
Submitted:
03 February 2025
Posted:
04 February 2025
You are already at the latest version
Abstract
Considering the importance of organic functionalization of MOFs, we here report a simple, tunable and efficient one-step post-modification procedure for introducing amino and carboxylic groups into the mesoporous metal-organic framework Al- and Cr-MIL-101-NH2 based on its reaction with alkyl bromides. This procedure allows also access to polyfunctionnalized MIL-101 decorated with both carboxylic and primary amino groups. Other chemical functions, such as alcohols and alkynes, were also successfully introduced by this method.
Keywords:
Metal-organic frameworks
; Post-synthetic modification
; PSM
; (Al)MIL101-NH2
; (Cr)MIL101-NH2
; alkylation
; alkyl halide
1. Introduction
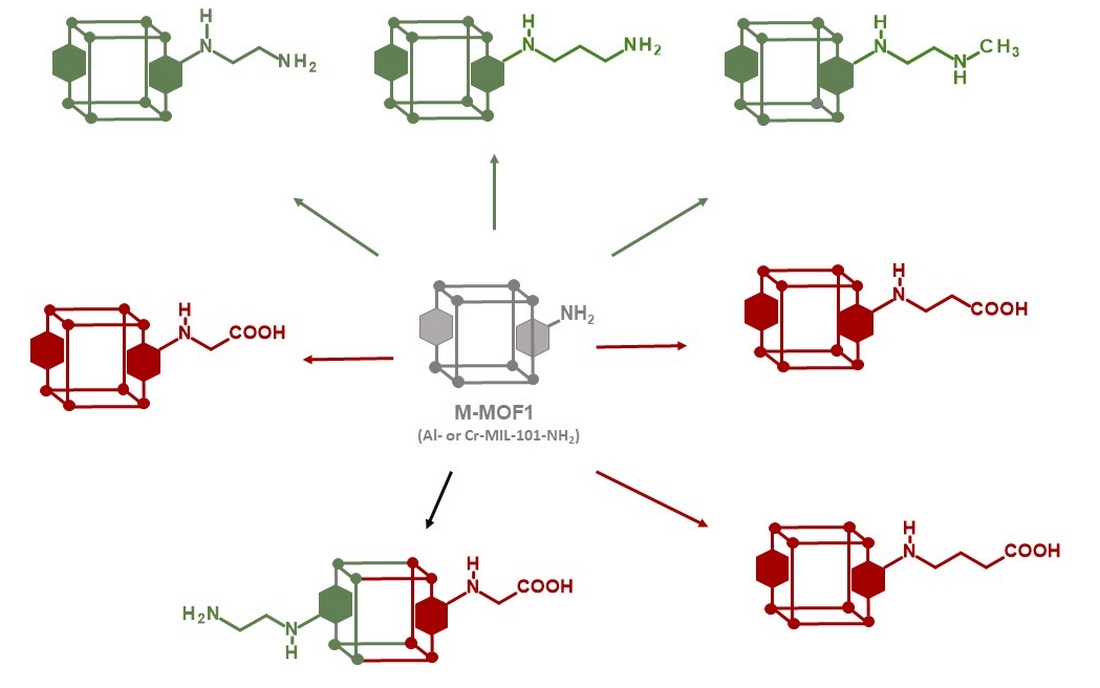
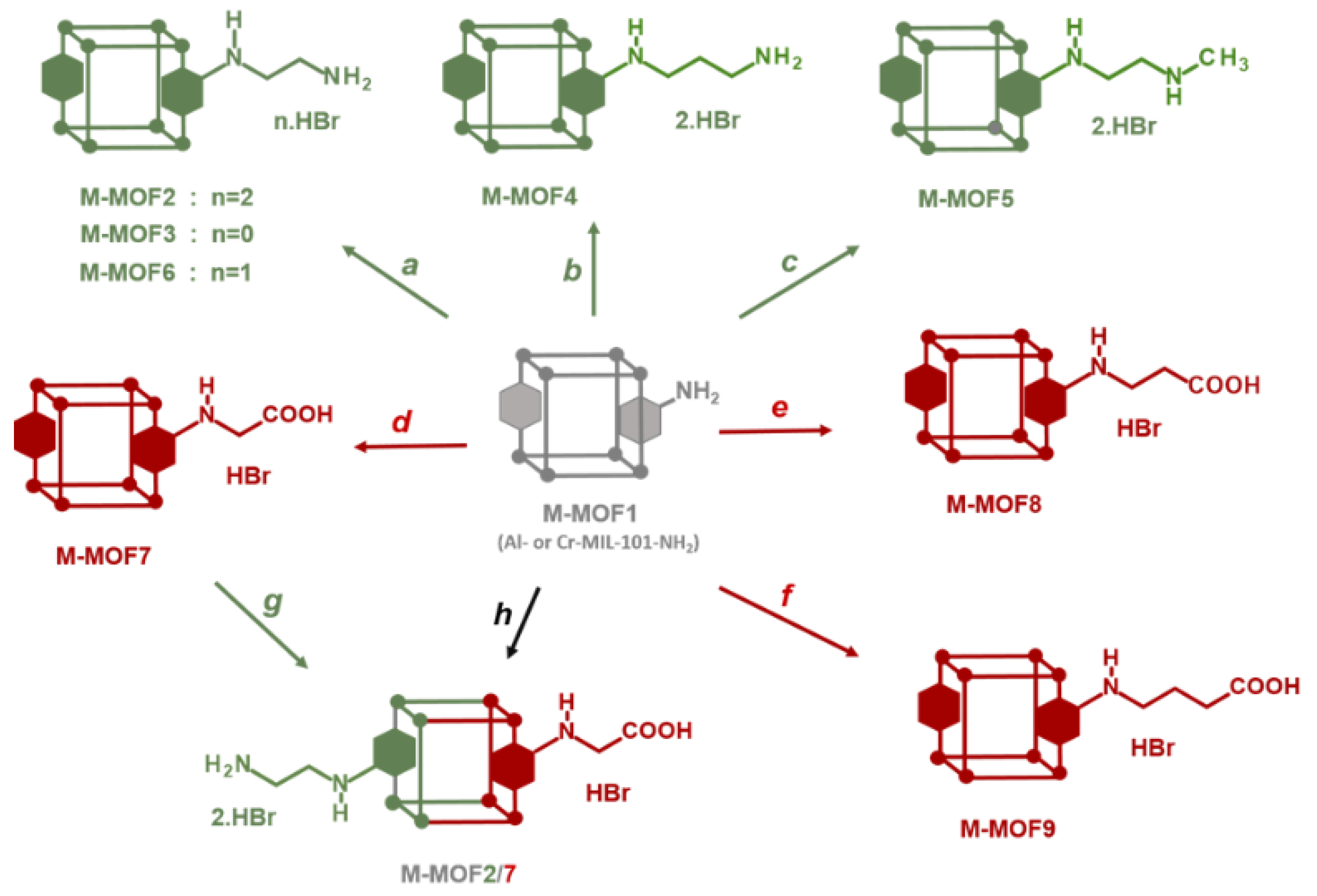
Post-synthetic modification (PSM) of metal organic frameworks (MOFs) is a process involving conversion of pendent groups located at the surface and in the pores of MOFs, aiming to change their physical or chemical properties [1,2,3,4,5,6]. This process thus gives access to grafted MOFs that could not be prepared by direct synthesis from a modified ligand. In most cases, PSM is achieved by a chemical reaction directly on the crystalline MOF powder. The resulting post-modified MOF should retain its crystallinity in order to ensure its topological integrity which can be confirmed by X-ray powder diffraction, in addition to other classical analysis methods such as N2 physisorption, TEM / SEM microscopy or NMR spectroscopies. In particular 1H- NMR, after MOF hydrolysis in acidic or basic conditions, is one of the most convenient method to identify the product(s) of PSMs, and also to calculate the yield and the percentage of modification. Amino-functionalized MOFs, such as MOFs synthesized from 1,4-benzenedicarboxylate (BDC) and post modified after nitration / reduction treatment [7], or MOFs directly synthesized from amino-1,4-benzenedicarboxylate (BDC-NH2) like MIL-101-NH2 [8] or IRMOF-3 [9], have often be used as starting materials for post-modifications, due to the reactivity of the aniline functional group towards a variety of electrophiles. On this basis, PSM have been carried out i) with alkyl / cyclic anhydrides or acyl chlorides to form amide bonds [10,11,12,13], ii) with diphosgene and thiophosgene to form isocyanates and isothiocyanates [13], then converted to ureas or carbamates by reacting with amine or alcohol derivatives, iii) with isocyanates to afford ureas [14,15], iv) with aldehydes to obtain imines [16,17] that can be then reduced to amines, or v) with tert-butyl nitrite and trimethylsilyl azide to convert the amino group to an azide group [18,19,20,21], that can subsequently be involved in a CuAAC reaction (“click” chemistry) by reacting with alkyne derivatives. Non-exhaustively, other reactants able to react with the amino group were used, such as cyanuric chloride [22], nitrogen monoxide [23], aziridine [24,25], 1,3-propane sultone [24,25], sodium nitrite in the presence of aqueous acid (diazotisation) [26], alkenes containing electron withdrawing groups [27], and other. Surprisingly, the use of alkyl halides, and in particular alkyl bromides, has been little exploited to post-modify amino-functionalized MOFs [28,29]. In this work, we show how this basic reaction between MIL-101-NH2 and alkyl bromides allows access to a series of post-modified MOFs, namely Cr- and Al-MOFn (n = 2-9) with diverse amino and carboxylic groups, and also to polyfunctionnalized MOFs (Cr- and Al-MOF2/7, Scheme 1) with both acid and primary amino groups.
Scheme 1.
Access to post-modified amino (●) and carboxylic (●) M-MOFs (M = Al or Cr).
