
Submitted:
12 October 2024
Posted:
15 October 2024
You are already at the latest version
Abstract
Febrile seizures (FSs) are defined as seizures occurring in children aged 6 months to 5 years with a background of elevated body temperature. It is one of the most common neurological disorders of childhood, emphasizing the importance of understanding the causes of FSs and their impact on the developing nervous system. However, there are significant limitations to the technologies currently available for studying the etiology and pathophysiology of seizures in humans. It is currently not possible to adequately capture the subtle molecular and structural rearrangements of the nervous system that can occur after seizures in humans. The use of animal models can be invaluable for these purposes. The most commonly used models in modern research are hyperthermic models in rats and mice aged 10-12 days. While these models can reproduce many of the characteristics of FSs, they have certain limitations. This review outlines the key considerations when working with models of FSs, provides an overview of current approaches to producing seizures in different model subjects, and presents a summary of key findings regarding morphological and functional changes in the brain, as well as behavioral alterations, that have been identified in studies using animal models of FSs.
Keywords:
epilepsy
; rat
; hippocampus
; animal model
; hyperthermia
1. Introduction
Febrile seizures (FSs) are a common neurological disorder affecting children between the ages of 6 months and 5 years, with a peak incidence during the second year of life [1]. The precise mechanism underlying FSs remains unclear. It presents when body temperature exceeds 38°C in the absence of a discernible inciting factor, such as trauma, infection, or intoxication of the central nervous system [1]. Epidemiological studies indicate that prevalence varies by geographical area, with rates ranging from 2 to 5% in the USA and Europe to 8 to 11% in East Asia [1,2,3,4]. There is still no consensus on whether FS causes brain damage and the development of neurological disorders later in life, or whether there was some brain dysfunction initially that caused the development of FS. The precise relationship between FSs and the subsequent development of a number of neurological and psychiatric disorders, including temporal lobe epilepsy, schizophrenia, anxiety disorders, and attention deficit hyperactivity disorder, remains unclear [5,6,7,8]. A substantial body of scientific literature is produced annually on this topic, encompassing both experimental studies and clinical data. For an up-to-date review of the effects of seizures, please refer to the recent paper by Yi et al. (2023) [9].
It is crucial for clinical practice to have a solid understanding of the mechanisms underlying seizures. However, it is not possible to fully investigate the underlying causes of seizure development, their associated consequences, and to pursue a means to anticipate and avert the onset of seizures in humans. The main reason for this is that FSs typically emerge abruptly and seldom manifest in circumstances where medical supervision is feasible. Moreover, the current methods do not allow for the study of molecular and structural changes in living human nervous tissue. It is therefore essential to develop reliable models that accurately replicate the characteristics of seizures in a child to study the effects of FSs on the developing brain.
The original FSs model, which involves raising the body temperature of immature rats using an infrared lamp, was first proposed in the early 1980s [10]. Subsequently, new methods for modeling hyperthermia, a condition that induces seizures in animals, were described using microwave radiation [11], hot air [12] and hot water [13]. The next significant advancement in the field of animal modeling of FSs was the initiative to develop a model based on fever rather than hyperthermia [14,15]. This approach more closely resembles the clinical characteristics of FSs. Additionally, the animal model, initially developed in rats, has been adapted for use in mice [16], Danio rerio [17] and Drosophila [18], opening new avenues for investigating genetic factors in the pathogenesis of FSs.
In this review article, we concentrate on several crucial criteria that must be considered when modelling FSs in animals. These issues were partially addressed in the review by Bender et al. (2004) [19]. This paper provides an update and expanded information on the key factors that need to be considered when working with an FSs model, describes the existing approaches for seizure generation in different model subjects, and summarizes the main results obtained so far in studies with animal models of FSs.
2. Key Features That Should Reproduce the FS MODEL
2.1. The Main Types of FSs
Currently, FSs are classified as simple or complex, depending on the duration and the presence of recurrent episodes. Simple FSs last less than 15 min and occur no more than once a day. It is the most common form of seizure in children (about 70% of all reported cases). Complex FSs include seizures lasting more than 15 min or recurrent episodes over a 24-h period. The most severe type of complex FSs is febrile status epilepticus - a seizure without recovery of consciousness lasting more than 30 min [1]. Cohort studies have shown that the risk of developing temporal lobe epilepsy is significantly higher in children who have had complex FSs than in those who have had simple FSs [5,6]. Therefore, when modelling FSs in animals, it is necessary to strictly control the duration of the seizures, which will allow us to talk about the consequences of simple, complex FSs or febrile status epilepticus.
2.2. Age-Related Characteristics of Febrile Seizures
FSs in children occur mainly between the ages of 6 months and 5 years. Therefore, when modelling this type of seizure, it is important to consider the age of the animals, which should correspond to the period of greatest susceptibility to seizures in children. Early studies looked at the nervous system as a whole and, based on the rate of brain growth and the process of myelination, suggested that the 5-7 day old rat might be comparable to a newborn human in terms of nervous system development [20,21]. More recent studies have compared the stages of maturation of individual structures, particularly the hippocampus and cerebral cortex, and these studies show that the first postnatal week of rat life is close to the third trimester of human prenatal development and the second postnatal week to the first year of human life [22,23]. There are notable discrepancies in the processes of neurogenesis and microgliogenesis in the rodent and human dentate gyrus (Figure 1). In humans, most granular cells in the dentate gyrus emerge by the 35th week of prenatal development. After this point, the rate of neurogenesis declines and maintains a consistent level throughout adulthood. In contrast, the main cohort of granular cells in the rat dentate gyrus emerge by the end of the first postnatal month [24]. Microglial cell progenitors appear at the earliest stages of prenatal development; however, whereas in humans microglial development and maturation occurs prenatally and microglial cells acquire a mature phenotype by the end of prenatal development, in rats microglia mature during the first weeks of postnatal development [25,26,27,28].
When assessing the susceptibility to hyperthermic convulsions in rats of different ages, it was found that the most appropriate age was 10-13 days of postnatal life, as this is the age at which convulsions occur at a temperature close to the threshold temperature for the development of FS in children, with convulsions developing in most animals and being stereotyped [12,35]. In rats less than 10 days, the threshold temperature for seizure onset and the pattern of seizure are highly variable [12]. As rats mature, the threshold temperature increases; in rats aged 10-12 days, body temperature must reach 39-41°C to develop seizures [12], at 15 days of age the threshold temperature is 43°C [36], and in rats aged three weeks, body temperature must exceed 44°C to develop seizures [36], which is significantly different from the clinical manifestation of FSs in humans.
Thus, animals of suitable age should be used in models, for rats and mice this is the middle of the second postnatal week.
2.3. Body Temperature
FSs develop at temperatures above 38°C, so animal models should replicate this while avoiding extremely high temperatures (> 42°C) that are life-threatening. Therefore, body temperature should be monitored continuously when modelling FSs. It is not possible to measure brain temperature directly without implanting a temperature sensor in the nerve tissue, but it has been shown that the temperature curves of the brain and the core are similar [37]. For this reason, the rectal temperature of animals is monitored when FSs are modelled. If the body temperature rises above 41°C, the animals should be placed on a cool surface to reduce the body temperature.
2.4. The Source of the Ictal Activity in the Brain during a Febrile Seizure
Clinical case reports have noted edema and hippocampal damage in children following FSs [38,39,40]. This may suggest that the hippocampus is the source of the abnormal activity, but the unpredictable onset of FSs makes this very difficult to confirm. To our knowledge, a single EEG recording obtained directly during a febrile seizure in an 11-month-old child has been described in which abnormal activity was recorded in the temporal regions [41]. The description of the behavior of children with febrile seizures also suggests a hippocampal origin of the seizures [42].
In a hyperthermic model of FSs in 10-day-old rats, rhythmic activity was recorded in the amygdala and dorsal hippocampus concurrent with behavioral manifestations of seizures. At the same time, only rare non-rhythmic discharges were observed on EEG recordings from the cerebral cortex [12].
3. Factors That Influence the Course of Febrile Seizures
3.1. Genetic Background
Significant differences in the incidence of FSs in different populations and geographical regions may indicate genetic involvement in its pathogenesis. In addition, it is now known that having a parent with epilepsy or childhood seizures increases the risk of a child developing FSs [1,43]. The search for possible genetic mutations that lead to an increased risk of seizures has focused on genes for fever response proteins and proteins involved in ion channel function, neurotransmitter release and binding, and vesicular transport (Figure 2).
It has been suggested that body temperature regulation may be different in children predisposed to FSs [58,59]. A genome-wide study supports this notion, identifying two novel loci associated with altered expression of the PTGER3 and IL10 genes [51]. PTGER3 encodes EP3, one of the four prostaglandin E2 receptors considered to be the major pyrogenic mediator of fever. [60]. Increased expression of the PTGER3 gene may lead to a more severe febrile response, which in turn may increase a child's risk of developing FSs. IL10 encodes the anti-inflammatory cytokine IL10, which acts as a central endogenous antipyretic agent in a complex cytokine signaling system [61]. Reduced IL10 levels may also lead to more severe fever, increasing the risk of FSs.
Also, the presence of certain alleles in the genes encoding interleukin-1beta IL-1β and interleukin-1 receptor antagonist IL-1RA also increases the risk of FSs [52,62]. IL-1β is a pro-inflammatory cytokine that triggers a defense response to pathogens, resulting in fever. IL-1RA, which binds to the IL-1 receptor, prevents the activation of intracellular signaling cascades of the pro-inflammatory cytokines IL-1α and IL-1β and plays the role of an anti-inflammatory cytokine. Administration of IL-1RA (anakinra) can attenuate epileptogenesis, as shown in a lithium-pilocarpine model of epilepsy in rats [63]. In vitro experiments have shown that polymorphism of the genes encoding IL-1β and IL-1RA affects the amount of cytokines produced [64], which in turn can lead to a more or less pronounced febrile response. It is true that children with FSs have elevated levels of IL-1β in serum taken within a few hours of the onset of the seizure [62,65].
In addition, mutations in the GABRG2 gene, which encodes the γ2 subunit of GABAA receptor, have been associated with the development of FSs [53]. GABRG2 variants are associated with a wide range of epileptic syndromes, from mild cases of FSs and childhood absence seizures to more severe forms of epilepsy such as Dravet syndrome [66,67]. The γ2 subunit is required for clustering and synaptic localization of GABAA receptors, and the mutant form of this subunit in the receptor pentamer may result in an altered rate of receptor activation [68,69]. Various mutations in the GABRG2 gene, may also alter the kinetics of the GABAA receptor and disrupt its assembly and trafficking [53,70]. Since GABAA receptors mediate most of the fast inhibitory neurotransmission in the central nervous system, a change in their functional properties can lead to increased excitability of neuronal circuits.
Mutations in genes encoding different subunits of the voltage-gated sodium channel have been described in families with generalized epilepsy with febrile seizures plus (GEFS+) [54,55,56]. This syndrome is characterized by the development of recurrent febrile seizures in children, which (unlike typical FSs) continue beyond the age of 6 years. In individuals diagnosed with Dravet syndrome, genetic mutations within the SCN1A gene, which encodes the NaV1.1 channel, have been identified. These pathogenic variants have been observed to be associated with a notable reduction in sodium current and action potential activity in GABAergic inhibitory interneurons [71].
Nevertheless, modeling of FSs has demonstrated that the majority of animals can be induced to develop such seizures at an early age, indicating that a genetic predisposition is not a prerequisite. Nevertheless, the presence of the aforementioned genetic mutations may elevate the risk of FSs or result in more severe manifestations.
3.2. Malformations of Cortical Development
The most severe and often pharmacoresistant forms of early-onset epilepsy, including a high likelihood of complex febrile seizures, are associated with congenital anomalies in the structure of the cerebral cortex [57,72,73]. Neocortical malformations represent a large and heterogeneous group of neuronal disorders. Developmental abnormalities associated with epilepsy include abnormal cell proliferation or apoptosis (micro-, macrocephaly), abnormal cell migration (lissencephaly, gray matter heterotopia) and abnormal postmigrational cortical development [57].
There are several models of cortical malformations caused by chemical or physical manipulation of pre- and neonatal animals [74]. In particular, administration of methylazoxymethanol acetate to female rats on day 15 of gestation impair the proliferation and migration of brain neurons in the offspring, which subsequently leads to cortical thinning, hippocampal and amygdala dysplasia, and enlargement of the lateral and third ventricles [75]. The administration of carmustine (1-3-bis-chloroethyl-nitrosourea, BCNU) to female rats on day 15 of gestation results in the disruption of the laminar structure of the cortex and the abnormal morphology of neurons in the offspring [76]. The focal freezing of the newborn rat cerebral cortex leads to the development of microgyria in this area [77].
This approach can be used to model a dual pathology: FSs on the background of a cortical malformation. In this case, temporal lobe epilepsy is highly likely to develop [78], making this model a valuable tool for studying epileptogenesis that occurs at an early age.
3.3. Sex-Related Characteristics of Febrile Seizures
Although FSs can occur in both boys and girls, epidemiological studies have identified some differences in the characteristics and risk factors associated with FSs in males and females. These studies have demonstrated a greater prevalence of FSs in boys compared to girls [79]. However, it is important to note that the exact reasons for this male predominance are not well understood. One possible cause is the different timing of the transition from GABAA-mediated excitation to inhibition in neurons in males and females [80,81]. During prenatal and early postnatal neurogenesis, GABA acts as an excitatory neurotransmitter, which is essential for neuronal growth and synapse formation [82]. Then, as development progresses, there is a decrease in the expression of the first isoform of the Na-K-Cl cotransporter (NKCC1), which in the immature brain ensures the accumulation of Cl¯ ions in the cell, leading to their release upon activation of GABAA receptors and, consequently, membrane depolarization. At the same time, the expression of the second isoform of the K-Cl cotransporter (KCC2) increases, which, on the contrary, ensures the removal of Cl¯ ions from the cell, resulting in a change in the equilibrium potential of this ion and a change in the function of the GABAA receptor to inhibitory [82]. It turns out that this process takes place at different times in female and male rats: in females, increased levels of KCC2 protein in the hippocampus are observed as early as 7 days after birth, whereas in males it is not observed until the end of the second week of life [81]. It has been suggested that the explanation for this sex difference may lie in the effect of sex hormones on brain development, including effects on the expression of genes encoding NKCC1 and KCC2 [83].
This suggests that FSs may occur differently in boys and girls due to different levels of excitation and inhibition in the brain in the early postnatal period. This, in turn, can have different effects on the developing nervous system depending on the sex. There are few studies on sex differences in the course and outcome of FSs. However, a recent study showed that sexually mature male rats had more severe cognitive impairment than females who had suffered prolonged FSs at an early age [84].
