
Submitted:
29 January 2025
Posted:
29 January 2025
You are already at the latest version
Abstract
While the beneficial effects of colchicine on inflammation and infarcted myocardium have been documented, its impact on cardiac fibroblast activation in the context of MI remains unknown. This study aimed to investigate the effect of colchicine on the regulation of NLRP3 inflammasome activation and IL-1β expression in fibroblasts. The 3T3 fibroblasts were exposed to 600 μM CoCl2 for 24 hours to simulate hypoxia, with normoxic cells as controls. Colchicine (1 μM) was administered for 24 hours. ASC-NLRP3 colocalization and IL-1β expression were evaluated using immunofluorescence and flow cytometry, respectively. Data were analyzed using t-tests and one-way ANOVA with post-hoc tests. Hypoxia treatment significantly induced ASC-NLRP3 colocalization (p < 0.05). Colchicine treatment of hypoxic 3T3 cells reduced ASC-NLRP3 colocalization, although this reduction was not statistically significant. Additionally, IL-1β expression was significantly inhibited in colchicine-treated hypoxic 3T3 cells compared to those treated with placebo (p < 0.05). The findings of this study indicate that colchicine treatment inhibits the activation of the NLRP3 inflammasome by disrupting the colocalization of ASC and NLRP3, thereby reducing IL-1β expression in CoCl2-treated 3T3 cells.
Keywords:
ASC-NLRP3
; inflammasome
; colchicine
; IL-1β
; ischemia
1. Introduction
Epidemiological data demonstrated that 58% of mortality associated with cardiovascular diseases (CVDs) occurred in Asia [1]. Myocardial infarction, commonly referred to as a heart attack, transpires when blood flow to the myocardium is obstructed, resulting in damage or necrosis of the cardiac muscle tissue [2]. Ischemic heart disease, encompassing acute myocardial infarction, represents a critical condition necessitating prompt medical intervention to mitigate further myocardial damage and to diminish the risk of complications such as heart failure and other potentially fatal outcome [3]. The onset of heart failure is associated with elevated morbidity and mortality rates, thereby exacerbating the social and economic burden [4]. In addition to myocardial necrosis, ventricular remodeling is intimately linked to the pathogenesis of heart failure in patients following myocardial infarction [5].
Inflammation, a multifaceted biological process, is integral to the pathogenesis of heart disease, including cardiac remodeling following MI [6,7]. Under ischemic conditions/hypoxia, reduced blood flow to the myocardium can cause significant tissue injury and initiate an inflammatory response [8]. While this inflammatory response is essential for cardiac repair following acute myocardial infarction, excessive inflammation can result in adverse ventricular remodeling [9,10]. This inflammatory activity constitutes a critical aspect of the pathophysiological mechanism in cardiac ischemia, involving various regulatory elements, including interleukin-1 beta (IL-1β) [11] and the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome complex [12], which serves as a principal mediator of sterile inflammation post-acute myocardial infarction [13]. This process culminates in the release of pro-inflammatory mediators that evoke pro-inflammatory responses and attract inflammatory cells to the infarcted area, contributing to ventricular remodeling [14]. Activation of the NLRP3 inflammasome leads to the production of pro-inflammatory cytokines, notably interleukin-1β (IL-1β), which contribute to tissue damage and adverse cardiac remodeling post-MI.
Colchicine, a natural alkaloid traditionally employed in the management of gout [15], has recently gained recognition as a potential therapeutic agent for ischemic heart disease [16]. As an anti-inflammatory compound, colchicine can inhibit the production of IL-1β and the activation of the NLRP3 inflammasome [17], thereby attracting considerable interest within the scientific community. In the context of acute myocardial infarction (AMI), colchicine administration has demonstrated efficacy in reducing infarct size, enhancing cardiac function, and modulating inflammatory responses [15]. Notably, the Colchicine Cardiovascular Outcomes Trial (COLCOT) reported a 23% reduction in the risk of adverse cardiovascular events among patients with acute coronary syndrome treated with colchicine, compared to those receiving a placebo [18]. While clinical studies have shown promising results in reducing cardiovascular events in patients with coronary artery disease, the precise mechanisms by which colchicine exerts its effects on cardiac inflammation, particularly in the context of ischemia, remain to be fully elucidated.
The speck itslef is not the NLRP3 inflammasome, but is instead a dynamic structure which may amplify the NLRP3 response to weak stimuli by facilitating the formation and release of small NLRP3:ASC complexes which in turn activate caspase-1 [19].
This study aims to investigate the effects of colchicine on NLRP3 inflammasome activation and IL-1β expression in a cellular model of hypoxia, using 3T3 fibroblasts. We hypothesize that colchicine may attenuate inflammasome activation by interfering with the assembly of its components, specifically the interaction between ASC and NLRP3 proteins.
2. Materials and Methods
2.1. Ethical statement
This study was approved by the Health Research Ethic Committee of the Faculty of Medicine, Universitas Brawijaya, Indonesia (Certificate No. 182/EC/KEPK-S3/07/2023) under the name of Tri Astuti as principal investigator. Trial registration: https://clinicaltrials.gov. Unique identifier: NCT06426537 (400/235/K.3/302/2020).
2.2. Study design
The 3T3 fibroblast cells were obtained from animal-derived, randomly allocated to different treatment groups, including control, hypoxia, colchicine, and combined colchicine-hypoxia groups. Randomization was performed using a random number generator to ensure unbiased allocation of cells into each experimental condition.
Cells were randomly allocated to four experimental groups: control (non-hypoxic cells), colchicine (non-hypoxic cells + colchicine for 24 hours), CoCl2 (hypoxic cells), and CoCl2 + colchicine (hypoxic cells + colchicine for 24 hours) [20].