2. Materials and Methods
2.1. Synthesis of Al-MIL-101-NH2 (Al-MOF1)
Al-MIL-101-NH2 was prepared according to a slightly modified procedure described by Hartmann and Fischer [30]: In a 100 mL round bottom flask 272 mg (1.5 mmol) of 2-aminoterephthalic acid were dissolved in 60 mL of DMF under magnetic stirring and heated to 110 °C (oil bath). To the solution, 724 mg (3 mmol) of AlCl3.6 H2O were added in 7 equal portions with a time delay of 15 min between two additions. After the last portion was added, the temperature was kept at 110 °C for 3 h under magnetic stirring, and for an additional 16 h without stirring. After cooling down to room temperature, the yellow solid was isolated by filtration on fritted-glass (porosity 3), washed with 20 mL of DMF and 20 mL of ethanol, and then washed (“evacuated”) by Soxhlet extraction with ethanol (130 mL) for 96 H. The yellow powder was then dried for 24h at 85°C to obtain Al-MOF1, which was stored in a desiccator until use.
2.2. Synthesis of Cr-MIL-101-NH2 (Cr-MOF1)
Cr-MIL-101-NH2 was prepared according a procedure described by Lin and coll. [31]: Chromium(III) nitrate nonahydrate (99%, 808 mg, 2 mmol), 2-aminoterephthalic acid (99, 365.9 mg, 2 mmol) and sodium hydroxide (200 mg, 5 mmol) were dispersed in deionized water (15 ml) in a 60 ml Schott vial. The solution was stirred for 5 minutes at room temperature, and then heated at 150 °C in an oven for 12 h. After cooling, the green precipitate was collected by centrifugation, washed with DMF (3 x 10 mL) at room temperature, and then with hot ethanol (30 mL) at 100° C for 24 h in a 60 ml Schott vial. The precipitate was filtered over fritted-glass (porosity 3), dried at 80° C in air and stored in a desiccator until use.
2.3. MOFs Post-Synthetic Modifications
All post-synthetic modified MOF were prepared using an Anton Paar Monowave 50 apparatus (see procedures, data and spectra in the Supporting Information). Al-MOF2 was also prepared following a classical procedure (see Supporting Information). PSM-yields were determined by NMR and LC-MS as follows: A MOF sample (≃ 10 mg) was suspended in D2O (1 mL), and a solution of 40 wt% NaOD in D2O (2 µL) was added. For aluminium MOFs (Al-MOF), the NaOD/D2O solution was allowed to stand at room temperature for 2 hours, and the terephthalate derivatives (deprotonated forms) were then analyzed by 1H NMR recorded on a Bruker Advance 300 spectrometer at 300 MHz. The residual proton signal of the deuterated water was used as an internal reference (δ = 4.79 ppm). For chromium MOFs (Cr-MOF), the NaOD/D2O solution was allowed to stand at room temperature for 16 hours, then centrifuged for 2 minutes at 10,000 rpm (Biofuge Fresco HeraeusTM centriguge), and filtered on a short column (through a Pasteur pipette) of alumina (aluminum oxide 90 active neutral from Merck) to remove paramagnetic chromium salts, and the terephthalate derivatives (deprotonated forms) were then analyzed by 1H NMR recorded on a Bruker Advance 300 spectrometer at 300 MHz. In most of the cases, yields were calculated by comparing the integrations of the more deshielded aromatic protons doublets of unmodified ligand (BDC2--NH2, δ = 7.69) and modified ligand, but integration of other aromatic protons can be considered (if overlapping for example) for yield determination.
2.4. LC-MS Yields Determination
NaOD/D2O solutions used for NMR analyses were also used for LC-MS: the samples were analyzed by analytical UPLC (UPLC Acquity Waters, PDA/MS detector) with a C18 column (BEH C18, 1.7 µm, 2.1 mm x 100 mm). Water with 0.1 % formic acid (A) and methanol with 0.1 % formic acid (B) were used as mobile phases, with a flow rate of 0.3 mL.min-1, using the following elution method: 0 min, 5 % B; 5 min, 100 % B; 7 min, 100 % B, then returned to initial conditions. The SQD was operated in an electrospray negative or positive ion mode by applying a voltage of 3.5 kV to the ESI capillary and the cone voltage was set at 30 V. The yields were calculated from integration of the peaks in the 254 nm chromatograms.
3. Results and Discussion
3.1. Postmodification with Amino Groups
The amino group, especially primary amino group, is probably the most important reactive group for further chemical modifications of MOFs. Reaction of Al-MIL-101-NH2 towards 2-bromoethylamine hydrobromide as alkyl halide was first investigated as initial reaction (path a, Scheme 1). The starting MOF Al-MIL-101-NH2 (Al-MOF1) was prepared from 2-aminoterephthalic acid (H2BDC-NH2) and aluminium chloride hexahydrate in DMF, evacuated and activated according to a slightly modified procedure [30], and characterized by powder X-ray diffraction, FT-IR and BET surface area measurement. BET surfaces of 2700 m2.g-1 were routinely obtained from independent experiments, values in good agreement with previous reported data [30]. Post-synthetic modification was then accomplished by reacting Al-MOF1 with 2-bromoethylamine hydrobromide in toluene as solvent. When performed at 110 °C (toluene reflux) under atmospheric pressure, followed by ethanol Soxhlet-extraction and drying, the expected ethyl amino-modified MOF, as di-hydrobromide salt (Al-MOF2) was obtained, with no noticeable secondary products. Conversion yields for post-modifications were estimated both from the LC-MS chromatogram of the NaOH-digested products of a sample of Al-MOF2 (Figure S7, ESI), and by 1H-NMR after its hydrolysis with aqueous sodium deuteroxide by comparing the ratio of aromatic protons doublets at δ =7.69 of unmodified ligand BDC2--NH2 and δ = 7.75 ppm of modified ligand 2-((2-aminoethyl) amino)terephthalate (BDC2--NH-(CH2)2-NH2) (Figure 1). Conversion of ≃ 35-40 % were routinely obtained from several independent experiments. Compared to the parent ligand, a deshielded chemical shift was observed for aromatic protons in ortho (Hc’) and meta (Ha’) position relative to the amino group in the modified ligand, while the para proton (Hb’) is slightly more shielded (Figure 1). In addition to the aromatic protons, two triplets at 2.82 and 3.27 ppm corresponding to the ethylene groups (Hd’ and He’) arise in the spectrum. For an unambiguous structure determination, a pure sample of the modified ligand 2-((2-aminoethyl)amino)terephthalic acid (H2BDC-NH-(CH2)2-NH2) was isolated from NaOH-digestion of approximatively 100 mg of Al-MOF2 by semi-preparative HPLC, and characterized by 1H-NMR, IR and MS analysis (Figure S1-S3, ESI).
The powder X-Ray diffraction (PXRD) pattern of the modified MOF Al-MOF2 was found in accordance with the basic framework of MIL-101-family structures, but with a severe loss in crystallinity compared to the starting MOF Al-MOF1 (Figure 2), as evidenced by the total signal loss in the 2-7 degree range. As expected, the ethylamino groups within the pores of Al-MOF2, combined with loss of crystallinity, lead to a marked decrease in its BET specific surface area as compared to Al-MOF1, from 2700 to 146 m2.g-1 (Figure S4, ESI). Upon a simple treatment of di-hydrobrominated Al-MOF2 with a solution of triethylamine in chloroform, free amino Al-MOF3 was obtained. When the yellow powder of the later MOF was suspended in an acetonic solution of ninhydrin, a chemical commonly used to detect primary and secondary amines, the color of the resulting still unsoluble powder changed gradually to deep-purple, the maximum coloration being reached within one hour (Figure S5, ESI), demonstrating the presence of reactive amino groups. In the same conditions, the color of Al-MIL-101-NH2 (Al-MOF1) remained unchanged, even after hours. To further demonstrate the reactivity of the primary amine groups in the post-modified MOF, Al-MOF3 was allow to react with p-tolylisocyanate in refluxing toluene, and, as expected, the corresponding urea formed with a rather good yield of 74 % (Figure S6, ESI).
For various potential applications of post-synthetically modified MOFs, it remains necessary to control the degree of rafting to accurately modulate the effectively available chemical functions. On this basis, reaction of Al-MIL-101-NH2, on a 100-mg MOF scale, with 2-bromoethylamine hydrobromide as model reaction was further investigated by varying the reaction temperature above toluene boiling point (110 °C) in synthesis reactors under generated pressure (performed in a Monowave 50 apparatus, Anton Paar). As expected, increased PSM-yields, from 57 % at 110 °C up to 88 % at 140 °C, were obtained by increasing temperature in toluene (Figure 3). All these reactions yielding to dihydrobrominated amino Al-MOF2 were found to be generally clean, even at high temperature, with no significant formation of polyalkylated or polymerized products. This absence of secondary products is explained by the fact that the process used, which consists in using 2-bromoethylamine hydrobromide without the addition of any base, releases during the reaction one equivalent of hydrobromic acid, which protonates the amino group of the terephthalate thus inhibiting any subsequent N-alkylation/polymerization reaction.
However, all the post-modified aluminium MOFs obtained at these different temperatures showed a strong loss of crystallinity, as attested by their PXRD spectra (not shown), similar or worse to that obtained at 110 °C (Figure 2), suggesting a marked degradation, or even collapse, of the framework. Consequently, the ethylamino post-synthetic modification starting from the same amino alkyl bromide derivative, 2-bromoethylamine hydrobromide, was carried out on the most rugged chromium MOF of the MIL family, i.e. Cr Al-MIL-101-NH2 (Cr-MOF 1). This Cr-MOF has a high porosity, unsaturated metal sites, and is, above all, known to be extremely stable compared to its iron and aluminum isoreticular MOFs, which are more prone to hydrolysis [32]. Cr-MOF1 was first prepared from 2-aminoterephthalic acid (H2BDC-NH2) and chromium nitrate in hydrothermal conditions, as already described [31]. Its crystalline structure was confirmed by PXRD (Figure 4), and from N2 sorption isotherm a BET specific surface of 1620 m2.g-1 was calculated in agreement with previous reported data [31,33]. The reaction of Cr-MOF1 with 2-bromoethylamine hydrobromide was then performed at 120 °C in toluene under generated pressure (in an Anton Paar Monowave 50 synthesis reactor) and the expected modified MOF, Cr-MOF2 was successfully obtained, with a PSM-yield estimated by 1H NMR in the range of 62-67 % from three independent experiments (Figure S17, ESI), that is, of the same order of magnitude than that obtained with the aluminium MOF (around 57 %). However, the NMR spectrum was found not as clean as that obtained with its aluminium counterpart, which could be due to amine grafting on coordinatively unsaturated chromium centers, or the formation of metal-catalyzed by-products. The PXRD pattern of Cr-MOF2 was found in agreement with the basic three-dimensional architecture of MIL101-family structures (Figure 4), also with a marked decrease in crystallinity, but not as great as that observed with aluminum MOF Al-MOF2 (Figure 2). The Cr-MOF samples exhibit a N2 sorption isotherm with firstly some degree of adsorption in the low-pressure domain, thus revealing some microporosity as in Type I isotherms, and secondly a narrow hysteresis loop at relative pressures from about 0.4 to 1.0 (Figure 5), which points to a Type IV isotherm. Therefore, the isotherm could be described as composite between Types I and IV, which are two of the six types of isotherms recognized by the IUPAC classification and characteristic of microporous (pore size below 2 nm) and mesoporous (pore size in the range 2-50 nm) adsorbents, respectively [34]. Despite a slight higher rate of grafting for the chromium MOF, the decrease in the BET specific surface area was found to be less marked compared to aluminium MOFs, from 1620 for Cr-MOF1 to 350 m2.g-1 for Cr-MOF2 (Figure 5).
