Submitted:
15 August 2024
Posted:
16 August 2024
Read the latest preprint version here
Abstract
The aim of this study was to investigate the differentially expressed genes associated with intramuscular fat deposition in the longissimus dorsi muscle of Xinjiang Brown Bulls. The longissimus dorsi muscles of 42 Xinjiang Brown Bulls were selected under the same feeding conditions. The intramuscular fat content of muscle samples were determined by Soxhlet extraction method, from which 5 samples with high intramuscular fat content (HIMF group) and 5 samples with low intramuscular fat content (LIMF group) were selected. It was found that the intramuscular fat content of HIMF group was 46.054% higher than that of LIMF group. Muscle samples produced by paraffin section for morphological observation. It was found that the fat richness of the HIMF group was better than that of the LIMF group. Transcriptome sequencing technology was used to analyze the gene expression differences of longissimus dorsi muscle. Through in-depth analysis of the longissimus dorsi muscle by transcriptome sequencing technology, we screened a total of 165 differentially expressed genes. The results of Gene Ontology (GO) enrichment analysis showed that the differentially expressed genes in the two groups were mainly clustered in biological pathways related to carbohydrate metabolic processes, redox processes and oxidoreductase activities. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis showed that the differentially expressed genes were significantly clustered in 15 metabolic pathways, which mainly covered fatty acid metabolism (related to lipid metabolism and glucose metabolism), pentose phosphate pathway, Peroxisome Proliferator-Activated Receptors (PPAR) signalling pathway and other important metabolic processes. The three genes that were predominantly enriched in the glycolipid metabolic pathway by analysis were SCD5, CPT1C, and FBP2, all of which directly or indirectly affect intramuscular fat deposition. In summary, the present study investigated the differences in gene expression between high and low intramuscular fat content in the longissimus dorsi muscle of Xinjiang Brown Bulls by transcriptome sequencing technology, and revealed the related signalling pathways. Therefore, we hypothesized that SCD5, CPT1C, and FBP2 were the key genes responsible for the significant differences in intramuscular fat content of the longissimus dorsi muscles in a population of Xinjiang Brown Bulls. We expect that these findings will provide fundamental support for subsequent studies exploring key genes affecting fat deposition characteristics in Xinjiang Brown Bulls.
Keywords:
longissimus dorsi muscle
; intramuscular fat
; differentially expressed gene
; transcriptome sequencing technology
; metabolic pathway
1. Introduction
Xinjiang Brown Cattle is an excellent local dairy and meat dual-use breed in Xinjiang, which is characterized by strong resistance and roughage tolerance. Xinjiang Brown Cattle is suitable for high-grade beef production because of its rich fat, bright meat color, low shear force, high water system and obvious marbling [1]. The study revealed that meat characteristics within the Xinjiang Brown Cattle breed showed diversity, including differences in intramuscular fat (IMF) content, under consistent rearing conditions. IMF is mainly distributed in the outer and inner muscle membranes as well as the fascia of the muscle, which has an important impact on the flavor, tenderness and nutritional value of beef [2,3]. Meanwhile IMF deposition is mainly regulated by genes related to fat metabolism. The process of lipid metabolism is a complex dynamic equilibrium involving the synthesis and catabolism of adipocytes, a process that is enabled by the co-regulation of multiple transcription factors [4]. Studies have shown that differences in key lipid-controlling genes in the beef cattle organism are a key factor contributing to individual differences in intramuscular fat deposition capacity between breeds [5]. For IMF deposition, the process of depositing large quantities of it requires the consumption of additional energy beyond the level necessary to maintain normal production activities [6,7]. Therefore, high energy levels are required for intramuscular fat deposition in adipose tissue deposition in the body.
With RNA-Seq (transcriptome sequencing) technology, it is possible to accurately capture and resolve the expression of all transcription products within a species under specific conditions [8]. The technology is capable of quantifying mRNA expression levels by means of high-throughput sequencing, and further combining it with bioinformatics analysis methods to study in-depth the characteristics of genes at the transcriptional level, including the amount of gene expression as well as the differences in the expression patterns among different samples. Using this technology, we were able to delve into differentially expressed genes in bovine muscle tissue, which may be closely related to quality characteristics such as fatty acid composition and intramuscular fat content of meat [9,10]. Cesar AS et al. [10] succeeded in revealing molecular mechanisms closely related to lipid metabolism by performing tissue transcriptome (mRNA sequencing) analyses and identified differentially expressed genes present within muscle tissues in Nile beef cattle populations. Zheng et al. [5] sequenced the transcriptome of longissimus dorsi muscle tissue samples from Angus and Chinese Simmental cattle and identified 17 genes that are closely related to muscle and fat metabolism. However, it is still unclear as to which genes regulate intramuscular fat deposition differences among individuals of Xinjiang Brown Cattle. Therefore, the main objective of this study was to determine the intramuscular fat content of the longissimus dorsi muscle of Xinjiang Brown Cattle, and then select tissue samples with high intramuscular fat content and low intramuscular fat content for transcriptome analysis. This process aims to screen out the differentially expressed genes and their related pathways that are closely related to fat deposition, thus providing a strong reference for further exploration of the key genes that regulate fat deposition.