To minimize bias during data analysis, the samples were labeled with coded identifiers that were unknown to the investigators until the analysis was completed. This procedure was maintained throughout the experiment to ensure objective evaluation of the results.
2.3. Cell culture and treatment
The 3T3 fibroblast cells used in this study were obtained from the European Collection of Authenticated Cell Cultures (ECACC, catalog no. 306-05A, RRID:CVCL_0594). The cells were authenticated using short tandem repeat (STR) profiling to confirm their identity, and they were regularly tested for mycoplasma contamination to ensure the integrity of the experimental results. The 3T3 cells were cultured in 6-well plates (80,000 cells/well) in Dulbecco's Modified Eagle Medium (DMEM, Gibco, catalog no. 11995065) supplemented with 10% fetal bovine serum (FBS, Gibco, catalog no. 16000044) and 1% penicillin-streptomycin (Gibco, catalog no. 15140122). Hypoxia was induced using 600 μM CoCl2 for 24 hours, while colchicine was administered at 1 μM for 24 hours [21].
Hypoxia Induction and Colchicine Treatment Hypoxic conditions were simulated using 600 μM Cobalt(II) Chloride Hexahydrate (CoCl2, Sigma-Aldrich, catalog no. C8661-25G, CAS: 7791-13-1) for 24 hours. The optimal concentration of 600 μM CoCl2 was established based on prior studies [22]. Colchicine (Sigma-Aldrich, catalog no. C9754, CAS: 64-86-8) was administered at a final concentration of 1 μM in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin, and the cells were subsequently incubated for 24 hours.
2.4. Statistical analysis
A priori power analysis was conducted using G*Power 3.1 software to determine the sample size. With an effect size of 0.8, α error probability of 0.05, and power (1-β error probability) of 0.8. We used 5 replicates per group to account for potential experimental loss. A power analysis was conducted to determine the appropriate number of replicates needed to detect significant differences between the treatment groups. The analysis suggested that a minimum of 5 replicates per group would be required to achieve a statistical power of 80% at a significance level of p < 0.05. This sample size was chosen to ensure that the experimental results were robust and reliable.
3. Results
3.1. NLRP3 Inflammasome
To evaluate the impact of colchicine treatment on the activation of the inflammasome pathway, a co-localization study of ASC and NLRP3 in 3T3 cells was conducted using the immunofluorescence method. Our data revealed that cobalt chloride treatment significantly induced co-localization of ASC and NLRP3 in 3T3 cells, as indicated by the presence of yellow dots or clusters within the cytoplasm (p<0.05). Additionally, colchicine administration resulted in a reduction in the percentage of 3T3 cells exhibiting positive ASC-NLRP3 co-localization, although this reduction was not statistically significant. Representative images of each group are presented in Figure 1, while the quantification of cells with positive ASC-NLRP3 co-localization is illustrated in Figure 2.
3.2. IL-1β Expression
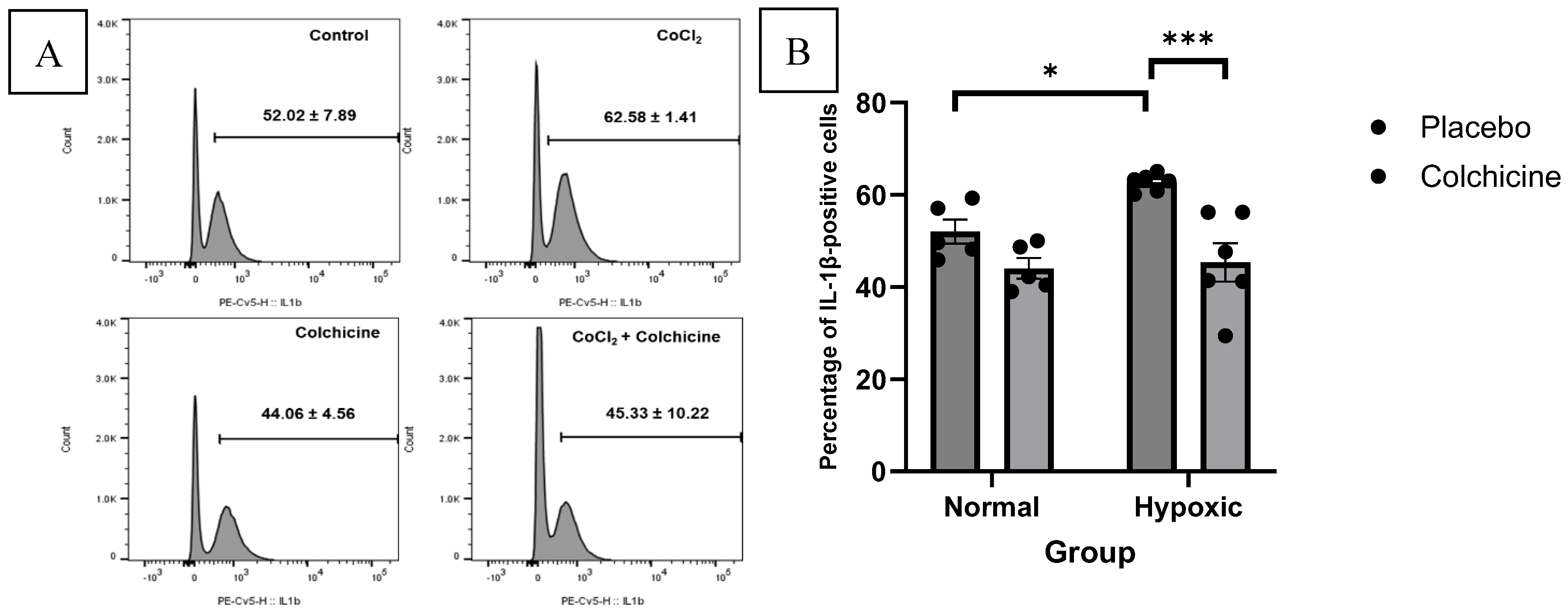
Our data showed that CoCl2 treatment significantly elevated the expression of IL-1β compared to normoxic 3T3 cells (62.58 ± 1.41% vs. 52.02 ± 7.89%, p < 0.05). Colchicine treatment significantly reduced IL-1β expression in hypoxic cells compared to placebo-treated hypoxic cells (45.33 ± 10.22% vs. 62.58 ± 1.41%, p < 0.001). In normoxic conditions, colchicine treatment showed a slight, non-significant reduction in IL-1β expression compared to untreated normoxic cells (44.06 ± 4.56% vs. 52.02 ± 7.89%, p > 0.05). These results suggest that colchicine's inhibitory effect on IL-1β expression is more pronounced under hypoxic conditions, aligning with its potential therapeutic benefit in ischemic scenarios. The calculation of IL-1β expression was depicted in the Figure 3.
