
Submitted:
16 April 2025
Posted:
18 April 2025
You are already at the latest version
Abstract
This study unveils a new class of spiro[1,2,4]triazolo[1,5-c]quinazoline derivatives as promising diacylglycerol kinase α (DGKα) modulators, a target implicated in cancer, neurological disorders, and immune dysfunction. Through structure-based computational design using the CB-Dock2 platform with human DGKα EF-hand domains (PDB ID: 6IIE), 40 novel compounds were systematically evaluated along established inhibitors (ritanserin, R59022, R59949, BMS502, and (5Z,2E)-CU-3) across five distinct binding pockets. Several compounds demonstrated remarkable binding profiles at the level or surpassing reference compounds. The physicochemical analysis revealed balanced drug-like properties with favorable molecular weights (252-412 g/mol) and appropriate three-dimensionality. Toxicological assessment indicated reassuring safety profiles with predicted LD50 values of 1000-2000 mg/kg and minimal hepatotoxicity, carcinogenicity, and mutagenicity potential. Notably, compound 33 (adamantyl-substituted) emerged as exceptionally promising, exhibiting strong binding affinity, moderate solubility, and selective CYP inhibition patterns that minimize drug-drug interaction risks. Detailed molecular interaction mapping identified critical binding determinants, including strategic hydrogen bonding with TRP151, GLU166, and ARG126. The multidimensional evaluation identified compounds 13, 18, 33, and 40 as particularly promising candidates that balance potent target engagement with favorable pharmaceutical profiles, establishing this scaffold as a valuable platform for developing next-generation therapeutics targeting DGKα-mediated signaling pathways.
Keywords:
diacylglycerol kinase
; molecular docking
; spirotriazoloquinazolines
; structure-activity relationships
; computational drug design
1. Introduction
Diacylglycerol kinase α (DGKα) has emerged as a promising therapeutic target, playing a crucial role in cell proliferation, T-cell responses, and viral infections. Recent studies have demonstrated that DGKα inhibition could enhance antitumor immunity and potentially serve as a strategy for treating viral infections such as HIV and HBV [1].
Mammalian diacylglycerol kinases (DGKs) comprise ten isoforms classified into five subfamilies with distinctive structural motifs [2]:
Type I DGKs (α, β, γ): Characterized by calcium-binding EF-hand motifs, with α-isoform demonstrating calcium-dependent membrane translocation and activation.
Type II DGKs (δ, η, κ): Feature pleckstrin homology (PH) domains and sterile alpha motifs (SAM), potentially enabling homo- and hetero-oligomerization.
Type III (ε): Uniquely characterized by substrate specificity, preferentially phosphorylating arachidonoyl-containing diacylglycerols.
Type IV (ζ, ι): Possess MARCKS-like phosphorylation domains and ankyrin repeats, facilitating protein-protein interactions.
Type V (θ): Contain three C1 domains and a PH domain with an embedded Ras association region.
The structural diversity of DGK isoforms underscores their multifaceted roles in cellular signaling. As highlighted by Topham & Epand [2], these enzymes are not merely metabolic regulators but sophisticated molecular switches capable of modulating complex signaling networks through precise spatial and temporal control of diacylglycerol and phosphatidic acid levels. Their activity is dynamically regulated through: calcium binding, phosphorylation by kinases like Src and PKC, interactions with scaffolding proteins, and modulation by anionic phospholipids.
Also, DGKs exhibit complex subcellular trafficking, translocating to specific cellular compartments like plasma membrane, nucleus, and cytoskeleton in response to cellular stimuli, mediated by domains such as MARCKS homology and PDZ binding regions. DGK isozymes have been reported to be involved in many physiological events, including cell proliferation and migration, glucose intake, immunity and neuronal network construction [3]. Emerging evidence suggests DGKs as potential therapeutic targets in: cancer immunomodulation, epilepsy, obsessive-compulsive disorder, bipolar disorder, fragile X syndrome, immunodeficiency, cardiac hypertrophy, hypertension, and type 2 diabetes.
Riese et al. [4] demonstrated that DGKα and DGKζ specifically regulate the pool of diacylglycerol generated as a second messenger after T cell receptor stimulation, with deletion of either isoform resulting in enhanced T cell activity against malignancy.
While early DGK inhibitors like R59022 (Figure 1) showed promise [5], their limitations have driven the search for more potent and selective compounds. A key finding from de Chaffoy de Courcelles et al. [5] was that R59022 treatment of thrombin-stimulated platelets resulted in marked elevation of diacylglycerol levels, decreased formation of phosphatidic acid, and increased protein kinase C activity compared with controls, establishing R59022’s role as a compound that potentiates the effect of diacylglycerol by preventing its rapid metabolism. Besides selectivity was demonstrated for R59022 with an IC50 of 2.8 ± 1.5 × 10-6 M [5] for the kinase acting on endogenous diacylglycerol. Afterwards it has been shown to inhibit DGK at concentrations of 50-100 μM in multiple systems, with physiologically relevant effects on both enzyme activity and cellular function [6,7].
Gómez-Merino et al. [6] demonstrated that R59022 differentially affects different DGK isoforms, with AtDGK2 being inhibited at lower concentrations (IC50 ≈ 50 μM) compared to AtDGK7, which was found to be affected by R59022 only at concentrations above 100 μM. Furthermore, R59022 at 80 μM inhibits root elongation and lateral root formation in Arabidopsis, providing physiologically relevant evidence for DGK involvement in plant development.
Jiang et al. [8] demonstrated that R59949 (Figure 1) inhibits DGK isoenzymes with remarkable selectivity, showing potent inhibition of Ca²⁺-activated DGKs while exhibiting only minimal effects on Ca²⁺-independent isoforms. And it is approximately 3-fold more potent than R59022 in inhibiting Ca²⁺-activated DGKs, which correlates well with their relative in vivo potencies. Besides, substrate kinetics studies have demonstrated that MgATP potentiates R59949 inhibition, with the inhibitor binding to the enzyme-MgATP complex 20-fold more tightly than to the free enzyme, indicating synergy between inhibitor and MgATP binding [8]. Moreover, Dominguez et al. [1], established DGKα as a critical signaling node in glioblastoma and other cancers. Their work established that attenuation of DGKα activity via siRNA by above-mentioned small-molecule inhibitors induced caspase-mediated apoptosis in glioblastoma cells while exhibiting minimal toxicity in non-cancerous cells. Moreover, Boroda et al. [9] demonstrated that ritanserin (Figure 1), a 5-HT2R antagonist with structural similarity to R59022, also functions as a DGKα inhibitor, while R59022 and R59949 exhibit 5-HT2R antagonist properties.
Our approach builds upon recent breakthroughs in DGK modulators, particularly the novel [1,2,4]triazolo [1,5-c]quinazoline scaffold disclosed in CN 115362003 B [10] (Figure 2). This patent reported compounds with potential applications in treating solid tumors and viral infections including HIV and HBV through DGK modulation. While the patent primarily explored N-substituted derivatives, our work systematically investigates the structure-activity relationships of spiro-fused analogs with varied ring sizes and heterocyclic substituents to optimize interactions with key binding regions of DGKα. This rational design strategy is informed by the work of Takahashi et al. [11], who reported the first crystal structure of human DGKα EF-hand domains (PDB ID: 6IIE), providing critical insights into calcium-dependent regulation of this enzyme and revealing key binding sites and conformational changes that can be exploited for targeted DGKα modulator design.
Figure 1 depicts the molecular structures of established DGK inhibitors that serve as reference compounds in this study. This diverse set of established inhibitors provides comprehensive benchmarks for evaluating the binding modes and potential efficacy of our novel spirotriazoloquinazoline derivatives (Figure 2).
In summary, this study presents a comprehensive computational investigation of novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives as potential DGKα modulators. By leveraging structure-based molecular docking, in silico physicochemical property analysis and ADME-Tox assessment, we aim to identify promising candidates, that combine strong target engagement with favorable pharmaceutical profiles. The following sections detail our methodological approach, results, and implications for developing next-generation therapeutics targeting DGKα-mediated signaling pathways in various pathological conditions.
2. Results and Discussion
2.1. Molecular Docking Studies
2.1.1. Structural and Functional Basis of DGKα as a Therapeutic Target
The selection of DGKα as the primary target is grounded in its well-characterized calcium-dependent regulatory mechanism and critical physiological roles. Topham & Epand [2], highlight that DGKα is uniquely characterized by two calcium-binding EF-hand motifs that dynamically modulate its enzymatic activity and subcellular localization. For instance, deletion studies with DGKα have shown that R59949 does not directly interact with the Ca²⁺-binding EF hand motifs, but rather binds to the catalytic domain, with inhibition being independent of Ca²⁺ concentration [8].
Specifically, DGKα demonstrates a sophisticated activation mechanism where:
- Calcium binding triggers conformational changes that promote membrane translocation
- The N-terminal recoverin homology domain works synergistically with EF-hand motifs to regulate enzyme activation
- Calcium-independent basal activity can be supported by specific lipid environments, including phosphatidylethanolamine and cholesterol
- Moreover, DGKα plays pivotal roles in critical cellular processes:
- Immune cell function, particularly T-cell responses
- Cell proliferation and migration
- Modulation of Rac activation and actin cytoskeleton remodeling
Moreover, enzyme’s regulatory complexity is further evidenced by multiple activation mechanisms, including: calcium-dependent membrane translocation, phosphorylation by Src tyrosine kinases, and interactions with phosphoinositide 3-kinase lipid products.
These multifaceted regulatory features make DGKα an exceptionally compelling target for detailed molecular investigation, offering insights into both enzymatic regulation and potential therapeutic interventions.
Recent work by Chupak et al. [12] demonstrated that dual inhibition of DGKα and DGKζ can enhance T-cell proliferation and cytokine production more effectively than targeting either isoform alone, suggesting potential synergistic effects of multi-isoform inhibition.
Dominguez et al. [1] studies revealed that R59022 exhibits promising blood-brain barrier penetration based on in silico models, making it particularly relevant for central nervous system applications. So, demonstrated that these small-molecule inhibitors selectively target DGKα and induce apoptosis in glioblastoma cells.
Moreover, Boroda et al. [9] demonstrated that treatment with DGKα inhibitors (R59022 and ritanserin) results in increased phosphorylation of PKC substrates, consistent with DAG accumulation following DGK inhibition.
The paper by Martinez et al. [13] provides important experimental evidence regarding DGK inhibition in human adipocytes. Their work demonstrated that R59022 attenuated inflammatory signaling pathways, supporting the critical role of DGKs in mediating cellular responses. Specifically, they found that R59022 attenuated MAPK activation (ERK, JNK) and calcium signaling, key pathways that are also implicated in the current investigation of triazoloquinazoline derivatives.
To systematically assess binding affinity pockets for reference compounds (Figure 2), we employed CB-Dock2 [14,15] to conduct a blind search. This initial analysis was essential for identifying and characterizing potential binding sites within the DGKα protein structure (RCSB PDB ID: 6IIE) [16]. The crystal structure of DGKα EF-hand domains confirms that DGKα contains a canonical pair of helix-loop-helix EF-hand motifs that bind two Ca²⁺ ions with different affinities (Kd¹ = 0.3 μM and Kd² = 2.3 μM) in a cooperative manner [11]. Isothermal titration calorimetry experiments demonstrated that Ca²⁺ binding is an exothermic process and that the two binding sites in EF1 and EF2 are asymmetric, with EF2 likely being the first Ca²⁺ binding site.
By examining interaction patterns across distinct binding cavities with varying volumes, we established a framework for comparing the binding behavior of both reference inhibitors and novel derivatives. Table 1 presents these cavity properties alongside binding affinity scores for each reference compound, revealing distinctive binding preferences that guided the subsequent investigation.
Five distinct binding cavities were identified with volumes ranging from 97 ų to 179 ų, each exhibiting unique geometric properties and interaction potentials. Notably, the 127 ų cavity demonstrated exceptional binding capability, consistently accommodating reference compounds with remarkably strong affinity scores, suggesting this pocket may represent the primary inhibitory binding site for DGK modulators. Takahashi et al. [11] demonstrated that Ca²⁺ binding induces substantial conformational changes in DGKα-EF, converting it from a more open, protease-susceptible conformation to a well-folded, compact monomeric structure. This conformational plasticity may influence the accessibility and properties of binding pockets identified in the computational analysis, particularly the 127 ų cavity which showed the strongest overall binding for the novel compounds.
Further examination of binding patterns across cavities revealed distinctive preferences for specific chemical scaffolds. The 179 ų cavity, representing the largest binding pocket identified, showed moderate to strong interactions with most reference compounds, with R59949 achieving a notable binding score of -7.7 kcal/mol. This cavity’s increased volume likely accommodates larger, more conformationally flexible ligands, potentially explaining its broader binding profile. In contrast, the more spatially constrained 97 ų cavity exhibited more selective binding behavior, with R59949 again demonstrating strong affinity (-7.6 kcal/mol), though slightly reduced compared to its interactions in the 127 ų pocket.
The binding profiles observed in the 121 ų and 146 ų cavities provide additional insights into structure-activity relationships for DGKα inhibitors. R59949 demonstrated the strongest binding in the 121 ų cavity (-8.1 kcal/mol), while binding affinities in the 146 ų cavity were generally more modest across all compounds, suggesting this pocket may be less favorable for inhibitor development. Interestingly, the differential performance of structurally related compounds (e.g., R59949 vs. R59022) across these distinct binding regions highlights the importance of specific molecular features in determining cavity preferences and binding modes.
The computational analysis aligns with the emerging understanding that DGK isoforms have nuanced, context-dependent roles beyond simple lipid metabolism. The observed structural variations resonate with Topham & Epand’s [2] framework, suggesting that each DGK subfamily might target distinct cellular processes through unique molecular interactions. The binding affinity observed for R59022 in the computational studies parallels experimental findings from Tu-Sekine et al. [7], who demonstrated that R59022 effectively inhibits purified DGK-θ in vitro at concentrations ≤1 μM under their conditions. Additionally, comparative analysis by Boroda et al. [9] revealed that R59022 and ritanserin are more potent inhibitors of DGKα than other DGK isoforms, with IC₅₀ values of approximately 25 μM and 15 μM, respectively.
The identification of multiple druggable cavities with distinct binding preferences provides a foundation for developing compounds with potentially improved selectivity profiles compared to existing DGKα inhibitors. Moreover, the strong binding displayed by reference compounds, particularly in the 127 ų cavity, establishes a clear threshold for identifying promising novel inhibitors worthy of further investigation.
The binding data presented in Table 2 reveals compounds with exceptional affinity profiles across multiple binding cavities (Table 2, Figure 3), establishing a rigorous threshold for assessing the novel compounds in this study.
The findings also align with previous experimental work by Martinez et al. [13], who demonstrated that R59022 effectively inhibits DGK activity and subsequent downstream signaling. Their study showed that R59022 attenuated intracellular calcium mobilization and MAPK activation, critical pathways also modulated by the novel spirotriazoloquinazoline derivatives. The similar inhibitory profiles suggest shared mechanisms of action, supporting the potential therapeutic applications of the compounds.
It is noteworthy that the optimized spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives differ structurally from those disclosed in CN 115362003 B [10], which primarily focused on N-alkyl and N-aryl substituted derivatives. The spiro-fused analogs represent a novel structural class within this scaffold, with the spiro-cycloalkane and heterocyclic modifications providing distinct binding profiles compared to the patented compounds.
Having completed this analysis of binding affinity, we next examined the spatial and molecular interactions of the most promising compounds within their respective binding cavities. This visual analysis provides critical insights into the structural basis for the exceptional binding properties observed in the computational studies and helps rationalize the structure-activity relationships identified through our evaluation.
2.1.2. Molecular Visualization and Key Interaction Analysis
The crystal structure of DGKα-EF reveals a large hydrophobic surface area clustered near the N- and C-termini when Ca²⁺ ions are bound [11]. This surface includes residues that may participate in inter-domain interactions within the full-length protein. The molecular docking results suggest, that leading compounds may interact with this hydrophobic region, potentially disrupting crucial protein-protein interactions necessary for DGKα activity. To characterize these interactions at the molecular level, we conducted a detailed analysis of the specific bonds formed between each compound and the surrounding amino acid residues. The visual representations in Figure 4 and Figure 5 provide a spatial understanding of how the most promising compounds interact within their respective binding pockets, highlighting their three-dimensional relationships with the DGKα protein structure.
Table 3 presents a systematic categorization of these binding interactions, including their types, distances, and the specific residues involved. This detailed characterization reveals the molecular mechanisms underlying the exceptional binding affinities observed in the computational analyses and provides critical insights for understanding structure-activity relationships at the atomic level. The comparison between reference inhibitors and the novel compounds within each cavity illuminates both conserved interaction patterns essential for DGKα binding and unique features that contribute to the enhanced potency of specific derivatives.
The molecular docking results presented in Table 3 and Figures 4 and 5 provide valuable insights into the binding mechanisms of both reference DGK inhibitors and the novel [1,2,4]triazolo [1,5-c]quinazoline derivatives. Sakane et al. [3] made an important observation regarding structural binding differences, noting that ‘unlike diacylglycerol-binding proteins, which have the common diacylglycerol-binding domain (the C1 domain), obviously common phosphatidic acid-binding motifs, like the C1 domain, have not been identified in phosphatidic acid-binding proteins.
Hence, in the result of the analysis of molecular interactions reveals that compounds forming stronger hydrogen bonds with key residues, particularly TRP151, GLU166, and ARG126, generally exhibit higher binding affinities.
