Submitted:
07 October 2024
Posted:
09 October 2024
Read the latest preprint version here
Abstract
The kidneys contribute to the overall health of the organism by maintaining systemic homeostasis. This process involves the regulation of various biological mechanisms, in which the Krüppel-like factor (KLF) transcription factors are essential for regulating development, differentiation, proliferation, and cellular apoptosis. They also play a role in the metabolic regulation of essential nutrients, such as glucose and lipids. The dysregulation of these transcription factors is associated with the development of various pathologies, which can ultimately lead to renal fibrosis, a process that severely compromises the function of these organs. In this context, the present article provides a comprehensive review of the existing literature, offering an enriching analysis of the findings related to the role of KLFs in the renal field, while also highlighting their potential therapeutic role in the treatment of renal diseases.
Keywords:
Krüppel-like factors
; Kidney
; Development
; Metabolism
; Disease
; Regulation
1. Introduction
The kidneys are intricate organs that have a vital function in maintaining an overall balance of waste and fluids in the body [1]. They preserve the body's electrolyte and acid-base balance by filtering and reabsorbing substances such as ions, glucose and amino acids from the bloodstream. In addition, they eliminate waste products produced by cellular metabolism and xenobiotics [2]. They additionally perform endocrine functions such as hormone production e.g., erythropoietin, 1-α hydroxylase, and renin, which are involved in erythropoiesis, calcitriol synthesis, and blood pressure regulation, respectively [3]. It is important to note that to maintain their proper cellular function, particular responses are determined by effectors such as transcriptions factors who respond to external signals resulting in the modulation of gene expression, thereby tightly controlling physiological function [4]. Amongst the responses from different transcription factors, in recent years the Krüppel-like factor (KLF) family of transcription factors have acquired much attention for their roles in development and functional maintenance of the kidney [5].
Briefly, the Krüppel-like factor (KLF) family is a 17-member family of transcription factors characterized by three zinc fingers (Cys2/His2) with highly conserved C-terminal domains [6]. Regarding renal function KLFs have been associated with several biological processes including differentiation, terminal maturation, as well as the maintenance of the structure and function of the glomerular filtration barrier, protecting glomerular endothelial cells and podocytes from inflammatory damage and the development of fibrosis [7]. Members such KLF10 as have been associated with the promotion of inflammation and cellular proliferation by positively regulating pro-inflammatory cytokines and activating pathways that induce cellular growth [8].
The structural variations in their regulatory domains determine the variety of roles that KLFs play in renal biological processes. Through their interactions with various coactivators and/or corepressors, these domains enable them activation or repression of the promoter activity of their target genes [9]. Interestingly, various dietary, hormonal, and neural signals can modulate KLF expression, particularly when in an altered state. This dysregulation in their expression affects a variety of functions in which they are involved. For this reason, the dysregulation of KLFs can lead to serious health issues [10].
This article aims to provide an overview, based on a comprehensive review, of the most relevant findings related to the involvement of KLFs in various biological processes of the kidney. We emphasize KLFs during renal development, their role in regulating metabolic processes of essential macronutrients such as glucose and lipids, as well as their implications in the onset and progression of significant renal diseases such as acute kidney injury (AKI), chronic kidney disease (CKD), and diabetic kidney disease (DKD), highlighting those KLFs that exhibit renal-protective effects.
2. Krüppel Like Factors
Krüppel-like factors make up a family of zinc-finger motif DNA-binding transcription regulators. Their name derives from the initial discovery of Krüppel protein in Drosophila melanogaster in 1993. A protein which participates in regulating fly embryo segmentation [11]. In humans, the KLFs are consisting of 17 members, which, as mentioned, exhibit three highly conserved Cys2His2-type zinc finger motifs in their carboxyl-terminal regions. Each zinc finger maintains a fixed length of 23 or 21 amino acids for their motifs [12]. The DNA-binding domain structure, evolutionarily conserved, shows structural homology with Specificity Proteins (SP), which leads to considering KLFs as a subgroup of the SP/KLF family [13]. These motifs allow binding to both the GC-rich proximal promoter regions and the CACCC elements (GT boxes) within the promoter regions of multiple genes [14].
The phylogenetic features that mark the evolutionary distance amongst KLF family members, coupled with specific structural traits in their less conserved amino-terminal regions, result in the division of KLFs into three distinct groups [15]. The first group is composed of KLF3, KLF8, and KLF12, which harbor a Pro-X-Asp-Leu-Ser (PXDLS) motif, where X represents a hydrophobic amino acid. This motif facilitates interaction with the amino-terminal substrate binding domain (SBD) of C-terminal binding proteins (CtBP), aiming to repress transcription [9,15,16,17]. The second group includes KLF1, KLF2, KLF4, KLF5, KLF6, and KLF7, who, except for KLF7, along with KLF8 from group 1 and KLF15 (the latter by homology), possess a transcriptional activation domain (TAD) in their N-terminal regions [9,16]. Both KLF1-2, KLF4-6, and KLF13 (the latter from group 3), can interact with histone acetyltransferase enzymes such as cAMP response element-binding protein (CREB) binding protein (CBP), p300, and p300/CBP-associated factor, facilitating chromatin remodeling and promoting transcriptional activity in DNA regions regulated by KLFs [9,16,18,19]. The binding of KLF1, KLF4, KLF5, and KLF11 (the latter from group 3) to histone deacetylase enzymes suppresses their transcriptional activity. Therefore, this second group of KLFs can modulate both activation and re-pression of gene expression at the transcriptional level depending on a biological context and the gene regulatory region through which they are operating [20]. Lastly, in the third group, members KLF9, KLF10, KLF11, KLF13, KLF14, and KLF16 contain a Cabut domain in their N-terminal section that encompasses a Sin3 interaction domain (SID) and their activities as transcriptional repressors [7]. Both KLF15 and KLF17 are not classified within these three phylogenetic groups because their protein interaction domains have not yet been determined. Finaly, researchers have identified that various KLFs also contain nuclear localization signals (NLS) and nuclear export signals (NES) which regulate their subcellular localization, as illustrated in Figure 1.
3. KLFs in Kidney Physiology
The kidneys play essential roles in maintaining homeostasis in the body, mediated by hormonal signaling processes and their interaction with transcription factors [8,21]. One family of transcription factors involved in regulating renal function is the KLFs [8,22,23], which are expressed in various parts of the nephron, contributing to both its structure and cellular composition [24]. The subsequently these two fundamental aspects of kidney's cellular biology provide a better understanding of the role of KLFs and their influence on determining cellular function and behavior.
Kidneys are divided into 2 specific regions: the renal cortex and medulla. Between these two regions are the nephrons, which are considered the functional and structural units of these organs, composed of the glomerulus and the tubular system [1,25].
Surrounded by a cup-shaped structure called Bowman's capsule, the glomerulus is where blood filtration occurs. The parietal epithelial layer is composed of parietal cells that play a structural maintenance role [1], while the visceral layer is formed by podocytes, which are perivascular cells enveloping the outer layer of the basal membrane of the glomerular capillaries (GC), together forming the glomerular filtration barrier (GFB) [26,27]. The integrity of this barrier, maintained by tight junctions between epithelial and endothelial cells, is the mechanism that ensures proper glomerular filtration. Occludin is one of the junction proteins expressed in podocytes, and its expression is downregulated by KLF2 to prevent its overexpression from altering the structure of the glomerular basement membrane (GBM) [28].
KLF4 collaborates with histone deacetylases (HDACs), specifically HDAC1 and HDAC3, to upregulate the expression of E-cadherin, podocin, and nephron [29]. E-cadherin is another adhesion molecule [30], while podocins and nephrin are crucial for the formation and maintenance of the slit diaphragm (SD), a specialized adhesion structure of podocytes [29,31,32]. KLF4 also induces the expression of cytokeratin 8 (K8) and K18, which, like podocins, participates in cytoskeletal organization [29]. Together, the expression of these proteins maintains the structure and functionality of podocytes by modulating cellular adhesion and polarity.
Maintaining cell adhesion and polarity is crucial to prevent acquisition of mesenchymal characteristics. KLF4 is also involved in downregulating mesenchymal markers, such as vimentin and α-smooth muscle actin (α-SMA) in podocytes [29]. Vimentin is expressed in mesenchymal cells, which generally lack both intercellular adhesion and polarity, providing resistance to migration-related stress [33]. In contrast, α-SMA contributes to motility and contraction of the cytoskeleton [34], allowing greater cell mobility and the ability to migrate to sites of injury or inflammation. Therefore, KLF4 prevents structural damage and the progression of diseases by inhibiting epithelial-to-mesenchymal transition (EMT).
In GC, which are made up of fenestrated endothelial cells (ECs) possessing transcellular pores of 60 to 100 nm in diameter, selective permeability for molecules according to their size and charge is achieved [1,35]. KLF2 contributes to regulating the size and distribution of these pores, preventing uncontrolled solute permeability by inhibiting the phosphorylation of the myosin light chain, thus avoiding the contraction of the cytoskeleton that would reduce the size of the ECs and lead to the formation of gaps between them [28]. Another way to maintain proper glomerular filtration is through the modulation of angiogenesis mediated by VEGF-A, where KLF2 downregulates its expression. This prevents the excess of unnecessary or dysfunctional blood vessels that could disrupt hemodynamic balance and affect blood pressure [36].
The role of KLF4 in ECs is related to mediating inflammation by downregulating adhesion molecules, such as VCAM1 induced by TNF-α. This is achieved by inhibiting the expression of the p65 subunit of NF-κB, which is required for the activation of this transcription factor and its binding to the VCAM1 promoter. In this way, KLF4 modulates the adhesion and recruitment of lymphocytes to endothelial cells, preventing chronic inflammation [37,38].
The GC also depends on mesangial cells to support their structure, as they lack an interstitial tissue lining [39]. These cells collaborate in regulating blood pressure and fluid balance because of their plasticity, which gives them the ability to produce renin and adapt to environmental changes [40]. In these cells, KLF4 attenuates the expansion of the mesangial matrix and their proliferation through the negative regulation of the mTOR pathway by downregulating the expression of phosphorylated (p) mTOR and pS6K proteins. This protects the kidney from mesangial cells, which produce extracellular matrix by secreting type IV and type V collagen and fibronectin from the mesangial matrix, from producing it in excess, which could lead to the development of fibrosis-related diseases [41].
Between the parietal layer and the visceral layer of Bowman’s capsule, a urinary space known as the "Bowman space" is formed. This space represents the beginning of the urinary system and is contiguous with the proximal convoluted tubule (PCT), in the renal cortex [42]. The PCT, which is the first segment of the nephron's tubular system, receives glomerular ultrafiltrate through epithelial cells (EpC) that allow permeability to both water and luminal fluid, reabsorbing glucose, amino acids, and minerals such as phosphate, chloride, and bicarbonate, as well as secreting hydrogen ions and toxins produced by cellular metabolism and xenobiotics present in the filtrate [2]. The expression of KLF4 and KLF11 mitigates inflammation and fibrosis by decreasing the expression of cytokines MCP-1, MIP-3α, and IL-8 induced by TGF-β1. This is possibly related to the phosphorylation of KLF4 that induces SMAD and p38/MAPK signaling pathways in vascular smooth muscle cells (VSMCs) [43,44] or through binding to p65 and inhibiting NF-kB signaling. The decrease of these inflammatory cytokines limits the production of collagen type I and tissue fibrosis [45].