4. Discussion
Our study employed 3T3 fibroblast cell culture, offering significant advantages over myocardial cells used in previous research. Fibroblasts, crucial for collagen and elastin production, exhibit unlimited proliferation and growth potential, enabling more extensive and reproducible experiments [23]. In contrast, myocardial cells have limited proliferation capacity and are challenging to maintain in culture, potentially compromising result consistency and scalability. Fibroblasts play a key role in cardiac remodelling post-myocardial infarction. Under hypoxic conditions, as simulated by CoCl2 in our study, these cells differentiate into myofibroblasts, contributing to scar formation and tissue remodelling [24]. This process is critical in the development of heart failure and ventricular dysfunction after infarction [25] by utilizing fibroblasts, our study provides valuable insights into post-ischemic remodelling mechanisms, particularly regarding inflammasome activation and colchicine's potential therapeutic effects. This approach enhances our understanding of the complex interactions between hypoxia, inflammation, and tissue remodelling in ischemic heart disease.
Our data demonstrated that induction of metabolic stress using CoCl2 induced the activation of the NLRP3 inflammasome in fibroblasts (3T3 cells), as evidenced by the increase in IL-1β expression. Colchicine’s inhibitory effect on inflammasome activation, especially its impact on ASC-NLRP3 colocalization, suggests a plausible mechanism of action. The 3T3 fibroblast cell line, derived from Swiss albino mouse embryos, was employed in the previous study by Tuncay et al [26]. 3T3 cells, responsible for the production of collagen and elastin, play crucial roles in maintaining connective tissue structure and strength. Notably, these cells exhibit advantages such as unlimited proliferation and growth potential [23].
Cobalt chloride is commonly employed to induce hypoxia in cells [27] by inhibiting hypoxia-inducible factor 1α (HIF-1α), an essential protein involved in the cellular responses to hypoxia [28]. HIF-1α activation stimulates the production of angiogenic factors, promoting neovascularization [29]. Therefore, inhibition of HIF-1α by CoCl2 diminishes the production of angiogenic factors, thereby impeding angiogenesis. Moreover, CoCl2 enhances the production of reactive oxygen species (ROS), which can induce cellular damage and apoptosis by activating caspase-3, an enzyme implicated in programmed cell death [30]. Extensive cell death mediated by caspase-3 activation may result in hypoxia. Activation of hypoxia-mediated signalling pathways recruits inflammatory cells crucial for infarct healing [9]. These inflammatory cells release various mediators such as cytokines and chemokines, thereby recruiting additional inflammatory cells to the infarct site [31]. While essential for initiating healing, uncontrolled inflammation can exacerbate tissue damage.
This study demonstrates that CoCl2 treatment significantly induces NLRP3 activation and IL-1β production in 3T3 cells. Targeting ASC-NLRP3 colocalization could offer a novel approach for anti-inflammatory therapy [32]. Apoptosis-associated speck-like protein containing a CARD (ASC) and NOD-like receptor protein 3 (NLRP3) are constituents of the inflammasome, a protein complex implicated in the inflammatory response and various diseases [33,34]. ASC functions as an adaptor in inflammasome assembly [35], while NLRP3 acts as a sensor detecting diverse stimuli such as oxidative stress, mitochondrial damage, or crystal accumulation, commonly observed in heart diseases like atherosclerosis, heart failure, or myocardial infarction [36]. Upon detection of these stimuli, NLRP3 interacts with ASC, leading to the activation of pro-caspase-1 and subsequent formation of the inflammasome complex [37]. Activated caspase-1 catalyses the processing of interleukin-1β and interleukin-18 precursors into their active forms [38] stimulate the inflammatory response in heart diseases [39].
ASC colocalization with NLRP3 serves as an indicator of inflammasome assembly and subsequent pro-inflammatory cytokine release [40]. A high colocalization coefficient between NLRP3 and ASC signifies a robust interaction between these proteins, thereby supporting NLRP3 inflammasome activation [41]. Consequently, the activation of inflammasomes through ASC and NLRP3 interaction may contribute to chronic inflammation, oxidative stress, and tissue damage observed in heart diseases [42].
In this study, colchicine administration significantly inhibited ASC-NLRP3 colocalization, thus markedly reduced IL-1β expression, suggesting that colchicine can mitigate inflammasome activation under ischemic conditions by impeding ASC and NLRP3 interaction or availability. Mechanistically, colchicine accomplishes this by interfering with speck formation, a crucial step in NLRP3 inflammasome activation, maybe through modulating the speck formation. Speck formation occurs when ASC oligomerizes and interacts with NLRP3 on the endoplasmic reticulum during inflammasome activation [41].
Colchicine further interferes with inflammasome assembly by disrupting the ASC-NLRP3 interaction, likely through its well-established effects on microtubule dynamics. Colchicine binds to tubulin, preventing microtubule formation, which is necessary for the trafficking of NLRP3 to interact with ASC [43]. This disruption could inhibit the movement and proximity of NLRP3 and ASC, essential for inflammasome assembly (44). Additionally, by affecting speck formation, colchicine might inhibit ASC oligomerization, a critical step in the activation of the inflammasome [43].