Cavity 127 ų: Ritanserin vs. Compound 3
In the 127 ų cavity, which consistently demonstrated the strongest binding for multiple compounds, ritanserin (Vina score: -9.3 kcal/mol) establishes a complex network of interactions. The reference inhibitor forms an attractive electrostatic interaction with GLU166, conventional hydrogen bonding with ALA146, and multiple halogen interactions through its fluorine atoms with MET142, ASP152, and LEU193. These interactions are complemented by several hydrophobic contacts, including π-sigma interactions with ALA146, MET163, and LEU193, and π-π stacked interactions with TRP151.
Compound 3, while showing slightly lower binding affinity (-7.1 kcal/mol), establishes a more focused interaction pattern dominated by a conventional hydrogen bond with TRP151 and multiple hydrophobic interactions. The π-π stacked interactions with TRP151 appear particularly important, occurring at multiple positions with distances ranging from 3.94 to 5.31 Å. The compound also forms several π-alkyl interactions with LEU156 and ALA146. This comparison suggests that while compound 3 lacks the electrostatic and halogen interactions of ritanserin, it compensates through optimized hydrophobic interactions with key aromatic residues.
Cavity 179 ų: R59949 vs. Compound 18
The comparison between R59949 (Vina score: -7.7 kcal/mol) and compound 18 (Vina score: -8.5 kcal/mol) in the 179 ų cavity reveals how structural modifications can enhance binding affinity. R59949 relies heavily on electrostatic attraction with GLU162 and multiple halogen interactions with ASP136, ASP168, GLY173, and SER174, complemented by hydrophobic contacts with LYS165 and VAL135.
In contrast, compound 18 establishes a stronger conventional hydrogen bond with SER132 (2.12 Å compared to no direct hydrogen bonding for R59949), coupled with π-anion interactions with ASP136 and hydrophobic interactions with LYS165 and VAL135. The superior binding affinity of compound 18 can be attributed to this strong hydrogen bonding and the optimized π-electron system that facilitates both electrostatic and hydrophobic interactions. This finding highlights the importance of balancing hydrogen bonding capability with appropriate hydrophobic features in the design of effective DGK inhibitors.
Cavity 97 ų: R59949 vs. Compound 40
The analysis of interactions in the 97 ų cavity provides particularly valuable insights, as compound 40 (-8.2 kcal/mol) significantly outperforms R59949 (-7.6 kcal/mol). R59949 establishes a complex network including attractive charge interaction with GLU166, conventional hydrogen bonding with ARG182 and GLU166, carbon hydrogen bonding with GLU166, and halogen interactions with ILE167 and GLU179.
Compound 40 achieves its superior binding primarily through a much stronger conventional hydrogen bond with GLU166 (1.85 Å compared to 3.74 Å for R59949), supplemented by π-anion interaction with the same residue. Additionally, compound 40 forms a π-donor hydrogen bond with THR187, π-sigma interactions with MET163 and ALA183, and an amide-π stacked interaction with ARG182 and ALA183. The strategic positioning of the spiro [1,2,4]triazolo [1,5-c]quinazoline scaffold appears to enable more favorable geometric alignment with GLU166, resulting in the formation of a stronger hydrogen bond that likely contributes significantly to the enhanced binding affinity.
Cavity 121 ų: R59949 vs. Compound 28
In the 121 ų cavity, R59949 (-8.1 kcal/mol) and compound 28 (-7.2 kcal/mol) exhibit different interaction profiles. R59949 primarily interacts through π-donor hydrogen bonds and π-π stacked interactions with TYR122, complemented by π-π stacked interaction with TYR148 and π-alkyl interactions with LEU121 and VAL145.
Compound 28, despite its lower overall binding score, establishes a stronger conventional hydrogen bond with GLN141 (3.16 Å) and forms a carbon hydrogen bond with VAL145, a π-cation interaction with ARG144, and a π-donor hydrogen bond with GLN141. The compound also engages in multiple hydrophobic interactions, including π-sigma interaction with VAL145, alkyl interactions with VAL145 and LEU149, and π-alkyl interactions with ARG144. This suggests that while compound 28 forms more diverse interactions, the aromatic stacking interactions of R59949 with tyrosine residues may contribute more significantly to binding affinity in this cavity.
Cavity 146 ų: R59022 vs. Compound 33
The comparison between R59022 (-7.1 kcal/mol) and compound 33 (-7.5 kcal/mol) in the 146 ų cavity demonstrates how structural modifications can improve binding through alternative interaction patterns. R59022 engages in multiple hydrogen bonds, including a conventional hydrogen bond with THR124 through its fluorine, carbon hydrogen bonds with LEU121, TYR122, and an intermolecular hydrogen bond. It also forms a π-cation interaction with LYS137, a π-donor hydrogen bond with THR124, multiple π-sigma interactions, and a π-alkyl interaction.
Compound 33, which shows slightly stronger binding, establishes a more focused interaction profile centered on a conventional hydrogen bond with ARG126 (3.20 Å) and multiple alkyl and π-alkyl interactions with LYS120 and ARG126. The superior binding of compound 33 despite its simpler interaction profile suggests that the strategic positioning of its hydrophobic groups optimally engages the lipophilic regions of the cavity, while the single, well-positioned hydrogen bond with ARG126 provides sufficient polar interaction to anchor the molecule.
2.1.3. Correlation Between Interaction Patterns and Binding Affinities
A comprehensive analysis of the interaction data reveals several key determinants of binding affinity across the novel [1,2,4]triazolo [1,5-c]quinazoline derivatives:
Hydrogen bonding strength: Compounds forming stronger hydrogen bonds generally exhibit higher binding affinities, as exemplified by compound 40 in the 97 ų cavity.
Aromatic interactions: π-π stacking interactions, particularly with tryptophan and tyrosine residues, contribute significantly to binding stability, as observed with compounds 3 and R59949 in the 127 ų and 121 ų cavities, respectively.
Electrostatic complementarity: The strategic positioning of charged or polarizable groups to engage in electrostatic interactions with complementary residues enhances binding affinity, as demonstrated by the π-anion interactions formed by 18 and 40.
Hydrophobic contact optimization: The most potent compounds establish multiple hydrophobic interactions that effectively engage lipophilic regions within the binding cavities, as seen with the alkyl and π-alkyl interactions formed by 33.
Interaction diversity vs. strength: While some compounds establish fewer interactions than reference inhibitors, the strategic optimization of these interactions can result in superior binding affinity, suggesting quality over quantity in interaction design.
The binding interactions observed with the most promising compounds (13, 18, 22, 28, 33, and 40) suggest that future design efforts should focus on incorporating heterocyclic systems that can engage in strong hydrogen bonding interactions while maintaining appropriate lipophilic characteristics to exploit the hydrophobic regions of the binding pockets. Additionally, the incorporation of polarizable groups capable of forming halogen bonds or π-electron interactions may further enhance binding affinity through engagement with aromatic residues present in multiple binding cavities.
Having established the binding profiles of these novel derivatives, we next investigated their physicochemical properties to assess their drug-like characteristics and potential for further development as pharmaceutical agents.
2.2. In Silico ADME Properties Analysis
2.2.1. Molecular Descriptors and Fundamental Property Analysis
To evaluate the drug-like characteristics of the novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives, a comprehensive analysis of their physicochemical properties was conducted via SwissADME website [17,18] along with all reference compounds, except (5Z,2E)-CU-3, which demonstrated the lowest results in affinity studies (Table S1).
The molecular weight of the compounds ranged from 252.31 g/mol (1) to 412.53 g/mol (28), with most derivatives falling within the desirable range for oral bioavailability (< 500 g/mol) according to Lipinski’s Rule of Five. The heavy atom count varied from 19 to 31, reflecting the structural diversity within the series, while aromatic heavy atoms ranged from 11 to 20, demonstrating varying degrees of aromaticity in the designed molecules.
Notably, compounds 10, 11, 20, 26, 32, and 33 exhibited the highest fraction of sp³ hybridized carbon atoms (Csp³ ≥ 0.58), suggesting enhanced three-dimensionality compared to their more planar counterparts. This three-dimensional character has been associated with improved selectivity profiles and reduced toxicity in drug candidates. The majority of compounds contained only one rotatable bond, with the exception of compounds 25-30, which contained two rotatable bonds due to the incorporation of the tert-butyl substituent on the cyclohexyl scaffold. This limited conformational flexibility may contribute to favorable binding entropy and potentially enhanced potency.
Hydrogen bonding capability analysis revealed that most compounds contained 2-4 Hydrogen bond acceptors and 1-2 Hydrogen bond donors, well within the desirable range for membrane permeability. Compounds 31-40, containing the piperidine scaffold, consistently demonstrated higher Hydrogen bond acceptor counts (3-4) compared to their cycloalkane counterparts, potentially enhancing their interaction with polar regions of the binding site.
The topological polar surface area (TPSA) values ranged from 42.74 Ų (1, 2, 9, 10, 11, 19, 20, 25, and 26) to 74.22 Ų (36). These values are significantly below the threshold of 140 Ų associated with poor membrane permeability, suggesting favorable passive diffusion properties for all compounds in the series. The compounds with sulfur-containing heterocycles (5, 14, 23, 29, and 36) exhibited notably higher TPSA values (70.98-74.22 Ų) compared to other derivatives.
Molecular refractivity, an indicator of molecular volume and polarizability, ranged from 76.70 (1) to 128.02 (28). Compounds with extended aromatic systems and benzofused heterocycles (22, 28, and 35) demonstrated the highest molecular refractivity values, consistent with their increased π-electron density.
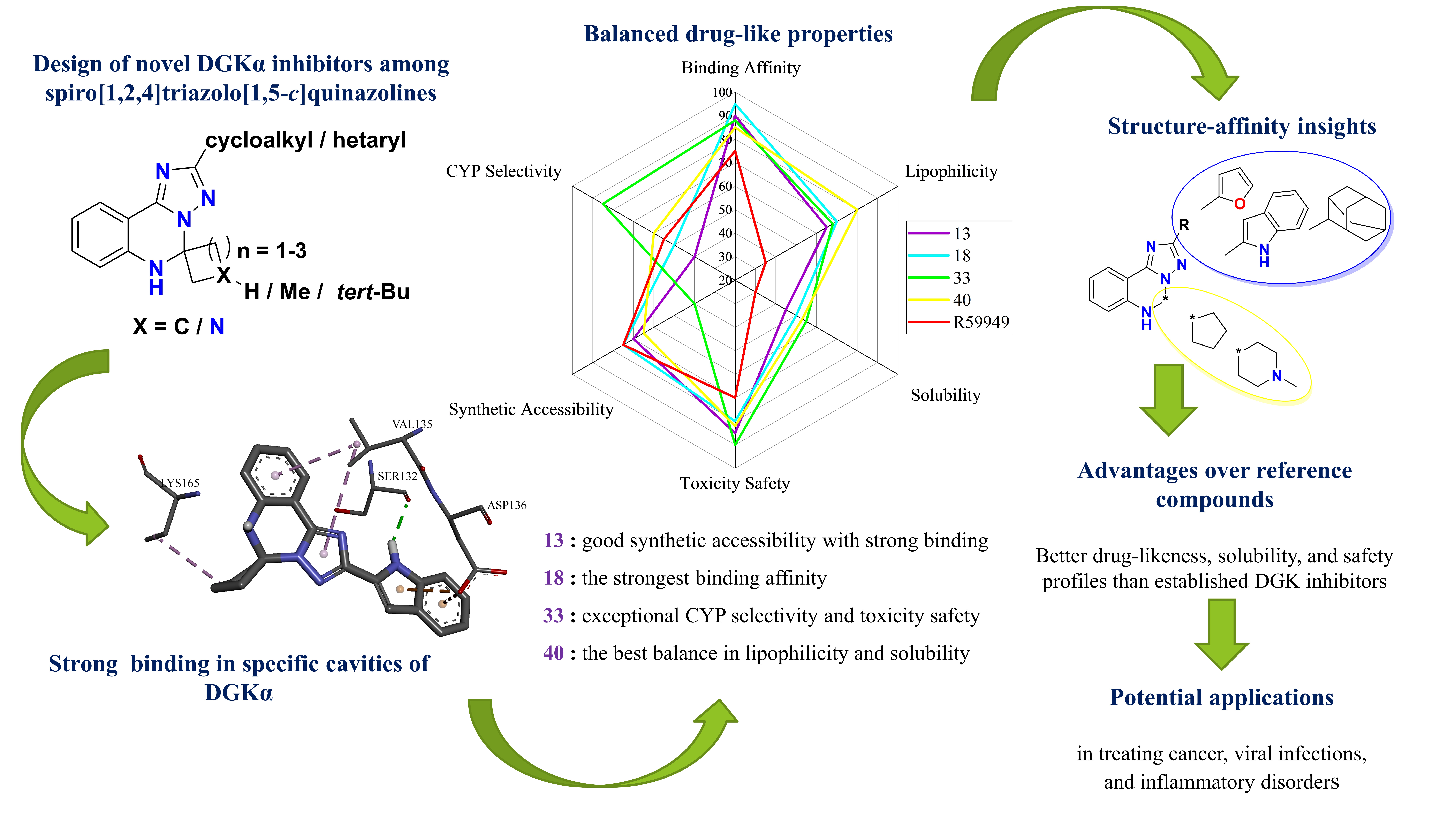
When comparing the physicochemical profiles of the most promising compounds based on binding affinity (13, 18, 22, 28, 33, and 40), all exhibited favorable drug-like properties. Compound 13 (MW: 342.39 g/mol, TPSA: 55.88 Ų) demonstrated balanced physicochemical parameters combined with excellent binding affinity, while compound 33 (MW: 389.54 g/mol, TPSA: 45.98 Ų) exhibited the highest Csp³ value (0.67) among these lead compounds, potentially contributing to its robust binding profile through enhanced three-dimensionality.
The superior binding affinity of compound 18 (MW: 341.41 g/mol, TPSA: 58.53 Ų) in the 179 ų cavity may be attributed to its optimal balance of aromaticity (20 aromatic heavy atoms) and hydrogen bonding capability (2 acceptors, 2 donors). Similarly, compound 40 (MW: 370.45 g/mol, TPSA: 61.77 Ų) exhibited exceptional binding in the 97 ų cavity, potentially facilitated by its favorable combination of structural features and hydrogen bonding pattern (3 acceptors, 2 donors).
This physicochemical analysis supports the potential of these novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives as promising candidates for further development, with compounds 13, 18, 22, 28, 33, and 40 demonstrating particularly favorable combinations of binding affinity and drug-like properties.
While binding affinity and favorable physicochemical profiles are essential, safety considerations are equally critical for drug development. Therefore, we conducted a toxicity assessment to evaluate potential liabilities of these compounds.
2.2.2. Toxicity
Table S2 summarizes the in silico oral toxicity predictions via ProTox-2 and 3 [19,20] for these compounds, providing valuable insights into their safety profiles and complementing the previously discussed physicochemical and binding characteristics.
All compounds demonstrated favorable toxicity indices, with predicted LD50 values predominantly in the range of 1000-2000 mg/kg, classifying them as Class IV (harmful if swallowed; 300 < LD50 ≤ 2000 mg/kg) or Class V (may be harmful if swallowed; 2000 < LD50 ≤ 5000 mg/kg) according to the Globally Harmonized System of Classification and Labeling of Chemicals. This finding suggests that these compounds possess acceptable safety margins that align with requirements for potential drug candidates.
The prediction accuracy percentages across multiple toxicity endpoints - hepatotoxicity (HT), carcinogenicity (CG), immunotoxicity (IT), mutagenicity (MG), and cytotoxicity (CT) provide a multidimensional assessment of toxicological liabilities. Notably, compounds exhibited high prediction accuracy (>90%) for immunotoxicity across the series, with values ranging from 80% to 99%, indicating reliable assessments for this endpoint. This observation is particularly relevant given the potential application of these compounds in immunomodulation through DGKα inhibition, as highlighted by Riese et al. [4] in their work on enhancing T-cell activity in cancer.
Compounds 25-40, which incorporate the tert-butyl substituent on the cyclohexyl scaffold or contain the piperidine structural element, demonstrated slightly more favorable toxicity profiles compared to compounds 1-24. This trend parallels their enhanced physicochemical properties, particularly their improved aqueous solubility and moderate lipophilicity profiles. Specifically, compounds 29, 33, 36, and 39, which were previously identified as possessing balanced pharmaceutical properties, also exhibit favorable toxicity predictions with relatively high LD50 values (1200-2000 mg/kg) and low probability values across multiple toxicity endpoints.
Hepatotoxicity predictions revealed probability values predominantly below 0.65, suggesting a low likelihood of liver toxicity for most compounds in the series. This favorable hepatic safety profile is particularly important considering that many drug candidates fail in clinical development due to liver toxicity concerns. The compounds with the lowest hepatotoxicity probability values (0.52-0.55) included those with furan (13), pyridine (30, 37-39), and benzofuran (22, 28) substituents, which had also demonstrated strong binding affinities in the computational docking studies.
Carcinogenicity and mutagenicity predictions showed consistently favorable profiles across the series, with probability values generally below 0.56 for carcinogenicity and below 0.57 for mutagenicity, indicating minimal genotoxic potential. This observation suggests that the spiro [1,2,4]triazolo [1,5-c]quinazoline scaffold does not introduce concerning structural alerts associated with DNA reactivity or carcinogenic potential.
Cytotoxicity predictions demonstrated some variability across the series, with probability values ranging from 0.53 to 0.60. Compounds 25-30, featuring the tert-butyl cyclohexyl scaffold, exhibited slightly elevated cytotoxicity probability values (0.59-0.60) compared to other derivatives, potentially reflecting their increased lipophilicity and subsequent higher membrane permeability. This observation aligns with their higher consensus logP values discussed previously.
Interestingly, compounds 13, 18, 33, and 40, which were identified as the most promising candidates based on their binding affinities and balanced pharmaceutical properties, also demonstrate acceptable toxicity profiles with LD50 values exceeding 1000 mg/kg and moderate probability values across toxicity endpoints. This correlation between favorable pharmacological properties and acceptable toxicity profiles further supports their potential as lead candidates for further development.