4. Models of Febrile Seizures Ex Vivo
The ex vivo model of FSs involves the study of excitability and cell morphology on slices or cultures of animal brains under hyperthermic conditions, allowing the changes in nervous tissue to be followed directly at elevated temperatures. In this approach, it is important to obtain brain tissue from animals of appropriate age.
Electrophysiological studies are performed at high temperatures of the perfusion solution with which the brain slice is washed. Under these conditions, when the temperature of the solution is above 38°C, a single stimulus induces an epileptiform response in brain slices from rats aged 4 to 28 days, while activity develops spontaneously in slices from animals aged 11 to 18 days [85]. Interestingly, in these experiments, the activity persists for an hour or more after the temperature has returned to its initial value (36°C). No single-stimulus induced epileptiform activity or spontaneous activity were recorded in slices obtained from animals less than 4 days old or more than 28 days old. [85].
Morphological studies of neural tissue in the in vitro model of febrile seizures are performed on organotypic cultures obtained from the brains of rat pups and incubated at high temperature. Under these conditions, expansion of the granular cell layer and its death in the dentate gyrus have been shown, accompanied by extensive activation of microglial cells [86].
However, when modeling FSs ex vivo, it is important to remember that high temperatures can increase tissue metabolism and reduce oxygen solubility, potentially leading to hypoxia in in vitro preparations where oxygenation may already be difficult. Particularly in electrophysiological experiments, the rate of perfusion solution should be chosen appropriately to minimize the effects of hypoxia, which may confound the results obtained [87].
5. Models of Febrile Seizures In Vivo
5.1. Species Used to Model FSs
The initial development of FSs models was conducted using rodents, with Sprague-Dawley and Wistar rats representing the most prevalent subjects. We did not reveal any obvious differences in the description of seizures across rat breeds when modelling FSs at 10-11 days of age [12,84,88], however, it is noteworthy that no direct comparisons have been made.
The accessibility of transgenic and viral genetic technologies, which are frequently employed in murine models, has prompted the adaptation of the rat model for studies in mice, including the assessment of the role of diverse genetic factors in the susceptibility to FSs. It has been demonstrated that C57BL/6J mice, a widely used inbred mouse line, are among the most susceptible to FSs. In this instance, the C3H/HeJ and A/J mouse lines were identified as the most resistant to FSs [16]. It is noteworthy that the A/J mouse line displays a greater susceptibility to chemically and electrically induced seizures in comparison to the C57BL/6J strain [89,90]. This may suggest the existence of disparate initiation mechanisms for distinct seizure types.
Furthermore, FSs models have been adapted for use with two-day-old Drosophila melanogaster and Danio rerio (zebrafish) larvae [17,18]. These model objects offer certain advantages over rodent models [91]. The small size, short life cycle, high reproductive capacity and ease of maintenance of Drosophila and Danio rerio make them excellent subjects for genetic studies. In particular, Drosophila lines [18] and Danio rerio [92] with mutations in gene of the alpha subunit SCN1A of the potential-dependent sodium channel have been obtained. In humans, this leads to the development of genetic epilepsy with febrile seizures plus (GEFS+) and Dravet syndrome [18,92]. Furthermore, the use of Danio rerio fish as a model subject for neonatal seizures allows for the relatively straightforward conduction of pharmacological studies to evaluate the efficacy of potential anticonvulsant drugs [17,92].
However, it should be noted that fish models have several limitations. The effects of potential drugs may differ significantly between species due to differences in the blood-brain barrier, metabolism and thermoregulation [93]. Furthermore, the extrapolation of data from Danio rerio may result in erroneous conclusions regarding the effects of seizures due to significant differences in the development and morphology of the central nervous system between fish and mammals [94,95]. Nevertheless, the similarities between the principal neurotransmitters, transporters and receptors of the CNS in fish and mammals render Danio rerio an effective model for investigating the etiology of seizure development during the early stages of organism development.
Table 1 provides a summary of representative studies performed on different animal species with a brief description of febrile seizures.
5.2. Hyperthermia-Based Model
Several models, mostly based on hyperthermia alone, have been developed to study the mechanisms and effects of FSs on the developing brain: the hair-dryer model [12], the heated chamber model with the infrared lamp [10], the model based on microwave [11], and the model involving immersion of animals in heated water heated to 45°C [13], which can also be referred to as the hot water reflex epilepsy model [98].
The most popular model for febrile seizures, because of its simplicity, is the hair-dryer model [12], in which a stream of warm air is generated over the chamber in which the animals are placed. This approach makes it easy to control the chamber temperature and to place and remove animals for frequent monitoring of body temperature. Seizures are modelled at a chamber air temperature of 45-46°C. Under such conditions, the course of convulsions in 10-11-day-old rats is stereotypical: in the first 10 minutes, the body temperature rises to 39-40°C, facial automatisms are observed, often accompanied by unilateral bending of the body, then myoclonic twitching of the hind limbs, followed by clonic convulsions. It should be noted that convulsions induced in rats using this model have been reproduced by several scientific groups [84,88,99], who also note that convulsions develop in an overwhelming number of animals in this model.
Electroencephalogram (EEG) recordings in animals have shown that facial automatisms characterized by forelimb biting and head scratching, observed within the first 10 minutes, coincide with activity in the amygdala and dorsal hippocampus [12]. An EEG recording of an 11-month-old child having a febrile seizure episode showed theta waves recorded in the temporal region [41], and descriptions of the behavior of children with FSs suggest a hippocampal origin of the seizures [42]. This brings this model of febrile seizures closer to the pathogenesis of seizures in children.
The advantage of all hyperthermia-based models is that the timing of the high temperature exposure can be tightly controlled, allowing the modelling of simple or complex febrile seizures or febrile status epilepticus.
5.3. Fever Model
The vast majority of animal models of FSs are based on hyperthermia rather than fever, which implies activation of the immune system, although in children FSs usually develop against a background of viral or bacterial infection [100]. Inflammatory processes and the release of pro-inflammatory cytokines are thought to be one of the causes of fever [101] and seizure development. In particular, IL-1β, acting through IL-1R1, promotes excitatory transmission by both increasing the conductance of NMDA receptors for Ca2+ ions through phosphorylation of GluN2A and GluN2B subunits [49] and decreasing GABAA receptor-mediated Cl- currents [50] (Figure 3).
Nevertheless, an elevation in body temperature has been demonstrated to result in an augmentation of the concentration of proinflammatory cytokines within the blood of animals [102]. The hypothesis concerning the significant function of cytokines in the pathogenesis of FSs is further supported by the observation that the threshold for the onset of seizures induced by hyperthermia is markedly elevated in mice with IL-1R1 receptor deficiency [103]. This also substantiates the notion that hyperthermia per se can lead to the activation of proinflammatory pathways.
The administration of lipopolysaccharide (LPS), a component of the cell wall of Gram-negative bacteria to immature animals does not allow to achieve a significant increase in body temperature [14], due to the imperfect thermoregulation in rats at this age [104]. However, even in the absence of convulsions, LPS administration induce alterations in the expression of ionotropic glutamate receptor genes and is accompanied by disturbances in long-term potentiation [105,106]. It is likely that the effect of FSs, occurring in the context of immune system activation, on the developing brain may be more pronounced. Consequently, the use of hyperthermia alone to model FSs may have inherent limitations. However, the current attempt to develop a model of fever-like symptoms using additional immune system activation has yet to yield successful results. A combination of LPS and kainic acid was proposed as a model of FSs [107], however, this approach does not result in the body temperature reaching the values characteristic of FSs.
5.3. Combination Models of FSs
The most approximate model may be a combination of LPS or pro-inflammatory cytokine injections against the background of a hyperthermic model, in which body temperature is raised by hot air [15]. This approach results in the sequential development of inflammation, fever and seizures. However, our observations suggest that the combined use of LPS or proinflammatory cytokines with a hyperthermic model results in animals that endure FSs significantly worse and mortality rates increase. Consequently, this approach, which may be the most closely aligned with the clinical course of FSs, necessitates the implementation of additional animal care measures.
5.4. Required Control Groups
It is incorrect to compare the results obtained in animals subjected to febrile seizure modeling with those obtained in intact animals only, since the animals are weaned from their dams at the time of modeling, which is a major stress at an early age and may also affect the further development of the nervous system [112,113,114]. Therefore, one of the control groups should be animals weaned from the female for the duration of the febrile seizure modelling but kept under normothermic conditions (Figure 4).
The effects of hyperthermia itself, which causes seizures, can also be very important. To assess which effects may be due to hyperthermia specifically and which hyperthermia and febrile seizure developed on its background, some trials have included another control group. Animals in this group have their body temperature raised, but the development of seizures is blocked by the administration of barbiturate [115,116]. However, the mere use of sedatives, including long-acting barbiturates, in immature animals leads to dysregulation of proteins associated with apoptosis, cell proliferation and synaptogenesis [117], as well as increased cell death and white matter damage [118,119,120]. In models of status epilepticus, phenobarbital has been shown to exacerbate seizure-induced neuronal damage in rats at an early age [121]. Therefore, short-acting barbiturates (e.g. pentobarbital) should be used in this approach, but do not rule out the possibility that any suppression of neuronal activity in the immature brain, even for a short period of time, may result in impaired cell maturation and synapse formation, as well as increased apoptosis of immature cells.
6. Main Results Obtained in Models of Febrile Seizures
6.1. Morphologic Changes in Neurons
Despite numerous studies, there is still no clear understanding of the morphological changes in the nervous system caused by FSs, which represents an area for further research (Table 2). Clinical studies have indicated that hippocampal edema may occur in children within 48 hours of prolonged FSs, with resolution occurring within five days [38]. Studies in animal models of complex FSs indicate that there is no significant loss of hippocampal neurons [122]. The CA1 region of the hippocampus is more susceptible to damage than other regions in the FSs model [123]. This may be explained by the later maturation of inhibitory synaptic transmission in CA1 neurons compared to CA3 neurons [124]. However, even in the absence of significant neuronal death, a considerable number of silver-stained "dark" neurons are observed in the hippocampus and amygdala [125]. This indicates that they are functionally damaged [126]. These cells are distinguished by a diminished volume without impairment to the plasma membrane, resulting in ultrastructural compaction (notably, there is a reduction in the volume of the cisternae of the endoplasmic reticulum) [127]. A distinctive attribute of these "dark" neurons is their capacity to recuperate their morphology [128,129,130], which may account for the minimal neuronal loss following FSs.
In the dentate gyrus, where postnatal neurogenesis occurs, one week after FSs, granular cells that appeared after seizures exhibited an increase in dendrite length. Four weeks later, the dendritic tree demonstrated increased complexity. After eight weeks, an additional increase in dendrite complexity was observed, accompanied by an increase in the number of mushroom spines [131]. These morphological alterations in the dendritic structure of granular cells may result in an augmented excitatory signal coming from the entorhinal cortex [132]. Furthermore, an overgrowth of mossy fibers has been documented three months following FSs, with no discernible differences from the control group observed 10 days after seizures [131]. Furthermore, two months following FSs, an increase in F-actin was observed in the presynaptic endings of mossy fiber synapses. This may result in an increase in presynaptic activity and an enhancement of excitatory synaptic transmission between the dentate gyrus and the CA3 region of the hippocampus [133].
The modeling of FSs in older animals (22-30 days) revealed the ultrastructural disruption of synaptic endings in both hippocampal neurons and temporal lobe cortical cells [134,135,136]. Twenty-four hours after the onset of seizures, a series of notable changes were observed in the hippocampal neurons of the CA1 region. These included mitochondrial swelling, degranulation of the rough endoplasmic reticulum, enlargement of the Golgi complex, enlargement of the synaptic gap, reduction in the length of the synaptic active zone, and reduction in postsynaptic density [136]. At three days post-FSs, in addition to the aforementioned ultrastructural disturbances, we also observed swelling of pre- and postsynaptic endings, which exhibited either optically empty fields or minimal residual microfibrillary material and enlarged mitochondria. Furthermore, a reduction in the number of synaptic vesicles was noted in the presynaptic endings [134,135].
6.2. Morphologic Changes in Glial Cells
Currently, there is no consensus regarding the occurrence of astroglial and microglial cell activation following FSs. Modeling febrile status epilepticus in 11-day-old rats, astroglial cell activation was observed in the CA3 region of the hippocampus and in the dentate gyrus 6 hours after seizures [137] and in the CA1, CA3 regions, and hilus of the hippocampus 24 hours after seizures [137,138]. Neither in the rat model of FSs 2 days after seizure nor in the mouse model after 5 days was astrogliosis detected in the hippocampus [96,139]. The evaluation of the area occupied by astrocytes in the CA1 and CA3 regions of the hippocampus 2 days and 11 days after FSs revealed that the age-related dynamics of astrocyte maturation is impaired in animals after seizures, which is accompanied by impaired synaptic plasticity [139]. In FSs modeling of 14-15 day old mice 5 days after seizures, a significant inhibition of inter-astrocytic gap junctions in the hippocampus is observed, which is accompanied by a decrease in the level of connexin 43, one of the gap junction proteins [96]. The disruption of astrocytic gap junctions can result in impaired clearance of excess potassium ions and glutamate from extracellular space, which may contribute to increased neuronal excitability [140,141]. Disruption of ion and molecule distribution through the astrocytic network can result in cell edema [141], as evidenced by an ultrastructural study of hippocampal and temporal lobe cortex astrocytes in a rat model of FSs three days after seizures [142]. In addition to edema, degenerative changes in astrocytes have been described, with the main manifestations being damage to the endoplasmic reticulum and mitochondria [142]. These changes can lead to disturbances in some biochemical processes, such as abnormal protein synthesis or inhibition of oxidative phosphorylation.
At one and three days following FSs, rats exhibit disruption of the blood-brain barrier [143,144], as evidenced by weakened and destroyed dense intercellular endothelial connections, frequent complete occlusion of the capillary lumen, caused both by endothelial cell damage and external pressure from markedly enlarged perivascular astrocytic processes [143].