Reactions of Al- and Cr-MIL-101-NH2 with other alkyl bromides bearing amino groups were also investigated (path a-c, Scheme 1). In our basic experimental conditions, i.e. toluene as solvent at 120 °C in Anton Paar Monowave 50 synthesis reactors under generated pressure, 3-bromopropylamine hydrobromide and N-methyl-2-bromoethylamine hydrobromide successfully reacted with Al- and Cr-MIL-101-NH2 affording the corresponding post-modified MOFs in approximatively 90% (Al-MOF 4), 80% (Al-MOF 5), 62% (Cr-MOF 4) and 70% (Cr-MOF 5) yields, as estimated from 1H-NMR spectra (Figure 6 and Figure S18-19, ESI) and LC-MS chromatograms (Figure S8-9, ESI).
In the same conditions, when Al- or Cr-MIL-101-NH2 was allow to react with the BOC-amino protected alkyl bromide N-Boc-2-bromoethylamine, MOFs Al-MOF 6 and Cr-MOF6 with the unblocked amine as sole product in approximatively 75% yield for both compounds (Figure 6 and Figure S20, ESI) were obtained, suggesting that a thermal, or an acid-catalyzed de-BOC reaction occured, probably due to an acid catalysis process generated by HBr. In that case it must be hypothesized that the post-modified amino-MOFs were not obtained as di-hydrobromide salt, like in former reactions, but as mono-hydrobromide salt. PXRD spectra show that there is a significant loss of MOF crystallinity for all amine modified Cr-MOFs (Figure 4), and even more for aluminum MOFs (not shown), confirming the partial degradation of the MOFs-network architecture in the presence of amine reagents. On the other hand, all amino Cr-MOFs were found to retain a significant porosity, the total pore volume ranging from of 0.15 to 0.25 cm3 g-1. The corresponding specific surface areas follow a similar evolution, from 166 to 374 m2.g-1 (Figure 5 and FigureS28, ESI).
3.2. Postmodification with Carboxylic Acids
While the amino group reacts with electrophiles, the carboxyl group reacts, once activated, with nucleophiles. The ability to introduce these two complementary groups into the pores of MOFs should therefore facilitate access to a wide variety of functionalized MOF materials. On this basis, reactions of Al- and Cr-MIL-101-NH2 with different commercially available bromocarboxylic acid derivatives, i.e. bromoacetic acid, 3-bromopropionic acid and 4-bromobutyric acid, were studied (path d-f, Scheme 1). When reactions starting from Al-MOF1 were run in toluene at 120 °C, the expected post-modified MOFs, Al-MOF 7 and Al-MOF 8, were obtained from bromoacetic acid (approx. 57% yield) and 3-bromopropionic acid (approx. 38 % yield), respectively, as revealed by 1H-NMR (Figure 7 and Figure S10-11, ESI). However, in the same conditions, no reaction occurred when Al-MOF 1 was allow to react with 4-bromobutyric acid, as evidenced by NMR. In this case, it can be hypothesized that the aromatic amine function of the MIL-101-NH2 framework catalyzes the cyclization reaction of the 4-bromobutyric acid into γ-butyrolactone, but no further experiments were run to verify the presence of the latter compound in the reaction mixture.
However, a modified MOF with a butyric chain, Al-MOF 9, that could not be prepared from 4-bromobutyric acid, was successfully obtained directly from the methyl ester derivative methyl-4-bromobutyrate. The acidity of the reaction mixture due to the release of hydrobromic acid is likely responsible for the hydrolysis of the ester function during the reaction process (Figure 7 and Figure S12, ESI). The same reactions starting from bromoacetic acid and 3-bromopropionic acid were then performed on the chromium MOF Cr-MOF1, and as expected, the corresponding modified MOFs, namely Cr-MOF7 and Cr-MOF8 were obtained with yields of 46 % and 50 % (Figure S21-22, ESI) and BET surfaces of 664 and 625 m2/g, respectively (Figure 5). On the other hand, reaction of Cr-MOF1 with methyl-4-bromobutyrate yielded the corresponding carboxylic modified MOF Cr-MOF9 in low yield (< 20%), and a secondary product (around 10%) as indicated by the aromatic part of the NMR spectrum (Figure S23, ESI). All acidic-modified Al- or Cr-MOFs showed a slight loss of crystallinity, but much lower than in the case of amino-modified Al- or Cr-MOFs, as attested by their PXRD spectra (Figure 4 for Cr-MOFs, not shown for Al-MOFs). Acidic Cr-MOFs were found to retain a significant porosity with a total pore volume in the 0.44-0.53 cm3 g-1 range, and specific surface areas from 664 to 847 m2.g-1 (Figure 5 and FigureS28, ESI).
3.3. Polyfunctionalization
Polyfunctionnalized MOFs would be also of interest for different purposes, for example as simple models of enzyme catalytic sites. MOF-pores decorated both with a primary amino group mimicking a lysine residue, and a carboxylate group found in glutamate or aspartate residues, could act as an enzyme active site mimic that may perform acid-base catalyzed reactions such as ester or amide hydrolysis [35,36]. On these bases, post-modified MOFs, Al-MOF2/7 and Cr-MOF2/7, have been prepared with both an ethyl amino group and an acetic group using two different methods: a one-pot procedure (path h, Scheme 1) by mixing at 120° C in toluene Al- or Cr-MOF1 [a] with N-Boc-2-bromoethylamine [b] and bromoacetic acid [c] using [a]/[b]/[c] proportions 1/3.45/7.20, and a sequential procedure (path d+g, Scheme 1), by reacting the carboxylic MOF Al- or Cr-MOF7 with 7.5 eq. of 2-(aminoethyl)bromide hydrobromide. As expected, the 1H-NMR spectra of a NaOD-digested sample of this trifunctionalized MOFs (i.e. primary amino, carboxylic and unreacted terephthalic amino groups) display signals of both M-MOF2 and M-MOF7 (M = Al or Cr), in addition to the signals derived from the unreacted aminoterphthalate (Figure 8 and Figure S24-25, ESI). These results were confirmed by LC-MS analyses of the Al-MOF2/7 digested products (Figure S13-14, ESI). In these experimental conditions, PSM-yields calculated from NMR spectra were approx. 14 % in amino groups and 76 % in carboxylic groups for Al-MOF2/7 prepared with the one-pot procedure, and 14 % (amino) and 46 % (carboxylic) for the MOF obtained by the sequential procedure. For Cr-MOF2/7, PSM-yields calculated from NMR spectra were approx. 33 % in amino groups and 45 % in carboxylic groups using the one-pot procedure, and 26 % (amino) and 33 % (carboxylic) for the MOF obtained by the sequential procedure (Figure S24-25, ESI). Obviously, it is likely that these yields as well as the amino/carboxylic groups’ proportion could be easily modulated by changing the experimental conditions, such as the temperature and the ratio of the starting alkyl bromide reactants.
3.4. Postmodification with Other Functional Groups
Following this simple procedure involving alkyl bromides, a terminal alkyne that may be used in the copper(I)-catalyzed azide–alkyne [3+2] cycloaddition (CuAAC, or “click chemistry”) to further connect other molecules, was also introduced. However, when propargyl bromide alone was used in our standard experimental conditions (toluene, 120 °C), the desired alkyne-modified MOF was not obtained. In this case, the only product formed was that resulting from the expected reaction (i.e. Al-MOF10) but the hydrobromic acid released during the reaction has added to the triple bond of Al-MOF10 leading finally to a vinyl bromide modified MOF. However by replacing toluene by N,N-dimethylformamide as solvent and in the presence of potassium carbonate to trap released hydrobromic acid, Al-MOF10 was obtained in 80 % yield (Scheme 2 and Figure S15, ESI). It should be noted that the presence of a small amount (approx. 4%) of a N-dialkylated product, i.e. the aniline amino group bearing two alkyne functions, was also identified in the LC-MS chromatogram of the digested products of Al-MOF10. In the same experimental conditions, Cr-MOF10 was obtained but with a rather low PSM-yield (30%, Figure S26, ESI), a low specific surface (149 m2/g, Figure S28, ESI) and a significant loss of cristallinity (Figure S29, ESI). On the other hand, no reaction occurred in our standard conditions when 4-bromo-1-butyne was used as alkyl bromide. Here, it can be hypothesized that the basic aromatic amino group of Al-MOF1 could catalyze the HBr elimination reaction from the starting bromo butyne, leading to 1-buten-3-yne.
Scheme 2.
Access to post-modified alkyne (●) and alcohol (●) M-MOFs (M = Al or Cr).
