
Submitted:
28 January 2025
Posted:
29 January 2025
You are already at the latest version
Abstract
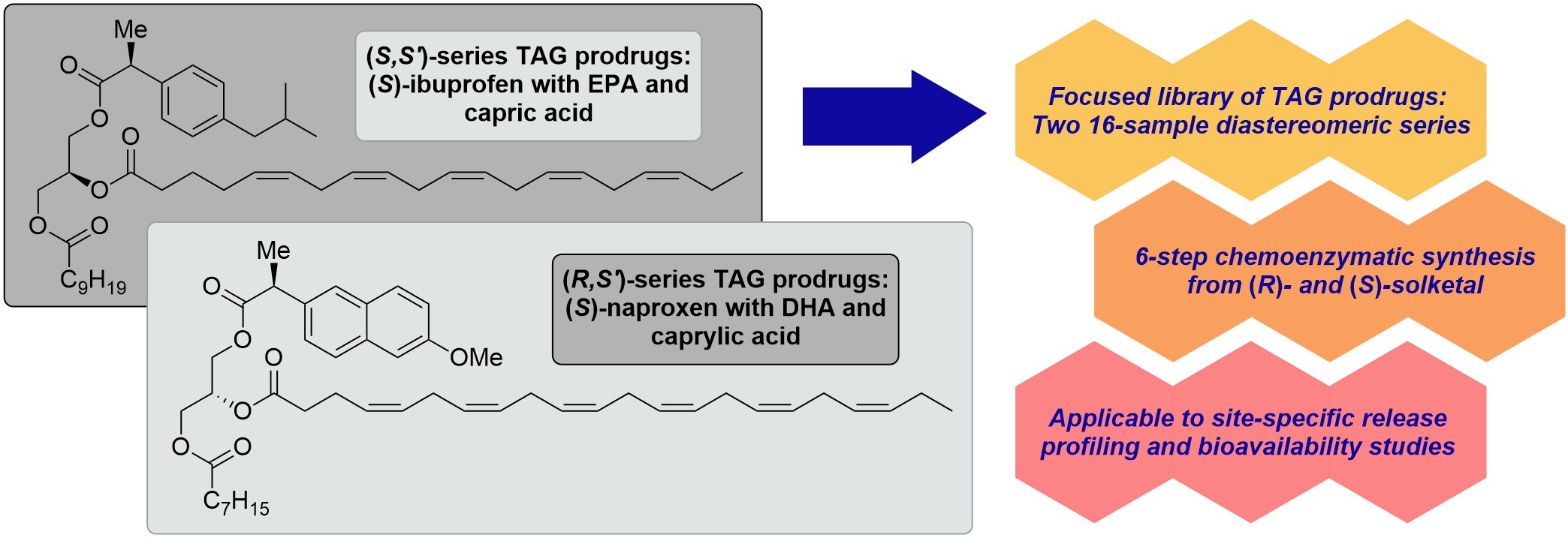
The current paper reports the asymmetric synthesis of a focused library of enantiostructured triacylglycerols (TAGs) constituting a potent drug of the NSAID type (ibuprofen or naproxen) along with a pure bioactive n-3 polyunsaturated fatty acid (EPA or DHA) intended as a novel type of prodrugs. In this second category TAG prodrugs one of the terminal sn-1 or sn-3 positions of the glycerol skeleton is acylated with a single saturated medium-chain fatty acid (C6, C8, C10 or C12), the remaining one with the drug entity, and the PUFA located in the sn-2 position. This was accomplished by a six-step chemoenzymatic approach, two of which promoted by a lipase, starting from enantiopure (R)- and (S)-solketals. The highly regioselective immobilized Candida antarctica lipase (CAL-B) played a crucial role in the regiocontrol of the synthesis. The most challenging key step involved the incorporation of the drugs activated as oxime esters by the lipase exclusively into the terminal position of glycerol protected as a benzyl ether. All combinations, the total of 32 such prodrug TAGs, were prepared, isolated and fully characterized, along with 40 acylglycerol intermediates, obtained in very high to excellent yields in majority of cases.
Keywords:
asymmetric synthesis
; enantiostructured triacylglycerols
; lipase
; n-3 PUFAs
; acylglycerol prodrugs
; (S)-ibuprofen
; (S)-naproxen
1. Introduction
In a very recent report, we proposed a novel type of lipid-based prodrugs possessing an active drug, a bioactive long-chain n-3 polyunsaturated fatty acid (PUFA) along with a saturated fatty acid, all attached to predetermined regio- and stereospecific positions of the glycerol backbone of triacylglycerol (TAG) molecular species (see Figure 1) [1]. Prodrug is a term used over a compound that delivers an active drug after undergoing a bioconversion (intra- or extracellular) within the body. The design of a prodrug aims at improving the bioavailability of a drug by exerting influence on its absorption, distribution, metabolism and excretion (ADME) [2-4]. Prodrugs based on lipids offer advantages including increased absorption through the intestines that may result in increased drug availability and targeting [5-8].
As was described in detail in the report the design of the TAG prodrugs is based on a combination of several important concepts and features associated with fatty acids and TAGs. The first is structured MLM (medium-long-medium) type TAGs possessing a medium-chain fatty acid (MCFA) located at the terminal positions of the glycerol backbone with a bioactive n-3 PUFA (EPA or DHA) located at the 2-position. Such structured TAGs have gained a growing interest among scientists because of their absorption properties in the digestive tract and nutritional value [9-11]. Here, we benefitted from our previous synthesis of such MLM type TAGs by a two-step chemoenzymatic synthesis starting from glycerol by aid of a highly regioselective immobilized Candida antarctica lipase (CAL-B) that introduced the MCFAs exclusively onto the terminal primary alcohol positions of glycerol [12,13].
The second one is the bioactive n-3 PUFAs EPA and DHA that are claimed to offer numerous beneficial effects on human health including cardiovascular disease, cognitive health, inflammatory diseases, and so forth [14-17]. EPA and DHA are also precursors to various highly potent lipid mediators such as the specialized pro-resolving mediators (SPMs) that display potent anti-inflammatory and pro-resolving activities and include resolvins, protectins and maresins [18-20]. As precursors to the SPMs EPA and DHA may themselves be regarded as anti-inflammatory prodrugs [21]. Furthermore, EPA and DHA as ethyl esters are also available as prescription drugs to treat hypertriglyceridemia [22] both as a mixture [23,24] and a pure EPA [25-27].
The third concept behind the prodrug design is termed as enantiostructured TAGs [1,28-30]. They are based on glycerol being prochiral and the consequent chirality of TAGs possessing selected fatty acyl groups occupying predetermined stereospecific positions [1,31,32] of their glycerol skeleton. Their involvement is predicated on our belief that the location of the active drug or the bioactive n-3 PUFAs, not only at the regiospecific (terminal versus mid positions) but also at stereospecific positions (sn-1, sn-2, sn-3) [1,31,32] within the TAGs, may influence the timing and site-specificity of their delivery from the proposed TAG prodrugs in or after the digestive tract as has been described in detail [1].
Finally, it was thought appropriate to base the demonstration of the idea of enantiostructured TAG prodrugs on the use of the non-stereoidal anti-inflammatory drugs (NSAIDs) (S)-ibuprofen and (S)-naproxen to which EPA and DHA may offer some synergistic effects as being precursors to the anti-inflammatory SPMs [1]. To our knowledge there are no reports on prodrug design that is based on acylglycerols constituting active drugs along with the n-3 PUFAs. The advantage offered by the presence of the MCFAs has also been addressed [1].
This resulted in the construction of two proposed enantiostructured TAG prodrug regioisomeric forms that are depicted in Figure 1. The first form is represented by structures 1a and 1b where the active drug (S)-ibuprofen is placed in the sn-2 position. In structure 1a the n-3 PUFA EPA is located at the sn-1 position along with capric acid (C10:0) in the sn-3 position. In structure 1b the positions of EPA and DHA have been interconverted. Consequently, structures 1a and 1b are diastereomers. The previous report [1] described the synthesis of TAG prodrugs that belong to this first category TAG prodrugs with all combinations of the two drugs (S)-ibuprofen and (S)-naproxen, EPA and DHA and the saturated fatty acids ranging from C6:0 to C16:0, the total of 48 such TAG prodrug molecular species (24 diastereomer pairs).
The current paper describes the corresponding asymmetric synthesis of the second category TAG prodrugs to which the diastereomeric structures 2a and 2b belong. In this category the drugs are placed in the terminal positions with the PUFAs in the sn-2 position. As noticed from Figure 1 (S)-naproxen represents the drugs, DHA the n-3 PUFAs and caprylic acid (C8:0) the MCFA in structures 2a and 2b where the acyl groups in the terminal positions have been swapped. It was decided to limit the task this time to MCFAs only, that is caproic, caprylic, capric and lauric acids (C6:0, C8:0, C10:0 and C12:0, respectively). All combinations of such TAG prodrug molecular species were prepared, the total of 32 TAG products (16 such diastereomeric pairs). This has resulted in our establishment of a large, focused library of enantiostructured TAG prodrugs that soon may be screened for some new and interesting properties to increase drug bioavailability and targeting.
2. Results and Discussion
A four-step chemoenzymatic approach was designed for the synthesis of the second category TAG prodrugs that is depicted in Figure 2 and involves two enzymatic steps. As before it is based on the use of 1-O-benzyl-sn-glycerol (prepared in two steps from (R)-solketal [1]) as a chiral precursor with the sn-1 position protected as a benzyl ether. The first step involves a lipase promoted regioselective acylation of the sn-3 hydroxyl group of the diol with the drug activated as an acetoxime ester. After removal of the benzyl protective group in the second step, the MCFA is introduced into the sn-1 position by use of lipase. The final step involves incorporation of the n-3 PUFA into the sn-2 position of the glycerol backbone brought about by a chemical coupling agent to complete the synthesis.
As can be noticed in Figure 2 the synthesis covers all combinations of the (S)-ibuprofen (Ibu) and (S)-naproxen (Nap) placed in the sn-3 position, with EPA and DHA in the sn-2 position and the four saturated medium-chain fatty acids, caproic, caprylic, capric and lauric acids (C6:0, C8:0, C10:0 and C12:0, respectively), located in the remaining sn-1 terminal position of the glycerol backbone. This results in a focused library of the total of 16 targeted enantiostructured TAG prodrugs (R,S')-11a–d – 14a–d also involving the total of 20 enantiopure acylglycerol intermediates.
The synthesis of the corresponding TAG prodrug diastereomers (S,S')-11a–d – 14a–d, where the enantiospecific location of the MCFAs and the drugs has been interchanged with the PUFAs still located at the sn-2 position, was of equal interest. Their synthetic route illustrated in Figure 3 is identical to the above one shown in Figure 2, this time starting from 3-O-benzyl-sn-gycerol (prepared in two steps from (S)-solketal [29]) as a chiral precursor.
2.1. The Enzymatic Coupling of the Drugs
The first step involved an enzymatic coupling of the drugs activated as acetoxime esters exclusively to the terminal position of the benzyl-protected glycerols. This was the main challenge to overcome in the second category TAG prodrug synthesis. Acetoxime esters have previously been used to activate esters [33-35], including the n-3 PUFAs EPA and DHA [36], to ensure faster reactions in biotransformations involving lipase. Faster reactions along with the mildness offered by lipase are the key parameters in controlling the regioselectivity of the lipase and to keep control on unwanted acyl migration [1,37] that is detrimental for the regioisomeric outcome of the reactions.
The acetoxime esters were prepared by chemical coupling of acetoxime to the drugs by use of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) in the presence of 4-dimethylaminopyridine (DMAP) in dichloromethane at r.t. by a previously reported protocol [36]. The reaction is shown for (S)-ibuprofen in Figure 4. The acetoxime ester of (S)-ibuprofen, (S)-3, was obtained as a liquid, whereas the corresponding ester of (S)-naproxen (S)-4 was obtained as a white solid with both reactions taking place in quantitative yields. Both derivatives underwent noteworthy changes in their specific optical activity values, (S)-Ibuprofen from +58.2 to -7.18 for its oxime ester, and (S)-naproxen from +78.3 to -12.3 for its corresponding ester.
Acetoxime esters have been successfully used to activate EPA and DHA [36], to accomplish excellent regioselectivity in their lipase promoted reactions involving the CAL-B with glycerol and 1-O-acylglycerol type ether lipids [36]. However, the oximes are clearly less reactive than the vinyl esters and their irreversibility not as explicit as when using the enol esters [30,36].
After the successful activation of the drugs as oxime esters the task was to have them incorporated exclusively to the terminal positions of the benzyl protected glycerols. As in our previous cases with EPA and DHA activated as oxime esters to acylate glycerol and ether lipids [36], we tried to perform the reaction in dry dichloromethane at room temperature by use of the immobilized Candida antarctica lipase B (CAL-B) with a 1.2-fold molar excess of the ibuprofen acetoxime (S)-3. There were no indications of a reaction taking place and the situation remained the same when increasing the temperature to 40 °C.
When, in one of the attempts, the solvent accidentally had evaporated off from the reaction mixture, we noticed that a reaction had indeed taken place. This prompted us to perform the reaction without a solvent at 40 °C. Since the starting material and the product displayed identical elution properties in all solvent systems tried, TLC was not an option to monitor the progress of the reaction. Instead, we had to depend on 1H NMR spectroscopy and the characteristic peaks belonging to the glyceryl protons of individual acylglycerol derivatives involved. This became of great use to monitor the progress of the reaction. This is illustrated in Figure S1 in the Supplementary Materials.
As is evident from the figure dramatic changes occurred in the glyceryl proton region of the spectra upon acylation with the drug entity. From our previous studies the typical patterns of all five glyceryl protons of the starting material and the product are easily recognised and assigned from which it is quite straight-forward to monitor the progress of the reaction. It is evident that already after 6 hours a substantial part of the starting material had been converted into the product. After 24 hours there was evidently some starting material present, but since virtually no changes took place in the spectrum after 30 hours it was decided to work up the reaction.
The product (R,S')-5 was furnished as a liquid in 90% yield after purification by flash column chromatography using boric acid impregnated silica gel to avoid acyl migration [1,30,38]. Similarly, the corresponding (S,S')-5 diastereomer was obtained from 3-O-benzyl-sn-glycerol in 92% yield. The yields along with the specific rotation of these intermediates are shown in Table 1. In the reactions involving the (S)-ibuprofen acetoxime (S)-3 and the benzyl protected glycerols there were no signs of any acyl migration taking place despite the heating at 40 °C. This may be surprising, but it should be pointed out that 1-O-alkylglycerols are less prone to undergo acyl migration than mono- and diacylated glycerols [36].