Furthermore, in the clinical setting, previous study showed that early initiation of colchicine (within 0-3 days post-myocardial infarction) demonstrated substantial benefits with a notable relative risk reduction in the primary endpoint, thereby advocating for colchicine initiation during hospitalization following myocardial infarction [15].
Colchicine mitigates inflammation triggered by ischemia by indirectly inhibiting NLRP3 oligomerization, which reduces the production of IL-1β and IL-18. Unlike direct NLRP3 inhibitors like MCC950, colchicine's effect on microtubule polymerization provides a unique mechanism of action [44]. This unique mechanism may contribute to colchicine's broader anti-inflammatory effects and established clinical safety profile. This finding is consistent with studies showing colchicine's ability to suppress NLRP3 inflammasome activation and decrease pro-inflammatory cytokines in cardiovascular contexts [45].
Colchicine's binding to tubulin prevents microtubule formation, inhibiting NLRP3 inflammasome translocation and reducing active inflammasome numbers [46,47]. This disruption also interferes with vesicular transport of pro-IL-1β, further decreasing IL-1β secretion [48]. Additionally, colchicine inhibits leukocyte recruitment and activation, crucial components of the inflammatory response [49]. Furthermore, colchicine modulates intracellular signalling pathways involved in inflammasome activation, including NF-κB, a key regulator of pro-inflammatory cytokine production and inflammasome gene expression [50]. It can also inhibit the TGF-β signalling pathway, either directly or by inhibiting TGF-β synthesis or secretion, resulting in decreased IL-1β production by myofibroblasts [51]. These multifaceted effects distinguish colchicine from more targeted NLRP3 inhibitors currently under development, potentially explaining its broader anti-inflammatory impact and well-established clinical safety profile.
Hypoxia-induced upregulation of TGF-β expression and activation stimulates fibroblast differentiation into myofibroblasts [24]. Excessive myofibroblast proliferation and differentiation may lead to undesirable scar tissue formation, disrupting normal organ or tissue function [52]. In the context of heart disease, myofibroblast proliferation and differentiation are associated with ventricular hypertrophy and myocardial fibrosis, contributing to decreased heart function and increased cardiovascular complications [25].
Our findings align with previous studies showing colchicine's anti-inflammatory effects in cardiovascular diseases [53,54,55,56]. However, while we observed a significant reduction in IL-1β expression with colchicine treatment, the effect on ASC-NLRP3 colocalization, although reduced, did not reach statistical significance. This discrepancy warrants further investigation and may suggest additional mechanisms by which colchicine modulates inflammasome activity. Colchicine's ability to disrupt ASC-NLRP3 colocalization and reduce IL-1β expression under hypoxic conditions provides a mechanistic explanation for its cardioprotective effects observed in clinical trials. For instance, the Colchicine Cardiovascular Outcomes Trial (COLCOT) demonstrated reduced cardiovascular events in patients with recent myocardial infarction treated with colchicine. Our results offer a cellular basis for these clinical observations, highlighting the potential of targeting inflammasome assembly as a therapeutic strategy in ischemic heart disease. By inhibiting inflammasome complex formation, colchicine decreases IL-1β production, potentially reducing inflammation and tissue damage associated with ischemia. Understanding the mechanisms of post-ischemic inflammation, particularly involving IL-1β and NLRP3, is crucial for developing effective therapies to mitigate tissue damage and improve prognosis in ischemic heart disease patients. This study lays the groundwork for further exploration of inflammasome colocalization in inflammation and the development of novel drugs targeting inflammasomes.
Future research should focus on elucidating colchicine's effects on inflammatory cells concerning NLRP3 inflammasome; assessing potential benefits and risks in managing ischemic heart disease and developing therapies targeting IL-1β and NLRP3 pathways or inhibiting inflammasome activity. These efforts may yield innovative and more efficacious treatments for ischemic heart disease.
Previous research has demonstrated the effectiveness of colchicine in reducing inflammation in cardiovascular diseases, particularly by decreasing myocardial damage and improving outcomes in patients with acute coronary syndrome [53,54,55,56]. However, the novelty of our study lies in elucidating the cellular mechanism by which colchicine attenuates NLRP3 inflammasome activation in fibroblasts under hypoxic conditions. Unlike previous studies focusing on clinical outcomes, this study provides direct evidence of colchicine's ability to disrupt ASC-NLRP3 colocalization at the cellular level, specifically in fibroblasts. This mechanism, targeting microtubule dynamics and inflammasome assembly, offers novel insights into colchicine's broader anti-inflammatory effects beyond clinical settings.
5. Conclusions
Our study demonstrates that colchicine treatment significantly reduces IL-1β expression in hypoxia-induced 3T3 fibroblasts, likely by disrupting NLRP3 inflammasome activation. While the effect on ASC-NLRP3 colocalization was not statistically significant, the trend towards reduced colocalization suggests this as a potential mechanism of action. These findings provide new insights into the cellular mechanisms underlying colchicine's anti-inflammatory effects in the context of ischemic heart disease. Future research should focus on validating these results in animal models of myocardial infarction and exploring the potential of targeting NLRP3 inflammasome assembly as a therapeutic strategy in cardiovascular diseases.