When contextualizing these findings within the broader literature, the observed toxicity profiles compare favorably with those reported for established DGK inhibitors. Boroda et al. [9] noted that ritanserin and R59022 exhibit manageable toxicity profiles at pharmacologically relevant concentrations, while Dominguez et al. [1] demonstrated that DGKα inhibitors showed minimal toxicity in non-cancerous cells despite inducing apoptosis in glioblastoma cells. The toxicity predictions for our novel derivatives align with these experimental observations, suggesting a potentially favorable therapeutic window.
2.2.3. Physicochemical Drug-likeness
To further evaluate the potential druggability of these compounds, a systematic assessment using established medicinal chemistry filters was conducted (Table S3). These filters—Lipinski’s Rule of Five, Ghose, Veber, Egan, and Muegge criteria—provide important benchmarks for predicting oral bioavailability and drug-likeness. The majority of the novel derivatives (compounds 1-10, 12-19, 21, 23, 24, 31-40) demonstrated excellent compliance with all five medicinal chemistry filters, suggesting favorable drug-like properties and potential for oral bioavailability.
Compounds 11, 25, and 29 passed four of the five filters, with a single violation of the Muegge filter (XLOGP3 > 5), indicating slightly elevated lipophilicity. Similarly, compounds 20, 22, 27, and 30 exhibited one Muegge violation while passing all other criteria. Despite these minor violations, compounds with a single deviation from the Muegge criteria may still exhibit adequate pharmacokinetic properties, as this filter is generally more stringent than other widely used criteria.
More significant deviations were observed with compounds 26 and 28, which violated multiple filters due to high lipophilicity. Compound 26 exceeded the acceptable lipophilicity threshold in three different logP calculation methods (MLOGP > 4.15, WLOGP > 5.6, and XLOGP3 > 5), while compound 28 additionally violated the Egan filter (WLOGP > 5.88). These compounds, despite their strong binding profiles, may require structural optimization to improve their pharmacokinetic properties.
Notably, among the six compounds identified as most promising based on binding affinity, four (13, 18, 22, and 40) passed all Lipinski criteria, indicating favorable drug-like properties. Compound 33, despite its excellent binding profile, had a single Muegge violation, while compound 28 exhibited multiple filter violations that may necessitate structural modifications to optimize its pharmacokinetic behavior.
The overall drug-likeness assessment suggests that most of the novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives possess favorable physicochemical profiles for further development as potential therapeutic agents. Particularly, compounds 13, 18, and 40 represent promising candidates that combine strong binding affinity with excellent compliance across all drug-likeness filters.
Further examination of the lipophilicity profiles of these compounds provides valuable insights into their potential membrane permeability and tissue distribution characteristics (Table S4). Lipophilicity, typically measured as the octanol-water partition coefficient (logP), is a critical parameter influencing absorption, distribution, blood-brain barrier penetration, and overall pharmacokinetic behavior.
The novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives demonstrated a wide range of lipophilicity values, with consensus logP values (arithmetic mean of five computational methods) spanning from 2.18 (37, 38, and 39) to 5.21 (28). This broad spectrum allows for potential optimization of compounds for specific target tissues and pharmacokinetic requirements.
Notably, compounds with pyridine rings (37, 38, and 39) exhibited the lowest lipophilicity values. These compounds, characterized by moderate lipophilicity, may achieve a balanced distribution between aqueous and lipid compartments in biological systems.
In contrast, benzofuran-substituted compounds with larger spiro rings (28 and 26) demonstrated significantly higher lipophilicity (consensus logP values of 5.21 and 5.14, respectively). While this enhanced lipophilicity might facilitate blood-brain barrier penetration—a desirable characteristic for CNS-targeted compounds—it also aligns with the observed violations of drug-likeness filters noted in Table B, potentially impacting aqueous solubility and overall pharmacokinetic behavior.
Among the compounds identified as most promising based on binding affinity, a range of lipophilicity profiles was observed. Compound 40 exhibited moderate lipophilicity (consensus logP = 3.07), while compounds 13, 18, 22, and 33 demonstrated progressively higher values (3.85, 3.71, 4.21, and 3.71, respectively). This distribution suggests that binding affinity is not strictly correlated with lipophilicity, and compounds with varied physicochemical profiles can achieve strong target engagement.
Comparison of different computational methods reveals some variability in predicted logP values, highlighting the importance of using consensus approaches for more reliable lipophilicity assessment. For instance, the XLOGP3 method consistently predicted higher values for compounds with extended aromatic systems compared to the iLOGP method, which may reflect differences in how these algorithms account for π-electron density and aromatic interactions.
Of particular interest for potential CNS applications, compounds with consensus logP values between 2-4 (such as 40, 35, 8, 2, and 18) may offer optimal blood-brain barrier penetration while maintaining acceptable aqueous solubility.
The analysis of compound 13, which demonstrated one of the strongest binding affinities in the 127 ų cavity, reveals a consensus logP of 3.85 - a value that suggests favorable membrane permeability while remaining below the problematic threshold for excessive lipophilicity. This balanced lipophilicity profile, combined with its strong binding affinity and favorable drug-likeness parameters, further supports compound 13 as a promising lead candidate for further development.
The comprehensive analysis of physicochemical properties, drug-likeness parameters, and lipophilicity profiles collectively provides a solid foundation for prioritizing compounds for synthesis and biological evaluation. Based on this integrated assessment, compounds 13, 18, and 40 (green dots) emerge as particularly promising candidates among several others (Figure 6), combining strong binding affinity with favorable physicochemical profiles conducive to drug development
In addition to lipophilicity, aqueous solubility represents a critical parameter for drug development, influencing formulation strategies, bioavailability, and dosing regimens. The solubility profiles of the novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives were evaluated using three complementary computational models: ESOL, Ali, and SILICOS-IT (Table S4).
The predicted solubility values demonstrated significant variability across the series, with compounds classified from “soluble” to “poorly soluble” depending on their structural features and the prediction model employed. This heterogeneity in solubility profiles suggests opportunities for structural optimization to enhance aqueous solubility while maintaining target binding.
The ESOL model, which typically provides reliable solubility estimates for drug-like molecules, predicted that compounds 1-11, 31, 34, 37-39 would exhibit “soluble” classification (10-4 to 10-3 mol/L). These compounds, characterized by nitrogen-containing heterocycles or smaller ring systems, demonstrated more favorable solubility profiles than their more lipophilic counterparts.
The Ali model generally predicted higher solubility values for most compounds, with 19 derivatives classified as “soluble.” This model indicated that pyridine-containing compounds (37-39) and those with piperidine scaffolds (31-40) demonstrated particularly favorable solubility characteristics, with predicted concentrations approaching 10-3 mol/L.
Conversely, the SILICOS-IT model provided more conservative solubility estimates, with the majority of compounds classified as “moderately soluble” or “poorly soluble.” This model identified compounds 26, 28, and 29 as particularly problematic from a solubility perspective, with predicted concentrations below 10-7 mol/L, suggesting potential challenges for formulation and bioavailability. All three models consistently identified compounds 26, 28, and 29 as having the poorest solubility profiles, which aligns with their high lipophilicity values noted in Table S3 and the drug-likeness violations observed in Table S6.
These compounds, characterized by extended aromatic systems and bulky substituents, may require solubility-enhancing formulation strategies or structural modifications to improve their pharmaceutical properties. Among the compounds identified as most promising based on binding affinity, varying solubility profiles were observed. Compound 40, despite its strong binding in the 97 ų cavity, exhibited moderate solubility according to ESOL and Ali models (10-5 mol/L) but poor solubility by SILICOS-IT prediction (8.95 × 10-8 mol/L). Similarly, compound 13 showed moderate solubility by ESOL and Ali models but poor solubility according to SILICOS-IT (4.49 × 10-8 mol/L). Compound 33, which combines strong binding with the adamantyl substituent, demonstrated moderate solubility across all three models (3.60 × 10-6 to 3.85 × 10-6 mol/L by ESOL and Ali), suggesting a more balanced physicochemical profile. In contrast, compound 22 exhibited borderline moderate to poor solubility, with values ranging from 1.65 × 10-6 mol/L (ESOL) to 2.42 × 10-8 mol/L (SILICOS-IT).
The discrepancies observed between different solubility prediction models highlight the importance of experimental verification in subsequent development stages. Nevertheless, the computational predictions provide valuable guidance for prioritizing compounds and identifying potential solubility challenges early in the development process. Based on this integrated assessment, compounds 13, 18, 33, and 40 emerge as particularly promising candidates, balancing strong binding affinity with acceptable physicochemical profiles conducive to drug development, though potential solubility challenges may necessitate appropriate formulation strategies.
2.2.4. Drug Development Considerations
Beyond physicochemical properties and solubility parameters, the prediction of pharmacokinetic behavior and potential drug-drug interactions provides critical insights for prioritizing compounds in the drug development pipeline. Figure 7 (Table S5) presents the predicted ADME (absorption, distribution, metabolism, and excretion) properties and potential cytochrome P450 (CYP) interactions for the novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives.
All compounds in the series were predicted to exhibit high gastrointestinal absorption, suggesting favorable oral bioavailability potential. This consistent prediction aligns with the moderate molecular weight range (252-412 g/mol) and appropriate lipophilicity profiles observed for most compounds in the series. However, significant variations were observed in other pharmacokinetic parameters, particularly regarding blood-brain barrier (BBB) penetration and CYP inhibition patterns. The majority of compounds (37 out of 40) were predicted to penetrate the blood-brain barrier, making them potentially suitable for targeting CNS disorders. Notable exceptions included compounds 28 and 29, which, despite their high lipophilicity, were predicted to have limited BBB penetration.
This unexpected finding suggests that structural features beyond simple lipophilicity, such as specific molecular shape or hydrogen bonding patterns, may influence BBB permeability for these compounds. Most compounds were predicted to be P-glycoprotein (P-gp) substrates, with exceptions including compounds 11 and 33, which share similar structural features including bulky hydrophobic substituents. P-gp substrates may experience reduced brain exposure due to active efflux transport at the BBB, potentially limiting their CNS efficacy. However, this property could be advantageous for targeting peripheral tissues while minimizing CNS side effects.
The analysis of potential cytochrome P450 interactions revealed diverse inhibition patterns across the series. Notably, compounds 13, 18, 23, 27, 30, and 36 were predicted to inhibit all five major CYP isoforms (1A2, 2C19, 2C9, 2D6, and 3A4), suggesting a high potential for drug-drug interactions. In contrast, compounds 11 and 33 demonstrated more selective CYP inhibition profiles, affecting only CYP2C19 and CYP3A4, which may indicate a reduced potential for drug-drug interactions. Skin permeability (Log Kp) values ranged from -3.85 cm/s (26) to -6.60 cm/s (38 and 39), with more negative values indicating reduced permeability. Compounds with higher lipophilicity generally exhibited less negative Log Kp values, consistent with enhanced membrane permeation. This parameter is particularly relevant for potential topical applications or assessing systemic exposure following dermal contact.
Among the compounds identified as most promising based on binding affinity, varying pharmacokinetic profiles were observed. Compound 13 exhibited BBB penetration capability and high gastrointestinal absorption but demonstrated inhibition of all five CYP isoforms, suggesting potential drug-drug interaction concerns. Similarly, compound 18 showed favorable BBB penetration but inhibited multiple CYP enzymes. Compound 40, which demonstrated strong binding in the 97 ų cavity, exhibited a more balanced profile with BBB penetration capability and inhibition of CYP2C9, 2D6, and 3A4, but not 1A2 or 2C19. This more selective CYP inhibition pattern may indicate a reduced potential for drug-drug interactions compared to compounds 13 and 18.
Perhaps most remarkably, compound 33 (adamantyl-substituted) exhibited a particularly favorable pharmacokinetic profile, with BBB penetration capability, absence of P-gp substrate properties, and selective inhibition of only CYP2C19 and 3A4. This profile, combined with its strong binding affinity and balanced physicochemical properties, further supports compound 33 as a particularly promising candidate for further development. Comparison with reference compounds provides additional context for interpreting these predictions.
The integrated analysis of binding affinity, physicochemical properties, and predicted pharmacokinetic parameters provides a foundation for prioritizing compounds for synthesis and biological evaluation. Based on this multidimensional assessment, compound 33 emerges as particularly promising, offering an optimal balance of strong binding affinity, favorable physicochemical properties, and a desirable pharmacokinetic profile with reduced potential for drug-drug interactions. Compounds 13, 18, and 40 also represent valuable candidates, though their broader CYP inhibition profiles may necessitate careful consideration of potential drug-drug interactions.
To complete the multidimensional pharmaceutical evaluation of these novel derivatives, an assessment of bioavailability, synthetic accessibility, and potential structural alerts was conducted (Table S6). The synthetic assessment of availability values, which estimate the relative ease of chemical synthesis, ranged from 3.50 (1) to 6.55 (33), with lower values indicating more favorable synthetic accessibility. Among the compounds identified as most promising based on binding affinity, varied synthetic complexity was observed. Compounds 13 and 18 demonstrated moderate synthetic complexity (3.88 and 3.77, respectively), suggesting reasonable synthetic feasibility. In contrast, compounds 22 and 40 exhibited slightly higher synthetic complexity (3.99 and 3.98), while compound 33 showed the highest synthetic complexity score (6.55) among all compounds, potentially reflecting challenges associated with incorporating the adamantyl moiety.
Notably, none of the compounds triggered Brenk or PAINS (Pan-Assay Interference Compounds) structural alerts. This finding is particularly significant, as it suggests the novel derivatives lack substructures associated with false positives in biochemical screening or problematic reactive functionalities, supporting their potential as reliable candidates for biological evaluation. The lead compound assessment revealed that 15 out of the 40 compounds (1, 3, 6, 7, 9, 12, 15-17, 31, 34, and 36-39) met all industry-standard drug-likeness criteria with zero violations. The remaining compounds showed violations primarily related to lipophilicity (XLOGP3 > 3.5) and, in some cases, molecular weight (MW > 350). These violations align with the observations from Table 4, reflecting the inherent lipophilicity of the spiro [1,2,4]triazolo [1,5-c]quinazoline scaffold and the substantial molecular weight of the more structurally complex derivatives.
Among the most promising compounds based on binding affinity, compound 40 showed two violations (MW > 350 and XLOGP3 > 3.5), compound 22 exhibited the same two violations, and compound 13 showed a single violation (XLOGP3 > 3.5). Compound 33, despite its exceptional binding profile and favorable pharmacokinetic properties, demonstrated two violations (MW > 350 and XLOGP3 > 3.5), reflecting its substantial molecular weight (389.54 g/mol) and high lipophilicity (XLOGP3 = 4.72).
The compounds with pyridine substituents (37-39) displayed the most favorable overall profiles in the lead compound assessment, meeting all drug-likeness criteria with zero violations while maintaining reasonable synthetic complexity (3.74-3.80) and moderate binding affinities. This finding suggests that nitrogen-containing heterocyclic substituents may offer an optimal balance of drug-like properties and synthetic feasibility within this chemical series.
The integrated analysis across Tables S1 through S6 provides a comprehensive, multidimensional evaluation of these novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives. Based on this holistic assessment, compounds 13, 18, and 40 emerge as balanced candidates for further development, offering strong binding affinity combined with acceptable pharmaceutical profiles despite minor drug-likeness violations. Compound 33, while exhibiting higher synthetic complexity, represents a particularly intriguing candidate due to its exceptional binding affinity, favorable pharmacokinetic profile, and the absence of P-gp substrate properties.
2.3. Comparative Analysis with Reference DGK Inhibitors
Our comparative analysis focuses on well-established DGK inhibitors (Figure 1) that serve as important benchmarks for evaluating novel derivatives. These reference compounds include ritanserin, a 5-HT2R antagonist with structural similarity to R59022 that also exhibits DGK inhibitory properties [9,21]; R59949, a more potent analog of R59022 [8]; R59022, one of the first reference DGK inhibitors [5,7,9,13]; (5Z,2E)-CU-3, a DGK-α selective inhibitor [22]; and BMS502, which targets multiple DGK isoforms including DGK-α, DGK-ζ, and DGK-ι [12]. Each of these compounds possesses distinctive structural features and inhibitory profiles that inform the design and evaluation of our novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives.
An examination of these novel derivatives in comparison with the established DGK inhibitors reveals significant distinctions across physicochemical properties, drug-likeness parameters, lipophilicity, solubility, pharmacokinetics, and binding characteristics that collectively inform their potential therapeutic applications.
Physicochemical property comparison. Reference compounds demonstrate substantially larger molecular frameworks (MW: 459.58-516.50 g/mol; heavy atoms: 33-38) compared to our novel derivatives (MW: 252.31-412.53 g/mol; heavy atoms: 19-31). This dimensional differential may confer advantages in membrane permeability and bioavailability to the spiro [1,2,4]triazolo [1,5-c]quinazoline series. Notably, reference inhibitors exhibit predominantly planar structures with limited three-dimensionality (Csp³: 0.21-0.26), while several novel compounds, particularly 10, 11, 20, 26, 32, and 33, demonstrate significantly higher Csp³ values (≥0.58). This enhanced three-dimensionality has been associated with improved selectivity profiles and reduced promiscuous binding in contemporary drug development paradigms.
The reference compounds’ conformational flexibility (5 rotatable bonds versus 1-2 in novel derivatives) and elevated hydrogen bond acceptor counts (4-8 versus 2-4) further distinguish these chemical classes. The topological polar surface area values of reference compounds (65.85-110.98 Ų) generally exceed those of the novel derivatives (42.74-74.22 Ų), with BMS502 (110.98 Ų) approaching the threshold associated with limited membrane permeability. This observation suggests that our novel compounds may demonstrate superior passive diffusion properties while maintaining adequate polarity for aqueous solubility.