The corresponding acylation of the benzyl protected glycerols with the acetoxime activated naproxen (S)-4 became far more of a challenge. The main reason was that (S)-4 unlike (S)-3 is a solid with melting point 42.3-43.4 °C and did not mix well with the glycerol substrate. This resulted in a significantly lower reaction rate as compared to the previous ibuprofen case even though the excess of the acetoxime was lowered to aid the solubility. An extreme care was also needed in terms of acyl migration that became noticeable before the reaction had proceeded to completion which resulted in significantly lower yields. This was clearly related to the prolonged heating at 40 °C over 40 hours.
As before, we were dependent on the 1H NMR spectroscopy to monitor the progress of the reaction. Figure S2 in the Supplementary Materials shows the progress of the reaction as based on the pattern of peaks characteristic of the glycerol derivatives involved. It took a long time to get the reaction to start and after 6 hours virtually no product had formed. The reaction mixture kept solidifying but as the reaction gradually got started this problem decreased. After 23 hours the conversion had reached 25% and after 31 hours the conversion reached 46%.
The conversion kept increasing as more of the acetoxime ester underwent reaction and after 47 hours it had reached 68% but at that stage an unwanted product of acyl migration started to appear in the spectrum as is clearly evident from the corresponding spectrum in Figure S2. Therefore, it was decided to terminate the reaction at this stage although it had not proceeded to completion. After work up and purification by column flash chromatography on silica gel impregnated with boric acid as before the product (R,S')-6 was afforded as a white solid free of the unwanted acyl migrated product in 69% yield. The corresponding diastereomer (S,S')-6 was similarly obtained as a white solid in 65% yield. The yields along with the specific rotation of these intermediates are shown in Table 1.
2.2. The Removal of the Benzyl Protective Group
In the second step of the second category prodrug synthesis all four ibuprofen and naproxen derivatives (R,S')-5, (S,S')-5, (R,S')-6 and (R,S')-6 were subjected to catalytic hydrogenolysis for removal of the benzyl protective group. A protocol identical to that of the previous synthesis of the first category TAG prodrugs was followed by use of a Pd/C catalyst in a mixture of THF and n-hexane under atmospheric pressure at r.t., using catalytic amount of perchloric acid to initiate the reaction [1].
The reactions proceeded very smoothly to afford the monoacylglycerol (MAG) products in excellent yields (93-99%) after only 12 min reaction time. The ibuprofen derivatives were obtained as liquids whereas the corresponding naproxen derivatives were obtained as crystalline material after purification by boric acid impregnated flash silica gel chromatography. The yields and specific optical rotation values are revealed in Table 2 for all four MAG products obtained from the deprotection reactions.
Despite the use of perchloric acid no acyl migration was observed to take place, but like before [1] care was taken in neutralising the reaction mixture with sodium bicarbonate after the reaction was completed. This is evident from the glyceryl proton region of the 1H NMR spectra in Figure S3 in the Supplementary Materials providing a comparison between the glyceryl proton region of the product (R,S')-7 and its precursor (R,S')-5 showing a spectrum typical of 1-MAGs with no signs of acyl migration.
2.3. The Enzymatic Coupling of the SFAs
The third step involved a second lipase promoted acylation of the medium-chain caproic, caprylic, capric and lauric acids (C6:0, C8:0, C10:0 and C12:0) activated as vinyl esters, onto the terminal position of the MAGs already acylated with the drugs obtained from the previous step. The advantages offered using vinyl esters as acylating agents in terms of faster irreversible reactions and milder conditions to maintain the excellent regioselectivity of the lipase and to avoid acyl migration have been discussed in detail in previous reports [1,30,36].
As anticipated the immobilized CAL-B acylated the drug derivatives (R,S')-6 and (S,S')-6, exclusively at the primary alcohol position to accomplish the ibuprofen containing products (R,S')-9a–d and (S,S')-9a–d as was confirmed by 1H NMR spectroscopy. Like before, the reactions were performed in dry dichloromethane at r.t., but it took the lipase significantly longer time to complete the reactions (4-6 hours) as compared to the previous case of the first category TAG prodrug synthesis [1] involving the benzyl protected glycerols (90 minutes). The yields after purification by boric acid impregnated silica gel flash chromatography were in most cases very high to excellent and varied from 74 – 97%. Table 3 shows the identity, yields and specific optical activity of the resulting 1,3-diacylglycerol (1,3-DAG) products involving ibuprofen in accordance with the reaction schemes in Figure 2 and Figure 3.
All ibuprofen containing products (R,S')-9a–c and (S,S')-9a–c were obtained as colourless oils, whereas (R,S')-9d and (S,S')-9d possessing the longest chain (C12:0) were obtained as crystalline material.
The corresponding reactions involving the naproxen derivatives (R,S')-7 and (S,S')-7 to accomplish the 1,3-DAG products (R,S')-10a–d and (S,S')-10a–d provided results quite comparable to those obtained for the ibuprofen 1,3-DAG products in terms of reaction time (4-6 hours) and yields (somewhat lower, 70 – 92%). In accordance with the reaction schemes in Figure 2 and Figure 3, the identity, yields and specific optical activity of the resulting 1,3-DAG products involving naproxen are shown in Table 4.
As in the case of the ibuprofen 1,3-DAG derivatives all the corresponding naproxen containing products (R,S')-10a–c and (S,S')-10a–c were obtained as colourless oils, whereas (R,S')-10d and (S,S')-10d possessing the longest saturated chain were obtained as crystalline material. It is evident that the task of the lipase to acylate the primary position of the 1-MAGs containing the drugs was more of a challenge as compared to the corresponding 1-O-benzylglycerols involved in the corresponding synthesis of the first category TAG prodrugs. This is clearly reflected in longer reaction time and lower yields.
The structures of the 1,3-DAGs were confirmed by the characteristic pattern for the glyceryl proton segment of their 1H-NMR spectra. Figure S4 in the Supplementary Materials presents a comparison of the glyceryl proton segment of the MAG starting material (R,S')-7 and the 1,3-DAG product (R,S')-9c. The characteristic pattern of peaks for the two types of acylglycerols are clear. Upon the second acylation the three protons belonging to the sn-1 and sn-2 positions have undergone a significant down-field shift merging with the sn-3 protons to form a multiplet at d 4.23-3.94 ppm that is characteristic of 1,3-DAGs. No sign of acyl migration was detected in the spectra that would certainly distort the peak pattern and result in additional peaks in the glyceryl proton region of these products.
2.4. The Coupling of the PUFA
The fourth and last step of the second category TAG prodrug synthesis involved a chemical coupling of EPA and DHA into the open sn-2 position of the 1,3-DAGs possessing the drug and the MCFA obtained from the previous step. Procedures already described from the synthesis of the first category TAG prodrugs were followed using approximately 5-10% excess of EPA and DHA, with EDCI as a coupling agent in the presence of DMAP in dichloromethane at r.t. As before, no acyl migration was observed to take place [12,13,29].
All TAG products were obtained as yellowish to yellow oils in very high to excellent yields in majority of cases. The reactions involving DHA were observed to require somewhat longer reaction time than those of EPA and afforded somewhat lower yields. Table 5, Table 6, Table 7 and Table 8 outline the identity, yields and the specific optical activity of the products in accordance with the reaction schemes in Figure 2 and Figure 3. The TAG prodrug products (R,S')-11a–d and (S,S')-11a–d possessing a MCFA, EPA and ibuprofen are shown in Table 5.
Similarly, the corresponding TAG prodrug products (R,S')-12a–d and (R,S')-12a–d possessing a MCFA, EPA and naproxen are shown in Table 6.
Table 7 outlines the TAG prodrug products (R,S')-13a–d and (S,S')-13a–d possessing a MCFA, DHA and ibuprofen.
Finally, the corresponding TAG prodrug products (R,S')-14a–d and (S,S')-14a–d possessing a MCFA, EPA and naproxen are shown in Table 8.
Figure S5 of the Supplementary Materials provides a comparison of the glyceryl proton region of the product (R,S')-11c and the precursor (R,S')-9c. As may be noticed changes anticipated for TAGs have taken place with a dramatic down-field shift of the protons belonging to the sn-2 position upon acylation into that position. The remaining sn-1 and sn-3 protons now resonate as two well dispersed doublets of doublets as is characteristic of TAGs.
As indicated earlier, the glyceryl proton segment of the 1H NMR spectra (δ 5.40-3.45 ppm) is of high utility to authenticate the structure and establish the purity of individual acylglycerol derivatives engaged in the TAG synthesis. This relies on the distinctive patterns of proton peaks representing the acylglycerols. This is also of uttermost importance for maintaining the regiocontrol by accurate detection of unwanted products related to acyl migration as has been discussed and described in detail in previous reports [1,12,13,36]. In the presented work we have benefited from the spectral details obtained from the 1H NMR and the 2D-NMR 1H-1H-COSY spectroscopy that has enabled a full assignment of the 1H NMR data to confirm the chemical purity of all intermediates and products involved.
3. Materials and Methods
3.1. General Information
The 1H- and 13C-NMR spectra were recorded on a 400 MHz Bruker Avance NEO 400 spectrometer (Bruker Switzerland AG, Faellanden, Switzerland). Chemical shifts (δ) are reported in parts per million (ppm) from tetramethylsilane with the solvent resonance used as an internal standard. In all cases, the solvent was deuterochloroform, which had been filtered through the aluminum oxide to rid of acid contamination. The coupling constants (J) are given in Hertz (Hz). The following abbreviations are used to describe the multiplicity: s, singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublets; dt, doublet of triplets; AB q, AB-quartet; and m, multiplet. For 13C-NMR, the number of carbon nuclei contributing to each signal is indicated in parentheses after the chemical shift value. Infrared spectra were recorded on a Nicolet Avatar FT-IR (E.S.P.) spectrometer (Thermo Scientific, Madison, WI, USA) using sodium chloride windows (NaCl) for liquid compounds or potassium bromide pellets (KBr) for solids. The following abbreviations are used to describe the peaks: s, strong; vs, very strong; m, medium; w, weak; and br, broad. The high-resolution mass spectra (HMRS) were recorded on a Bruker OTOF-Q Compact ESI mass spectrometer (Bruker Daltonic, Bremen, Germany). The optical activity was measured on an Autopol V automatic Polarimeter from Rudolph Research Analytical (Hacketstown, NJ, USA) using a 40T-2.5-100-0.7 TempTrol polarimetric cell with 2.5 mm inside diameter, 100 mm optical length and 0.7 mL volume with c (concentration) referring to g sample/100mL. Melting points were determined using a Büchi m-560 melting point apparatus (Uster, Switzerland). TLC monitoring was done on silica plates from SiliCycle (Québec, QC, Canada) and the plates were developed in 4% PMA solution in methanol. Boric acid-impregnated silica gel was prepared by dissolving 4 g of boric acid in 100 mL methanol and then adding 55 g of silica and swirling the resulting slurry for a few minutes. The methanol was then evaporated off, and the silica was dried in vacuo for 6 h at 40◦C.
All chemicals and solvents were used without further purification unless otherwise stated. All solvents used, deuterated chloroform (99.8% D), diethyl ether (≥99.8%), ethyl acetate (≥99.7%), dichloromethane (99.8%), ethanol (≥99.8%), hexane (>99%), methanol (99.9%) and tetrahydrofuran (99.9%), were from Sigma-Aldrich (Steinheim, Germany). Tetrahydrofuran was dried over natrium wire in the presence of benzophenone under a dry nitrogen atmosphere prior to use. Dichloromethane was stored over molecular sieves under nitrogen after being taken to use. All of the following chemicals were obtained from Sigma-Aldrich: acetone oxime (98%), boric acid (≥99.5%), hydrochloric acid (37%), magnesium sulfate (≥99.5%), phosphomolybdic acid, sodium bicarbonate (≥99.0%), sodium hydride (60% dispersion in mineral oil), sodium sulfate (≥99%), (R)-solketal (98%, 98% ee), (S)-solketal (98%, 99% ee), S)-ibuprofen (99%), vinyl dodecanoate (≥99%), palladium on carbon catalyst, perchloric acid (>70%), benzyl bromide (98%), EDCI (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, >99%) and DMAP (4-dimethylaminopyridine, >99%). Vinyl hexanoate (>99%), vinyl octanoate (>99%) and vinyl decanoate (>99%) were purchased from TCI Europe (Zwinderecht, Belgium). The immobilized Candida antarctica lipase B (CAL-B, Novozym 435) was obtained as a gift from Novozymes Denmark (Bagsvaerd, Denmark). EPA (98%) and DHA (≥95%) were obtained as ethyl esters from Pronova Biopharma (Sandefjord, Norway) and were hydrolyzed to their corresponding free acids [1]. (S)-Naproxen was acquired from Prof. Thorsteinn Loftsson at the Faculty of Pharmaceutical Sciences at the University of Iceland (Reykjavik, Iceland). The silica gel for the chromatography (40–63 µm, 0.060–0.300, F60) was obtained from SiliCycle. The TLC plates were dipped into a methanol solution of phosphomolybdic acid (PMA) to develop the spots.
3.2. Activation of Drugs as Oximes
3.2.1. Synthesis of (S)-propan-2-one-O-(2-(4-isobutylphenyl)propanoyl Oxime, (S)-3
To a solution of (S)-ibuprofen (94 mg, 0.465 mmol), DMAP (16 mg, 0.131 mmol) and EDCI (105 mg, 0.553 mmol) in CH2Cl2 (2 mL) were added acetoxime (34 mg, 0.465 mmol) and the solution stirred on a magnetic stirrer at room temperature for 3-4 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by use of ethyl acetate/petroleum ether (3:2) as eluent. The solvent was removed in vacuo on a rotary evaporator and the crude product applied to a silica gel flash chromatography using ethyl acetate/petroleum ether (1:1) as eluent, which afforded the product (S)-3 as a slightly yellow liquid in a quantitative yield (115 mg, 0.465 mmol). [α]20D = -7.18 (c. 7.3, CH2Cl2). IR (NaCl, νmax / cm-1): 3058 (vs), 2958 (vs), 2931 (vs), 2853 (vs), 1758 (vs), 1653 (s). 1H NMR (400 MHz, CDCl3) δH: 7.27-7.17 (m, 2H, Ibu-2,6), 7.14-7.01 (m, 2H, Ibu-3,5), 3.79 (q, J=7.2 Hz, 1H, CHCH3), 2.44 (d, J=7.2 Hz, 2H, CH2CH), 1.99 (s, 3H, NC(CH3)2), 1.83 (nonet, J=6.7 Hz, 1H, CH(CH3)2), 1.83 (s, 3H, NC(CH3)2), 1.56 (d, J=7.2 Hz, 3H, CHCH3), 0.88 (d, J=6.7 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 171.9, 164.4 (N=C), 140.7, 137.5 (2), 129.4 (2), 127.3, 45.1, 44.3, 30.3, 22.5 (2), 22.1, 18.5, 16.9 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C16H23NO2Na 284.1621; found, 284.1618.