Extensive microgliosis was demonstrated by in vitro experiments on organotypic cultures of hippocampal slices, which were subjected to heat shock [86]. The results of some in vivo experiments align with this finding. The activation of microglial cells is observed in the rat hippocampus 24 hours after FSs [28], with the most significant changes occurring in FSs modeling at 15 days of age in comparison to the microglial response in FSs modeling at 5, 10, or 20 days of age. These changes include an increased number of microglial cells and morphological alterations in individual cells, such as the emergence of short, thick outgrowths and a distinct large soma [28]. In mice aged 14-15 days, microglial cell activation is observed within 1 to 7 days after seizures [96,145]. However, when modeling FSs on animals aged 10-11 days, no increase in the total number of microglial cells is observed. However, there is a significant increase in the proportion of activated microglia with an amoeboid shape [138]. Recently, microglial cell activation has been considered as two polar states: classical (the M1 phenotype) or alternative (the M2 phenotype). The M1 phenotype has been demonstrated to release pro-inflammatory factors and free radicals, which have been shown to impair the repair and regeneration of brain tissue. In contrast, M2 microglia facilitate brain repair and regeneration by enhancing phagocytosis, releasing trophic factors, and eliminating inflammation [146,147]. In a mouse model of FS, it has been demonstrated that hyperthermia activates microglial TRPV1, which subsequently suppresses microglia activation via the M2 pathway [145]. Inhibition of microglial polarization through the alternative pathway can result in the predominance of the M1 phenotype and the subsequent release of proinflammatory cytokines, including interleukins-1β, interleukin-6, and tumor necrosis factor-α (TNFα), whose increase in the brains of experimental animals after FS has been confirmed in several studies [28,138,148,149].
6.3. Changes in Neuronal Properties and Synaptic Transmission
The use of ex vivo model of FSs allows us to directly record changes in the biophysical properties of neurons and synaptic transmission in brain slices as temperature rises, which may provide valuable information about the causes of seizure activity and the search for targets to stop it. Thus, on hippocampal slices from 13-16 day old mice showed that the high temperature bath solution (41°C) led to increased excitability, decreased input resistance and spontaneous activity of both excitatory pyramidal neurons and inhibitory interneurons in the hippocampus [150]. The increase in neuronal excitability at elevated temperature is associated with a calcium current mediated by the Cav1.2 subunit of L-type calcium channels. At the same time, the selective L-type calcium channel blocker nimodipine reduces the frequency and duration of seizures in animals in an experimental model of FSs in vivo [47]. Using this model, it has also been shown that increasing temperature increases the sodium current through the non-selective cation channel NALCN, which also leads to depolarization of the nerve cell membrane [47].
The use of in vivo models of FSs makes it possible to assess the short- and long-term effects of FSs. Many studies have focused on neuronal excitability and synaptic transmission at different times after febrile seizures. Immediately after FSs, a decrease in synaptic transmission efficiency and a decrease in calcium conductance through calcium-permeable AMPA receptors are observed in rat hippocampal neurons [151]. This is thought to prevent excessive excitotoxicity, and no significant neuronal death is observed in this model. Two days after seizures in rats, a decrease in synaptic transmission efficiency, changes in short-term plasticity in hippocampal CA3-CA1 neurons and a decrease in the frequency of miniature excitatory synaptic currents in hippocampal CA1 field neurons were observed. In general, these data indicate a decrease in the probability of mediator release [123]. Furthermore, an additional neuroprotective mechanism has been delineated in the context of augmented of glutamate transporter 1 (GLT-1) levels following hyperthermia-induced seizures [152]. This may contribute to a reduction in the concentration of glutamate within synaptic clefts [153], which was validated in the rat cerebral cortex two days following FSs [152].
One week after seizures in rats, stimulation of Schaffer collaterals with a single stimulus resulted in a decrease in the amplitude of population spikes in CA1 neurons as a result of increased inhibitory postsynaptic currents in the hippocampus mediated by GABAA receptors [154]. However, when Schaffer collaterals are stimulated with a series of stimuli, epileptiform activity develops only in slices obtained from animals one week after FSs compared to the control, indicating increased excitability of hippocampal neuronal circuits [155]. This contradiction can be explained by the fact that increased inhibition can reduce activity in response to a single stimulus, but when stimulated by a series of pulses, increased GABAA conductance leads to accumulation of Cl- in neurons, strong activation of the KCC2 transporter and an increase in K+ ions in the extracellular space, resulting in a shift of the neuronal resting potential towards depolarization and increased cell excitability [156,157]. Increased expression of the GABAA receptor in granular cells born after FSs has also been demonstrated in the dentate gyrus [158]. In addition, increased Na+/K+-ATPase activity was found in rat cortical membranes 20 days and 2 months after hyperthermia-induced seizures, which may also represent a compensatory mechanism for a decrease in the extracellular concentration of K+ ions and, consequently, a decrease in the excitability of cortical cells [159]. However, 20 days after the seizures an increase in metabotropic glutamate receptors 5 (mGluR5) levels [152]. Activation of mGluR5, a member of group I metabotropic glutamate receptors, leads to cell depolarization and increased neuronal excitability, as well as modulation of NMDA receptors [160,161]. Although the causal relationship between group I mGluRs and susceptibility to seizures has not yet been fully established, high expression of mGluR1 and mGluR5 is considered as a potential mechanism of epileptogenesis [162]. Consequently, the increase in mGluR5 levels observed in the FSs model may lead to an elevated risk of recurrent seizure episodes.
6.4. Changes in Synaptic Plasticity
In examining the effects of FSs on synaptic plasticity, a number of studies have reported conflicting results: increased long-term potential (LTP) and decreased long-term depression (LTD) in CA3-CA1 synapses of the hippocampus 1 month after complex FSs [163] and, conversely, decreased LTP in animals 11 days and 1.5 months after complex FSs [88,139], and, in addition to decreased LTP, increased LTD 1 month after complex FSs [164], indicating the need for additional studies. In examining LTP in rats of varying ages, it was shown that FSs might impede the functional maturation of the hippocampus. This is because rats at three weeks of age exhibit an unstable LTP [139], a phenomenon observed in younger animals (less than two weeks old) [165,166,167,168].
One potential mechanism underlying the impairment of synaptic plasticity following complex FSs may be a reduction in tyrosine phosphorylation of the GluN2A subunit of NMDA receptors [164]. Additionally, it was demonstrated that the reduction in LTP observed in rats following FSs may be attributable to a heightened desensitization of NMDA receptors [88]. The absence of changes in NMDA receptor subunit composition after FSs [88,164] and the restoration of the level of LTP to control values with the NMDA receptor glycine site coagonist D-serine [88,139] indicate that the observed pronounced desensitization may be due to insufficient activation of the NMDA receptor glycine site [169,170]. Furthermore, the restoration of LTP levels in the presence of D-serine may suggest impaired interactions between neurons and glial cells, as astrocytes have been demonstrated to regulate NMDA-dependent plasticity by Ca+2-dependent release of D-serine [171].
6.5. A Predisposition to Recurrent Seizures and Epilepsy
When evaluating the seizure threshold in rats after FSs, mixed results were obtained. An increase in the threshold for pentylenetetrazol-induced seizures has been shown [115], as well as an increase in the maximal electroshock seizure threshold [123]. Meanwhile, other authors have demonstrated an increased susceptibility to seizures in rats when kainate was used [155] and electroencephalographically recorded epileptiform discharges in the limbic system [172]. Such differences in results could arise, firstly, from the different ages of seizure susceptibility assessment. Thus, an increase in seizure threshold has been shown in younger animals (<2.5 months). Secondly, all the studies used different approaches to study the threshold of seizure development, in particular, the use of pentylenetetrazole, which is an antagonist of GABAA receptors, resulted in an increase in the threshold of seizure development, while the use of kainic acid, which is an agonist of kainate and AMPA receptors and increases excitatory transmission, led, on the contrary, to a decrease in the seizure threshold. These data show the need for a more detailed study of the effect of FS on the probability of recurrent episodes of seizures and epilepsy.
6.6. Behavioural Disorders after Febrile Seizures
A number of studies have described memory impairment in children following prolonged exposure to FSs [173,174]. A substantial body of experimental evidence from animal models of FS has also demonstrated cognitive impairment in both immature [175] and adult [84,139,176,177] animals. This is evidenced by impaired spatial memory in Morris and Barnes mazes [139,175,176,177], reduced latency period in the inhibitory avoidance task [177], and increased electrocution in the active avoidance test [84].
In addition to cognitive impairment in animals, depressive-like behavior was noted in models of FSs, manifested by decreased sucrose preference and increased immobility during the forced swim test in rats at 37 and 60 days of age [178]. However, it should be noted that in this study febrile convulsions were modeled by administration of LPS and kainic acid. This leads to the development of convulsions against a background of activation of inflammatory cascades, although no significant increase in body temperature necessary for the development of febrile convulsions is observed in this model. But other hyperthermia-based models have also shown depressive-like behavior and increased levels of anxiety-like behavior in adult animals [179,180]. A recent study in a febrile seizure model described animal behavior similar to autism spectrum disorders, including social novelty deficits, repetitive behavior and hyperlocomotion [149].
Thus, to date, there is considerable evidence that FSs adversely affect the development of the nervous system, leading to a wide range of different neurological disorders.
7. Conclusions. Limitations and Future Directions
As FSs remains one of the most common childhood neurological disorders, it is important to find ways to predict its development, suppress seizure activity and prevent the development of subsequent neurological disorders. These questions can only be answered by a combination of long-term clinical observation of patients and studies in experimental animal models. The advantage of animal models over clinical studies of FSs is the ability to either exclude the influence of any factors, such as pre-existing CNS pathology, genetic predisposition or drug side effects, or to simulate dual pathology. In addition, working with experimental models allows strict control of the timing of hyperthermia, the duration and the nature of the seizures, which can be a key indicator for assessing the results.
As is the case with numerous animal models of diverse diseases, the FSs model has certain limitations. The main challenges associated with modeling FSs in animals and subsequent evaluation of the outcomes are due to the fact that FSs manifest at an early age, when the maturation of the nervous system is actively occurring. However, the development of different brain regions in humans and mice or rats occurs at varying rates, and these processes are not necessarily parallel in humans and rodents. This discrepancy may result in erroneous conclusions when attempting to extrapolate findings from rodent models to humans.
Another limitation is the difficulty in modeling the cause of fever and the inflammatory response, which is the most common cause of hyperthermia and FSs in children. Today, models based on hyperthermia are most commonly used, although it is known that temperature elevation itself also leads to the release of proinflammatory cytokines [102,103]. However, in this case, hyperthermia occurs first, followed by an inflammatory response of the nervous system, whereas in clinical cases, inflammation develops first, then fever, then seizures. This initial inflammation already affects synaptic transmission [49,50], so models based on hyperthermia alone may not fully reflect the course of FSs in children.
However, despite their limitations, existing animal models remain a valuable tool for studying the etiology and pathophysiology of FSs. The emergence of new transgenic animal lines, the development of viral genetic constructs and new experimental approaches can provide much more information about the development of the animal brain after FSs than we can obtain in humans.