Drug-likeness. Examination of drug-likeness criteria reveals substantial differences in compliance profiles between reference and novel compounds. While most novel derivatives (compounds 1-10, 12-19, 21, 23, 24, 31-40) demonstrated excellent adherence to all five medicinal chemistry filters, the reference compounds exhibited multiple violations. BMS502 failed to meet two Ghose criteria (MW>480, MR>130) and violated Lipinski’s Rule of Five (MW>500). Similarly, R59022, R59949, and Ritanserin each violated multiple filters, particularly relating to lipophilicity and molecular refractivity thresholds.
Most notably, R59949 violated three Ghose criteria (MW>480, WLOGP>5.6, MR>130), exceeded Egan’s lipophilicity threshold (WLOGP>5.88), and failed Muegge’s filter (XLOGP3>5). This contrasts sharply with the novel derivatives 13, 18, and 40, which demonstrated complete compliance across all drug-likeness filters while maintaining strong binding affinities. The significantly improved drug-likeness profiles of the novel compounds suggest enhanced developability potential compared to existing DGK inhibitors.
Lipophilicity profiles. Lipophilicity comparison reveals distinctive patterns between reference and novel compounds. Reference inhibitors displayed notably high consensus logP values (BMS502: 3.13; R59022: 5.28; R59949: 5.92; Ritanserin: 5.60) compared to many novel derivatives. This elevated lipophilicity in reference compounds, particularly evident in the SILICOS-IT method (R59949: 7.57; Ritanserin: 7.08), suggests potential issues with aqueous solubility and excessive tissue distribution.
In contrast, several high-affinity novel compounds demonstrated more balanced lipophilicity profiles: compound 13 (consensus logP: 3.85), compound 18 (consensus logP: 3.71), and compound 40 (consensus logP: 3.07). These moderate lipophilicity values, approaching the generally accepted optimal range of 2-3.5 for orally administered drugs, suggest more favorable pharmacokinetic behavior without compromising membrane permeability. The outlier compound 33, despite its excellent binding profile, exhibited a higher consensus logP (3.71) that remains significantly lower than reference inhibitors.
Solubility characteristics. Solubility prediction by three complementary models (ESOL, Ali, and SILICOS-IT) revealed substantial differences between reference and novel compounds. The reference compounds consistently demonstrated poor solubility classifications, with particularly problematic profiles for R59949 and Ritanserin. R59949 was classified as “poorly soluble” across all three models, with predicted concentrations ranging from 1.05 × 10-7 mol/L (ESOL) to 3.09 × 10-10 mol/L (SILICOS-IT), suggesting significant formulation challenges.
In contrast, several novel derivatives, particularly those with pyridine substituents (37-39) and piperidine scaffolds (31-40), exhibited more favorable solubility characteristics. Compound 33, despite incorporating the adamantyl moiety, maintained moderate solubility across all prediction models (3.60 × 10-6 to 3.85 × 10-6 mol/L by ESOL and Ali), suggesting a more balanced physicochemical profile than reference inhibitors. The improved solubility characteristics of selected novel compounds may confer advantages in formulation flexibility and absorption consistency.
Pharmacokinetic parameters. Comparison of predicted ADME properties reveals significant distinctions in potential pharmacokinetic behavior. While all compounds (reference and novel) were predicted to exhibit high gastrointestinal absorption, notable differences emerged in other parameters. Reference compounds BMS502, R59022, and ritanserin were predicted to penetrate the blood-brain barrier, but R59949 was not, despite its high lipophilicity.
More striking differences appeared in cytochrome P450 inhibition profiles. Reference compounds demonstrated specific inhibition patterns: BMS502 inhibited CYP1A2, CYP2C19, and CYP3A4; R59022 inhibited CYP1A2, CYP2C19, and CYP3A4; R59949 inhibited CYP1A2; and Ritanserin inhibited CYP1A2, CYP2C19, and CYP3A4. In contrast, compound 33 exhibited a more selective inhibition profile (affecting only CYP2C19 and CYP3A4) compared to most other compounds in both reference and novel series.
Skin permeability coefficients (Log Kp) for reference compounds (-6.88 to -4.81 cm/s) generally indicated reduced permeability compared to several novel derivatives, particularly those with high lipophilicity. The collective pharmacokinetic analysis suggests that while reference compounds demonstrate established DGK inhibitory activity, selected novel derivatives, particularly compound 33, may offer improved pharmacokinetic profiles with reduced drug-drug interaction potential.
Toxicity classification difference. Reference compounds generally show LD₅₀ values in the range of 900-1600 mg/kg, placing them predominantly in Class IV toxicity. This is comparable to many novel derivatives, though some novel compounds demonstrated potentially improved safety profiles with higher LD50 values.
Organ-specific toxicity profiles. The reference compounds display distinctive toxicological signatures compared to the novel derivatives:
- R59949 shows higher mutagenicity probability (0.75) compared to most novel compounds (typically <0.61).
- Ritanserin and R59022 both exhibit cytotoxicity probabilities (0.67) that exceed those of the novel derivatives (0.53-0.60).
- BMS502 uniquely demonstrates higher mutagenicity probability (0.91/yes) compared to all novel compounds, which consistently showed “no” predictions for mutagenicity.
Balanced toxicity parameters. While reference compounds show strong binding affinity, they generally demonstrate less favorable toxicological profiles across multiple endpoints. The novel derivatives, particularly compounds 13, 18, 33, and 40, maintain comparable binding affinity while exhibiting improved safety profiles with lower probabilities across hepatotoxicity, carcinogenicity, and mutagenicity endpoints.
Prediction accuracy considerations. The toxicity predictions for reference compounds benefit from their established pharmacological profiles and extensive experimental data, potentially increasing prediction reliability. Nevertheless, the consistent prediction patterns observed for novel derivatives suggest reliable safety projections that merit experimental verification.
Binding affinity comparison. The most significant comparative dimension emerges in the analysis of binding affinities across multiple cavities. While reference inhibitors demonstrated strong binding in specific cavities (Ritanserin: -9.3 kcal/mol in 127 ų; R59022: -9.2 kcal/mol in 127 ų; R59949: -8.7 kcal/mol in 127 ų, -8.1 kcal/mol in 121 ų), several novel derivatives exhibited comparable or superior binding in alternative cavities.
Compound 18 achieved exceptional binding in the 179 ų cavity (-8.5 kcal/mol), surpassing all reference compounds in this specific binding region. Similarly, compound 40 demonstrated superior affinity in the 97 ų cavity (-8.2 kcal/mol), outperforming all reference compounds in this pocket. Compounds 13, 22, and 33 (all -8.3 to -8.4 kcal/mol in the 127 ų cavity) approached the potency of reference inhibitors while maintaining improved physicochemical profiles.
The enhanced binding of novel derivatives in specific cavities, coupled with their improved drug-like properties, suggests potential advantages in selectivity and reduced off-target effects compared to reference inhibitors. The differential binding patterns across cavities further indicate opportunities for developing cavity-specific inhibitors with optimized pharmacological profiles.
Synthetic accessibility. Assessment of synthetic feasibility revealed substantial differences between reference and novel compounds. Reference inhibitors demonstrated moderate synthetic complexity (BMS502: 3.81; R59022: 3.73; R59949: 3.67; Ritanserin: 3.73), comparable to many novel derivatives. However, significant variations existed within the novel series, with compounds 1 and 7 offering the most favorable synthetic routes (scores of 3.50) and compound 33 presenting greater synthetic challenges (score of 6.55).
Importantly, several compounds with excellent binding profiles (13: 3.88; 18: 3.77; 40: 3.98) demonstrated synthetic complexity comparable to reference inhibitors, suggesting feasible synthetic approaches for these promising candidates. This analysis provides valuable guidance for prioritizing compounds based on the balance between binding potency and synthetic accessibility.
Integrated comparative assessment. The multidimensional comparative analysis reveals that while reference DGK inhibitors (BMS502, R59022, R59949, and Ritanserin) demonstrate established activity, they exhibit several pharmaceutical limitations, including excessive molecular weight, high lipophilicity, poor predicted solubility, and multiple drug-likeness violations. In contrast, selected novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives, particularly compounds 13, 18, 33, and 40, offer a more balanced profile combining strong binding affinity with improved physicochemical and pharmacokinetic properties.
Compound 33 emerges as especially noteworthy, demonstrating exceptional binding affinity (-8.3 kcal/mol in 127 ų cavity), favorable pharmacokinetic profile (selective CYP inhibition, absence of P-gp substrate properties), moderate solubility, and enhanced three-dimensionality (Csp³: 0.67) despite its synthetic complexity. Similarly, compound 13 offers an optimal balance of strong binding (-8.4 kcal/mol in 127 ų cavity), favorable drug-likeness, acceptable lipophilicity (consensus logP: 3.85), and reasonable synthetic accessibility (score: 3.88).
This integrated comparison suggests that the novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives represent a substantial advancement over existing DGK inhibitors, offering improved pharmaceutical profiles while maintaining comparable or superior binding capabilities. These findings provide a robust foundation for the further development of these compounds as next-generation DGK modulators with enhanced therapeutic potential.
2.4. Structure-Activity Relationships
The systematic computational analysis revealed critical structural determinants that significantly influence binding affinity, physicochemical properties, and pharmacokinetic parameters. Figure 8 illustrates the identified structure-activity relationships (SAR).
The [1,2,4]triazolo [1,5-c]quinazoline core scaffold provides the essential framework for DGKα binding, while three key regions allow for strategic modifications: the spiro ring size, the scaffold type (piperidine or cyclohexane), and the heterocyclic substituent. Our analysis demonstrates that each of these regions contributes distinctively to the overall molecular properties and target engagement.
The spiro ring size emerged as a critical determinant of binding affinity. Derivatives containing an n = 2 system (cyclopentane, compounds 9-17) consistently exhibited superior binding compared to their n = 1 (cyclobutane) or n = 3 (cyclohexane) counterparts. This optimal ring size likely provides the ideal spatial orientation for engaging key binding residues within the DGKα protein structure, particularly in the 127 ų and 179 ų cavities that showed the strongest binding potential.
The heterocyclic substituent significantly influenced both binding affinity and physicochemical properties. Oxygen-containing heterocycles demonstrated exceptional binding characteristics, with the furan-containing compound 13 exhibiting the strongest binding among novel derivatives in the 127 ų cavity (-8.4 kcal/mol) and benzofuran derivatives (22 and 28) showing excellent binding across multiple cavities. Nitrogen-containing heterocycles provided distinctive binding profiles, with the indole-substituted compound 18 achieving the highest binding affinity in the 179 ų cavity (-8.5 kcal/mol) and pyridine-containing derivatives (30 and 33) showing favorable binding coupled with improved physicochemical properties. The adamantyl group (33) produced one of the most promising candidates, combining strong binding affinity with selective CYP inhibition profiles.
Modification of the piperidine/cyclohexane scaffold revealed that the presence of a tert-butyl group enhanced binding through additional hydrophobic interactions, while piperidine-containing compounds (31-40) demonstrated higher hydrogen bond acceptor capacities that influenced both binding and solubility profiles.
Detailed molecular interaction analysis identified key residues critical for high-affinity binding. Compounds forming stronger hydrogen bonds with TRP151, GLU166, and ARG126 generally exhibited superior binding profiles. Notably, compound 40 achieved its exceptional binding in the 97 ų cavity through a much stronger conventional hydrogen bond with GLU166 (1.85 Å) compared to reference inhibitors, highlighting the importance of optimal hydrogen bonding networks.
The interplay between structural modifications and physicochemical properties revealed important trends. Pyridine-substituted derivatives demonstrated improved solubility and lower lipophilicity (consensus logP = 2.18), while benzofuran-substituted compounds showed higher binding affinity but increased lipophilicity (logP > 5). Compounds with higher fractions of sp³ hybridized carbon atoms (Csp³ ≥ 0.58), particularly compounds 10, 11, 20, 26, 32, and 33, exhibited enhanced three-dimensionality, which has been associated with improved selectivity profiles.
Pharmacokinetic analysis revealed that most compounds were predicted to penetrate the blood-brain barrier, with the notable exception of compounds 28 and 29 despite their high lipophilicity. Compound 33 emerged as particularly promising, demonstrating selective CYP inhibition (affecting only CYP2C19 and CYP3A4) and absence of P-gp substrate properties, suggesting reduced potential for drug-drug interactions.
Based on this SAR analysis, compounds 13, 18, 33, and 40 represent the most promising candidates for further development, offering a balanced profile of strong binding affinity and favorable physicochemical and pharmacokinetic properties. These findings provide a solid foundation for the rational design of next-generation DGKα modulators with optimized therapeutic potential.
2.5. Limitations
Our computational approach to evaluating novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives as DGKα modulators has several key limitations:
1. Methodological
- Comprehensive structural modeling: The integration of complete protein structure beyond the EF-hand domains (PDB ID: 6IIE) may reveal additional interaction sites, particularly within the catalytic domain where inhibitors like R59949 demonstrate binding affinity.
- Implementation of dynamic models: Application of molecular dynamics simulations could potentially capture protein flexibility and conformational changes during ligand binding, offering more nuanced insights than static docking approaches.
2. Mechanistic Validation Possibilities
- Experimental assay integration: Development of biochemical assays could provide critical validation parameters (IC50, Ki values, binding kinetics) that may confirm or refine computational predictions.
- Isoform selectivity assessment: Comprehensive profiling across DGK isoforms might identify compounds with optimal selectivity profiles, potentially minimizing off-target effects that commonly limit kinase inhibitor utility.
- Structure-activity relationship development: Systematic structural modifications could potentially establish clear correlations between molecular features and both binding affinity and selectivity, guiding rational design iterations.
3. Translational Research Opportunities
- Expanded pharmacokinetic evaluation: Detailed investigation of physiological stability, metabolic pathways, and bioavailability profiles could identify candidates with favorable drug-like properties.
- Enhanced CNS penetration analysis: Advanced modeling and experimental validation of blood-brain barrier permeability might identify compounds suitable for neurological applications, particularly through refined QSPR analyses.
- Comprehensive interaction profiling: Broader assessment of potential drug-drug interactions, including transporter effects and pharmacodynamic consequences, could identify compounds with favorable clinical profiles.
4. Technical Advancement Possibilities
- Scoring function diversification: Application of multiple complementary evaluation methods might yield more robust binding predictions by mitigating algorithm-specific biases.
- Advanced simulation implementation: Extended molecular dynamics with enhanced sampling techniques could potentially provide deeper insights into the stability and kinetics of predicted protein-ligand complexes.
- Quantum mechanical modeling integration: Incorporation of QM/MM approaches might more accurately represent electronic effects in critical binding interactions, particularly for compounds with complex electronic distributions.
2.6. Future Directions
Dose-response relationship investigation. A critical next step in developing these DGKα modulators will be to conduct dose-response studies to establish optimal therapeutic windows. This is particularly important given that DGK inhibition may exhibit bell-shaped dose-response curves, where moderate inhibition may enhance therapeutic effects while complete inhibition could lead to adverse outcomes. We propose investigating concentration ranges from 1 nM to 10 µM across multiple cell lines to determine IC50 values and establish structure-activity relationships in cellular contexts [7,8].
Additional validation models. Our findings require validation across multiple experimental models. We propose:
- Ex vivo organoid models to evaluate compound efficacy in more complex tissue environments
- Animal models of cancer and inflammatory disorders to validate in vivo efficacy [9].
Specific receptor binding assays. Rather than broadly suggesting further studies, we propose specific binding assays to clarify our compounds’ mechanism of action:
- Radioligand binding assays using [³H]phorbol 12-myristate 13-acetate (PMA) displacement to determine direct interaction with the DGKα catalytic domain
- Surface plasmon resonance experiments to measure binding kinetics and affinity constants
- Thermal shift assays to confirm physical interaction with the target protein [11]
- Competitive binding studies with established inhibitors like R59949 to determine binding site overlap [8]
Structural optimization roadmap. We propose a logical progression for compound optimization:
- For compounds showing the highest binding affinity (e.g., 18 and 40), introduce modifications to improve solubility while maintaining target engagement
- For compounds with balanced pharmacokinetic profiles (e.g., 33), explore bioisosteric replacements of the adamantyl group to maintain binding while reducing synthetic complexity
- Develop hybrid structures incorporating the indole moiety from compound 18 with the spiro-piperidine scaffold from compound 33 to potentially combine superior binding with improved pharmacokinetics
Broader therapeutic applications. Beyond cancer immunotherapy and viral infections mentioned in the original patent (CN 115362003 B) [10], our novel DGKα modulators may have significant therapeutic potential in:
- Neurodegenerative disorder [2], where abnormal lipid signaling contributes to disease progression
- Autoimmune disorders [4], where modulation of T-cell responses could restore immune homeostasis
- Cardiac hypertrophy and heart failure [3], where DGKα plays a role in pathological remodeling
- Metabolic disorders, particularly type 2 diabetes [2], where altered diacylglycerol signaling affects insulin response
Besides, the novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives have already demonstrated significant potential for addressing complex neurological and neuropsychiatric conditions through multifaceted molecular mechanisms [25]. The molecular profile suggests potential therapeutic utility in: anxiety disorder management, cognitive dysfunction rehabilitation, potential anti-depressant interventions, and mitigation of stress-induced neurological impairments.
These expanded applications would significantly broaden the impact of our structural approach to developing DGKα modulators and provide additional avenues for therapeutic development.
3. Materials and Methods
3.1. Molecular Docking Studies
3.1.1. Computational Framework of Structure-Based Blind Docking Analysis
The computational approach was informed by the structural complexity of DGK isoforms, recognizing that each subfamily possesses unique regulatory domains that may influence binding interactions. Following Topham & Epand, [2] framework of DGK structural classification, was potential domain-specific interactions carefully considered during the docking simulations.