3.2.2. Synthesis of (S)-propan-2-one-O-(2-(6-methoxynaphthalen-2-yl)propanoyl Oxime, (S)-4
The same procedure was followed as described for (S)-3 using (S)-naproxen (105 mg, 0.456 mmol), DMAP (14 mg, 0.115 mmol) and EDCI (109 mg, 0.569 mmol) in CH2Cl2 (2 mL) were added acetoxime (33 mg, 0.451 mmol). Purification on silica gel flash chromatography using ethyl acetate/petroleum ether (1:1) as eluent, followed by recrystallization from n-hexane, afforded the product (S)-4 as a white solid in a quantitative yield (130 mg, 0.456 mmol). M.p. 42.3-43.4°C. [α]20D = -12.3 (c. 16.5, CH2Cl2). IR (NaCl, νmax / cm-1): 3052 (vs), 2956 (vs), 2932 (vs), 2863 (vs), 2848 (vs), 1753 (vs), 1652 (s). 1H NMR (400 MHz, CDCl3) δH: : 7.72-7.67 (m, 3H, Nap-1,4,8), 7.44 (dd, J=8.5, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J=8.9, 2.5 Hz, 1H, Nap-7), 7.11 (d, J=2.5 Hz, 1H, Nap-5), 3.96 (q, J=7.2 Hz, 1H, CHCH3), 3.91 (s, 3H, OCH3), 1.99 (s, 3H, NC(CH3)2), 1.83 (s, 3H, NC(CH3)2), 1.65 (d, J=7.2 Hz, 3H, CHCH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 171.9, 164.4, 157.8, 135.4, 133.8, 129.4, 129.0, 127.3, 126.4, 126.1, 119.1, 105.7, 55.4, 44.6, 22.1, 18.7, 17.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C17H19NO3Na 308.1257; found, 308.1251.
3.3. Enzymatic Coupling of the Drugs: Synthesis of (R,S')-5, (S,S')-5, (R,S')-6 and (S,S')-6
3.3.1. Synthesis of 1-O-benzyl-3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (R,S')-5
To a mixture of 1-O-benzyl-sn-glycerol (100 mg, 0.549 mmol) and ibuprofen acetoxime ester (S)-3 (163 mg, 0.659 mmol), immobilized CAL-B (40 mg) was added and the resulting mixture stirred at 40 °C for 31 h under nitrogen atmosphere. The lipase preparation was separated by filtration and the solvent removed in vacuo on a rotary evaporator. The concentrate was applied to a 4% boric acid impregnated flash silica gel chromatography using petroleum ether/ethyl acetate (3:2) as an eluent. The first fraction from the column was contaminated with some oxime starting material and required a repeated chromatography. The combined fractions afforded the product (R,S')-5 as a colorless liquid in 90% yield (175 mg, 0.472 mmol). [α]20D = +25.0 (c. 14.0, CH2Cl2). IR (NaCl, νmax / cm-1): 3458 (br s), 3089 (s), 3462 (br s), 3089 (s), 3062 (s), 3028 (s), 2954 (vs), 2925 (vs), 2868 (vs), 1736 (vs), 1607. 1H NMR (400 MHz, CDCl3) δH: 7.38-7.27 (m, 5H, Ph-H), 7.21-7.16 (m, 2H, Ibu-2,6), 7.10-7.06 (m, 2H, Ibu-3,5), 4.47 (s, 2H, CH2Ph), 4.18 (dd, J=11.4, 4.7 Hz, 1H, CH2 sn-3), 4.13 (dd, J=11.4, 6.1 Hz, 1H, CH2 sn-3), 3.99-3.95 (m, 1H, CH sn-2), 3.72 (q, J=7.2 Hz, 1H, CHCH3), 3.44 (dd, J=9.6, 4.5 Hz, 1H, CH2 sn-1), 3.37 (dd, J=9.6, 5.9 Hz, 1H, CH2 sn-1), 2.44 (d, J=7.2 Hz, 2H, CH2CH(CH3)2), 1.84 (nonet, J=6.8 Hz, 1H, CH(CH3)2), 1.49 (d, J=7.2 Hz, 3H, CHCH3), 0.89 (d, J=6.8 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.9, 140.8, 137.8, 137.7, 129.5 (2), 128.6 (2), 128.0 (2), 127.8 (2), 127.3, 73.6, 70.8, 69.0, 65.7, 45.2, 30.3, 22.5, 18.53 (2), 18.47 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C23H30O4Na 393.2036; found, 393.2030.
3.3.2. Synthesis of 3-O-benzyl-1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (S,S')-5
The same procedure was followed as described for (R,S')-5 using 3-O-benzyl-sn-glycerol (100 mg, 0.549 mmol), ibuprofen acetoxime ester (S)-3 (163 mg, 0.659 mmol), and immobilized CAL-B (45 mg). Purification on a 4% boric acid impregnated flash silica gel column using pet. ether/ethyl acetate (3:2) as eluent afforded the product (S,S')-5 as a colorless liquid in 92% yield (187 mg, 0.505 mmol). As before, the first fraction from the column was contaminated with some oxime starting material and required a repeated chromatography. [α]20D = +20.7 (c. 11.0, CH2Cl2). IR (NaCl, νmax / cm-1): 3458 (br s), 3089 (s), 3062 (s), 3028 (s), 2954 (vs), 2925 (vs), 2865 (vs), 1740 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.37-7.27 (m, 5H, Ph-H), 7.18 (d, J=8.1 Hz, 2H, Ibu-2,6), 7.08 (d, J=8.1 Hz, 2H, Ibu-3,5), 4.47 (s, 2H, CH2Ph), 4.16 (d, J=5.2 Hz, 2H, CH2 sn-1), 3.98-3.94 (m, 1H, CH sn-2), 3.72 (q, J=7.2 Hz, 1H, CHCH3), 3.42 (dd, J=9.6, 4.5 Hz, 1H, CH2 sn-3), 3.36 (dd, J=9.6, 5.9 Hz, 1H, CH2 sn-3), 2.44 (d, J=7.2 Hz, 2H, CH2CH(CH3)2), 1.84 (nonet, J=6.8 Hz, 1H, CH(CH3)2), 1.49 (d, J=7.2 Hz, 3H, CHCH3), 0.89 (d, J=6.8 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.9, 140.8, 137.9, 137.7, 129.5 (2), 128.6 (2), 128.0 (2), 127.9 (2), 127.3, 73.6, 70.8, 69.0, 65.6, 45.2, 30.3, 22.5, 18.52 (2), 18.46 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C23H30O4Na 393.2036; found, 393.2031.
3.3.3. Synthesis of 1-O-benzyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol, (R,S')-6
The same procedure was followed as described for (R,S')-5 using 1-O-benzyl-sn-glycerol (100 mg, 0.549 mmol), naproxen acetoxime ester (S)-4 (172 mg, 0.604 mmol), and immobilized CAL-B (38 mg). Purification on a 4% boric acid impregnated flash silica gel column using pet. ether/ethyl acetate (3:2) as eluent resulted in a first fraction contaminated with the starting material that as before required repeated chromatography. Recrystallization of the combined fractions from n-hexane afforded the product (R,S')-6 as a white solid in 69% yield (149 mg, 0.378 mmol). M.p. 51.7-52.1°C. [α]20D = +53.8 (c. 1.9, CH2Cl2). IR (NaCl, νmax / cm-1): 3538 (br s), 3057 (s), 2973 (vs), 2936 (vs), 2909 (vs), 2864 (vs), 1719 (vs), 1632 (s), 1605 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.71-7.65 (m, 3H, Nap-1,4,8), 7.38 (dd, J=8.4, 1.9 Hz, 1H, Nap-3), 7.34-7.22 (m, 5H, Ph-H), 7.14 (dd, J=8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J=2.5 Hz, 1H, Nap-5), 4.40 (s, 2H, CH2Ph), 4.19 (dd, J=11.5, 4.8 Hz, 1H, CH2 sn-3), 4.14 (dd, J=11.5, 6.0 Hz, 1H, CH2 sn-3), 4.00-3.87 (m, 1H, CH sn-2), 3.91 (s, 3H, OCH3), 3.87 (q, J=7.2 Hz, 1H, CHCH3), 3.40 (dd, J=9.6, 4.4 Hz, 1H, CH2 sn-1), 3.32 (dd, J=9.6, 6.0 Hz, 1H, CH2 sn-1), 2.31 (d, J=5.2 Hz, 1H, OH), 1.58 (d, J=7.2 Hz, 3H, CHCH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.8, 157.9, 137.8, 135.6, 133.9, 129.4, 129.1, 128.6, 128.0 (2), 127.8 (2), 127.4, 126.3, 129.1, 119.2, 105.8, 73.6, 70.9, 69.0, 65.8, 55.5, 45.5, 18.5 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C24H26O5Na 417.1672; found, 417.1663.
3.3.4. Synthesis of 3-O-benzyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol, (S,S')-6
The same procedure was followed as described for (R,S')-5 using 3-O-benzyl-sn-glycerol (76 mg, 0.417 mmol), naproxen acetoxime ester (S)-4 (172 mg, 0.439 mmol), and immobilized CAL-B (42 mg). Purification on a 4% boric acid impregnated flash silica gel column using pet. ether/ethyl acetate (3:2) as eluent resulted in a first fraction contaminated with the starting material that as before required repeated chromatography. Recrystallization of the combined fractions from n-hexane afforded the product (S,S')-6 as a white solid in 65% yield (107 mg, 0.272 mmol). M.p. 63.2-63.5°C. [α]20D = +47.7 (c. 1.7, CH2Cl2). IR (NaCl, νmax / cm-1): 3540 (br s), 3053 (s), 2972 (vs), 2940 (vs), 2904 (vs), 2862 (vs), 1718 (vs), 1630 (s), 1607 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.71-7.64 (m, 3H, Nap-1,4,8 ), 7.38 (dd, J=8.5, 1.9 Hz, 1H, Nap-3), 7.34-7.21 (m, 5H, Ph-H), 7.14 (dd, J=8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J=2.5 Hz, 1H, Nap-5), 4.37 (s, 2H, CH2Ph), 4.17 (d, J=5.5 Hz, 2H, CH2 sn-1), 4.00-3.90 (m, 1H, CH sn-2), 3.91 (s, 3H, OCH3), 3.88 (q, J=7.2 Hz, 1H, CHCH3), 3.37 (dd, J=9.6, 4.4 Hz, 1H, CH2 sn-3), 3.31 (dd, J=9.6, 6.2 Hz, 1H, CH2 sn-3), 2.32 (d, J=4.8 Hz, 1H, OH), 1.58 (d, J=7.2 Hz, 3H, CHCH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.8, 157.8, 137.8, 135.6, 133.9, 129.4, 129.1, 128.6, 128.0 (2), 127.8 (2), 127.4, 126.3, 126.1, 119.2, 105.8, 73.5, 70.8, 68.9, 65.7, 55.5, 45.5, 18.5 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C24H26O5Na 417.1672; found, 417.1671.
3.4. Removal of the benzyl protective group: Synthesis of (R,S')-7, (S,S')-7, (R,S')-8 and (S,S')-8
3.4.1. Synthesis of 3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (R,S')-7
Pd/C catalyst (8 mg) was placed into a 25 mL flame-dried two-necked round-bottom flask equipped with a magnetic stirrer under nitrogen atmosphere at room temperature and the flask sealed with a septum. A solution of 1-O-benzyl-3-[(S)-2-(4-isobutylphenyl)-propanoyl]-sn-glycerol (R,S')-5 (40 mg, 0.108 mmol) dissolved in dry THF (3.2 mL) was added with a syringe, followed by n-hexane (5.2 mL). A balloon filled with hydrogen gas was then mounted on a syringe and stuck through the septum. The mixture was stirred while the hydrogen gas was blown through the flask to replace the nitrogen atmosphere with hydrogen. Then a tiny drop of perchloric acid was added and the solution stirred vigorously at room temperature while being monitored with TLC. When the reaction came to completion according to the TLC (approximately 12 minutes) the flask was promptly opened and the acid neutralized by adding NaHCO3 (s). Then the solution was filtered, and the solvent removed in vacuo on a rotary evaporator. The crude product was applied to a 4% boric acid impregnated flash silica gel chromatography using petroleum ether/ethyl acetate (2:3) as eluent, wich afforded the product (R,S')-7 as a pale-yellow oil, in 98% yield (30 mg, 0.107 mmol). [α]20D = +42.9 (c. 3.5, CH2Cl2). IR (NaCl, νmax / cm-1): 3423 (br s), 3063 (s), 3025 (s), 2954 (vs), 2923 (vs), 2867 (vs), 1740 (vs) 1H NMR (400 MHz, CDCl3) δH: 7.19 (d, J=8.1 Hz, 2H, Ibu-2,6), 7.10 (d, J=8.1 Hz, 2H, H-4,6 Ibu), 4.22 (dd, J=11.6, 4.6 Hz, 1H, CH2 sn-3), 4.10 (dd, J=11.4, 6.1 Hz, 1H, CH2 sn-3), 3.87-3.80 (m, 1H, CH sn-2), 3.74 (q, J=7.2 Hz, 1H, CHCH3), 3.57 (dd, J=11.5, 4.0 Hz, 1H, CH2 sn-1), 3.45 (dd, J=11.5, 5.6 Hz, 1H, CH2 sn-1), 3.10-2.75 (bm, 2H, OH), 2.45 (d, J=7.2 Hz, 2H, CH2CH(CH3)2), 1.85 (nonet, J=6.8 Hz, 1H, CH(CH3)2), 1.51 (d, J=7.2 Hz, 3H, CHCH3), 0.89 (d, J=6.8 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 175.2, 140.8, 137.4, 129.5 (2), 127.1 (2), 70.2, 65.4, 63.2, 45.1, 45.1, 30.2, 22.4 (2), 18.4 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C16H24O4Na 303.1567; found, 303.1563.
3.4.2. Synthesis of 1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (S,S')-7
The same procedure was followed as described for (R,S')-7 using Pd/C (15 mg), 3-O-benzyl-1-[(S)-2-(4-isobutylphenyl)-propanoyl]-sn-glycerol (S,S')-5 (50 mg, 0.135 mmol), THF (4.0 mL) and n-hexane (6.5 mL). Purification on a 4% boric acid impregnated flash silica gel column using pet. ether/ethyl acetate (2:3) as eluent afforded the product (S,S')-7 as a colorless liquid in 93% yield (35 mg, 0.125 mmol). [α]20D = +33.9 (c. 2.0, CH2Cl2). IR (NaCl, νmax / cm-1): 3455 (br s), 3060 (s), 3028 (s), 2954 (vs), 2925 (vs), 2865 (vs), 1742 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.19 (d, J=8.1 Hz, 2H, Ibu-2,6), 7.10 (d, J=8.1 Hz, 2H, H-4,6 Ibu), 4.22 (dd, J=11.6, 4.6 Hz, 1H, CH2 sn-1), 4.10 (dd, J=11.4, 6.2 Hz, 1H, CH2 sn-1), 3.87-3.80 (m, 1H, CH sn-2), 3.74 (q, J=7.2 Hz, 1H, CHCH3), 3.57 (dd, J=11.5, 4.0 Hz, 1H, CH2 sn-3), 3.45 (dd, J=11.5, 5.6 Hz, 1H, CH2 sn-3), 2.45 (d, J=7.2 Hz, 2H, CH2CH(CH3)2), 2.35-2.19 (bs, 1H, OH), 1.82-1.92 (bs, 1H, OH), 1.85 (nonet, J=6.8 Hz, 1H, CH(CH3)2), 1.51 (d, J=7.2 Hz, 3H, CHCH3), 0.89 (d, J=6.8 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 175.2, 140.8, 137.5, 129.5 (2), 127.1 (2), 70.2, 65.4, 63.2, 45.1, 45.1, 30.2, 22.4 (2), 18.4 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C16H24O4Na 303.1567; found, 303.1569.