Author Contributions
Conceptualization, T.A. and M.S.R..; methodology, T.A. and T.W.; software, A.E. and E.T.; validation, M.S.R., T.W., H.S., D.S., D.O., B.L.; formal analysis, R.A.N.; investigation, T.A.; resources, T.A., A.E., E.T.; data curation, T.A.; writing—original draft preparation, R.A.N.; writing—review and editing, M.S.R., T.W., H.S., D.S., D.O., B.L.; visualization, A.E. and E.T.; supervision, M.S.R., T.W., H.S., D.S., D.O., B.L.; project administration, T.A.; funding acquisition, T.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study protocol was approved by the Health Research Ethic Committee of the Faculty of Medicine, Universitas Brawijaya, Indonesia (Certificate No. 182/EC/KEPK-S3/07/2023) on 1st July 2023 under the name of Tri Astuti as principal investigator.
Informed Consent Statement
Not applicable.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials.
Acknowledgments
We would like to express our sincere appreciation to the Doctoral Program of Medical Science, Faculty of Medicine, University of Brawijaya, Malang, for providing invaluable educational support and research opportunities. We extend our gratitude to Doctor Iskak General Hospital, Tulungagung, for their ongoing support of the lead author's doctoral studies. Our thanks also go to the Laboratory of Animal Physiology, Structure, and Development, Faculty of Mathematics and Natural Sciences, Brawijaya University, for granting access to their research facilities. This collaborative effort has been instrumental in the completion of this study.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| AMI | acute myocardial infarction |
| ASC | caspase recruitment domain |
| CVDs | cardiovascular diseases |
| IL-1β | Interleukin-1β |
| COLCOT | Colchicine Cardiovascular Outcomes Trial |
| ECACC | European Collection of Authenticated Cell Cultures |
| HIF-1α | hypoxia-inducible factor 1α |
| NLRP3 | NOD-like receptor protein 3 |
| ROS | reactive oxygen species |
| STR | short tandem repeat |
| TGF-β | Transforming Growth Factor-β |
References
- Zhao D (2021) Epidemiological Features of Cardiovascular Disease in Asia. JACC Asia 1:1–13. [CrossRef]
- Heusch G (2022) Coronary blood flow in heart failure: cause, consequence and bystander. Basic Res Cardiol 117:1. [CrossRef]
- Roth GA, Mensah GA, Johnson CO, et al (2020) Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019. J Am Coll Cardiol 76:2982–3021. [CrossRef]
- Galli A, Lombardi F (2016) Postinfarct Left Ventricular Remodelling: A Prevailing Cause of Heart Failure. Cardiol Res Pract 2016:1–12. [CrossRef]
- Choe JC, Cha KS, Yun EY, et al (2018) Reverse Left Ventricular Remodelling in ST-Elevation Myocardial Infarction Patients Undergoing Primary Percutaneous Coronary Intervention: Incidence, Predictors, and Impact on Outcome. Heart Lung Circ 27:154–164. [CrossRef]
- Bartekova M, Radosinska J, Jelemensky M, Dhalla NS (2018) Role of cytokines and inflammation in heart function during health and disease. Heart Fail Rev 23:733–758. [CrossRef]
- Amin MN, Siddiqui SA, Ibrahim M, et al (2020) Inflammatory cytokines in the pathogenesis of cardiovascular disease and cancer. SAGE Open Med 8:205031212096575. [CrossRef]
- Gourdin MJ, Bree B, De Kock M (2009) The impact of ischaemia–reperfusion on the blood vessel: Eur J Anaesthesiol 26:537–547. [CrossRef]
- Frangogiannis NG (2014) The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 11:255–265. [CrossRef]
- Fang, L, Moore, X, Dart, A.M, Wang, L (2015) Systemic inflammatory response following acute myocardial infarction. Journal of Geriatric Cardiology 12:305−312.
- Napodano C, Carnazzo V, Basile V, et al (2023) NLRP3 Inflammasome Involvement in Heart, Liver, and Lung Diseases—A Lesson from Cytokine Storm Syndrome. Int J Mol Sci 24:16556. [CrossRef]
- Abderrazak A, Syrovets T, Couchie D, et al (2015) NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol 4:296–307. [CrossRef]
- Gao R, Shi H, Chang S, et al (2019) The selective NLRP3-inflammasome inhibitor MCC950 reduces myocardial fibrosis and improves cardiac remodeling in a mouse model of myocardial infarction. Int Immunopharmacol 74:105575. [CrossRef]
- Ong S-B, Hernández-Reséndiz S, Crespo-Avilan GE, et al (2018) Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther 186:73–87. [CrossRef]
- Kurup R, Galougahi KK, Figtree G, et al (2021) The Role of Colchicine in Atherosclerotic Cardiovascular Disease. Heart Lung Circ 30:795–806. [CrossRef]
- Zhang F, He Q, Qin CH, et al (2022) Therapeutic potential of colchicine in cardiovascular medicine: a pharmacological review. Acta Pharmacol Sin 43:2173–2190. [CrossRef]
- Martínez GJ, Robertson S, Barraclough J, et al (2015) Colchicine Acutely Suppresses Local Cardiac Production of Inflammatory Cytokines in Patients With an Acute Coronary Syndrome. J Am Heart Assoc 4:e002128. [CrossRef]
- Bouabdallaoui N, Tardif J-C, Waters DD, et al (2020) Time-to-treatment initiation of colchicine and cardiovascular outcomes after myocardial infarction in the Colchicine Cardiovascular Outcomes Trial (COLCOT). Eur Heart J 41:4092–4099. [CrossRef]
- Nagar A, Rahman T and Harton JA (2021) The ASC Speck and NLRP3 Inflammasome Function Are Spatially and Temporally Distinct. Front. Immunol. 12:752482. doi: 10.3389/fimmu.2021.752482.