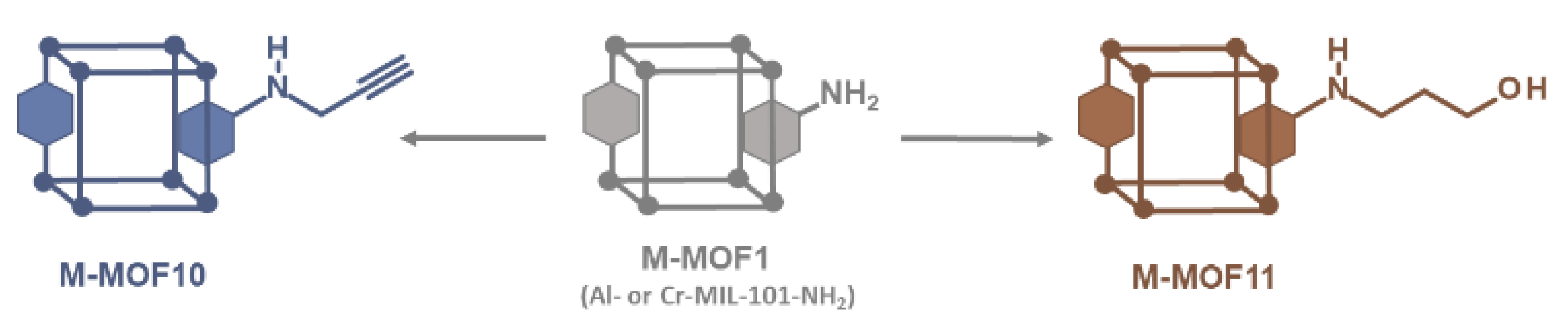
Modification of MOF with alcohol functions is of interest to further covalently link other organic molecules, but also to modify the inner and outer hydrophilic properties of MOF pores. From 3-bromo-1-propanol in toluene at 120 °C, post-modified Al-MOF11 was obtained, but as a mixture of polypropyl ether derivatives, which is not devoid of interest, as revealed by the LC-MS chromatogram of the digested products (Figure S16, ESI). The expected post-synthetic modification leading to the amine-linked propanol chain was obtained in 22% yield, but was accompanied by other modifications leading to the formation of two, three, and four consecutive ether chains in 12 %, 3 %, and 1% yield, respectively. In contrast to previous reactions with amino alkyl bromides, which lead to unpolymerizable protonated amino functions, reactions with alcoholic alkyl bromides give primary hydroxylated MOFs that retain their nucleophilic properties, thus leading to polymerization reactions. The same reaction on Cr-MOF1 gave the analogous chromium MOF Cr-MOF11 with a PSM-overall yield of 40% (Figure S27, ESI), a BET specific surface of 713 m2/g (Figure S28, ESI), and a good crystallinity retention (Figure S29, ESI).
4. Conclusions
We have reported an effective, modulable and versatile methodology for postsynthetically introducing amino or carboxylic groups into MOFs of the MIL-101-NH2 family. This method, based on condensation reactions with alkyl bromides without addition of base, releases one or two equivalents of hydrobromic acid, thus avoiding undesirable polyalkylation and polymerisation reactions. Our results indicate that the chromium MOF is less affected by hydrolysis or degradation than its aluminum counterpart under all post-modification reaction conditions, and that amines, although protonated, are more damaging for the framework than carboxylic acids. This strategy also allows the two functional groups to be introduced into the MOF pores in varying proportions. The amino and carboxylic groups can be retained as is, for example to act as mimics of enzyme amino-acid residues, or can be modified independently of each other for other purposes. By using the same approach, it is also possible to introduce other functional groups such as alcohol and alkyne, but the corresponding alkylation are accompanied by secondary modifications resulting from dialkylation or polymerization reactions that are difficult to control.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., post-modified MOFs syntheses, preparative purification and characterization of 2-((2-aminoethyl)amino) terephthalic acid, NMR spectra, LC-MS chromatograms, N2-adsorption-desorption isotherms, PXRD spectra, and other data.
Author Contributions
IF and MCB: Resources. AV, CL and CL: Conceptualization, Investigation, Writing – review & editing. PH: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the “Centre National de la Recherche Scientifique” (CNRS, France) and by the “University Toulouse 3 Paul Sabatier” (France).
Acknowledgments
The authors thank Fernanda Goncalves for the chromatographic analyses recorded on an UPC² which is part of the Integrated Screening Platform of Toulouse (PICT, IBISA). The authors thank Benjamin Duployer (CIRIMAT) for his help with the PXRD analyses. Technical assistance provided by the Institut de Chimie de Toulouse platform (Université Toulouse 3 Paul Sabatier, CNRS, France- www.ict.ups-tlse.fr) is also gratefully acknowledged.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tanabe, K.K.; Cohen, S.M. Postsynthetic Modification of Metal–Organic Frameworks—a Progress Report. Chem. Soc. Rev. 2011, 40, 498–519. [Google Scholar] [CrossRef] [PubMed]
- Razavi, S.A.A.; Morsali, A. Linker Functionalized Metal-Organic Frameworks. Coord. Chem. Rev. 2019, 399, 213023. [Google Scholar] [CrossRef]
- Yin, Z.; Wan, S.; Yang, J.; Kurmoo, M.; Zeng, M.-H. Recent Advances in Post-Synthetic Modification of Metal–Organic Frameworks: New Types and Tandem Reactions. Coord. Chem. Rev. 2019, 378, 500–512. [Google Scholar] [CrossRef]
- Mandal, S.; Natarajan, S.; Mani, P.; Pankajakshan, A. Post-Synthetic Modification of Metal–Organic Frameworks Toward Applications. Adv. Funct. Mat. 2021, 31, 2006291. [Google Scholar] [CrossRef]
- Cohen, S.M.; Rosi, N.L. Postsynthetic Modification of Metal−Organic Frameworks. Inorg. Chem. 2021, 60, 11703–11705. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Lv, D.; Guan, Y.; Yu, S. Post-Synthesis Modification of Metal–Organic Frameworks: Synthesis, Characteristics, and Applications. J. Mater. Chem. A 2023, 11, 24519–24550. [Google Scholar] [CrossRef]
- Bernt, S.; Guillerm, V.; Serre, C.; Stock, N. Direct Covalent Post-Synthetic Chemical Modification of Cr-MIL-101 Using Nitrating Acid. Chem. Commun. 2011, 47, 2838–2840. [Google Scholar] [CrossRef]
- Bauer, S.; Serre, C.; Devic, T.; Horcajada, P.; Marrot, J.; Férey, G.; Stock, N. High-Throughput Assisted Rationalization of the Formation of Metal Organic Frameworks in the Iron(III) Aminoterephthalate Solvothermal System. Inorg. Chem. 2008, 47, 7568–7576. [Google Scholar] [CrossRef]
- Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.M. Systematic Design of Pore Size and Functionality in Isoreticular MOFs and their Application in Methane Storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Cohen, S.M. Tandem Modification of Metal-Organic Frameworks by a Postsynthetic Approach. Angew. Chem. Int. Ed. 2008, 47, 4699–4702. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Cohen, S.M. Postsynthetic Covalent Modification of a Neutral Metal−Organic Framework. J. Am. Chem. Soc. 2007, 129, 12368–12369. [Google Scholar] [CrossRef]
- Seo, J.S.; Whang, D.; Lee, H.; Jun, S.I.; Oh, J.; Jeon, Y.J.; Kim, K. A Homochiral Metal-Organic Porous Material for Enantioselective Separation and Catalysis. Nature 2000, 404, 982–986. [Google Scholar] [CrossRef]