3.4.3. Synthesis of 3-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol, (R,S')-8
The same procedure was followed as described for (R,S')-7 using 1-O-benzyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol (R,S')-6 (130 mg, 0.330 mmol), THF (9.5 mL), n-hexane (16.5 mL) and Pd/C catalyst (25 mg). Purification on a 4% boric acid impregnated flash silica gel column using pet. ether/ethyl acetate (2:3) as eluent, followed by recrystallization from n-hexane, afforded the product (R,S')-8 as white, thin, needle-like crystals in 93% yield (93 mg, 0.306 mmol). M.p. 42.7-43.4°C. [α]20D = +43.5 (c. 2.2, CH2Cl2). IR (NaCl, νmax / cm-1): 3459 (br), 3058 (vs), 2980 (vs), 2940 (vs), 2878 (vs), 1732 (vs), 1634 (s), 1606 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.71-7.61 (m, 3H, Nap-1,4,8), 7.38 (dd, J=8.6, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J=8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J=2.5 Hz, 1H, Nap-5), 4.23-4.07 (m, 2H, CH2 sn-3), 3.90 (s, 3H, OCH3), 3.96-3.84 (m, 1H, CH sn-2), 3.81 (q, J=7.2 Hz, 1H, CHCH3), 3.57 (m, 1H, CH2 sn-1), 3.45 (m, 1H, CH2 sn-1), 1.59 (d, J=7.2 Hz, 3H, CHCH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 175.2, 157.9, 135.4, 133.9, 129.4, 129.0, 127.4, 126.1 (2), 119.3, 105.8, 70.2, 65.7, 63.3, 55.4, 45.5, 18.5 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C17H20O5Na 327.1203; found, 327.1201.
3.4.4. Synthesis of 1-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol, (S,S')-8
The same procedure was followed as described for (R,S')-7 using 3-O-benzyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol (S,S')-6 (126 mg, 0.319 mmol), THF (9.5 mL), n-hexane (15 mL) and Pd/C catalyst (17 mg). Purification on a 4% boric acid impregnated flash silica gel column using pet. ether/ethyl acetate (2:3) as eluent, followed by recrystallization from n-hexane, afforded the product (S,S')-8 as white solid in 99% yield (96 mg, 0.315 mmol). M.p. 58.9-59.7°C. [α]20D = +34.3 (c. 1.0, CH2Cl2). IR (NaCl, νmax / cm-1): 3455 (br s), 3056 (vs), 2982 (vs), 2945 (vs), 2874 (vs), 1734 (vs), 1633 (s), 1605 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.71-7.61 (m, 3H, Nap-1,4,8), 7.39 (dd, J=8.6, 1.5 Hz, 1H, Nap-3), 7.15 (dd, J=8.9, 2.4 Hz, 1H, Nap-7), 7.12 (d, J=2.4 Hz, 1H, Nap-5), 4.18 (d, J=4.9 Hz, 2H, CH2 sn-1), 3.92 (s, 3H, OCH3), 3.92-3.88 (m, 1H, CHCH3), 3.88-3.82 (m, 1H, CH sn-2), 3.56 (dd, J=11.1, 4.9 Hz, 1H, CH2 sn-3), 3.45 (dd, J=11.1, 5.6 Hz, 1H, CH2 sn-3), 2.30-184 (m, 2H, OH), 1.59 (d, J=7.2 Hz, 3H, CHCH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 175.2, 157.9, 135.4, 133.9, 129.4, 129.1, 127.5, 126.1 (2), 119.3, 105.8, 70.2, 65.7, 63.3, 55.5, 45.5, 18.5 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C17H20O5Na 327.1203; found, 327.1212.
3.5. The enzymatic coupling of the MCFAs: Synthesis of (R,S')-9a, (S,S')-9a, (R,S')-10a and (S,S')-10a
For synthesis of (R,S')-9b–d, (S,S')-9b–d, (R,S')-10b–d and (S,S')-10b–d see Supplementary Materials
3.5.1. Synthesis of 1-hexanoyl-3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (R,S')-9a
Immobilized CAL-B (18 mg) was added to a solution of 3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol (R,S')-7 (37 mg, 0.132 mmol), and vinyl hexanoate (21 mg, 0.145 mmol) in CH2Cl2 (3.5 mL). The resulting mixture was stirred at room temperature for 7 h. The lipase preparation was separated by filtration and the solvent removed in vacuo on rotary evaporator. The concentrate was applied to a 4% boric acid impregnated flash silica gel chromatography using petroleum ether/ethyl acetate (7:3) as eluent. This afforded the product (R,S')-9a as a colorless liquid in 80% yield (40 mg, 0.106 mmol). [α]20D = +22.7 (c. 3.0, CH2Cl2). IR (NaCl, νmax / cm-1): 3321 (br s), 2956 (vs), 2931 (vs), 2870 (vs), 1740 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.19 (d, J=8.1 Hz, 2H, Ibu-2,6), 7.10 (d, J=8.1 Hz, 2H, H-4,6 Ibu), 4.21-3.94 (m, 5H, CH2 sn-1/3, CH sn-2), 3.74 (q, J=7.2 Hz, 1H, CHCH3), 2.44 (d, J=6.8 Hz, 2H, CH2CH(CH3)2), 2.34-2.27 (m, 2H, CH2COO SFA), 1.84 (nonet, J=6.8 Hz, 1H, CH(CH3)2), 1.67-1.57 (m, 2H, CH2CH2COO), 1.50 (d, J=7.2 Hz, 3H, CHCH3), 1.37-1.22 (m, 4H, CH2), 0.90 (t, J=6.9 Hz, 3H, CH2CH3), 0.89 (d, J=6.8 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.9 (C=O Ibu), 174.0 (C=O SFA), 140.9, 137.6, 129.6 (2), 127.2 (2), 68.5, 65.5, 65.0, 45.2 (2), 34.2, 31.4, 30.3, 24.7, 22.5 (2), 22.4, 18.5, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C22H34O5Na 401.2298; found, 401.2300.
3.5.2. Synthesis of 3-hexanoyl-1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (S,S')-9a
Immobilized CAL-B (17 mg) was added to a solution of 1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol (S,S')-7 (25 mg, 0.089 mmol), and vinyl hexanoate (14 mg, 0.098 mmol) in CH2Cl2 (2 mL). The resulting mixture was stirred at room temperature for 7 h. The lipase preparation was separated by filtration and the solvent removed in vacuo on rotary evaporator. The concentrate was applied to a 4% boric acid impregnated flash silica gel chromatography using petroleum ether/ethyl acetate (4:1) as eluent. This afforded the product (S,S')-9a as a colorless liquid in 94% yield (32 mg, 0.085 mmol). [α]20D = +21.0 (c. 0.4, CH2Cl2). IR (NaCl, νmax / cm-1): 3465 (br s), 2975 (vs), 2941 (vs), 2864 (vs), 2834 (vs), 1738 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.20 (d, J=8.1 Hz, 2H, Ibu-2,6), 7.10 (d, J=8.1 Hz, 2H, H-4,6 Ibu), 4.21-3.97 (m, 5H, CH2 sn-1/3, CH sn-2), 3.74 (q, J=7.2 Hz, 1H, CHCH3), 2.44 (d, J=6.8 Hz, 2H, CH2CH(CH3)2), 2.39-2.28 (m, 1H, OH), 2.34-2.27 (m, 2H, CH2COO SFA), 1.84 (nonet, J=6.8 Hz, 1H, CH(CH3)2), 1.68-1.55 (m, 2H, CH2CH2COO), 1.51 (d, J=7.2 Hz, 3H, CHCH3), 1.38-1.24 (m, 4H, CH2), 0.90 (t, J=6.9 Hz, 3H, CH2CH3), 0.89 (d, J=6.8 Hz, 6H, CH(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.9 (C=O Ibu), 174.0 (C=O SFA), 140.9, 137.6, 129.6 (2), 127.2 (2), 68.5, 65.4, 65.0, 45.2, 45.2, 34.2, 31.4, 30.3, 24.7, 22.5 (2), 22.4, 18.5, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C22H34O5Na 401.2298; found, 401.2294.
3.5.3. Synthesis of 1-hexanoyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol, (R,S')-10a
Immobilized CAL-B (15 mg) was added to a solution of 3-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol (R,S')-8 (27 mg, 0.089 mmol), and vinyl hexanoate (14 mg, 0.098 mmol) in CH2Cl2 (2.4 mL). The resulting mixture was stirred at room temperature for 2 h after which more CAL-B (5 mg) was added to speed up the reaction. After further 3.5 h reaction TLC monitoring indicated a complete reaction. The lipase preparation was separated by filtration and the solvent removed in vacuo on rotary evaporator. The concentrate was applied to a 4% boric acid impregnated flash silica gel chromatography using petroleum ether/ethyl acetate (7:3) as eluent. This afforded the product (R,S')-10a as a colorless liquid in 92% yield (33 mg, 0.082 mmol). [α]20D = +22.6 (c. 2.5, CH2Cl2). IR (NaCl, νmax / cm-1): 3459 (br s), 2946 (vs), 2930 (vs), 2870 (vs), 1740 (vs), 1635 (s), 1605 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.86-7.58 (m, 3H, Nap-1,4,8), 7.47-7.30 (m, 1H, Nap-3), 7.19-7.04 (m, 2H, Nap-5,7), 4.33-3.62 (m, 9H, CH2 sn-1/3, CH sn-2, OCH3, CHCH3), 2.24-2.19 (m, 3H, OH, CH2COO), 1.61-1.52 (m, 5H, CH2CH2COO, CHCH3), 1.32-1.23 (m, 4H, CH2), 0.87 (t, J=6.7 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Nap), 173.3 (C=O SFA), 157.9, 135.4, 133.9, 129.4, 129.1, 127.4, 126.10, 126.07, 119.2, 105.8, 68.4, 65.6, 65.0, 55.4, 45.4, 34.1, 31.4, 24.7, 22.4, 18.5, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C23H30O6Na 425.1935; found, 425.1933.
3.5.4. Synthesis of 3-hexanoyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol, (S,S')-10a
Immobilized CAL-B (15 mg) was added to a solution of 1-[(S)-2-(6-methoxynaphthalen-2-yl)]-sn-glycerol (S,S')-8 (33 mg, 0.108 mmol), and vinyl hexanoate (28 mg, 0.198 mmol) in CH2Cl2 (3 mL). The resulting mixture was stirred at room temperature for 3 h after which more CAL-B (5 mg) was added to speed up the reaction. After further 5.5 h reaction TLC monitoring indicated a complete reaction. The lipase preparation was separated by filtration and the solvent removed in vacuo on rotary evaporator. The concentrate was applied to a 4% boric acid impregnated flash silica gel chromatography using petroleum ether/ethyl acetate (7:3) as eluent. This afforded the product (S,S')-10a as a colorless liquid in 70% yield (30 mg, 0.075 mmol). [α]20D = +21.5 (c. 0.6, CH2Cl2). IR (NaCl, νmax / cm-1): 3466 (br s), 2969 (vs), 2972 (vs), 1735 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.73-7.68 (m, 2H, Nap-4,8), 7.66 (d, J=1.9 Hz, 1H, Nap-1), 7.39 (dd, J=8.5, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J=8.9, 2.5 Hz, 1H, Nap-7), 7.11 (d, J=2.5 Hz, 1H, Nap-5), 4.21-3.98 (m, 5H, CH2 sn-1/3, CH sn-2), 3.91 (s, 3H, OCH3), 3.90 (q, J=7.2 Hz, 1H, CHCH3), 2.28 (t, J=7.6 Hz, 2H, CH2COO), 1.61-1.57 (m, 2H, CH2CH2COO), 1.59 (d, J=7.2 Hz, 3H, CHCH3), 1.32-1.23 (m, 4H, CH2), 0.88 (t, J=6.9 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Nap), 174.8 (C=O Nap), 174.0 (C=O SFA), 157.9, 135.4, 133.9, 129.4, 129.1, 127.5, 126.2, 126.1, 119.3, 105.8, 68.5, 65.6, 65.0, 55.5, 45.5, 34.2, 31.4, 24.7, 22.4, 18.6, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C23H30O6Na 425.1935; found, 425.1939.
3.6. Coupling of EPA: Synthesis of (R,S')-11a, (S,S')-11a, (R,S')-12a and (S,S')-12a
For synthesis of (R,S')-11b–d, (S,S')-11b–d, (R,S')-12b–d and (S,S')-12b–d see Supplementary Materials
3.6.1. Synthesis of 2-[5Z,8Z,11Z,14Z,17Z)-eicosa-5,8,11,14,17-pentaenoyl]-1-hexanoyl-3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (R,S')-11a
To a solution of 1-hexanoyl-3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol (R,S')-9a (15 mg, 0.040 mmol) and EPA as a free acid (13 mg, 0.044 mmol) in CH2Cl2 (2 mL) were added DMAP (6 mg, 0.043 mmol) and EDCI (12 mg, 0.058 mmol). The solution was stirred on a magnetic stirrer at room temperature for 23 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by use of Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (9:1) as eluent, which afforded the product (R,S')-11a as a yellow oil, in 96% yield (26 mg, 0.039 mmol). [α]20D = +8.29 (c. 2.8, CH2Cl2). IR (NaCl, νmax / cm-1): 3012 (vs), 2958 (vs), 2927 (vs), 2871 (vs), 1744 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.18 (d, J=8.1 Hz, 2H, Ibu-2,6), 7.08 (d, J=8.1 Hz, 2H, Ibu-3,5), 5.40-5.28 (m, 10H, =CH), 5.23-5.17 (m, 1H, CH sn-2), 4.29 (dd, J=11.9, 4.4 Hz, 1H, CH2 sn-1/3), 4.21 (dd, J=11.9, 5.4 Hz, 1H, CH2 sn-1/3), 4.14 (dd, J=11.9, 5.8 Hz, 1H, CH2 sn-1/3), 4.00 (dd, J=11.9, 5.9 Hz, 1H, CH2 sn-1/3), 3.70 (q, J=7.1 Hz, 1H, CHCH3), 2.89-2.77 (m, 8H, =CHCH2CH=), 2.44 (d, J=7.2 Hz, 2H, CH2CH(CH3)2), 2.31-2.20 (m, 4H, CH2COO EPA, CH2COO SFA), 2.13-2.03 (m, 4H, CH2CH2CH= and =CHCH2CH3), 1.84 (nonet, J=6.9 Hz, 1H, CH(CH3)2), 1.69-1.62 (m, 2H, CH2CH2COO EPA), 1.62-1.55 (m, 2H, CH2CH2COO SFA), 1.49 (d, J=7.1 Hz, 3H, CHCH3), 1.33-1.21 (m, 4H, CH2), 0.97 (t, J=7.5 Hz, 3H, CH3 EPA), 0.89 (d, J=6.7 Hz, 6H, CH(CH3)2), 0.88 (t, J=7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Ibu), 173.3 (C=O SFA), 172.6 (C=O EPA), 140.8, 137.4, 132.2, 129.5 (2), 129.1, 129.0, 128.7, 128.5, 128.4, 128.3, 128.2, 128.0, 127.3 (2), 127.2, 69.1, 62.4, 62.1, 45.2, 45.1, 34.7, 34.1, 33.7, 31.4, 26.7, 26.4 (3), 25.8, 24.8, 24.7, 22.5 (2), 22.4, 20.7, 18.4, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C42H62O6Na 685.4439; found, 685.4412.