KLF15 in these EpC can decrease the expression of these fibrogenic components by negatively regulating the MAPK pathways, which, when activated, contribute to the production of TGF-β1 and other pro-fibrogenic factors. Thus, KLF15 also modulates the fibrogenic response and helps prevent the accumulation of an extracellular matrix [46].
The specific expression of genes within each segment of the renal tubule determines its respective role [24], unfortunately, given what we know, the expression and function of KLFs in the subsequent segments of the nephron's tubular system remain incompletely understood. Following the proximal PCT, there are several segments, one of which is the loop of Henle. The Henle's loop aids in the concentration of urine by passively reabsorbing water into the medullary interstitium. It reabsorbs approximately 25% of sodium chloride, chloride, and potassium, all of which are later expelled alongside hydrogen ions. The distal convoluted tubule performs the functions of reabsorbing sodium, potassium, and chloride, besides secreting hydrogen and potassium ions. Finally, the cortical collecting tubule, a specific region of the collecting duct, connects to a collecting duct that drains into the renal papillae. This segment reabsorbs the same electrolytes as the distal convoluted tubule [1,42].
Therefore, the main function of KLFs in the kidneys can be briefly summarized as the preservation of structure, cell adhesion, the glomerular filtration barrier, and regulating the extracellular matrix (ECM) and inflammation of the cell lineage that composes them. This has the effect of modulating glomerular filtration, secretion, and elimination of unnecessary toxins from the body. Figure 2 illustrates the basic structure of the nephron, along with the location of its main cell groups that integrate the specific gene expression of KLFs within it. Table 1 summarizes the specific functions of each of the KLFs involved in the kidney's functional regulation processes.
3.1. Klf in Kidney Development
The process of organogenesis involves the division and organization of cells to establish the foundational structures of the embryo. Cell cycle arrest plays a crucial role in this process as it enables the progression towards terminal differentiation, which is a vital step in acquiring specialized functions [47].
The intricate progression of the cell cycle and cellular differentiation at a molecular level is regulated by signaling pathways and transcription factors [48]. This principle highlights the importance of the KLF family in contributing to kidney development. In embryonic cell cycle regulation, in vitro studies suggest Klf5 promotes podocyte survival by blocking the mitogen-activated protein (MAP) kinase ERK/p38 pathway, as well as decreasing the expression of apoptosis-related proteins such as Bax, caspase-3, caspase-8, and caspase-9, while increasing the expression of the antiapoptotic protein Bcl-2, which inhibits both cell cycle arrest and apoptosis by preventing Bax from translocating to the mitochondria [49,50,51]. In contrast, the inhibition of Klf5, along with other proteins such as Stat5a/b and ICAM-1 through pharmacological agents, has been associated with a positive regulation of the Bax/Bcl-2 ratio. This relationship regulates apoptosis; specifically, the inhibition of Klf5 induces an increase in the expression of Bax, which translocate to the mitochondria, promoting the release of cytochrome c and consequently the activation of caspase-3 and PARP, which are key proteins in the apoptotic process. The expression of Bcl-2, is not significantly affected during KLF5 downregulation [52]. KLF6 has been associated with maintaining mitochondrial function and preventing cell death in podocytes. This is because of KLF6's ability to bind to the promoter region of the cytochrome c oxidase assembly gene (SCO2) and positively regulate it. Continuous expression of SCO2, a metallo-chaperone, is crucial for transporting copper ions to electron carriers in the mitochondrial electron transport chain. This process, including cytochrome c, is vital for preventing the activation of the intrinsic apoptotic pathway in podocytes. By doing so, it avoids any harm to the glomerulus and maintains its filtration capacity [23].
The complete maturation of the nephrons occurs postnatally and is essential for the kidney to develop its maximum urinary concentrating ability. Various mechanisms are outlined below to illustrate how both KLF12 and KLF15 are fundamental in this process. KLF15 acts as a negative regulator of the chloride channel ClC-K1, which is expressed in the EpC thin ascending limb of the loop of Henle during postnatal development. This KLF15/ClC-K1 repression prevents the formation of channels that facilitate the passage of chloride into the urine, avoiding dysregulation in the organism's electrolyte balance, since the concentration of chloride in urine directly affects its osmolarity [22]. KLF12 is also overexpressed between 15 and 22 days after birth in the EpC of the medullary collecting ducts (IMCD) undergoing maturation. The specificity of KLF12 expression was determined by comparing it with the expression of aquaporin 2 (AQP2), which is upregulated in the IMCD and shares similar DNA-binding sites to those of the KLFs. The co-localization of KLF12 and AQP2 suggests KLF12 plays a key role in the positive regulation of AQP2 expression and its target gene, the urea transporter (UT-A1) [53,54]. This positive regulation of KLF12/AP-2/UT-A1 influences the transport of water and urea into the IMCD, resulting in the accumulation of urea necessary for proper urine concentration [55]. Figure 3 illustrates the involvement of KLFs in regulating renal organogenesis and postnatal maturation of nephrons.
KLF15 has been also associated with podocyte differentiation, since it upregulates the expression of nephrin, podocin, synaptopodin, and Wt1; all essential genes for maintaining a differentiated phenotype and preventing the loss and detachment of podocytes [26,56]. Meanwhile, KLF4 regulates the differentiation of specific nephron segments and individual cell types by cooperating with p53 and CREB, as evidenced by the p53-CRE-KLF binding sites in the promoter regions of renal function genes, such as AQP2, bradykinin receptor B2 (B2R) and epithelial sodium channel (ENaC) during terminal nephron differentiation [8,48,57].
To summarize, KLFs are vital for the proper development, structure, and function of the glomerular filtration barrier. They safeguard endothelial cells (ECs), facilitate the differentiation of podocytes, maintain their specialized integrity, and regulate cell cycle and apoptosis.
3.2. Klf in Kidney Metabolism
Metabolic processes enable the kidney to use, produce, and reabsorb nutrients, fulfilling its own energy demands while also ensuring homeostasis. To regulate these metabolic processes effectively, transcription factors and coactivators are involved in modulating the expression of genes that code for the enzymes involved. This ensures the proper occurrence of these processes.
Transcription factors such as KLFs have been closely associated with regulating metabolism, especially in the hepatic context [58,59,60,61,62,63,64], where they regulate genes involved in lipid metabolism such as CREB, Carbohydrate Response Element Binding Protein (ChREBP), Sterol Regulatory Element Binding Protein 1 (SREBP-1), Peroxisome Proliferator-Activated Receptor (PPAR). Moreover, they further regulate genes involved in glycolytic metabolism such as Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1 Alpha (PGC-1α) [58,65,66,67] because they share the same DNA binding sites in conserved CACCC sequences and GC-rich elements [68].
The variations in metabolic activity among KLFs remain unexplored. However, in vitro studies have shown that KLF6 overexpression in proximal tubular cells (Hk-2) exposed to high glucose concentrations increases the expression of the protein that interacts with thioredoxin (Txnip) [69]. This protein is particularly intriguing due to its involvement in glucose oxidation, which is vital for energy acquisition. One of the most significant factors is related to the glucose transporter GLUT1. It is regulated by Txnip and could influence its localization by directly binding to the transporter. This binding induces internalization through clathrin-coated pits [70]. Therefore, its expression suggests that peripheral glucose uptake and utilization of this substrate could be compromised, leading to metabolic disturbances in conditions of high and persistent glucose consumption, such as in diabetes mellitus. Thus, silencing KLF6 may represent a therapeutic target, as its inhibition significantly attenuates Txnip expression and mitigates metabolic consequences.
Within the Hk-2 cellular model, KLF14 is an important player in lipid metabolism. The lack of KLF14 has been linked to a decrease in mitochondrial activity by reducing the expression of PPARα, resulting in the suppression of target genes involved in fatty acid transport. One example of such a gene is FATP1, which is found in the cell membrane and helps in the absorption and incorporation of lipids. Furthermore, it inhibits the expression of CPT1, an enzyme that facilitates the transfer of acyl groups from acyl-CoA to carnitine in the intermembrane space of the mitochondria, leading to the production of acyl-carnitine esters. By utilizing this process, the inner mitochondrial membrane allows for the transportation of long-chain fatty acids. In the next step, CPT2 takes over and transfers the acyl groups from acyl-carnitine back to CoA, ultimately leading to the restoration of acyl-CoA. Subsequently, this acyl-CoA can be metabolized through a series of enzymatic reactions within the β-oxidation pathway of fatty acids [71,72].
Thus, the downregulation of PPARα causes the accumulation of lipids inside cells, a process that can occur in a wide range of renal cell types, including mesangial cells, podocytes, and proximal PCT [73]. The latter cells, which rely on fatty acid oxidation for energy production, are the ones that this applies to [74]. Renal damage can occur when lipids accumulate in cells that are not meant for storage. The build-up of this substance promotes lipoperoxidation, which generates reactive oxygen species (ROS), leading to oxidative stress and contributing to the development of different diseases like acute kidney injury (AKI) and fibrosis [75]. Nevertheless, the overexpression of KLF14 counteracts these effects by enhancing mitochondrial activity, reducing lipogenesis, and decreasing lipid accumulation. In experiments conducted on living subjects, it has been found that KLF15 can positively regulate both CPT1 and Acyl-CoA Acyltransferase 2 (ACAA2) by tightly binding to the PPARα binding sites [76]. The closeness of DNA suggests that KLF15 and PPARα may coordinate in governing the expression of these genes.
Thus, it is crucial to have high levels of both KLF14 and KLF15 to ensure sufficient energy supply, specifically to the proximal tubular cells, by utilizing lipids through fatty acid oxidation. Figure 4 illustrates the implication of KLFs in renal metabolism both under healthy conditions and their effect on states of dysregulation.
3.3. Klfs in Kidney Disease
When the expression of KLFs is dysregulated, it can disrupt various physiological processes in which they are involved. This disruption can cause poor management of inflammation, tissue repair, regeneration, and other cellular adaptations to stress.
Throughout this review, it has been discussed that KLFs demonstrate distinct patterns of expression in different renal cells, greatly influencing their various functions in maintaining overall balance within the body [77]. The dysregulation of KLFs is associated with kidney diseases, including CKD, AKI, and DKD, where the pathophysiology affects the balance of glomerular, tubular, and inflammatory functions, and this variability is commonly observed. Take into consideration that specific KLFs have protective properties for the kidneys and blocking them frequently leads to the deterioration and advancement of the disease. Conversely, the disease can also be exacerbated by the excessive expression of other KLFs.
