
Submitted:
19 February 2025
Posted:
19 February 2025
You are already at the latest version
Abstract
Mitochondrial dynamics, governed by fusion and fission, are crucial for maintaining cellular homeostasis, energy production, and stress adaptation. MFN2 and OPA1, key regulators of mitochondrial fusion, play essential roles beyond their structural functions, influencing bioenergetics, intracellular signalling, and quality control mechanisms such as mitophagy. Disruptions in these processes, often caused by MFN2 or OPA1 mutations, are linked to neurodegenerative diseases like Charcot-Marie-Tooth disease type 2A (CMT2A) and Autosomal Dominant Optic Atrophy (ADOA). This review explores the molecular mechanisms underlying mitochondrial fusion, the impact of MFN2 and OPA1 dysfunction on oxidative phosphorylation and autophagy, and their role in disease progression. Additionally, we discuss the divergent cellular responses to MFN2 and OPA1 mutations, particularly in terms of proliferation, senescence, and metabolic signalling. Finally, we highlight emerging therapeutic strategies to restore mitochondrial integrity, including mTOR modulation and autophagy-targeted approaches, with potential implications for neurodegenerative and metabolic disorders.
Keywords:
mitochondria
; mitochondrial dynamics
; neurodegenerative disases
; autophagy
; mitophagy
; oxidative phosphorilation
; proliferation
; senescence
; mTOR signalling
1. Introduction
Mitochondria are dynamic organelles constantly undergoing fusion and fission to maintain cellular balance and adapt to metabolic needs. These processes, known as mitochondrial dynamics, are essential for preserving the mitochondrial structure, optimizing energy production, and supporting intracellular signalling. By responding to environmental and physiological signals, mitochondria adjust their shape to help cells cope with stress and energy fluctuations [1,2]. Fusion is mediated by GTPase proteins, including mitofusins (MFN1 and MFN2) in the outer mitochondrial membrane (OMM) and optic atrophy 1 (OPA1) in the inner mitochondrial membrane (IMM) [3,4]. These proteins maintain mitochondrial elongation, preserve membrane potential, and regulate oxidative phosphorylation (OxPhos) and cellular quality control mechanisms like mitophagy. These processes are particularly crucial in high-energy tissues such as the brain, where neuronal health and function heavily depend on mitochondrial dynamics [5,6]. Conversely, fission is primarily driven by dynamin-related protein 1 (Drp1) [7,8]. Beyond their well-characterized roles in mitochondrial fusion, MFN2 and OPA1 influence broader cellular processes [9,10,11]. Disruptions in mitochondrial dynamics, often caused by mutations in MFN2 (OMIM #608507) or OPA1 (OMIM #605290), are associated with several neurodegenerative diseases and hereditary neuropathies, such as Charcot-Marie-Tooth disease type 2A (CMT2A, OMIM #609260) and Autosomal Dominant Optic Atrophy (ADOA, OMIM #165500). This review explores the structural and functional complexities of MFN2 and OPA1, focusing on their roles in mitochondrial quality control and bioenergetics and their involvement in cellular differentiation and metabolic signalling. By highlighting emerging therapeutic strategies targeting these proteins, we aim to enlighten their potential to restore mitochondrial integrity and cellular homeostasis in the context of neurodegenerative diseases.
2. Structural insights into MFN2 and OPA1
MFN2 and OPA1 possess sophisticated structures and undergo precise regulation, enabling their diverse functions.
MFN2: Structure and Function. MFN2 is a nuclear-encoded protein localized to the outer mitochondrial membrane (OMM) [12]. The MFN2 gene (OMIM *603560) resides on chromosome 1 at position 36.22, encompassing 19 exons and producing two transcript variants. These variants encode two protein isoforms: Isoform 1 (757aa; 86,402Da) and Isoform 2 (436aa; 50,041Da). MFN2 contains several conserved functional domains arranged sequentially from the N-terminal to the C-terminal: a GTPase domain, two coiled-coil domains (HR1 and HR2), a proline-rich domain, and two transmembrane domains [13]. The GTPase domain is critical for mediating the fusion process, with motifs G1 to G5 forming the catalytic centre and coordinating GTP binding and hydrolysis [14,15]. The HR2 domain enables tethering of adjacent mitochondria through an antiparallel coiled-coil dimeric structure, forming both homotypic (MFN1–MFN1 or MFN2–MFN2) and heterotypic dimers (MFN1–MFN2) [16,17]. Additionally, MFN2 possesses a unique N-terminal P21 Ras-binding domain, distinguishing it functionally from its homolog MFN1 [18]. While MFN1 is predominantly expressed in the heart and testis, MFN2 is abundant in the brain, underscoring its specialized role in neural tissue [19].
OPA1: Isoforms and Functional Domains. OPA1, a nuclear-encoded protein located in the inner mitochondrial membrane (IMM), is encoded by the OPA1 gene (OMIM #605290) on chromosome 3q29. This gene spans over 60 kb and produces eight isoforms through alternative splicing, primarily involving exons 4, 4b, and 5b [20]. Key isoforms include Isoform 1: 960aa (111,631Da); Isoform 2: 997aa (115,884Da); Isoform 3: 961aa (111,822Da). OPA1 contains several functional regions, including an N-terminal mitochondrial targeting sequence (MTS), a GTPase domain, two coiled-coil domains, and a GTPase effector domain (GED). These regions are critical for anchoring OPA1 to the IMM, mediating protein-protein interactions, and enabling GTP-dependent fusion activity [21]. After mitochondrial import, OPA1 undergoes proteolytic cleavage at specific sites: under stress conditions, OMA1 cleaves at site S1 (corresponding to exon 5). YME1L cleaves at site S2 (corresponding to exon 5b) during normal conditions [22]. This processing generates two OPA1 forms: long OPA1 (l-OPA1) and short OPA1 (s-OPA1). While l-OPA1 anchors to the IMM, s-OPA1 resides in the intermembrane space (IMS). Both forms are essential for mitochondrial fusion, with l-OPA1 facilitating homotypic interactions and s-OPA1 enhancing the fusion process [23].
3. MFN2 and OPA1 in the Mitochondrial Fusion Process
Mitochondrial fusion involves several key proteins and interactions across the outer and inner mitochondrial membranes (OMM and IMM) [6,24]. The fusion of the OMM is initiated by the mitofusins, MFN1 and MFN2, which tether two mitochondria through interactions between their HR2 domains. This tethering is followed by GTP hydrolysis-induced conformational changes, driving membrane fusion. While MFN1 is primarily responsible for completing OMM fusion, MFN2 plays a supportive role, including mediating OMM fusion and regulating mitochondria–endoplasmic reticulum (mito-ER) contact sites, also known as the ER mitochondria-associated membranes (MAMs). Several processes known to be regulated or directly occurring at MAM have been reported to be influenced by the presence of MFN2 (see paragraphs 4 and 6). Recent structural studies of MFN1 and MFN2 have provided mechanistic insights into their roles in mitochondrial fusion [25,26]. However, the physiological relevance remains under investigation due to the absence of the transmembrane domain in these structures and uncertainties surrounding their topology [27]. Once OMM fusion is complete, IMM undergoes fusion, a process mediated by OPA1. OPA1 exists in two forms: long OPA1 (L-OPA1) and short OPA1 (S-OPA1), generated through proteolytic cleavage. L-OPA1 facilitates homotypic interactions between membranes but requires S-OPA1 to complete the fusion process. Studies have demonstrated that L-OPA1 interacts with cardiolipin, a mitochondrial-specific phospholipid, to form trans complexes capable of driving membrane fusion. S-OPA1 enhances this process, indicating that both isoforms are essential for IMM fusion [23,28]. The coordination of OMM and IMM fusion remains an unresolved aspect of mitochondrial dynamics. One model suggests that MFN1-mediated GTP hydrolysis stimulates the oligomerization of OMM fusion pores, subsequently facilitating IMM fusion. OPA1 may then form right-turned helical assemblies, potentially activated by MFN1, to drive IMM fusion [29]. This coordinated interaction ensures that mitochondrial fusion occurs seamlessly, maintaining organelle integrity and function. Mitochondrial fusion is crucial in maintaining mitochondrial quality by selectively preventing the reintegration of defective mitochondria with lower membrane potential [30]. Additionally, it facilitates the exchange of mitochondrial DNA (mtDNA) and proteins, diluting damage and supporting mitochondrial health [31]. In the presence of mtDNA mutations, fusion helps mitigate defects in respiratory chain proteins, highlighting its importance in sustaining bioenergetic efficiency [32]. Disruption of this process due to the loss of key fusion regulators, such as MFN1, MFN2, or OPA1, leads to a decrease in mtDNA copy numbers and an increase in mitochondrial dysfunction [33].
4. Mitochondrial Fusion Proteins in the Regulation of Oxidative Phosphorylation
The efficiency of OxPhos—the process of producing ATP via the electron transport chain—is tightly regulated by mitochondrial dynamics. MFN2 and OPA1, key proteins governing mitochondrial fusion and cristae structure—invaginations of the inner mitochondrial membrane where respiratory chain complexes are located—directly influence bioenergetic efficiency and cellular metabolic states. Elongated mitochondria, formed through fusion mediated by MFN2 and OPA1, enhance OxPhos efficiency and ATP production [34,35]. In contrast, fragmented mitochondria often exhibit impaired OxPhos, reduced mitochondrial DNA (mtDNA) content, and elevated reactive oxygen species (ROS) levels, contributing to cellular dysfunction [36,37,38]. Recent research highlights the adaptability of mitochondria under metabolic stress. During nutrient deficiency, mitochondria dynamically segregate into specialized subpopulations: ATP-producing units and biosynthetic hubs. One subpopulation, devoid of cristae but enriched in pyrroline-5-carboxylate synthase (P5CS), supports proline and ornithine biosynthesis, optimizing metabolic flexibility through fusion-fission cycles [39]. This segregation exemplifies how mitochondrial dynamics align with cellular metabolic demands.
MFN2: A Key Regulator of Fusion, Bioenergetics and Beyond. MFN2 is a critical regulator of mitochondrial fusion and bioenergetics, and its role extends beyond mere mitochondrial dynamics. Alterations in mitochondrial fusion, often caused by impaired MFN2-GTPase activity, lead to severe metabolic dysfunctions, including suppressed OxPhos, diminished ATP production, and mtDNA instability. Such dysfunctions are implicated in pathological conditions such as neurodegeneration and metabolic syndromes [37]. As a tethering factor, MFN2 facilitates direct communication between the mitochondria and the ER [40,41]. This interaction is crucial in maintaining cellular bioenergetics by limiting ROS production, alleviating ER stress, and facilitating calcium transfer from the ER to mitochondria. These processes sustain NADH levels, which are critical for OxPhos [42,43]. Furthermore, MFN2-mediated fusion enhances OxPhos during periods of high metabolic demand, such as cell proliferation, though proliferative cells often shift to glycolysis to meet biosynthetic needs [34,44]. Despite the central role of MFN2 in tethering, conflicting findings exist regarding the effects of MFN2 loss on mito-ER contacts. Some studies suggest that the loss of MFN2 reduces these contacts, impairing mitochondrial function and increasing cellular stress [45]. In contrast, other studies suggest that the absence of MFN2 may increase these contacts, complicating the interpretation of its precise role in metabolic regulation [46]. A detailed discussion of this controversy is beyond the scope of this review and can be found in other literature [47,48]. In addition to its functions in mitochondrial dynamics, MFN2 is involved in mitochondrial transport along axons, a process essential for ensuring neurons meet their energetic and metabolic demands [49]. MFN2 facilitates the attachment of mitochondria to microtubules via interactions with motor proteins like Miro and Milton, ensuring efficient mitochondrial distribution [50]. Interestingly, while MFN2 knockout impairs mitochondrial transport along axons, similar defects are not observed in OPA1-deficient neurons, despite both conditions causing comparable mitochondrial fragmentation [10,51]. This distinction highlights MFN2's multifaceted role, extending beyond mitochondrial fusion to influence a broader spectrum of cellular processes that support cellular metabolism and energy production [9].
OPA1: Master Regulator of Cristae Structure and Supercomplexes Assembly. OPA1 plays a central role in maintaining mitochondrial cristae structure organizing respiratory chain complexes (RCCs) into supercomplexes (RCS). These supercomplexes optimize electron transfer and maximize OxPhos' efficiency, highlighting the importance of cristae integrity in bioenergetics [52,53]. Overexpression of OPA1 in mice enhances cristae integrity, improving OxPhos and ATP production, while its knockdown disrupts mitochondrial membrane potential and respiration, underscoring its essential role in mitochondrial function [53]. OPA1 acts as a metabolic sensor, interacting with solute transporters in the inner mitochondrial membrane (IMM) to regulate cristae remodelling in response to cellular energy demands. This remodelling is independent of OPA1’s fusion activity and is crucial for maintaining respiration and cell survival during metabolic stress, such as nutrient deprivation [53]. Studies suggest OPA1 interacts with mitochondrial solute carriers (i.e., SLC25A), sensing substrate availability and adjusting mitochondrial function, accordingly, thus bridging bioenergetics and metabolic signalling [54]. Modulating these proteins could enhance OxPhos efficiency, reduce ROS production, and improve cellular resilience under metabolic stress. Additionally, understanding the role of mitochondrial solute carriers in OPA1-mediated cristae remodelling offers new avenues for designing interventions tailored to specific metabolic conditions.