3.6.2. Synthesis of 2-[5Z,8Z,11Z,14Z,17Z)-eicosa-5,8,11,14,17-pentaenoyl]-3-hexanoyl-1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (S,S')-11a
To a solution of 3-hexanoyl-1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol (S,S')-9a (11 mg, 0.029 mmol) and EPA as a free acid (10 mg, 0.032 mmol) in CH2Cl2 (1.5 mL) were added DMAP (4 mg, 0.031 mmol) and EDCI (8 mg, 0.042 mmol). The solution was stirred on a magnetic stirrer at room temperature for 25 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by use of Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (4:1) as eluent, which afforded the product (S,S')-11a as a yellow oil, in 89% yield (17 mg, 0.026 mmol). [α]20D = +8.27 (c. 2.2, CH2Cl2). IR (NaCl, νmax / cm-1): 3013 (vs), 2970 (vs), 2873 (vs), 2829 (vs), 1744 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.18 (d, J=8.1 Hz, 2H, Ibu-2,6), 7.08 (d, J=8.1 Hz, 2H, Ibu-3,5), 5.40-5.26 (m, 10H, =CH), 5.23-5.17 (m, 1H, CH sn-2), 4.29 (dd, J=11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.19 (dd, J=11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.12 (dd, J=11.9, 6.1 Hz, 1H, CH2 sn-1/3), 4.05 (dd, J=11.9, 5.9 Hz, 1H, CH2 sn-1/3), 3.70 (q, J=7.1 Hz, 1H, CHCH3), 2.87-2.77 (m, 8H, =CHCH2CH=), 2.44 (d, J=7.2 Hz, 2H, CH2CH(CH3)2), 2.37-2.20 (m, 4H, CH2COO EPA, CH2COO SFA), 2.13-2.02 (m, 4H, CH2CH2CH= and =CHCH2CH3), 1.85 (nonet, J=6.9 Hz, 1H, CH(CH3)2), 1.70-1.54 (m, 4H, CH2CH2COO EPA, CH2CH2COO SFA), 1.49 (d, J=7.1 Hz, 3H, CHCH3), 1.33-1.21 (m, 4H, CH2), 0.97 (t, J=7.5 Hz, 3H, CH3 EPA), 0.89 (d, J=6.8 Hz, 6H, CH(CH3)2), 0.88 (t, J=7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Ibu), 173.3 (C=O SFA), 172.7 (C=O EPA), 140.8, 137.5, 132.2, 129.5 (2), 129.1, 129.0, 128.7, 128.5, 128.4, 128.3, 128.2, 128.0, 127.3 (2), 127.2, 69.0, 62.6, 62.1, 45.2, 45.1, 34.7, 34.1, 33.7, 31.4, 26.7, 25.8 (3), 25.7, 24.9, 24.7, 22.5 (2), 22.4, 20.7, 18.4, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C42H62O6Na 685.4439; found, 685.4436.
3.6.3. Synthesis of 2-[5Z,8Z,11Z,14Z,17Z)-eicosa-5,8,11,14,17-pentaenoyl]-1-hexanoyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol, (R,S')-12a
To a solution of 1-hexanoyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol (R,S')-10a (10 mg, 0.025 mmol) and EPA as a free acid (8 mg, 0.027 mmol) in CH2Cl2 (1.3 mL) were added DMAP (3 mg, 0.027 mmol) and EDCI (8 mg, 0.037 mmol). The solution was stirred on a magnetic stirrer at room temperature for 24 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by use of Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (4:1) as eluent, which afforded the product (R,S')-12a as a yellow oil, in 80% yield (14 mg, 0.020 mmol). [α]20D = +9.29 (c. 1.4, CH2Cl2). IR (NaCl, νmax / cm-1): 3013 (vs), 2970 (vs), 2940 (vs), 2853 (vs), 1743 (vs), 1635 (s), 1607 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.72-7.66 (m, 2H, Nap-4,8), 7.64 (d, J=1.9 Hz, 1H, Nap-1), 7.37 (dd, J=8.5, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J=8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J=2.5 Hz, 1H, Nap-5), 5.48-5.27 (m, 10H, =CH), 5.20 (m, 1H, CH sn-2), 4.30 (dd, J=11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.22 (dd, J=11.9, 4.4 Hz, 1H CH2 sn-1/3), 4.16 (dd, J=11.9, 6.0 Hz, 1H, CH2 sn-1/3), 4.03 (dd, J=11.9, 5.8 Hz, 1H, CH2 sn-1/3), 3.90 (s, 3H, OCH3), 3.86 (q, J=7.2 Hz, 1H, CHCH3), 2.90-2.75 (m, 8H, =CHCH2CH=), 2.24 (t, J=7.5 Hz, 2H, CH2COO EPA), 2.12-2.04 (m, 2H, CH2COO SFA), 2.08 (td, J=7.4, 1.6 Hz, 2H, CH2CH2CH=), 2.05-1.96 (m, 2H, =CHCH2CH3), 1.83-1.75 (m, 2H, CH2CH2COO EPA), 1.60-1.51 (m, 5H, CH2CH2COO SFA and CHCH3), 1.34-1.21 (m, 4H, CH2), 0.98 (t, J=7.5 Hz, 3H, CH3 EPA), 0.88 (t, J=7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.2 (C=O Nap), 173.3 (C=O SFA), 172.6 (C=O EPA), 157.9, 135.3, 133.9, 132.2, 129.4, 129.1, 129.0, 128.7, 128.6, 128.5, 128.4, 128.3, 128.2, 128.0, 127.3, 127.2, 126.3, 126.1, 119.2, 105.7, 69.0, 62.5, 62.1, 55.4, 45.5, 34.1, 33.6, 31.4, 26.4, 25.8 (3), 25.7, 24.6, 24.2, 22.4, 20.7, 18.4, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C43H58O7Na 709.4075; found, 709.4059.
3.6.4. Synthesis of 2-[5Z,8Z,11Z,14Z,17Z)-eicosa-5,8,11,14,17-pentaenoyl]-3-hexanoyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol, (S,S')-12a
To a solution of 3-hexanoyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol (S,S')-10a (11 mg, 0.027 mmol) and EPA as a free acid (9 mg, 0.030 mmol) in CH2Cl2 (1.3 mL) were added DMAP (4 mg, 0.029 mmol) and EDCI (8 mg, 0.040 mmol). The solution was stirred on a magnetic stirrer at room temperature for 30 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by use of Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (4:1) as eluent, which afforded the product (S,S')-12a as a yellow oil, in 95% yield (18 mg, 0.026 mmol). [α]20D = +12.4 (c. 1.5, CH2Cl2). IR (NaCl, νmax / cm-1): 3012 (vs), 2962 (vs), 2934 (vs), 2873 (vs), 1743 (vs), 1635 (s), 1607 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.72-7.66 (m, 2H, Nap-4,8), 7.65 (d, J=1.9 Hz, 1H, Nap-1), 7.37 (dd, J=8.5, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J=8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J=2.5 Hz, 1H, Nap-5), 5.44-5.28 (m, 10H, =CH), 5.24 (m, 1H, CH sn-2), 4.30 (dd, J=11.9, 4.1 Hz, 1H, CH2 sn-1/3), 4.20 (dd, J=11.9, 4.4 Hz, 1H CH2 sn-1/3), 4.13 (dd, J=11.9, 6.3 Hz, 1H, CH2 sn-1/3), 4.06 (dd, J=11.9, 5.8 Hz, 1H, CH2 sn-1/3), 3.91 (s, 3H, OCH3), 3.86 (q, J=7.2 Hz, 1H, CHCH3), 2.89-2.75 (m, 8H, =CHCH2CH=), 2.24 (t, J=7.5 Hz, 2H, CH2COO EPA), 2.19-2.11 (m, 2H, CH2COO SFA), 2.07 (td, J=7.4, 1.4 Hz, 2H, CH2CH2CH=), 2.05-1.97 (m, 2H, =CHCH2CH3), 1.85-1.71 (m, 2H, CH2CH2COO EPA), 1.60-1.53 (m, 2H, CH2CH2COO SFA), 1.58 (d, J=7.2 Hz, 3H, CHCH3), 1.34-1.21 (m, 4H, CH2), 0.98 (t, J=7.5 Hz, 3H, CH3 EPA), 0.88 (t, J=6.9 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.2 (C=O Nap), 173.3 (C=O SFA), 172.6 (C=O EPA), 157.9, 135.4, 133.9, 132.2, 129.4, 129.1, 129.0, 128.44, 128.36, 128.3, 128.2, 128.0, 127.3, 127.2, 126.3, 126.1, 119.2, 105.7, 69.0, 62.7, 62.1, 55.4, 45.4, 34.1, 33.6, 31.4, 29.9, 26.6, 25.78, 25.75 (3), 25.7, 24.8, 24.6, 22.4, 20.7, 18.5, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C43H58O7Na 709.4059; found, 709.4059.
3.7. Coupling of DHA: Synthesis of (R,S')-13a, (S,S')-13a, (R,S')-14a and (S,S')-14a
For synthesis of (R,S')-13b–d, (S,S')-13b–d, (R,S')-14b–d and (S,S')-14b–d see Supplementary Materials
3.7.1. Synthesis of 2-[4Z,7Z,10Z,13Z,16Z,19Z)-docosa-4,7,10,13,16,19-hexaenoyl]-1-hexanoyl-3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (R,S')-13a
To a solution of 1-hexanoyl-3-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol (R,S')-9a (15 mg, 0.040 mmol) and DHA as a free acid (15 mg, 0.044 mmol) in CH2Cl2 (2 mL) were added DMAP (6 mg, 0.043 mmol) and EDCI (12 mg, 0.058 mmol). The solution was stirred on a magnetic stirrer at room temperature for 23 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by use of Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (9:1) as eluent, which afforded the product (R,S')-13a as a yellow oil, in 79% yield (22 mg, 0.032 mmol). [α]20D = +6.90 (c. 1.0, CH2Cl2). IR (NaCl, νmax / cm-1): 3013 (vs), 2954 (vs), 2925 (vs), 2854 (vs), 1743 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.18 (d, J=7.8 Hz, 2H, Ibu-2,6), 7.08 (d, J=7.8 Hz, 2H, Ibu-3,5), 5.50-5.24 (m, 12H, =CH), 5.23-5.17 (m, 1H, CH sn-2), 4.29 (dd, J=11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.21 (dd, J=11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.14 (dd, J=11.9, 5.7 Hz, 1H, CH2 sn-1/3), 4.01 (dd, J=11.9, 5.9 Hz, 1H, CH2 sn-1/3), 3.70 (q, J=7.2 Hz, 1H, CHCH3), 2.89-2.79 (m, 10H, =CHCH2CH=), 2.44 (d, J=7.2 Hz, 2H, CH2CH(CH3)2), 2.37-2.19 (m, 6H, CH2CH2COO DHA, CH2COO SFA), 2.08 (quint., J=7.6 Hz, 2H, =CHCH2CH3), 1.83 (nonet, J=6.8 Hz, 1H, CH(CH3)2), 1.62-1.56 (m, 2H, CH2CH2COO SFA), 1.49 (d, J=7.2 Hz, 3H, CHCH3), 1.36-1.13 (m, 4H, CH2), 0.97 (t, J=7.5 Hz, 3H, CH3 DHA), 0.89 (d, J=6.4 Hz, 6H, CH(CH3)2), 0.88 (t, J=7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Ibu), 173.3 (C=O SFA), 172.9 (C=O DHA), 140.8, 137.4, 132.2, 129.5 (2), 128.7, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.6 (2), 127.3, 127.2, 69.00, 62.5, 62.1, 45.2, 45.1, 34.2, 34.1, 31.4, 30.3, 25.8 (3), 25.7, 25.5, 24.7, 22.7, 22.5 (2), 22.4, 20.7, 18.4, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C44H64O6Na 711.4595; found, 711.4577.
3.7.2. Synthesis of 2-[4Z,7Z,10Z,13Z,16Z,19Z)-docosa-4,7,10,13,16,19-hexaenoyl]-3-hexanoyl-1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol, (S,S')-13a
To a solution of 3-hexanoyl-1-[(S)-2-(4-isobutylphenyl)propanoyl]-sn-glycerol (S,S')-9a (11 mg, 0.029 mmol) and DHA as a free acid (11 mg, 0.032 mmol) in CH2Cl2 (1.5 mL) were added DMAP (4 mg, 0.031 mmol) and EDCI (8 mg, 0.042 mmol). The solution was stirred on a magnetic stirrer at room temperature for 25 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by use of Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (4:1) as eluent, which afforded the product (S,S')-13a as a yellow oil, in 65% yield (13 mg, 0.019 mmol). [α]20D = +8.22 (c. 0.9, CH2Cl2). IR (NaCl, νmax / cm-1): 3013 (vs), 2972 (vs), 2874 (vs), 1748 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.18 (d, J=7.8 Hz, 2H, Ibu-2,6), 7.08 (d, J=7.8 Hz, 2H, Ibu-3,5), 5.46-5.27 (m, 12H, =CH), 5.23 (tt, J=6.0, 4.3 Hz, 1H, CH sn-2), 4.29 (dd, J=11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.19 (dd, J=11.9, 4.3 Hz, 1H, CH2 sn-1/3), 4.12 (dd, J=11.9, 6.1 Hz, 1H, CH2 sn-1/3), 4.05 (dd, J=11.9, 5.9 Hz, 1H, CH2 sn-1/3), 3.70 (q, J=7.2 Hz, 1H, CHCH3), 2.90-2.80 (m, 10H, =CHCH2CH=), 2.44 (d, J=7.2 Hz, 2H, CH2CH(CH3)2), 2.39-2.18 (m, 6H, CH2CH2COO DHA, CH2COO SFA), 2.13-2.03 (m, 2H, =CHCH2CH3), 1.84 (nonet, J=6.8 Hz, 1H, CH(CH3)2), 1.64-1.57 (m, 2H, CH2CH2COO SFA), 1.49 (d, J=7.2 Hz, 3H, CHCH3), 1.36-1.23 (m, 4H, CH2), 0.97 (t, J=7.5 Hz, 3H, CH3 DHA), 0.89 (d, J=6.4 Hz, 6H, CH(CH3)2), 0.88 (t, J=7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Ibu), 173.3 (C=O SFA), 172.2 (C=O DHA), 140.9, 137.5, 132.2, 129.5 (2), 128.7, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.6 (2), 127.3, 127.2, 69.1, 62.5, 62.1, 45.2, 45.1, 34.2, 34.1, 31.4, 30.3, 25.8 (3), 25.8, 25.7, 24.7, 22.8, 22.5 (2), 22.4, 20.7, 18.4, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C44H64O6Na 711.4595; found, 711.4581.