It is important to mention that AKI, which is characterized by a sudden decrease in kidney function, has the potential to progress to chronic injury and eventually develop into CKD [78]. In this context, KLF2, KLF4, KLF9, KLF10, and KLF15 have beneficial effects [36,79,80,81,82] while KLF5 promotes cell proliferation, tubular damage, and inflammation [83]. Various pathological conditions, such as diabetic nephropathy, which is characterized by albuminuria and progressive renal insufficiency [84], precipitate the development of CKD. Here, the KLFs that stand out are primarily KLF3 and KLF6, which induce inflammation and fibrosis and facilitate epithelial-mesenchymal transition, respectively. Ultimately, all these diseases converge on a common outcome of renal fibrosis, characterized by excessive deposition of ECM [85,86]. The activity of each KLF in these three diseases is detailed in Table 2.
5. Perspectives
Ongoing research is advancing our understanding of KLFs and how they affect the development of the kidneys, metabolic activity, and kidney diseases. With increasing understanding of these factors, it is apparent that a more thorough exploration of the cellular functions in which KLFs might be involved is necessary. Thus, it is of utmost importance to examine the specific interactions of KLFs with other transcription factors and signaling pathways. Understanding how these factors regulate gene expression in different contexts can provide valuable insights into their role in renal homeostasis and response to injury. Moreover, a more detailed approach is required regarding the effects of KLFs on regulating renal metabolism. Given that the kidneys are crucial organs for the metabolism of nutrients such as glucose and lipids, investigating how KLFs modulate these metabolic pathways could reveal new therapeutic strategies for diseases like diabetes and chronic kidney disease.
Developing experimental models that permit the study of KLF function in pathological conditions is also of great significance. The creation of in vivo and in vitro models will facilitate the identification of biomarkers and the evaluation of therapeutic interventions that could improve renal health. Ultimately, incorporating cutting-edge technologies like genetic editing and systems biology will enhance our understanding of the intricate relationship between KLFs and renal physiology. This could open new avenues for identifying therapeutic targets and intervention strategies in kidney diseases.
In summary, studying KLFs has the potential to significantly expand our knowledge of renal development and disease. A multidisciplinary approach will be key to unlocking these mechanisms and contributing to the development of new therapies that improve renal health.
Author Contributions
Conceptualization: ISS-C, AEE-R, investigation: ISS-C, MAJ-G, GGG, DFB-C, writing—original draft preparation: ISS-C, AEE-R, DAG-K, writing—review editing: MGS-S ENG-T GRP-R JFI, supervision: J.F.I.
Funding
This research received no external funding.
Acknowledgments
The authors acknowledge mutual collaboration of all involved institutions.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wallace, M.A. Anatomy and Physiology of the Kidney. AORN J 1998, 68, 799–820. [CrossRef]
- Levassort, H.; Essig, M. Le Rein, Son Anatomie et Ses Grandes Fonctions. Soins Gerontol 2024, 29, 10–20. [CrossRef]
- Acharya, V.; Olivero, J. The Kidney as an Endocrine Organ. Methodist Debakey Cardiovasc J 2018, 14, 305. [CrossRef]
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A Census of Human Transcription Factors: Function, Expression and Evolution. Nat Rev Genet 2009, 10, 252–263. [CrossRef]
- Chiplunkar, A.R.; Lung, T.K.; Alhashem, Y.; Koppenhaver, B.A.; Salloum, F.N.; Kukreja, R.C.; Haar, J.L.; Lloyd, J.A. Krüppel-Like Factor 2 Is Required for Normal Mouse Cardiac Development. PLoS One 2013, 8, e54891. [CrossRef]
- Oishi, Y.; Manabe, I. Krüppel-Like Factors in Metabolic Homeostasis and Cardiometabolic Disease. Front Cardiovasc Med 2018, 5, 1–14. [CrossRef]
- Rane, M.J.; Zhao, Y.; Cai, L. Krϋppel-like Factors (KLFs) in Renal Physiology and Disease. EBioMedicine 2019, 40, 743–750. [CrossRef]
- Li, J.; Liu, L.; Zhou, W.; Cai, L.; Xu, Z.; Rane, M.J. Roles of Krüppel-like Factor 5 in Kidney Disease. J Cell Mol Med 2021, 25, 2342–2355. [CrossRef]
- Swamynathan, S.K. Krüppel-like Factors: Three Fingers in Control. Hum Genomics 2010, 4, 263–270. [CrossRef]
- Fletcher, B.R.; Damery, S.; Aiyegbusi, O.L.; Anderson, N.; Calvert, M.; Cockwell, P.; Ferguson, J.; Horton, M.; Paap, M.C.S.; Sidey-Gibbons, C.; et al. Symptom Burden and Health-Related Quality of Life in Chronic Kidney Disease: A Global Systematic Review and Meta-Analysis. PLoS Med 2022, 19, e1003954. [CrossRef]
- Zhang, J.; Li, G.; Feng, L.; Lu, H.; Wang, X. Krüppel-like Factors in Breast Cancer: Function, Regulation and Clinical Relevance. Biomedicine & Pharmacotherapy 2020, 123, 109778. [CrossRef]
- Wang, H.; Han, J.; Dmitrii, G.; Ning, K.; Zhang, X. KLF Transcription Factors in Bone Diseases. J Cell Mol Med 2024, 28. [CrossRef]
- Santoyo-Suarez, M.G.; Mares-Montemayor, J.D.; Padilla-Rivas, G.R.; Delgado-Gallegos, J.L.; Quiroz-Reyes, A.G.; Roacho-Perez, J.A.; Benitez-Chao, D.F.; Garza-Ocañas, L.; Arevalo-Martinez, G.; Garza-Treviño, E.N.; et al. The Involvement of Krüppel-like Factors in Cardiovascular Diseases. Life 2023, 13, 420. [CrossRef]
- García-Loredo, J.A.; Santoyo-Suarez, M.G.; Rodríguez-Nuñez, O.; Benitez Chao, D.F.; Garza-Treviño, E.N.; Zapata-Morin, P.A.; Padilla-Rivas, G.R.; Islas, J.F. Is the Cis-Element CACCC-Box a Master Regulatory Element during Cardiovascular Disease? A Bioinformatics Approach from the Perspective of the Krüppel-like Family of Transcription Factors. Life 2024, 14, 493. [CrossRef]
- Abe, M.; Saeki, N.; Ikeda, Y.; Ohba, S. Kruppel-like Factors in Skeletal Physiology and Pathologies. Int J Mol Sci 2022, 23, 15174. [CrossRef]
- Pearson, R.; Fleetwood, J.; Eaton, S.; Crossley, M.; Bao, S. Krüppel-like Transcription Factors: A Functional Family. International Journal of Biochemistry and Cell Biology 2008, 40, 1996–2001. [CrossRef]
- Pollak, N.M.; Hoffman, M.; Goldberg, I.J.; Drosatos, K. Krüppel-Like Factors: Crippling and Uncrippling Metabolic Pathways. JACC Basic Transl Sci 2018, 3, 132–156. [CrossRef]
- Dang, D.T.; Pevsner, J.; Yang, V.W. The Biology of the Mammalian Krüppel-like Family of Transcription Factors. Int J Biochem Cell Biol 2000, 32, 1103–1121. [CrossRef]
- Kaczynski, J.; Cook, T.; Urrutia, R. Sp1- and Krüppel-like Transcription Factors. Genome Biol 2003, 4, 1–8. [CrossRef]
- Andrés García-Loredo, J.; Santoyo-Suarez, M.G.; Rodríguez-Núñez, O.; Francisco Benitez Chao, D.; Garza-Treviño, E.N.; Zapata-Morin, P.A.; Padilla-Rivas, G.R.; Islas, J.F. In the Cis-Element CACCC-Box a Master Regulatory Element during Cardiovascular Disease? A Bioinformatics Approach from the Perspective of the Kruppel-like Family of Transcription Factors. 2024. [CrossRef]
- Butterworth, M.B. MicroRNAs and the Regulation of Aldosterone Signaling in the Kidney. American Journal of Physiology-Cell Physiology 2015, 308, C521–C527. [CrossRef]
- Uchida, S.; Tanaka, Y.; Ito, H.; Saitoh-Ohara, F.; Inazawa, J.; Yokoyama, K.K.; Sasaki, S.; Marumo, F. Transcriptional Regulation of the CLC-K1 Promoter by Myc-Associated Zinc Finger Protein and Kidney-Enriched Krüppel-Like Factor, a Novel Zinc Finger Repressor. Mol Cell Biol 2000, 20, 7319–7331. [CrossRef]
- Mallipattu, S.K.; Horne, S.J.; D’Agati, V.; Narla, G.; Liu, R.; Frohman, M.A.; Dickman, K.; Chen, E.Y.; Ma’ayan, A.; Bialkowska, A.B.; et al. Krüppel-like Factor 6 Regulates Mitochondrial Function in the Kidney. Journal of Clinical Investigation 2015, 125, 1347–1361. [CrossRef]
- Lee, J.W.; Chou, C.-L.; Knepper, M.A. Deep Sequencing in Microdissected Renal Tubules Identifies Nephron Segment–Specific Transcriptomes. Journal of the American Society of Nephrology 2015, 26, 2669–2677. [CrossRef]
- Balzer, M.S.; Rohacs, T.; Susztak, K. How Many Cell Types Are in the Kidney and What Do They Do? Annu Rev Physiol 2022, 84, 507–531. [CrossRef]
- Müller-Deile, J.; Schiffer, M. Podocytes from the Diagnostic and Therapeutic Point of View. Pflugers Arch 2017, 469, 1007–1015. [CrossRef]