- Garibay, S.J.; Wang, Z.; Tanabe, K.K.; Cohen, S.M. Postsynthetic Modification: A Versatile Approach Toward Multifunctional Metal-Organic Frameworks. Inorg. Chem. 2009, 48, 7341–7349. [Google Scholar] [CrossRef] [PubMed]
- Volkringer, C.; Cohen, S.M. Generating Reactive MILs: Isocyanate- and Isothiocyanate-Bearing MILs through Postsynthetic Modification. Angew. Chem. Int. Ed. 2010, 49, 4644–4648. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, T.; Siegel, R.; Reimer, N.; Milius, W.; Stock, N.; Senker, J. Enhancing the Water Stability of Al-MIL-101-NH2 via Postsynthetic Modification. Chem. Eur. J. 2015, 21, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Ingleson, M.J.; Perez Barrio, J.; Guilbaud, J.-B.; Khimyak, Y.Z.; Rosseinsky, M.J. Framework Functionalisation Triggers Metal Complex Binding. Chem. Commun. 2008. [Google Scholar] [CrossRef]
- Garzón-Tovar, L.; Rodríguez-Hermida, S.; Imaz, I.; Maspoch, D. Spray Drying for Making Covalent Chemistry: Postsynthetic Modification of Metal–Organic Frameworks. J. Am. Chem. Soc. 2017, 139, 897–903. [Google Scholar] [CrossRef]
- Savonnet, M.; Bazer-Bachi, D.; Bats, N.; Perez-Pellitero, J.; Jeanneau, E.; Lecocq, V.; Pinel, C.; Farrusseng, D. Generic Postfunctionalization Route from Amino-Derived Metal−Organic Frameworks. J. Am. Chem. Soc. 2010, 132, 4518–4519. [Google Scholar] [CrossRef]
- Wu, S.; Chen, L.; Yin, B.; Li, Y. “Click” Post-Functionalization of a Metal–Organic Framework for Engineering Active Single-Site Heterogeneous Ru(III) Catalysts. Chem. Commun. 2015, 51, 9884–9887. [Google Scholar] [CrossRef]
- Legrand, A.; Pastushenko, A.; Lysenko, V.; Geloen, A.; Quadrelli, E.A.; Canivet, J.; Farrusseng, D. Enhanced Ligand-Based Luminescence in Metal–Organic Framework Sensor. ChemNanoMat. 2016, 2, 866–872. [Google Scholar] [CrossRef]
- Ma, W.; Xu, L.; Li, Z.; Sun, Y.; Bai, Y.; Liu, H. Post-Synthetic Modification of an Amino-Functionalized Metal–Organic Framework for Highly Efficient Enrichment of N-Linked Glycopeptides. Nanoscale 2016, 8, 10908–10912. [Google Scholar] [CrossRef]
- Yoo, Y.; Jeong, H.-K. Generation of Covalently Functionalized Hierarchical IRMOF-3 by Post-Synthetic Modification. Chem. Eng. J. 2012, 181-182, 740–745. [Google Scholar] [CrossRef]
- Nguyen, J.G.; Tanabe, K.K.; Cohen, S.M. Postsynthetic Diazeniumdiolate Formation and NO Release from MOFs. CrystEngComm. 2010, 12, 2335–2338. [Google Scholar] [CrossRef]
- Britt, D.; Lee, C.; Uribe-Romo, F.J.; Furukawa, H.; Yaghi, O.M. Ring-Opening Reactions within Porous Metal−Organic Frameworks. Inorg. Chem. 2010, 49, 6387–6389. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xi, F.-G.; Sun, W.; Yang, N.-N.; Gao, E.-Q. Amino- and Sulfo-Bifunctionalized Metal–Organic Frameworks: One-Pot Tandem Catalysis and the Catalytic Sites. Inorg. Chem. 2016, 55, 5753–5755. [Google Scholar] [CrossRef]
- Jiang, D.; Keenan, L.L.; Burrows, A.D.; Edler, K.J. Synthesis and Post-Synthetic Modification of MIL-101(Cr)-NH2 via a Tandem Diazotisation Process. Chem. Commun. 2012, 48, 12053–12055. [Google Scholar] [CrossRef] [PubMed]
- Hamzah, H.A.; Crickmore, T.S.; Rixson, D.; Burrows, A.D. Post-Synthetic Modification of Zirconium Metal–Organic Frameworks by Catalyst-Free Aza-Michael Additions. Dalton Trans. 2018, 47, 14491–14496. [Google Scholar] [CrossRef]
- Ma, D.; Li, B.; Liu, K.; Zhang, X.; Zou, W.; Yang, Y.; Li, G.; Shi, Z.; Feng, S. Bifunctional MOF Heterogeneous Catalysts Based on the Synergy of Dual Functional Sites for Efficient Conversion of CO2 Under Mild and Co-catalyst Free Conditions. J. Mater. Chem. A. 2015, 3, 23136–23142. [Google Scholar] [CrossRef]
- Li, B.; Ma, D.; Li, Y.; Zhang, Y.; Li, G.; Shi, Z.; Feng, S.; Zaworotko, M.J.; Ma, S. Dual Functionalized Cages in Metal–Organic Frameworks via Stepwise Postsynthetic Modification. Chem. Mater. 2016, 28, 4781–4786. [Google Scholar] [CrossRef]
- Hartmann, M.; Fischer, M. Amino-Functionalized Basic Catalysts with MIL-101 Structure. Microporous Mesoporous Mat. 2012, 164, 38–43. [Google Scholar] [CrossRef]
- Lin, Y.; Kong, C.; Chen, L. Direct Synthesis of Amine-Functionalized MIL-101(Cr) Nanoparticles and Application for CO2 Capture. RSC Adv. 2012, 2, 6417–6419. [Google Scholar] [CrossRef]
- Zou, M.; Dong, M.; Zhao, T. Advances in Metal-Organic Frameworks MIL-101(Cr). Int. J. Mol. Sci. 2022, 23, 9396. [Google Scholar] [CrossRef] [PubMed]
- Babaee, S.; Zarei, M.; Sepehrmansourie, H.; Zolfigol, M.A.; Rostamnia, S. Synthesis of Metal–Organic Frameworks MIL-101(Cr)-NH2 Containing Phosphorous Acid Functional Groups: Application for the Synthesis of N-Amino-2-pyridone and Pyrano [2,3-c]pyrazole Derivatives via a Cooperative Vinylogous Anomeric-Based Oxidation. ACS Omega 2020, 5, 6240–6249. [Google Scholar] [CrossRef] [PubMed]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Kuah, E.; Toh, S.; Yee, J.; Ma, Q.; Gao, Z. Enzyme Mimics: Advances and Applications. Chem. Eur. J. 2016, 22, 8404–8430. [Google Scholar] [CrossRef]
- Nothling, M.D.; Xiao, Z.; Bhaskaran, A.; Blyth, M.T.; Bennett, C.W.; Coote, M.L.; Connal, L.A. Synthetic Catalysts Inspired by Hydrolytic Enzymes. ACS Catal. 2019, 9, 168–187. [Google Scholar] [CrossRef]
Figure 1.
1H-NMR spectra (300 MHz) of the NaOD/D2O-digested Al-MOF2 prepared from Al-MOF1 and 2-bromoethylamine hydrobromide in toluene at 110 °C.
Figure 1.
1H-NMR spectra (300 MHz) of the NaOD/D2O-digested Al-MOF2 prepared from Al-MOF1 and 2-bromoethylamine hydrobromide in toluene at 110 °C.
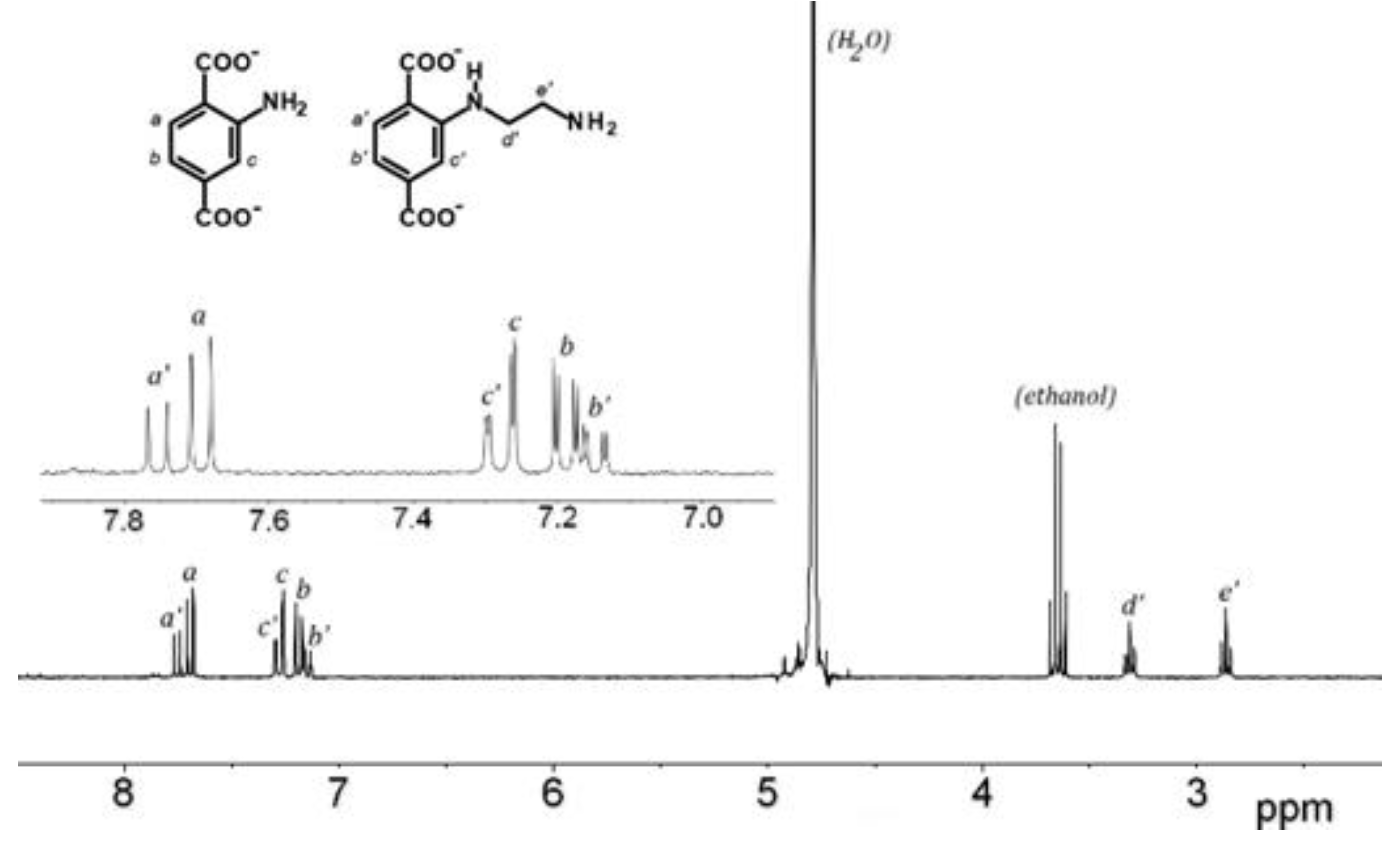
Figure 2.
PXRD patterns simulated (1.54056 Å) from the crystal data of Cr-MIL-101, and recorded for a sample of Al-MOF1 (Al-MIL-101-NH2) and post-modified Al-MOF2.
Figure 2.
PXRD patterns simulated (1.54056 Å) from the crystal data of Cr-MIL-101, and recorded for a sample of Al-MOF1 (Al-MIL-101-NH2) and post-modified Al-MOF2.
