistance. Nat Commun 2016, 7, 13435. [Google Scholar] [CrossRef] [PubMed]
- Boyes, D.; Nam, J.; Dangl, J. The Arabidopsis Thaliana RPM1 Disease Resistance Gene Product Is a Peripheral Plasma Membrane Protein That Is Degraded Coincident with the Hypersensitive Response. Proceedings of the National Academy of Sciences of the United States of America 1998, 95 26, 15849–15854. [Google Scholar] [CrossRef]
- Zhan, P.; Ma, S.; Xiao, Z.; Li, F.; Wei, X.; Lin, S.; Wang, X.; Ji, Z.; Fu, Y.; Pan, J.; et al. Natural Variations in Grain Length 10 (GL10) Regulate Rice Grain Size. Journal of Genetics and Genomics 2022, 49, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Tamiru, M.; Abe, A.; Utsushi, H.; Yoshida, K.; Takagi, H.; Fujisaki, K.; Undan, J.; Rakshit, S.; Takaichi, S.; Jikumaru, Y.; et al. The Tillering Phenotype of the Rice Plastid Terminal Oxidase (PTOX) Loss-of-Function Mutant Is Associated with Strigolactone Deficiency. The New phytologist 2014, 202 1, 116–131. [Google Scholar] [CrossRef]
- Vij, S.; Giri, J.; Dansana, P.K.; Kapoor, S.; Tyagi, A. The Receptor-like Cytoplasmic Kinase (OsRLCK) Gene Family in Rice: Organization, Phylogenetic Relationship, and Expression during Development and Stress. Molecular plant 2008, 1 5, 732–750. [Google Scholar] [CrossRef]
- Kim, S.; Park, S.-I.; Kwon, H.; Cho, M.; Kim, B.-G.; Chung, J.; Nam, M.; Song, J.S.; Kim, K.-H.; Yoon, I. The Rice Abscisic Acid-Responsive RING Finger E3 Ligase OsRF1 Targets OsPP2C09 for Degradation and Confers Drought and Salinity Tolerance in Rice. Frontiers in Plant Science 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Gong, Y.; Chong, K.; Xu, Y. Phosphatase OsPP2C27 Directly Dephosphorylates OsMAPK3 and OsbHLH002 to Negatively Regulate Cold Tolerance in Rice. Plant, cell & environment, 2020. [Google Scholar] [CrossRef]
- Loutre, C.; Wicker, T.; Travella, S.; Galli, P.; Scofield, S.; Fahima, T.; Feuillet, C.; Keller, B. Two Different CC-NBS-LRR Genes Are Required for Lr10-Mediated Leaf Rust Resistance in Tetraploid and Hexaploid Wheat. The Plant journal : for cell and molecular biology, 2009; 60 6, 1043–1054. [Google Scholar] [CrossRef]
- Luo, H.; Song, F.; Goodman, R.M.; Zheng, Z. Up-Regulation of OsBIHD1, a Rice Gene Encoding BELL Homeodomain Transcriptional Factor, in Disease Resistance Responses. Plant biology 2005, 7 5, 459–468. [Google Scholar] [CrossRef]
- Wang, A.; Shu, X.; Jing, X.; Jiao, C.; Chen, L.; Zhang, J.; Ma, L.; Jiang, Y.; Yamamoto, N.; Li, S.; et al. Identification of Rice ( Oryza Sativa L.) Genes Involved in Sheath Blight Resistance via a Genome-wide Association Study. Plant Biotechnology Journal 2021, 19, 1553–1566. [Google Scholar] [CrossRef]
- Wang, L.-L.; Jin, J.-J.; Li, L.-H.; Qu, S.-H. Long Non-Coding RNAs Responsive to Blast Fungus Infection in Rice. Rice 2020, 13, 77. [Google Scholar] [CrossRef]
- Jain, P.; Sharma, V.; Dubey, H.; Singh, P.K.; Kapoor, R.; Kumari, M.; Singh, J.; Pawar, D.V.; Bisht, D.; Solanke, A.U.; et al. Identification of Long Non-Coding RNA in Rice Lines Resistant to Rice Blast Pathogen Maganaporthe Oryzae. Bioinformation 2017, 13, 249–255. [Google Scholar] [CrossRef]
- Metraux, J. Recent Breakthroughs in the Study of Salicylic Acid Biosynthesis. Trends in plant science 2002, 7 8, 332–334. [Google Scholar] [CrossRef]
- Zhao, Y. Auxin Biosynthesis: A Simple Two-Step Pathway Converts Tryptophan to Indole-3-Acetic Acid in Plants. Molecular plant 2012, 5 2, 334–338. [Google Scholar] [CrossRef]
- Huot, B.; Yao, J.; Montgomery, B.L.; He, S.Y. Growth-Defense Tradeoffs in Plants: A Balancing Act to Optimize Fitness. Mol Plant 2014, 7, 1267–1287. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, C.; Zhao, T.; Yang, L.; Shang, Q.; Wang, G.; Liu, Z.; Gai, Y.; Ji, X. Integrated Analysis of lncRNAs and mRNAs Reveals Complex Gene Network Mediated by lncRNAs and Regulatory Function of MuLRR-RLK-AS in Response to Phytoplasma Infection in Mulberry. Biomolecules 2024, 14, 308. [Google Scholar] [CrossRef] [PubMed]
- Franco-Zorrilla, J.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; García, J.; Paz-Ares, J. Target Mimicry Provides a New Mechanism for Regulation of microRNA Activity. Nature Genetics 2007, 39, 1033–1037. [Google Scholar] [CrossRef]
- Cui, J.; Jiang, N.; Hou, X.; Wu, S.; Zhang, Q.; Meng, J.; Luan, Y. Genome-Wide Identification of lncRNAs and Analysis of ceRNA Networks During Tomato Resistance to Phytophthora Infestans. Phytopathology 2020, 110, 456–464. [Google Scholar] [CrossRef]
- Sun, X.; Zheng, H.; Li, J.; Sui, N. Comparative Transcriptome Analysis of Sweet Sorghum Provides Insights into New lncRNAs Acting as ceRNAs during Salt Responses. 2019. [Google Scholar] [CrossRef]
- Kajala, K.; Gouran, M.; Shaar-Moshe, L.; Mason, G.A.; Rodriguez-Medina, J.; Kawa, D.; Pauluzzi, G.; Reynoso, M.; Canto-Pastor, A.; Manzano, C.; et al. Innovation, Conservation, and Repurposing of Gene Function in Root Cell Type Development. Cell 2021, 184, 3333–3348.e19. [Google Scholar] [CrossRef]