- Tian, X.; Bunda, P.; Ishibe, S. Podocyte Endocytosis in Regulating the Glomerular Filtration Barrier. Front Med (Lausanne) 2022, 9. [CrossRef]
- Lin, Z.; Natesan, V.; Shi, H.; Dong, F.; Kawanami, D.; Mahabeleshwar, G.H.; Atkins, G.B.; Nayak, L.; Cui, Y.; Finigan, J.H.; et al. Kruppel-Like Factor 2 Regulates Endothelial Barrier Function. Arterioscler Thromb Vasc Biol 2010, 30, 1952–1959. [CrossRef]
- Hayashi, K.; Sasamura, H.; Nakamura, M.; Azegami, T.; Oguchi, H.; Sakamaki, Y.; Itoh, H. KLF4-Dependent Epigenetic Remodeling Modulates Podocyte Phenotypes and Attenuates Proteinuria. Journal of Clinical Investigation 2014, 124, 2523–2537. [CrossRef]
- Lee, S.-Y.; Han, S.M.; Kim, J.-E.; Chung, K.-Y.; Han, K.-H. Expression of E-Cadherin in Pig Kidney. J Vet Sci 2013, 14, 381. [CrossRef]
- Li, X.; He, J.C. An Update: The Role of Nephrin inside and Outside the Kidney. Sci China Life Sci 2015, 58, 649–657. [CrossRef]
- Huber, T.B.; Schermer, B.; Benzing, T. Podocin Organizes Ion Channel-Lipid Supercomplexes: Implications for Mechanosensation at the Slit Diaphragm. Nephron Exp Nephrol 2007, 106, e27–e31. [CrossRef]
- Kokkinos, M.I.; Wafai, R.; Wong, M.K.; Newgreen, D.F.; Thompson, E.W.; Waltham, M. Vimentin and Epithelial-Mesenchymal Transition in Human Breast Cancer – Observations in Vitro and in Vivo. Cells Tissues Organs 2007, 185, 191–203. [CrossRef]
- Yuan, S.-M. α-Smooth Muscle Actin and ACTA2 Gene Expressions in Vasculopathies. Revista Brasileira de Cirurgia Cardiovascular 2015. [CrossRef]
- Scott, R.P.; Quaggin, S.E. The Cell Biology of Renal Filtration. Journal of Cell Biology 2015, 209, 199–210. [CrossRef]
- Zhong, F.; Lee, K.; He, J.C. Role of Krüppel-like Factor-2 in Kidney Disease. Nephrology 2018, 23, 53–56. [CrossRef]
- Yoshida, T.; Yamashita, M.; Iwai, M.; Hayashi, M. Endothelial Krüppel-Like Factor 4 Mediates the Protective Effect of Statins against Ischemic AKI. Journal of the American Society of Nephrology 2016, 27, 1379–1388. [CrossRef]
- Zhang, X.; Wang, L.; Han, Z.; Dong, J.; Pang, D.; Fu, Y.; Li, L. KLF4 Alleviates Cerebral Vascular Injury by Ameliorating Vascular Endothelial Inflammation and Regulating Tight Junction Protein Expression Following Ischemic Stroke. J Neuroinflammation 2020, 17, 1–16. [CrossRef]
- Pollak, M.R.; Quaggin, S.E.; Hoenig, M.P.; Dworkin, L.D. The Glomerulus. Clinical Journal of the American Society of Nephrology 2014, 9, 1461–1469. [CrossRef]
- Gomez, R.A.; Lopez, M.L.S.S. Plasticity of Renin Cells in the Kidney Vasculature. Curr Hypertens Rep 2017, 19, 14. [CrossRef]
- Zhao, J.-H. Mesangial Cells and Renal Fibrosis. In; 2019; pp. 165–194. [CrossRef]
- Falkson, S.R.; Bordoni, B. Anatomy, Abdomen and Pelvis: Bowman Capsule; 2024;
- Kimura, T.; Isaka, Y.; Yoshimori, T. Autophagy and Kidney Inflammation. Autophagy 2017, 13, 997–1003. [CrossRef]
- Mreich, E.; Chen, X.; Zaky, A.; Pollock, C.A.; Saad, S. The Role of Krüppel-like Factor 4 in Transforming Growth Factor- β –Induced Inflammatory and Fibrotic Responses in Human Proximal Tubule Cells. Clin Exp Pharmacol Physiol 2015, 42, 680–686. [CrossRef]
- De Lorenzo, S.B.; Vrieze, A.M.; Johnson, R.A.; Lien, K.R.; Nath, K.A.; Garovic, V.D.; Khazaie, K.; Grande, J.P. KLF11 Deficiency Enhances Chemokine Generation and Fibrosis in Murine Unilateral Ureteral Obstruction. PLoS One 2022, 17, e0266454. [CrossRef]
- Gao, X.; Wu, G.; Gu, X.; Fu, L.; Mei, C. Kruppel-Like Factor 15 Modulates Renal Interstitial Fibrosis by ERK/MAPK and JNK/MAPK Pathways Regulation. Kidney Blood Press Res 2013, 37, 631–640. [CrossRef]
- Zhu, L.; Skoultchi, A.I. Coordinating Cell Proliferation and Differentiation. Curr Opin Genet Dev 2001, 11, 91–97. [CrossRef]
- El-Dahr, S.S.; Aboudehen, K.; Saifudeen, Z. Transcriptional Control of Terminal Nephron Differentiation. American Journal of Physiology-Renal Physiology 2008, 294, F1273–F1278. [CrossRef]
- Rao, L.; White, E. Bcl-2 and the ICE Family of Apoptotic Regulators: Making a Connection. Curr Opin Genet Dev 1997, 7, 52–58. [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and Functions of P38 MAPK Signalling. Biochemical Journal 2010, 429, 403–417. [CrossRef]
- Zhao, W.; Ma, L.; Cai, C.; Gong, X. Caffeine Inhibits NLRP3 Inflammasome Activation by Suppressing MAPK/NF-ΚB and A2aR Signaling in LPS-Induced THP-1 Macrophages. Int J Biol Sci 2019, 15, 1571–1581. [CrossRef]
- ZHANG, Y.; LIU, K.; ZHANG, Y.; QI, J.; LU, B.; SHI, C.; YIN, Y.; CAI, W.; LI, W. ABL-N May Induce Apoptosis of Human Prostate Cancer Cells through Suppression of KLF5, ICAM-1 and Stat5b, and Upregulation of Bax/Bcl-2 Ratio: An in Vitro and in Vivo Study. Oncol Rep 2015, 34, 2953–2960. [CrossRef]
- Lamontagne, J.O.; Zhang, H.; Zeid, A.M.; Strittmatter, K.; Rocha, A.D.; Williams, T.; Zhang, S.; Marneros, A.G. Transcription Factors AP-2α and AP-2β Regulate Distinct Segments of the Distal Nephron in the Mammalian Kidney. Nat Commun 2022, 13, 2226. [CrossRef]
- Suda, S.; Rai, T.; Sohara, E.; Sasaki, S.; Uchida, S. Postnatal Expression of KLF12 in the Inner Medullary Collecting Ducts of Kidney and Its Trans-Activation of UT-A1 Urea Transporter Promoter. Biochem Biophys Res Commun 2006, 344, 246–252. [CrossRef]
- Chou, C.-L.; Hwang, G.; Hageman, D.J.; Han, L.; Agrawal, P.; Pisitkun, T.; Knepper, M.A. Identification of UT-A1- and AQP2-Interacting Proteins in Rat Inner Medullary Collecting Duct. American Journal of Physiology-Cell Physiology 2018, 314, C99–C117. [CrossRef]
- Guo, Y.; Pace, J.; Li, Z.; Ma’ayan, A.; Wang, Z.; Revelo, M.P.; Chen, E.; Gu, X.; Attalah, A.; Yang, Y.; et al. Podocyte-Specific Induction of Krüppel-Like Factor 15 Restores Differentiation Markers and Attenuates Kidney Injury in Proteinuric Kidney Disease. Journal of the American Society of Nephrology 2018, 29, 2529–2545. [CrossRef]
- Saifudeen, Z.; Dipp, S.; Fan, H.; El-Dahr, S.S. Combinatorial Control of the Bradykinin B2 Receptor Promoter by P53, CREB, KLF-4, and CBP: Implications for Terminal Nephron Differentiation. American Journal of Physiology-Renal Physiology 2005, 288, F899–F909. [CrossRef]
- Sun, N.; Shen, C.; Zhang, L.; Wu, X.; Yu, Y.; Yang, X.; Yang, C.; Zhong, C.; Gao, Z.; Miao, W.; et al. Hepatic Krüppel-like Factor 16 (KLF16) Targets PPARα to Improve Steatohepatitis and Insulin Resistance. Gut 2021, 70, 2183–2195. [CrossRef]
- Cuthbert, C.E.; Foster, J.E.; Ramdath, D.D. A Maternal High-Fat, High-Sucrose Diet Alters Insulin Sensitivity and Expression of Insulin Signalling and Lipid Metabolism Genes and Proteins in Male Rat Offspring: Effect of Folic Acid Supplementation. British Journal of Nutrition 2017, 118, 580–588. [CrossRef]
- Oishi, Y.; Manabe, I.; Tobe, K.; Ohsugi, M.; Kubota, T.; Fujiu, K.; Maemura, K.; Kubota, N.; Kadowaki, T.; Nagai, R. SUMOylation of Krüppel-like Transcription Factor 5 Acts as a Molecular Switch in Transcriptional Programs of Lipid Metabolism Involving PPAR-δ. Nat Med 2008, 14, 656–666. [CrossRef]
- Chen, J.-L.; Lu, X.-J.; Zou, K.-L.; Ye, K. Krüppel-like Factor 2 Promotes Liver Steatosis through Upregulation of CD36. J Lipid Res 2014, 55, 32–40. [CrossRef]
- Zhang, H.; Chen, Q.; Jiao, T.; Cui, A.; Sun, X.; Fang, W.; Xie, L.; Liu, Y.; Fang, F.; Chang, Y. Involvement of KLF11 in Hepatic Glucose Metabolism in Mice via Suppressing of PEPCK-C Expression. PLoS One 2014, 9, e89552. [CrossRef]
- Zheng, D.; Hong, X.; He, X.; Lin, J.; Fan, S.; Wu, J.; Liang, Z.; Chen, S.; Yan, L.; Ren, M.; et al. Intermittent Fasting–Improved Glucose Homeostasis Is Not Entirely Dependent on Caloric Restriction in Db/Db Male Mice. Diabetes 2024, 73, 864–878. [CrossRef]
- Yu, S.; Meng, S.; Xiang, M.; Ma, H. Phosphoenolpyruvate Carboxykinase in Cell Metabolism: Roles and Mechanisms beyond Gluconeogenesis. Mol Metab 2021, 53, 101257. [CrossRef]
- Rowe, G.C.; Arany, Z. Genetic Models of PGC-1 and Glucose Metabolism and Homeostasis. Rev Endocr Metab Disord 2014, 15, 21–29. [CrossRef]
- Iizuka, K.; Takeda, J.; Horikawa, Y. Krüppel-like Factor-10 Is Directly Regulated by Carbohydrate Response Element-Binding Protein in Rat Primary Hepatocytes. Biochem Biophys Res Commun 2011, 412, 638–643. [CrossRef]
- Tang, Y.; Li, K.; Hu, B.; Cai, Z.; Li, J.; Tao, H.; Cao, J. Fatty Acid Binding Protein 5 Promotes the Proliferation, Migration, and Invasion of Hepatocellular Carcinoma Cells by Degradation of Krüppel-like Factor 9 Mediated by MiR-889-5p via CAMP-Response Element Binding Protein. Cancer Biol Ther 2022, 23, 424–438. [CrossRef]