5. Dynamic Changes in Mitochondrial Structure and Quality Control
Dynamic changes in mitochondrial structure are central to quality control mechanisms that maintain cellular and mitochondrial health [55]. These mechanisms enable the mitochondrial network to adapt to challenges such as protein misfolding, oxidative stress, and organelle damage [56,57]. Mitochondrial quality control encompasses transcriptional responses mediated by factors that shuttle between mitochondria and the nucleus—such as those involved in retrograde signalling and the mitochondrial unfolded protein response (mtUPR)—as well as stress-induced mitochondrial hyperfusion (SIMH), which enhances mitochondrial resilience under acute stress. These mechanisms also involve the degradation of misfolded proteins, regulating mitochondrial proteostasis, and the selective removal of damaged mitochondria through processes like mitophagy and autophagy [58,59].
Mitochondrial Retrograde Signaling. Mitochondria contain multiple copies of a small genome, the mitochondrial DNA (mtDNA), with most of the mitochondrial proteome encoded by nuclear genes regulated by transcription factors (TFs) and their associated cofactors [60,61,62,63]. Thus, maintaining mitochondrial homeostasis critically depends on nuclear transcription and signalling pathways that inform the nucleus of mitochondrial dysfunctions and changes in cellular metabolism [64,65]. This regulation occurs partly through nuclear TFs and cofactors modulated by cytosolic mediators such as ROS and the NAD/NADH ratio [66,67]. When mitochondrial dysfunction occurs, retrograde signalling pathways convey mitochondrial distress to the nucleus, triggering adaptive changes in nuclear gene expression and cellular physiology [68,69].
Stress-Induced Mitochondrial Hyperfusion (SIMH). Acute stressors, such as oxidative damage or starvation, trigger SIMH, a protective response characterized by elongated mitochondrial networks. SIMH enhances bioenergetic efficiency, ATP production, and cellular resilience under stress conditions [70,71]. This response is regulated by the inhibition of Drp1, suppressing mitochondrial fission, and the activation of OPA1 and MFN1, promoting fusion [35,71]. Notably, SIMH prevents mitophagy during nutrient deprivation, preserving mitochondrial function and sustaining energy production [35,72]. Additionally, it enhances calcium buffering, reduces ROS, and supports oxidative metabolism [70]. In contrast, nutrient excess induces mitochondrial fragmentation, shifting metabolism from oxidative phosphorylation to glycolysis, leading to lipid accumulation and metabolic dysfunction [1]. Fragmented mitochondria produce excessive ROS, contributing to oxidative stress and cellular damage [73,74]. One proposed mechanism is that excessive fragmentation disrupts IMM and cristae structure, impairing electron transport chain (ETC) complexes and hindering electron transfer and proton transport [75,76,77]. This electron and proton leakage decreases ATP production while increasing ROS levels, and activates Drp1, further promoting mitochondrial fission and perpetuating oxidative stress in a positive feedback loop [78,79].
Integrated Stress Response (ISR) as a Key Regulator of Stress Adaptation. Mitochondria play a pivotal role in the Integrated Stress Response (ISR), a conserved mechanism that adjusts metabolism and protein synthesis under stress conditions. Triggered by mitochondrial dysfunction, nutrient deprivation, or oxidative stress, the ISR downregulates general protein synthesis while selectively upregulating stress-adaptive genes to conserve energy and enhance cellular resilience [80,81]. For instance, OPA1 deficiency in muscle cells activates the ISR, leading to mitochondrial fragmentation, suppression of mTORC1 signalling, and stimulation of AMP-activated protein kinase (AMPK) activity [72]. This dual regulation reduces anabolic processes like protein synthesis while promoting catabolic pathways such as fatty acid oxidation to restore energy balance [82]. Moreover, genetic inhibition of mitochondrial function or treatment with mitochondria-targeted antioxidants can activate AMPK signalling, triggering mitochondrial fragmentation and inducing catabolic pathways as a survival mechanism [83]. In response to mitochondrial injury, AMPK activation can also increase the expression of fusion proteins such as MFN1 and OPA1, promoting a hyper-tubular mitochondrial state that enhances OxPhos and ATP production under stress conditions [37,84,85]. The role of AMPK in mitochondrial dynamics varies depending on the cellular context, the type of stress, the metabolic state, and specific cellular demands. Notably, chronic ISR activation and impaired mitochondrial fusion are linked to neurodegenerative diseases such as Alzheimer’s and Parkinson’s [86,87,88,89,90]. These mechanisms highlight the intricate relationship between mitochondrial dynamics and stress adaptation, underscoring their critical role in maintaining cellular function and homeostasis.
The mitochondrial unfolded protein response (mtUPR) exemplifies a crucial pathway for maintaining mitochondrial proteostasis [91,92]. Protein homeostasis depends on a balance between unfolded proteins and the folding capacity of cellular compartments. Mitochondria possess their chaperones and proteases for quality control; however, when misfolded proteins exceed this capacity, mitochondrial dysfunction ensues, triggering mtUPR to enhance protein folding and degradation [93,94]. This retrograde signalling activates the transcription of nuclear-encoded mitochondrial chaperones and proteases, sustaining mitochondrial proteostasis. While not strictly a quality control mechanism, retrograde communication mitigates mitochondrial dysfunction, preventing further cellular damage [92,95].
Proteolytic Systems and Mitochondrial Proteostasis. The ubiquitin-proteasome system (UPS) plays a crucial role in maintaining mitochondrial proteostasis by clearing defective proteins and facilitating the replacement of non-functional components [92,96]. In addition to its degradative function, the UPS supports protein transport across the mitochondrial outer membrane by targeting import-deficient polypeptides for ubiquitination, marking them for proteasomal degradation [92,97]. Recently, it has been observed that disruption of mitochondrial protein import triggers a quality control mechanism in which YME1L1, the ATP-dependent protease involved in OPA1 cleavage, degrades unassembled translocase components, preventing the precursor stalling in the translocase of the outer membrane complex. This process is a protective response to maintain mitochondrial quality control and cellular viability. Import plugging leads to a cell growth defect and the loss of YME1L1 yeast exacerbates this inhibition, underscoring the protective role of YME1L1 [98].
6. Autophagy/Mitophagy and Mitochondrial Dynamics
Beyond the coordinated action of chaperones and proteases, mitochondrial quality control relies on autophagy, a fundamental mechanism for degrading dysfunctional organelles and protein aggregates. Autophagy can be non-selective, ensuring survival during nutrient deprivation, or selective, targeting specific cellular components such as mitochondria—a process known as mitophagy [99,100,101,102]. This process begins with phagophore formation, likely originating from the ER, trans-Golgi, or endosomes, which expands to engulf damaged components, forming an autophagosome that subsequently fuses with a lysosome to create an autolysosome, where cargo degradation occurs through hydrolases [103,104]. Mitophagy plays a crucial role in mitochondrial turnover and proteostasis, preventing the accumulation of dysfunctional organelles that could otherwise compromise cellular homeostasis [105,106]. A key regulator of this pathway is the PINK1/Parkin system, where PINK1 accumulates on OMM of damaged mitochondria, recruiting Parkin, an E3 ubiquitin ligase, which ubiquitinates MFNs, leading to mitochondrial fragmentation and clearance [107,108,109,110,111,112,113]. Unfolded mitochondrial proteins can trigger mtUPR, and activate PINK1/Parkin-dependent mitophagy, reinforcing mitochondrial quality control [99]. Additionally, PINK1-phosphorylated MFN2 serves as a Parkin receptor, marking mitochondria for degradation [10], while Parkin-mediated MFN2 inhibition regulates ER-mitochondria crosstalk, influencing autophagic signalling [110,114]. This integrated network of mtUPR, UPS, autophagy, and mitophagy ensures mitochondrial function is preserved, preventing cellular damage and dynamically adapting mitochondrial activity to metabolic demands under physiological and stress conditions [105,106]. Mitochondrial dynamics dictate whether mitochondria undergo fusion, preserving their function, or fragmentation, facilitating mitophagy [115]. During mitophagy, fragmentation exposes “eat-me” signals, promoting autophagosome formation and lysosomal degradation [116]. In contrast, fusion prevents degradation during periods of nutrient scarcity, forming interconnected mitochondrial networks that enhance ATP production [117]. OPA1 is a crucial regulator of this balance, interacting with FUNDC1 and BNIP3 to mediate autophagy and mitophagy [118,119]. Moreover, mitochondrial-derived vesicles (MDVs) selectively remove damaged mitochondrial components, predominantly occurring at MAMs, where MFN2 localises and orchestrates mitochondrial fusion and autophagosome formation [103,120,121]. MFN2 depletion disrupts MAM integrity, impairing starvation-induced autophagy and hindering autophagosome formation [121,122]. Furthermore, MFN2 deletion in cardiomyocytes and mouse embryonic fibroblasts leads to autophagosome accumulation, as it blocks autophagosome-lysosome fusion, a crucial step in autophagic degradation, ultimately resulting in metabolic dysfunction, defective lipid metabolism, and reduced mitochondrial oxidative phosphorylation [123,124,125]. These findings highlight the critical role of MFN2-Rab7 interactions, which regulate autophagosome-lysosome fusion, further linking mitochondrial dynamics to autophagic regulation [123]. More recently, MFN2 has been identified as an interactor with novel autophagy-related partners, including RAB5C, a key endosomal regulator of mitochondrial homeostasis, and SLC27A2 [126].
7. Metabolic Cues Driving Mitochondrial Fusion
Mitochondrial bioenergetics is essential for cellular function, linking metabolic signals to regulate mitochondrial fusion and fission [24,127]. This dynamic nature allows mitochondria to adapt to fluctuating cellular demands by integrating their activity with physiological processes and shaping metabolism. Under nutrient scarcity, mitochondria undergo significant remodelling, characterized by reduced cristae density, respiratory capacity, and size, alongside increased contact with the ER. These changes facilitate calcium and lipid flux, enabling the cell to balance energy under metabolic stress [35,128]. Proteolytic inactivation of OPA1, mediated by MFN2, further promotes mito-ER contact formation, underscoring the importance of mitochondrial dynamics in stress adaptation [128,129].
Regulation by Metabolic Sensor Kinases and Post-Translational Modifications. Metabolic sensor kinases, including mTOR, AMPK, and PKA, dynamically regulate mitochondrial fusion and fission by integrating extracellular signals such as nutrients, oxygen, and growth factors [130,131]. For example, mTOR signalling enhances mitochondrial fusion by modulating OPA1 expression and activity, while AMPK activation during nutrient deprivation suppresses mTORC1 and promotes fusion, increasing energy efficiency [35,131,132,133]. Post-translational modifications (PTMs), such as phosphorylation, ubiquitylation, and sumoylation, provide an additional layer of regulation. Phosphorylation of Drp1 at Ser637 by PKA inhibits its fission activity, promoting mitochondrial hyperfusion [134]. Meanwhile, ubiquitylation of MFN2 by E3 ligases modulates its fusion activity in response to specific stimuli, enabling precise adaptation of mitochondrial dynamics to cellular needs [135].
8. Mitochondrial Dynamics in Neuronal Differentiation and Bioenergetics
Mitochondrial dynamics are essential for neuronal differentiation, given the high energy demands of neurons. These processes support critical functions such as synaptic transmission, axonal transport, signal propagation, calcium flux, and glutamate cycling [136]. Unlike astrocytes, which predominantly rely on glycolysis, neurons depend on oxidative phosphorylation, highlighting the metabolic diversity within the nervous system [137,138]. Proteomic analyses further confirm this distinction, showing that neuronal mitochondria possess specialized respiratory chain complexes adapted for oxidative metabolism, whereas astrocytic mitochondria exhibit a reduced OxPhos capacity. During neuronal differentiation, mitochondria undergo significant remodelling, which includes alterations in their abundance, localization, and metabolic activity. This process is tightly regulated by MFN2 and OPA1, which facilitate the metabolic transition from glycolysis to oxidative phosphorylation (OxPhos), a crucial step for differentiation [139,140,141]. MFN2 plays a particularly critical role by optimizing bioenergetics, maintaining mitochondrial integrity, and regulating key signalling pathways, such as PI3K-Akt. Silencing MFN2 disrupts mitochondrial function and differentiation, while its overexpression enhances mitochondrial efficiency and promotes differentiation in neural progenitor cells [142,143]. Interestingly, silencing MFN2 paradoxically enhances neural differentiation by increasing Akt phosphorylation in embryonic stem cells, suggesting a context-dependent regulatory role [144]. A similar phenomenon occurs during cardiomyocyte differentiation, where elevated MFN2 and reduced OPA1 levels promote mitochondrial elongation, enhancing OxPhos to meet the increased energy demands of mature cells [145]. This highlights the direct influence of mitochondrial morphology on cellular bioenergetics during differentiation. The structure of mitochondrial cristae, regulated by OPA1, plays a pivotal role in neuronal differentiation. Cristae support respiratory chain complex (RCC) organization, facilitating efficient electron transfer and ATP production. OPA1 remodelling is crucial for meeting the increased OxPhos demands of differentiating neurons, while OPA1 dysfunction impairs respiration and reduces differentiation efficiency, underscoring its significance in neuronal function and pathology [146,147].