Author Contributions
Conceptualization, T.P. and A.Z.; writing—original draft preparation, A.G; writing—review and editing, funding acquisition, Y.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Science Foundation, grant number 23-25-00143.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Leung, A.K.; Hon, K.L.; Leung, T.N.H. Febrile Seizures: An Overview. Drugs Context 2018, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Jung, J.Y.; Kim, J.H.; Kwon, H.; Park, J.W.; Kwak, Y.H.; Kim, D.K.; Lee, J.H. Febrile Seizures: Are They Truly Benign? Longitudinal Analysis of Risk Factors and Future Risk of Afebrile Epileptic Seizure Based on the National Sample Cohort in South Korea, 2002–2013. Seizure 2019, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Sugai, K. Current Management of Febrile Seizures in Japan: An Overview. Brain Dev. 2010, 32, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Verity, C.M.; Butler, N.R.; Golding, J. Febrile Convulsions in a National Cohort Followed up from Birth. I--Prevalence and Recurrence in the First Five Years of Life. BMJ 1985, 290, 1307–1310. [Google Scholar] [CrossRef]
- Civan, A.B.; Ekici, A.; Havali, C.; Kiliç, N.; Bostanci, M. Evaluation of the Risk Factors for Recurrence and the Development of Epilepsy in Patients with Febrile Seizure. Arq. Neuropsiquiatr. 2022, 80, 779–785. [Google Scholar] [CrossRef]
- Nelson, K.B.; Ellenberg, J.H. Predictors of Epilepsy in Children Who Have Experienced Febrile Seizures. N. Engl. J. Med. 1976, 295, 1029–1033. [Google Scholar] [CrossRef]
- Salehi, B.; Yousefichaijan, P.; Safi Arian, S.; Ebrahimi, S.; Naziri, M. Comparison of Relation between Attention Deficit Hyperactivity Disorder in Children with and without Simple Febrile Seizure Admitted in Arak Central Iran. Iran. J. child Neurol. 2016, 10, 56–61. [Google Scholar]
- Dreier, J.W.; Pedersen, C.B.; Cotsapas, C.; Christensen, J. Childhood Seizures and Risk of Psychiatric Disorders in Adolescence and Early Adulthood: A Danish Nationwide Cohort Study. Lancet. Child Adolesc. Heal. 2019, 3, 99–108. [Google Scholar] [CrossRef]
- Yi, Y.; Zhong, C.; Wei-Wei, H. The Long-Term Neurodevelopmental Outcomes of Febrile Seizures and Underlying Mechanisms. Front. cell Dev. Biol. 2023, 11, 1186050. [Google Scholar] [CrossRef]
- Holtzman, D.; Obana, K.; Olson, J. Hyperthermia-Induced Seizures in the Rat Pup: A Model for Febrile Convulsions in Children. Science 1981, 213, 1034–1036. [Google Scholar] [CrossRef]
- Hjeresen, D.L.; Guy, A.W.; Petracca, F.M.; Diaz, J. A Microwave-hyperthermia Model of Febrile Convulsions. Bioelectromagnetics 1983, 4, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Baram, T.Z.; Gerth, A.; Schultz, L. Febrile Seizures: An Appropriate-Aged Model Suitable for Long-Term Studies. Brain Res. Dev. Brain Res. 1997, 98, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Duong, T.M.; de Lanerolle, N.C. The Neuropathology of Hyperthermic Seizures in the Rat. Epilepsia 1999, 40, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Heida, J.G.; Boissé, L.; Pittman, Q.J. Lipopolysaccharide-Induced Febrile Convulsions in the Rat: Short-Term Sequelae. Epilepsia 2004, 45, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Eun, B.L.; Abraham, J.; Mlsna, L.; Kim, M.J.; Koh, S. Lipopolysaccharide Potentiates Hyperthermia-Induced Seizures. Brain Behav. 2015. [Google Scholar] [CrossRef]
- van Gassen, K.L.I.; Hessel, E.V.S.; Ramakers, G.M.J.; Notenboom, R.G.E.; Wolterink-Donselaar, I.G.; Brakkee, J.H.; Godschalk, T.C.; Qiao, X.; Spruijt, B.M.; van Nieuwenhuizen, O.; et al. Characterization of Febrile Seizures and Febrile Seizure Susceptibility in Mouse Inbred Strains. Genes. Brain. Behav. 2008, 7, 578–586. [Google Scholar] [CrossRef]
- Hunt, R.F.; Hortopan, G.A.; Gillespie, A.; Baraban, S.C. A Novel Zebrafish Model of Hyperthermia-Induced Seizures Reveals a Role for TRPV4 Channels and NMDA-Type Glutamate Receptors. Exp. Neurol. 2012, 237, 199–206. [Google Scholar] [CrossRef]
- Sun, L.; Gilligan, J.; Staber, C.; Schutte, R.J.; Nguyen, V.; O’Dowd, D.K.; Reenan, R. A Knock-in Model of Human Epilepsy in Drosophila Reveals a Novel Cellular Mechanism Associated with Heat-Induced Seizure. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 14145–14155. [Google Scholar] [CrossRef]
- Bender, R.A.; Dubé, C.; Baram, T.Z. Febrile Seizures and Mechanisms of Epileptogenesis: Insights from an Animal Model. Adv. Exp. Med. Biol. 2004, 548, 213–225. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Quantitative Growth and Development of Human Brain. Arch. Dis. Child. 1973, 48, 757–767. [Google Scholar] [CrossRef]
- Gottlieb, A. , Keydar, I. K., & Epstein, H.T. Rodent Brain Growth Stages: An Analytical Review. Neonatology 1977, 6, 166–176. [Google Scholar]
- Avishai-Eliner, S.; Brunson, K.L.; Sandman, C.A.; Baram, T.Z. Stressed-out, or in (Utero)? Trends Neurosci. 2002, 25, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Herschkowitz, N.; Kagan, J.; Zilles, K. Neurobiological Bases of Behavioral Development in the First Year. Neuropediatrics 1997, 28, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Bayer, S.A. Development of the Hippocampal Region in the Rat I. Neurogenesis Examined with 3 H-thymidine Autoradiography. J. Comp. Neurol. 1980, 190, 87–114. [Google Scholar] [CrossRef] [PubMed]
- Menassa, D.A.; Gomez-Nicola, D. Microglial Dynamics During Human Brain Development. Front. Immunol. 2018, 9, 1014. [Google Scholar] [CrossRef]
- Esiri, M.M.; al Izzi, M.S.; Reading, M.C. Macrophages, Microglial Cells, and HLA-DR Antigens in Fetal and Infant Brain. J. Clin. Pathol. 1991, 44, 102–106. [Google Scholar] [CrossRef]
- Dermitzakis, I.; Manthou, M.E.; Meditskou, S.; Tremblay, M.-È.; Petratos, S.; Zoupi, L.; Boziki, M.; Kesidou, E.; Simeonidou, C.; Theotokis, P. Origin and Emergence of Microglia in the CNS-An Interesting (Hi)Story of an Eccentric Cell. Curr. Issues Mol. Biol. 2023, 45, 2609–2628. [Google Scholar] [CrossRef]
- Kim, I.; Mlsna, L.M.; Yoon, S.; Le, B.; Yu, S.; Xu, D.; Koh, S. A Postnatal Peak in Microglial Development in the Mouse Hippocampus Is Correlated with Heightened Sensitivity to Seizure Triggers. Brain Behav. 2015, 5, e00403. [Google Scholar] [CrossRef]
- Lu, D.; He, L.; Xiang, W.; Ai, W.-M.; Cao, Y.; Wang, X.-S.; Pan, A.; Luo, X.-G.; Li, Z.; Yan, X.-X. Somal and Dendritic Development of Human CA3 Pyramidal Neurons from Midgestation to Middle Childhood: A Quantitative Golgi Study. Anat. Rec. (Hoboken). 2013, 296, 123–132. [Google Scholar] [CrossRef]
- Degl’Innocenti, E.; Dell’Anno, M.T. Human and Mouse Cortical Astrocytes: A Comparative View from Development to Morphological and Functional Characterization. Front. Neuroanat. 2023, 17, 1130729. [Google Scholar] [CrossRef]
- Vivi, E.; Di Benedetto, B. Brain Stars Take the Lead during Critical Periods of Early Postnatal Brain Development: Relevance of Astrocytes in Health and Mental Disorders. Mol. Psychiatry 2024. [Google Scholar] [CrossRef] [PubMed]
- Jakovcevski, I.; Filipovic, R.; Mo, Z.; Rakic, S.; Zecevic, N. Oligodendrocyte Development and the Onset of Myelination in the Human Fetal Brain. Front. Neuroanat. 2009, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Shimizu, T.; Sherafat, A.; Richardson, W.D. Life-Long Oligodendrocyte Development and Plasticity. Semin. Cell Dev. Biol. 2021, 116, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Kracht, L.; Borggrewe, M.; Eskandar, S.; Brouwer, N.; Chuva de Sousa Lopes, S.M.; Laman, J.D.; Scherjon, S.A.; Prins, J.R.; Kooistra, S.M.; Eggen, B.J.L. Human Fetal Microglia Acquire Homeostatic Immune-Sensing Properties Early in Development. Science 2020, 369, 530–537. [Google Scholar] [CrossRef]
- Hjeresen, D.L.; Diaz, J. Ontogeny of Susceptibility to Experimental Febrile Seizures in Rats. Dev. Psychobiol. 1988, 21, 261–275. [Google Scholar] [CrossRef]
- Morimoto, T.; Nagao, H.; Sano, N.; Takahashi, M.; Matsuda, H. Hyperthermia-Induced Seizures with a Servo System: Neurophysiological Roles of Age, Temperature Elevation Rate and Regional GABA Content in the Rat. Brain Dev. 1990, 12, 279–283. [Google Scholar] [CrossRef]
- Sundgren-Andersson, A.K.; Östlund, P.; Bartfai, T. Simultaneous Measurement of Brain and Core Temperature in the Rat during Fever, Hyperthermia, Hypothermia and Sleep. Neuroimmunomodulation 1998, 5, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.C. Hippocampal Abnormalities after Prolonged Febrile Convulsion: A Longitudinal MRI Study. Brain 2003, 126, 2551–2557. [Google Scholar] [CrossRef]
- Sokol, D.K.; Demyer, W.E.; Edwards-Brown, M.; Sanders, S.; Garg, B. From Swelling to Sclerosis: Acute Change in Mesial Hippocampus after Prolonged Febrile Seizure. Seizure 2003, 12, 237–240. [Google Scholar] [CrossRef]
- Peng, S.-J.; Hsieh, K.L.-C.; Lin, Y.-K.; Tsai, M.-L.; Wong, T.-T.; Chang, H. Febrile Seizures Reduce Hippocampal Subfield Volumes but Not Cortical Thickness in Children with Focal Onset Seizures. Epilepsy Res. 2021, 179, 106848. [Google Scholar] [CrossRef]
- Hamelin, S.; Vercueil, L. A Simple Febrile Seizure with Focal Onset. Epileptic Disord. 2014, 16, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Neville, B.; Gindner, D. Febrile Seizures Are a Syndrome of Secondarily Generalized Hippocampal Epilepsy. Dev. Med. Child Neurol. 2010, 52, 1151–1153. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.T.; Shinnar, S.; Shapiro, E.D.; Salomon, M.E.; Crain, E.F.; Hauser, W.A. Risk Factors for a First Febrile Seizure: A Matched Case-Control Study. Epilepsia 1995, 36, 334–341. [Google Scholar] [CrossRef]
- Schuchmann, S.; Hauck, S.; Henning, S.; Grüters-Kieslich, A.; Vanhatalo, S.; Schmitz, D.; Kaila, K. Respiratory Alkalosis in Children with Febrile Seizures. Epilepsia 2011, 52, 1949–1955. [Google Scholar] [CrossRef]
- Schuchmann, S.; Schmitz, D.; Rivera, C.; Vanhatalo, S.; Salmen, B.; Mackie, K.; Sipilä, S.T.; Voipio, J.; Kaila, K. Experimental Febrile Seizures Are Precipitated by a Hyperthermia-Induced Respiratory Alkalosis. Nat. Med. 2006, 12, 817–823. [Google Scholar] [CrossRef]
- Balestrino, M.; Somjen, G.G. Concentration of Carbon Dioxide, Interstitial PH and Synaptic Transmission in Hippocampal Formation of the Rat. J. Physiol. 1988, 396, 247–266. [Google Scholar] [CrossRef]
- Radzicki, D.; Yau, H.J.; Pollema-Mays, S.L.; Mlsna, L.; Cho, K.; Koh, S.; Martina, M. Temperature-Sensitive Cav1.2 Calcium Channels Support Intrinsic Firing of Pyramidal Neurons and Provide a Target for the Treatment of Febrile Seizures. J. Neurosci. 2013, 33, 9920–9931. [Google Scholar] [CrossRef]
- Ziemann, A.E.; Schnizler, M.K.; Albert, G.W.; Severson, M.A.; Howard, M.A.; Welsh, M.J.; Wemmie, J.A. Seizure Termination by Acidosis Depends on ASIC1a. Nat. Neurosci. 2008, 11, 816–822. [Google Scholar] [CrossRef]
- Viviani, B.; Bartesaghi, S.; Gardoni, F.; Vezzani, A.; Behrens, M.M.; Bartfai, T.; Binaglia, M.; Corsini, E.; Di Luca, M.; Galli, C.L.; et al. Interleukin-1β Enhances NMDA Receptor-Mediated Intracellular Calcium Increase through Activation of the Src Family of Kinases. J. Neurosci. 2003, 23, 8692–8700. [Google Scholar] [CrossRef]