2. Materials and Methods
2.1. Animals
In this experiment, 42 healthy Xinjiang Brown Cattle with the same genetic background and raised under the same feeding conditions were carefully selected and fattened to 26 months of age at the Xinjiang Yili New Brown Breeding Farm. The diets are formulated according to the NRC (National Research Council) Nutrient Requirements for Beef Cattle and include whole plant silage corn, corn stover, alfalfa, wheat straw, corn kernels, cottonseed meal, soybean meal and bran. This study was approved by the Animal Ethics Committee of Xinjiang Agricultural University.
2.2. Sample Collection
All Xinjiang Brown Cattles were humanely slaughtered at the Shengyuan Cattle and Sheep Designated Slaughterhouse in Changji, Xinjiang. Longissimus dorsi muscle samples were collected immediately, and cut into small pieces using surgical scissors carefully, and then quickly placed the samples into freezing tubes and put them into liquid nitrogen for cryopreservation. Intramuscular fat (IMF) content was determined by Soxhlet extraction method, and we designated 21 Xinjiang Brown Cattle with higher IMF content among them as the test group (HIMF group), while the remaining 21 Xinjiang Brown Cattle were used as the control group (LMIF group). Five muscle samples were selected from each of the HIMF and LIMF groups, and these samples were divided equally into two parts. One part of sample was immersed in 4% formalin to make paraffin sections for morphological observation, and the other was frozen in liquid nitrogen for transcriptome sequencing technology to analyze the gene expression differences of the longissimus dorsi muscle between the two groups.
2.3. Determination of IMF Content
IMF content was determined by Soxhlet extraction. 3 grams sample of the longissimus dorsi muscle was stirred into a dried paper packet and weighed. After continuous drying in an oven at 65 ℃ for 15 hours or more, the paper packs were then placed in a desiccator to cool for 10 minutes and subsequently weighed. Subsequently, the paper packets were transferred to a Soxhlet extractor and soaked in ether overnight. Afterwards, they were refluxed in a water bath heated to 75 ℃ for 10 hours and then placed in a well-ventilated area to allow the ether to evaporate naturally. Finally, the samples were dried in an oven at 105 ℃ until their weight no longer changed, i.e., a constant weight state was reached, and subsequently weighed, from which the IMF content was calculated.
2.4. Histology
Five samples of the longissimus dorsi muscle were selected from the HIMF and LIMF groups, respectively, and subsequently cut along the natural texture of the muscle fibres into muscle tissue blocks, each measuring 1 cm × 1 cm × 0.5 cm. The tissues were immersed in 4% paraformaldehyde fixative (Wuhan Carnoss Technology Co., LTD, Wuhan, China) for a period of 24 hours to complete the tissue fixation process. They were then placed in a series of alcohol (Xinjiang Aidil Biotechnology Co., Ltd, Xinjiang, China) solutions at increasingly concentrated levels to progressively remove water from the tissue samples. Finally, the tissue samples were transferred to xylene (Heng Xing Reagent, Tianjing, China) for hyalinization. The samples were immersed in a paraffin (Shanghai Yi Yang Instrument Co. Shanghai, China) solution for the purpose of embedding, after which they were left to cool and solidify naturally. Subsequently, they were sectioned. The cut paraffin sections were placed on slides and deparaffinised using xylene (Heng Xing Reagent, Tianjing, China). Next, the samples were stained with hematoxylin (Zhuhai Beso Biotech Co., Ltd, Zhuhai, China) and slightly washed with running water. They were then differentiated with 0.1% hydrochloric acid (concentrated hydrochloric acid:anhydrous ethanol=1:1) in ethanol and washed with water. Subsequently, the sections were subjected to staining with eosin (Zhuhai Beso Biotech Co., Ltd, Zhuhai, China), followed by a washing and dehydration step involving alcohol. Ultimately, the slices were treated with xylene transparency and sealed with resin (Shanghai Yi Yang Instrument Co., Shanghai, China), thus completing the preparation process. The tissue sections were then were then examined in morphological detail by FEI Quanta 250 Field Emission Environmental Scanning Electron Microscope.