9. Pathological Implications of MFN2 and OPA1 Mutations
Neurons are particularly vulnerable to mitochondrial dynamics defects due to their reliance on properly positioned mitochondria for ATP production and calcium buffering [9]. MFN2 is essential for mitochondrial transport along axons, while OPA1 is critical for respiratory efficiency and mitochondrial genome stability. Although both proteins regulate mitochondrial fusion, their mutations lead to distinct pathologies due to their unique roles and the specific vulnerabilities of neuronal subtypes [13].
MFN2 Mutations and Charcot-Marie-Tooth Disease Type 2A (CMT2A). MFN2 mutations are primarily associated with CMT2A, an autosomal dominant axonal neuropathy characterized by distal limb weakness, muscle atrophy, and sensory loss [148,149]. Over 60 pathogenic variants have been identified, often affecting the GTPase or coiled-coil domains (Figure 1). These mutations disrupt mitochondrial fusion, trafficking, and bioenergetics, leading to neuronal energy deficits and impaired calcium homeostasis [148,150]. Dysfunctional MFN2 impairs axonal transport, causing mitochondrial aggregation in the soma and reduced distribution to distal axons [151]. Both retrograde and anterograde transport are affected, leading to mislocalized mitochondria, and compromised neuronal survival [125,152]. Loss-of-function MFN2 mutations result in fragmented mitochondrial networks with reduced OxPhos activity, while gain-of-function mutations may lead to abnormal mitochondrial morphology or toxic effects on neurons [153]. A dominant-negative mechanism often exacerbates dysfunction by interfering with wild-type MFN2 activity [154]. Additionally, mutant MFN2 proteins disrupt interactions with motor proteins, impairing mito-ER connectivity, calcium signalling, and lipid synthesis [155,156]. Interestingly, motor neurons expressing mutant MFN2 show apoptosis resistance due to p53 suppression, which enhances mitochondrial turnover as a compensatory mechanism [157,158] (Table 1). Potential therapeutic approaches for CMT2A focus on improving mitochondrial function and axonal transport. Strategies include modulating tubulin and Miro acetylation to facilitate mitochondrial motility and reducing stress-induced transcriptional changes affecting axonal organelle transport pathways [144].
OPA1 Mutations and Autosomal Dominant Optic Atrophy (ADOA). Mutations in OPA1 are associated with autosomal dominant optic atrophy (ADOA), the most common mitochondrial optic neuropathy, characterized by retinal ganglion cell degeneration and progressive vision loss [159]. Over 370 pathogenic variants have been identified, including splicing mutations, frameshift deletions, and missense variants in the GTPase domain (Figure 1). These mutations disrupt inner membrane fusion and cristae organization, leading to respiratory dysfunction and mitochondrial fragmentation [160,161]. OPA1 haploinsufficiency reduces protein expression, sometimes triggering enhanced mitophagy and autophagy as compensatory responses to respiratory dysfunction [160,162,163,164,165,166,167,168]. Paradoxically, inhibiting autophagy in these cases has been shown to protect certain neuronal populations in mouse models [167,168]. However, no major alterations in the mitochondrial network were detected in other cases, correlating with low mitophagic/autophagic activity [161,169]. Some missense mutations operate through a dominant-negative mechanism, leading to excessive mitochondrial fragmentation despite normal OPA1 expression levels [161] (Table 2). Additionally, OPA1-deficient neurons exhibit decreased BNIP3 expression, a key regulator of mitophagy. Notably, restoring BNIP3 levels reactivates autophagic and mitophagic activity, highlighting its therapeutic potential [169,170]. Several studies have investigated therapeutic methods to restore mitochondrial fusion, regulate autophagy and mitophagy, and target compensatory mechanisms such as the downregulation of BNIP3. One potential strategy involves small molecules that enhance mitochondrial fusion and bioenergetics. For instance, Wang et al. (2012) identified M1, a compound that increased mitochondrial fusion and respiratory complex expression, offering a potential treatment avenue [171]. Additionally, Mdivi-1, a well-known DRP1 inhibitor, has been shown to increase OPA1 expression in cells carrying OPA1 variants, reducing excessive mitophagy and significantly improving cellular function [172]. Another approach focuses on direct OPA1 GTPase activation, which can promote mitochondrial fusion as a protective mechanism in various diseases [173]. Altogether, these therapeutic strategies hold promise for mitigating neurodegenerative disease progression and improving neuronal survival and function in disorders such as CMT2A and ADOA [10,156]. For further insights into therapeutic developments, readers are referred to recent comprehensive reviews [174,175,176].
10. Distinct Cellular Outcomes of MFN2 and OPA1 Mutations
Recent studies (Zanfardino et al., 2022; 2024) have revealed distinct cellular effects of MFN2 and OPA1 mutations, particularly concerning mitochondrial dynamics, autophagy, and cellular phenotypes. Researchers analysing primary fibroblasts carrying the MFN2C217F and OPA1H42Y mutations found significant mitochondrial fragmentation, depolarization, and impaired respiration, primarily due to decreased activity of respiratory chain complexes. These findings suggest a dominant-negative effect of the mutations, highlighting their detrimental impact on mitochondrial quality control [177,178,179]. Both MFN2 and OPA1 mutations reduced the formation of autophagosomes, indicating a shared defect in autophagy regulation. Interestingly, despite the accumulation of damaged and fragmented mitochondria, autophagy was not activated in either mutant fibroblast line. The mitophagy machinery remained intact, suggesting that the observed defects arise from impaired signalling or autophagosome biogenesis, rather than a failure of the mitophagy process itself [177,180]. Transcriptomic profiling uncovered differential cellular responses between MFN2 and OPA1 mutations. In MFN2-mutated cells, genes associated with the PI3K/AKT signalling pathway and cell proliferation were upregulated, correlating with increased mTORC2/AKT activation, accelerated cell division, and reduced autophagy—consistent with the anabolic effects of PI3K/AKT/mTOR signalling [181,182] (Figure 2). These findings align with previous research demonstrating that MFN2 suppresses mTORC2 activity, influencing AKT phosphorylation at Ser473, a mechanism implicated in cancer growth and metastasis [183]. Additionally, in pancreatic cancer cells, MFN2 enhances autophagy by inhibiting PI3K/AKT/mTOR signalling, underscoring its role in maintaining MAM integrity and energy balance [178,184]. Conversely, OPA1-mutated fibroblasts exhibited a distinct profile, characterized by p21WAF1/CIP1 and p53 pathway activation, and downregulation of mitotic cell cycle genes. This expression pattern suggests an early senescent phenotype, confirmed by elevated SA-β-Gal activity. Additionally, these cells showed increased ROS levels, indicating oxidative stress and mitochondrial dysfunction, which may drive senescence [185,186,187,188]. In OPA1 mutants, impaired mTORC2 activation, cellular senescence, and weakened autophagic responses were observed. Researchers suggested that mitochondrial dysfunction in OPA1-deficient cells contributes to premature ageing by suppressing AMPK responsiveness, a key regulator of autophagy and metabolic balance and preventing mTORC1 inhibition [180,189] (Figure 2). In support of this, previous studies in mice demonstrated that OPA1 deletion in muscle cells induces premature senescence and early death, further linking OPA1 mutations, mitochondrial dysfunction, and senescence [72,190]. Moreover, OPA1 overexpression in Chang cells has been linked to mitochondrial fusion and senescence markers, reinforcing its role in ageing [191]. Similarly, aged mesenchymal stromal/stem cells (MSCs) and diseased postmitotic cells accumulate dysfunctional mitochondria with altered OPA1 levels, further emphasizing its role in aging and cellular decline [190,192,193].
Therapeutic Implications. The PI3K/AKT/mTOR pathway has emerged as a key regulator of the divergent outcomes observed in MFN2 and OPA1 mutants. Pharmacological modulation of this pathway effectively normalized cellular phenotypes in mutant cell models. In MFN2-mutated cells, mTOR inhibition with torin1 restored normal cell proliferation, while AKT inhibition with miransertib improved both autophagy and cell proliferation [177,179]. In OPA1-mutated cells, mTORC1 inhibition with everolimus counteracted senescence and restored autophagy [180] (Figure 2). These findings underscore the crucial link between mitochondrial dysfunction, organelle morphology, and cellular signalling. The PI3K/AKT/mTOR pathway serves as a key sensor of mitochondrial distress, orchestrating the subsequent cellular adaptations [144]. Targeting this pathway presents a promising therapeutic strategy to counteract the pathological consequences of MFN2 and OPA1 mutations and restore mitochondrial function [177,178,179,180].
Phosphatidylinositol 3-kinase (PtdIns3K) catalyzes the phosphorylation of phosphatidylinositol 4,5-bisphosphate (PtdIns2P) to phosphatidylinositol-3,4,5-trisphosphate (PtdIns3P), recruiting proteins such as 3-phosphoinositide-dependent kinase (PDK1). PDK1 phosphorylates AKT at Thr(308), while second phosphorylation at Ser(473) by mTORC2 fully activates AKT, promoting cellular proliferation. MFN2, localized at the outer mitochondrial membrane (OMM) or mitochondria-associated ER membranes (MAMs), suppresses mTORC2 kinase activity, thereby inhibiting AKT.
Mutant MFN2 (red lightning) loses this inhibitory function, leading to hyperactivated AKT, reduced autophagy, and increased proliferation. AKT promotes proliferation directly or via mTORC1 and inhibits autophagy through both mTORC1-dependent and independent mechanisms. Miransertib (violet), an AKT inhibitor, restores autophagy and normalizes proliferation (left panel).
AMPK, a serine-threonine kinase, acts as an energy sensor, maintaining cellular homeostasis by promoting survival and autophagy through mTORC1 suppression (right panel). OPA1, located in the inner mitochondrial membrane (IMM), regulates mitochondrial function. Mutant OPA1 (blue emoticon) induces mitochondrial depolarization (green mitochondrion), increases ROS production, and impairs the electron transfer chain (ETC). These dysfunctions lead to premature senescence and a decline in AMPK responsiveness (dotted line), reducing autophagy initiation and cell survival.
OPA1-driven mitochondrial dysfunction contributes to p53 activation, which regulates stress response genes, including the cell cycle inhibitor p21. Increased p53 and p21 expression in OPA1-mutated cells promotes senescence. However, senescence and autophagy defects can be reversed by inhibiting mTORC1 using everolimus (blue) (right panel).