- Zhu, W.; Xu, J.; Chen, S.; Chen, J.; Liang, Y.; Zhang, C.; Li, Q.; Lai, J.; Li, L. Large-Scale Translatome Profiling Annotates the Functional Genome and Reveals the Key Role of Genic 3’ Untranslated Regions in Translatomic Variation in Plants. Plant Commun 2021, 2, 100181. [Google Scholar] [CrossRef]
- Reynoso, M.A.; Borowsky, A.T.; Pauluzzi, G.C.; Yeung, E.; Zhang, J.; Formentin, E.; Velasco, J.; Cabanlit, S.; Duvenjian, C.; Prior, M.J.; et al. Gene Regulatory Networks Shape Developmental Plasticity of Root Cell Types under Water Extremes in Rice. Developmental Cell 2022, 57, 1177–1192.e6. [Google Scholar] [CrossRef]
- Yang, X.; Song, B.; Cui, J.; Wang, L.; Wang, S.; Luo, L.; Gao, L.; Mo, B.; Yu, Y.; Liu, L. Comparative Ribosome Profiling Reveals Distinct Translational Landscapes of Salt-Sensitive and -Tolerant Rice. BMC Genomics 2021, 22, 612. [Google Scholar] [CrossRef]
- Reynoso, M.A.; Kajala, K.; Bajic, M.; West, D.A.; Pauluzzi, G.; Yao, A.I.; Hatch, K.; Zumstein, K.; Woodhouse, M.; Rodriguez-Medina, J.; et al. Evolutionary Flexibility in Flooding Response Circuitry in Angiosperms. Science 2019, 365, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, Q.; Chen, Z.; Yue, Y.; Liu, Y.; Zhao, Y.; Zhou, D.-X. Histone Deacetylases Control Lysine Acetylation of Ribosomal Proteins in Rice. Nucleic Acids Res 2021, 49, 4613–4628. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-L.; Jin, J.-J.; Li, L.-H.; Qu, S.-H. Long Non-Coding RNAs Responsive to Blast Fungus Infection in Rice. Rice (N Y) 2020, 13, 77. [Google Scholar] [CrossRef]
- Fan, J.; Quan, W.; Li, G.-B.; Hu, X.-H.; Wang, Q.; Wang, H.; Li, X.-P.; Luo, X.; Feng, Q.; Hu, Z.-J.; et al. circRNAs Are Involved in the Rice-Magnaporthe Oryzae Interaction1[OPEN]. Plant Physiol 2020, 182, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Tsunematsu, H.; Yanoria, M.J.T.; Ebron, L.A.; Hayashi, N.; Ando, I.; Kato, H.; Imbe, T.; Khush, G.S. Development of Monogenic Lines of Rice for Blast Resistance. Breeding Science 2000, 50, 229–234. [Google Scholar] [CrossRef]
- Wingett, S.; Andrews, S. FastQ Screen: A Tool for Multi-Genome Mapping and Quality Control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality Control of RNA-Seq Experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat Biotechnol 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat Biotechnol 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Res 2020, 9, ISCB. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded Coverage of Metagenomic, Viral and microRNA Families. Nucleic Acids Res 2021, 49, D192–D200. [Google Scholar] [CrossRef]
- Kang, Y.-J.; Yang, D.-C.; Kong, L.; Hou, M.; Meng, Y.-Q.; Wei, L.; Gao, G. CPC2: A Fast and Accurate Coding Potential Calculator Based on Sequence Intrinsic Features. Nucleic Acids Res 2017, 45, W12–W16. [Google Scholar] [CrossRef]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A Tool for Predicting Long Non-Coding RNAs and Messenger RNAs Based on an Improved k-Mer Scheme. BMC Bioinformatics 2014, 15, 311. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing Sequence Intrinsic Composition to Classify Protein-Coding and Long Non-Coding Transcripts. Nucleic Acids Research 2013, 41, e166. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Fukunaga, T.; Hamada, M. RIblast: An Ultrafast RNA-RNA Interaction Prediction System Based on a Seed-and-Extension Approach. Bioinformatics 2017, 33, 2666–2674. [Google Scholar] [CrossRef] [PubMed]
- Golicz, A.A.; Bhalla, P.L.; Edwards, D.; Singh, M.B. Rice 3D Chromatin Structure Correlates with Sequence Variation and Meiotic Recombination Rate. Commun Biol 2020, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Yuan, C.; Zhang, J.; Zhang, Z.; Bai, L.; Meng, Y.; Chen, L.-L.; Chen, M. PlantNATsDB: A Comprehensive Database of Plant Natural Antisense Transcripts. Nucleic Acids Res 2012, 40, D1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Zhang, Z.; Ling, Y.; Xu, W.; Su, Z. PNRD: A Plant Non-Coding RNA Database. Nucleic Acids Res 2015, 43, D982–989. [Google Scholar] [CrossRef]
- RNAcentral Consortium RNAcentral 2021: Secondary Structure Integration, Improved Sequence Search and New Member Databases. Nucleic Acids Res 2021, 49, D212–D220. [CrossRef]
- Zhao, L.; Wang, J.; Li, Y.; Song, T.; Wu, Y.; Fang, S.; Bu, D.; Li, H.; Sun, L.; Pei, D.; et al. NONCODEV6: An Updated Database Dedicated to Long Non-Coding RNA Annotation in Both Animals and Plants. Nucleic Acids Res 2021, 49, D165–D171. [Google Scholar] [CrossRef]
- Szcześniak, M.W.; Bryzghalov, O.; Ciomborowska-Basheer, J.; Makałowska, I. CANTATAdb 2.0: Expanding the Collection of Plant Long Noncoding RNAs. Methods Mol Biol 2019, 1933, 415–429. [Google Scholar] [CrossRef]
- Di Marsico, M.; Paytuvi Gallart, A.; Sanseverino, W.; Aiese Cigliano, R. GreeNC 2.0: A Comprehensive Database of Plant Long Non-Coding RNAs. Nucleic Acids Res 2022, 50, D1442–D1447. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Research 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2. WIREs Computational Statistics 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation (Camb) 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- xuzhougeng Xuzhougeng/Org. Osativa.Eg.Db v0.01 2019.
- Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Jin, J.; Gao, G. PlantRegMap: Charting Functional Regulatory Maps in Plants. Nucleic Acids Research 2020, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Wu, L.; Wang, L.; Hu, S.; Tang, G.; Chen, J.; Yi, Y.; Xie, H.; Lin, J.; Wang, M.; Wang, D.; et al. RNALocate v3.0: Advancing the Repository of RNA Subcellular Localization with Dynamic Analysis and Prediction. Nucleic Acids Research 2024, gkae872. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proceedings of the National Academy of Sciences 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A Plant Small RNA Target Analysis Server (2017 Release). Nucleic Acids Res 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, J.; Xu, Q.; Dong, J.; Song, L.; Wang, W.; Shen, F. Long Noncoding RNA lncRNA354 Functions as a Competing Endogenous RNA of miR160b to Regulate Genes in Response to Salt Stress in Upland Cotton. Plant, Cell & Environment 2021, 44, 3302–3321. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New Capabilities and Interfaces. Nucleic Acids Res 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A Tool to Design Target-Specific Primers for Polymerase Chain Reaction. BMC Bioinformatics 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Bioinformatics pipeline for lncRNA identification and visualization of disease-resistance-associated lncRNAs in rice. (A) Comprehensive analysis workflow: Sequential steps from left to right, illustrating the construction of the "CodingRNA" dataset, criteria and processes for identifying non-coding RNAs (including lncRNAs), and strategies for screening disease-resistance-associated lncRNAs. (B-D) Venn diagrams: Evaluation of coding potential of transcripts using multiple tools and databases, including Pfam, Rfam, Nr, PLEK, CPC2, and CNCI. (E) Pie chart: Classification of the final identified lncRNAs.
Figure 1.
Bioinformatics pipeline for lncRNA identification and visualization of disease-resistance-associated lncRNAs in rice. (A) Comprehensive analysis workflow: Sequential steps from left to right, illustrating the construction of the "CodingRNA" dataset, criteria and processes for identifying non-coding RNAs (including lncRNAs), and strategies for screening disease-resistance-associated lncRNAs. (B-D) Venn diagrams: Evaluation of coding potential of transcripts using multiple tools and databases, including Pfam, Rfam, Nr, PLEK, CPC2, and CNCI. (E) Pie chart: Classification of the final identified lncRNAs.
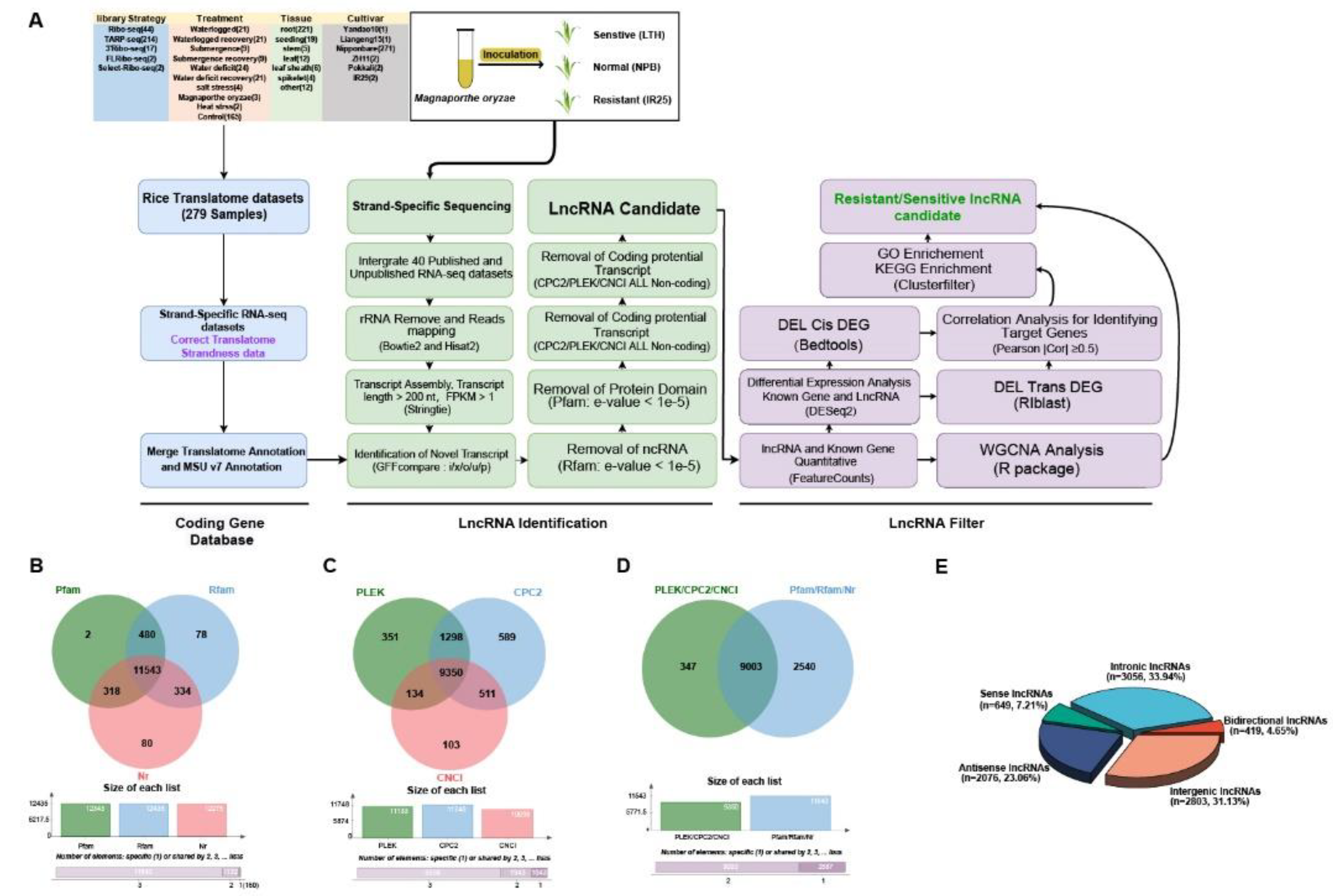
Figure 2.
Comparative analysis of the basic characteristics of lncRNAs and mRNAs. (A) Circos plot of the trans-target genes of four specific lncRNAs. From innermost to outermost: the first ring represents chromosomes; the second ring shows GC content (outer side for lncRNAs, inner side for mRNAs); the third ring displays heatmaps of gene expression levels and line graphs of gene density (outer side for lncRNAs, inner side for mRNAs). Trans-target gene connections are represented by lines. (B) Transcript length distribution. (C) Normalized expression level distribution, displayed as violin plots. (D) Distribution of the number of exons per transcript. (E) Distribution of the number of splice variants per gene locus. Red represents lncRNAs, and blue represents mRNAs.
Figure 2.
Comparative analysis of the basic characteristics of lncRNAs and mRNAs. (A) Circos plot of the trans-target genes of four specific lncRNAs. From innermost to outermost: the first ring represents chromosomes; the second ring shows GC content (outer side for lncRNAs, inner side for mRNAs); the third ring displays heatmaps of gene expression levels and line graphs of gene density (outer side for lncRNAs, inner side for mRNAs). Trans-target gene connections are represented by lines. (B) Transcript length distribution. (C) Normalized expression level distribution, displayed as violin plots. (D) Distribution of the number of exons per transcript. (E) Distribution of the number of splice variants per gene locus. Red represents lncRNAs, and blue represents mRNAs.