3.7.3. Synthesis of 2-[4Z,7Z,10Z,13Z,16Z,19Z)-docosa-4,7,10,13,16,19-hexaenoyl]-1-hexanoyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol, (R,S')-14a
To a solution of 1-hexanoyl-3-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol (R,S')-10a (10 mg, 0.025 mmol) and DHA as a free acid (9 mg, 0.027 mmol) in CH2Cl2 (1.3 mL) were added DMAP (3 mg, 0.027 mmol) and EDCI (8 mg, 0.037 mmol). The solution was stirred on a magnetic stirrer at room temperature for 24 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by use of Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (4:1) as eluent, which afforded the product (R,S')-14a as a yellow oil, in 84% yield (15 mg, 0.021 mmol). [α]20D = +8.00 (c. 1.5, CH2Cl2). IR (NaCl, νmax / cm-1): 3009 (vs), 2979 (vs), 2941 (vs), 2837 (vs), 1740 (vs), 1634 (s), 1609 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.72-7.66 (m, 2H, Nap-4,8), 7.65 (d, J=1.9 Hz, 1H, Nap-1), 7.37 (dd, J=8.5, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J=8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J=2.5 Hz, 1H, Nap-5), 5.44-5.24 (m, 12H, =CH), 5.21 (tt, J=5.9, 4.5 Hz, 1H, CH sn-2), 4.30 (dd, J=11.9, 4.4 Hz, 1H, CH2 sn-1/3), 4.21 (dd, J=11.9, 4.4 Hz, 1H, CH2 sn-1/3), 4.16 (dd, J=11.9, 5.9 Hz, 1H, CH2 sn-1/3), 4.03 (dd, J=11.9, 5.9 Hz, 1H, CH2 sn-1/3), 3.91 (s, 3H, OCH3), 3.87 (q, J=7.2 Hz, 1H, CHCH3), 2.89-2.76 (m, 10H, =CHCH2CH=), 2.31-2.16 (m, 6H, CH2CH2COO DHA, =CHCH2CH3), 2.13-2.03 (m, 2H, CH2COO SFA), 1.63-1.50 (m, 2H, CH2CH2COO SFA), 1.58 (d, J=7.1 Hz, 3H, CHCH3), 1.33-1.20 (m, 4H, CH2), 0.97 (t, J=7.5 Hz, 3H, CH3 DHA), 0.88 (t, J=7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.2 (C=O Nap), 173.3 (C=O SFA), 172.1 (C=O DHA), 157.9, 135.3, 133.9, 132.2, 129.5, 129.4, 129.1, 128.7, 128.47 (2), 128.45, 128.4, 128.3, 128.23, 128.16, 128.0, 127.8, 127.3, 127.2, 126.3, 126.1, 119.2, 105.8, 69.2, 62.5, 62.1, 55.5, 45.5, 34.1, 34.0, 31.4, 25.8 (3), 25.73, 25.70, 24.6, 22.7, 22.4, 20.7, 18.4, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C45H60O7Na 735.4231; found, 735.4210.
3.7.4. Synthesis of 2-[4Z,7Z,10Z,13Z,16Z,19Z)-docosa-4,7,10,13,16,19-hexaenoyl]-3-hexanoyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol, (S,S')-14a
To a solution of 3-hexanoyl-1-[(S)-2-(6-methoxynaphthalen-2-yl)propanoyl]-sn-glycerol (S,S')-10a (11 mg, 0.027 mmol) and DHA as a free acid (10 mg, 0.030 mmol) in CH2Cl2 (1.3 mL) were added DMAP (4 mg, 0.029 mmol) and EDCI (8 mg, 0.037 mmol). The solution was stirred on a magnetic stirrer at room temperature for 30 h. The reaction was disconnected by passing the reaction mixture through a short column packed with silica gel by use of Et2O/CH2Cl2 (1:9). The solvent was removed in vacuo on a rotary evaporator. The residue was applied to a silica gel chromatography using petroleum ether/ethyl acetate (4:1) as eluent, which afforded the product (S,S')-14a as a yellow oil, in 79% yield (15 mg, 0.021 mmol). [α]20D = +12.6 (c. 0.5, CH2Cl2). IR (NaCl, νmax / cm-1): 3012 (vs), 2977 (vs), 2941 (vs), 2878 (vs), 2834 (vs), 1741 (vs), 1635 (s), 1607 (vs). 1H NMR (400 MHz, CDCl3) δH: 7.72-7.66 (m, 2H, Nap-4,8), 7.65 (d, J=1.9 Hz, 1H, Nap-1), 7.37 (dd, J=8.5, 1.9 Hz, 1H, Nap-3), 7.14 (dd, J=8.9, 2.5 Hz, 1H, Nap-7), 7.10 (d, J=2.5 Hz, 1H, Nap-5), 5.44-5.24 (m, 12H, =CH), 5.21 (tt, J=6.0, 4.4 Hz, 1H, CH sn-2), 4.30 (dd, J=11.9, 4.2 Hz, 1H, CH2 sn-1/3), 4.19 (dd, J=11.9, 4.5 Hz, 1H, CH2 sn-1/3), 4.13 (dd, J=11.9, 5.9 Hz, 1H, CH2 sn-1/3), 4.03 (dd, J=11.9, 5.9 Hz, 1H, CH2 sn-1/3), 3.91 (s, 3H, OCH3), 3.85 (q, J=7.1 Hz, 1H, CHCH3), 2.89-2.77 (m, 10H, =CHCH2CH=), 2.30-2.16 (m, 6H, CH2CH2COO DHA, =CHCH2CH3), 2.10-2.03 (m, 2H, CH2COO SFA), 1.60-1.52 (m, 2H, CH2CH2COO SFA), 1.58 (d, J=7.1 Hz, 3H, CHCH3), 1.33-1.20 (m, 4H, CH2), 0.97 (t, J=7.5 Hz, 3H, CH3 DHA), 0.88 (t, J=7.0 Hz, 3H, CH3 SFA) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 174.3 (C=O Nap), 173.3 (C=O SFA), 172.2 (C=O DHA), 157.9, 135.3, 133.9, 132.2, 129.5, 129.4, 129.1, 128.7, 128.5 (2), 128.5, 128.4, 128.3, 128.2, 128.0, 127.8, 127.4, 127.2, 126.3, 126.1, 119.2, 105.8, 69.1, 62.7, 62.1, 55.5, 45.4, 34.1, 34.0, 31.4, 25.8 (3), 25.74, 25.71, 24.6, 22.7, 22.4, 20.7, 18.5, 14.4, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C45H60O7Na 735.4231; found, 735.4217.
4. Conclusions
The successful asymmetric synthesis of a focused library of two 16-sample diastereomeric series of enantiostructured TAGs constituting a MCFA, a bioactive PUFA, and a potent drug has been completed by a six-step chemoenzymatic approach. All combinations of MCFAs ranging from C6:0 to C12:0, EPA and DHA, and (S)-ibuprofen and (S)-naproxen, were prepared. They belong to the second category enantiostructured TAG prodrugs with the MCFA and the drug attached to each of the terminal positions and the PUFA to the mid position of the glycerol skeleton of the molecule.
All the TAG products (32) and intermediates (40) were isolated, purified and fully characterized, and accomplished in a high chemical, regio- and stereoisomeric purity, in high to excellent yields in most cases. They add to the corresponding first category enantiostructured TAG focused library of 48 TAG molecular species that was recently reported. It is anticipated that the resulting enantiostructured TAG library may find use as an interesting and novel type of prodrugs applicable to site-specific release profiling and bioavailability studies.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Progress of the lipase promoted acylation of 1-O-benzyl-sn-glycerol with (S)-3 as monitored by 1H NMR spectroscopy (pS1); Figure S2: Progress of the lipase promoted acylation of 1-O-benzyl-sn-glycerol with (S)-4 as monitored by 1H NMR spectroscopy (pS2); Figure S3: Comparison of the glyceryl proton region of the 1H NMR spectra for the drug adduct (R,S’)-5 starting material and the deprotected monoacylglycerol (R,S’)-7 (pS3); Figure S4: Comparison of the glyceryl proton region of the 1H NMR spectra for (R,S’)-7 and (R,S')-9c possessing (S)-ibuprofen (pS4); Figure S5: Comparison of the glyceryl proton region of the 1H NMR spectra for (R,S')-9c and its acylated product (R,S')-11c (pS5); Experimental Information: pS6-S19; NMR spectra (1H and 13C NMR for (S)-3 and (S)-4; 1H and 13C NMR, 1H-1H COSY and 13C-1H HSQC shown for all compounds belonging to 5 – 8 (except (S,S’)-8, where there is only 1H NMR available), 9a and 10a, and (S,S’)-11c – (S,S’)-14c): pS20-S52.
Author Contributions
Conceptualization, G.G.H.; methodology, G.G.H.; validation, G.G.H.; formal analysis, L.R.J. and G.G.H.; investigation, L.R.J. and G.G.H.; resources, G.G.H.; data curation, L.R.J. and G.G.H.; writing—original draft preparation, L.R.J. and G.G.H.; writing—review and editing, G.G.H. and L.R.J.; supervision, G.G.H.; project administration, G.G.H.; funding acquisition, G.G.H. (a grant from Lysi hf. in Iceland and the University of Iceland Research Fund). Both authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Lysi hf in Iceland and the University of Iceland Research Fund.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data underlying this study are available in the published article and its online supplementary material.
Acknowledgments
Sigrídur Jónsdóttir University of Iceland is acknowledged for NMR and accurate MS measurements, Novozymes in Bagsvaerd, Denmark for the lipase, Olav Thorstad and Elin Kulas at Pronova Biopharma in Sandefjord, Norway for EPA and DHA, and Prof. Thorsteinn Loftsson at the Faculty of Pharmaceutical Sciences, University of Iceland for (S)-naproxen. Financial support from the University of Iceland Research Fond and Lysi hf. in Iceland is highly appreciated (LRJ).
Conflicts of Interest
The authors declare that this study received funding from Lysi hf. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
References
- Jónsdóttir, L.R.; Haraldsson, G.G. Synthesis of enantiostructured triacylglycerols possessing a saturated fatty acid, a polyunsaturated fatty acid and an active drug intended as novel prodrugs. Molecules 2024, 29, 5745. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.W. A new classification of prodrugs: Regulatory perspectives. Pharmaceuticals 2009, 2, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: design and clinical applications. Nature Reviews. Drug Discovery 2008, 7, 255–270. [Google Scholar] [CrossRef]
- Rautio, J.; Meanwell, N.A.; Di, L.; Hageman, M.J. The expanding role of prodrugs in contemporary drug design and development. Nature Reviews. Drug Discovery 2018, 17, 559–587. [Google Scholar] [CrossRef]
- Lambert, D.M. Rationale and application of lipids as prodrug carriers. Eur. J. Pharmaceut. Sci. 2000, 11, Suppl. 2, S15–S27. [Google Scholar] [CrossRef] [PubMed]
- Zaro, J.L. Lipid-based carriers for prodrugs to enhance delivery. AAPS J. 2015, 17, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Nguyen, M.N.; Watts, G.F.; Ooi, E.M.; Barrett, P.H.R. Effects of atorvastatin and n-3 fatty acid supplementation on VLDL apolipoprotein C-III kinetics in men with abdominal obesity. Am. J. Clin. Nutr. 2010, 91, 900–906. [Google Scholar] [CrossRef]
- Savelieva, I.; Kourliouros, A.; Camm, J. Primary and secondary prevention of atrial fibrillation with statins and polyunsaturated fatty acids: review of evidence and clinical relevance. Naunyn Schmiedebergs Arch. Pharmacol. 2010, 381, 1–13. [Google Scholar] [PubMed]
- Zhou, J. , Lee, Y.-Y.; Mao, Y.; Wang, Y.; Zhang, Z. Future of structured lipids: Enzymatic synthesis and their new applications in food systems. Foods 2022, 11, 2400. [Google Scholar] [CrossRef]
- Hong, C.R.; Jeon, B.J.; Park, K.-M.; Lee, E.H.; Hong, S.-C.; Choi, S.J. An overview of structured lpids in food science: Synthesis, methods, applications and future prospects. J. Chem. 2023, 1222373. [Google Scholar]
- Lopes, P.A.; Alfaia, C.M.; Pestana, J.M.; Prates, J.A.M. Structured lipids engineering for health: Novel formulations enriched in n-3 long-chain polyunsaturated fatty acids with potential nutritional benefits. Metabolites 2023, 13, 1060. [Google Scholar] [CrossRef]
- Halldorsson, A.; Magnusson, C.D.; Haraldsson, G.G. Chemoenzymatic synthesis of structured triacylglycerols by highly regioselective acylation. Tetrahedron 2003, 59, 9101–9109. [Google Scholar] [CrossRef]
- Magnusson, C.D.; Haraldsson, G.G. Chemoenzymatic synthesis of symmetrically structured triacylglycerols possessing short-chain fatty acids. Tetrahedron 2010, 66, 2728–2731. [Google Scholar] [CrossRef]
- Lembke, P. Omega-3 Fatty Acids. A Scientific Approach to Healthy Aging and Optimized Nutrition; Academic Press: London, UK, 2024; p. 296. [Google Scholar]
- Dawczynski, C.; Dittrich, M.; Neumann, T.; Goetze, K.; Welzel, A.; Oelzner, P.; Völker, S.; Schaible, A.M.; Troisi, F.; Thomas, L.; Pace, S.; Koeberle, A.; Werz, O.; Schlattmann, P.; Lorkowski, S.; Jahreis, G. Docosahexaenoic acid in the treatment of rheumatoid arthritis: A double-blind, placebo-controlled, randomized cross-over study with microalgae vs. sunflower oil. Clin. Nutr. 2018, 37, 494–504. [Google Scholar] [CrossRef]
- Innes, J.K.; Calder, P.C. Marine omega-3 (n-3) fatty acids for cardiovascular health: An update for 2020. Intern. J. Mol. Sci. 2020, 21, 1362. [Google Scholar] [CrossRef]
- Calder, P.C. Health benefits of omega-3 fatty acids. In Omega-3 Delivery Systems. Production, Physical Characterization and Oxidative Stability, Garcia-Moreno, P.J., Jacobsen, C., Moltke Sörensen, A.-D., Yesiltas, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2021: pp. 25-53.