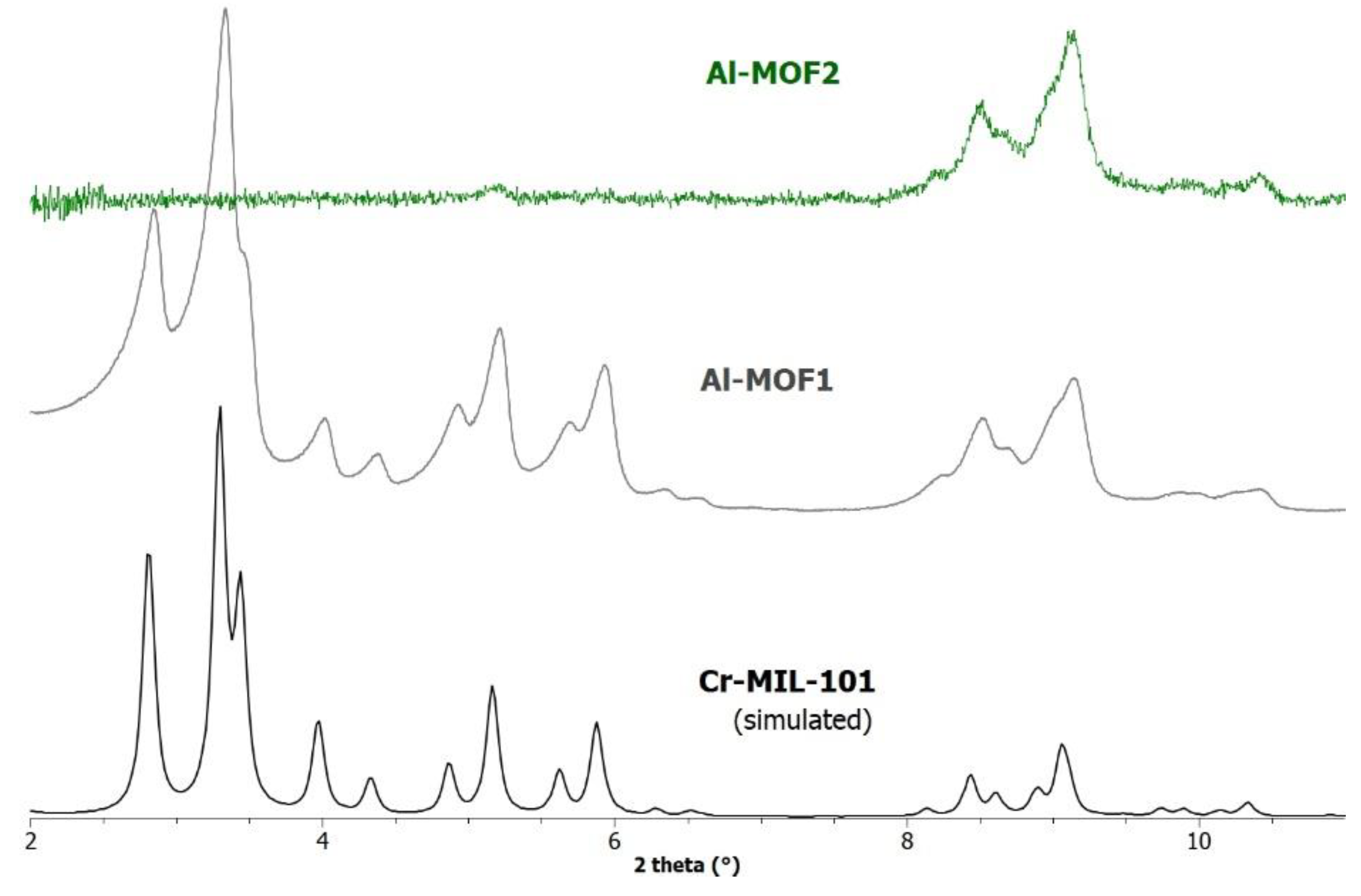
Figure 3.
(Left panel) 7.0-8.0 ppm-range 1H-NMR spectra (300 MHz) of the digested Al-MOF2 prepared from Al-MOF1 and 2-bromoethylamine hydrobromide in toluene at different temperatures. (Right panel) PSM-yields as a function of temperature: (green) yields calculated by NMR from the ratio of doublets at δ =7.69 ppm of unmodified ligand BDC2--NH2 and δ = 7.75 ppm of modified ligand BDC2--NH-(CH2)2-NH2; (blue) yields calculated by UPLC from peak area integration of crude mixtures chromatograms.
Figure 3.
(Left panel) 7.0-8.0 ppm-range 1H-NMR spectra (300 MHz) of the digested Al-MOF2 prepared from Al-MOF1 and 2-bromoethylamine hydrobromide in toluene at different temperatures. (Right panel) PSM-yields as a function of temperature: (green) yields calculated by NMR from the ratio of doublets at δ =7.69 ppm of unmodified ligand BDC2--NH2 and δ = 7.75 ppm of modified ligand BDC2--NH-(CH2)2-NH2; (blue) yields calculated by UPLC from peak area integration of crude mixtures chromatograms.
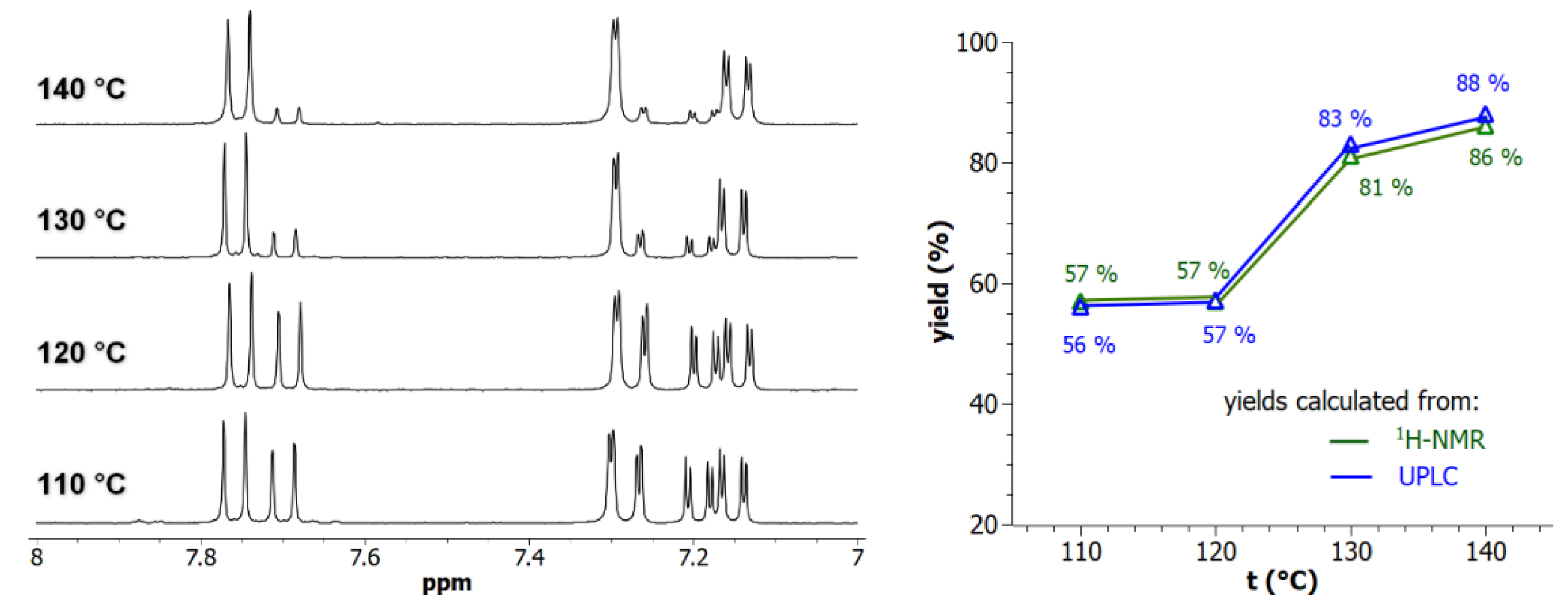
Figure 4.
PXRD patterns recorded for a sample of Cr-MOF1 (Cr-MIL-101-NH2), amino (Cr-MOF2 and Cr-MOF4-6) and carboxylic (Cr-MOF7-9) post-modified MOFs.
Figure 4.
PXRD patterns recorded for a sample of Cr-MOF1 (Cr-MIL-101-NH2), amino (Cr-MOF2 and Cr-MOF4-6) and carboxylic (Cr-MOF7-9) post-modified MOFs.
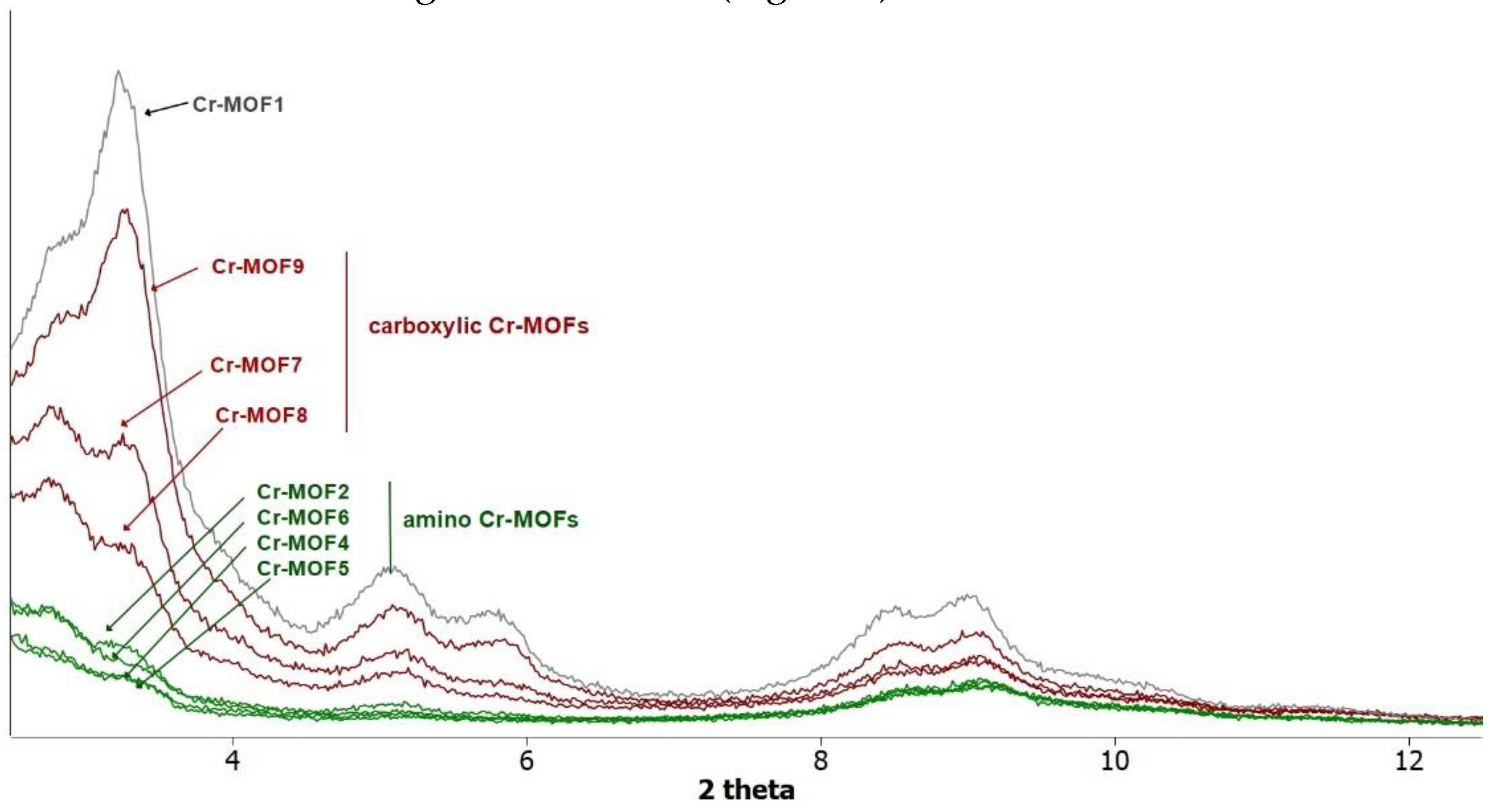
Figure 5.