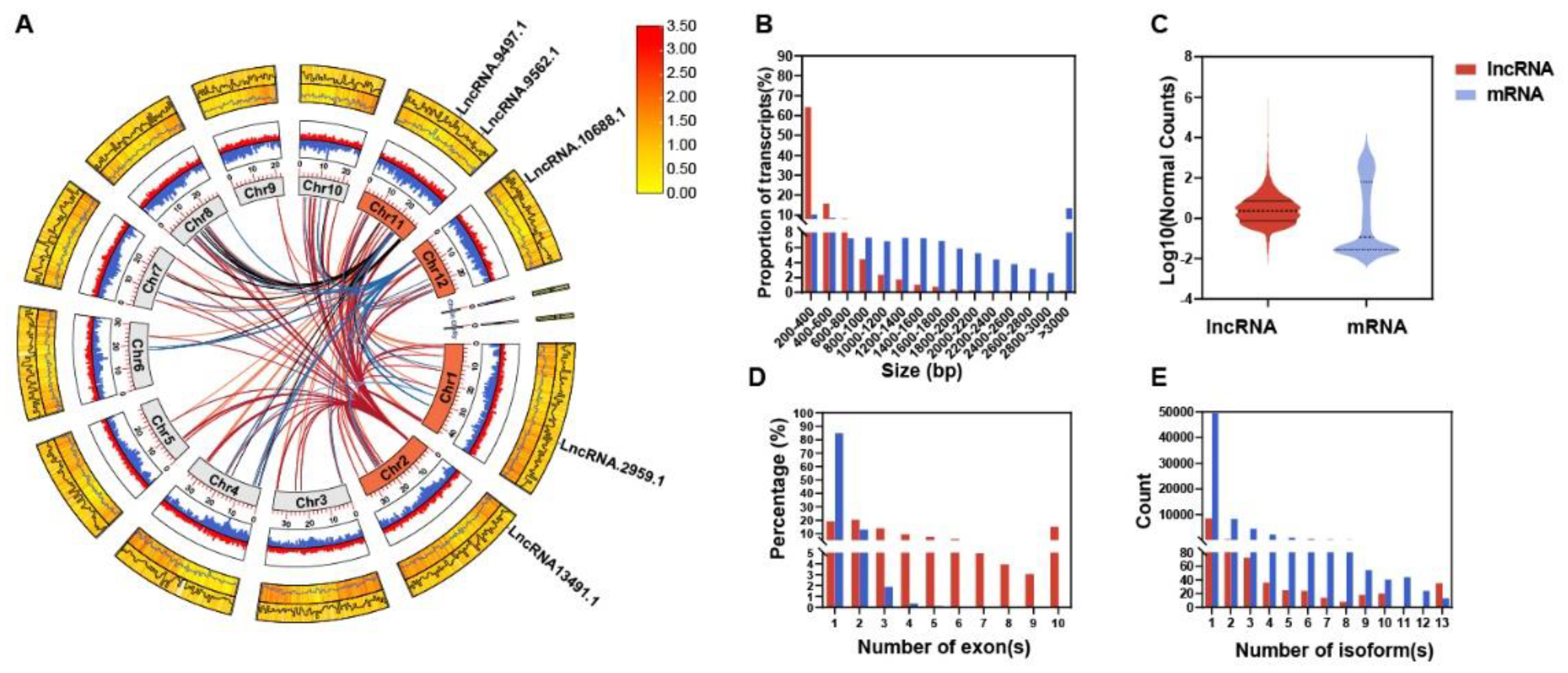
Figure 3.
Functional enrichment analysis of DELs and their target DEGs in IR25 and LTH after rice blast infection. (A) Bar charts showing the number of DEGs and differentially expressed lncRNAs (DELs), with upregulated genes in red and downregulated genes in blue. (B) Venn diagrams of differentially expressed lncRNAs (DELs) between LTH and IR25 before and after inoculation (Fold change > 1.5, p-value < 0.05). (C-E) Functional analysis of target genes predicted for uniquely expressed DELs in IR25 and LTH, including GO enrichment analysis (D, F) and KEGG pathway enrichment analysis. The top 20 significant GO terms were selected based on a p.adjust < 0.05 cutoff, and the top 10 KEGG pathways were selected with the same significance threshold.
Figure 3.
Functional enrichment analysis of DELs and their target DEGs in IR25 and LTH after rice blast infection. (A) Bar charts showing the number of DEGs and differentially expressed lncRNAs (DELs), with upregulated genes in red and downregulated genes in blue. (B) Venn diagrams of differentially expressed lncRNAs (DELs) between LTH and IR25 before and after inoculation (Fold change > 1.5, p-value < 0.05). (C-E) Functional analysis of target genes predicted for uniquely expressed DELs in IR25 and LTH, including GO enrichment analysis (D, F) and KEGG pathway enrichment analysis. The top 20 significant GO terms were selected based on a p.adjust < 0.05 cutoff, and the top 10 KEGG pathways were selected with the same significance threshold.