- Brey, C.W.; Nelder, M.P.; Hailemariam, T.; Gaugler, R.; Hashmi, S. Krüppel-like Family of Transcription Factors: An Emerging New Frontier in Fat Biology. Int J Biol Sci 2009, 622–636. [CrossRef]
- Qi, W.; Chen, X.; Holian, J.; Tan, C.Y.R.; Kelly, D.J.; Pollock, C.A. Transcription Factors Krüppel-Like Factor 6 and Peroxisome Proliferator-Activated Receptor-γ Mediate High Glucose-Induced Thioredoxin-Interacting Protein. Am J Pathol 2009, 175, 1858–1867. [CrossRef]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.-H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-Dependent Degradation of TXNIP upon Energy Stress Leads to Enhanced Glucose Uptake via GLUT1. Mol Cell 2013, 49, 1167–1175. [CrossRef]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [CrossRef]
- Schlaepfer, I.R.; Joshi, M. CPT1A-Mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161. [CrossRef]
- Gai, Z.; Wang, T.; Visentin, M.; Kullak-Ublick, G.; Fu, X.; Wang, Z. Lipid Accumulation and Chronic Kidney Disease. Nutrients 2019, 11, 722. [CrossRef]
- Gewin, L.S. Sugar or Fat? Renal Tubular Metabolism Reviewed in Health and Disease. Nutrients 2021, 13, 1580. [CrossRef]
- Jang, H.-S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front Med (Lausanne) 2020, 7. [CrossRef]
- Piret, S.E.; Attallah, A.A.; Gu, X.; Guo, Y.; Gujarati, N.A.; Henein, J.; Zollman, A.; Hato, T.; Ma’ayan, A.; Revelo, M.P.; et al. Loss of Proximal Tubular Transcription Factor Krüppel-like Factor 15 Exacerbates Kidney Injury through Loss of Fatty Acid Oxidation. Kidney Int 2021, 100, 1250–1267. [CrossRef]
- Ghajar-Rahimi, G.; Agarwal, A. Endothelial KLF11 as a Nephroprotectant in AKI. Kidney360 2022, 3, 1302–1305. [CrossRef]
- Niculae, A.; Gherghina, M.-E.; Peride, I.; Tiglis, M.; Nechita, A.-M.; Checherita, I.A. Pathway from Acute Kidney Injury to Chronic Kidney Disease: Molecules Involved in Renal Fibrosis. Int J Mol Sci 2023, 24, 14019. [CrossRef]
- Wang, Z.; Zhou, Z.; Zhang, Y.; Zuo, F.; Du, J.; Wang, M.; Hu, M.; Sun, Y.; Wang, X.; Liu, M.; et al. Diacylglycerol Kinase Epsilon Protects against Renal Ischemia/Reperfusion Injury in Mice through Krüppel-like Factor 15/Klotho Pathway. Ren Fail 2022, 44, 902–913. [CrossRef]
- Sadrkhanloo, M.; Paskeh, M.D.A.; Hashemi, M.; Raesi, R.; Bahonar, A.; Nakhaee, Z.; Entezari, M.; Beig Goharrizi, M.A.S.; Salimimoghadam, S.; Ren, J.; et al. New Emerging Targets in Osteosarcoma Therapy: PTEN and PI3K/Akt Crosstalk in Carcinogenesis. Pathol Res Pract 2023, 251, 154902. [CrossRef]
- Zhengbiao, Z.; Liang, C.; Zhi, Z.; Youmin, P. Circular RNA_HIPK3-Targeting MiR-93-5p Regulates KLF9 Expression Level to Control Acute Kidney Injury. Comput Math Methods Med 2023, 2023. [CrossRef]
- Zhao, J.; Wang, X.; Wu, Y.; Zhao, C. Krüppel-like Factor 4 Modulates the MiR-101/COL10A1 Axis to Inhibit Renal Fibrosis after AKI by Regulating Epithelial–Mesenchymal Transition. Ren Fail 2024, 46. [CrossRef]
- Liu, Y.; Wang, Y.; Xu, C.; Zhang, Y.; Wang, Y.; Qin, J.; Lan, H.-Y.; Wang, L.; Huang, Y.; Mak, K.K.; et al. Activation of the YAP/KLF5 Transcriptional Cascade in Renal Tubular Cells Aggravates Kidney Injury. Molecular Therapy 2024, 32, 1526–1539. [CrossRef]
- Gupta, S.; Dominguez, M.; Golestaneh, L. Diabetic Kidney Disease. Medical Clinics of North America 2023, 107, 689–705. [CrossRef]
- Nastase, M. V.; Zeng-Brouwers, J.; Wygrecka, M.; Schaefer, L. Targeting Renal Fibrosis: Mechanisms and Drug Delivery Systems. Adv Drug Deliv Rev 2018, 129, 295–307. [CrossRef]
- Li, Q.; Liu, J.; Su, R.; Zhen, J.; Liu, X.; Liu, G. Small Extracellular Vesicles-Shuttled MiR-23a-3p from Mesenchymal Stem Cells Alleviate Renal Fibrosis and Inflammation by Inhibiting KLF3/STAT3 Axis in Diabetic Kidney Disease. Int Immunopharmacol 2024, 139, 112667. [CrossRef]
- Zhong, F.; Mallipattu, S.K.; Estrada, C.; Menon, M.; Salem, F.; Jain, M.K.; Chen, H.; Wang, Y.; Lee, K.; He, J.C. Reduced Krüppel-Like Factor 2 Aggravates Glomerular Endothelial Cell Injury and Kidney Disease in Mice with Unilateral Nephrectomy. Am J Pathol 2016, 186, 2021–2031. [CrossRef]
- Wen, Y.; Lu, X.; Ren, J.; Privratsky, J.R.; Yang, B.; Rudemiller, N.P.; Zhang, J.; Griffiths, R.; Jain, M.K.; Nedospasov, S.A.; et al. KLF4 in Macrophages Attenuates TNFα-Mediated Kidney Injury and Fibrosis. Journal of the American Society of Nephrology 2019, 30, 1925–1938. [CrossRef]
- Gujarati, N.A.; Frimpong, B.O.; Zaidi, M.; Bronstein, R.; Revelo, M.P.; Haley, J.D.; Kravets, I.; Guo, Y.; Mallipattu, S.K. Podocyte-Specific KLF6 Primes Proximal Tubule CaMK1D Signaling to Attenuate Diabetic Kidney Disease. Nat Commun 2024, 15, 8038. [CrossRef]
- Wang, L.; Lin, W.; Chen, J. Krüppel-like Factor 15: A Potential Therapeutic Target For Kidney Disease. Int J Biol Sci 2019, 15, 1955–1961. [CrossRef]
- Gao, X.; Wu, G.; Gu, X.; Fu, L.; Mei, C. Kruppel-Like Factor 15 Modulates Renal Interstitial Fibrosis by ERK/MAPK and JNK/MAPK Pathways Regulation. Kidney Blood Press Res 2013, 37, 631–640. [CrossRef]
- Zhao, J.; Wang, X.; Wu, Y.; Zhao, C. Krüppel-like Factor 4 Modulates the MiR-101/COL10A1 Axis to Inhibit Renal Fibrosis after AKI by Regulating Epithelial–Mesenchymal Transition. Ren Fail 2024, 46. [CrossRef]
- Li, Z.; Wang, B.; Lv, L.; Tang, T.; Wen, Y.; Cao, J.; Zhu, X.; Feng, S.; Crowley, S.D.; Liu, B. FIH-1-Modulated HIF-1α C-TAD Promotes Acute Kidney Injury to Chronic Kidney Disease Progression via Regulating KLF5 Signaling. Acta Pharmacol Sin 2021, 42, 2106–2119. [CrossRef]
- Zou, H.; Zhu, S.; Chen, Y.; Cai, N.; Xu, C.; Tu, W.; Qin, X. Kruppel Like Factor 5 Enhances High Glucose-Induced Renal Tubular Epithelial Cell Transdifferentiation in Diabetic Nephropathy. Crit Rev Eukaryot Gene Expr 2022, 32, 35–45. [CrossRef]
- Suzuki, N.; Kanai, A.; Suzuki, Y.; Ogino, H.; Ochi, H. Adrenergic Receptor Signaling Induced by Klf15, a Regulator of Regeneration Enhancer, Promotes Kidney Reconstruction. Proceedings of the National Academy of Sciences 2022, 119. [CrossRef]
- Hung, P.-H.; Hsu, Y.-C.; Chen, T.-H.; Lin, C.-L. Recent Advances in Diabetic Kidney Diseases: From Kidney Injury to Kidney Fibrosis. Int J Mol Sci 2021, 22, 11857. [CrossRef]
- Chen, S.; Lv, L.; Liu, B.; Tang, R. Crosstalk between Tubular Epithelial Cells and Glomerular Endothelial Cells in Diabetic Kidney Disease. Cell Prolif 2020, 53. [CrossRef]
- Jiang, S.; Luo, M.; Bai, X.; Nie, P.; Zhu, Y.; Cai, H.; Li, B.; Luo, P. Cellular Crosstalk of Glomerular Endothelial Cells and Podocytes in Diabetic Kidney Disease. J Cell Commun Signal 2022, 16, 313–331. [CrossRef]
- Gong, J.; Zhan, H.; Li, Y.; Zhang, W.; Jin, J.; He, Q. Krüppel-like factor 4 Ameliorates Diabetic Kidney Disease by Activating Autophagy via the MTOR Pathway. Mol Med Rep 2019. [CrossRef]
- Zhang, X.; Chen, J.; Lin, R.; Huang, Y.; Wang, Z.; Xu, S.; Wang, L.; Chen, F.; Zhang, J.; Pan, K.; et al. Lactate Drives Epithelial-Mesenchymal Transition in Diabetic Kidney Disease via the H3K14la/KLF5 Pathway. Redox Biol 2024, 75, 103246. [CrossRef]
- Chen, W.-C.; Lin, H.-H.; Tang, M.-J. Matrix-Stiffness–Regulated Inverse Expression of Krüppel-Like Factor 5 and Krüppel-Like Factor 4 in the Pathogenesis of Renal Fibrosis. Am J Pathol 2015, 185, 2468–2481. [CrossRef]
- Holian, J.; Qi, W.; Kelly, D.J.; Zhang, Y.; Mreich, E.; Pollock, C.A.; Chen, X.-M. Role of Krüppel-like Factor 6 in Transforming Growth Factor-Β1-Induced Epithelial-Mesenchymal Transition of Proximal Tubule Cells. American Journal of Physiology-Renal Physiology 2008, 295, F1388–F1396. [CrossRef]
- Hsu, Y.-C.; Ho, C.; Shih, Y.-H.; Ni, W.-C.; Li, Y.-C.; Chang, H.-C.; Lin, C.-L. Knockout of KLF10 Ameliorated Diabetic Renal Fibrosis via Downregulation of DKK-1. Molecules 2022, 27, 2644. [CrossRef]
- Lin, C.; Hsu, Y.; Huang, Y.; Shih, Y.; Wang, C.; Chiang, W.; Chang, P. A KDM6A–KLF10 Reinforcing Feedback Mechanism Aggravates Diabetic Podocyte Dysfunction. EMBO Mol Med 2019, 11. [CrossRef]
- Mou, X.; Zhou, D.; Liu, Y.; Liu, K.; Zhou, D. Identification of Potential Therapeutic Target Genes in Mouse Mesangial Cells Associated with Diabetic Nephropathy Using Bioinformatics Analysis. Exp Ther Med 2019. [CrossRef]
Figure 1.