N2-adsorption (ø) –desorption (ø) isotherms of Cr-MOF2, Cr-MOF4, Cr-MOF5, Cr-MOF7, Cr-MOF8 and Cr-MOF2/7. The BET specific surface areas and pore volumes (Vp) were calculated from the adsorption isotherms. BET specific surface areas calculated from adsorption isotherms of Cr-MOF6, Cr-MOF9, Cr-MOF10 and Cr-MOF11 were 222, 847, 149 and 713 m2/g, respectively, with pore volumes of 0.16, 0.53, 0.11 and 0.47 cm3.g-1, respectively (FigureS28, ESI).
Figure 5.
N2-adsorption (ø) –desorption (ø) isotherms of Cr-MOF2, Cr-MOF4, Cr-MOF5, Cr-MOF7, Cr-MOF8 and Cr-MOF2/7. The BET specific surface areas and pore volumes (Vp) were calculated from the adsorption isotherms. BET specific surface areas calculated from adsorption isotherms of Cr-MOF6, Cr-MOF9, Cr-MOF10 and Cr-MOF11 were 222, 847, 149 and 713 m2/g, respectively, with pore volumes of 0.16, 0.53, 0.11 and 0.47 cm3.g-1, respectively (FigureS28, ESI).
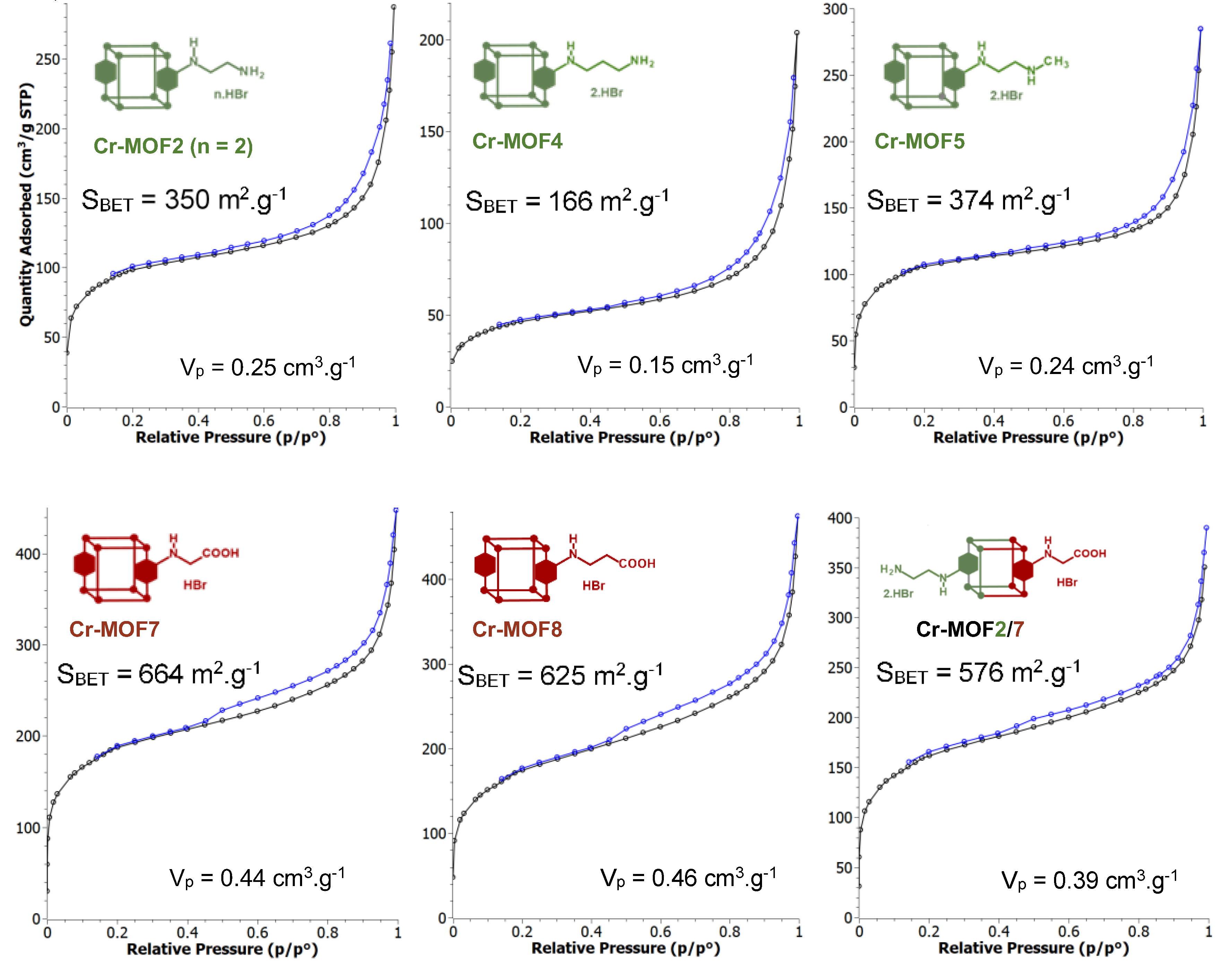
Figure 6.
NMR of amino MOFs. 6.95-7.85 ppm-range 1H-NMR spectra (300 MHz) of digested Al-MOF1 and amino post-modified MOFs prepared from Al-MOF1 and 2-bromoethylamine hydrobromide (Al-MOF2), N-Boc-2-bromoethylamine (Al-MOF6), 3-bromopropylamine hydrobromide (Al-MOF4) and N-methyl-3-bromoethylamine hydrobromide (Al-MOF5). All reactions were carried out in toluene at 120°C, except for Al-MOF2 (125 °C).
Figure 6.
NMR of amino MOFs. 6.95-7.85 ppm-range 1H-NMR spectra (300 MHz) of digested Al-MOF1 and amino post-modified MOFs prepared from Al-MOF1 and 2-bromoethylamine hydrobromide (Al-MOF2), N-Boc-2-bromoethylamine (Al-MOF6), 3-bromopropylamine hydrobromide (Al-MOF4) and N-methyl-3-bromoethylamine hydrobromide (Al-MOF5). All reactions were carried out in toluene at 120°C, except for Al-MOF2 (125 °C).
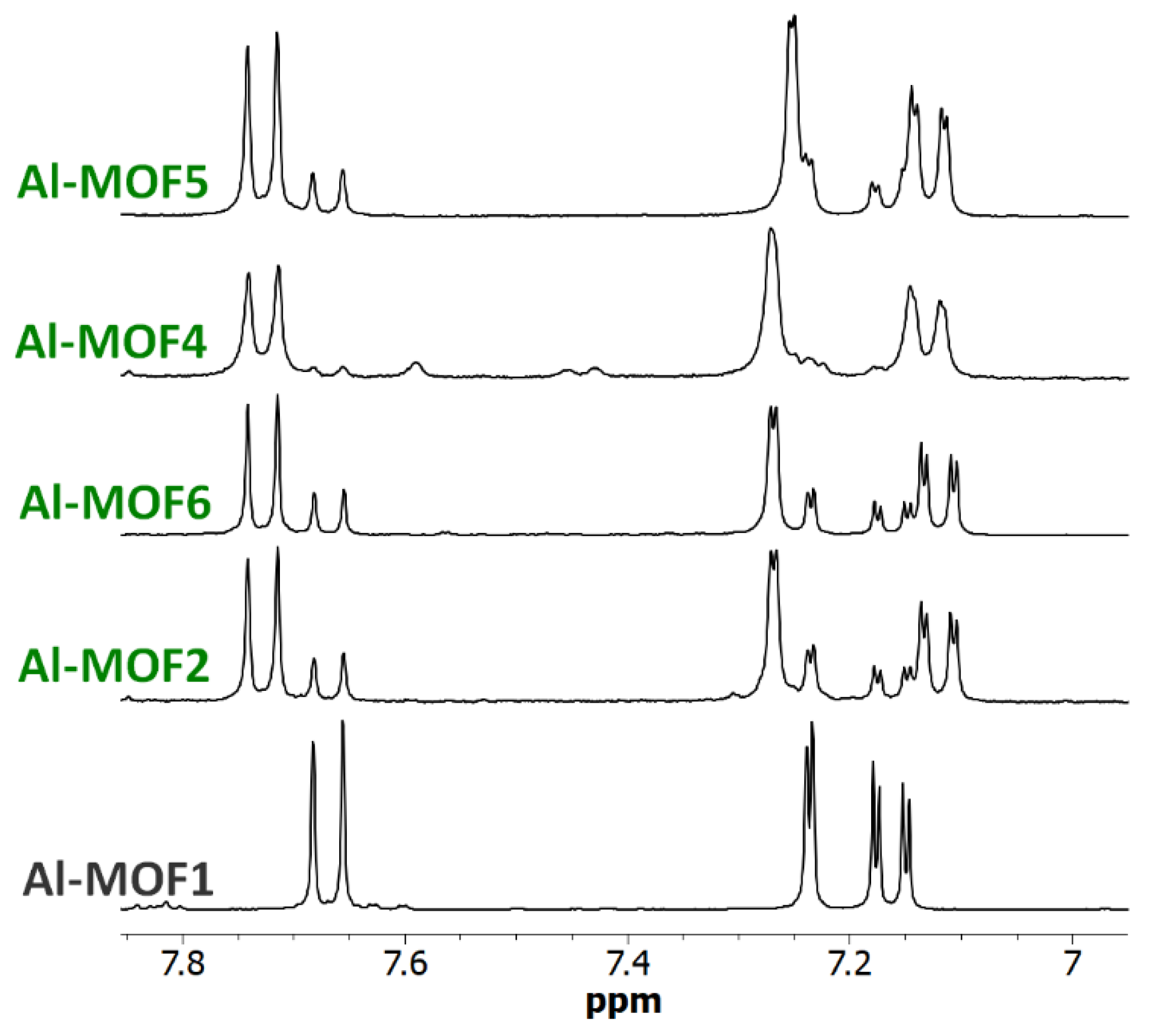
Figure 7.