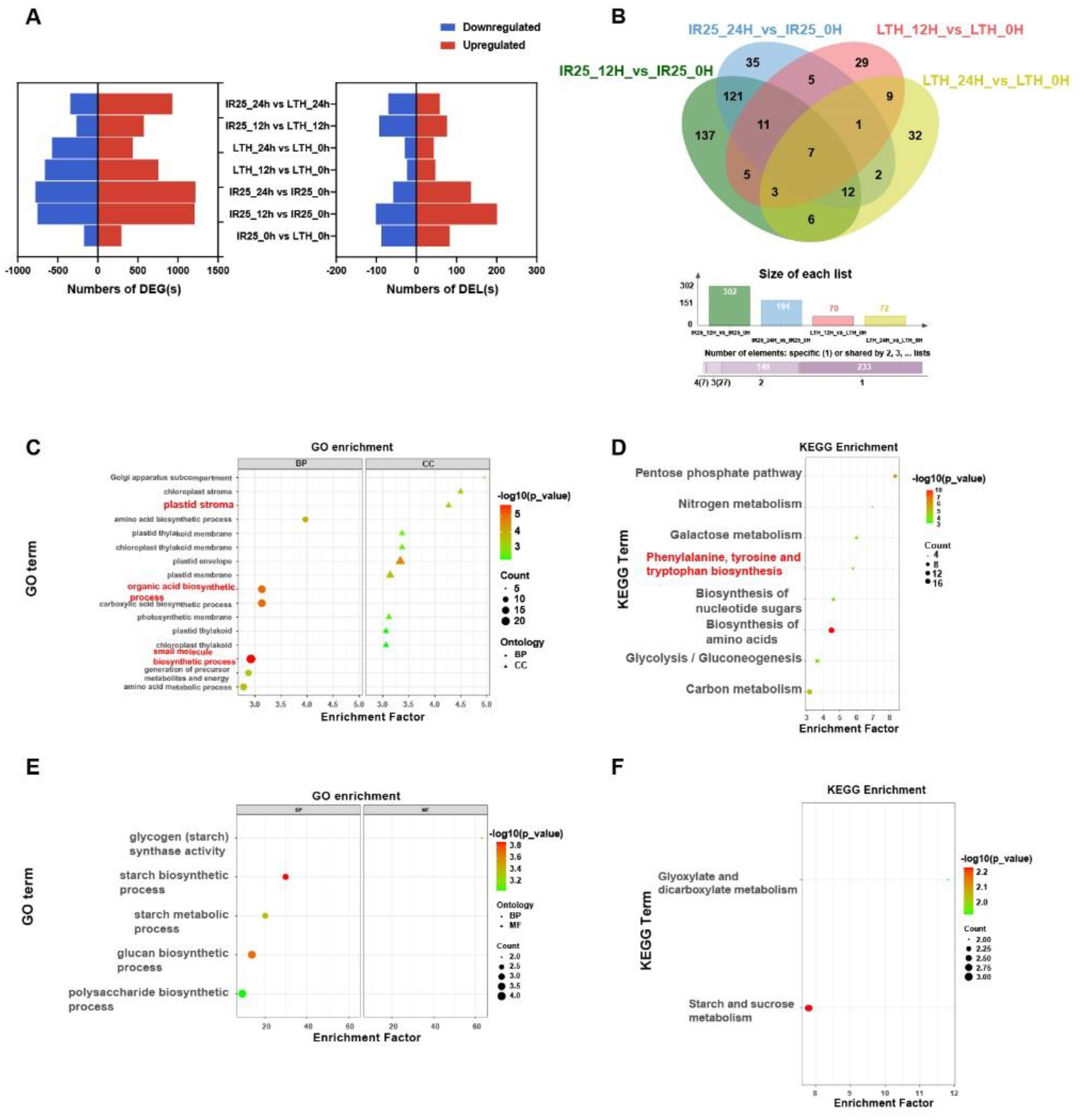
Figure 4.
Co-expression ceRNA network of lncRNAs, miRNAs, and mRNAs. Inverted triangles represent miRNAs (purple), triangles represent lncRNAs (red), ovals represent mRNAs, and rectangles represent plant hormones, disease resistance-related genes, and transcription factors (orange). Solid lines indicate connections between miRNAs and mRNAs, while dashed lines represent interactions between miRNAs and lncRNAs. The line color corresponds to the expectation values predicted by psRNAtarget.
Figure 4.
Co-expression ceRNA network of lncRNAs, miRNAs, and mRNAs. Inverted triangles represent miRNAs (purple), triangles represent lncRNAs (red), ovals represent mRNAs, and rectangles represent plant hormones, disease resistance-related genes, and transcription factors (orange). Solid lines indicate connections between miRNAs and mRNAs, while dashed lines represent interactions between miRNAs and lncRNAs. The line color corresponds to the expectation values predicted by psRNAtarget.
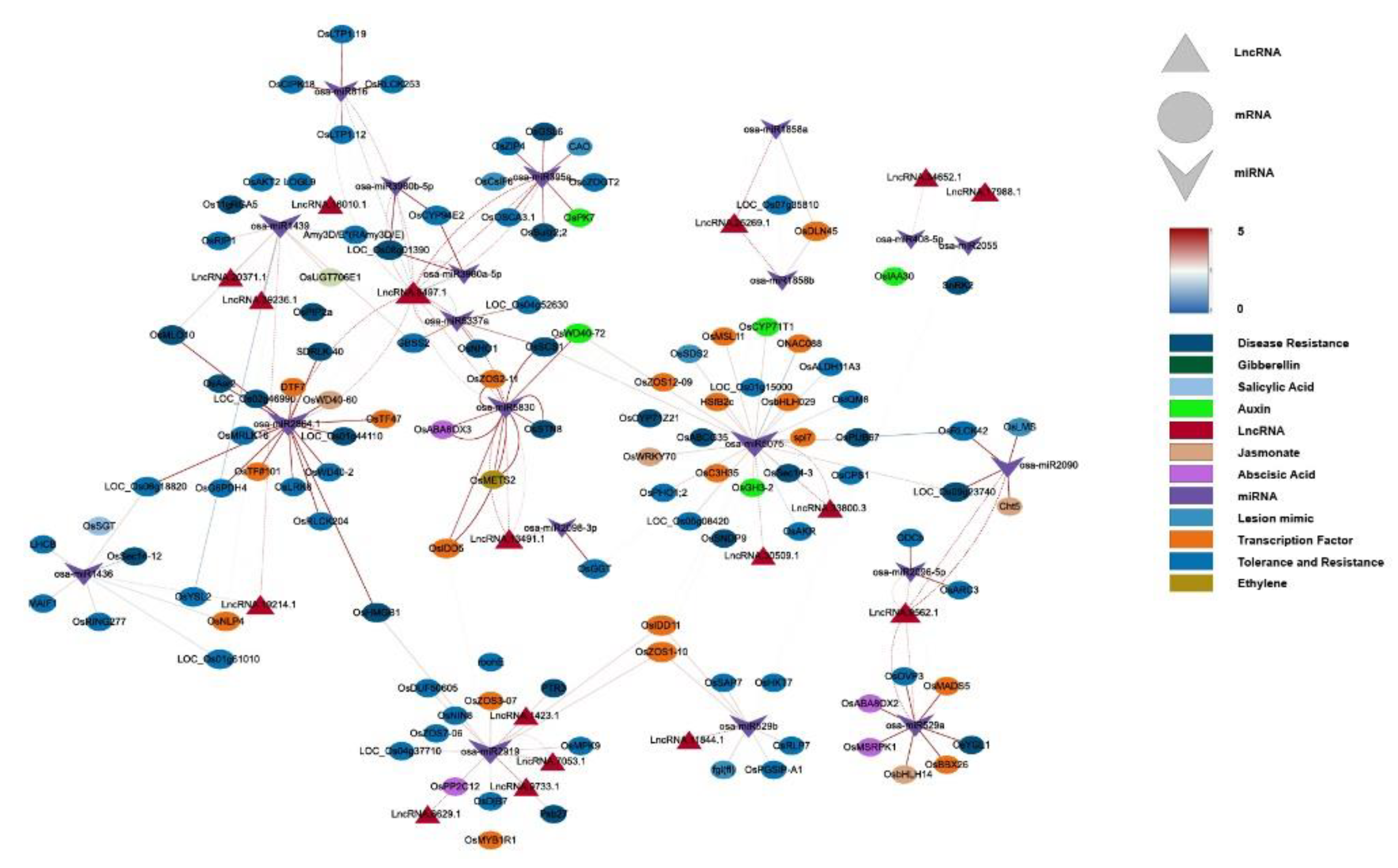
Figure 5.