Different groups of KLFs based on their phylogenetic and structural characteristics of the amino-terminal region. Group 1 includes KLF3, KLF8, and KLF12, whose primary function is transcriptional repression through interaction with the C-terminal binding protein (CtBP). Group 2 comprises KLF1, KLF2, KLF4, KLF5, KLF6, and KLF7; these KLFs exhibit diverse activities and can function as either transcriptional activators or repressors. Finally, Group 3 includes KLF9, KLF10, KLF11, KLF13, KLF14, and KLF16, which exert repressive effects similar to those of Group 1, but their action depends on their interaction with the transcriptional corepressor Sin3A. KLF15 and KLF17 are not assigned to a specific group because their protein interaction mechanisms are not yet fully explained, and they are also phylogenetically more distantly related.
Figure 1.
Different groups of KLFs based on their phylogenetic and structural characteristics of the amino-terminal region. Group 1 includes KLF3, KLF8, and KLF12, whose primary function is transcriptional repression through interaction with the C-terminal binding protein (CtBP). Group 2 comprises KLF1, KLF2, KLF4, KLF5, KLF6, and KLF7; these KLFs exhibit diverse activities and can function as either transcriptional activators or repressors. Finally, Group 3 includes KLF9, KLF10, KLF11, KLF13, KLF14, and KLF16, which exert repressive effects similar to those of Group 1, but their action depends on their interaction with the transcriptional corepressor Sin3A. KLF15 and KLF17 are not assigned to a specific group because their protein interaction mechanisms are not yet fully explained, and they are also phylogenetically more distantly related.
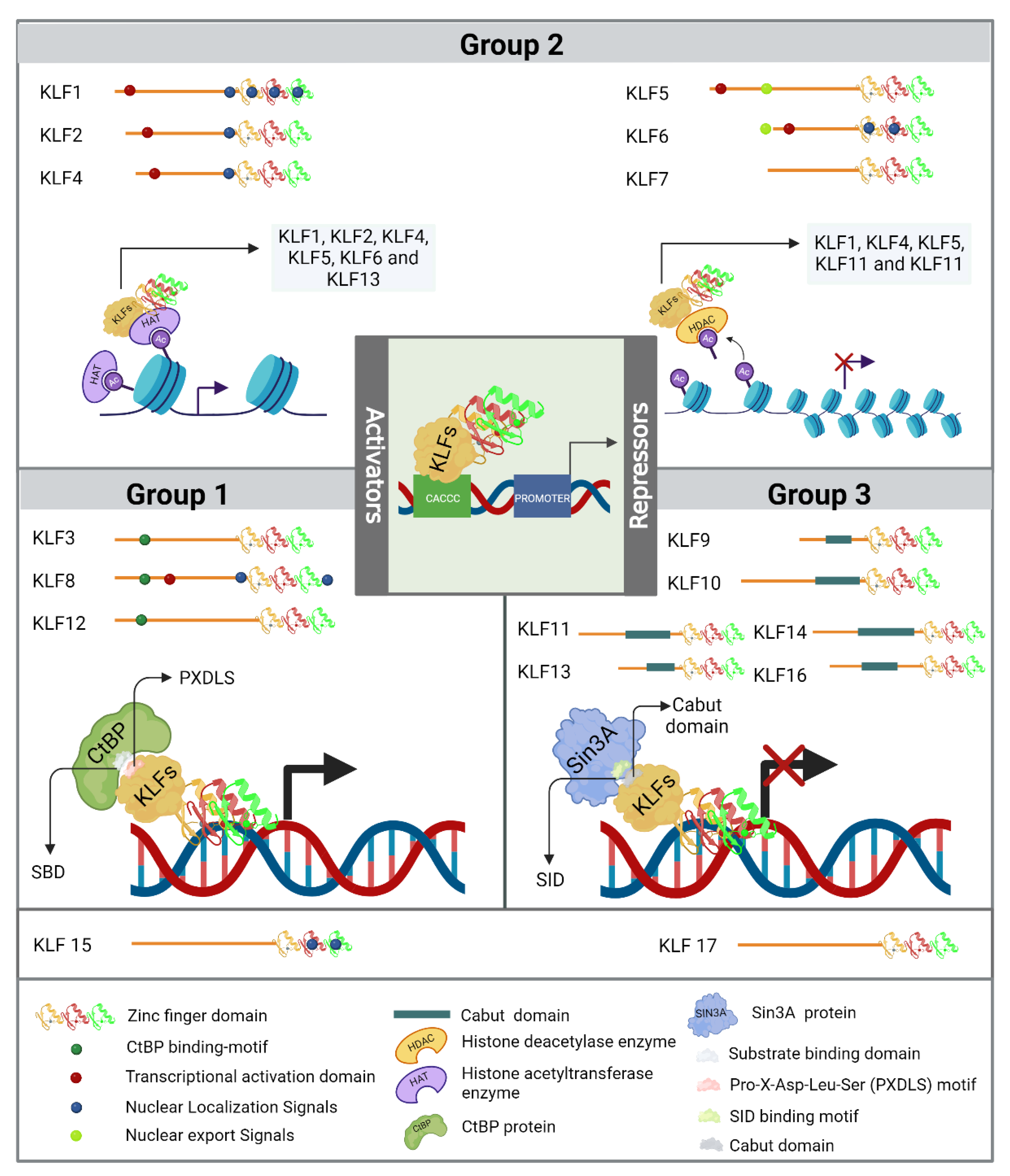
Figure 2.
The basic structure of the nephron consists of the glomerulus and the tubule. The tubule is divided into several segments: the proximal tubule, where early solute reabsorption occurs to prevent accumulation and nephrotoxicity; the loop of Henle; the distal convoluted tubule; and finally, the collecting duct, where urine is concentrated through coordinated processes of reabsorption and secretion. The location of the main cellular groups within the nephron is crucial for accurately identifying the specific gene expression of KLFs. It has been shown that KLF10 is the most prevalent transcription factor in the glomerulus, inner medulla (LDLIM), thin ascending limb of the loop of Henle, and inner medullary collecting duct (IMCD). KLF4 follows KLF10, being detected in large quantities in both the glomerulus and loop of Henle. Additionally, KLF2, KLF4, and KLF11 are expressed in renal endothelial cells.
Figure 2.
The basic structure of the nephron consists of the glomerulus and the tubule. The tubule is divided into several segments: the proximal tubule, where early solute reabsorption occurs to prevent accumulation and nephrotoxicity; the loop of Henle; the distal convoluted tubule; and finally, the collecting duct, where urine is concentrated through coordinated processes of reabsorption and secretion. The location of the main cellular groups within the nephron is crucial for accurately identifying the specific gene expression of KLFs. It has been shown that KLF10 is the most prevalent transcription factor in the glomerulus, inner medulla (LDLIM), thin ascending limb of the loop of Henle, and inner medullary collecting duct (IMCD). KLF4 follows KLF10, being detected in large quantities in both the glomerulus and loop of Henle. Additionally, KLF2, KLF4, and KLF11 are expressed in renal endothelial cells.
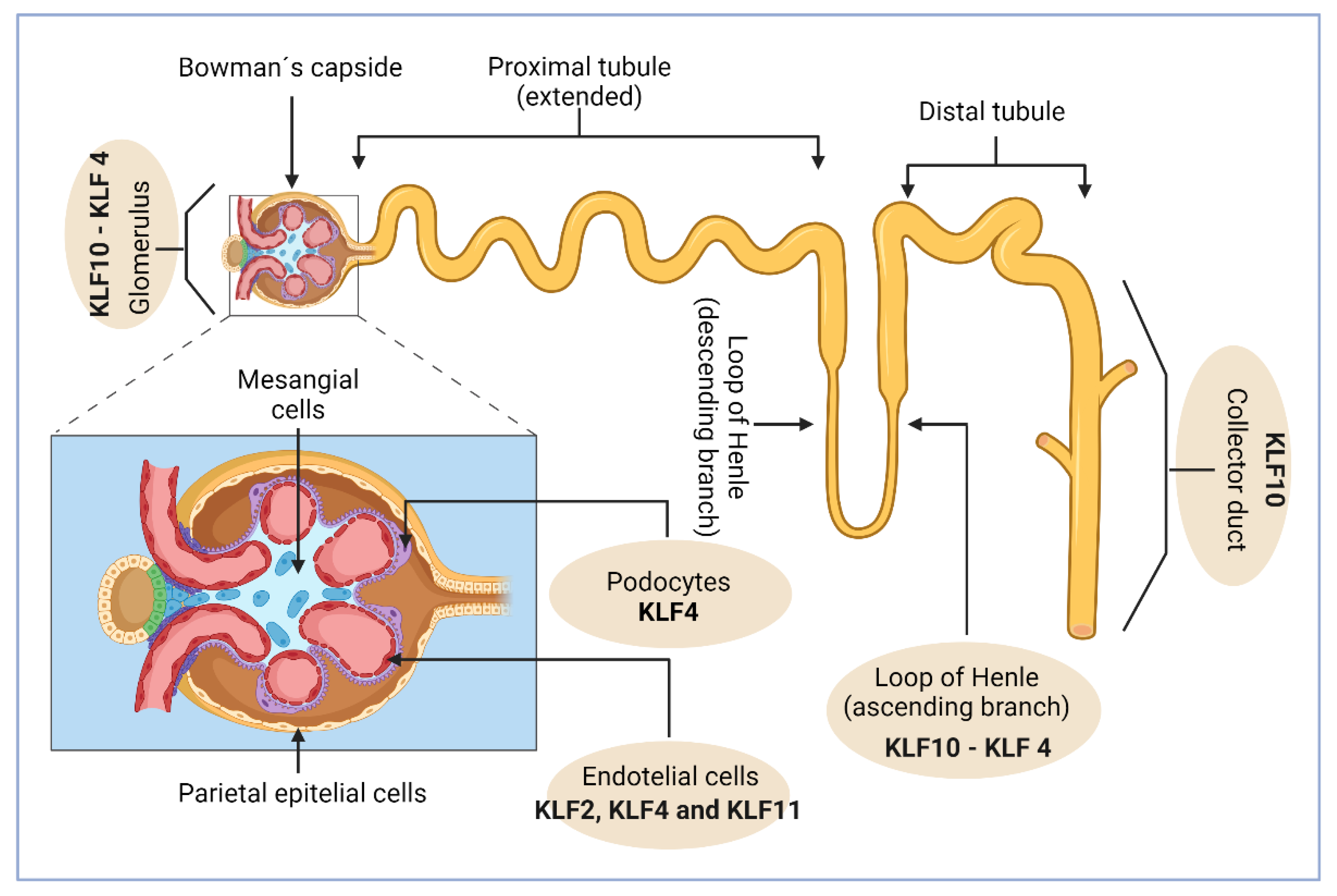
Figure 3.