- Astiawati T, Rohman MS, Wihastuti TA, et al (2024) Antifibrotic effects of colchicine on 3T3 cell line ischemia to mitigate detrimental remodeling. J Pharm Pharmacogn Res 12:296–302. [CrossRef]
- Dhurandhar EJ, Dubuisson O, Mashtalir N, et al (2011) E4orf1: A Novel Ligand That Improves Glucose Disposal in Cell Culture. PLoS ONE 6:e23394. [CrossRef]
- Handari SD, Rohman MS, Sargowo D, Aulanni'am, Nugraha RA, Lestari B, Oceandy D. Novel Impact of Colchicine on Interleukin-10 Expression in Acute Myocardial Infarction: An Integrative Approach. J Clin Med. 2024 Aug 7;13(16):4619. doi: 10.3390/jcm13164619.
- Habanjar O, Diab-Assaf M, Caldefie-Chezet F, Delort L (2021) 3D Cell Culture Systems: Tumor Application, Advantages, and Disadvantages. Int J Mol Sci 22:12200. [CrossRef]
- Liang M, Si L, Yu Z, et al (2023) Intermittent hypoxia induces myofibroblast differentiation and extracellular matrix production of MRC5s via HIF-1α-TGF-β/Smad pathway. Sleep Breath. [CrossRef]
- Schlittler M, Pramstaller PP, Rossini A, De Bortoli M (2023) Myocardial Fibrosis in Hypertrophic Cardiomyopathy: A Perspective from Fibroblasts. Int J Mol Sci 24:14845. [CrossRef]
- Tuncay S, Senol H, Guler EM, et al (2018) Synthesis of Oleanolic Acid Analogues and Their Cytotoxic Effects on 3T3 Cell Line. Med Chem 14:617–625. [CrossRef]
- Tripathi VK, Subramaniyan SA, Hwang I (2019) Molecular and Cellular Response of Co-cultured Cells toward Cobalt Chloride (CoCl 2 )-Induced Hypoxia. ACS Omega 4:20882–20893. [CrossRef]
- Tsuzuki T, Okada H, Cho H, et al (2012) Hypoxic stress simultaneously stimulates vascular endothelial growth factor via hypoxia-inducible factor-1 and inhibits stromal cell-derived factor-1 in human endometrial stromal cells. Hum Reprod 27:523–530. [CrossRef]
- Zou D, Zhang Z, He J, et al (2012) Blood vessel formation in the tissue-engineered bone with the constitutively active form of HIF-1α mediated BMSCs. Biomaterials 33:2097–2108. [CrossRef]
- Cheng B-C, Chen J-T, Yang S-T, et al (2017) Cobalt chloride treatment induces autophagic apoptosis in human glioma cells via a p53-dependent pathway. Int J Oncol 50:964–974. [CrossRef]
- Liu J, Wang H, Li J (2016) Inflammation and Inflammatory Cells in Myocardial Infarction and Reperfusion Injury: A Double-Edged Sword. Clin Med Insights Cardiol 10:CMC.S33164. [CrossRef]
- He X, Zeng Y, Li G, et al (2020) Extracellular ASC exacerbated the recurrent ischemic stroke in an NLRP3-dependent manner. J Cereb Blood Flow Metab 40:1048–1060. [CrossRef]
- Wang Z, Zhang S, Xiao Y, et al (2020) NLRP3 Inflammasome and Inflammatory Diseases. Oxid Med Cell Longev 2020:1–11. [CrossRef]
- Fusco R, Siracusa R, Genovese T, et al (2020) Focus on the Role of NLRP3 Inflammasome in Diseases. Int J Mol Sci 21:4223. [CrossRef]
- Protti MP, De Monte L (2020) Dual Role of Inflammasome Adaptor ASC in Cancer. Front Cell Dev Biol 8:40. [CrossRef]
- Rahman T, Nagar A, Duffy EB, et al (2020) NLRP3 Sensing of Diverse Inflammatory Stimuli Requires Distinct Structural Features. Front Immunol 11:1828. [CrossRef]
- Zheng D, Liwinski T, Elinav E (2020) Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov 6:36. [CrossRef]
- Karaba AH, Figueroa A, Massaccesi G, et al (2020) Herpes simplex virus type 1 inflammasome activation in proinflammatory human macrophages is dependent on NLRP3, ASC, and caspase-1. PLOS ONE 15:e0229570. [CrossRef]
- Zhang H, Dhalla NS (2024) The Role of Pro-Inflammatory Cytokines in the Pathogenesis of Cardiovascular Disease. Int J Mol Sci 25:1082. [CrossRef]
- Chen H, Zhang X, Liao N, et al (2018) Enhanced Expression of NLRP3 Inflammasome-Related Inflammation in Diabetic Retinopathy. Investig Opthalmology Vis Sci 59:978. [CrossRef]
- Nagar A, Rahman T, Harton JA (2021) The ASC Speck and NLRP3 Inflammasome Function Are Spatially and Temporally Distinct. Front Immunol 12:752482. [CrossRef]