WGCNA of genes and lncRNAs in rice after M. oryzae infection. (A-B) Soft threshold selection for network construction. The optimal soft threshold was chosen based on scale-free topology fit (A) and mean connectivity (B). (C) Hierarchical clustering tree of transcripts in different modules. The dendrogram shows the clustering of transcripts into modules, with each module labeled by a different color. (D) Module-trait relationship. The heatmap shows the correlation between module eigengenes and traits. The correlation coefficients and p-values indicate the strength and significance of the relationship. (E) Scatter plots of transcript significance (TS) versus module membership (MM) for the salt-associated module MEgreys60.
Figure 5.
WGCNA of genes and lncRNAs in rice after M. oryzae infection. (A-B) Soft threshold selection for network construction. The optimal soft threshold was chosen based on scale-free topology fit (A) and mean connectivity (B). (C) Hierarchical clustering tree of transcripts in different modules. The dendrogram shows the clustering of transcripts into modules, with each module labeled by a different color. (D) Module-trait relationship. The heatmap shows the correlation between module eigengenes and traits. The correlation coefficients and p-values indicate the strength and significance of the relationship. (E) Scatter plots of transcript significance (TS) versus module membership (MM) for the salt-associated module MEgreys60.
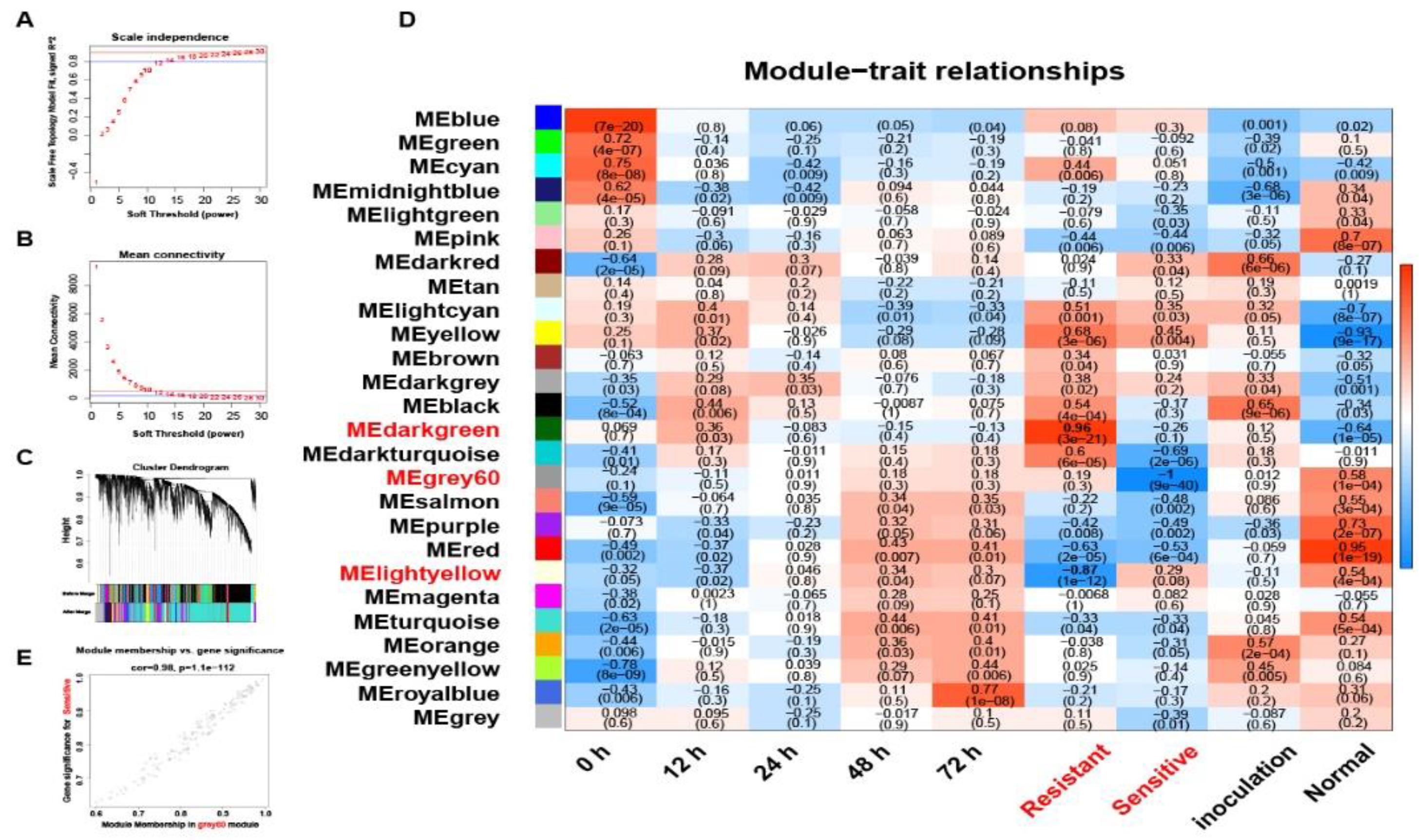
Figure 6.
WGCNA network plot showing the correlation between the three modules strongly associated with susceptible and resistant phenotypes. (A) Darkgreen module is significantly positively correlated with resistance traits (r = 0.96, p = 3 × 10^-21).(B) Grey60 module is significantly negatively correlated with susceptibility traits (r = -1.0, p = 9 × 10^-40).(C) Lightyellow module is significantly negatively correlated with resistance traits (r = -0.87, p = 1 × 10^-12). The annotations for these related genes are provided in Table S14.
Figure 6.
WGCNA network plot showing the correlation between the three modules strongly associated with susceptible and resistant phenotypes. (A) Darkgreen module is significantly positively correlated with resistance traits (r = 0.96, p = 3 × 10^-21).(B) Grey60 module is significantly negatively correlated with susceptibility traits (r = -1.0, p = 9 × 10^-40).(C) Lightyellow module is significantly negatively correlated with resistance traits (r = -0.87, p = 1 × 10^-12). The annotations for these related genes are provided in Table S14.