KLFs involvement in regulating renal organogenesis and postnatal maturation of nephrons. The survival of podocytes mediated by KLF5, through the inhibition of the mitogen-activated protein (MAP) kinase pathway, may seem like a contradictory description, as ERK has been shown to positively induce the expression of anti-apoptotic proteins such as Bcl-2. Therefore, ERK is typically associated with promoting cellular survival. However, its interaction with p38 can lead to apoptosis under severe stress conditions. For this reason, KLF5 may influence the balance between the activation of ERK and p38, so that, by modulating this signaling, cellular survival is favored.
Figure 3.
KLFs involvement in regulating renal organogenesis and postnatal maturation of nephrons. The survival of podocytes mediated by KLF5, through the inhibition of the mitogen-activated protein (MAP) kinase pathway, may seem like a contradictory description, as ERK has been shown to positively induce the expression of anti-apoptotic proteins such as Bcl-2. Therefore, ERK is typically associated with promoting cellular survival. However, its interaction with p38 can lead to apoptosis under severe stress conditions. For this reason, KLF5 may influence the balance between the activation of ERK and p38, so that, by modulating this signaling, cellular survival is favored.
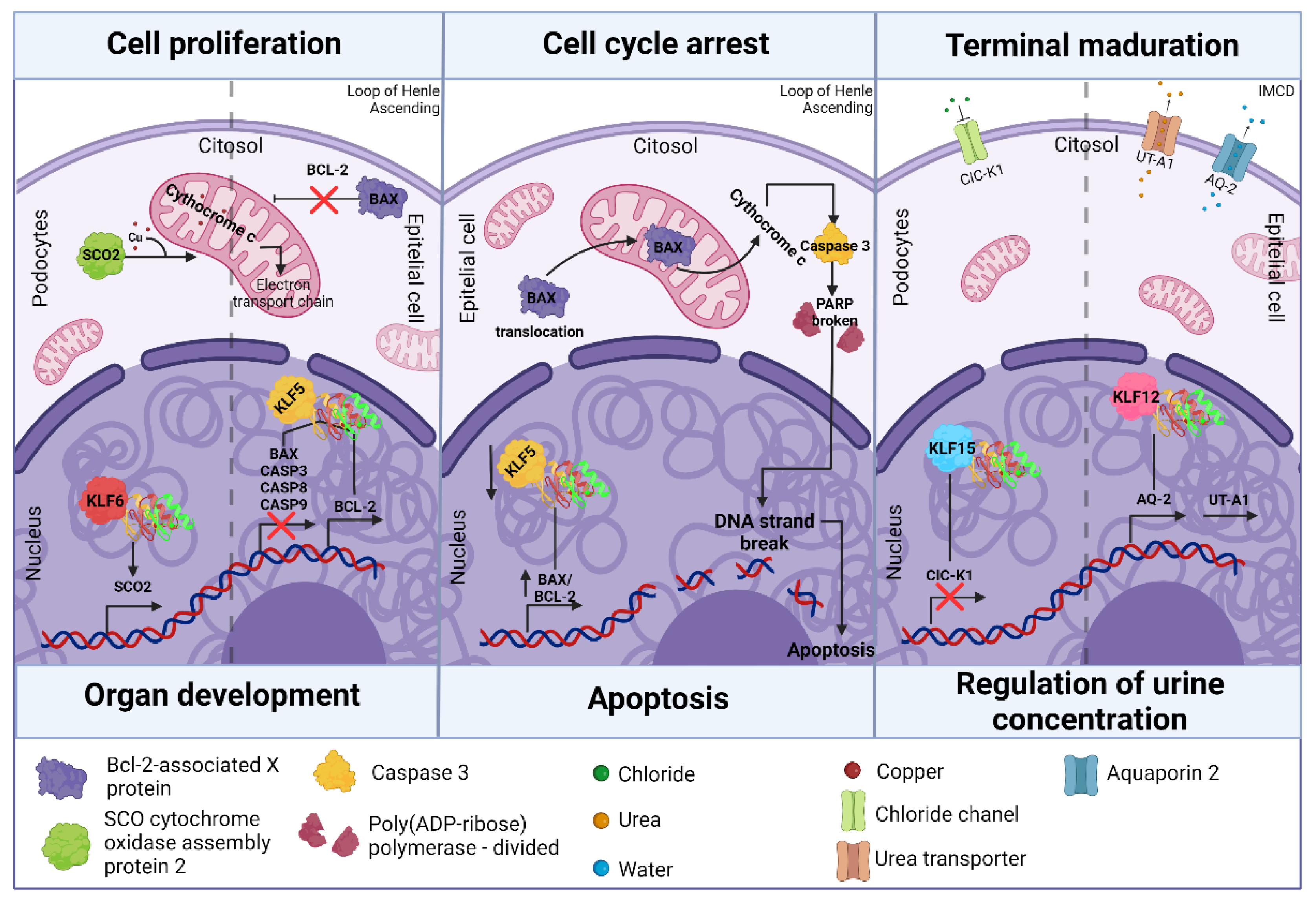
Figure 4.
Implication of KLFs in renal metabolism both under healthy conditions and their effect in states of dysregulation.
Figure 4.
Implication of KLFs in renal metabolism both under healthy conditions and their effect in states of dysregulation.
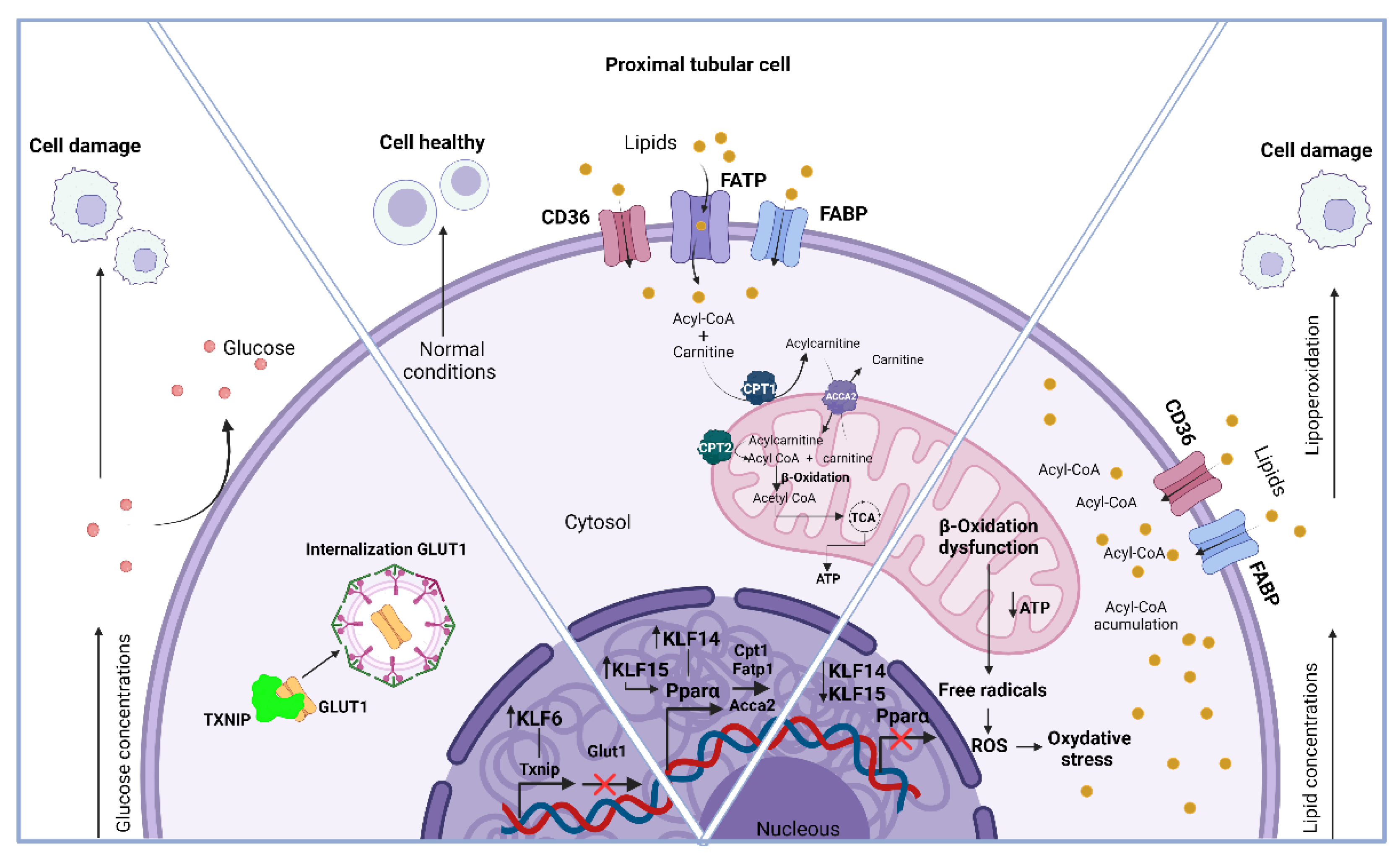

Table 1.
Briefly shows the role of KLF in nephron cells and its implication in kidney physiology. -.
Table 1.