We employed the CB-Dock2 web server platform [12,13] to conduct structure-based blind docking analyses. This computational approach integrates two complementary strategies: a curvature-based algorithm that detects potential binding cavities on the protein surface when no suitable templates are available, with subsequent molecular docking via AutoDock Vina (v1.2.0); and a template-based approach that leverages homologous structural information when compounds with significant topological similarity (FP2 similarity score ≥ 0.4) are identified in the BioLip structural repository (2021.09.15 release). The template-based approach incorporates the hierarchical FitDock algorithm to optimize ligand placement and generate refined binding poses. This dual-pathway methodology enhances binding site prediction accuracy by integrating evolutionary information when available while maintaining robust performance for novel protein-ligand interactions.
3.1.2. Application to DGKα Inhibitor Analysis
So, a blind search using CB-Dock2 was conducted to identify and characterize potential binding sites within the DGKα protein structure (RCSB PDB ID: 6IIE) [14]. Five distinct binding cavities were identified with volumes ranging from 97 ų to 179 ų, each exhibiting unique geometric properties and interaction potentials (Table 1). The crystal structure of DGKα EF-hand domains confirms [11] that DGKα contains a canonical pair of helix-loop-helix EF-hand motifs that bind two Ca²⁺ ions with different affinities (Kd¹ = 0.3 μM and Kd² = 2.3 μM) in a cooperative manner. By examining interaction patterns across these cavities with varying volumes, a framework was established for comparing the binding behavior of both reference inhibitors and the novel derivatives.
All molecular structures were processed using standardized mol format inputs, while the protein target was analyzed in pdb format. BIOVIA Discovery Studio 2017 R2 was used for visualization and analysis of the resulting binding poses.
3.2. Physicochemical and ADME Property Prediction
The physicochemical property analysis was conducted using SwissADME [15,16], an integrated web tool for medicinal chemistry and drug discovery applications. Molecular structures of all compounds were submitted in SMILES format to calculate key physicochemical parameters including molecular weight, heavy atom count, aromatic heavy atom count, fraction of sp³ hybridized carbon atoms, rotatable bonds, hydrogen bond acceptors and donors, molecular refractivity, and topological polar surface area.
SwissADME was also employed to assess drug-likeness according to multiple industry-standard filters (Lipinski, Ghose, Veber, Egan, and Muegge) and to calculate lipophilicity parameters using five distinct computational methods: iLOGP (internal VCCLAB method), XLOGP3 (atom-based method), WLOGP (atomistic method based on fragmental data), MLOGP (topological method), and SILICOS-IT (hybrid method). Consensus lipophilicity values were calculated as the arithmetic mean of these five predictions.
Solubility was evaluated using three complementary computational models: ESOL, Ali, and SILICOS-IT, with results reported in both mg/ml and mol/L concentrations. Compounds were classified as highly soluble, soluble, moderately soluble, or poorly soluble based on established threshold values for each model.
Pharmacokinetic property prediction was performed using both SwissADME and ProTox-2 and 3 [17,18] web servers. SwissADME was used to predict gastrointestinal absorption, blood-brain barrier permeation, P-glycoprotein substrate status, and skin permeability coefficient (Log Kp). ProTox provided complementary toxicity predictions and CYP inhibition profiles for five major cytochrome P450 isoforms: CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4. Synthetic accessibility was assessed using SwissADME’s built-in synthetic assessment algorithm, which estimates the relative ease of chemical synthesis on a scale from 1 (very easy to synthesize) to 10 (very difficult to synthesize).
Structural alerts were evaluated using both SwissADME (for Brenk and PAINS alerts) and ProTox for additional toxicophoric identification. Bioavailability was estimated using Abbott’s bioavailability score as implemented in SwissADME, with values closer to 1 indicating higher probability of >10% oral bioavailability in humans.
All computational predictions were performed with default parameters as implemented in the respective web servers. The combined use of multiple prediction methodologies for key parameters such as lipophilicity and solubility provided a more robust assessment of physicochemical and pharmacokinetic properties, reducing potential bias associated with individual computational models.
3.3. Reference Compounds
Several established DGK inhibitors were used as reference compounds for comparative analysis (Figure 1). Ritanserin (6-[2-[4-[bis(4-fluorophenyl)methylene]-1-piperidinyl]ethyl]-7-methyl-5H-thiazolo [3,2-a]pyrimidin-5-one), originally developed as a serotonin receptor antagonist, was later found to inhibit DGK [9,19]. R59949 (3-[2-[4-[bis(4-fluorophenyl)methylene]-1-piperidinyl]ethyl]-2,3-dihydro-2-thioxo-4(1H)-quinazolinone), a more potent analog of R59022 [8], and R59022 (6-[2-[4-[(4-fluorophenyl)phenylmethylene]-1-piperidinyl]ethyl]-7-methyl-5H-thiazolo [3,2-a]pyrimidin-5-one), which is considered one of the first reference DGK inhibitors [5,7,9,20], were included in the analysis. Additionally, we examined (5Z,2E)-CU-3 (DGK-α, N-[(5Z)-5-[(2E)-3-(2-furanyl)-2-propen-1-ylidene]-4-oxo-2-thioxo-3-thiazolidinyl]-benzenesulfonamide) [21] and BMS502 (8-[4-[bis(4-fluorophenyl)methyl]-1-piperazinyl]-5,6-dihydro-5-methyl-7-nitro-6-oxo-1,5-naphthyridine-2-carbonitrile), which targets DGK-α, DGK-ζ, and DGK-ι [22].
4. Conclusions
This study presents a computational investigation of novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives as potential diacylglycerol kinase α (DGKα) modulators. Through structure-based blind docking analysis using the CB-Dock2 platform, among 40 novel derivatives, compounds 13, 18, 22, 28, 33, and 40 demonstrated strong binding affinities across five distinct binding cavities of human DGKα EF-hand domains (PDB ID: 6IIE).
Molecular interaction analysis identified critical binding determinants, with oxygen- and nitrogen-containing heterocycles providing superior binding profiles. Strong hydrogen bonding with key residues (TRP151, GLU166, ARG126) significantly enhanced binding affinity. The optimal spiro ring size (n=2, cyclopentane derivatives) consistently demonstrated superior binding compared to n=1 or n=3 systems.
Toxicity assessment revealed favorable safety profiles across the series, with predicted LD₅₀ values of 1000-2000 mg/kg (Class IV-V, GHS classification), low hepatotoxicity probabilities (<0.65), and minimal genotoxic potential. Leading compounds maintained these reassuring toxicity characteristics while exhibiting no concerning structural alerts.
Physicochemical characterization revealed appropriate drug-like properties for most compounds, though several high-affinity derivatives exhibited elevated lipophilicity. All compounds demonstrated high predicted gastrointestinal absorption, with most expected to penetrate the blood-brain barrier. Compound 33 (adamantyl-substituted) displayed a particularly favorable pharmacokinetic profile with selective CYP inhibition (affecting only CYP2C19 and CYP3A4) and absence of P-glycoprotein substrate properties, suggesting reduced drug-drug interaction potential.
The multidimensional evaluation across binding affinity, physicochemical properties, solubility, pharmacokinetics, and synthetic accessibility identified above-mentioned 13, 18, 33, and 40 as the most promising candidates for further studies. These compounds effectively balance potent target engagement with favorable pharmaceutical profiles, though solubility challenges may necessitate appropriate formulation strategies.
This study establishes the spiro [1,2,4]triazolo [1,5-c]quinazoline scaffold as a valuable platform for developing novel DGKα modulators with potential applications in cancer, viral infections, and inflammatory disorders. The identified structure-activity relationships and critical binding interactions provide a rational framework for designing optimized compounds targeting DGK-mediated signaling pathways. With experimental validation and further optimization, these derivatives represent promising candidates for therapeutic intervention in various pathological conditions where DGKα plays a significant role.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Table S1: Physicochemical properties and molecular descriptors. Table S2: In silico oral toxicity predictions for spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives using Protox-2 and 3. Table S3: Lipophilicity parameters using different methods. Table S4: Solubility analysis in different solvent systems. Table S5: Pharmacokinetic and enzyme inhibitory properties compounds. Table S6: Bioavailability assessment and drug-likeness parameters.
Author Contributions
L.A., K.S., O.A.: Conceptualization, Methodology, Investigation, Formal Analysis, Software, Writing—Original Draft, Visualization, Writing—Review and Editing; M.A., O.K., V.O., S.K.: Resources, Project administration, Supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The Supplementary Materials are available free of charge on the MDPI Publication website.
Acknowledgments
Authors gratefully acknowledge Armed Forces of Ukraine with Territorial Defense Forces of the Armed Forces of Ukraine for preparing this paper in the safe conditions of Zaporizhzhia, Ukraine; and assistance of large language model Claude 3.7 by Anthropic during manuscript preparation in English language.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| DGKα | Diacylglycerol kinase alpha |
| PDB | Protein Data Bank |
| EF | EF-hand (calcium-binding motif) |
| DAG | Diacylglycerol |
| PA | Phosphatidic acid |
| PKC | Protein kinase C |
| PH | Pleckstrin homology (domain) |
| SAM | Sterile alpha motif |
| MARCKS | Myristoylated alanine-rich C-kinase substrate |
| PDZ | Post synaptic density protein, Drosophila disc large tumor suppressor, Zonula occludens-1 protein (domain) |
| HIV | Human immunodeficiency virus |
| HBV | Hepatitis B virus |
| 5-HT2R | Serotonin 2 receptor |
| IC50 | Half maximal inhibitory concentration |
| Kd | Dissociation constant |
| ADME | Absorption, Distribution, Metabolism, Excretion |
| TPSA | Topological polar surface area |
| ESOL | Estimated aqueous solubility |
| MW | Molecular weight |
| MR | Molecular refractivity |
| logP | Partition coefficient (octanol-water) |
| PAINS | Pan-Assay Interference Compounds |
| BBB | Blood-brain barrier |
| P-gp | P-glycoprotein |
| CYP | Cytochrome P450 |
| LD50 | Median lethal dose |
| HT | Hepatotoxicity |
| CG | Carcinogenicity |
| IT | Immunotoxicity |
| MG | Mutagenicity |
| CT | Cytotoxicity |
| SAR | Structure-activity relationship |
| CNS | Central nervous system |
| PMA | Phorbol 12-myristate 13-acetate |
| GHS | Globally Harmonized System (of Classification and Labeling of Chemicals) |
References
- Dominguez, C.L.; Floyd, D.H.; Xiao, A.; Mullins, G.R.; Kefas, B.A.; Xin, W.; et al. Diacylglycerol kinase α is a critical signaling node and novel therapeutic target in glioblastoma and other cancers. Cancer Discov 2013, 3, 782–797. [Google Scholar] [CrossRef] [PubMed]
- Topham, M.K.; Epand, R.M. Mammalian diacylglycerol kinases: molecular interactions and biological functions of selected isoforms. Biochim Biophys Acta 2009, 1790, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Sakane, F.; Hoshino, F.; Murakami, C. New era of diacylglycerol kinase, phosphatidic acid and phosphatidic acid-binding protein. Int J Mol Sci 2020, 21, 6794. [Google Scholar] [CrossRef] [PubMed]
- Riese, M.J.; Moon, E.K.; Johnson, B.D.; Albelda, S.M. Diacylglycerol kinases (DGKs): novel targets for improving T cell activity in cancer. Front Cell Dev Biol 2016, 4, 108. [Google Scholar] [CrossRef] [PubMed]
- de Chaffoy de Courcelles D, Roevens P, Van Belle H. R 59022, a diacylglycerol kinase inhibitor. Its effect on diacylglycerol and thrombin-induced C kinase activation in the intact platelet. J Biol Chem 1985, 260, 15762–15770. [CrossRef] [PubMed]
- Gómez-Merino, F.C.; Arana-Ceballos, F.A.; Trejo-Téllez, L.I.; Skirycz, A.; Brearley, C.A.; Dörmann, P.; et al. Arabidopsis AtDGK7, the smallest member of plant diacylglycerol kinases (DGKs), displays unique biochemical features and saturates at low substrate concentration: the DGK inhibitor R59022 differentially affects AtDGK2 and AtDGK7 activity in vitro and alters plant growth and development. J Biol Chem 2005, 280, 34888–34899. [Google Scholar] [PubMed]
- Tu-Sekine, B.; Goldschmidt, H.; Petro, E.; Raben, D.M. Diacylglycerol kinase θ: regulation and stability. Adv Biol Regul 2013, 53, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sakane, F.; Kanoh, H.; Walsh, J.P. Selectivity of the diacylglycerol kinase inhibitor 3-[2-[4-[bis-(4-fluorophenyl)methylene]-1-piperidinyl]ethyl]-2,3-dihydro-2-thioxo-4(1H)quinazolinone (R59949) among diacylglycerol kinase subtypes. Biochem Pharmacol 2000, 59, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Boroda, S.; Niccum, M.; Raje, V.; Purow, B.W.; Harris, T.E. Dual activities of ritanserin and R59022 as DGKα inhibitors and serotonin receptor antagonists. Biochem Pharmacol 2017, 123, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Kalna Bioscience and Gilead Sciences. Diacylglycerol kinase modulating compounds. CN 115362003 B, 22 March 2024.
- Takahashi, D.; Suzuki, K.; Sakamoto, T.; Iwamoto, T.; Murata, T.; Sakane, F. Crystal structure and calcium-induced conformational changes of diacylglycerol kinase α EF-hand domains. Protein Sci 2019, 28, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Chupak, L.; Wichroski, M.; Zheng, X.; Ding, M.; Martin, S.; Allard, C.; et al. Discovery of potent, dual-inhibitors of diacylglycerol kinases alpha and zeta guided by phenotypic optimization. ACS Med Chem Lett 2023, 14, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Martinez, K.; Shyamasundar, S.; Kennedy, A.; Chuang, C.C.; Marsh, A.; Kincaid, J.; et al. Diacylglycerol kinase inhibitor R59022 attenuates conjugated linoleic acid-mediated inflammation in human adipocytes. J Lipid Res 2013, 54, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.-X.; Cao, Y. CB-Dock2: improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res 2022, 50, W284–W293. [Google Scholar] [CrossRef] [PubMed]
- Yang, X. CB-Dock2: Cavity detection guided blind docking. Available online: https://cadd.labshare.cn/cb-dock2/index.php (accessed on 7 January 2025).
- RCSB PDB - 6IIE: Crystal structure of human diacylglycerol kinase alpha EF-hand domains bound to Ca2+. RCSB PDB: Homepage. Available online: https://www.rcsb.org/structure/6IIE (accessed on 17 February 2025).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- SwissADME Available online: http://www.swissadme.ch/ (accessed on 16 May 2024; 23 March 2025).
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res 2024, 52, W440–W445. [Google Scholar] [CrossRef] [PubMed]
- ProTox-2 and Protox-3 - Prediction of Toxicity of Chemicals. Available online: https://tox-new.charite.de/protox_II/index.php?site=home.-do (accessed on 11 May 2024; 25 March 2025).
- Janssen, P.A. Pharmacology of potent and selective S2-serotonergic antagonists. J Cardiovasc Pharmacol 1985, 7 (Suppl 7), S2–S11. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kunii, N.; Sakuma, M.; Yamamoto, A.; Mizuno, S.; Sato, Y.; et al. A novel diacylglycerol kinase α-selective inhibitor, CU-3, induces cancer cell apoptosis and enhances immune response. J Lipid Res 2016, 57, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Tice, C.M.; Singh, S.B. The use of spirocyclic scaffolds in drug discovery. Bioorg Med Chem Lett 2014, 24, 3673–3682. [Google Scholar] [CrossRef] [PubMed]
- Batista, V.F.; Pinto, D.C.G.A.; Silva, A.M.S. Recent in vivo advances of spirocyclic scaffolds for drug discovery. Expert Opin Drug Discov 2022, 17, 603–618. [Google Scholar] [CrossRef] [PubMed]
- Shabelnyk, K.; Antypenko, L.; Bohdan, N.; Ryzhenko, V.; Belenichev, I.; Kamyshnyi, O.; et al. Search for potential neuroprotectors for correction of cognitive and behavioral disorders after ketamine anesthesia among 2’-R-6’H-spiro(cycloalkyl-, heterocyclyl)[1,2,4]triazolo [1,5-c]quinazolines: fragment-oriented design, molecular docking, ADMET, synthesis and in vivo study. 2025, under submission.
Figure 1.
Molecular structures of reference diacylglycerol kinase (DGK) inhibitors used in this study. Ritanserin (serotonin receptor antagonist with DGK inhibitory properties), R59022 (first-generation DGK inhibitor), R59949 (potent analog of R59022), (5Z,2E)-CU-3 (DGK-α selective inhibitor), and BMS502 (multi-isoform DGK inhibitor targeting DGK-α, DGK-ζ, and DGK-ι). These compounds served as comparative standards for evaluating the binding affinities of novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives and identifying shared pharmacophore features essential for DGK modulation.
Figure 1.
Molecular structures of reference diacylglycerol kinase (DGK) inhibitors used in this study. Ritanserin (serotonin receptor antagonist with DGK inhibitory properties), R59022 (first-generation DGK inhibitor), R59949 (potent analog of R59022), (5Z,2E)-CU-3 (DGK-α selective inhibitor), and BMS502 (multi-isoform DGK inhibitor targeting DGK-α, DGK-ζ, and DGK-ι). These compounds served as comparative standards for evaluating the binding affinities of novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives and identifying shared pharmacophore features essential for DGK modulation.