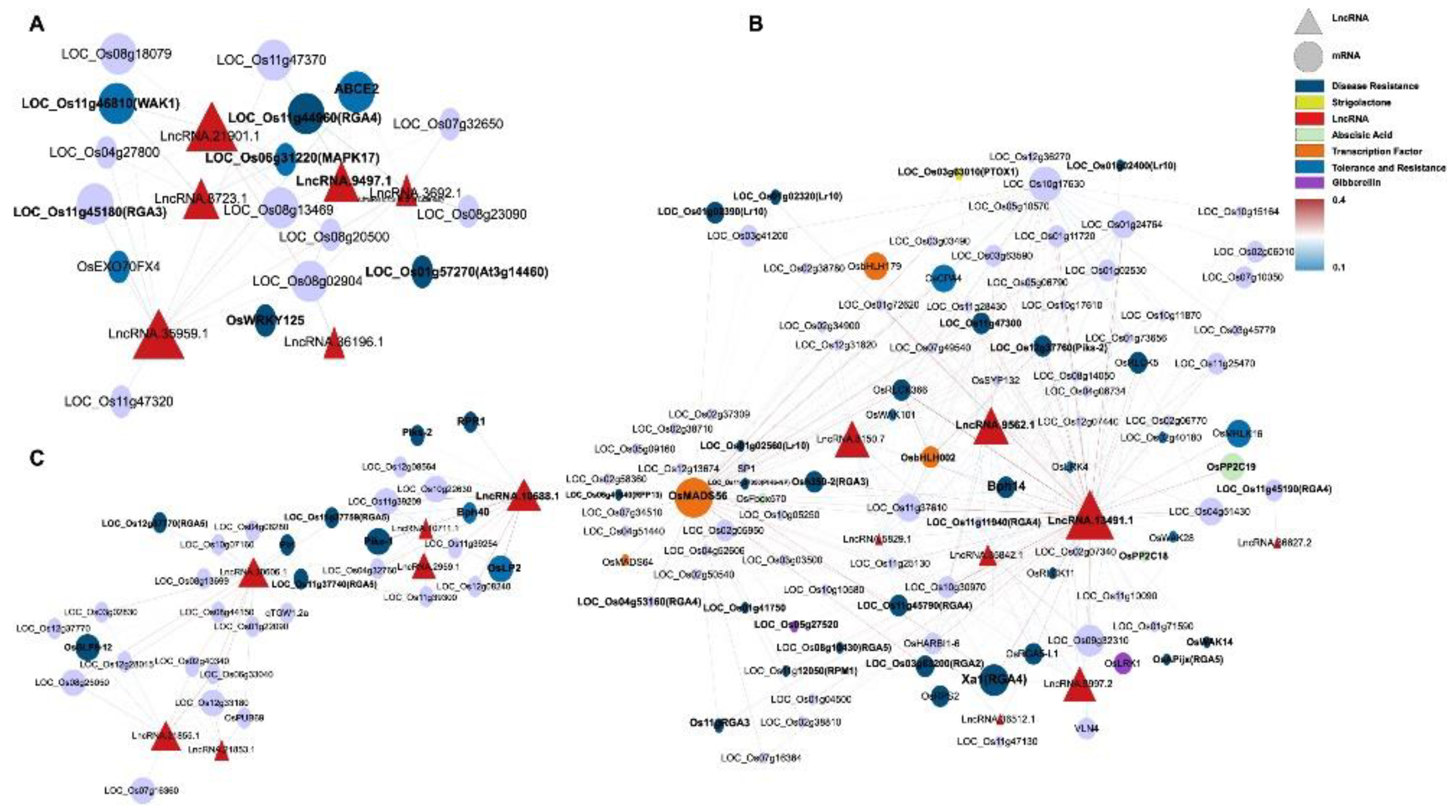
Figure 7.
GSEA enrichment analysis of gene sets based on four lncRNAs in GO and KEGG pathways. (A) lncRNA.9497.1, (B) lncRNA.9562.1, (C) lncRNA.10688.1, and (D) lncRNA.13491.1: Enrichment analysis of the gene sets in GO and KEGG pathways for each of the four lncRNAs. The x-axis represents the enrichment score (ES), while the color gradient indicates the adjusted p-value (p.adjust), reflecting the significance level of the enrichment.
Figure 7.
GSEA enrichment analysis of gene sets based on four lncRNAs in GO and KEGG pathways. (A) lncRNA.9497.1, (B) lncRNA.9562.1, (C) lncRNA.10688.1, and (D) lncRNA.13491.1: Enrichment analysis of the gene sets in GO and KEGG pathways for each of the four lncRNAs. The x-axis represents the enrichment score (ES), while the color gradient indicates the adjusted p-value (p.adjust), reflecting the significance level of the enrichment.