- Wang, S.; Cheng, Q.; Malik, S.; Yang, J. Interleukin-1beta Inhibits Gamma-Aminobutyric Acid Type A (GABA(A)) Receptor Current in Cultured Hippocampal Neurons. J. Pharmacol. Exp. Ther. 2000, 292, 497–504. [Google Scholar]
- Skotte, L.; Fadista, J.; Bybjerg-Grauholm, J.; Appadurai, V.; Hildebrand, M.S.; Hansen, T.F.; Banasik, K.; Grove, J.; Albiñana, C.; Geller, F.; et al. Genome-Wide Association Study of Febrile Seizures Implicates Fever Response and Neuronal Excitability Genes. Brain 2022, 145, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Virta, M.; Hurme, M.; Helminen, M. Increased Frequency of Interleukin-1beta (-511) Allele 2 in Febrile Seizures. Pediatr. Neurol. 2002, 26, 192–195. [Google Scholar] [CrossRef]
- Hernandez, C.C.; Shen, Y.; Hu, N.; Shen, W.; Narayanan, V.; Ramsey, K.; He, W.; Zou, L.; Macdonald, R.L. GABRG2 Variants Associated with Febrile Seizures. Biomolecules 2023, 13. [Google Scholar] [CrossRef]
- Wallace, R.H.; Wang, D.W.; Singh, R.; Scheffer, I.E.; George, A.L.; Phillips, H.A.; Saar, K.; Reis, A.; W. johnson, E.; Sutherland, G.R.; et al. Febrile Seizures and Generalized Epilepsy Associated with a Mutation in the Na+-Channel Β1 Subunit Gene SCN1B. Nat. Genet. 1998, 19, 366–370. [Google Scholar] [CrossRef]
- Audenaert, D.; Claes, L.; Ceulemans, B.; Löfgren, A.; Van Broeckhoven, C.; De Jonghe, P. A Deletion in SCN1B Is Associated with Febrile Seizures and Early-Onset Absence Epilepsy. Neurology 2003, 61, 854–856. [Google Scholar] [CrossRef]
- Liao, W.-P.; Shi, Y.-W.; Long, Y.-S.; Zeng, Y.; Li, T.; Yu, M.-J.; Su, T.; Deng, P.; Lei, Z.-G.; Xu, S.-J.; et al. Partial Epilepsy with Antecedent Febrile Seizures and Seizure Aggravation by Antiepileptic Drugs: Associated with Loss of Function of Na(v) 1.1. Epilepsia 2010, 51, 1669–1678. [Google Scholar] [CrossRef]
- Barkovich, A.J.; Dobyns, W.B.; Guerrini, R. Malformations of Cortical Development and Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022392. [Google Scholar] [CrossRef] [PubMed]
- Rantala, H.; Uhari, M.; Hietala, J. Factors Triggering the First Febrile Seizure. Acta Paediatr. 1995, 84, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.E.; Dooley, J.M.; Wood, E.P.; Bethune, P. Is Temperature Regulation Different in Children Susceptible to Febrile Seizures? Can. J. Neurol. Sci. 2009, 36, 192–195. [Google Scholar]
- Evans, S.S.; Repasky, E.A.; Fisher, D.T. Fever and the Thermal Regulation of Immunity: The Immune System Feels the Heat. Nat. Rev. Immunol. 2015, 15, 335–349. [Google Scholar] [CrossRef]
- Roth, J. Endogenous Antipyretics. Clin. Chim. Acta 2006, 371, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Al Morshedy, S.; Elsaadany, H.F.; Ibrahim, H.E.; Sherif, A.M.; Farghaly, M.A.A.; Allah, M.A.N.; Abouzeid, H.; Elashkar, S.S.A.; Hamed, M.E.; Fathy, M.M.; et al. Interleukin-1β and Interleukin-1receptor Antagonist Polymorphisms in Egyptian Children with Febrile Seizures. Medicine (Baltimore). 2017, 96, e6370. [Google Scholar] [CrossRef]
- Dyomina, A. V.; Zubareva, O.E.; Smolensky, I. V.; Vasilev, D.S.; Zakharova, M. V.; Kovalenko, A.A.; Schwarz, A.P.; Ischenko, A.M.; Zaitsev, A. V. Anakinra Reduces Epileptogenesis, Provides Neuroprotection, and Attenuates Behavioral Impairments in Rats in the Lithium–Pilocarpine Model of Epilepsy. Pharmaceuticals 2020, 13, 340. [Google Scholar] [CrossRef] [PubMed]
- Santtila, S.; Savinainen, K.; Hurme, M. Presence of the IL-1RA Allele 2 (IL1RN*2) Is Associated with Enhanced IL-1beta Production in Vitro. Scand. J. Immunol. 1998, 47, 195–198. [Google Scholar] [CrossRef]
- Tütüncüoğlu, S.; Kütükçüler, N.; Kepe, L.; Coker, C.; Berdeli, A.; Tekgül, H. Proinflammatory Cytokines, Prostaglandins and Zinc in Febrile Convulsions. Pediatr. Int. 2001, 43, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Niu, X.; Cheng, M.; Zeng, Q.; Deng, J.; Tian, X.; Wang, Y.; Yu, J.; Shi, W.; Wu, W.; et al. Phenotypic Spectrum and Prognosis of Epilepsy Patients With GABRG2 Variants. Front. Mol. Neurosci. 2022, 15. [Google Scholar] [CrossRef]
- Kang, J.Q.; MacDonald, R.L. Molecular Pathogenic Basis for GABRG2Mutations Associatedwith a Spectrum of Epilepsy Syndromes, from Generalized Absence Epilepsy to Dravet Syndrome. JAMA Neurol. 2016, 73, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Essrich, C.; Lorez, M.; Benson, J.A.; Fritschy, J.M.; Lüscher, B. Postsynaptic Clustering of Major GABAA Receptor Subtypes Requires the Gamma 2 Subunit and Gephyrin. Nat. Neurosci. 1998, 1, 563–571. [Google Scholar] [CrossRef]
- Haas, K.F.; Macdonald, R.L. GABA(A) Receptor Subunit Γ2 and δ Subtypes Confer Unique Kinetic Properties on Recombinant GABA(A) Receptor Currents in Mouse Fibroblasts. J. Physiol. 1999, 514, 27–45. [Google Scholar] [CrossRef]
- Huang, X.; Hernandez, C.C.; Hu, N.; Macdonald, R.L. Three Epilepsy-Associated GABRG2 Missense Mutations at the Γ+/β- Interface Disrupt GABAA Receptor Assembly and Trafficking by Similar Mechanisms but to Different Extents. Neurobiol. Dis. 2014, 68, 167–179. [Google Scholar] [CrossRef]
- Catterall, W.A. Dravet Syndrome: A Sodium Channel Interneuronopathy. Curr. Opin. Physiol. 2018, 2, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Fauser, S.; Huppertz, H.-J.; Bast, T.; Strobl, K.; Pantazis, G.; Altenmueller, D.-M.; Feil, B.; Rona, S.; Kurth, C.; Rating, D.; et al. Clinical Characteristics in Focal Cortical Dysplasia: A Retrospective Evaluation in a Series of 120 Patients. Brain 2006, 129, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Hesdorffer, D.C.; Chan, S.; Tian, H.; Allen Hauser, W.; Dayan, P.; Leary, L.D.; Hinton, V.J. Are MRI-Detected Brain Abnormalities Associated with Febrile Seizure Type? Epilepsia 2008, 49, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Luhmann, H.J. Models of Cortical Malformation--Chemical and Physical. J. Neurosci. Methods 2016, 260, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-H.; Yum, M.-S.; Lee, M.; Kim, E.-J.; Shim, W.-H.; Ko, T.-S. A New Rat Model of Epileptic Spasms Based on Methylazoxymethanol-Induced Malformations of Cortical Development. Front. Neurol. 2017, 8, 271. [Google Scholar] [CrossRef]
- Aquiles, A.; Fiordelisio, T.; Luna-Munguia, H.; Concha, L. Altered Functional Connectivity and Network Excitability in a Model of Cortical Dysplasia. Sci. Rep. 2023, 13, 12335. [Google Scholar] [CrossRef]
- Dvorák, K.; Feit, J. Migration of Neuroblasts through Partial Necrosis of the Cerebral Cortex in Newborn Rats-Contribution to the Problems of Morphological Development and Developmental Period of Cerebral Microgyria. Histological and Autoradiographical Study. Acta Neuropathol. 1977, 38, 203–212. [Google Scholar] [CrossRef]
- Scantlebury, M.H.; Gibbs, S.A.; Foadjo, B.; Lema, P.; Psarropoulou, C.; Carmant, L. Febrile Seizures in the Predisposed Brain: A New Model of Temporal Lobe Epilepsy. Ann. Neurol. 2005, 58, 41–49. [Google Scholar] [CrossRef]
- Forsgren, L.; Sidenvall, R.; Blomquist, H.K.; Heijbel, J. A Prospective Incidence Study of Febrile Convulsions. Acta Paediatr. Scand. 1990, 79, 550–557. [Google Scholar] [CrossRef]
- Galanopoulou, A.S. Dissociated Gender-Specific Effects of Recurrent Seizures on GABA Signaling in CA1 Pyramidal Neurons: Role of GABAA Receptors. J. Neurosci. 2008, 28, 1557–1567. [Google Scholar] [CrossRef]
- Nunez, J.L.; McCarthy, M.M. Evidence for an Extended Duration of GABA-Mediated Excitation in the Developing Male Versus Female Hippocampus. Dev. Neurobiol. 2007, 14, 1879–1890. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Excitatory Actions of GABA during Development: The Nature of the Nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef]
- Kight, K.E.; McCarthy, M.M. Androgens and the Developing Hippocampus. Biol. Sex Differ. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Kloc, M.L.; Marchand, D.H.; Holmes, G.L.; Pressman, R.D.; Barry, J.M. Cognitive Impairment Following Experimental Febrile Seizures Is Determined by Sex and Seizure Duration. Epilepsy Behav. 2022, 126, 108430. [Google Scholar] [CrossRef] [PubMed]
- Tancredi, V.; D’Arcangelo, G.; Zona, C.; Siniscalchi, A.; Avoli, M. Induction of Epileptiform Activity by Temperature Elevation in Hippocampal Slices from Young Rats: An In Vitro Model for Febrile Seizures? Epilepsia 1992, 33, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Weninger, J.; Meseke, M.; Rana, S.; Förster, E. Heat-Shock Induces Granule Cell Dispersion and Microgliosis in Hippocampal Slice Cultures. Front. Cell Dev. Biol. 2021, 9, 1–16. [Google Scholar] [CrossRef]
- Hájos, N.; Mody, I. Establishing a Physiological Environment for Visualized in Vitro Brain Slice Recordings by Increasing Oxygen Supply and Modifying ACSF Content. J. Neurosci. Methods 2009, 183, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Postnikova, T.Y.; Griflyuk, A. V.; Amakhin, D. V.; Kovalenko, A.A.; Soboleva, E.B.; Zubareva, O.E.; Zaitsev, A. V. Early Life Febrile Seizures Impair Hippocampal Synaptic Plasticity in Young Rats. Int. J. Mol. Sci. 2021, 22, 8218. [Google Scholar] [CrossRef]
- Ferraro, T.N.; Golden, G.T.; Smith, G.G.; DeMuth, D.; Buono, R.J.; Berrettini, W.H. Mouse Strain Variation in Maximal Electroshock Seizure Threshold. Brain Res. 2002, 936, 82–86. [Google Scholar] [CrossRef]
- Kosobud, A.E.; Crabbe, J.C. Genetic Correlations among Inbred Strain Sensitivities to Convulsions Induced by 9 Convulsant Drugs. Brain Res. 1990, 526, 8–16. [Google Scholar] [CrossRef]
- Yushko, L. V.; Kotova, M.M.; Vyunova, T. V.; Kalueff, A. V. Experimental Zebrafish Models of Mitochondrial Dysfunction in the Pathogenesis of CNS Diseases. J. Evol. Biochem. Physiol. 2023 596 2024, 59, 2114–2128. [Google Scholar] [CrossRef]
- Dinday, M.T.; Baraban, S.C. Large-Scale Phenotype-Based Antiepileptic Drug Screening in a Zebrafish Model of Dravet Syndrome. eNeuro 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Adhish, M.; Manjubala, I. Effectiveness of Zebrafish Models in Understanding Human Diseases—A Review of Models. Heliyon 2023, 9, e14557. [Google Scholar] [CrossRef] [PubMed]
- Meshalkina, D.A.; Kysil, E. V.; Warnick, J.E.; Demin, K.A.; Kalueff, A. V. Adult Zebrafish in CNS Disease Modeling: A Tank That’s Half-Full, Not Half-Empty, and Still Filling. Lab Anim. (NY). 2017, 46, 378–387. [Google Scholar] [CrossRef]
- Stewart, A.M.; Ullmann, J.F.P.; Norton, W.H.J.; Parker, M.O.; Brennan, C.H.; Gerlai, R.; Kalueff, A. V Molecular Psychiatry of Zebrafish. Mol. Psychiatry 2015, 20, 2–17. [Google Scholar] [CrossRef]
- Khan, D.; Dupper, A.; Deshpande, T.; Graan, P.N.E.D.; Steinhäuser, C.; Bedner, P. Experimental Febrile Seizures Impair Interastrocytic Gap Junction Coupling in Juvenile Mice. J. Neurosci. Res. 2016, 94, 804–813. [Google Scholar] [CrossRef]
- Nolte, C.; Matyash, M.; Pivneva, T.; Schipke, C.G.; Ohlemeyer, C.; Hanisch, U.-K.; Kirchhoff, F.; Kettenmann, H. GFAP Promoter-Controlled EGFP-Expressing Transgenic Mice: A Tool to Visualize Astrocytes and Astrogliosis in Living Brain Tissue. Glia 2001, 33, 72–86. [Google Scholar] [CrossRef]
- Hanci, F.; Türay, S.; Balci, P.; Kabakuş, N. Reflex Epilepsy with Hot Water: Clinical and EEG Findings, Treatment, and Prognosis in Childhood. Neuropediatrics 2020, 51, 336–340. [Google Scholar] [CrossRef]