2.5. Total RNA Extraction, Library Construction and Sequencing
The muscle samples were carefully ground into powder form, from which total RNA was extracted using TRIzol reagent (Beijing Adderall Biotechnology Co., Ltd, Beijing, China). mRNA molecules with polyA tails were specifically enriched using Oligo(dT) magnetic bead technology. The integrity of mRNA was accurately examined using an Agilent 2100 Bioanalyzer.
A library was constructed using 5 μg of RNA, and the first strand of cDNA was synthesized using fragmented mRNA as a template and random oligonucleotides (Guangzhou Reebok Biotechnology Co., Guangzhou, China) as primers in the M-MuLV reverse transcriptase system. The RNA strand is degraded using RNaseH (Shanghai Lianmai Biological Engineering Co., Shanghai,China) and the second strand of cDNA is subsequently synthesized using dNTPs in a DNA polymerase I system. The purified double-stranded cDNA undergoes end repair, addition of an A-tail and ligation of sequencing junctions. Afterwards, PCR amplification is performed and the PCR products are purified again using AMPure XP beads (Beckman Coulter, Brea, CA) to produce the final library.
The libraries were initially quantified using a Qubit 2.0 Fluorometer and subsequently diluted to a concentration of 1.5 ng/ul. Next, the insert size of the libraries was examined using an Agilent 2100 bioanalyzer. After confirming that the insert sizes were as expected, the libraries were pooled according to the effective concentration of each library and the target sequencing data volume for Illumina sequencing. In the flow cell for sequencing, we add four kinds of fluorescent labeled dNTP, DNA polymerase and junction primers for amplification. The sequencer captures these fluorescent signals and converts them into sequencing peaks through the computer software to obtain the sequence information of the DNA fragments to be sequenced.
2.6. Real-time quantitative PCR (RT-qPCR)
RNA samples from transcriptome sequencing were taken for RT-qPCR validation with three biological replicates for each sample, and cDNA was obtained by operating according to the instructions of the Reverse Transcription Kit (Nanjing Novozymes Biotechnology Co., Ltd., Nanjing, China). Data on 10 differential genes associated with fat metabolism among individuals of Xinjiang Brown Cattle were identified using an ABI SteponePlus instrument according to the protocol outlined in the Fluorescence Quantification Kit for Genetically Modified Organisms Technology (Nanjing Novozymes Biotechnology Co., Ltd., Nanjing, China). Primer sequences were designed using Primer 5 (Table 1) and the internal reference gene was β-actin. The RT-qPCR amplification program was set to 95 ℃ for 5 min, 95 ℃ for 10 s and 60 ℃ for 30 s (40 cycles), and the results were analyzed by 2-ΔΔCT method.
2.7. Data Processing
2.7.1. Quality Control, Transcript Assembly and Splicing
Sequenced fragments are converted into reads by CASAVA base recognition of the image data measured by the high-throughput sequencer, and the files are in fastq format. Filter from raw read length to net read length. Filtering content: removal of reads with adapters (connectors), removal of reads containing N (N indicates that base information could not be determined), and removal of low-quality reads (reads where the number of bases with Qphred<=5 accounted for more than 50% of the entire read length). Q20, Q30, and GC content were calculated for the net data. The reference genome and gene annotation file (Bos_taurus.ARS-UCD1.2) were downloaded from the ensemb database, and the Clean Reads were quickly and accurately compared with the reference genome using HISAT2 software. Based on the comparison information, StringTie software was applied to assemble the new transcripts.
2.7.2. Sequence Data Mining and Analysis
Expression levels of genes were expressed as transcript fragments per kilobase per million mapped read values. Differentially expressed m RNAs were obtained from the expression level analysis of m RNAs, and transcripts were analyzed for differences using the negative binomial distribution of DESeq2 (1.20.0). P-adjust≤0.05 and |log2(Fold Change)| ≥1 were set to screen for differentially expressed genes.
2.7.3. GO and KEGG Enrichment Analysis of Differentially Expressed Genes
The differential gene sets were analyzed for GO functional enrichment and KEGG pathway enrichment using clusterProfiler software, and both GO functional enrichment and KEGG (Kanehisa et al., 2000) pathway enrichment were analyzed with P < 0.05 as the threshold for significant enrichment.