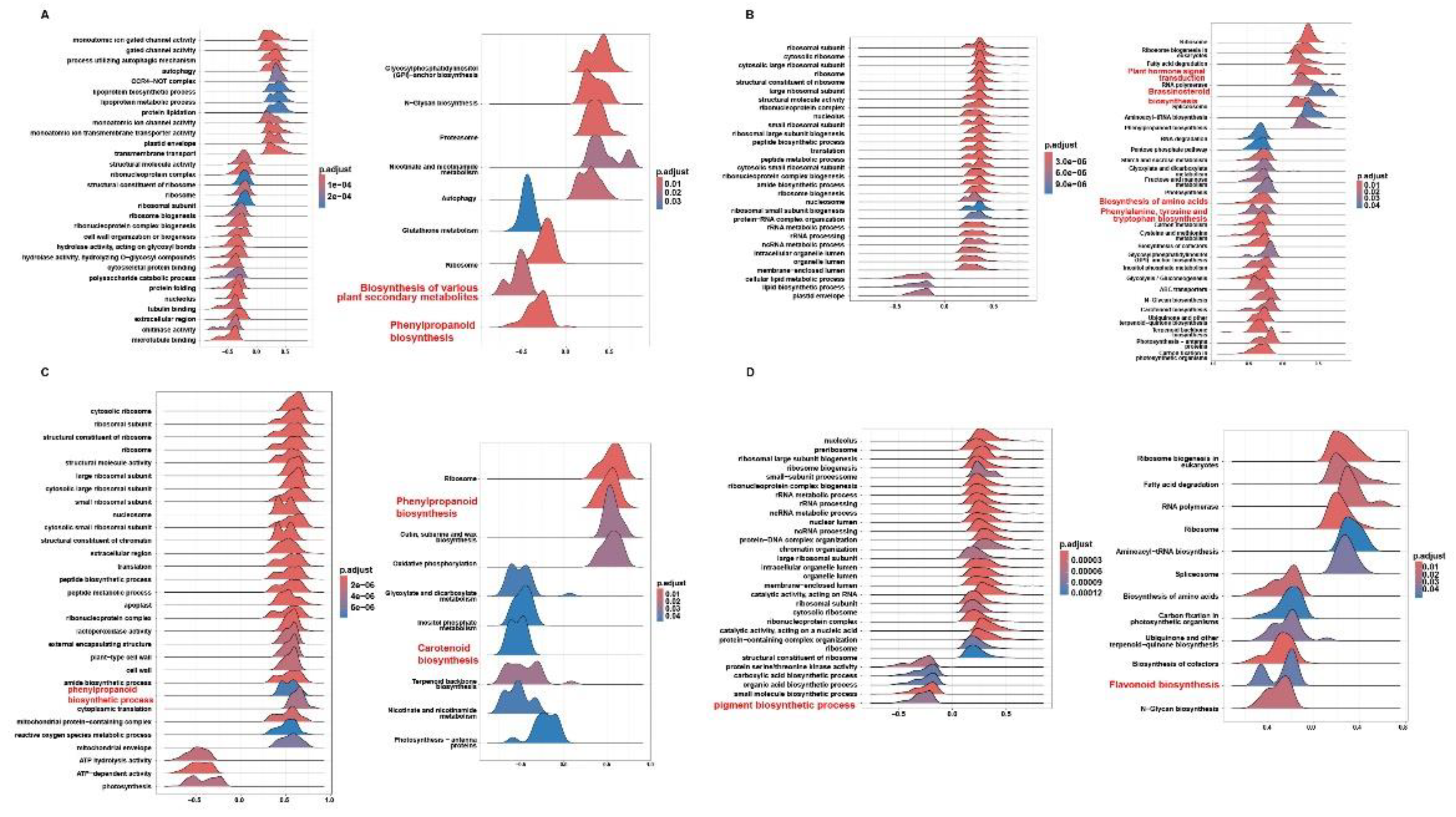
Figure 8.
Verification of RNA-seq data by qRT-PCR for DELs and their target genes responsive to M. oryzae. The yellow line with solid circles represents RNA-seq results, and the blue line with solid squares represents qRT-PCR results. Error bars indicate the standard error of three replicates. (First and third rows): Expression levels of lncRNAs at different time points.(Second and fourth rows): Corresponding target gene expression levels at the same time points.
Figure 8.
Verification of RNA-seq data by qRT-PCR for DELs and their target genes responsive to M. oryzae. The yellow line with solid circles represents RNA-seq results, and the blue line with solid squares represents qRT-PCR results. Error bars indicate the standard error of three replicates. (First and third rows): Expression levels of lncRNAs at different time points.(Second and fourth rows): Corresponding target gene expression levels at the same time points.
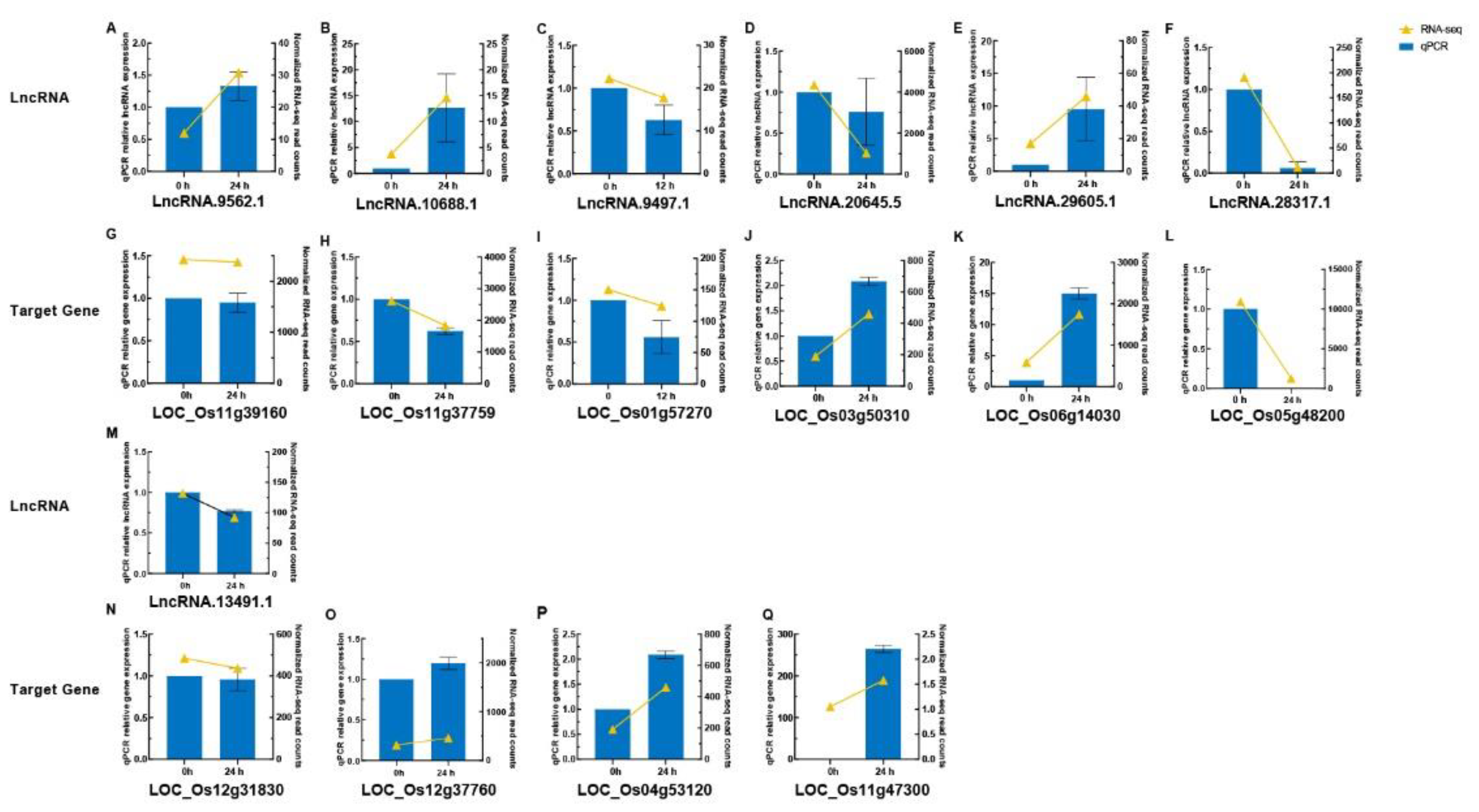
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