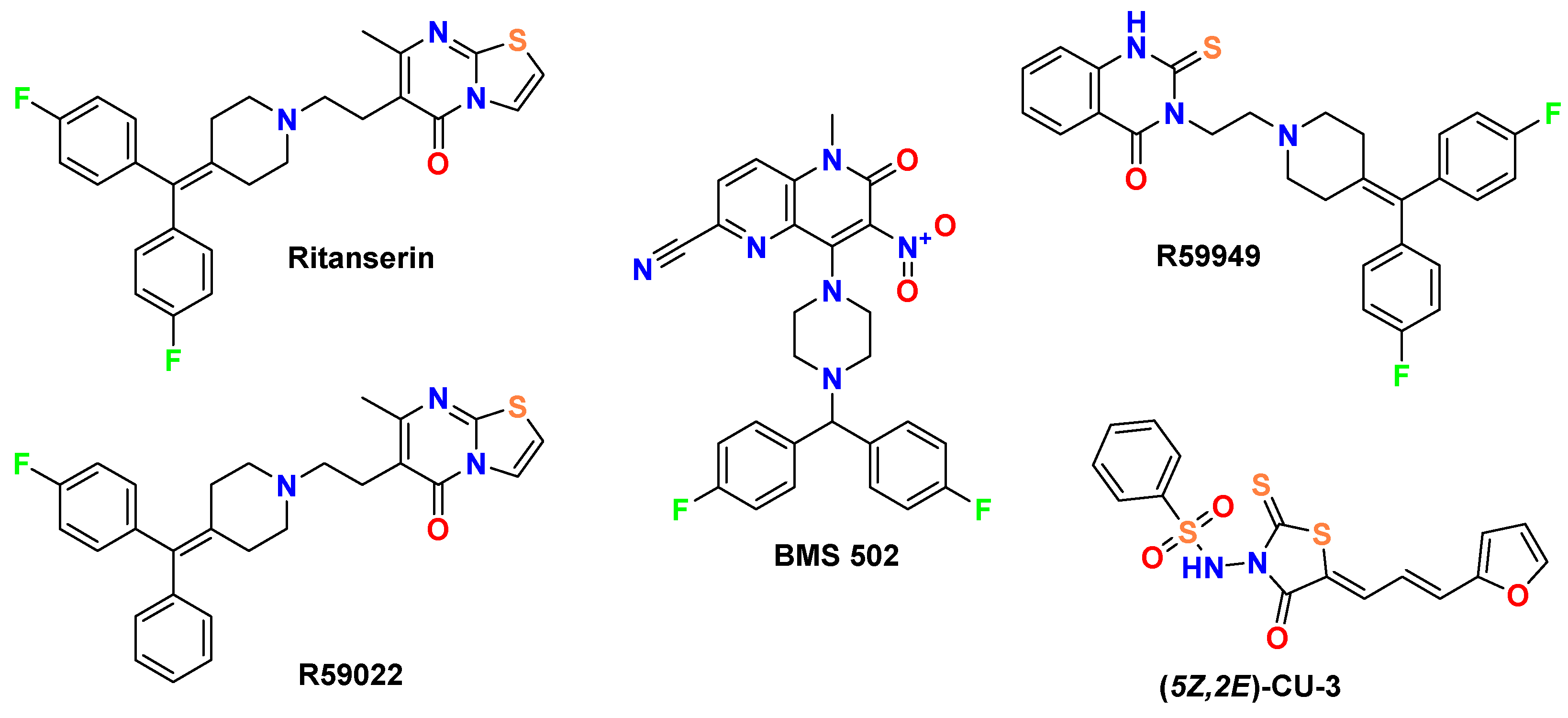
Figure 2.
Chemical structures and design strategy of investigated diacylglycerol kinase α (DGKα) modulators. Previously patented [1,2,4]triazolo [4,3-a]quinazolin-5-amine derivatives that served as inspiration for the design approach. And novel 1-methyl/4-(tert-butyl)-2’-(cycloalkyl/hetaryl)-6’H-spiro[piperidine/cycloalkane-4,5’/1,5’-[1,2,4]triazolo [1,5-c]quinazolines] (1-40) proposed and evaluated in this study, illustrating key structural modifications: varying spiro ring sizes (n = 1, 2, 3), cyclohexyl vs. piperidine scaffolds, and diverse heterocyclic substituents strategically positioned to enhance binding interactions with DGKα.
Figure 2.
Chemical structures and design strategy of investigated diacylglycerol kinase α (DGKα) modulators. Previously patented [1,2,4]triazolo [4,3-a]quinazolin-5-amine derivatives that served as inspiration for the design approach. And novel 1-methyl/4-(tert-butyl)-2’-(cycloalkyl/hetaryl)-6’H-spiro[piperidine/cycloalkane-4,5’/1,5’-[1,2,4]triazolo [1,5-c]quinazolines] (1-40) proposed and evaluated in this study, illustrating key structural modifications: varying spiro ring sizes (n = 1, 2, 3), cyclohexyl vs. piperidine scaffolds, and diverse heterocyclic substituents strategically positioned to enhance binding interactions with DGKα.
Figure 3.
Comparative analysis of binding affinities across multiple DGKα binding sites. The graph displays Vina scores (kcal/mol) for seven top-performing [1,2,4]triazolo [1,5-c]quinazoline derivatives (compounds 13, 18, 22, 28, 33, 35, and 40) compared against established reference inhibitors (ritanserin, R59022, R59949, BMS502, and (5Z,2E)-CU-3). For each compound, the two highest affinity scores found across different binding cavities are shown, highlighting their preferred binding sites. The 127 ų and 179 ų cavities emerge as particularly favorable binding regions for most compounds, while the 146 ų cavity showed consistently lower binding affinity and is therefore not represented. More negative Vina scores indicate stronger predicted binding, with scores below -8.0 kcal/mol suggesting high potential for biological activity. This comparison reveals that several novel compounds (particularly 18, 28, and 33) demonstrate binding affinities comparable to or exceeding those of established DGK inhibitors in specific cavities, supporting their potential as promising candidates for experimental validation and further optimization.
Figure 3.
Comparative analysis of binding affinities across multiple DGKα binding sites. The graph displays Vina scores (kcal/mol) for seven top-performing [1,2,4]triazolo [1,5-c]quinazoline derivatives (compounds 13, 18, 22, 28, 33, 35, and 40) compared against established reference inhibitors (ritanserin, R59022, R59949, BMS502, and (5Z,2E)-CU-3). For each compound, the two highest affinity scores found across different binding cavities are shown, highlighting their preferred binding sites. The 127 ų and 179 ų cavities emerge as particularly favorable binding regions for most compounds, while the 146 ų cavity showed consistently lower binding affinity and is therefore not represented. More negative Vina scores indicate stronger predicted binding, with scores below -8.0 kcal/mol suggesting high potential for biological activity. This comparison reveals that several novel compounds (particularly 18, 28, and 33) demonstrate binding affinities comparable to or exceeding those of established DGK inhibitors in specific cavities, supporting their potential as promising candidates for experimental validation and further optimization.
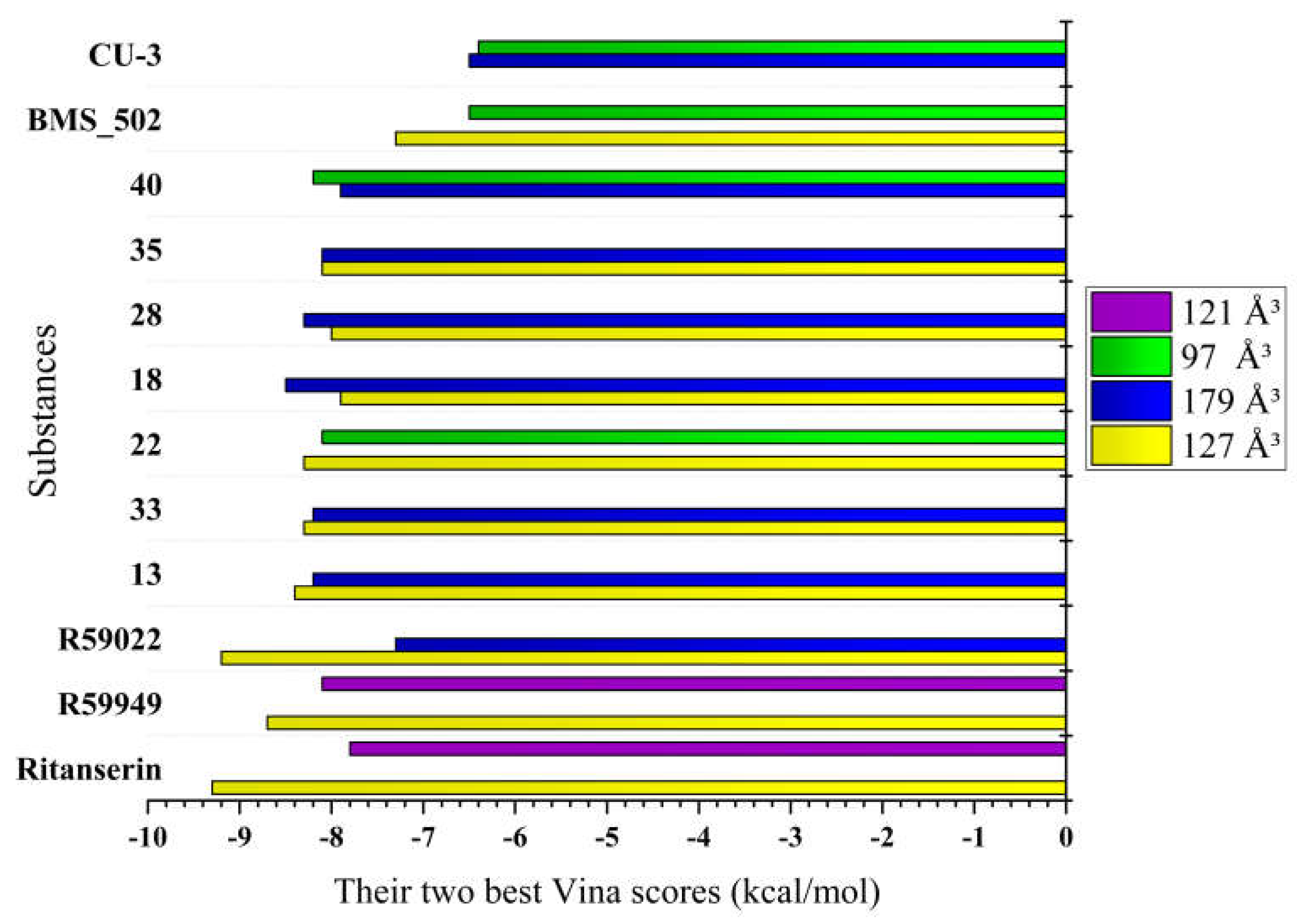
Figure 4.
Comparative binding pose analysis of reference inhibitors and top-performing novel compounds in three distinct DGKα binding cavities. The figure illustrates molecular docking results with key interacting residues labeled and colored according to interaction type. (A) Ritanserin versus compound 3 in the 127 ų cavity, highlighting shared interactions with TRP151 and distinct binding modes at LEU156 and ALA146. (B) R59949 versus compound 18 (yellow) in the 179 ų cavity, demonstrating how compound 18 achieves superior binding through stronger hydrogen bonding with SER132. (C) R59949 versus compound 40 in the 97 ų cavity, showing the enhanced GLU166 interaction of compound 40 that contributes to its improved binding affinity. Oxygen atoms are shown in red, nitrogen in blue, sulfur in yellow, and fluorine in cyan. Hydrogen bonds are represented as green dashed lines, π-interactions as purple dashed lines, and hydrophobic interactions as pink dashed lines.
Figure 4.
Comparative binding pose analysis of reference inhibitors and top-performing novel compounds in three distinct DGKα binding cavities. The figure illustrates molecular docking results with key interacting residues labeled and colored according to interaction type. (A) Ritanserin versus compound 3 in the 127 ų cavity, highlighting shared interactions with TRP151 and distinct binding modes at LEU156 and ALA146. (B) R59949 versus compound 18 (yellow) in the 179 ų cavity, demonstrating how compound 18 achieves superior binding through stronger hydrogen bonding with SER132. (C) R59949 versus compound 40 in the 97 ų cavity, showing the enhanced GLU166 interaction of compound 40 that contributes to its improved binding affinity. Oxygen atoms are shown in red, nitrogen in blue, sulfur in yellow, and fluorine in cyan. Hydrogen bonds are represented as green dashed lines, π-interactions as purple dashed lines, and hydrophobic interactions as pink dashed lines.
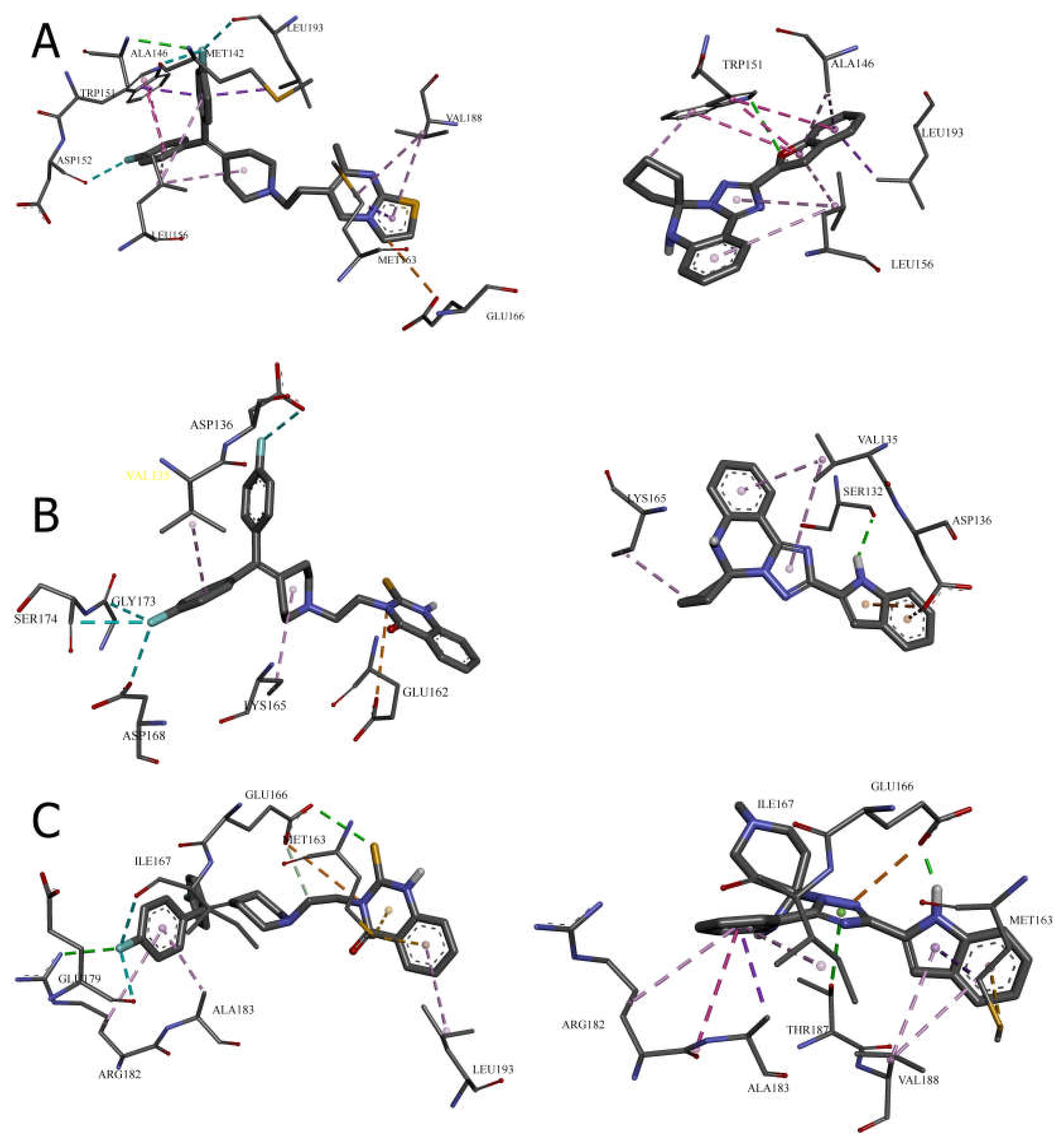
Figure 5.
Molecular docking analysis comparing binding poses of reference inhibitors and top-performing novel compounds in the remaining two DGKα binding cavities. (A) R59949 versus compound 28 in the 121 ų cavity, illustrating how compound 28 establishes a conventional hydrogen bond with GLN141 and forms π-cation interaction with ARG144, creating a binding mode distinct from R59949’s tyrosine-centered interactions. (B) R59022 versus compound 33 in the 146 ų cavity, demonstrating compound 33’s superior binding through optimal hydrogen bonding with ARG126 and extensive hydrophobic interactions with LYS120. Key atomic features are colored consistently (oxygen in red, nitrogen in blue, sulfur in yellow, fluorine in cyan), and critical interactions are represented as dashed lines (hydrogen bonds in green, π-interactions in purple, hydrophobic interactions in pink).
Figure 5.
Molecular docking analysis comparing binding poses of reference inhibitors and top-performing novel compounds in the remaining two DGKα binding cavities. (A) R59949 versus compound 28 in the 121 ų cavity, illustrating how compound 28 establishes a conventional hydrogen bond with GLN141 and forms π-cation interaction with ARG144, creating a binding mode distinct from R59949’s tyrosine-centered interactions. (B) R59022 versus compound 33 in the 146 ų cavity, demonstrating compound 33’s superior binding through optimal hydrogen bonding with ARG126 and extensive hydrophobic interactions with LYS120. Key atomic features are colored consistently (oxygen in red, nitrogen in blue, sulfur in yellow, fluorine in cyan), and critical interactions are represented as dashed lines (hydrogen bonds in green, π-interactions in purple, hydrophobic interactions in pink).