2.7.4. Statistical Analysis
The data were organized using Microsoft Office Excel (version 2016, Microsoft Corporation, USA). The data of IMF content and RT-qPCR were statistically analyzed by independent t test using SPSS (version 20.0, IBM, USA), and results were expressed as mean ± standard deviation. Difference multiplicity plots were plotted using Origin (version 7.5, OriginLab Corporation, USA).
3. Results
3.1. Comparison of IMF Content and Morphological Observation of the Longissimus Dorsi Muscle in Two Groups
The IMF content in the longissimus dorsi muscle of the 42 Xinjiang Brown Cattle in the experiment differed among individuals, with the overall IMF content in the HIMF group being 2.819% higher than that in the LIMF group (P = 0.0001) (Table 2). Transverse sections of longissimus dorsi muscle of both groups are shown in Figure 1. Five muscle samples were randomly selected for slicing from each of the HIMF and LIMF groups, and it was clearly observed that the texture and abundance of fat in the five sliced samples from the HIMF group was superior to that of the LIMF group.
3.2. Transcriptome Sequencing Data Analysis
For the longissimus dorsi muscle samples of Xinjiang Brown Cattle, an average of 556,876,666,000 raw read lengths were obtained for each sample, and an average of 43,276,318,600 filtered read lengths were obtained after raw data filtering. All of them were below 0.03% (Q20 ≥ 97.81%, Q30 ≥ 93.85%) as checked by the sequencing error rate, indicating that the sequencing data were valid (Table 3). When analyzed against the bovine reference genome (Table 4), Clean reads maintained an overall match rate of more than 93.10% and a unique match rate of more than 90.04%. Only a small number of readers matched to multiple locations in the reference genome (≤3.41%), and the vast majority of reads matched uniquely to the reference genome.
3.3. Quantitative Analysis of Transcriptome Sequencing
According to the Venn diagram, the number of expressed genes in the HIMF group was 12625, while the number of expressed genes in the LIMF group was 11796. There were 11,486 co-expressed gene numbers in both groups (Figure 2A). Component analysis of the 10 samples showed that both groups were clustered with good inter-sample reproducibility (Fig. 2B). In terms of the distribution of gene expression in the samples, the mRNA expression levels were relatively homogeneous in the two groups of samples (Figure 2C). The correlation of gene expression levels between samples is an important indicator to test the reliability of the experiment and whether the sample selection is reasonable. The closer its correlation coefficient is to 1, the higher the similarity of expression patterns between samples. According to the inter-sample correlation heatmap, it can be seen that the R2 of the samples in this test were all greater than 0.94, indicating a high inter-sample correlation coefficient (Figure 2D).
3.4. Transcriptome Sequencing of Differentially Expressed Genes
A total of 165 differential genes were screened in the two groups of longissimus dorsi muscle samples, including 101 up-regulated genes and 64 down-regulated genes (Figure 3A). The distribution of differential genes among samples can be visualized from the volcano plot (Figure 3B). As shown by the heat map of differentially expressed gene clustering, the two groups of differentially expressed genes were well clustered (Figure 3C).
3.5. GO Enrichment Analysis
The 30 most significant terms from the GO enrichment analysis results were selected to be plotted in a scatter plot for presentation (Figure 4). Among them, most of the differentially upregulated genes were enriched in 18 terms, and most of the differentially downregulated genes were enriched in 16 terms. There are four processes that are co-enriched by both: xidation-reduction process、carbohydrate metabolic process、non-membrane-bounded organelle and oxidoreductase activity. According to Figure 4, the term involved in oxidation-reduction reaction in biological processes was the most significant (P = 0.0036), followed by the term involved in carbohydrate metabolic processes (P = 0.0305). In terms of molecular function the main focus is on the term of oxidoreductase activity, which includes the action of oxidoreductase activity on the aldehyde or oxygen group of the donor, the CH-OH group, the CH-OH group as a donor, and the NAD or NADP as an acceptor.