11. Concluding Remarks
MFN2 and OPA1 are crucial regulators of mitochondrial dynamics, playing key roles in balancing fusion and fission to maintain mitochondrial structure, quality control, and energy production in response to cellular conditions. MFN2 primarily functions at the outer mitochondrial membrane, while OPA1 operates at the inner membrane mitochondrial fusion plays a vital role in the exchange of mitochondrial components, which helps to prevent the buildup of defective proteins and mutated mtDNA and allows for functional complementation. During periods of metabolic stress, such as starvation or cellular senescence, mitochondrial fusion is enhanced and forms elongated networks. These networks support oxidative phosphorylation (OxPhos) and help cells evade mitophagy. In contrast, severe mitochondrial damage, excessive nutrient levels, or increased cell proliferation trigger mitochondrial fission, resulting in smaller mitochondria. This fission process facilitates the redistribution of mitochondria during mitosis and helps isolate damaged mitochondria for degradation through mitophagy (Figure 3). Although mitophagy is a specialized form of autophagy, it is independently regulated in response to mitochondrial damage. Despite this, both processes can interact to maintain cellular homeostasis. For instance, autophagy complements mitophagy by responding to broader metabolic cues, ensuring the health of the cell. Central to the coordination of these processes is the mTOR/AKT pathway, which integrates signals related to nutrient availability, energy status, mitochondrial integrity, and even cellular differentiation. This coordination between mitochondrial dynamics and cellular function is also reflected in the dynamic regulation of mitochondrial morphology. The balance between fusion and fission is critical for metabolic adaptation, as elongated mitochondria, regulated by OPA1 and MFN2, enhance OxPhos, whereas fragmented mitochondria favor glycolysis. These shifts are particularly evident during cellular reprogramming. For instance, the generation of induced pluripotent stem cells (iPSCs) is linked to a fragmented mitochondrial network and increased glycolysis. In contrast, differentiation restores mitochondrial elongation and enhances OxPhos. Disruptions in the balance of mitochondrial function, such as those caused by mutations in the MFN2 and OPA1 genes, can negatively impact cellular health, especially in neurons. The initiation of mitophagy, the process where damaged mitochondria are removed, is determined by the extent of mitochondrial damage or dysfunction and the associated energy status of the cell [194]. When mitochondria are not significantly harmed, with respect to the high-energy costs, the cell tends to avoid their removal through mitophagy. The cell can recognize a decline in energy production resulting from mitochondrial changes. In response, it may initiate nonselective autophagy to acquire the nutrients needed for vital biosynthetic processes [195]. Research has indicated that mitochondrial dysfunction is associated with increased autophagic markers across different cellular disease models. For instance, in fibroblasts deficient in OxPhos, mitophagy is not increased but there is an accumulation of autophagosomes, lysosomes and late autophagic vacuoles [196]. This accumulation is thought to result from a trigger of nonselective autophagy, which occurs in response to a pseudo-starvation state. When mitochondria are significantly impaired, cells can trigger mitophagy and activate a signalling pathway that encourages nonselective autophagy. Many studies highlight the role of specific mitophagy factors in regulating the overall autophagic response to mitochondrial damage [197]. Investigating how the absence of these factors affects autophagy initiation, rate, extent, and overall cell survival would provide valuable insights. Research conducted in yeast demonstrates that mitophagy is consistently accompanied by nonselective autophagy, indicating a strong connection between mitochondrial status and autophagic regulation [197]. However, it remains unclear whether mitochondrial fission is universally required for mitophagy or if only necessary under specific conditions. General autophagy-inducing situations, such as starvation, also trigger mitophagy, but the mechanisms governing selective mitochondrial degradation in these contexts remain uncertain. Furthermore, it is possible to identify conditions that allow the induction of mitophagy while non-selective autophagy is not triggered simultaneously. For example, in mammalian cells experiencing mild oxidative stress, DRP1-dependent mitophagy occurs without the activation of nonselective autophagy, suggesting that mitophagy can be regulated independently [198]. Additionally, it's important to note that mitochondrial fragmentation due to MFN2 and OPA1 mutations does not always lead to autophagy, as certain factors, such as cell proliferation (as seen in CMT2A) or premature senescence (as in ADOA), can suppress the autophagic process (Figure 3). MFN2 and OPA1 have differing effects on these cellular processes through their influence on mTOR/AKT signalling, a key regulator in either cancer or neurodegeneration. In CMT2A, dysfunction of MFN2 disrupts mitochondrial transport and bioenergetics, leading to a suppression of autophagy and an increase in cellular proliferation, potentially promoting excessive survival of neuronal stem cells. Conversely, mutations in OPA1 impair mitochondrial quality control, driving senescence and worsening neurodegeneration in ADOA by failing to efficiently clear damaged mitochondria. Although MFN2 and OPA1 have overlapping functions, their distinct roles contribute to the specific disease phenotypes observed in CMT2A and ADOA. Both conditions involve mitochondrial fragmentation and dysfunction; however, their impact on the regulation of cellular function differs, which in turn shapes disease progression in unique ways.
This illustration depicts the contrasting effects of different cellular conditions on mitochondrial dynamics and homeostasis.
-
Mitochondrial Hyperfusion:
- -
- Triggered by cellular senescence, amino acid starvation, loss of fission factors, mild mitochondrial stress, or mitochondrial DNA (mtDNA) mutations.
- -
- Supports oxidative phosphorylation (OxPhos) and ATP production.
- -
- Prevents autophagic clearance of damaged mitochondria by mitophagy despite cellular stressors.
-
Mitochondrial Fragmentation:
- -
- Induced by cellular proliferation, nutrient excess, loss of fusion factors (like MFN2 and OPA1), and severe mitochondrial dysfunction.
- -
- Promotes selective autophagic clearance (mitophagy) of dysfunctional mitochondria.
Mitophagy typically occurs due to mitochondrial dysfunction, autophagy can also respond to severe cellular stress, driven by factors like energy depletion, nutrient starvation, and metabolic stress.
- 3.
-
Autophagy Regulation:
- -
- There are instances when, despite mitochondrial fragmentation, autophagy may not be activated, particularly in contexts such as cell proliferation (as seen in Charcot-Marie-Tooth disease type 2A) or premature senescence (observed in Autosomal Dominant Optic Atrophy).
- -
- Under mild oxidative stress, mitophagy is activated; however, the induction of non-selective autophagy may not occur simultaneously.
This graphical abstract illustrates the delicate interplay between mitochondrial dynamics and cellular homeostasis and the implications that disruptions in this balance can have on cellular health and disease.
12. Future Directions
The divergent effects of MFN2 and OPA1 mutations raise critical questions about their shared and distinct roles in disease progression:
- Could the proliferative changes observed in CMT2A2 fibroblasts and cancer models of MFN2 dysfunction also occur in neuronal stem cells affected by CMT2A?
- Does senescence caused by OPA1 deficiency directly contribute to neurodegeneration in ADOA, and is this related to impaired autophagy?
- How does metabolic reprogramming influence the progression of mitochondrial diseases, and can interventions be implemented to restore the balance of autophagy and mitophagy?
To address these questions, future research should concentrate on advanced neuronal stem cell models, co-cultures, and 3D organoids that better replicate in vivo environments. Investigating the interactions between mitochondrial dysfunction, metabolic shifts, and mTOR/AKT signalling within these systems will provide essential insights into disease mechanisms. Furthermore, targeting autophagy and mitophagy pathways therapeutically presents promising opportunities for intervention. The potential of mTOR inhibitors (such as everolimus and torin1) and modulators of MAM integrity should be examined for their ability to restore autophagic flux, enhance mitochondrial quality control, and prevent neurodegeneration. Understanding the relationship between mitochondrial dysfunction, autophagy, metabolism, and signalling pathways is crucial for developing novel therapies. These advancements could benefit not only those with mitochondrial and neurodegenerative diseases but also have wider implications in the fields of ageing and cancer.
Author Contributions
Conceptualization, P.Z. and V.P.; writing—original draft preparation, P.Z. and V.P.; writing, review, supervision and editing, P.Z., A.A., M.P. and V.P.; supervision, V.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from donations of Parents’ Associations (to VP). The authors alone are responsible for the content and writing.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank the patients’ association MITOCON and UILDM (Unione Italiana Lotta alla Distrofia Muscolare) and Fondazione Opera Pia Monte di Pietà e Confidenze, Molfetta (Bari).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. 2013, 17, 491–506. [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [CrossRef]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [CrossRef]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [CrossRef]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [CrossRef]
- Chan, D.C. Mitochondrial Fusion and Fission in Mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [CrossRef]
- Gomes, L. C.; Scorrano, L. Mitochondrial Morphology in Mitophagy and Macroautophagy. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2013, 1833 (1), 205–212. [CrossRef]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Molecular Cell 2016, 61 (5), 683–694. [CrossRef]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: from functions to disease. Cell Death Dis. 2018, 9, 1–13. [CrossRef]
- Gilkerson, R.; De La Torre, P.; Vallier, S.S. Mitochondrial OMA1 and OPA1 as Gatekeepers of Organellar Structure/Function and Cellular Stress Response. Front. Cell Dev. Biol. 2021, 9. [CrossRef]
- Rojo, M.; Legros, F.; Chateau, D.; Lombès, A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 2002, 115, 1663–1674. [CrossRef]
- Chandhok, G.; Lazarou, M.; Neumann, B. Structure, Function, and Regulation of Mitofusin-2 in Health and Disease. Biological Reviews 2018, 93 (2), 933–949. [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature 1990, 348, 125–132. [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [CrossRef]
- Chen, H.C.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural Basis of Mitochondrial Tethering by Mitofusin Complexes. Science 2004, 305, 858–862. [CrossRef]
- Chen, K.-H.; Guo, X.; Ma, D.; Guo, Y.; Li, Q.; Yang, D.; Li, P.; Qiu, X.; Wen, S.; Xiao, R.-P.; et al. Dysregulation of HSG triggers vascular proliferative disorders. Nat. Cell Biol. 2004, 6, 872–883. [CrossRef]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two Mitofusin Proteins, Mammalian Homologues of FZO, with Distinct Functions Are Both Required for Mitochondrial Fusion. J. Biochem. 2003, 134, 333–344. [CrossRef]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [CrossRef]
- Amati-Bonneau, P.; Milea, D.; Bonneau, D.; Chevrollier, A.; Ferré, M.; Guillet, V.; Gueguen, N.; Loiseau, D.; Crescenzo, M.-A. P. D.; Verny, C.; Procaccio, V.; Lenaers, G.; Reynier, P. OPA1-Associated Disorders: Phenotypes and Pathophysiology. The International Journal of Biochemistry & Cell Biology 2009, 41 (10), 1855–1865. [CrossRef]
- Baker, M.J.; Lampe, P.A.; Stojanovski, D.; Korwitz, A.; Anand, R.; Tatsuta, T.; Langer, T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014, 33, 578–593. [CrossRef]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [CrossRef]
- Van Der Bliek, A. M.; Shen, Q.; Kawajiri, S. Mechanisms of Mitochondrial Fission and Fusion. Cold Spring Harbor Perspectives in Biology 2013, 5 (6), a011072–a011072. [CrossRef]
- Li, Y.-J.; Cao, Y.-L.; Feng, J.-X.; Qi, Y.; Meng, S.; Yang, J.-F.; Zhong, Y.-T.; Kang, S.; Chen, X.; Lan, L.; et al. Structural insights of human mitofusin-2 into mitochondrial fusion and CMT2A onset. Nat. Commun. 2019, 10, 1–14. [CrossRef]
- Yan, L.; Qi, Y.; Huang, X.; Yu, C.; Lan, L.; Guo, X.; Rao, Z.; Hu, J.; Lou, Z. Structural basis for GTP hydrolysis and conformational change of MFN1 in mediating membrane fusion. Nat. Struct. Mol. Biol. 2018, 25, 233–243. [CrossRef]
- Mattie, S.; Riemer, J.; Wideman, J.G.; McBride, H.M. A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J. Cell Biol. 2017, 217, 507–515. [CrossRef]
- Duvezin-Caubet, S.; Jagasia, R.; Wagener, J.; Hofmann, S.; Trifunovic, A.; Hansson, A.; Chomyn, A.; Bauer, M.F.; Attardi, G.; Larsson, N.-G.; et al. Proteolytic Processing of OPA1 Links Mitochondrial Dysfunction to Alterations in Mitochondrial Morphology. J. Biol. Chem. 2006, 281, 37972–37979. [CrossRef]
- Brandt, T.; Cavellini, L.; Kühlbrandt, W.; Cohen, M.M. A mitofusin-dependent docking ring complex triggers mitochondrial fusion in vitro. eLife 2016, 5. [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [CrossRef]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial Fusion Is Required for mtDNA Stability in Skeletal Muscle and Tolerance of mtDNA Mutations. Cell 2010, 141, 280–289. [CrossRef]
- Chen, H.; Chan, D.C. Physiological functions of mitochondrial fusion. Ann. New York Acad. Sci. 2010, 1201, 21–25. [CrossRef]
- Rodríguez-Nuevo, A.; Díaz-Ramos, A.; Noguera, E.; Díaz-Sáez, F.; Duran, X.; Muñoz, J.P.; Romero, M.; Plana, N.; Sebastián, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018, 37, e96553. [CrossRef]
- Yao, C.-H.; Wang, R.; Wang, Y.; Kung, C.-P.; Weber, J.D.; Patti, G.J. Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. eLife 2019, 8, e41351. [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [CrossRef]
- Molina, A.J.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial Networking Protects β-Cells From Nutrient-Induced Apoptosis. Diabetes 2009, 58, 2303–2315. [CrossRef]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Signal Transduct. Target. Ther. 2023, 8, 1–25. [CrossRef]
- Jheng, H.-F.; Tsai, P.-J.; Guo, S.-M.; Kuo, L.-H.; Chang, C.-S.; Su, I.-J.; Chang, C.-R.; Tsai, Y.-S. Mitochondrial Fission Contributes to Mitochondrial Dysfunction and Insulin Resistance in Skeletal Muscle. Mol. Cell. Biol. 2012, 32, 309–319. [CrossRef]
- Ryu, K.W.; Fung, T.S.; Baker, D.C.; Saoi, M.; Park, J.; Febres-Aldana, C.A.; Aly, R.G.; Cui, R.; Sharma, A.; Fu, Y.; et al. Cellular ATP demand creates metabolically distinct subpopulations of mitochondria. Nature 2024, 635, 746–754. [CrossRef]
- De Brito, O. M.; Scorrano, L. Mitofusin 2 Tethers Endoplasmic Reticulum to Mitochondria. Nature 2008, 456 (7222), 605–610. [CrossRef]
- Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernández, V.; Vasconcelos, N.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895.e17. [CrossRef]
- Zorzano, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Muñoz, J.P. Mitofusin 2 as a Driver That Controls Energy Metabolism and Insulin Signaling. Antioxidants Redox Signal. 2015, 22, 1020–1031. [CrossRef]
- Kaufman, R.J.; Malhotra, J.D. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2014, 1843, 2233–2239. [CrossRef]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U. E.; Flomenberg, N.; Birbe, R.; Witkiewicz, A. K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R. G.; Broda, P.; Minetti, C.; Lisanti, M. P.; Sotgia, F. Hyperactivation of Oxidative Mitochondrial Metabolism in Epithelial Cancer Cells in Situ: Visualizing the Therapeutic Effects of Metformin in Tumor Tissue. Cell Cycle 2011, 10 (23), 4047–4064. [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum–mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [CrossRef]
- Naon, D.; Scorrano, L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2014, 1843, 2184–2194. [CrossRef]
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 2017, 62, 1–15. [CrossRef]
- Lin, M.-Y.; Sheng, Z.-H. Regulation of mitochondrial transport in neurons. Exp. Cell Res. 2015, 334, 35–44. [CrossRef]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 Is Necessary for Transport of Axonal Mitochondria and Interacts with the Miro/Milton Complex. J. Neurosci. 2010, 30, 4232–4240. [CrossRef]
- Mou, Y.; Dein, J.; Chen, Z.; Jagdale, M.; Li, X.-J. MFN2 Deficiency Impairs Mitochondrial Transport and Downregulates Motor Protein Expression in Human Spinal Motor Neurons. Front. Mol. Neurosci. 2021, 14. [CrossRef]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, M.A.; et al. Supercomplex Assembly Determines Electron Flux in the Mitochondrial Electron Transport Chain. Science 2013, 340, 1567–1570. [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. 2013, 155, 160–171. [CrossRef]
- A Patten, D.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014, 33, 2676–2691. [CrossRef]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [CrossRef]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620–e21620. [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [CrossRef]
- Mottis, A.; Jovaisaite, V.; Auwerx, J. The mitochondrial unfolded protein response in mammalian physiology. Mamm. Genome 2014, 25, 424–433. [CrossRef]
- Fan, W.; Evans, R. PPARs and ERRs: molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [CrossRef]
- Cardamone, M.D.; Tanasa, B.; Cederquist, C.T.; Huang, J.; Mahdaviani, K.; Li, W.; Rosenfeld, M.G.; Liesa, M.; Perissi, V. Mitochondrial Retrograde Signaling in Mammals Is Mediated by the Transcriptional Cofactor GPS2 via Direct Mitochondria-to-Nucleus Translocation. Mol. Cell 2018, 69, 757–772.e7. [CrossRef]
- Hock, M.B.; Kralli, A. Transcriptional Control of Mitochondrial Biogenesis and Function. Annu. Rev. Physiol. 2009, 71, 177–203. [CrossRef]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. 2012, 23, 459–466. [CrossRef]
- Finley, L.W.; Haigis, M.C. The coordination of nuclear and mitochondrial communication during aging and calorie restriction. 2009, 8, 173–188. [CrossRef]
- Guha, M.; Avadhani, N.G. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion 2013, 13, 577–591. [CrossRef]
- Bohovych, I.; Khalimonchuk, O. Sending Out an SOS: Mitochondria as a Signaling Hub. Front. Cell Dev. Biol. 2016, 4, 109. [CrossRef]
- Chandel, N.S. Evolution of Mitochondria as Signaling Organelles. Cell Metab. 2015, 22, 204–206. [CrossRef]
- Butow, R. A.; Avadhani, N. G. Mitochondrial Signaling. Molecular Cell 2004, 14 (1), 1–15.