- Serhan, C.N.; Petasis, N.A. Resolvins and protectins in inflammation resolution. Chem. Rev. 2012, 111, 5922–5943. [Google Scholar] [CrossRef]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel macrophage mediators with potent antiinflammatory and proreseolving actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Novel pro-resolving lipid mediators in inflammation are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Omega-3 polyunsaturated fatty acids and inflammatory processes: Nutrition or pharmacology? Br. J. Clin. Pharmacol. 2012, 75, 645–662. [Google Scholar] [CrossRef] [PubMed]
- Skulas-Ray, A.C.; Wilson, P.W.F.; Harris, W. S.; Brinton, E.A.; Kris-Etherton, P.M.; Richter, C.K.; Jacobson, T.A.; Engler, M.B.; Miller, M.; Robinson, J.G.; Blum, C.B.; Rodriguez-Leyva, D.; de Ferranti, S.D.; Welty, F.K. Omega-3 fatty acids for the management of hypertriglyceridemia. A science advisory from the American Heart Association. Circulation. 2019, 140, e673–e691. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M.; Keating, G.M. Omega-3 ethylester concentrate: A review of its use in secondary prevention post-myocardial infarction and the treatment of hypertriglyceridaemia. Drugs 2009, 69, 1077–1105. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, T.A.; Glickstein, S.B.; Rowe, J.D.; Soni, P.N. Effects of eicosapentaenoic acid and docosahexaenoic acid on low-density lipoprotein cholesterol and other lipids: A review. J. Clin. Lipidol. 2012, 6, 5–18. [Google Scholar] [CrossRef]
- Tatsuno, I.; Saito, Y.; Kudou, K.; Ootake, J. Efficacy and safety of TAK-085 compared with eicosapentaenoic acid in Japanese subjects with hypertriglyceridemia undergoing lifestyle modification: The omega-3 fatty acids randomized long-term (ORL) study. J. Clin. Lipidol. 2013, 7, 199–207. [Google Scholar] [CrossRef]
- Tatsuno, I.; Saito, Y.; Kudou, K.; Ootake, J. Long-term safety and efficacy of TAK-085 in Japanese subjects with hypertriglyceridemia undergoing lifestyle modification: The omega-3 fatty acids randomized double-blind (ORL) study. J. Clin. Lipidol. 2013, 7, 615–625. [Google Scholar] [CrossRef]
- Bays, H.E.; Braeckman, R.A.; Ballantyne, C.M.; Kastelein, J.J.; Otvos, J.D.; Stirtan, W.G.; Soni, P.N. Icosapent ethyl, a pure EPA omega-3 fatty acid: Effects on lipoprotein particle concentration and size in patients with very high triglyceride levels (the MARINE study). J. Clin. Lipidol. 2012, 6, 565–572. [Google Scholar] [CrossRef]
- Gudmundsson, H. G.; Linderborg, K. M.; Kallio, H.; Yang, B.; Haraldsson, G. G. Synthesis of Enantiopure ABC-Type Triacylglycerols. Tetrahedron 2020, 76, 130813. [Google Scholar] [CrossRef]
- Kalpio, M.; Magnússon, J. D.; Gudmundsson, H. G.; Linderborg, K. M.; Kallio, H.; Haraldsson, G. G.; Yang, B. Synthesis and enantiospecific analysis of enantiostructured triacylglycerols containing n-3 polyunsaturated fatty acids. Chem. Phys. Lipids 2020, 231, 104937. [Google Scholar] [CrossRef] [PubMed]
- Haraldsdottir, H.; Gudmundsson, H. G.; Linderborg, K. M.; Yang, B.; Haraldsson, G. G. Chemoenzymatic synthesis of ABC-type enantiostructured triacylglycerols by the use of the p-methoxybenzyl protective group. Molecules 2024, 29, 1633. [Google Scholar] [CrossRef]
- Gunstone, F. D. The Chemistry of Oils and Fats. Sources, Composition, Properties and Uses; Blackwell Publishing: Oxford, UK, 2004; pp. 50–75. [Google Scholar]
- IUPAC-IUB Commission on Biochemical Nomenclature, 1977. The nomenclature of lipids, Eur J. Biochem. 1977, 79, 11–21. [CrossRef]
- Ghogare, A.; Kumar, G.S. Oxime esters as novel irreversible acyl transfer agents for lipase catalysis in organic media. J. Chem. Soc., Chem. Commun. 1989, 1533–1535. [Google Scholar] [CrossRef]
- Chenevert, R.; Pelchat, N.; Jacques, F. Stereoselective enzymatic acylations (transesterifications). Curr. Org. Chem. 2006, 10, 1067–1094. [Google Scholar] [CrossRef]
- Faber, K. 3.1.1 Ester synthesis. In Biotransformations in Organic Chemistry; Faber, K., Ed.; Springer-Verlag: Berlin, 2004; pp. 344–349. [Google Scholar]
- Magnusson, C.D.; Haraldsson, G.G. Activation of n-3 polyunsaturated fatty acids as oxime esters: a novel approach for their exclusive incorporation into the primary alcoholic positions of the glycerol moiety by lipase. Chem. Phys. Lipids 2012, 165, 712–720. [Google Scholar] [CrossRef]
- Christie, W.W. Lipid Analysis. Isolation, Separation, Identification and Structural Analysis of Lipids, 3rd ed.; The oily press: Bridgwater, UK, 2003; Chapter 4; pp. 105–135. [Google Scholar]
- Kodali, D.R.; Tercyak, A.; Fahey, D.A.; Small, D.M. Acyl migration in 1,2-dipalmitoyl-sn-glycerol. Chem. Phys. Lipids 1990, 52, 163–170. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The structure of TAG prodrug diastereomers 1a and 1b belonging to the first category prodrugs, and TAG prodrug diastereomers 2a and 2b belonging to the second category prodrugs.
Figure 1.
The structure of TAG prodrug diastereomers 1a and 1b belonging to the first category prodrugs, and TAG prodrug diastereomers 2a and 2b belonging to the second category prodrugs.
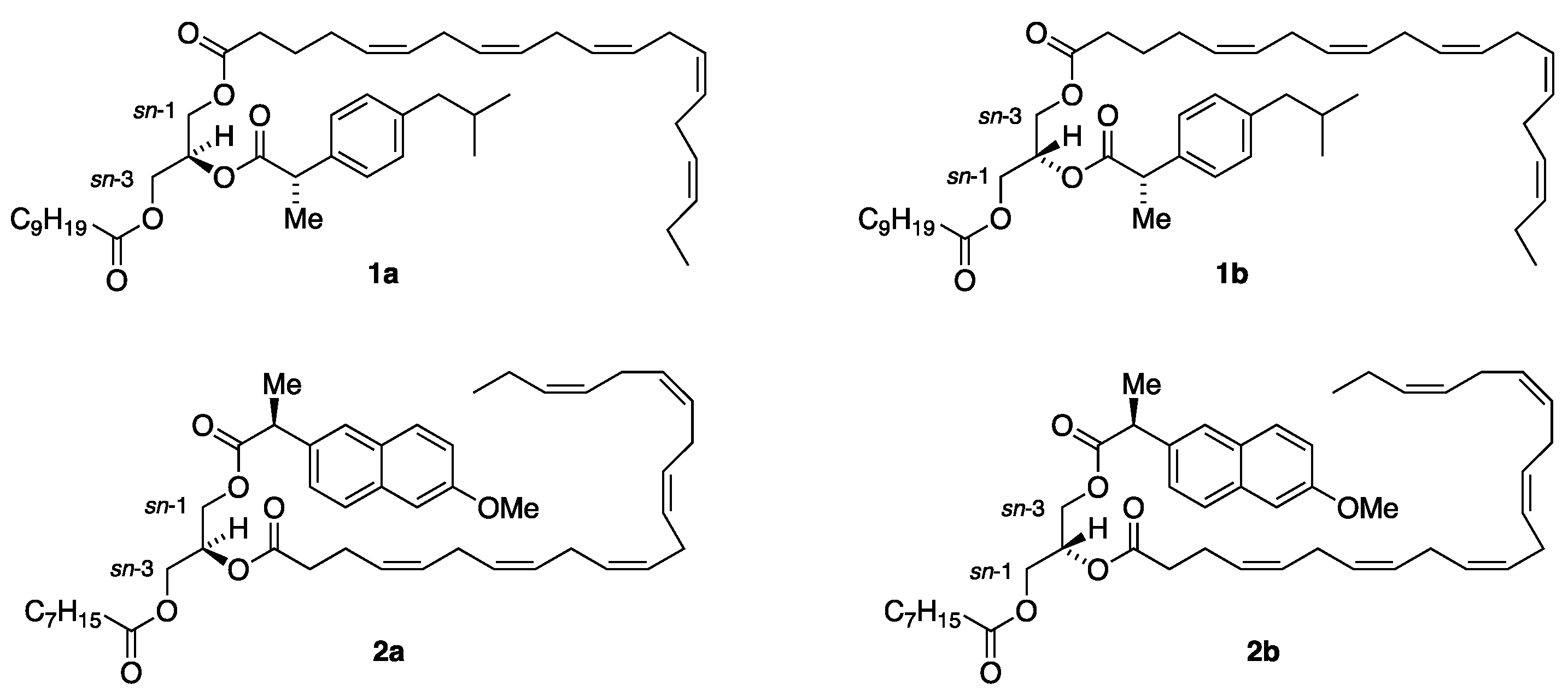
Figure 2.
Chemoenzymatic synthesis of the second category TAG prodrug diastereomer series (R,S')-11a–d – 14a–d, starting from 1-O-benzyl-sn-glycerol. In the scheme MCFA-CO-, PUFA-CO- and Drug-CO- refer to the corresponding saturated medium-chain fatty acyl, polyunsaturated fatty acyl and drug acyl group substituents, respectively. In box: (S')-ibuprofen and (S')-naproxen attached as esters to acylglycerols (AG).
Figure 2.
Chemoenzymatic synthesis of the second category TAG prodrug diastereomer series (R,S')-11a–d – 14a–d, starting from 1-O-benzyl-sn-glycerol. In the scheme MCFA-CO-, PUFA-CO- and Drug-CO- refer to the corresponding saturated medium-chain fatty acyl, polyunsaturated fatty acyl and drug acyl group substituents, respectively. In box: (S')-ibuprofen and (S')-naproxen attached as esters to acylglycerols (AG).
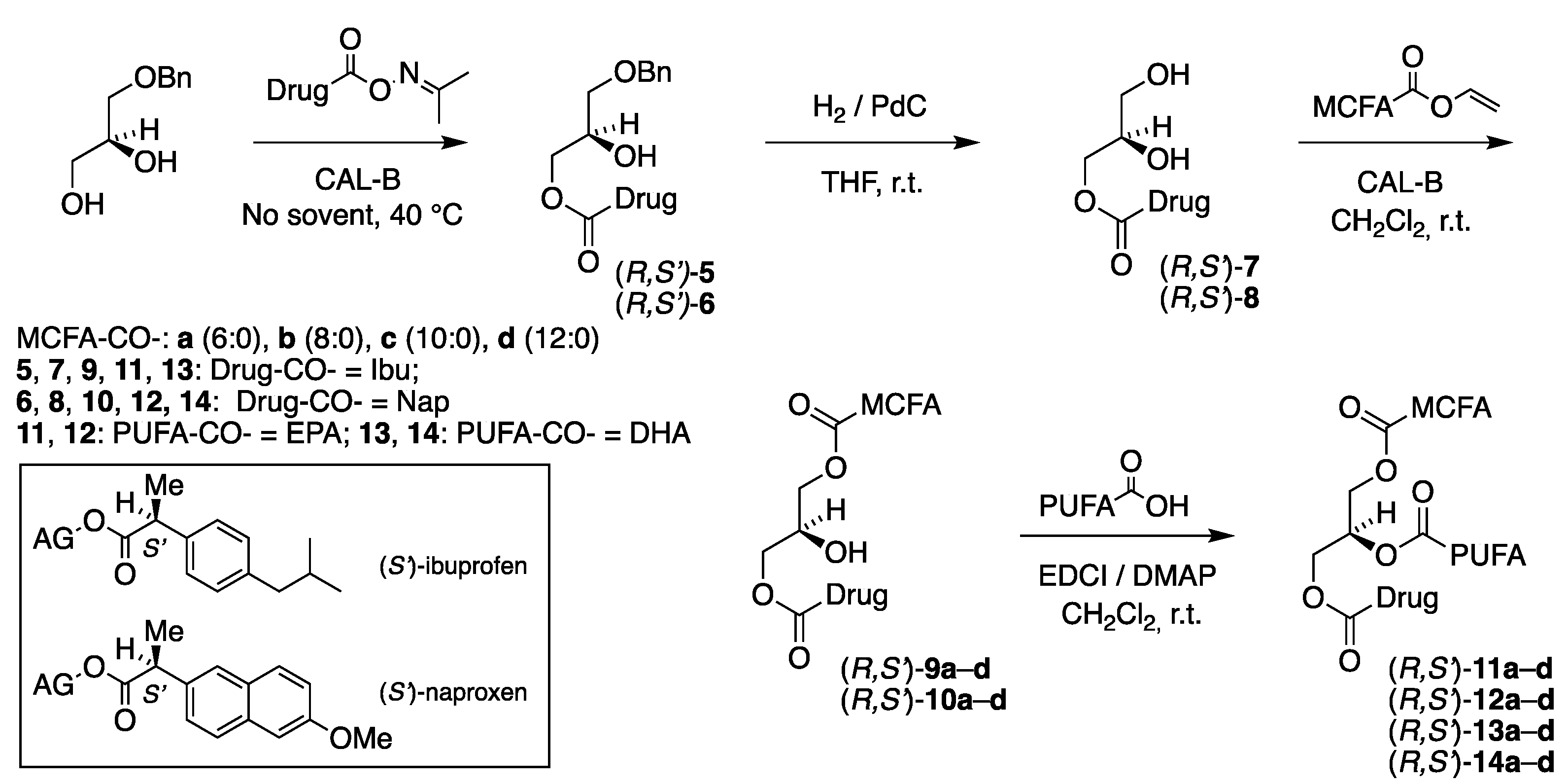
Figure 3.