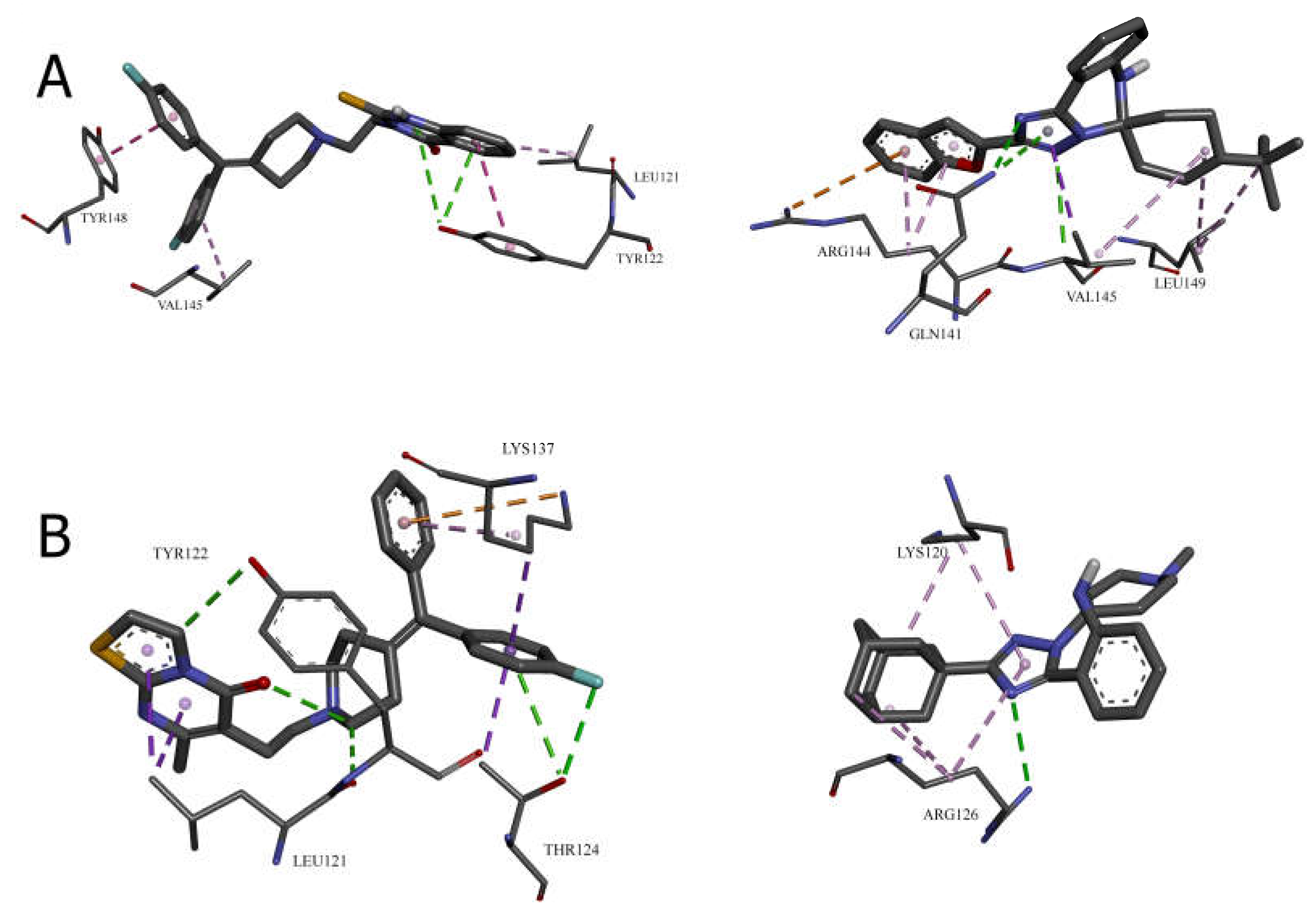
Figure 6.
Structure-property relationships of novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives and reference DGK inhibitors. The scatter plot illustrates the relationship between binding affinity (kcal/mol) and lipophilicity (consensus logP), with point size representing topological polar surface area (TPSA). High-affinity compounds (13, 18, 22, 28, 33, and 40, highlighted in green) demonstrate balanced physicochemical profiles compared to reference inhibitors (orange). Blue shades for other compounds based on TPSA values. The vertical dashed line indicates optimal logP range (3.5) for drug-like molecules, while the horizontal line at -8.0 kcal/mol indicates the threshold for strong binding affinity.
Figure 6.
Structure-property relationships of novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives and reference DGK inhibitors. The scatter plot illustrates the relationship between binding affinity (kcal/mol) and lipophilicity (consensus logP), with point size representing topological polar surface area (TPSA). High-affinity compounds (13, 18, 22, 28, 33, and 40, highlighted in green) demonstrate balanced physicochemical profiles compared to reference inhibitors (orange). Blue shades for other compounds based on TPSA values. The vertical dashed line indicates optimal logP range (3.5) for drug-like molecules, while the horizontal line at -8.0 kcal/mol indicates the threshold for strong binding affinity.
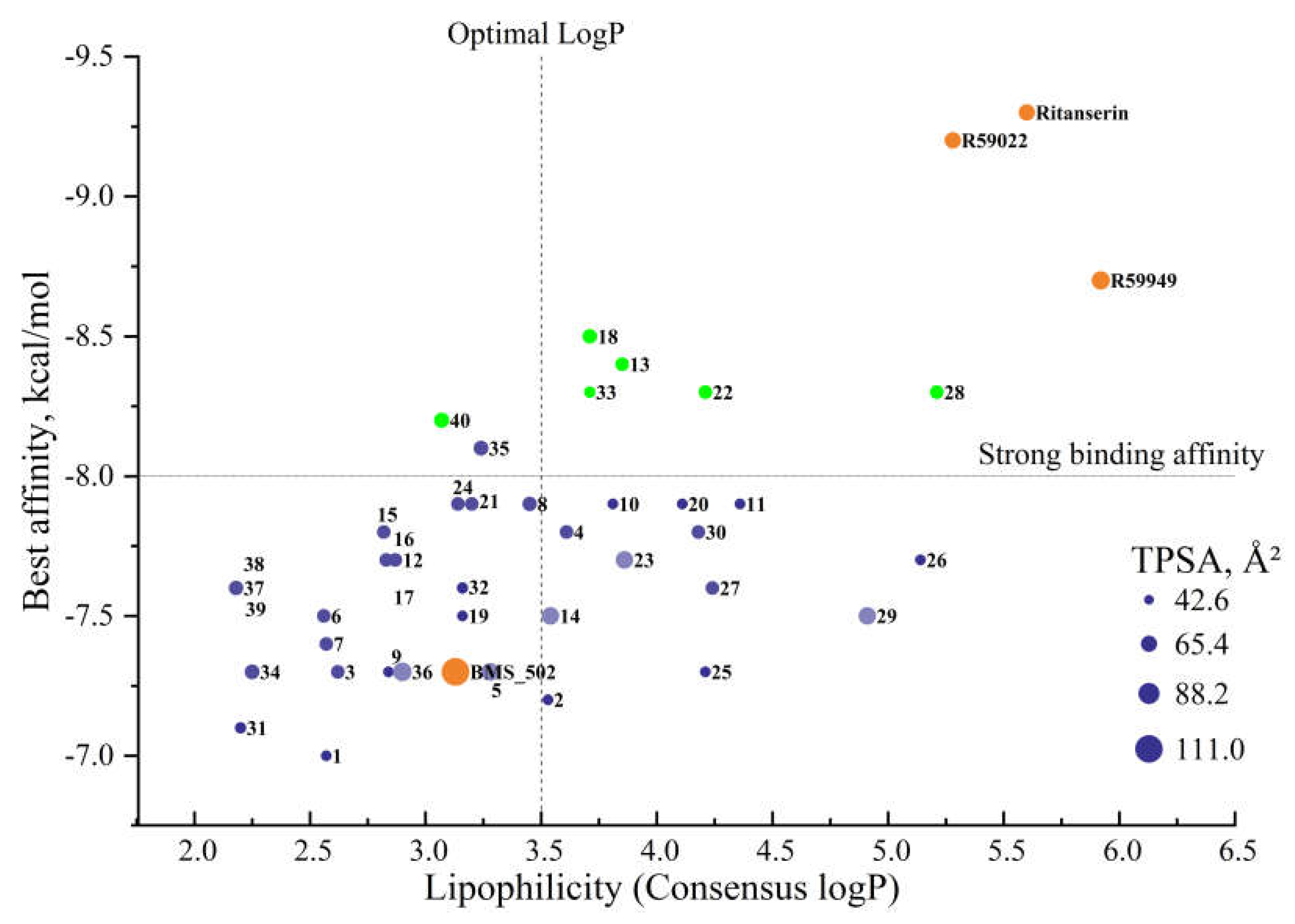
Figure 7.
Comparative pharmacokinetic (P-gp substrate status, blood-brain barrier penetration, and CYP inhibition profiles) alongside with ESOL solubility profiles of novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives and reference DGK inhibitors (BMS502, R59022, Ritanserin, and R59949).
Figure 7.
Comparative pharmacokinetic (P-gp substrate status, blood-brain barrier penetration, and CYP inhibition profiles) alongside with ESOL solubility profiles of novel spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives and reference DGK inhibitors (BMS502, R59022, Ritanserin, and R59949).
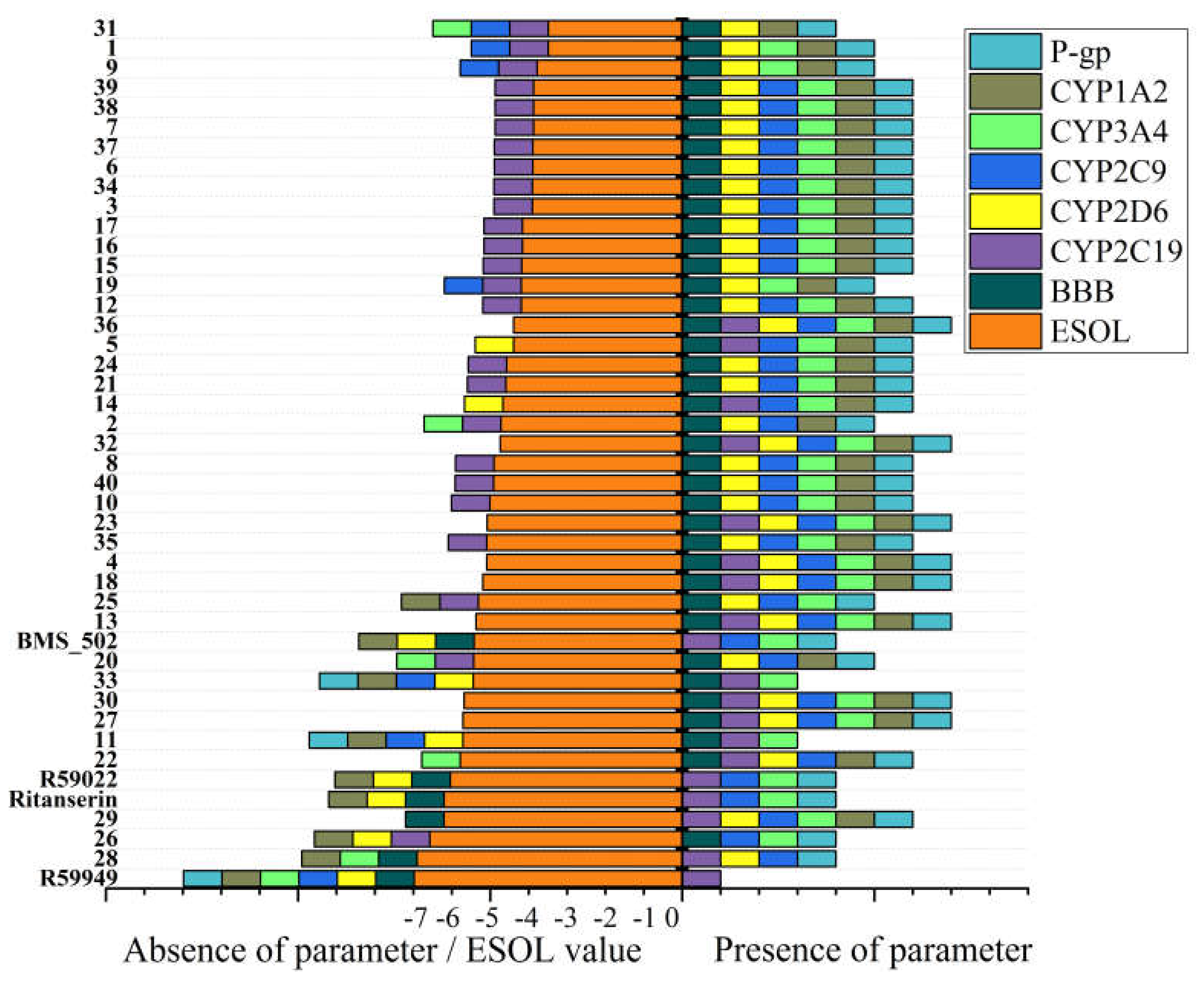
Figure 8.
Structure-activity relationships of spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives as DGKα modulators: key structural features and binding determinants.
Figure 8.
Structure-activity relationships of spiro [1,2,4]triazolo [1,5-c]quinazoline derivatives as DGKα modulators: key structural features and binding determinants.


Table 1.
Comparative analysis of binding cavity properties and affinity scores (kcal/mol) of reference DGKα inhibitors across five distinct binding sites in the DGKα structure (RCSB PDB ID: 6IIE).
Table 1.
Comparative analysis of binding cavity properties and affinity scores (kcal/mol) of reference DGKα inhibitors across five distinct binding sites in the DGKα structure (RCSB PDB ID: 6IIE).
| Volume, ų | Center (x, y, z) | Docking size (x, y, z) |
Ritanserin | R59022 | R59949 | BMS502 | (5Z,2E)-CU-3 |
|---|---|---|---|---|---|---|---|
| 127 | -27.971, 12.861, -7.859 | 21, 21, 21 | -9.3 | -9.2 | -8.7 | -7.3 | -6.3 |
| 179 | -16.001, 4.140, 1.890 | 21, 21, 21 | -7.1 | -7.3 | -7.7 | -6.4 | -6.5 |
| 97 | -12.904, 16.082, -0.064 | 21, 21, 21 | -7.5 | -7.3 | -7.6 | -6.5 | -6.4 |
| 121 | -29.580, 15.955, 7.175 | 21, 21, 21 | -7.8 | -6.8 | -8.1 | -6.2 | -5.7 |
| 146 | -21.667, 10.869, 16.559 | 21, 21, 21 | -6.9 | -7.1 | -6.9 | -6.2 | -6.2 |
* Cavity volumes are reported in cubic Angstroms (ų) and represent the accessible volume of each binding pocket as calculated by the CB-Dock2 algorithm. Center (x, y, z) coordinates define the geometric center of each cavity in the protein structure coordinate system (RCSB PDB ID: 6IIE). Docking size (x, y, z) parameters represent the dimensions of the search box used for molecular docking in each direction, optimized to encompass the entire binding pocket. Vina scores are reported in kcal/mol, with more negative values indicating stronger predicted binding affinity. Typical interpretations consider scores below -8.0 kcal/mol as strong binding, -6.5 to -8.0 kcal/mol as moderate binding, and above -6.5 kcal/mol as weak binding. Cavities are ranked in order of the sum of binding affinities across all reference compounds, with the 127 ų cavity showing the strongest overall binding potential, suggesting it may represent the primary inhibitor binding site. The reference compounds represent structurally diverse DGK inhibitors, allowing assessment of binding pocket preferences across different chemical scaffolds.
Table 2.
Binding affinity analysis of novel [1,2,4]triazolo [1,5-c]quinazoline derivatives and reference DGKα inhibitors across five distinct binding cavities, ranked by binding strength.
Table 2.