- Liu, Z.; Butow, R.A. Mitochondrial Retrograde Signaling. Annu. Rev. Genet. 2006, 40, 159–185. [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. Essential amino acids and glutamine regulate induction of mitochondrial elongation during autophagy. Cell Cycle 2011, 10, 2635–2639. [CrossRef]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [CrossRef]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389.e6. [CrossRef]
- Yoon, Y.; Galloway, C. A.; Jhun, B. S.; Yu, T. Mitochondrial Dynamics in Diabetes. Antioxidants & Redox Signaling 2011, 14 (3), 439–457.
- Yoon, Y.; Yu, T.; Wang, L. Morphological control of mitochondrial bioenergetics. Front. Biosci. 2015, 20, 229–246. [CrossRef]
- Lee, H.; Smith, S.B.; Yoon, Y. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J. Biol. Chem. 2017, 292, 7115–7130. [CrossRef]
- Sessions, D.T.; Kim, K.-B.; Kashatus, J.A.; Churchill, N.; Park, K.-S.; Mayo, M.W.; Sesaki, H.; Kashatus, D.F. Opa1 and Drp1 reciprocally regulate cristae morphology, ETC function, and NAD+ regeneration in KRas-mutant lung adenocarcinoma. Cell Rep. 2022, 41, 111818–111818. [CrossRef]
- Frezza, C.; Cipolat, S.; de Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [CrossRef]
- Hu, J.; Zhang, Y.; Jiang, X.; Zhang, H.; Gao, Z.; Li, Y.; Fu, R.; Li, L.; Li, J.; Cui, H.; et al. ROS-mediated activation and mitochondrial translocation of CaMKII contributes to Drp1-dependent mitochondrial fission and apoptosis in triple-negative breast cancer cells by isorhamnetin and chloroquine. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [CrossRef]
- Steinberg, G.R.; Hardie, D.G. New insights into activation and function of the AMPK. Nat. Rev. Mol. Cell Biol. 2022, 24, 255–272. [CrossRef]
- Lu, H.-J.; Koju, N.; Sheng, R. Mammalian integrated stress responses in stressed organelles and their functions. Acta Pharmacol. Sin. 2024, 45, 1095–1114. [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1–AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [CrossRef]
- Zhao, B.; Qiang, L.; Joseph, J.; Kalyanaraman, B.; Viollet, B.; He, Y.-Y. Mitochondrial dysfunction activates the AMPK signaling and autophagy to promote cell survival. Genes Dis. 2016, 3, 82–87. [CrossRef]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [CrossRef]
- Kang, S.W.S.; Haydar, G.; Taniane, C.; Farrell, G.; Arias, I.M.; Lippincott-Schwartz, J.; Fu, D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLOS ONE 2016, 11, e0165638. [CrossRef]
- Hoozemans, J.J.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [CrossRef]
- Ma, T.; Trinh, M. A.; Wexler, A. J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D. R.; Klann, E. Suppression of eIF2α Kinases Alleviates Alzheimer’s Disease–Related Plasticity and Memory Deficits. Nat Neurosci 2013, 16 (9), 1299–1305. [CrossRef]
- Millot, P.; Pujol, C.; Paquet, C.; Mouton-Liger, F. Impaired mitochondrial dynamics in the blood of patients with Alzheimer’s disease and Lewy body dementia. Alzheimer's Dement. 2023, 19. [CrossRef]
- Bilen, M.; Benhammouda, S.; Slack, R.S.; Germain, M. The integrated stress response as a key pathway downstream of mitochondrial dysfunction. Curr. Opin. Physiol. 2022, 27. [CrossRef]
- Oliveira, M.M.; Lourenco, M.V. Integrated Stress Response: Connecting ApoE4 to Memory Impairment in Alzheimer's Disease. J. Neurosci. 2016, 36, 1053–1055. [CrossRef]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2017, 19, 109–120. [CrossRef]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 2020, 22, 54–70. [CrossRef]
- Vannuvel, K.; Renard, P.; Raes, M.; Arnould, T. Functional and morphological impact of ER stress on mitochondria. J. Cell. Physiol. 2013, 228, 1802–1818. [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [CrossRef]
- Arnould, T.; Michel, S.; Renard, P. Mitochondria Retrograde Signaling and the UPRmt: Where Are We in Mammals? IJMS 2015, 16 (8), 18224–18251. [CrossRef]
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The Ubiquitin-Proteasome System. J. Biosci. 2006, 31 (1), 137–155. [CrossRef]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [CrossRef]
- Hsu, M.-C.; Kinefuchi, H.; Lei, L.; Kikuchi, R.; Yamano, K.; Youle, R.J. Mitochondrial YME1L1 governs unoccupied protein translocase channels. Nat. Cell Biol. 2025, 1–13. [CrossRef]
- Jin, S. M.; Youle, R. J. The Accumulation of Misfolded Proteins in the Mitochondrial Matrix Is Sensed by PINK1 to Induce PARK2/Parkin-Mediated Mitophagy of Polarized Mitochondria. Autophagy 2013, 9 (11), 1750–1757. [CrossRef]
- Mohan, J.; Wollert, T. Human ubiquitin-like proteins as central coordinators in autophagy. Interface Focus 2018, 8, 20180025. [CrossRef]
- Chang, C.; Jensen, L.E.; Hurley, J.H. Autophagosome biogenesis comes out of the black box. Nat. Cell Biol. 2021, 23, 450–456. [CrossRef]
- Ganley, I.G.; Simonsen, A. Diversity of mitophagy pathways at a glance. J. Cell Sci. 2022, 135. [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [CrossRef]
- Simonsen, A.; Tooze, S.A. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell Biol. 2009, 186, 773–782. [CrossRef]
- Markaki, M.; Tsagkari, D.; Tavernarakis, N. Mitophagy mechanisms in neuronal physiology and pathology during ageing. Biophys. Rev. 2021, 13, 955–965. [CrossRef]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.-W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 1–28. [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [CrossRef]
- Gegg, M. E.; Cooper, J. M.; Chau, K.-Y.; Rojo, M.; Schapira, A. H. V.; Taanman, J.-W. Mitofusin 1 and Mitofusin 2 Are Ubiquitinated in a PINK1/Parkin-Dependent Manner upon Induction of Mitophagy. Human Molecular Genetics 2010, 19 (24), 4861–4870. [CrossRef]
- Ziviani, E.; Tao, R.N.; Whitworth, A.J. Drosophila Parkin requires PINK1 for mitochondrial translocation and ubiquitinates Mitofusin. Proc. Natl. Acad. Sci. USA 2010, 107, 5018–5023. [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [CrossRef]
- Cohen, M. M. J.; Leboucher, G. P.; Livnat-Levanon, N.; Glickman, M. H.; Weissman, A. M. Ubiquitin–Proteasome-Dependent Degradation of a Mitofusin, a Critical Regulator of Mitochondrial Fusion. MBoC 2008, 19 (6), 2457–2464. [CrossRef]
- Sugiura, A.; Nagashima, S.; Tokuyama, T.; Amo, T.; Matsuki, Y.; Ishido, S.; Kudo, Y.; McBride, H.M.; Fukuda, T.; Matsushita, N.; et al. MITOL Regulates Endoplasmic Reticulum-Mitochondria Contacts via Mitofusin2. Mol. Cell 2013, 51, 20–34. [CrossRef]
- Kissová, I.; Deffieu, M.; Manon, S.; Camougrand, N. Uth1p Is Involved in the Autophagic Degradation of Mitochondria. J. Biol. Chem. 2004, 279, 39068–39074. [CrossRef]
- Princely Abudu, Y.; Pankiv, S.; Mathai, B. J.; Håkon Lystad, A.; Bindesbøll, C.; Brenne, H. B.; Yoke Wui Ng, M.; Thiede, B.; Yamamoto, A.; Mutugi Nthiga, T.; Lamark, T.; Esguerra, C. V.; Johansen, T.; Simonsen, A. NIPSNAP1 and NIPSNAP2 Act as “Eat Me” Signals for Mitophagy. Developmental Cell 2019, 49 (4), 509-525.e12. [CrossRef]
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Wu, H.; Gong, G. Mitochondrial autophagy: molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022, 13, 1–15. [CrossRef]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [CrossRef]
- Landes, T.; Emorine, L.J.; Courilleau, D.; Rojo, M.; Belenguer, P.; Arnauné-Pelloquin, L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. Embo Rep. 2010, 11, 459–465. [CrossRef]
- Sugiura, A.; McLelland, G.-L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria Supply Membranes for Autophagosome Biogenesis during Starvation. Cell 2010, 141, 656–667. [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nature 2013, 495, 389–393. [CrossRef]
- Zhao, T.; Huang, X.; Han, L.; Wang, X.; Cheng, H.; Zhao, Y.; Chen, Q.; Chen, J.; Cheng, H.; Xiao, R.; et al. Central Role of Mitofusin 2 in Autophagosome-Lysosome Fusion in Cardiomyocytes. J. Biol. Chem. 2012, 287, 23615–23625. [CrossRef]
- Ding, Y.; Gao, H.; Zhao, L.; Wang, X.; Zheng, M. Mitofusin 2-Deficiency Suppresses Cell Proliferation through Disturbance of Autophagy. PLOS ONE 2015, 10, e0121328–e0121328. [CrossRef]
- Pich, S.; Bach, D.; Briones, P.; Liesa, M.; Camps, M.; Testar, X.; Palacín, M.; Zorzano, A. The Charcot–Marie–Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 2005, 14, 1405–1415. [CrossRef]
- Gordaliza-Alaguero, I.; Sànchez-Fernàndez-De-Landa, P.; Radivojevikj, D.; Villarreal, L.; Arauz-Garofalo, G.; Gay, M.; Martinez-Vicente, M.; Seco, J.; Martín-Malpartida, P.; Vilaseca, M.; et al. Endogenous interactomes of MFN1 and MFN2 provide novel insights into interorganelle communication and autophagy. Autophagy 2024, 1–22. [CrossRef]