- Pellegrini C, Martelli A, Antonioli L, et al (2021) NLRP3 inflammasome in cardiovascular diseases: Pathophysiological and pharmacological implications. Med Res Rev 41:1890–1926. [CrossRef]
- Zheng Y, Xu L, Dong N, Li F (2022) NLRP3 inflammasome: The rising star in cardiovascular diseases. Front Cardiovasc Med 9:927061. [CrossRef]
- Hu J, Xu J, Zhao J, et al (2024) Colchicine ameliorates short-term abdominal aortic aneurysms by inhibiting the expression of NLRP3 inflammasome components in mice. Eur J Pharmacol 964:176297. [CrossRef]
- Pagliaro P, Penna C (2023) Inhibitors of NLRP3 Inflammasome in Ischemic Heart Disease: Focus on Functional and Redox Aspects. Antioxidants 12:1396. [CrossRef]
- Naaz F, Haider MR, Shafi S, Yar MS (2019) Anti-tubulin agents of natural origin: Targeting taxol, vinca, and colchicine binding domains. Eur J Med Chem 171:310–331. [CrossRef]
- Banco D, Mustehsan M, Shah B (2024) Update on the Role of Colchicine in Cardiovascular Disease. Curr Cardiol Rep 26:191–198. [CrossRef]
- Sitia R, Rubartelli A (2018) The unconventional secretion of IL-1β: Handling a dangerous weapon to optimize inflammatory responses. Semin Cell Dev Biol 83:12–21. [CrossRef]
- Meyer-Lindemann U, Mauersberger C, Schmidt A-C, et al (2022) Colchicine Impacts Leukocyte Trafficking in Atherosclerosis and Reduces Vascular Inflammation. Front Immunol 13:898690. [CrossRef]
- Olcum M, Tastan B, Ercan I, et al (2020) Inhibitory effects of phytochemicals on NLRP3 inflammasome activation: A review. Phytomedicine 75:153238. [CrossRef]
- Kim S-K (2022) The Mechanism of the NLRP3 Inflammasome Activation and Pathogenic Implication in the Pathogenesis of Gout. J Rheum Dis 29:140–153. [CrossRef]
- Schuster R, Younesi F, Ezzo M, Hinz B (2023) The Role of Myofibroblasts in Physiological and Pathological Tissue Repair. Cold Spring Harb Perspect Biol 15:a041231. [CrossRef]
- Bonaventura A, Abbate A (2023) Colchicine for cardiovascular prevention: the dawn of a new era has finally come. Eur Heart J 44:3303–3304. [CrossRef]
- Muñoz-Sánchez J, Chánez-Cárdenas ME (2019) The use of cobalt chloride as a chemical hypoxia model. J Appl Toxicol 39:556–570. [CrossRef]
- Yuan X, Bhat OM, Zou Y, et al (2022) Endothelial Acid Sphingomyelinase Promotes NLRP3 Inflammasome and Neointima Formation During Hypercholesterolemia. J Lipid Res 63:100298. [CrossRef]
- Cullen SP, Kearney CJ, Clancy DM, Martin SJ (2015) Diverse Activators of the NLRP3 Inflammasome Promote IL-1β Secretion by Triggering Necrosis. Cell Rep 11:1535–1548. [CrossRef]
Figure 1.
This is a figure illustrates immunofluorescence staining on 3T3 cells following various exposures i.e. (A) normal condition treated with vehicle; (B) normal condition treated with colchicine; (C) ischaemic condition treated with vehicle; and (D) ischaemic condition treated with colchicine. ASC (labelled in red) and NLRP3 (labelled in green) protein co-localization configured yellow specks signifying ASC-dependent inflammasome activation. The bottom images are identical to the top images, with additional highlights on the yellow specks (indicated by yellow arrows) identified during the speck intensity measurement. .
Figure 1.
This is a figure illustrates immunofluorescence staining on 3T3 cells following various exposures i.e. (A) normal condition treated with vehicle; (B) normal condition treated with colchicine; (C) ischaemic condition treated with vehicle; and (D) ischaemic condition treated with colchicine. ASC (labelled in red) and NLRP3 (labelled in green) protein co-localization configured yellow specks signifying ASC-dependent inflammasome activation. The bottom images are identical to the top images, with additional highlights on the yellow specks (indicated by yellow arrows) identified during the speck intensity measurement. .
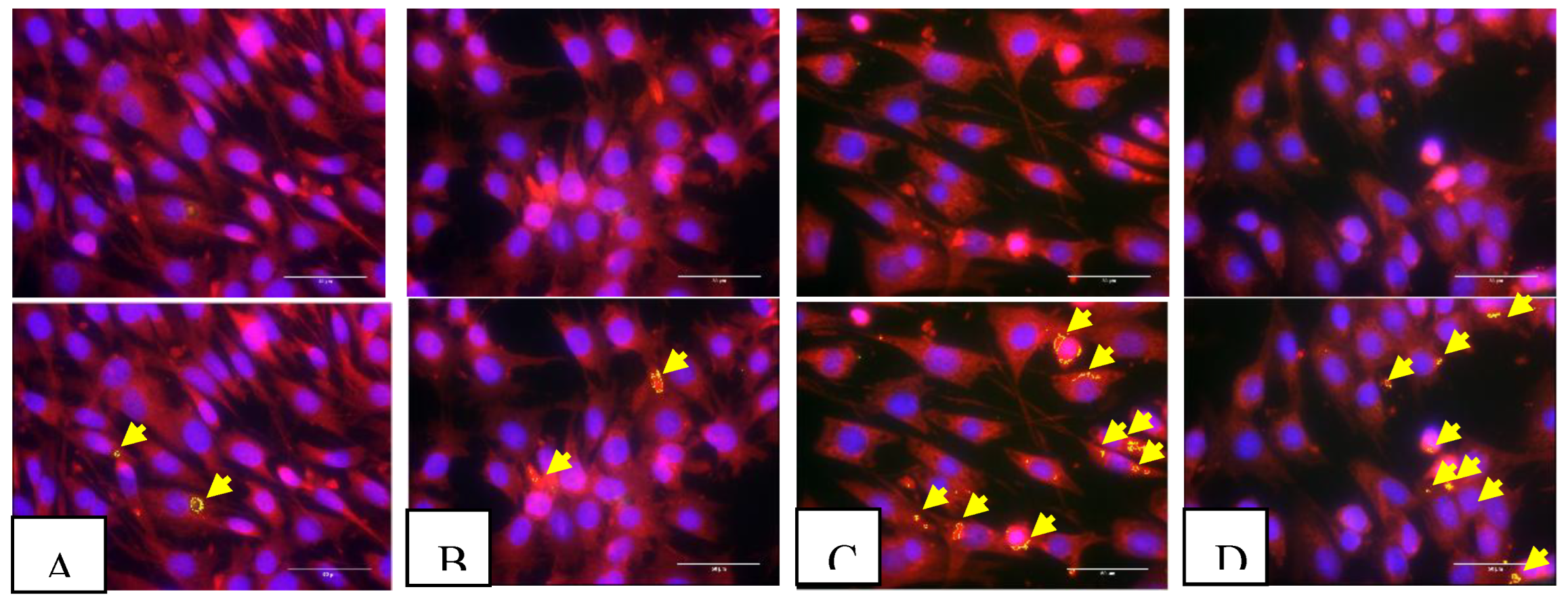
Figure 2.