Chemoenzymatic synthesis of the first category TAG prodrug diastereomer series (S,S')- 11a–d – 14a–d, starting from 3-O-benzyl-sn-glycerol. In the scheme SFA-CO-, PUFA-CO- and Drug-CO- refer to the corresponding saturated medium-chain fatty acyl, polyunsaturated fatty acyl and drug acyl group substituents, respectively. In box: (S')-ibuprofen and (S')-naproxen attached as esters to acylglycerols (AG).
Figure 3.
Chemoenzymatic synthesis of the first category TAG prodrug diastereomer series (S,S')- 11a–d – 14a–d, starting from 3-O-benzyl-sn-glycerol. In the scheme SFA-CO-, PUFA-CO- and Drug-CO- refer to the corresponding saturated medium-chain fatty acyl, polyunsaturated fatty acyl and drug acyl group substituents, respectively. In box: (S')-ibuprofen and (S')-naproxen attached as esters to acylglycerols (AG).
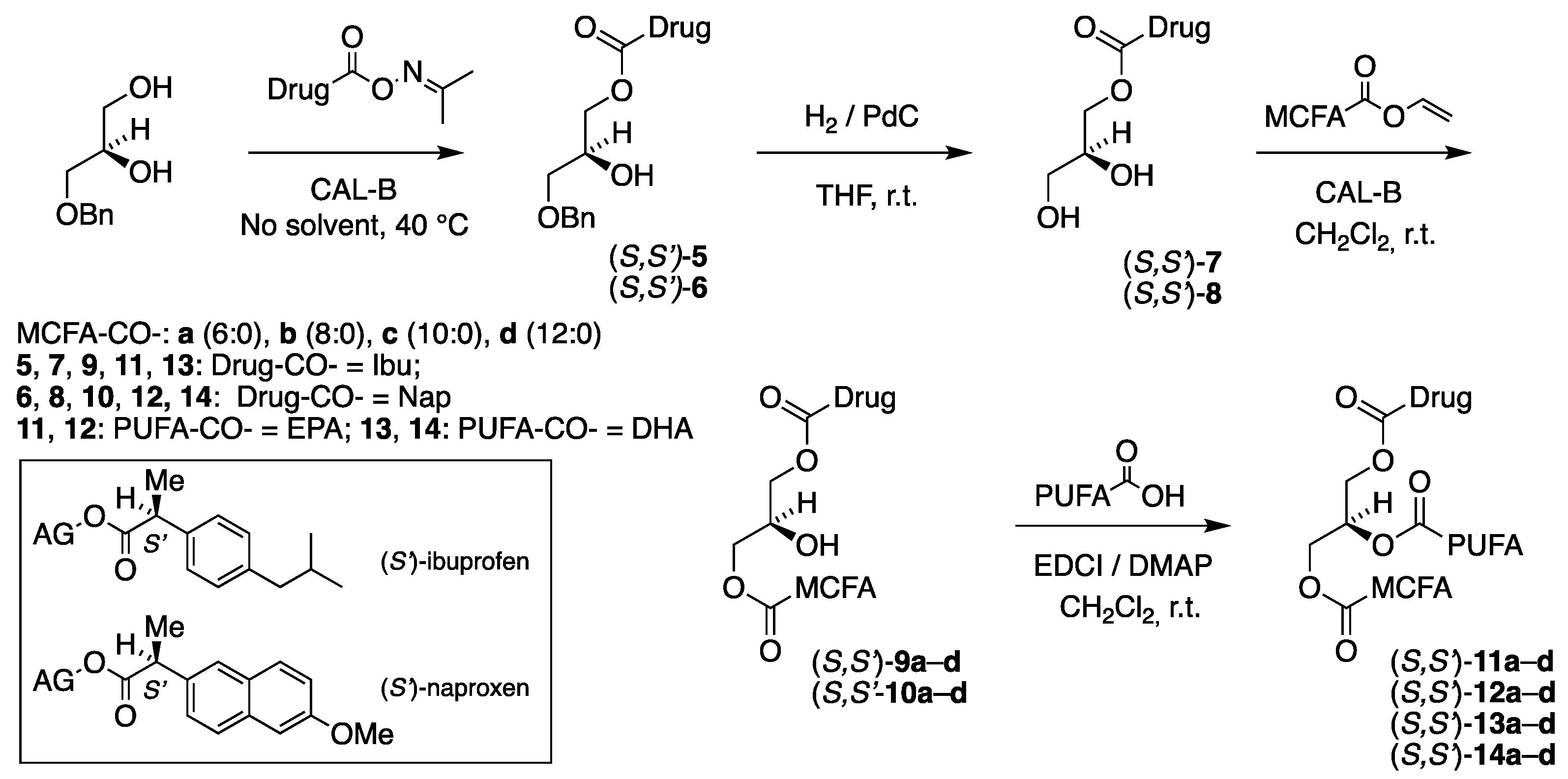
Figure 4.
Preparation of an activated acetoxime ester (S)-3 of ibuprofen by chemical coupling of acetoxime to (S)-ibuprofen.
Figure 4.
Preparation of an activated acetoxime ester (S)-3 of ibuprofen by chemical coupling of acetoxime to (S)-ibuprofen.
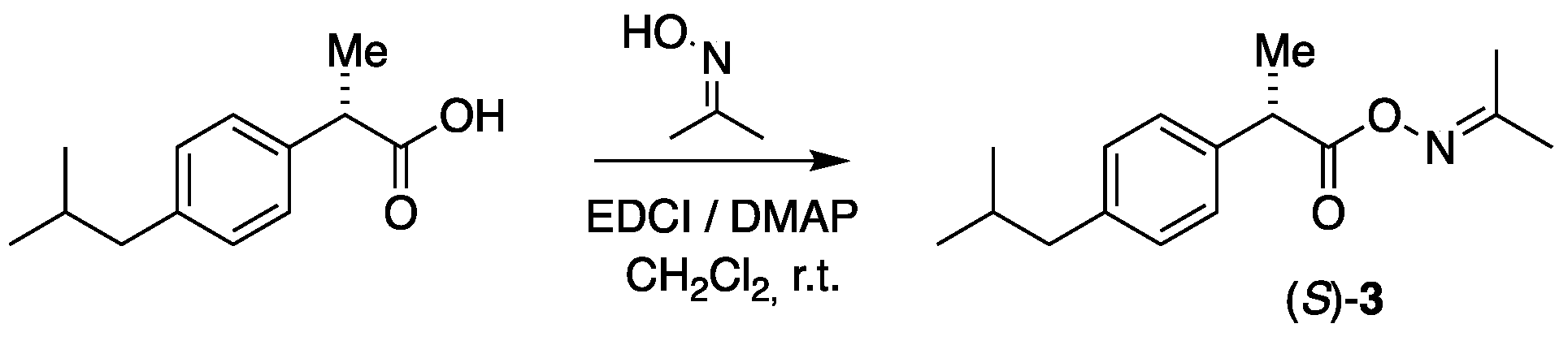

Table 1.
Summary of the yields and specific rotation of the intermediates (R,S')-5, (S,S')-5, (R,S')-6 and (S,S')-6 obtained from the lipase promoted acylation with the drugs.
Table 1.
Summary of the yields and specific rotation of the intermediates (R,S')-5, (S,S')-5, (R,S')-6 and (S,S')-6 obtained from the lipase promoted acylation with the drugs.
| Compound | sn-1 | sn-2 | sn-3 | Yields | [α]20D |
|---|---|---|---|---|---|
| (R,S')-5 | OBn | OH | Ibu | 90% | +25.0 |
| (S,S')-5 | Ibu | OH | OBn | 92% | +20.7 |
| (R,S')-6 | OBn | OH | Nap | 69% | +53.8 |
| (S,S')-6 | Nap | OH | OBn | 65% | +47.7 |
Table 2.
Summary of the yields and specific rotation of the intermediates (R,S')-7, (S,S')-7, (R,S')-8 and (S,S')-8 obtained from the debenzylation reaction.
Table 2.
Summary of the yields and specific rotation of the intermediates (R,S')-7, (S,S')-7, (R,S')-8 and (S,S')-8 obtained from the debenzylation reaction.
| Compound | sn-1 | sn-2 | sn-3 | Yields | [α]20D |
|---|---|---|---|---|---|
| (R,S')-7 | OH | OH | Ibu | 98% | +42.9 |
| (S,S')-7 | Ibu | OH | OH | 93% | +33.9 |
| (R,S')-8 | OH | OH | Nap | 93% | +43.5 |
| (S,S')- 8 | Nap | OH | OH | 99% | +34.3 |
Table 3.
Summary of the yields and specific rotation of the intermediates (R,S')-9a–d and (S,S')-9a–d obtained from the second lipase promoted reaction.
Table 3.
Summary of the yields and specific rotation of the intermediates (R,S')-9a–d and (S,S')-9a–d obtained from the second lipase promoted reaction.
| Compound | sn-1 | sn-2 | sn-3 | Yields | [α]20D |
|---|---|---|---|---|---|
| (R,S')-9a | C6:0 | OH | Ibu | 80% | +52.3 |
| (R,S')-9b | C8:0 | OH | Ibu | 80% | +42.0 |
| (R,S')-9c | C10:0 | OH | Ibu | 88% | +39.2 |
| (R,S')- 9d | C12:0 | OH | Ibu | 88% | +35.3 |
| (S,S')-9a | Ibu | OH | C6:0 | 94% | +21.0 |
| (S,S')-9b | Ibu | OH | C8:0 | 94% | +21.2 |
| (S,S')-9c | Ibu | OH | C10:0 | 74% | +22.6 |
| (S,S')-9d | Ibu | OH | C12:0 | 97% | +24.2 |
Table 4.
Summary of the yields and specific rotation of the intermediates (R,S')-10a–d and (S,S')-10a–d obtained from the second lipase promoted reaction.
Table 4.
Summary of the yields and specific rotation of the intermediates (R,S')-10a–d and (S,S')-10a–d obtained from the second lipase promoted reaction.
| Compound | sn-1 | sn-2 | sn-3 | Yields | [α]20D |
|---|---|---|---|---|---|
| (R,S')-10a | C6:0 | OH | Nap | 92% | +22.6 |
| (R,S')-10b | C8:0 | OH | Nap | 91% | +38.0 |
| (R,S')-10c | C10:0 | OH | Nap | 90% | +22.9 |
| (R,S')- 10d | C12:0 | OH | Nap | 83% | +27.5 |
| (S,S')-10a | Nap | OH | C6:0 | 70% | +21.5 |
| (S,S')-10b | Nap | OH | C8:0 | 70% | +23.1 |
| (S,S')-10c | Nap | OH | C10:0 | 75% | +23.6 |
| (S,S')-10d | Nap | OH | C12:0 | 91% | +25.5 |
Table 5.
Summary of the yields and specific rotation of the TAG prodrug products (R,S')-11a–d and (S,S')-11a–d.
Table 5.
Summary of the yields and specific rotation of the TAG prodrug products (R,S')-11a–d and (S,S')-11a–d.
| Compound | sn-1 | sn-2 | sn-3 | Yields | [α]20D |
|---|---|---|---|---|---|
| (R,S')-11a | C6:0 | EPA | Ibu | 96% | +8.29 |
| (R,S')-11b | C8:0 | EPA | Ibu | 88% | +12.0 |
| (R,S')-11c | C10:0 | EPA | Ibu | 83% | +12.2 |
| (R,S')-11d | C12:0 | EPA | Ibu | 87% | +8.35 |
| (S,S')-11a | Ibu | EPA | C6:0 | 89% | +8.27 |
| (S,S')-11b | Ibu | EPA | C8:0 | 90% | +8.60 |
| (S,S')-11c | Ibu | EPA | C10:0 | 94% | +9.74 |
| (S,S')-11d | Ibu | EPA | C12:0 | 89% | +10.4 |
Table 6.
Summary of the yields and specific rotation of the TAG prodrug products (R,S')-12a–d and (S,S')-12a–d.
Table 6.
Summary of the yields and specific rotation of the TAG prodrug products (R,S')-12a–d and (S,S')-12a–d.
| Compound | sn-1 | sn-2 | sn-3 | Yields | [α]20D |
|---|---|---|---|---|---|
| (R,S')-12a | C6:0 | EPA | Nap | 80% | +9.29 |
| (R,S')-12b | C8:0 | EPA | Nap | 78% | +9.17 |
| (R,S')-12c | C10:0 | EPA | Nap | 77% | +9.62 |
| (R,S')-12d | C12:0 | EPA | Nap | 86% | +5.38 |
| (S,S')-12a | Nap | EPA | C6:0 | 95% | +12.4 |
| (S,S')-12b | Nap | EPA | C8:0 | 95% | +11.2 |
| (S,S')-12c | Nap | EPA | C10:0 | 86% | +10.5 |
| (S,S')-12d | Nap | EPA | C12:0 | 86% | +9.60 |
Table 7.
Summary of the yields and specific rotation of the TAG prodrug products (R,S')-13a–d and (S,S')-13a–d.
Table 7.
Summary of the yields and specific rotation of the TAG prodrug products (R,S')-13a–d and (S,S')-13a–d.
| Compound | sn-1 | sn-2 | sn-3 | Yields | [α]20D |
|---|---|---|---|---|---|
| (R,S')-13a | C6:0 | DHA | Ibu | 79% | +16.9 |
| (R,S')-13b | C8:0 | DHA | Ibu | 85% | +5.77 |
| (R,S')-13c | C10:0 | DHA | Ibu | 72% | +7.91 |
| (R,S')-13d | C12:0 | DHA | Ibu | 84% | +5.42 |
| (S,S')-13a | Ibu | DHA | C6:0 | 65% | +8.22 |
| (S,S')-13b | Ibu | DHA | C8:0 | 77% | +8.57 |
| (S,S')-13c | Ibu | DHA | C10:0 | 80% | +9.60 |
| (S,S')-13d | Ibu | DHA | C12:0 | 79% | +9.67 |
Table 8.
Summary of the yields and specific rotation of the TAG prodrug products (R,S')-14a–d and (S,S')-14a–d.
Table 8.
Summary of the yields and specific rotation of the TAG prodrug products (R,S')-14a–d and (S,S')-14a–d.
| Compound | sn-1 | sn-2 | sn-3 | Yields | [α]20D |
|---|---|---|---|---|---|
| (R,S')-14a | C6:0 | DHA | Nap | 84% | +8.00 |
| (R,S')-14b | C8:0 | DHA | Nap | 83% | +8.37 |
| (R,S')-14c | C10:0 | DHA | Nap | 91% | +4.20 |
| (R,S')-14d | C12:0 | DHA | Nap | 86% | +6.71 |
| (S,S')-14a | Nap | DHA | C6:0 | 79% | +12.6 |
| (S,S')-14b | Nap | DHA | C8:0 | 68% | +11.8 |
| (S,S')-14c | Nap | DHA | C10:0 | 79% | +10.8 |
| (S,S')-14d | Nap | DHA | C12:0 | 83% | +10.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