- Sauvanet, C.; Duvezin-Caubet, S.; di Rago, J.-P.; Rojo, M. Energetic requirements and bioenergetic modulation of mitochondrial morphology and dynamics. Semin. Cell Dev. Biol. 2010, 21, 558–565. [CrossRef]
- Sood, A.; Jeyaraju, D.V.; Prudent, J.; Caron, A.; Lemieux, P.; McBride, H.M.; Laplante, M.; Tóth, K.; Pellegrini, L. A Mitofusin-2–dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc. Natl. Acad. Sci. 2014, 111, 16017–16022. [CrossRef]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.-C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [CrossRef]
- Morita, M.; Prudent, J.; Basu, K.; Goyon, V.; Katsumura, S.; Hulea, L.; Pearl, D.; Siddiqui, N.; Strack, S.; McGuirk, S.; et al. mTOR Controls Mitochondrial Dynamics and Cell Survival via MTFP1. Mol. Cell 2017, 67, 922–935.e5. [CrossRef]
- Parra, V.; Verdejo, H. E.; Iglewski, M.; Del Campo, A.; Troncoso, R.; Jones, D.; Zhu, Y.; Kuzmicic, J.; Pennanen, C.; Lopez-Crisosto, C.; Jaña, F.; Ferreira, J.; Noguera, E.; Chiong, M.; Bernlohr, D. A.; Klip, A.; Hill, J. A.; Rothermel, B. A.; Abel, E. D.; Zorzano, A.; Lavandero, S. Insulin Stimulates Mitochondrial Fusion and Function in Cardiomyocytes via the Akt-mTOR-NFκB-Opa-1 Signaling Pathway. Diabetes 2014, 63 (1), 75–88. [CrossRef]
- Cai, W.J.; Chen, Y.; Shi, L.X.; Cheng, H.R.; Banda, I.; Ji, Y.H.; Wang, Y.T.; Li, X.M.; Mao, Y.X.; Zhang, D.F.; et al. AKT-GSK3βSignaling Pathway Regulates Mitochondrial Dysfunction-Associated OPA1 Cleavage Contributing to Osteoblast Apoptosis: Preventative Effects of Hydroxytyrosol. Oxidative Med. Cell. Longev. 2019, 2019, 1–20. [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. Embo Rep. 2007, 8, 939–944. [CrossRef]
- Escobar-Henriques, M.; Joaquim, M. Mitofusins: Disease Gatekeepers and Hubs in Mitochondrial Quality Control by E3 Ligases. Front. Physiol. 2019, 10, 517. [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab. 2011, 14, 724–738. [CrossRef]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C–Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [CrossRef]
- Bittner, C.X.; Valdebenito, R.; Ruminot, I.; Loaiza, A.; Larenas, V.; Sotelo-Hitschfeld, T.; Moldenhauer, H.; Martín, A.S.; Gutiérrez, R.; Zambrano, M.; et al. Fast and Reversible Stimulation of Astrocytic Glycolysis by K+and a Delayed and Persistent Effect of Glutamate. J. Neurosci. 2011, 31, 4709–4713. [CrossRef]
- Choi, H.W.; Kim, J.H.; Chung, M.K.; Hong, Y.J.; Jang, H.S.; Seo, B.J.; Jung, T.H.; Kim, J.S.; Chung, H.M.; Byun, S.J.; et al. Mitochondrial and Metabolic Remodeling During Reprogramming and Differentiation of the Reprogrammed Cells. Stem Cells Dev. 2015, 24, 1366–1373. [CrossRef]
- Prigione, A.; Fauler, B.; Lurz, R.; Lehrach, H.; Adjaye, J. The Senescence-Related Mitochondrial/Oxidative Stress Pathway is Repressed in Human Induced Pluripotent Stem Cells. STEM CELLS 2010, 28, 721–733. [CrossRef]
- Kasahara, A.; Cipolat, S.; Chen, Y.; Dorn, G.W.; Scorrano, L. Mitochondrial Fusion Directs Cardiomyocyte Differentiation via Calcineurin and Notch Signaling. Science 2013, 342, 734–737. [CrossRef]
- Yi, S.; Cui, C.; Huang, X.; Yin, X.; Li, Y.; Wen, J.; Luan, Q. MFN2 silencing promotes neural differentiation of embryonic stem cells via the Akt signaling pathway. J. Cell. Physiol. 2019, 235, 1051–1064. [CrossRef]
- Fang, D.; Yan, S.; Yu, Q.; Chen, D.; Yan, S.S. Mfn2 is Required for Mitochondrial Development and Synapse Formation in Human Induced Pluripotent Stem Cells/hiPSC Derived Cortical Neurons. Sci. Rep. 2016, 6, 31462. [CrossRef]
- Van Lent, J.; Verstraelen, P.; Asselbergh, B.; Adriaenssens, E.; Mateiu, L.; Verbist, C.; De Winter, V.; Eggermont, K.; Bosch, L.V.D.; De Vos, W.H.; et al. Induced pluripotent stem cell-derived motor neurons of CMT type 2 patients reveal progressive mitochondrial dysfunction. Brain 2021, 144, 2471–2485. [CrossRef]
- Pellegrini, L.; Scorrano, L. A cut short to death: Parl and Opa1 in the regulation of mitochondrial morphology and apoptosis. Cell Death Differ. 2007, 14, 1275–1284. [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [CrossRef]
- Youle, R. J.; Van Der Bliek, A. M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337 (6098), 1062–1065.
- Züchner, S.; De Jonghe, P.; Jordanova, A.; Claeys, K.G.; Guergueltcheva, V.; Cherninkova, S.; Hamilton, S.R.; Van Stavern, G.; Krajewski, K.M.; Stajich, J.; et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann. Neurol. 2006, 59, 276–281. [CrossRef]
- Vallat, J.-M.; Ouvrier, R.A.; Pollard, J.D.; Magdelaine, C.; Zhu, D.; Nicholson, G.A.; Grew, S.; Ryan, M.M.; Funalot, B. Histopathological Findings in Hereditary Motor and Sensory Neuropathy of Axonal Type With Onset in Early Childhood Associated WithMitofusin 2Mutations. J. Neuropathol. Exp. Neurol. 2008, 67, 1097–1102. [CrossRef]
- Verhoeven, K. MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain 2006, 129, 2093–2102. [CrossRef]
- Baloh, R.H.; Schmidt, R.E.; Pestronk, A.; Milbrandt, J. Altered Axonal Mitochondrial Transport in the Pathogenesis of Charcot-Marie-Tooth Disease from Mitofusin 2 Mutations. J. Neurosci. 2007, 27, 422–430. [CrossRef]
- Detmer, S.A.; Chan, D.C. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 2007, 176, 405–414. [CrossRef]
- Chen, H.; Chan, D.C. Critical dependence of neurons on mitochondrial dynamics. Curr. Opin. Cell Biol. 2006, 18, 453–459. [CrossRef]
- Feely, S.; Laura, M.; Siskind, C.; Sottile, S.; Davis, M.; Gibbons, V.; Reilly, M.; Shy, M. MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology 2011, 76, 1690–1696. [CrossRef]
- Larrea, D.; Pera, M.; Gonnelli, A.; Quintana-Cabrera, R.; Akman, H.O.; Guardia-Laguarta, C.; Velasco, K.R.; Area-Gomez, E.; Dal Bello, F.; De Stefani, D.; et al. MFN2 mutations in Charcot–Marie–Tooth disease alter mitochondria-associated ER membrane function but do not impair bioenergetics. Hum. Mol. Genet. 2019, 28, 1782–1800. [CrossRef]
- Dorn, G.W. Mitofusin 2 Dysfunction and Disease in Mice and Men. Front. Physiol. 2020, 11, 782. [CrossRef]
- Rizzo, F.; Ronchi, D.; Salani, S.; Nizzardo, M.; Fortunato, F.; Bordoni, A.; Stuppia, G.; Del Bo, R.; Piga, D.; Fato, R.; et al. Selective mitochondrial depletion, apoptosis resistance, and increased mitophagy in human Charcot-Marie-Tooth 2A motor neurons. Hum. Mol. Genet. 2016, 25, 4266–4281. [CrossRef]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc. Natl. Acad. Sci. 2014, 111, 3116–3121. [CrossRef]
- Yang, T.-H.; Kang, E.Y.-C.; Lin, P.-H.; Yu, B.B.-C.; Wang, J.H.-H.; Chen, V.; Wang, N.-K. Mitochondria in Retinal Ganglion Cells: Unraveling the Metabolic Nexus and Oxidative Stress. Int. J. Mol. Sci. 2024, 25, 8626. [CrossRef]
- Carelli, V.; Musumeci, O.; Caporali, L.; Zanna, C.; La Morgia, C.; Del Dotto, V.; Porcelli, A. M.; Rugolo, M.; Valentino, M. L.; Iommarini, L.; Maresca, A.; Barboni, P.; Carbonelli, M.; Trombetta, C.; Valente, E. M.; Patergnani, S.; Giorgi, C.; Pinton, P.; Rizzo, G.; Tonon, C.; Lodi, R.; Avoni, P.; Liguori, R.; Baruzzi, A.; Toscano, A.; Zeviani, M. Syndromic Parkinsonism and Dementia Associated with OPA 1 Missense Mutations. Annals of Neurology 2015, 78 (1), 21–38. [CrossRef]
- Kane, M. S.; Alban, J.; Desquiret-Dumas, V.; Gueguen, N.; Ishak, L.; Ferre, M.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Lenaers, G.; Reynier, P.; Chevrollier, A. Autophagy Controls the Pathogenicity of OPA 1 Mutations in Dominant Optic Atrophy. J Cellular Molecular Medi 2017, 21 (10), 2284–2297. [CrossRef]
- Sun, S.; Erchova, I.; Sengpiel, F.; Votruba, M. Opa1 Deficiency Leads to Diminished Mitochondrial Bioenergetics With Compensatory Increased Mitochondrial Motility. Investig. Opthalmology Vis. Sci. 2020, 61, 42–42. [CrossRef]
- Sun, C.; Wu, X.; Bai, H.-X.; Wang, C.; Liu, Z.; Yang, C.; Lu, Y.; Jiang, P. OPA1 haploinsufficiency due to a novel splicing variant resulting in mitochondrial dysfunction without mitochondrial DNA depletion. Ophthalmic Genet. 2021, 42, 45–52. [CrossRef]
- Liao, C.; Ashley, N.; Diot, A.; Morten, K.; Phadwal, K.; Williams, A.; Fearnley, I.; Rosser, L.; Lowndes, J.; Fratter, C.; et al. Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology 2017, 88, 131–142. [CrossRef]