NMR of carboxylic Al-MOFs. 6.95-7.90 ppm-range 1H-NMR spectra (300 MHz) of digested Al-MOF1 and carboxylic post-modified MOFs prepared from Al-MOF1 and bromoacetic acid (Al-MOF7), 3-bromopropionic acid (Al-MOF8) and methyl-4-bromobutyrate (Al-MOF9).
Figure 7.
NMR of carboxylic Al-MOFs. 6.95-7.90 ppm-range 1H-NMR spectra (300 MHz) of digested Al-MOF1 and carboxylic post-modified MOFs prepared from Al-MOF1 and bromoacetic acid (Al-MOF7), 3-bromopropionic acid (Al-MOF8) and methyl-4-bromobutyrate (Al-MOF9).
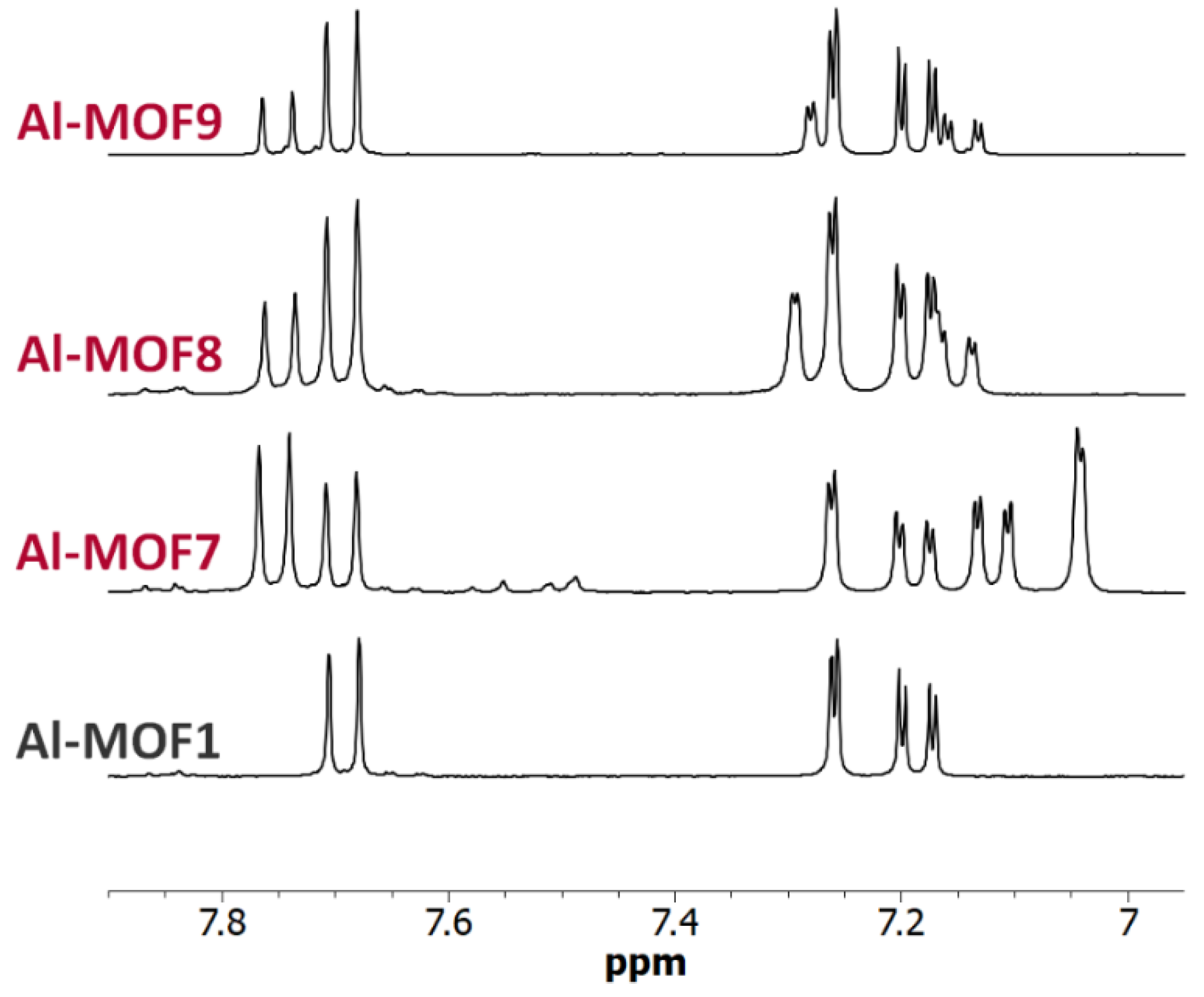
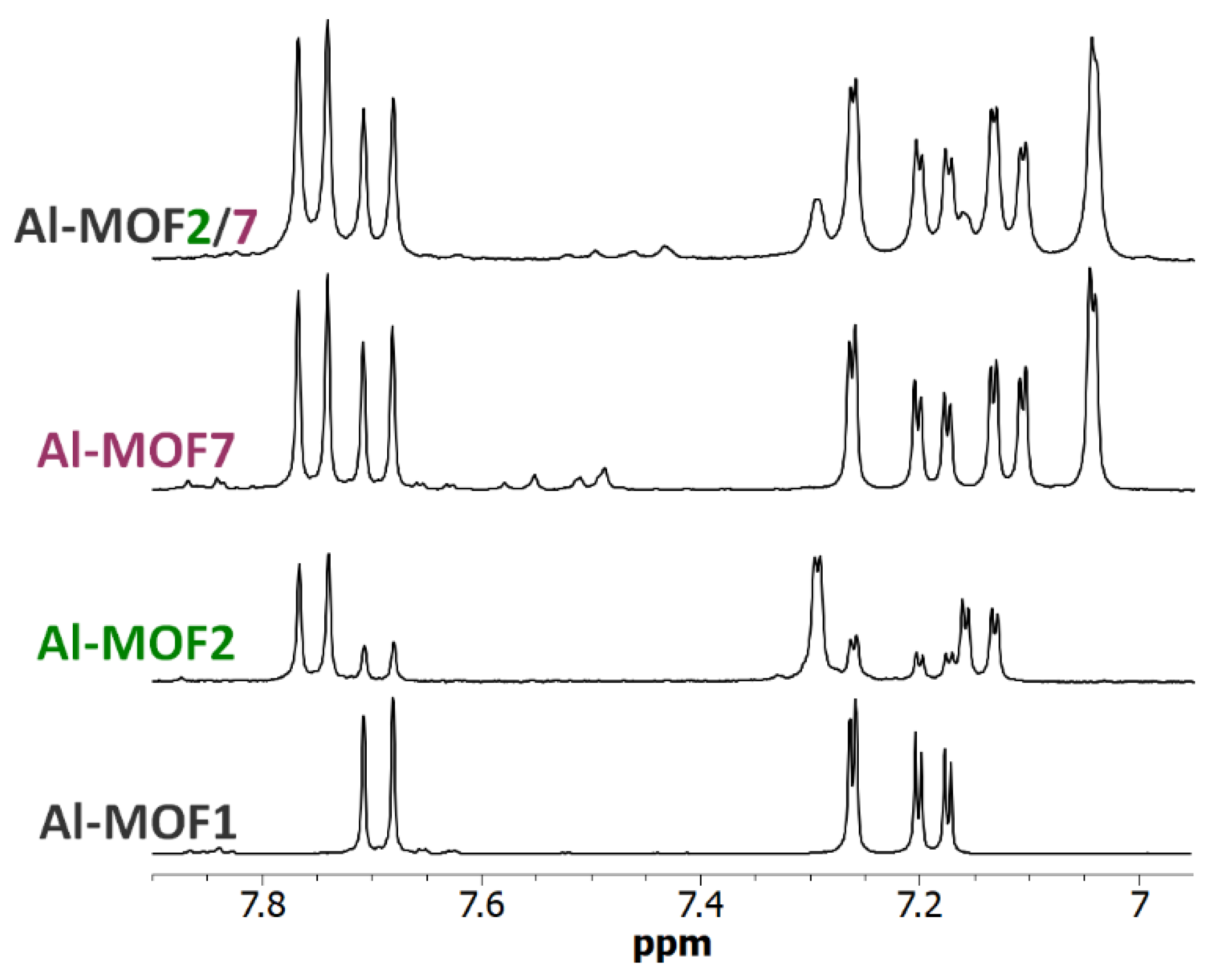
Figure 8.
6.95-7.90 ppm-range 1H-NMR spectra (300 MHz) of digested Al-MOF1 and post-modified MOFs prepared from Al-MOF1 and 2-bromoethylamine hydrobromide (Al-MOF2), from Al-MOF1 and bromoacetic acid (Al-MOF7), and from Al-MOF7 and 2-bromoethylamine hydrobromide (Al-MOF2/7) containing both ethyl amino and acetic groups.
Figure 8.
6.95-7.90 ppm-range 1H-NMR spectra (300 MHz) of digested Al-MOF1 and post-modified MOFs prepared from Al-MOF1 and 2-bromoethylamine hydrobromide (Al-MOF2), from Al-MOF1 and bromoacetic acid (Al-MOF7), and from Al-MOF7 and 2-bromoethylamine hydrobromide (Al-MOF2/7) containing both ethyl amino and acetic groups.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