Binding affinity analysis of novel [1,2,4]triazolo [1,5-c]quinazoline derivatives and reference DGKα inhibitors across five distinct binding cavities, ranked by binding strength.
| Cavity volume / Substance / Vina score, kcal/mol | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 127 ų | 179 ų | 97 ų | 121 ų | 146 ų | |||||
| Ritans. | -9.3 | 18 | -8.5 | 40 | -8.2 | R59949 | -8.1 | 33 | -7.5 |
| R59022 | -9.2 | 28 | -8.3 | 22 | -8.1 | Ritans. | -7.8 | 11 | -7.3 |
| R59949 | -8.7 | 13 | -8.2 | 8 | -7.9 | 28 | -7.2 | 28 | -7.3 |
| 13 | -8.4 | 33 | -8.2 | 18 | -7.9 | 33 | -7.2 | 22 | -7.1 |
| 22 | -8.3 | 35 | -8.1 | 28 | -7.8 | 40 | -7.1 | R59022 | -7.1 |
| 33 | -8.3 | 10 | -7.9 | 35 | -7.8 | 11 | -7.0 | 18 | -6.9 |
| 35 | -8.1 | 11 | -7.9 | 30 | -7.7 | 13 | -6.9 | Ritans. | -6.9 |
| 28 | -8.0 | 22 | -7.9 | R59949 | -7.6 | 35 | -6.8 | R59949 | -6.9 |
| 11 | -7.9 | 40 | -7.9 | 4 | -7.5 | R59022 | -6.8 | 4 | -6.8 |
| 18 | -7.9 | 4 | -7.8 | 13 | -7.5 | 30 | -6.7 | 13 | -6.8 |
| 20 | -7.9 | 8 | -7.8 | Ritans. | -7.5 | 18 | -6.6 | 35 | -6.8 |
| 21 | -7.9 | 15 | -7.8 | 29 | -7.4 | 22 | -6.6 | 40 | -6.8 |
| 24 | -7.9 | 30 | -7.8 | 24 | -7.3 | 26 | -6.6 | 8 | -6.7 |
| 10 | -7.8 | 12 | -7.7 | 27 | -7.3 | 4 | -6.4 | 24 | -6.6 |
| 40 | -7.8 | 16 | -7.7 | R59022 | -7.3 | 8 | -6.4 | 26 | -6.6 |
| 23 | -7.7 | 17 | -7.7 | 33 | -7.2 | 10 | -6.3 | 30 | -6.6 |
| 26 | -7.7 | R59949 | -7.7 | 11 | -7.1 | 29 | -6.3 | 27 | -6.5 |
| 4 | -7.6 | 20 | -7.6 | 15 | -7.1 | 24 | -6.2 | 29 | -6.5 |
| 8 | -7.6 | 26 | -7.6 | 21 | -7.1 | 32 | -6.2 | 32 | -6.5 |
| 32 | -7.6 | 27 | -7.6 | 25 | -7.0 | BMS502 | -6.2 | 15 | -6.3 |
| 37 | -7.6 | 37 | -7.6 | 37 | -7 | 2 | -6.1 | 20 | -6.3 |
| 6 | -7.5 | 38 | -7.6 | 12 | -6.9 | 16 | -6 | 23 | -6.3 |
| 19 | -7.5 | 39 | -7.6 | 26 | -6.9 | 27 | -6 | 16 | -6.2 |
| 7 | -7.4 | 14 | -7.5 | 31 | -6.9 | 38 | -6 | 17 | -6.2 |
| 15 | -7.4 | 29 | -7.5 | 34 | -6.9 | 15 | -5.9 | 21 | -6.2 |
| 30 | -7.4 | 6 | -7.4 | 16 | -6.8 | 17 | -5.9 | BMS502 | -6.2 |
| 27 | -7.3 | 7 | -7.4 | 3 | -6.6 | 20 | -5.9 | CU-3 | -6.2 |
| 29 | -7.3 | 19 | -7.4 | 6 | -6.6 | 37 | -5.9 | 6 | -6.1 |
| BMS502 | -7.3 | 24 | -7.4 | 7 | -6.6 | 39 | -5.9 | 10 | -6.1 |
| 16 | -7.2 | 32 | -7.4 | 9 | -6.6 | 7 | -5.8 | 12 | -6.1 |
| 17 | -7.2 | 3 | -7.3 | 14 | -6.6 | 34 | -5.8 | 37 | -6.1 |
| 3 | -7.1 | 5 | -7.3 | 17 | -6.6 | 6 | -5.7 | 38 | -6.1 |
| 5 | -7.1 | 9 | -7.3 | 23 | -6.6 | 21 | -5.7 | 39 | -6.1 |
| 12 | -7.1 | 21 | -7.3 | 38 | -6.6 | 25 | -5.7 | 14 | -6 |
| 25 | -7.1 | 25 | -7.3 | 10 | -6.5 | 36 | -5.7 | 25 | -5.9 |
| 38 | -7.1 | 34 | -7.3 | 36 | -6.5 | CU-3 | -5.7 | 31 | -5.9 |
| 1 | -7.0 | 36 | -7.3 | 39 | -6.5 | 9 | -5.6 | 34 | -5.9 |
| 2 | -7.0 | R59022 | -7.3 | BMS502 | -6.5 | 12 | -5.6 | 2 | -5.8 |
| 14 | -7 | 2 | -7.2 | 1 | -6.4 | 14 | -5.6 | 7 | -5.8 |
| 39 | -7 | 31 | -7.1 | 20 | -6.4 | 19 | -5.6 | 19 | -5.8 |
| 34 | -6.9 | Ritans. | -7.1 | CU-3 | -6.4 | 23 | -5.6 | 36 | -5.8 |
| 9 | -6.8 | 1 | -7.0 | 5 | -6.3 | 31 | -5.5 | 1 | -5.7 |
| 36 | -6.7 | 23 | -7.0 | 19 | -6.3 | 5 | -5.4 | 3 | -5.7 |
| 31 | -6.6 | CU-3 | -6.5 | 32 | -6.2 | 1 | -5.2 | 5 | -5.7 |
| CU-3 | -6.3 | BMS502 | -6.4 | 2 | -6 | 3 | -5.2 | 9 | -5.6 |
* Compounds are presented in descending order of binding affinity (Vina score) within each cavity, allowing direct comparison of relative binding potency across all tested compounds. Reference compounds: Ritanserin, R59022, R59949, BMS502, and (5Z,2E)-CU-3. Vina scores are reported in kcal/mol with more negative values indicating stronger predicted binding affinity. Scores below -8.0 kcal/mol represent compounds with particularly strong binding potential. The highest scoring compounds in each cavity (e.g., ritanserin in 127 ų, 18 in 179 ų, 40 in 97 ų, R59949 in 121 ų, and 33 in 146 ų) represent lead candidates for targeting these specific binding sites. Consistent high-scoring compounds across multiple cavities (e.g., 13, 22, 28, 33, and 35) suggest molecular features that enable adaptable binding to different protein environments. Reference inhibitors (particularly ritanserin and R59022) establish performance benchmarks, with novel compounds scoring within 1.0 kcal/mol of these references considered to have comparable predicted binding potency. The 127 ų cavity shows the highest overall binding scores, particularly for reference compounds, suggesting it may represent the primary inhibitory binding site for DGK modulators. Some compounds (e.g., 18, 40) outperform reference inhibitors in specific cavities, indicating their potential as selective DGK modulators targeting these distinct binding regions.
Table 3.
Detailed molecular interaction analysis of top-performing reference compounds and novel [1,2,4]triazolo [1,5-c]quinazoline derivatives in each DGKα binding cavity, characterizing interaction types, distances, and specific amino acid residues involved in stabilizing ligand-protein complexes.
Table 3.
Detailed molecular interaction analysis of top-performing reference compounds and novel [1,2,4]triazolo [1,5-c]quinazoline derivatives in each DGKα binding cavity, characterizing interaction types, distances, and specific amino acid residues involved in stabilizing ligand-protein complexes.
| Amino acid residue | Distance, ų | Bond category | Bond type |
|---|---|---|---|
| Ritanserin in cavity 127 ų | |||
| GLU166 | 5.29773 | Electrostatic | Attractive Charge |
| ALA146 | 3.65885 |
Hydrogen Bond; Halogen |
Conventional Hydrogen Bond; Halogen (Fluorine) |
| MET142 | 3.19895 | Halogen | Halogen (Fluorine) |
| ASP152 | 3.68042 | Halogen | Halogen (Fluorine) |
| LEU193 | 2.63495 | Halogen | Halogen (Fluorine) |
| ALA146 | 3.9193 | Hydrophobic | Pi-Sigma |
| MET163 | 3.94403 | Hydrophobic | Pi-Sigma |
| LEU193 | 3.93009 | Hydrophobic | Pi-Sigma |
| MET163 | 3.77279 | Other | Pi-Sulfur |
| TRP151 | 4.06681 | Hydrophobic | Pi-Pi Stacked |
| TRP151 | 4.54522 | Hydrophobic | Pi-Pi Stacked |
| LEU156 | 5.35624 | Hydrophobic | Alkyl |
| LEU156 | 5.08272 | Hydrophobic | Pi-Alkyl |
| LEU156 | 5.06585 | Hydrophobic | Pi-Alkyl |
| VAL188 | 4.92692 | Hydrophobic | Pi-Alkyl |
| VAL188 | 4.81405 | Hydrophobic | Pi-Alkyl |
| Substance 3 in cavity 127 ų | |||
| TRP151 | 3.05971 | Hydrogen Bond | Conventional Hydrogen Bond |
| LEU193 | 3.8508 | Hydrophobic | Pi-Sigma |
| TRP151 | 3.94194 | Hydrophobic | Pi-Pi Stacked |
| TRP151 | 5.00543 | Hydrophobic | Pi-Pi Stacked |
| TRP151 | 5.31489 | Hydrophobic | Pi-Pi Stacked |
| TRP151 | 4.69541 | Hydrophobic | Pi-Alkyl |
| LEU156 | 5.44551 | Hydrophobic | Pi-Alkyl |
| LEU156 | 5.2451 | Hydrophobic | Pi-Alkyl |
| ALA146 | 4.39575 | Hydrophobic | Pi-Alkyl |
| LEU156 | 5.32117 | Hydrophobic | Pi-Alkyl |
| ALA146 | 4.32019 | Hydrophobic | Pi-Alkyl |
| R59949 in cavity 179 ų | |||
| GLU162 | 4.64611 | Electrostatic | Attractive Charge |
| ASP136 | 3.18667 | Halogen | Halogen (Fluorine) |
| ASP168 | 3.5015 | Halogen | Halogen (Fluorine) |
| GLY173 | 3.68482 | Halogen | Halogen (Fluorine) |
| SER174 | 3.40189 | Halogen | Halogen (Fluorine) |
| LYS165 | 4.83406 | Hydrophobic | Alkyl |
| VAL135 | 4.64342 | Hydrophobic | Pi-Alkyl |
| Substance 18 in cavity 179 ų | |||
| SER132 | 2.12242 | Hydrogen Bond | Conventional Hydrogen Bond |
| ASP136 | 3.67142 | Electrostatic | Pi-Anion |
| ASP136 | 3.69271 | Electrostatic | Pi-Anion |
| LYS165 | 4.12278 | Hydrophobic | Alkyl |
| VAL135 | 4.17437 | Hydrophobic | Pi-Alkyl |
| VAL135 | 4.98862 | Hydrophobic | Pi-Alkyl |
| R59949 in cavity 97 ų | |||
| GLU166 | 4.80272 | Electrostatic | Attractive Charge |
| ARG182 | 3.12515 |
Hydrogen Bond; Halogen |
Conventional Hydrogen Bond; Halogen (Fluorine) |
| GLU166 | 3.7357 | Hydrogen Bond | Conventional Hydrogen Bond |
| GLU166 | 3.22464 | Hydrogen Bond | Carbon Hydrogen Bond |
| ILE167 | 3.31873 | Halogen | Halogen (Fluorine) |
| GLU179 | 3.25959 | Halogen | Halogen (Fluorine) |
| MET163 | 3.82172 | Other | Pi-Sulfur |
| MET163 | 3.91728 | Other | Pi-Sulfur |
| LEU193 | 5.31213 | Hydrophobic | Pi-Alkyl |
| ARG182 | 5.20158 | Hydrophobic | Pi-Alkyl |
| ALA183 | 4.29659 | Hydrophobic | Pi-Alkyl |
| Substance 40 in cavity 97 ų | |||
| GLU166 | 1.85385 | Hydrogen Bond | Conventional Hydrogen Bond |
| GLU166 | 3.92296 | Electrostatic | Pi-Anion |
| THR187 | 3.27552 | Hydrogen Bond | Pi-Donor Hydrogen Bond |
| MET163 | 3.86247 | Hydrophobic | Pi-Sigma |
| ALA183 | 3.7804 | Hydrophobic | Pi-Sigma |
| MET163 | 4.04032 | Other | Pi-Sulfur |
| ARG182, ALA183 | 4.76159 | Hydrophobic | Amide-Pi Stacked |
| ILE167 | 5.38614 | Hydrophobic | Pi-Alkyl |
| ARG182 | 4.85254 | Hydrophobic | Pi-Alkyl |
| VAL188 | 4.54636 | Hydrophobic | Pi-Alkyl |
| VAL188 | 5.13921 | Hydrophobic | Pi-Alkyl |
| R59949 in cavity 121 ų | |||
| TYR122 | 4.03824 | Hydrogen Bond | Pi-Donor Hydrogen Bond |
| TYR122 | 3.51351 | Hydrogen Bond | Pi-Donor Hydrogen Bond |
| TYR122 | 4.19688 | Hydrophobic | Pi-Pi Stacked |
| TYR148 | 3.86497 | Hydrophobic | Pi-Pi Stacked |
| LEU121 | 5.26796 | Hydrophobic | Pi-Alkyl |
| VAL145 | 4.71276 | Hydrophobic | Pi-Alkyl |
| Substance 28 in cavity 121 ų | |||
| GLN141 | 3.15959 | Hydrogen Bond | Conventional Hydrogen Bond |
| VAL145 | 3.68904 | Hydrogen Bond | Carbon Hydrogen Bond |
| ARG144 | 4.65278 | Electrostatic | Pi-Cation |
| GLN141 | 3.87404 | Hydrogen Bond | Pi-Donor Hydrogen Bond |
| VAL145 | 3.69747 | Hydrophobic | Pi-Sigma |
| VAL145 | 5.03024 | Hydrophobic | Alkyl |
| LEU149 | 5.09337 | Hydrophobic | Alkyl |
| LEU149 | 5.04472 | Hydrophobic | Alkyl |
| ARG144 | 3.97846 | Hydrophobic | Pi-Alkyl |
| ARG144 | 4.07433 | Hydrophobic | Pi-Alkyl |
| R59022 in cavity 146 ų | |||
| THR124 | 2.99368 |
Hydrogen Bond; Halogen |
Conventional Hydrogen Bond; Halogen (Fluorine) |
| LEU121 | 3.16241 | Hydrogen Bond | Carbon Hydrogen Bond |
| intermolecular | 3.25258 | Hydrogen Bond | Carbon Hydrogen Bond |
| TYR122 | 3.48925 | Hydrogen Bond | Carbon Hydrogen Bond |
| LYS137 | 4.93829 | Electrostatic | Pi-Cation |
| THR124 | 4.06017 | Hydrogen Bond | Pi-Donor Hydrogen Bond |
| LEU121 | 3.60325 | Hydrophobic | Pi-Sigma |
| LEU121 | 3.94699 | Hydrophobic | Pi-Sigma |
| THR124 | 3.40756 | Hydrophobic | Pi-Sigma |
| LYS137 | 3.22303 | Hydrophobic | Pi-Sigma |
| LYS137 | 4.00354 | Hydrophobic | Pi-Alkyl |
| Substance 33 in cavity 146 ų | |||
| ARG126 | 3.19588 | Hydrogen Bond | Conventional Hydrogen Bond |
| LYS120 | 3.8771 | Hydrophobic | Alkyl |
| ARG126 | 4.81701 | Hydrophobic | Alkyl |
| ARG126 | 4.62315 | Hydrophobic | Alkyl |
| LYS120 | 4.5638 | Hydrophobic | Pi-Alkyl |
| ARG126 | 4.95772 | Hydrophobic | Pi-Alkyl |
* Bond distances are reported in Angstroms (Å) and represent the shortest atomic distance between the interacting groups. Smaller distances generally indicate stronger interactions, particularly for hydrogen bonds where distances below 3.0 Å typically represent strong binding. Electrostatic interactions include attractive charges (between oppositely charged groups) and π-anion/π-cation interactions (between aromatic systems and charged residues). These interactions can contribute significantly to binding affinity, especially in polar binding pockets. Hydrogen bonds are classified as conventional (between typical hydrogen bond donors and acceptors), carbon hydrogen bonds (weaker interactions involving C-H as donors), and π-donor hydrogen bonds (where aromatic systems serve as acceptors). Conventional hydrogen bonds with distances under 3.0 Å provide substantial binding energy. Halogen interactions involve fluorine atoms functioning as weak hydrogen bond acceptors or participating in halogen bonding. Though individually weaker than hydrogen bonds, multiple halogen interactions can collectively contribute significant binding energy. Hydrophobic interactions include alkyl (aliphatic-aliphatic), π-alkyl (aromatic-aliphatic), π-sigma, and π-π stacked (aromatic-aromatic) contacts. These non-polar interactions are particularly important in binding pockets with hydrophobic characteristics. Pi-sulfur interactions involve the interaction between aromatic π-systems and sulfur-containing residues (typically methionine), representing a specialized type of non-covalent interaction that can significantly enhance binding stability.
Table 4.
Drug-likeness assessment according to common medicinal chemistry filters.
| Sub | Lipinski | Ghose | Veber | Egan | Muegge |
|---|---|---|---|---|---|
| 1-10, 12-19, 21, 23, 24, 31-40 | Yes; 0 violation | Yes | Yes | Yes | Yes |
| 11, 25, 29 | Yes; 1 violation: MLOGP>4.15 | No; 1 violation: XLOGP3>5 | |||
| 20, 22, 27, 30 | Yes; 0 violation | No; 1 violation: XLOGP3>5 | |||
| 26 | Yes; 1 violation: MLOGP>4.15 | No; 1 violation: WLOGP>5.6 | No; 1 violation: XLOGP3>5 | ||
| 28 | Yes; 1 violation: MLOGP>4.15 | No; 1 violation: WLOGP>5.6 | No; 1 violation: WLOGP>5.88 | No; 1 violation: XLOGP3>5 | |
| BMS502 | Yes; 1 violation: MW>500 | No; 2 violations: MW>480, MR>130 | Yes | ||
| R59022 | Yes; 1 violation: MLOGP>4.15 | No; 1 violation: MR>130 | Yes | No; 1 violation: XLOGP3>5 | |
| R59949 | Yes; 1 violation: MLOGP>4.1 | No; 3 violations: MW>480, WLOGP>5.6, MR>130 | Yes | No; 1 violation: WLOGP>5.88 | No; 1 violation: XLOGP3>5 |
| Ritanserin | Yes; 1 violation: MLOGP>4.15 | No; 2 violations: WLOGP>5.6, MR>130 | Yes | No; 1 violation: WLOGP>5.88 | No; 1 violation: XLOGP3>5 |
* “Yes” indicates the compound passes all criteria for the respective filter, while “No” indicates violation. The specific violated parameter is indicated after each violation. MLOGP, WLOGP, and XLOGP3 represent different computational methods for calculating the compound’s octanol-water partition coefficient (logP), a measure of lipophilicity. MW = Molecular Weight; MR = Molar Refractivity. Compounds with 0-1 violations of Lipinski’s Rule of Five are considered to have favorable drug-like properties.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