- Sarzi, E.; Angebault, C.; Seveno, M.; Gueguen, N.; Chaix, B.; Bielicki, G.; Boddaert, N.; Mausset-Bonnefont, A.-L.; Cazevieille, C.; Rigau, V.; et al. The human OPA1delTTAG mutation induces premature age-related systemic neurodegeneration in mouse. Brain 2012, 135, 3599–3613. [CrossRef]
- Diot, A.; Agnew, T.; Sanderson, J.; Liao, C.; Carver, J.; Neves, R. P. D.; Gupta, R.; Guo, Y.; Waters, C.; Seto, S.; Daniels, M. J.; Dombi, E.; Lodge, T.; Morten, K.; Williams, S. A.; Enver, T.; Iborra, F. J.; Votruba, M.; Poulton, J. Validating the RedMIT/GFP-LC3 Mouse Model by Studying Mitophagy in Autosomal Dominant Optic Atrophy Due to the OPA1Q285STOP Mutation. Front. Cell Dev. Biol. 2018, 6, 103. [CrossRef]
- Zaninello, M.; Palikaras, K.; Naon, D.; Iwata, K.; Herkenne, S.; Quintana-Cabrera, R.; Semenzato, M.; Grespi, F.; Ross-Cisneros, F.N.; Carelli, V.; et al. Inhibition of autophagy curtails visual loss in a model of autosomal dominant optic atrophy. Nat. Commun. 2020, 11, 1–12. [CrossRef]
- Zaninello, M.; Palikaras, K.; Sotiriou, A.; Tavernarakis, N.; Scorrano, L. Sustained intracellular calcium rise mediates neuronal mitophagy in models of autosomal dominant optic atrophy. Cell Death Differ. 2021, 29, 167–177. [CrossRef]
- Moulis, M.F.; Millet, A.M.; Daloyau, M.; Miquel, M.-C.; Ronsin, B.; Wissinger, B.; Arnauné-Pelloquin, L.; Belenguer, P. OPA1 haploinsufficiency induces a BNIP3-dependent decrease in mitophagy in neurons: relevance to Dominant Optic Atrophy. J. Neurochem. 2017, 140, 485–494. [CrossRef]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, Å.B. Microtubule-associated Protein 1 Light Chain 3 (LC3) Interacts with Bnip3 Protein to Selectively Remove Endoplasmic Reticulum and Mitochondria via Autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [CrossRef]
- Wang, D.; Wang, J.; Bonamy, G.M.C.; Meeusen, S.; Brusch, R.G.; Turk, C.; Yang, P.; Schultz, P.G. A Small Molecule Promotes Mitochondrial Fusion in Mammalian Cells. Angew. Chem. Int. Ed. Engl. 2012, 51, 9302–9305. [CrossRef]
- Lin, Y.; Wang, D.; Li, B.; Wang, J.; Xu, L.; Sun, X.; Ji, K.; Yan, C.; Liu, F.; Zhao, Y. Targeting DRP1 with Mdivi-1 to correct mitochondrial abnormalities in ADOA+ syndrome. J. Clin. Investig. 2024, 9. [CrossRef]
- Szabo, A.; Sumegi, K.; Fekete, K.; Hocsak, E.; Debreceni, B.; Setalo, G.; Kovacs, K.; Deres, L.; Kengyel, A.; Kovacs, D.; et al. Activation of mitochondrial fusion provides a new treatment for mitochondria-related diseases. Biochem. Pharmacol. 2018, 150, 86–96. [CrossRef]
- Alberti, C.; Rizzo, F.; Anastasia, A.; Comi, G.; Corti, S.; Abati, E. Charcot-Marie-tooth disease type 2A: An update on pathogenesis and therapeutic perspectives. Neurobiol. Dis. 2024, 193, 106467. [CrossRef]
- Ng, W.S.V.; Trigano, M.; Freeman, T.; Varrichio, C.; Kandaswamy, D.K.; Newland, B.; Brancale, A.; Rozanowska, M.; Votruba, M. New avenues for therapy in mitochondrial optic neuropathies. Ther. Adv. Rare Dis. 2021, 2. [CrossRef]
- Kumar, V.; Kodam, P.; Maity, S. Targeting protein interaction networks in mitochondrial dynamics for neurodegenerative diseases. J. Proteins Proteom. 2024, 15, 309–328. [CrossRef]
- Zanfardino, P.; Longo, G.; Amati, A.; Morani, F.; Picardi, E.; Girolamo, F.; Pafundi, M.; Cox, S.N.; Manzari, C.; Tullo, A.; et al. Mitofusin 2 mutation drives cell proliferation in Charcot-Marie-Tooth 2A fibroblasts. Hum. Mol. Genet. 2022, 32, 333–350. [CrossRef]
- Zanfardino, P.; Amati, A.; Petracca, E.A.; Santorelli, F.M.; Petruzzella, V. Torin1 restores proliferation rate in Charcot-Marie-Tooth disease type 2A cells harbouring MFN2 (mitofusin 2) mutation. 2022, 41, 201–206. [CrossRef]
- Zanfardino, P.; Petruzzella, V. Autophagy and proliferation are dysregulated in Charcot-Marie-Tooth disease type 2A cells harboring MFN2 (mitofusin 2) mutation. Autophagy Rep. 2022, 1, 537–541. [CrossRef]
- Zanfardino, P.; Amati, A.; Doccini, S.; Cox, S.N.; Tullo, A.; Longo, G.; D’erchia, A.; Picardi, E.; Nesti, C.; Santorelli, F.M.; et al. OPA1 mutation affects autophagy and triggers senescence in autosomal dominant optic atrophy plus fibroblasts. Hum. Mol. Genet. 2024, 33, 768–786. [CrossRef]
- Shimobayashi, M.; Hall, M.N. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [CrossRef]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 51. [CrossRef]
- Xu, K.; Chen, G.; Li, X.; Wu, X.; Chang, Z.; Xu, J.; Zhu, Y.; Yin, P.; Liang, X.; Dong, L. MFN2 Suppresses Cancer Progression through Inhibition of mTORC2/Akt Signaling. Sci Rep 2017, 7 (1), 41718.
- Xue, R.; Meng, Q.; Lu, D.; Liu, X.; Wang, Y.; Hao, J. Mitofusin2 Induces Cell Autophagy of Pancreatic Cancer through Inhibiting the PI3K/Akt/mTOR Signaling Pathway. Oxidative Med. Cell. Longev. 2018, 2018, 2798070. [CrossRef]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 1–19. [CrossRef]
- Balaban, R. S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120 (4), 483–495.
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD + elevation. Aging Cell 2016, 15, 416–427. [CrossRef]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2016, 13, 99–113. [CrossRef]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal. 2019, 12. [CrossRef]
- Romanello, V.; Scalabrin, M.; Albiero, M.; Blaauw, B.; Scorrano, L.; Sandri, M. Inhibition of the Fission Machinery Mitigates OPA1 Impairment in Adult Skeletal Muscles. Cells 2019, 8, 597. [CrossRef]
- Lee, S.; Jeong, S.-Y.; Lim, W.-C.; Kim, S.; Park, Y.-Y.; Sun, X.; Youle, R.J.; Cho, H. Mitochondrial Fission and Fusion Mediators, hFis1 and OPA1, Modulate Cellular Senescence. J. Biol. Chem. 2007, 282, 22977–22983. [CrossRef]
- Stab, B.R.; Martinez, L.; Grismaldo, A.; Lerma, A.; Gutiérrez, M.L.; Barrera, L.A.; Sutachan, J.J.; Albarracín, S.L. Mitochondrial Functional Changes Characterization in Young and Senescent Human Adipose Derived MSCs. Front. Aging Neurosci. 2016, 8, 299. [CrossRef]
- Navratil, M.; Terman, A.; Arriaga, E.A. Giant mitochondria do not fuse and exchange their contents with normal mitochondria. Exp. Cell Res. 2008, 314, 164–172. [CrossRef]
- Bhatia-Kiššová, I.; Camougrand, N. Mitophagy is not induced by mitochondrial damage but plays a role in the regulation of cellular autophagic activity. Autophagy 2013, 9, 1897–1899. [CrossRef]
- Eskelinen, E.-L. Maturation of Autophagic Vacuoles in Mammalian Cells. Autophagy 2005, 1, 1–10. [CrossRef]
- Morán, M.; Delmiro, A.; Blázquez, A.; Ugalde, C.; Arenas, J.; Martín, M. A. Bulk Autophagy, but Not Mitophagy, Is Increased in Cellular Model of Mitochondrial Disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2014, 1842 (7), 1059–1070. [CrossRef]
- Deffieu, M.; Bhatia-Kiššová, I.; Salin, B.; Klionsky, D.J.; Pinson, B.; Manon, S.; Camougrand, N. Increased levels of reduced cytochrome b and mitophagy components are required to trigger nonspecific autophagy following induced mitochondrial dysfunction. J. Cell Sci. 2013, 126, 415–426. [CrossRef]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2012, 1823, 2297–2310. [CrossRef]
Figure 1.
Secondary structure and commonly reported variants of MFN2 and OPA1 proteins. The schematic representation of MFN2 (above) highlights its structural domains: Helic bundle domain, p21 RAS domain, GTPase domain, heptad repeat domain 1 (HR1), a proline-rich domain, transmembrane domain, and heptad repeat domain 2 (HR2), each depicted in distinct colours. Below, OPA1 is illustrated with its key structural features: MTS (Mitochondrial Targeting Sequence), a transmembrane domain, Sp1, heptad repeat domain 1, GTPase domain, middle domain, and GED (GTPase Effector Domain), also represented with corresponding colours. The most reported variants for both proteins are indicated in their respective domains.
Figure 1.
Secondary structure and commonly reported variants of MFN2 and OPA1 proteins. The schematic representation of MFN2 (above) highlights its structural domains: Helic bundle domain, p21 RAS domain, GTPase domain, heptad repeat domain 1 (HR1), a proline-rich domain, transmembrane domain, and heptad repeat domain 2 (HR2), each depicted in distinct colours. Below, OPA1 is illustrated with its key structural features: MTS (Mitochondrial Targeting Sequence), a transmembrane domain, Sp1, heptad repeat domain 1, GTPase domain, middle domain, and GED (GTPase Effector Domain), also represented with corresponding colours. The most reported variants for both proteins are indicated in their respective domains.
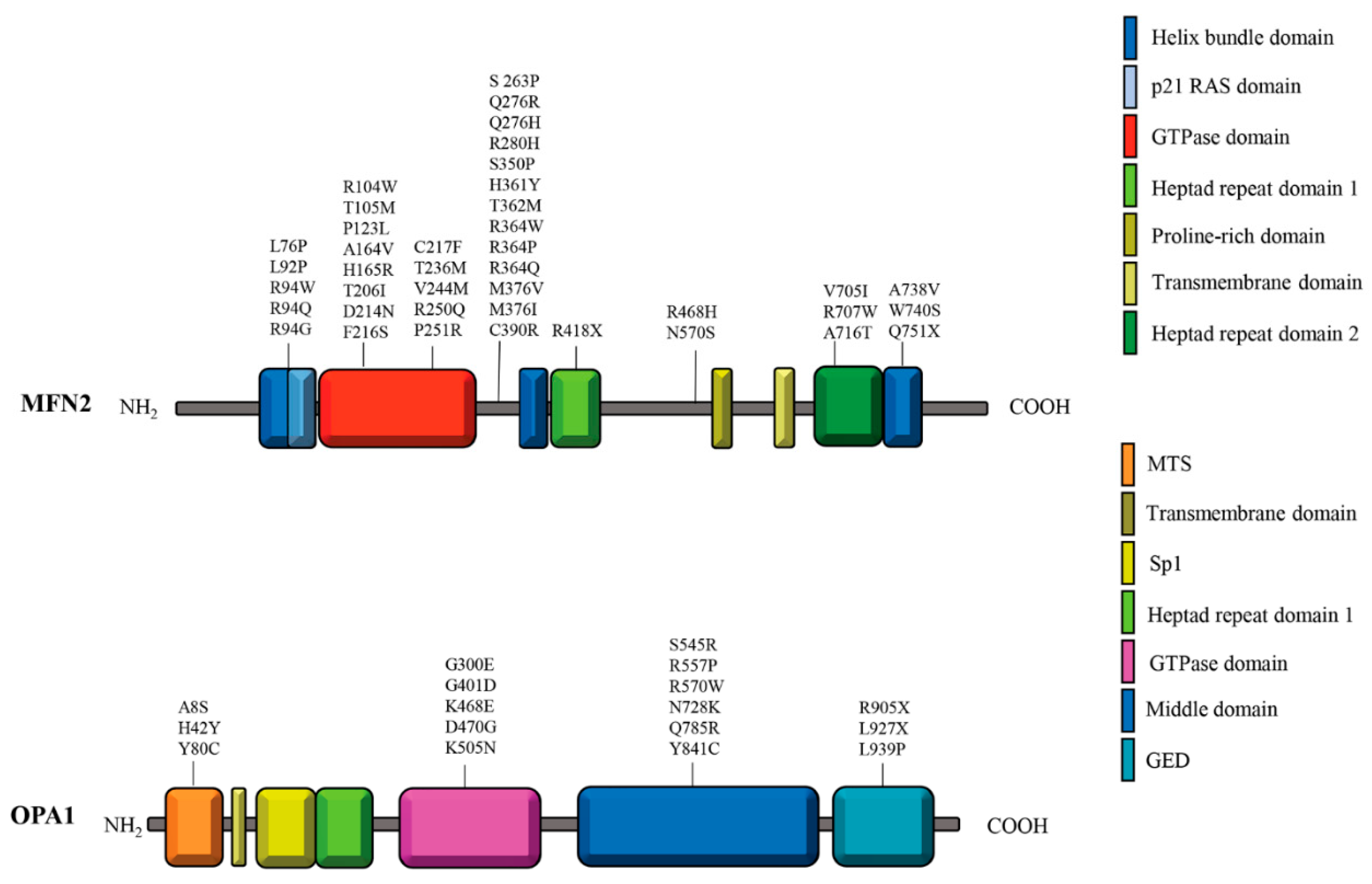
Figure 2.
Schematic representation of altered processes in CMT2A and ADOA fibroblasts.