- Hoogland, G.; Raijmakers, M.; Clynen, E.; Brône, B.; Rigo, J.-M.; Swijsen, A. Experimental Early-Life Febrile Seizures Cause a Sustained Increase in Excitatory Neurotransmission in Newborn Dentate Granule Cells. Brain Behav. 2022, 12, e2505. [Google Scholar] [CrossRef]
- Keum, H.R.; Lee, S.J.; Kim, J.M.; Kim, S.W.; Baek, H.S.; Byun, J.C.; Kim, Y.K.; Kim, S.; Lee, J.M. Seasonal Trend of Viral Prevalence and Incidence of Febrile Convulsion: A Korea Public Health Data Analysis. Child. (Basel, Switzerland) 2023, 10. [Google Scholar] [CrossRef]
- Leon, L.R. Invited Review: Cytokine Regulation of Fever: Studies Using Gene Knockout Mice. J. Appl. Physiol. 2002, 92, 2648–2655. [Google Scholar] [CrossRef] [PubMed]
- Haveman, J.; Geerdink, A.G.; Rodermond, H.M. Cytokine Production after Whole Body and Localized Hyperthermia. Int. J. Hyperth. 1996, 12, 791–800. [Google Scholar] [CrossRef]
- Dubé, C.; Vezzani, A.; Behrens, M.; Bartfai, T.; Baram, T.Z. Interleukin-1β Contributes to the Generation of Experimental Febrile Seizures. Ann. Neurol. 2005, 57, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Oladehin, A.; Blatteis, C.M. Induction of Fos Protein in Neonatal Rat Hypothalami Following Intraperitoneal Endotoxin Injection. Ann. N. Y. Acad. Sci. 1997, 813, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Zubareva, O.E.; Postnikova, T.Y.; Grifluk, A. V.; Schwarz, A.P.; Smolensky, I. V.; Karepanov, A.A.; Vasilev, D.S.; Veniaminova, E.A.; Rotov, A.Y.; Kalemenev, S. V.; et al. Exposure to Bacterial Lipopolysaccharide in Early Life Affects the Expression of Ionotropic Glutamate Receptor Genes and Is Accompanied by Disturbances in Long-Term Potentiation and Cognitive Functions in Young Rats. Brain. Behav. Immun. 2020, 90, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Postnikova, T.Y.; Griflyuk, A. V.; Ergina, J.L.; Zubareva, O.E.; Zaitsev, A. V. Administration of Bacterial Lipopolysaccharide during Early Postnatal Ontogenesis Induces Transient Impairment of Long-Term Synaptic Plasticity Associated with Behavioral Abnormalities in Young Rats. Pharmaceuticals 2020, 13, 48. [Google Scholar] [CrossRef]
- Heida, J.G.; Teskey, G.C.; Pittman, Q.J. Febrile Convulsions Induced by the Combination of Lipopolysaccharide and Low-Dose Kainic Acid Enhance Seizure Susceptibility, Not Epileptogenesis, in Rats. Epilepsia 2005, 46, 1898–1905. [Google Scholar] [CrossRef]
- Banks, W. a; Robinson, S.M. Minimal Penetration of LPS across the Murine BBB. Brain Behav. Immunol. 2010, 24, 102–109. [Google Scholar] [CrossRef]
- Singh, A.K.; Jiang, Y. How Does Peripheral Lipopolysaccharide Induce Gene Expression in the Brain of Rats? Toxicology 2004, 201, 197–207. [Google Scholar] [CrossRef]
- Lucile, Capuron; Miller, A.H. Immune System to Brain Signaling. Pharmacol. Ther. 2011, 130, 226–238. [Google Scholar] [CrossRef]
- Perry, V.H. The Influence of Systemic Inflammation on Inflammation in the Brain: Implications for Chronic Neurodegenerative Disease. Brain. Behav. Immun. 2004, 18, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Litvin, Y.; Tovote, P.; Pentkowski, N.S.; Zeyda, T.; King, L.B.; Vasconcellos, A.J.; Dunlap, C.; Spiess, J.; Blanchard, D.C.; Blanchard, R.J. Maternal Separation Modulates Short-Term Behavioral and Physiological Indices of the Stress Response. Horm. Behav. 2010, 58, 241–249. [Google Scholar] [CrossRef]
- Abraham, M.; Schmerder, K.; Hedtstück, M.; Bösing, K.; Mundorf, A.; Freund, N. Maternal Separation and Its Developmental Consequences on Anxiety and Parvalbumin Interneurons in the Amygdala. J. Neural Transm. 2023, 130, 1167–1175. [Google Scholar] [CrossRef]
- Kim, E.G.; Chang, W.; Shin, S.Y.; Adhikari, A.S.; Seol, G.H.; Song, D.Y.; Min, S.S. Maternal Separation in Mice Leads to Anxiety-like/Aggressive Behavior and Increases Immunoreactivity for Glutamic Acid Decarboxylase and Parvalbumin in the Adolescence Ventral Hippocampus. Korean J. Physiol. Pharmacol. 2023, 27, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ramirez, M.; Salgado-Ceballos, H.; Orozco-Suarez, S.A.; Rocha, L. Hyperthermic Seizures and Hyperthermia in Immature Rats Modify the Subsequent Pentylenetetrazole-Induced Seizures. Seizure 2009, 18, 533–536. [Google Scholar] [CrossRef]
- Brewster, A.; Bender, R.A.; Chen, Y.; Dube, C.; Eghbal-Ahmadi, M.; Baram, T.Z. Developmental Febrile Seizures Modulate Hippocampal Gene Expression of Hyperpolarization-Activated Channels in an Isoform- and Cell-Specific Manner. J. Neurosci. 2002, 22, 4591–4599. [Google Scholar] [CrossRef]
- Kaindl, A.M.; Koppelstaetter, A.; Nebrich, G.; Stuwe, J.; Sifringer, M.; Zabel, C.; Klose, J.; Ikonomidou, C. Brief Alteration of NMDA or GABAA Receptor Mediated Neurotransmission Has Long Term Effects on the Developing Cerebral Cortex. Mol. Cell. Proteomics 2008, 7, 2293–2310. [Google Scholar] [CrossRef]
- Huizenga, M.N.; Forcelli, P.A. Neuroprotective Action of the CB1/2 Receptor Agonist, WIN 55,212-2, against DMSO but Not Phenobarbital-Induced Neurotoxicity in Immature Rats. Neurotox. Res. 2020, 35, 173–182. [Google Scholar] [CrossRef]
- Kaushal, S.; Tamer, Z.; Opoku, F.; Forcelli, P.A. Anticonvulsant Drug-Induced Cell Death in the Developing White Matter of the Rodent Brain. Epilepsia 2016, 57, 727–734. [Google Scholar] [CrossRef]
- Ikonomidou, C. Triggers of Apoptosis in the Immature Brain. Brain Dev. 2009, 31, 488–492. [Google Scholar] [CrossRef]
- Torolira, D.; Suchomelova, L.; Wasterlain, C.G.; Niquet, J. Phenobarbital and Midazolam Increase Neonatal Seizure-Associated Neuronal Injury. Ann. Neurol. 2017, 82, 115–120. [Google Scholar] [CrossRef]
- Bender, R.A.; Dubé, C.; Gonzalez-Vega, R.; Mina, E.W.; Baram, T.Z. Mossy Fiber Plasticity and Enhanced Hippocampal Excitability, without Hippocampal Cell Loss or Altered Neurogenesis, in an Animal Model of Prolonged Febrile Seizures. Hippocampus 2003, 13, 399–412. [Google Scholar] [CrossRef]
- Griflyuk, A. V.; Postnikova, T.Y.; Malkin, S.L.; Zaitsev, A. V. Alterations in Rat Hippocampal Glutamatergic System Properties after Prolonged Febrile Seizures. Int. J. Mol. Sci. 2023, 24, 16875. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.W.; Brady, R.J.; Martin, D.L. Postnatal Development of GABA-Mediated Synaptic Inhibition in Rat Hippocampus. Neuroscience 1989, 28, 551–561. [Google Scholar] [CrossRef]
- Toth, Z.; Yan, X.-X.X.; Haftoglou, S.; Ribak, C.E.; Baram, T.Z. Seizure-Induced Neuronal Injury: Vulnerability to Febrile Seizures in an Immature Rat Model. J. Neurosci. 1998, 18, 4285–4294. [Google Scholar] [CrossRef]
- Gallyas, F.; Güldner, F.H.; Zoltay, G.; Wolff, J.R. Golgi-like Demonstration of “Dark” Neurons with an Argyrophil III Method for Experimental Neuropathology. Acta Neuropathol. 1990, 79, 620–628. [Google Scholar] [CrossRef]
- Gallyas, F.; Farkas, O.; Mázló, M. Gel-to-Gel Phase Transition May Occur in Mammalian Cells: Mechanism of Formation of “Dark” (Compacted) Neurons. Biol. cell 2004, 96, 313–324. [Google Scholar] [CrossRef]
- Gallyas, F.; Csordás, A.; Schwarcz, A.; Mázló, M. “Dark” (Compacted) Neurons May Not Die through the Necrotic Pathway. Exp. brain Res. 2005, 160, 473–486. [Google Scholar] [CrossRef]
- Tóth, A.; Kátai, E.; Kálmán, E.; Bogner, P.; Schwarcz, A.; Dóczi, T.; Sík, A.; Pál, J. In Vivo Detection of Hyperacute Neuronal Compaction and Recovery by MRI Following Electric Trauma in Rats. J. Magn. Reson. Imaging 2016, 44, 814–822. [Google Scholar] [CrossRef]
- Vasilev, D.S.; Tumanova, N.L.; Kim, K.K.; Lavrentyeva, V. V.; Lukomskaya, N.Y.; Zhuravin, I.A.; Magazanik, L.G.; Zaitsev, A. V. Transient Morphological Alterations in the Hippocampus After Pentylenetetrazole-Induced Seizures in Rats. Neurochem. Res. 2018, 43, 1671–1682. [Google Scholar] [CrossRef]
- Raijmakers, M.; Clynen, E.; Smisdom, N.; Nelissen, S.; Brône, B.; Rigo, J.M.; Hoogland, G.; Swijsen, A. Experimental Febrile Seizures Increase Dendritic Complexity of Newborn Dentate Granule Cells. Epilepsia 2016, 57, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.L.; Pun, R.Y.K.; Yin, H.; Faulkner, C.R.; Loepke, A.W.; Danzer, S.C. Heterogeneous Integration of Adult-Generated Granule Cells into the Epileptic Brain. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Chen, Y.-B.; Zhang, Y.-F. The Rearrangement of Actin Cytoskeleton in Mossy Fiber Synapses in a Model of Experimental Febrile Seizures. Front. Neurol. 2023, 14, 1107538. [Google Scholar] [CrossRef] [PubMed]
- Łotowska, J.; Sobaniec, P.; Sobaniec-Łotowska, M.; Szukiel, B.; Łukasik, M.; Łotowska, S. Effects of Topiramate on the Ultrastructure of Synaptic Endings in the Hippocampal CA1 and CA3 Sectors in the Rat Experimental Model of Febrile Seizures: The First Electron Microscopy Report. Folia Neuropathol. 2019, 57, 267–276. [Google Scholar] [CrossRef]
- Sobaniec, P.; Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Zochowska-Sobaniec, M. Influence of Topiramate on the Synaptic Endings of the Temporal Lobe Neocortex in an Experimental Model of Hyperthermia-Induced Seizures: An Ultrastructural Study. Brain Sci. 2021, 11. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, F.; Zhang, J.; Gao, H.; Yang, Y.; Fu, R. Repeated Febrile Convulsions Impair Hippocampal Neurons and Cause Synaptic Damage in Immature Rats: Neuroprotective Effect of Fructose-1,6-Diphosphate. Neural Regen. Res. 2014, 9, 937–942. [Google Scholar] [CrossRef]
- Dubé, C.M.; Ravizza, T.; Hamamura, M.; Zha, Q.; Keebaugh, A.; Fok, K.; Andres, A.L.; Nalcioglu, O.; Obenaus, A.; Vezzani, A.; et al. Epileptogenesis Provoked by Prolonged Experimental Febrile Seizures: Mechanisms and Biomarkers. J. Neurosci. 2010, 30, 7484–7494. [Google Scholar] [CrossRef]
- Patterson, K.P.; Brennan, G.P.; Curran, M.; Kinney-Lang, E.; Dubé, C.; Rashid, F.; Ly, C.; Obenaus, A.; Baram, T.Z. Rapid, Coordinate Inflammatory Responses after Experimental Febrile Status Epilepticus: Implications for Epileptogenesis. eNeuro 2015, 2, 1–13. [Google Scholar] [CrossRef]
- Griflyuk, A. V.; Postnikova, T.Y.; Zaitsev, A. V. Prolonged Febrile Seizures Impair Synaptic Plasticity and Alter Developmental Pattern of Glial Fibrillary Acidic Protein (GFAP)-Immunoreactive Astrocytes in the Hippocampus of Young Rats. Int. J. Mol. Sci. 2022, 23, 12224. [Google Scholar] [CrossRef]
- Shen, W.; Pristov, J.B.; Nobili, P.; Nikolić, L. Can Glial Cells Save Neurons in Epilepsy? Neural Regen. Res. 2023, 18, 1417–1422. [Google Scholar] [CrossRef]
- Pannasch, U.; Vargová, L.; Reingruber, J.; Ezan, P.; Holcman, D.; Giaume, C.; Syková, E.; Rouach, N. Astroglial Networks Scale Synaptic Activity and Plasticity. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 8467–8472. [Google Scholar] [CrossRef] [PubMed]
- Łotowska, J.M.; Sobaniec-Łotowska, M.E.; Sobaniec, W. Ultrastructural Features of Astrocytes in the Cortex of the Hippocampal Gyrus and in the Neocortex of the Temporal Lobe in an Experimental Model of Febrile Seizures and with the Use of Topiramate. Folia Neuropathol. 2009, 47, 268–277. [Google Scholar] [PubMed]