This is a figure illustrates colocalization speck intensity as a ratio of ASC fluorescence intensity, revealing ASC-NLRP3 complex formation. There is no significant difference of ASC-NLRP3 colocalization in colchicine vs vehicle treatments within the same condition related to ischaemic condition (cobalt chloride exposure). The statistical significance was identified in the control group under ischaemic condition compared to normal condition treated with either vehicle or colchicine (p < 0.05).
Figure 2.
This is a figure illustrates colocalization speck intensity as a ratio of ASC fluorescence intensity, revealing ASC-NLRP3 complex formation. There is no significant difference of ASC-NLRP3 colocalization in colchicine vs vehicle treatments within the same condition related to ischaemic condition (cobalt chloride exposure). The statistical significance was identified in the control group under ischaemic condition compared to normal condition treated with either vehicle or colchicine (p < 0.05).
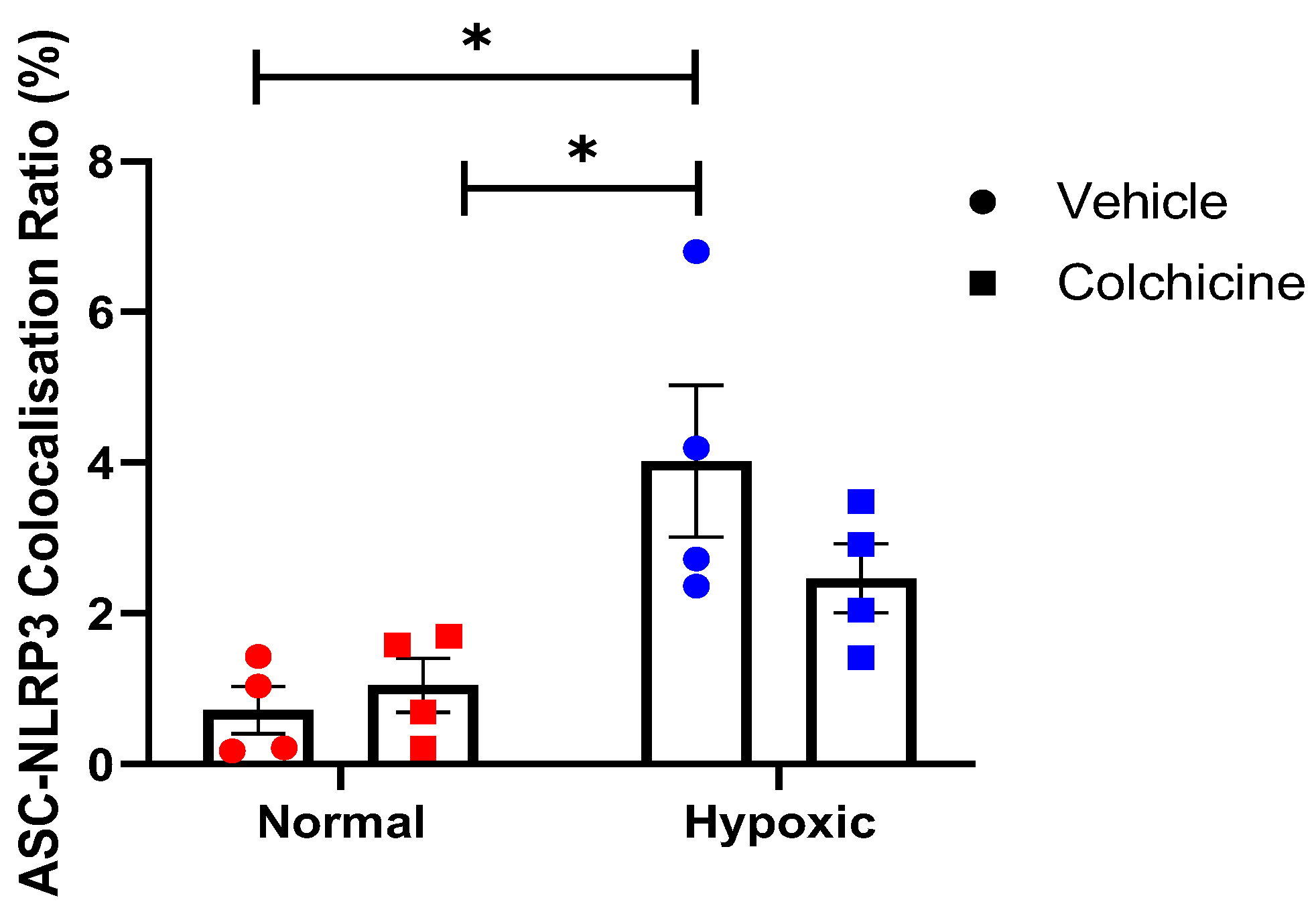
Figure 3.
This is a figure illustrates the effect of colchicine treatment in normoxic and hypoxic 3T3 cells. The IL-1β expression was significantly reduced in colchicine-treated hypoxic cells compared to placebo-treated hypoxic cells (62.58 ± 1.41% vs. 45.33 ± 10.22%, p < 0.001).
Figure 3.
This is a figure illustrates the effect of colchicine treatment in normoxic and hypoxic 3T3 cells. The IL-1β expression was significantly reduced in colchicine-treated hypoxic cells compared to placebo-treated hypoxic cells (62.58 ± 1.41% vs. 45.33 ± 10.22%, p < 0.001).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