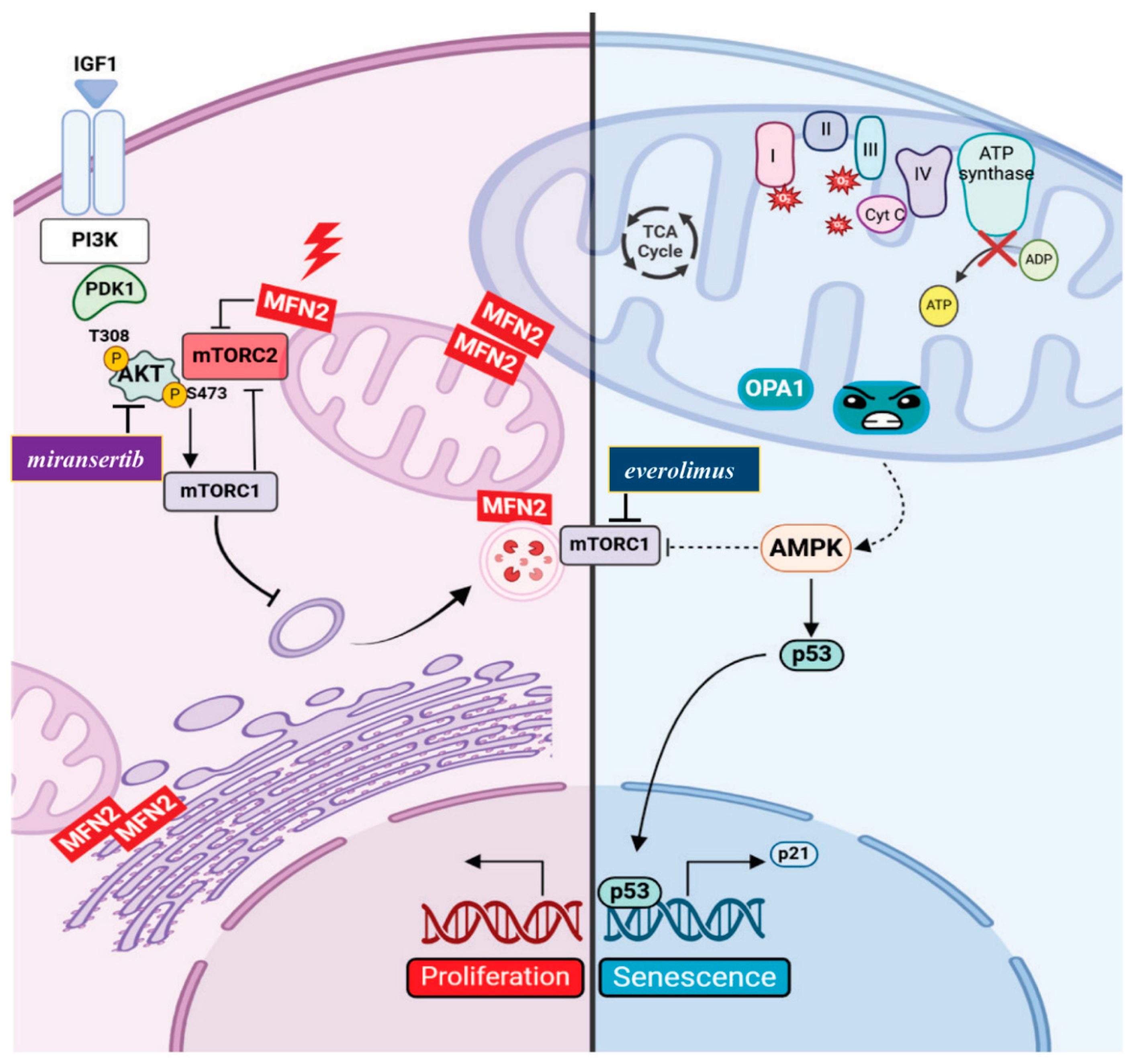
Figure 3.
The Balance Between Mitochondrial Dynamics and Cellular Homeostasis.
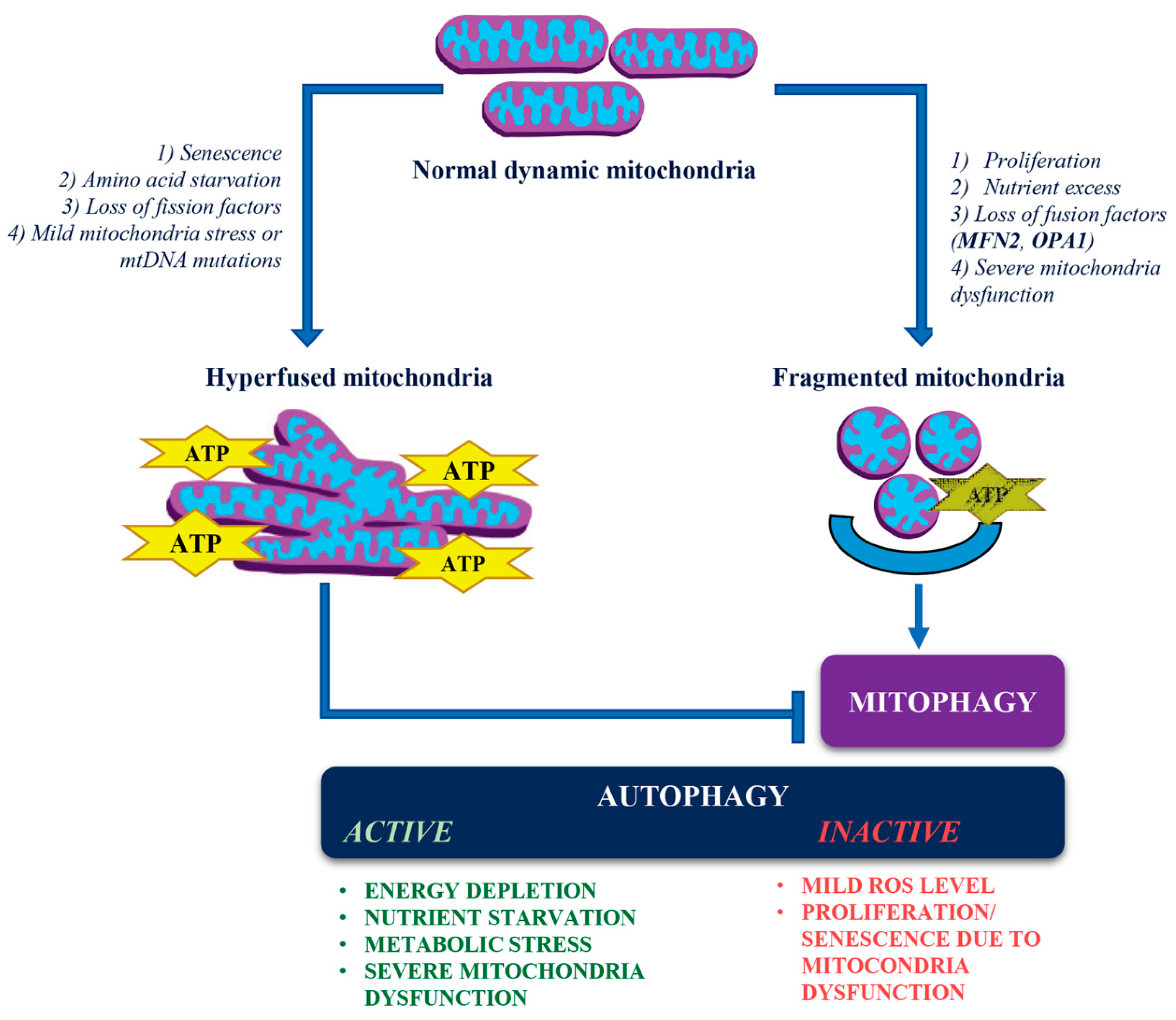

Table 1.
Mitochondrial alterations in CMT2A2 cellular models.
| Cell Type | MFN2 mutation | Remarks | References |
| Skin fibroblasts from patients with CMT2A | 1) P123L 2) L92P |
1) Mitochondrial fragmentation and abnormal mitochondrial accumulation around the nucleus 2) Mitochondrial fragmentation |
Verhoeven K. et al., 2006 |
| Skin fibroblasts from 4 patients with CMT2A2 | M21V R364Q A166T |
Reduced cellular respiration: uncoupling causing decreased ATP/O Reduced ΔѰm |
Loiseau D. et al., 2007 |
| Skin fibroblasts from patients with CMT2A | T105M I213T V273G |
Mitochondrial morphology, mtDNA integrity and and respiratory enzyme activities are unchanged Extensive mitochondrial fusion |
Amiott E. A. et al., 2008 |
| Skin fibroblasts from patients with CMT2A | p.D210V | Respiratory chain defects Multiple mtDNA deletions Defect in mtDNA damage repair system Fragmentation of the mitochondrial network |
Rouzier C. et al., 2012 |
| Skin fibroblasts derived from 4 patients with CMT2A | M376V R707P V226_S229del Q74R |
Mitochondrial respiratory chain dysfunction Decreased mtDNA copy number High levels of mtDNA depletion |
Vielhaber S. et al., 2013 |
| Motor neurons derived from iPSCs of patients with CMT2A | R364W | Reduced mitochondrial trafficking with slower anterograde and retrograde velocities along axons. Electrophysiological impairments including increased excitability, higher sodium current density, and reduced inactivation of voltage-dependent sodium and calcium channel |
Saporta et al., 2015 |
| Motor neurons derived from iPSCs of patients with CMT2A |
A383V | Decreased respiratory chain activity: complexes II and III Reduced mitochondrial mass and mtDNA content No mtDNA alterations Decrease of mitochondrial trafficking leading to perinuclear aggregation No survival and morphometric defects Increased resistance to apoptosis Increased autophagic and mitophagic flux |
Rizzo F. et al., 2016 |
| Motor neurons derived from iPSCs of patients with CMT2A | R94Q | Abnormal mitochondrial morphology: shorter mitochondrial length within neurites, presence of abnormal mitochondria with loss of crista Reduction in the percentage of moving mitochondria Decrease in ATP levels in neurites Increased toxicity sensitivity to vincristine and paclitaxel |
Ohara et al., 2017 |
| Motor neurons derived from iPSCs of patients with CMT2A | R94Q | Hyper-connectivity: increase in burst rate Alterations in mitochondrial morphology: reduced mitochondrial elongation and increase in circularity Impairment in axonal transport: decrease in the speed of mitochondria and lysosomes and the proportion of active mitochondria and lysosomes moving within the cells Defects in OxPhos: decrease in mitochondrial basal respiration. Transcriptomic analysis: enrichment in “PI3K-AKT signalling” and respiratory chain pathway |
Van Lent et 2021 |
| Skin fibroblasts derived from CMT2A patients | R364W M376V W740S |
Mitochondrial mass and mtDNA levels are unchanged Moderate disturbances in Ca2+ homeostasis Reduced ER-mitochondria contacts |
Larrea D. et al., 2019 |
| Skin fibroblasts from CMT2A patient | C217F | Mitochondrial mass and mtDNA levels and integrity unchanged Mitochondrial fragmentation Reduced ΔѰm Reduction of respiratory chain complexes activity Transcriptomic analysis: enrichment in “PI3K-AKT signalling” Reduced autophagy and increased cellular proliferation (mTORC2/AKT activation) |
Zanfardino P. et al., 2022 |
Table 2.
Mitochondrial Alterations in ADOA/ADOA Plus Cellular Models.
| Cell Type | OPA1 mutation | Remarks | References |
| Fibroblasts from 3 patients with ADOA plus | V903Gfs3, E221K QT86Sfs15, H957Y QT86Sfs*15, H957Y |
Increased mitochondrial fragmentation Depletion of mtDNA Altered mitochondrial localization Increased mitophagy flux |
Liao C. et al., 2017 |
| Fibroblasts from 7 ADOA patients |
|
1) and 2) Fragmented and punctiform mitochondria 3) Mild mitochondrial fragmentation 1), 2), and 3) Loss of mitochondrial volume 1) and 2) Mild uncoupling of oxidative phosphorylation 4) and 6) Severe defects in oxidative phosphorylation Altered autophagy 4) and 5) Mild mtDNA depletion |
Kane M. S. et al., 2017 |
| Lymphoblastoid cells derived from ADOA patients | P400A | Decreased mtDNA copy number Reduced levels of 4 mtDNA-encoded polypeptides Respiratory capacity defects ATP synthesis defects Altered mitochondrial membrane potential Increased ROS production Increased apoptosis Mitochondrial morphological defects (fragmentation and swelling) |
Zhang et al., 2017 |
| Lymphoblastoid cell lines from ADOA patient | c.1444–2A>C (splicing variants, deletion of the 15th exon in mRNA transcript, low protein levels) | Respiratory chain activity defects More punctate mitochondria clustered in the perinuclear region, No marked depletion of mtDNA or mitochondrial mass Reduced ATP synthesis Reduced ΔѰm Increased ROS production Increased mitophagy |
Sun C. et al., 2021 |
| Skin fibroblasts from ADOA plus patient | H42Y | Mitochondrial mass unchanged mtDNA levels slighly increased mtDNA integrity unchanged Mitochondrial fragmentation Reduced ΔѰm Reduction of respiratory chain complexes activity Increased ROS production Transcriptomic analysis: enrichment in “p21WAF1/CIP1 and p53 pathways” along with downregulation of mitotic cell cycle genes Reduced autophagy and increased expression of senescence markers (SA-β galactosidase; p53 and p21) associated with lower mTORC2 activity |
Zanfardino P. et al., 2024 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