- Łotowska, J.M.; Sobaniec-Łotowska, M.E.; Sendrowski, K.; Sobaniec, W.; Artemowicz, B. Ultrastructure of the Blood-Brain Barrier of the Gyrus Hippocampal Cortex in an Experimental Model of Febrile Seizures and with the Use of a New Generation Antiepileptic Drug--Topiramate. Folia Neuropathol. 2008, 46, 57–68. [Google Scholar] [PubMed]
- Garcia-Curran, M.M.; Hall, A.M.; Patterson, K.P.; Shao, M.; Eltom, N.; Chen, K.; Dubé, C.M.; Baram, T.Z. Dexamethasone Attenuates Hyperexcitability Provoked by Experimental Febrile Status Epilepticus. eNeuro 2019, 6. [Google Scholar] [CrossRef]
- Kong, W.; Wang, X.; Yang, X.; Huang, W.; Han, S.; Yin, J.; Liu, W.; He, X.; Peng, B. Activation of TRPV1 Contributes to Recurrent Febrile Seizures via Inhibiting the Microglial M2 Phenotype in the Immature Brain. Front. Cell. Neurosci. 2019, 13, 442. [Google Scholar] [CrossRef]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and Macrophage Polarization—New Prospects for Brain Repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Zaniani, N.R.; Roohbakhsh, A.; Moghimi, A.; Mehri, S. Protective Effect of Toll-like Receptor 4 Antagonist on Inflammation, EEG, and Memory Changes Following Febrile Seizure in Wistar Rats. Behav. Brain Res. 2022, 420, 113723. [Google Scholar] [CrossRef]
- Yu, Y.H.; Kim, S.-W.; Im, H.; Lee, Y.R.; Kim, G.W.; Ryu, S.; Park, D.-K.; Kim, D.-S. Febrile Seizure Causes Deficit in Social Novelty, Gliosis, and Proinflammatory Cytokine Response in the Hippocampal CA2 Region in Rats. Cells 2023, 12. [Google Scholar] [CrossRef]
- Kim, J.A.; Connors, B.W. High Temperatures Alter Physiological Properties of Pyramidal Cells and Inhibitory Interneurons in Hippocampus. Front. Cell. Neurosci. 2012, 6, 1–12. [Google Scholar] [CrossRef]
- Postnikova, T.Y.; Griflyuk, A. V; Zhigulin, A.S.; Soboleva, E.B.; Barygin, O.I.; Amakhin, D. V; Zaitsev, A. V Febrile Seizures Cause a Rapid Depletion of Calcium-Permeable AMPA Receptors at the Synapses of Principal Neurons in the Entorhinal Cortex and Hippocampus of the Rat. Int. J. Mol. Sci. 2023, 24, 12621. [Google Scholar] [CrossRef]
- Crespo, M.; León-Navarro, D.A.; Martín, M. Glutamatergic System Is Affected in Brain from an Hyperthermia-Induced Seizures Rat Model. Cell. Mol. Neurobiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zaitsev, A. V.; Smolensky, I. V.; Jorratt, P.; Ovsepian, S. V. Neurobiology, Functions, and Relevance of Excitatory Amino Acid Transporters (EAATs) to Treatment of Refractory Epilepsy. CNS Drugs 2020, 34, 1089–1103. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Baram, T.Z.; Soltesz, I. Febrile Seizures in the Developing Brain Result in Persistent Modification of Neuronal Excitability in Limbic Circuits. Nat. Med. 1999, 5, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Dube, C.; Chen, K.; Eghbal-Ahmadi, M.; Brunson, K.; Soltesz, I.; Baram, T.Z.; Eghbal-Ahmadi, M.; Brunson, K.; Soltesz, I.; Baram, T.Z.; et al. Prolonged Febrile Seizures in the Immature Rat Model Enhance Hippocampal Excitability Long Term. Ann. Neurol. 2000, 47, 336–344. [Google Scholar] [CrossRef]
- Lillis, K.P.; Kramer, M.A.; Mertz, J.; Staley, K.J.; White, J.A. Pyramidal Cells Accumulate Chloride at Seizure Onset. Neurobiol. Dis. 2012, 47, 358–366. [Google Scholar] [CrossRef]
- Librizzi, L.; Losi, G.; Marcon, I.; Sessolo, M.; Scalmani, P.; Carmignoto, G.; de Curtis, M. Interneuronal Network Activity at the Onset of Seizure-Like Events in Entorhinal Cortex Slices. J. Neurosci. 2017, 37, 10398–10407. [Google Scholar] [CrossRef]
- Swijsen, A.; Brône, B.; Rigo, J.-M.; Hoogland, G. Long-Lasting Enhancement of GABA(A) Receptor Expression in Newborn Dentate Granule Cells after Early-Life Febrile Seizures. Dev. Neurobiol. 2012, 72, 1516–1527. [Google Scholar] [CrossRef]
- Crespo, M.; León-Navarro, D.A.; Martín, M. Na(+)/K(+)- and Mg(2+)-ATPases and Their Interaction with AMPA, NMDA and D(2) Dopamine Receptors in an Animal Model of Febrile Seizures. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Gregory, K.J.; Goudet, C. International Union of Basic and Clinical Pharmacology. CXI. Pharmacology, Signaling, and Physiology of Metabotropic Glutamate Receptors. Pharmacol. Rev. 2021, 73, 521–569. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic Glutamate Receptors: Physiology, Pharmacology, and Disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xiao, W.; Wang, Y.; Li, J.; Gong, J.; Tu, E.; Long, L.; Xiao, B.; Yan, X.; Wan, L. Metabotropic Glutamate Receptors (MGluRs) in Epileptogenesis: An Update on Abnormal MGluRs Signaling and Its Therapeutic Implications. Neural Regen. Res. 2024, 19, 360–368. [Google Scholar] [CrossRef]
- Notenboom, R.G.E.; Ramakers, G.M.J.J.; Kamal, A.; Spruijt, B.M.; De Graan, P.N.E. Long-Lasting Modulation of Synaptic Plasticity in Rat Hippocampus after Early-Life Complex Febrile Seizures. Eur. J. Neurosci. 2010, 32, 749–758. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Kuo, Y.-M.; Huang, A.-M.; Huang, C.-C. Repetitive Febrile Seizures in Rat Pups Cause Long-Lasting Deficits in Synaptic Plasticity and NR2A Tyrosine Phosphorylation. Neurobiol. Dis. 2005, 18, 466–475. [Google Scholar] [CrossRef]
- Harris, K.M.; Teyler, T.J. Developmental Onset of Long-Term Potentiation in Area CA1 of the Rat Hippocampus. J. Physiol. 1984, 346, 27–48. [Google Scholar] [CrossRef]
- Jackson, P.S.; Suppes, T.; Harris, K.M. Stereotypical Changes in the Pattern and Duration of Long-Term Potentiation Expressed at Postnatal Days 11 and 15 in the Rat Hippocampus. J. Neurophysiol. 1993, 70, 1412–1419. [Google Scholar] [CrossRef]
- Muller, D.; Oliver, M.; Lynch, G. Developmental Changes in Synaptic Properties in Hippocampus of Neonatal Rats. Brain Res. Dev. Brain Res. 1989, 49, 105–114. [Google Scholar] [CrossRef]
- Cao, G.; Harris, K.M. Developmental Regulation of the Late Phase of Long-Term Potentiation (L-LTP) and Metaplasticity in Hippocampal Area CA1 of the Rat. J. Neurophysiol. 2012, 107, 902–912. [Google Scholar] [CrossRef]
- Benveniste, M.; Clements, J.; Vyklický, L.; Mayer, M.L. A Kinetic Analysis of the Modulation of N-Methyl-D-Aspartic Acid Receptors by Glycine in Mouse Cultured Hippocampal Neurones. J. Physiol. 1990, 428, 333–357. [Google Scholar] [CrossRef]
- Cummings, K.A.; Popescu, G.K. Glycine-Dependent Activation of NMDA Receptors. J. Gen. Physiol. 2015, 145, 513–527. [Google Scholar] [CrossRef]
- Henneberger, C.; Papouin, T.; Oliet, S.H.R.; Rusakov, D.A. Long-Term Potentiation Depends on Release of d-Serine from Astrocytes. Nature 2010. [Google Scholar] [CrossRef] [PubMed]
- Dubé, C.; Richichi, C.; Bender, R.A.; Chung, G.; Litt, B.; Baram, T.Z. Temporal Lobe Epilepsy after Experimental Prolonged Febrile Seizures: Prospective Analysis. Brain 2006, 129, 911–922. [Google Scholar] [CrossRef]
- Kipp, K.H.; Mecklinger, A.; Becker, M.; Reith, W.; Gortner, L. Infant Febrile Seizures: Changes in Declarative Memory as Revealed by Event-Related Potentials. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2010, 121, 2007–2016. [Google Scholar] [CrossRef] [PubMed]
- Martinos, M.M.; Yoong, M.; Patil, S.; Chin, R.F.M.; Neville, B.G.; Scott, R.C.; de Haan, M.; Haan, M. De Recognition Memory Is Impaired in Children after Prolonged Febrile Seizures. Brain 2012, 135, 3153–3164. [Google Scholar] [CrossRef] [PubMed]
- Yagoubi, N.; Jomni, Y.; Sakly, M. Hyperthermia-Induced Febrile Seizures Have Moderate and Transient Effects on Spatial Learning in Immature Rats. Behav. Neurol. 2015, 2015, 924303. [Google Scholar] [CrossRef] [PubMed]
- Dubé, C.M.; Zhou, J.-L.; Hamamura, M.; Zhao, Q.; Ring, A.; Abrahams, J.; McIntyre, K.; Nalcioglu, O.; Shatskih, T.; Baram, T.Z.; et al. Cognitive Dysfunction after Experimental Febrile Seizures. Exp. Neurol. 2009, 215, 167–177. [Google Scholar] [CrossRef]
- Dai, Y.; Wu, D.; Feng, B.; Chen, B.; Tang, Y.; Jin, M.; Zhao, H.; Dai, H.; Wang, Y.; Chen, Z. Prolonged Febrile Seizures Induce Inheritable Memory Deficits in Rats through DNA Methylation. CNS Neurosci. Ther. 2019, 25, 601–611. [Google Scholar] [CrossRef]
- Alese, O.O.; Mabandla, M. V Transgenerational Deep Sequencing Revealed Hypermethylation of Hippocampal MGluR1 Gene with Altered MRNA Expression of MGluR5 and MGluR3 Associated with Behavioral Changes in Sprague Dawley Rats with History of Prolonged Febrile Seizure. PLoS One 2019, 14, e0225034. [Google Scholar] [CrossRef]
- Yu, Y.H.; Kim, S.-W.; Im, H.; Song, Y.; Kim, S.J.; Lee, Y.R.; Kim, G.W.; Hwang, C.; Park, D.-K.; Kim, D.-S. Febrile Seizures Cause Depression and Anxiogenic Behaviors in Rats. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Crespo, M.; León-Navarro, D.A.; Martín, M. Early-Life Hyperthermic Seizures Upregulate Adenosine A2A Receptors in the Cortex and Promote Depressive-like Behavior in Adult Rats. Epilepsy Behav. 2018, 86, 173–178. [Google Scholar] [CrossRef]
Figure 1.
Dynamics of key developmental processes of the human and rodent hippocampus during prenatal and early postnatal development (except for gliogenesis and myelination in humans, these data are presented for the cerebral cortex) [22,26,27,28,29,30,31,32,33,34].
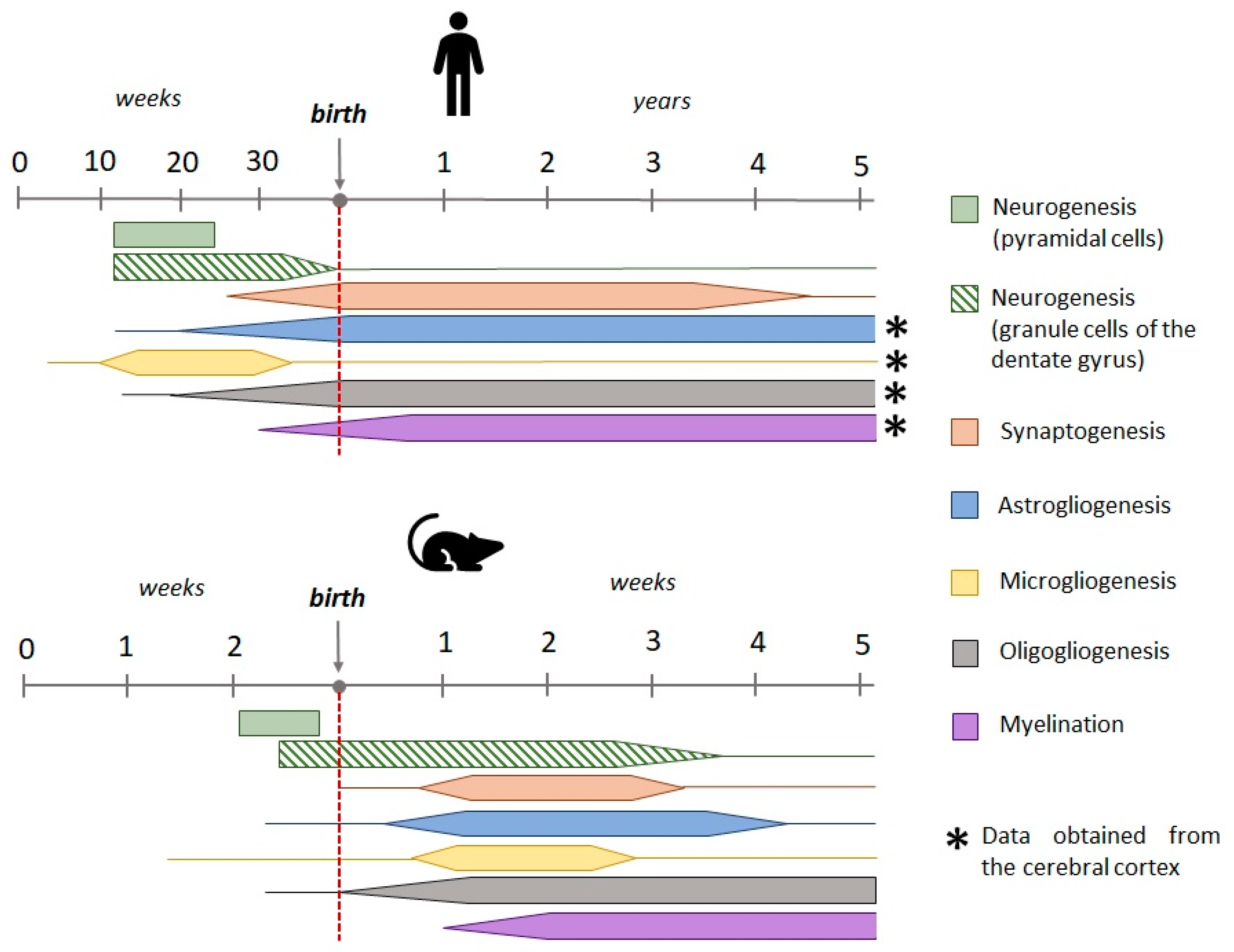
Figure 2.
The etiology of FSs. The most common cause of FSs is a viral or bacterial infection, which results in the release of pro-inflammatory cytokines and fever. Both pro-inflammatory cytokines (paragraph 5.3) and elevated brain temperature (paragraph 5.2) result in enhanced neuronal excitability. An increase in blood pH, which is indicative of respiratory alkalosis, has been observed in individuals experiencing fever [44,45]. This phenomenon has also been linked to an elevation in neuronal excitability [46]. Specific genetic mutations may elevate the probability of developing FSs or increase the likelihood of FSs becoming more severe (paragraph 3.1). An elevation in body temperature that is not infectious in nature also results in the release of pro-inflammatory cytokines (paragraph 5.2). Cortical malformations have been identified as a potential cause of both FSs and epilepsy (paragraph 3.2). [47,48,49,50,51,52,53,54,55,56,57]. EP3 - prostaglandin E2 receptor 3, IL10 - Interleukin 10, IL-1β - Interleukin-1 beta.
Figure 2.
The etiology of FSs. The most common cause of FSs is a viral or bacterial infection, which results in the release of pro-inflammatory cytokines and fever. Both pro-inflammatory cytokines (paragraph 5.3) and elevated brain temperature (paragraph 5.2) result in enhanced neuronal excitability. An increase in blood pH, which is indicative of respiratory alkalosis, has been observed in individuals experiencing fever [44,45]. This phenomenon has also been linked to an elevation in neuronal excitability [46]. Specific genetic mutations may elevate the probability of developing FSs or increase the likelihood of FSs becoming more severe (paragraph 3.1). An elevation in body temperature that is not infectious in nature also results in the release of pro-inflammatory cytokines (paragraph 5.2). Cortical malformations have been identified as a potential cause of both FSs and epilepsy (paragraph 3.2). [47,48,49,50,51,52,53,54,55,56,57]. EP3 - prostaglandin E2 receptor 3, IL10 - Interleukin 10, IL-1β - Interleukin-1 beta.
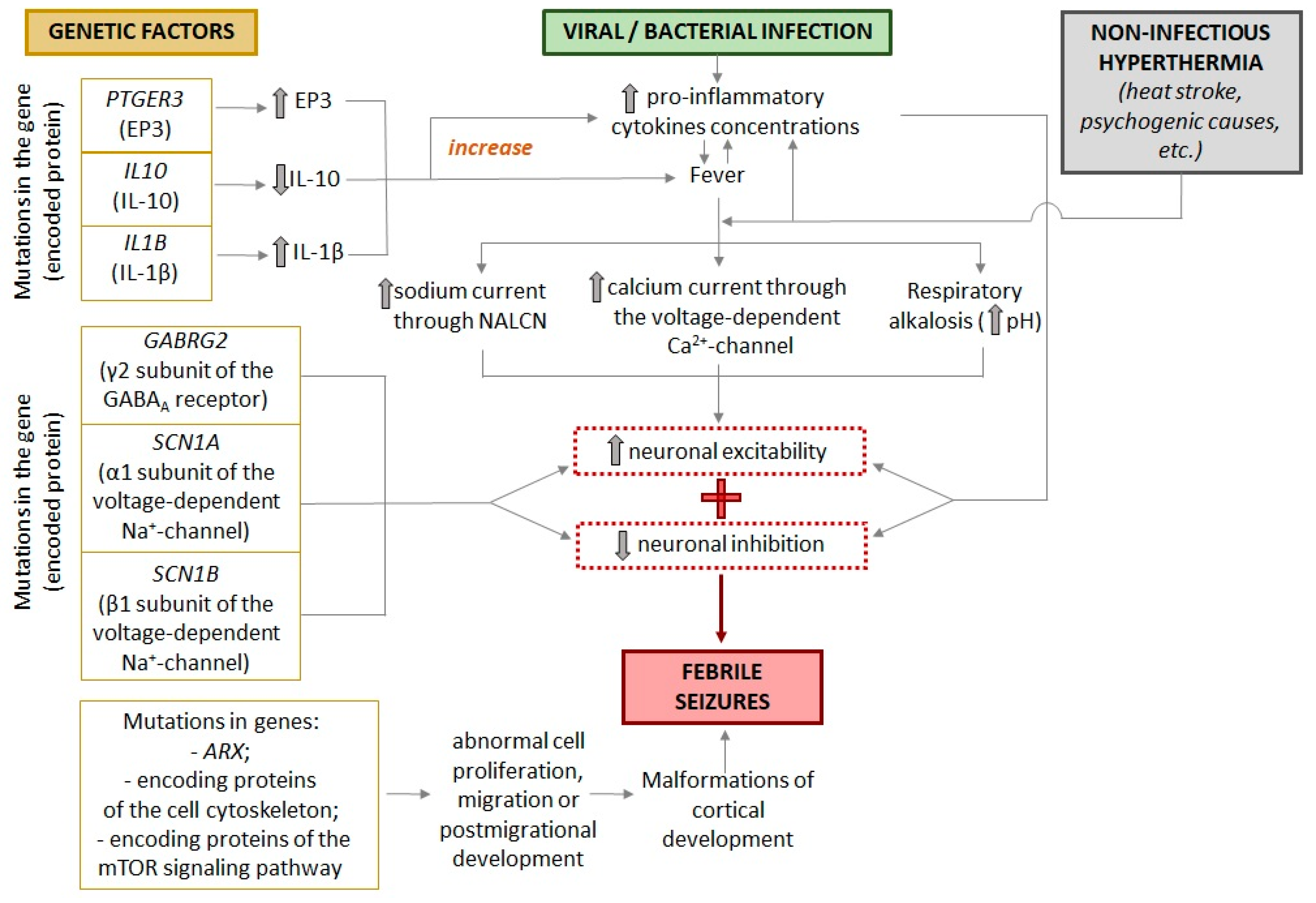
Figure 3.