Briefly shows the role of KLF in nephron cells and its implication in kidney physiology. -.
| Cell of nephron | KLF | Role | Reference |
|---|---|---|---|
| Podocytes | KLF2 | Downregulates the expression of occludin, preventing its overexpression from altering the structure of the GBM. | [26] |
| KLF4 | Induces positive expression of E-cadherin, podocin, and nephrin through interactions with HDACs, for the maintenance of tight junctions and the slit diaphragm. ____________________________________________________ Induces the expression of cytokeratins (K8 and K18) that help in the cytoskeleton's organization. _____________________________________________________ Downregulates mesenchymal markers such as vimentin and α-SMA, preventing EMT and structural damage. |
[29] | |
| Glomerular endothelial cells | KLF2 | Regulates the size and distribution of transcellular pores in the ECs by inhibiting the phosphorylation of the myosin light chain. ____________________________________________________________ Modulates VEGF-A-mediated angiogenesis by downregulating its expression, preventing an excess of blood vessels. |
[28,36] |
| KLF4 | Mediates inflammation by downregulating VCAM1 induced by TNF-α, inhibiting the p65 subunit of NF-κB. | [37] | |
| Mesangial cells | KLF4 | Attenuates the expansion of the mesangial matrix and its proliferation by negatively regulating the mTOR pathway, downregulating the expression of phosphorylated (p) mTOR and p S6K proteins, preventing excessive extracellular ECM production, | [41] |
| Proximal tubule cells | KLF4 | Mitigates inflammation and fibrosis by decreasing the expression of pro-inflammatory cytokines, such as MCP-1, MIP-3α, and IL-8. | [44] |
| KLF11 | Like KLF4, it participates in the mitigation of inflammation and fibrosis by reducing the expression of the same pro-inflammatory cytokines. | [43] | |
| KLF15 | Decreases the expression of fibronectin by negatively regulating the MAPK pathways. | [46] |
Table 2.
shows the role of KLF’s on kidney disease.
| Disease | Group Klf | Role | Reference |
|---|---|---|---|
| Chronic kidney disease | Group 1 (Klf 3, 8 and 12) |
Not Available | |
| Group 2 (Klf 1, 2, 4, 5, 6 and 7) |
KLF2 protects endothelial cell injury through anti-inflammatory, anti-thrombotic, and anti-angiogenic effects, as it maintains the proper function of glomerular endothelial cells. Its deficiency has been shown to lead to the progression of renal disease | [36,87] | |
| KLF4 suppression causes the polarization of infiltrating macrophages into myeloid cells that accumulate in the glomerulus and tubular interstitium in CKD to shift to an M1 phenotype. The M1 phenotype of macrophages promotes the production of pro-inflammatory cytokines, such as TNFα and IL-1β. These cytokines exacerbate renal parenchymal injury and accelerate disease progression. Conversely, KLF4 expression suppresses the differentiation of infiltrating macrophages, mitigating renal damage by inhibiting TNFα expression in myeloid cells. Thus, KLF4 is considered a protective transcription factor. In addition, KLF4 mitigates inflammation and fibrosis caused by the TGF-β1-induced release of cytokines MCP-1, MIP-3α and IL-8 in human proximal tubule cells, possibly relating to the phosphorylation of KLF4 that TGF-β1 induces via SMAD and p38/MAPK signaling in vascular smooth muscle cells (VSMCs). It has even been linked to the inhibition of podocyte apoptosis through regulating the mTOR signaling pathway, which is involved in regulating cell growth, proliferation, and survival. | [44,88] | ||
| KLF5 participates in the initiation and progression of tubulointerstitial inflammation, and its expression is increased in proliferating renal tubule cells in the cortex and medulla of fibrotic kidneys. KLF5 regulates renal fibrosis through activation of HIF-1α-KLF5-TGF-β1 pathway, renal cell proliferation through activation of ERK/YAP1/KLF5/cyclin D1 pathway, and tubulointerstitial inflammation with upregulation of pro-inflammatory cytokines which promotes kidney injury. | [7,8] | ||
| KLF6 triggers the release of Apolipoprotein J/Clusterin (Apoj) in podocytes. Apoj activates the calcium/calmodulin-dependent protein kinase 1D (CaMK1D) signaling in neighboring proximal tubular cells. This is crucial because CaMK1D can attenuate mitochondrial fission and restore mitochondrial function under diabetic conditions [7]. | [89] | ||
| Group 3 (Klf 9, 10, 11, 13, 14 and 16) |
KLF11 deficiency is associated with increased renal atrophy, fibrosis, and interstitial inflammation in a mouse model of chronic renal obstruction (UUO). In KLF11 KO-UUO mice, this deficiency is linked to the upregulation of genes such as collagen type I, fibronectin, TGF-β1, as well as IL-6 and TNF-α. These genes are associated with TGF-β signaling, fibrosis, and inflammation. |
[45] | |
| No group (15 and 17) |
KLF15 is downregulated by TGF-β1, which activates multiple intracellular signal transduction systems and MAPK pathways, including ERK and JNK, leading to renal fibrosis. Thus, KLF15 may play an anti-fibrotic factor in renal interstitial fibrosis by decreasing extracellular matrix fibronectin, type III collagen and CTGF expression in renal fibroblast. KLF15 Prevents fibrosis by inhibiting the Wnt/β-catenin pathways and suppress the recruitment of P/CAF to the CTGF promoter in mesangial cells. | [90,91] | |
| Acute kidney injury | Group 1 (Klf 3, 8 and 12) |
Not Available | |
| Group 2 (Klf 1, 2, 4, 5, 6 and 7) |
Overexpression of KLF4 in proximal tubular cells (HK-2) upregulates the expression of miR-101. This increase in miR-101, downregulates the expression of COL10A1, thereby suppressing EMT and renal fibrosis during the pathogenic process of renal fibrosis associated with acute kidney injury. In contrast, the inhibition of KLF4 expression, directly mediated by epigenetic regulatory enzymes such as DNA methyltransferase 1 (Dnmt1), which hypermethylates the KLF4 promoter region, contributes to the progression of EMT in renal EpC. | [92] | |
| KLF5 is regulated by YAP and promotes the expression of Mst1/2, which are proteins involved in the Hippo signaling pathway. Activation of this pathway leads to over proliferation of tubular cells, tubular injury, and inflammation. KLF5 can be upregulated in severe acute kidney injury because of the activation of HIF-1α, which facilitates the transition to chronic kidney disease. The overexpression of KLF5 promotes renal fibrosis and tubular dysfunction, exacerbating acute kidney injury. Another mechanism by which KLF5 is attributed the ability to drive the transdifferentiation of renal tubular EpC is that, in a hypercaloric state, KLF5 binds to the HMGB1 promoter, thereby promoting the transcription of High Mobility Group Box 1 protein. | [83,93,94] | ||
| Group 3 (Klf 9, 10, 11, 13, 14 and 16) |
KLF9, which is upregulated by miR-93-5p, inhibited the expression of circHIPK3, leading to alleviation of oxidative stress and apoptosis in an in vivo model of AKI established by ischemia/reperfusion (I/R) in C57BL/6 mice or hypoxia/reoxygenation (H/R) in HK-2 cells. The circular RNA HIPK3 (circHIPK3), derived from the HIPK3 gene, is important because of its pro-inflammatory activity. | [81] | |
| KLF10 is downregulated in tubular cells during acute kidney injury. This finding suggests that KLF10 acts as a renoprotective protein and provides protection against acute kidney injury, as its induction improves tubular regeneration through the ZBTB7A-KLF10-PTEN axis. PTEN is important because it can inhibit the PI3K/Akt pathway, which regulates cell growth, death, migration, and differentiation. | [80] | ||
| No group (15 and 17) |
KLF15 acts as a bridge connecting the signaling of diacylglycerol kinase epsilon (DGKE) and Klotho. This DGKE/KLF15/Klotho pathway protects against renal ischemia/reperfusion injury (IRI) and AKI in a murine model. In a Xenopus laevis model, it was showed that KLF15 directly binds to enhancers and stimulates the expression of regenerative genes, including adrenoreceptor α 1A (adra1α), suggesting that KLF15 might even promote the regeneration of nephric tubules. As KLF15 attenuates damage and development of glomerulosclerosis, tubulointerstitial fibrosis, inflammation, and stabilizes the actin cytoskeleton, thereby improving renal function. | [79,95] | |
| Diabetic kidney disease | Group 1 (Klf 3, 8 and 12) |
KLF3 directly regulates the transcription of STAT3. In proximal tubular cells (HK-2) exposed to high glucose concentrations, the suppression of KLF3 mediated by miR-23a-3p resulted in the inhibition of STAT3, a protein crucial for regulating inflammation and fibrosis associated with metabolic diseases. Thus, the inhibition of KLF3 leads to a protective effect in renal disease. | [86] |
| Group 2 (Klf 1, 2, 4, 5, 6 and 7) |
KLF2 is upregulated by insulin treatment and downregulated by high glucose concentrations in cultured endothelial cells from KLF2 KO diabetic mice. This effect was showed through FOXO1-dependent transcriptional silencing, which led to glomerular endothelial damage and podocyte injury. This inhibition of KLF2 by FOXO1 has been shown to decrease the expression of the genes nephrin, podocin, and synaptopodin, which are important for the structure and function of podocytes. The deletion of KLF2 (knockout, KO) in the glomeruli reduces the expression of several of its target genes, including endothelial nitric oxide synthase (eNOS), zonula occludens-1 (ZO-1), the glycocalyx, fms-related tyrosine kinase 1 (Flt1), tyrosine kinase with immunoglobulin-like and EGF-like domains 2 (Tie2), and angiopoietin 1 (Angpt1). These genes are primarily involved in the function and integrity of the vascular endothelium, which is why KLF2 is considered a vasoprotective factor. |
[7,96,97,98] |
|
| KLF4 overexpression induces podocyte autophagy, protecting the tissue from damage in DKD. Suppresses cell proliferation and differentiation during fibrosis and inhibits EMT processes. Hyperglycemia also decreases KLF4 expression and increases TGF-β expression leading to unregulated inflammation in renal tissue. | [7,96,97,99] | ||
| KLF5 is overexpressed in the collecting duct EpC found in diabetic kidney and tubulointerstitial disease and associated with alterations like an expansion of mesangial matrix and tubular interstitial space, podocyte damage, and glomerular basement membrane thickening, showing that KLF5 plays a pivotal role in the initiation and progression of renal inflammation. In fact, the inverse expression of KLF4 and KLF5 in the pathogenesis of renal fibrosis is modulated by a matrix stiffness-regulated extracellular signal-regulated kinase (ERK), which increases the protein level and nuclear translocation of mechanosensitive YAP1, preventing the degradation of KLF5. KLF5 is upregulated under hyperglycemic conditions through lactylation of lysine 14 on histone H3 (H3K14la). KLF5 binds to the promoter of the gene encoding E-cadherin (Cadherin 1, cdh1) and inhibits its transcription, promoting disease progression. This lactylation results from the accumulation of lactate because of the metabolic reprogramming that renal PCT undergo in a hyperglycemic state, specifically the shift from oxidative phosphorylation (OXPHOS) to glycolysis. | [8,100,101] | ||
| KLF6, under conditions that promote renal damage and fibrosis, such as diabetic nephropathy, its overexpression enables TGF-β1 to induce the loss of E-cadherin, gain in vimentin expression, and EMT of proximal tubule cells. In CKD, TGF-β promotes renal fibrosis by enhancing matrix formation, cell proliferation, and cell migration via MAPK, phosphatidylinositol 3-kinase/protein kinase B, and Smad2/3/4 pathways, subsequently elevating fibronectin, collagen, and α-SMA. | [102] | ||
| Group 3 Klf 9, 10, 11, 13, 14 and 16) |
KLF 10 Activates KDM6A and induces proteinuria, kidney damage and fibrosis under diabetic conditions. Represses nephrin, WT1, podocin, and synaptophysin in podocytes. Increases expression of type I and III collagen, fibronectin, and metalloproteinases. | [96,103,104] | |
| No group and (KLF15 and KLF17) |
KLF15 modulates mitochondrial biogenesis and homeostasis through the SIRT1-PGC-1α pathway in mouse mesangial cells associated with diabetic nephropathy. This finding was determined through enrichment analysis, which identifies KLF15 as a therapeutic target. | [105] |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