A model of bacterial infection during lipopolysaccharide (LPS) administration. There is minimal direct penetration of LPS across the blood-brain barrier. The effect of LPS on the brain is primarily mediated by pro-inflammatory cytokines, which are secreted by macrophages in response to LPS binding to receptors. Subsequently, the cytokines act via perivascular endothelial cells and afferent nerve fibers in the vagus nerve. This results in the stimulation of the enzyme cyclooxygenase-2, which converts arachidonic acid to prostaglandin E2, thereby inducing fever. Concurrently, glial cells within the brain also generate pro-inflammatory cytokines that regulate neurotransmission and may be implicated in the generation of seizures during fever [108,109,110,111].
Figure 3.
A model of bacterial infection during lipopolysaccharide (LPS) administration. There is minimal direct penetration of LPS across the blood-brain barrier. The effect of LPS on the brain is primarily mediated by pro-inflammatory cytokines, which are secreted by macrophages in response to LPS binding to receptors. Subsequently, the cytokines act via perivascular endothelial cells and afferent nerve fibers in the vagus nerve. This results in the stimulation of the enzyme cyclooxygenase-2, which converts arachidonic acid to prostaglandin E2, thereby inducing fever. Concurrently, glial cells within the brain also generate pro-inflammatory cytokines that regulate neurotransmission and may be implicated in the generation of seizures during fever [108,109,110,111].
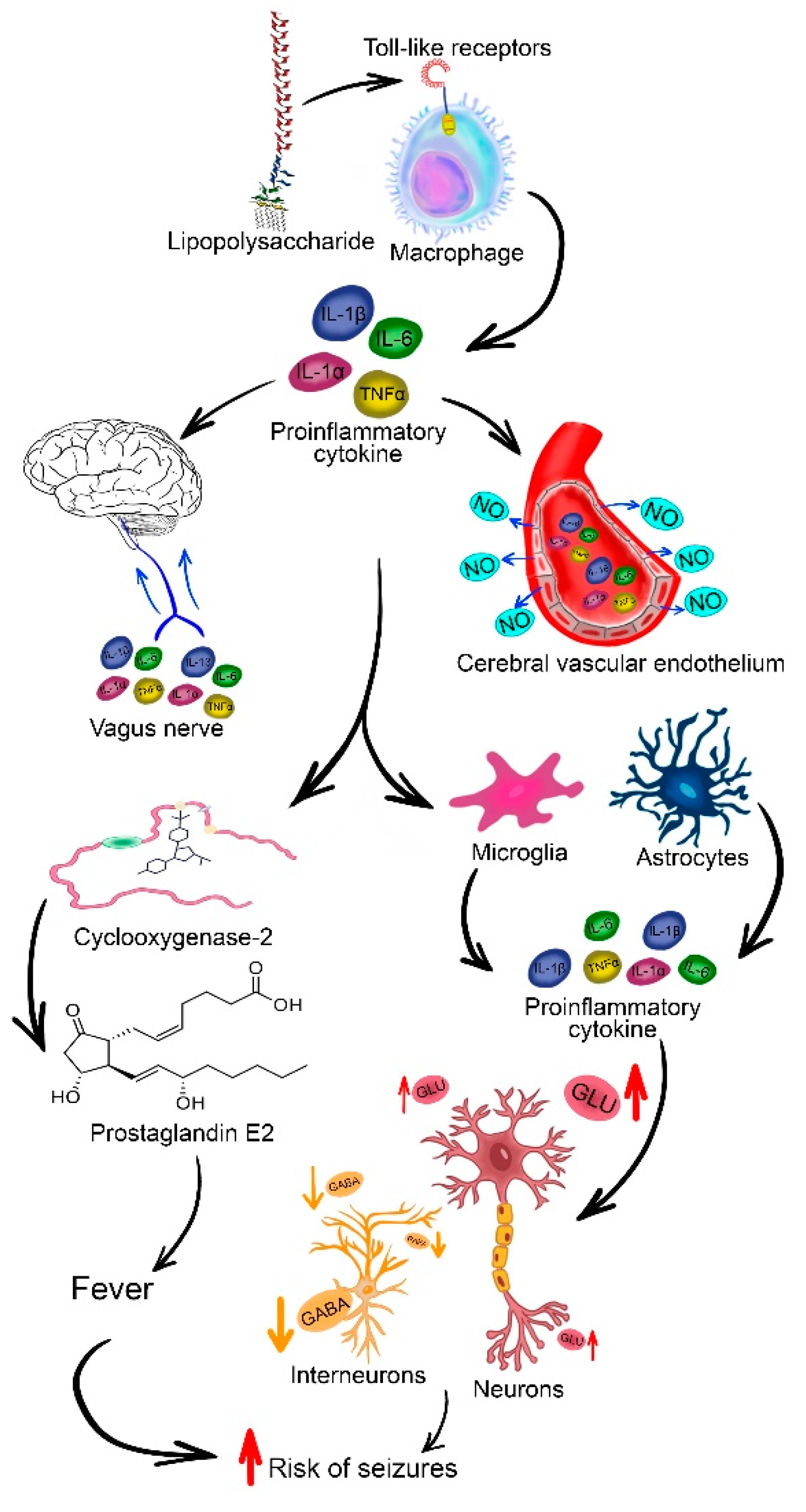
Figure 4.
Required control groups. The stress of weaning a pup from the nest can be assessed in isolated animals. The contribution of hyperthermia itself can be assessed by using barbiturates to block seizures.
Figure 4.
Required control groups. The stress of weaning a pup from the nest can be assessed in isolated animals. The contribution of hyperthermia itself can be assessed by using barbiturates to block seizures.
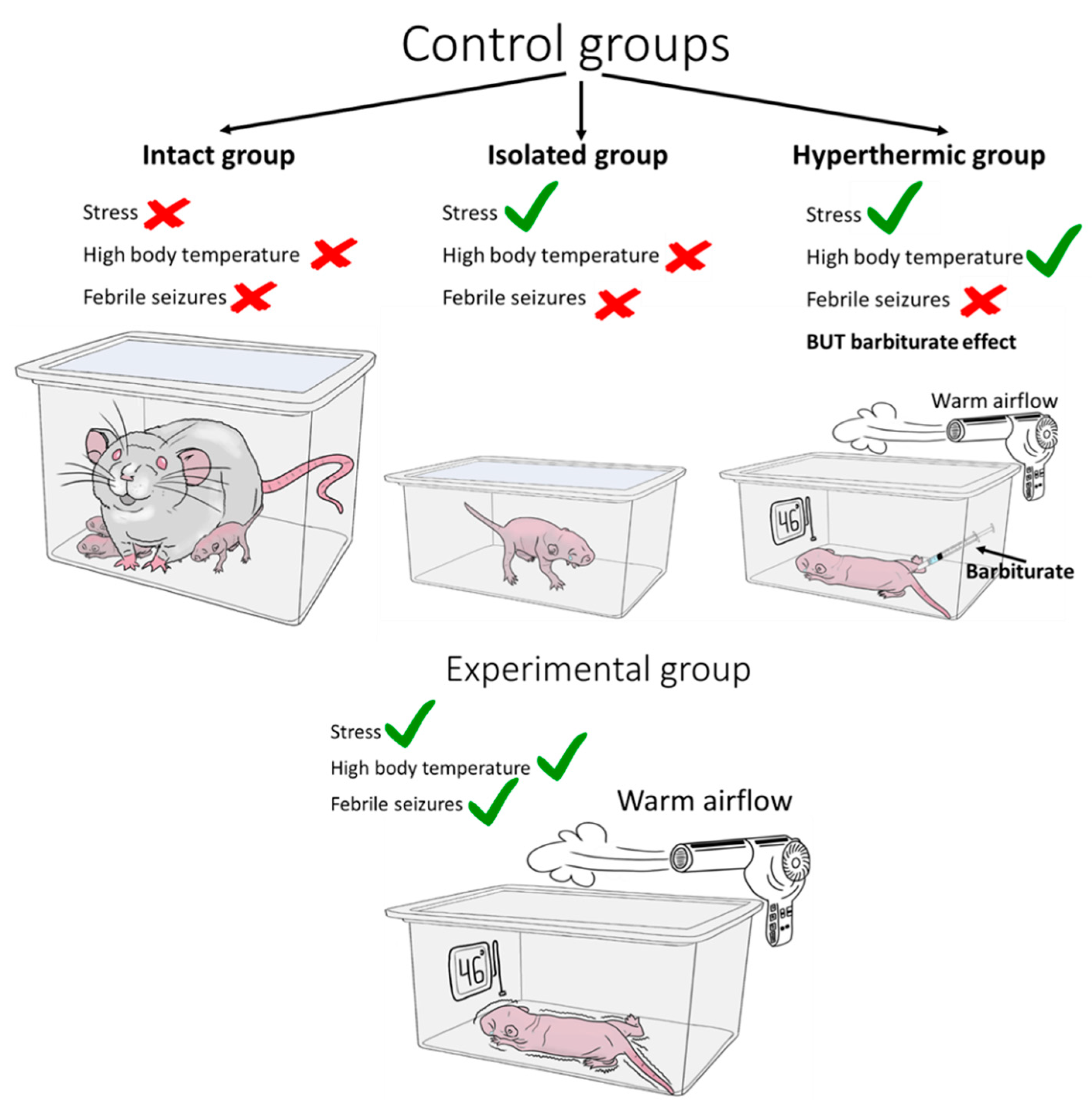

Table 1.
The animals employed as subjects in FSs models, along with their principal characteristics of seizures.
Table 1.
The animals employed as subjects in FSs models, along with their principal characteristics of seizures.
| Model organism |
Line / breed | Age | The characteristics of the convulsive episode | Source |
|---|---|---|---|---|
| Rat | Sprague-Dawley | P6-P8 | Body temperature rises to 40°C. Freezing, rare facial automatisms | [12] |
| Sprague-Dawley Wistar |
P10-P11 | Body temperature rises to 39°-40°C. Facial automatisms, often accompanied by unilateral bending of the body. Myoclonic twitching of the hind limbs. Clonic convulsions. | [12,84,88] | |
| Wistar | P20 | Threshold temperature 44°C. Generalised clonic convulsions. The duration of convulsions is significantly longer than in rats of other ages. | [36] | |
| Mouse | C57BL/6J | P10 | Similar to the course of FSs in rats at the age of P10 | [96] |
| transgenic mice with human GFAP (hGFAP) promoter-controlled expression of EGFРP (based on the FVB/N line) | P14-P15 | Several episodes of sudden cessation of activity and prolonged immobility with reduced responsiveness. Facial automatisms (chewing). Clonic or tonic–clonic seizures are not observed. | [96], [97] | |
| Drosophila | 2-day-old fly | Brief leg twitches, followed by failure to maintain standing posture, with wing flapping, leg twitching, and sometimes abdominal curling. | [18] | |
| Danio rerio | larval aged 3 to 7 days post-fertilization | After implantation of a glass microelectrode into the forebrain of the larva, abnormal electrographic convulsive activity is recorded when the temperature in the bath where the larvae are kept rises. | [17] | |
Table 2.
Main findings obtained from febrile seizure models following various time periods.
| 1 week after FSs | 1 to 4 weeks after FSs | > 1 month after FSs | ||
|---|---|---|---|---|
| Morphology | Neurons | There is minimal cell death in the initial hours following FSs [125] Significant populations of silver-stained 'dark' neurons in discrete hippocampal and amygdala regions [125] A decrease in the number of neurons in the hippocampal CA1 region, hilus and dentate gyrus [123] The ultrastructural disruption of synaptic endings in both hippocampal neurons and temporal lobe cortical cells [134,135,136] |
A decrease in the number of neurons in the hippocampal CA1 region [123] Increased dendrite length of granular cells that appeared after seizures [131] |
A decrease in the number of neurons in the hippocampal CA1 region [123] Excessive growth of mossy fibers [122] Increase in F-actin in the presynaptic endings of mossy fiber synapses [133] Increased dendritic complexity and number of mushroom-like spines in dentate gyrus cells [131] |
| Glial cells | Activation of astroglial cells [137,138] Reduction of astrocytic gap junction coupling, decrease in the level of connexin 43 [96] Edema in astrocytes, damage to the endoplasmic reticulum and mitochondria [142]. Disruption of the blood-brain barrier [143,144] Activation of microglial cells [28,96,138,145] |
Activation of astrocytic and microglial cells in the hippocampal CA2 region [149] Impaired of the age-related dynamics of astrocyte maturation [139] |
||
|
Synaptic transmission Synaptic plasticity |
Decrease in synaptic transmission efficiency and a decrease in calcium conductance through calcium-permeable AMPA receptors [151] Decrease in synaptic transmission efficiency, changes in short-term plasticity in hippocampal CA3-CA1 neurons and a decrease in the frequency of miniature excitatory synaptic currents [123] Increased GLT-1 levels, reduced glutamate concentration, and unchanged mGlu5R levels in the cortex brain [152] |
Increased inhibitory postsynaptic currents in the hippocampus mediated by GABAA receptors [154] Increased Na+/K+-ATPase activity in cortical plasma membranes [159] Decrease in the level of LTP [139,164] Disturbance of age-related changes in the magnitude and character of LTP [139] Heightened desensitization of NMDA receptors [88] Reduction in tyrosine phosphorylation of the GluN2A subunit of NMDA receptors [164] Increase in the threshold for pentylenetetrazol-induced seizures [115] Decrease in GLT-1 levels [152] Increase in mGlu5R levels [152] |
Increased expression of the GABAA receptor in granular cells born after FSs in the dentate gyrus [158] Increased Na+/K+-ATPase activity in cortical plasma membranes [159] Decrease in the level of the LTP [139] Increase in the maximal electroshock seizure threshold [123] Increased susceptibility to seizures in rats when kainate was used [155] Electroencephalographically recorded epileptiform discharges in the limbic system [172] |
|
| Behavior | Cognitive impairment [175] Depressive-like behavior [178,179] Anxiogenic behaviors [179] Social novelty deficits, repetitive behavior and hyperlocomotion [149] |
Cognitive impairment [84,139,176,177] Depressive-like behavior [178,179,180] Anxiogenic behaviors [179] Social novelty deficits, repetitive behavior [149] |
||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