3.6. KEGG Pathway Analysis
The 20 KEGG pathways with the most significant KEGG enrichment results were selected to be plotted in a scatter plot for presentation (Figure 5). Most of the differentially upregulated genes were highly significantly enriched in Glucagon signaling pathway (P = 0.0072), C-type lectin receptor signaling pathway (P = 0.0083), Pyruvate metabolism (P = 0.0047) and PPAR signaling pathway (P = 0.0026), as well as significantly enriched in Tyrosine metabolism (P = 0.0280), Propanoate metabolism (P = 0.0264), and Fatty acid metabolism (P = 0.0118) signaling pathway. Most of the differentially downregulated genes were highly significantly enriched in Glycolysis/Gluconeogenesis (P<0.0001) and HIF-1 signaling pathway (P = 0.0018), as well as significantly enriched in Fatty acid degradation (P = 0.0417), Fructose and mannose metabolism (P = 0.0312), Biosynthesis of amino acids (P = 0.0222), Pentose phosphate pathway (P = 0.0205), and Carbon metabolism signaling pathways (P = 0.0129). According to Table 5, it can be seen that SCD5 among the differentially upregulated genes was enriched in 4 pathways, CPT1C was enriched in 6 pathways, and FBP2 among the differentially downregulated genes was enriched in 6 pathways. Meanwhile the three were co-enriched in the AMPK signaling pathway.
3.7. RT-qPCR Validation of RNA-Seq results
Ten genes were screened in the transcriptome sequencing results for RT-qPCR validation, and the results showed that the transcriptome sequencing data were consistent with the data expression results of RT-qPCR (Figure 6). It proved the accuracy and reliability of the transcriptome sequencing results.
4. Discussion
In this study, by measuring the intramuscular fat (IMF) content of the longissimus dorsi muscle of Xinjiang Brown Cattle, it was found that there were differences IMF content between samples. After tissue transcriptomics analysis, significant enrichment of SCD5, CPT1C and FBP2 genes was mainly found in the KEGG pathway closely related to glycolipid metabolism, especially in the metabolic pathway of PPAR signaling pathway, Pyruvate metabolism, Fatty acid metabolism, and Fatty acid degradation, which may play key roles in the process of IMF deposition. The deposition of fat in the animal organism is the result of the dynamic balance between fat synthesis and catabolism, as well as the equilibrium between the intake of energy-supplying substances and the energy consumption of the organism, whereas fat is deposited within the muscle to form IMF. It has been shown that IMF improves meat tenderness due to the fact that entrapment in the perimuscular connective tissue weakens the association between collagen fibers and reduces the force required to break down the connective tissue [11]. IMF deposition plays an important role in meat quality improvement, and the longissimus dorsi muscle of cattle is the most susceptible to fat deposition. In this study, we further explored the related genes affecting IMF deposition by comparatively analyzing the transcriptomic data of the longissimus dorsi muscle of Xinjiang Brown Cattle in the high intramuscular fat content group and the low intramuscular fat content group.
In GO functional enrichment, differentially upregulated genes were most prominent in biological processes such as redox reactions, followed by carbohydrate metabolism processes. At the molecular functional level these genes were then mainly focused on oxidoreductase activity. In biological organisms, oxidation-reduction reactions are ubiquitous, occurring between various metabolic pathways. Studies have shown that a wide range of physiological processes within the cells of living organisms involve and depend on the conduct of various redox reactions. These include energy production, signaling, enzyme-catalyzed reactions, and complex activities such as cell division, proliferation, and differentiation, and even the processes of cellular autophagy, apoptosis, and necrosis, which are critical in physiological and pathological responses [12,13].
Upon analysis of KEGG-enriched pathways, it was shown that the PPAR signaling pathway, Pyruvate metabolism, Fatty acid metabolism, Fatty acid degradation, and Fructose and mannose metabolism pathways are all closely linked to the process of fat synthesis and metabolism. Among them, the PPAR signaling pathway covers three isoforms, PPARα, PPARβ/δ, and PPARγ, while the peroxisome proliferator-activated receptor γ (PPARγ) signaling pathway shows high expression in adipose tissue [14]. It plays an important role in early lipogenesis and is able to regulate fatty acid production by modulating phosphoenolpyruvate. The fatty acid metabolic pathway encompasses the key processes of fatty acid synthesis (i.e., the fatty acid synthesis pathway) as well as fatty acid β-oxidation (also known as the fatty acid degradation pathway). Fatty acid synthesis is the result of a dynamic balance between triglyceride catabolism and re-esterification processes [15,16]. However, re-esterification of triglycerides requires the release of cytoplasmic phosphoenolpyruvate carboxylase (PEPCK-C), the major glycolytic enzyme of adipose tissue [17].
In this study, the SCD5 gene belongs to the differentially upregulated genes, which are located in the PPAR signaling pathway and Fatty acid metabolism signaling pathway. SCD (including two isoforms, SCD1 and SCD5) was found to be the main enzyme regulating the conversion of saturated fatty acids to unsaturated fatty acids, which is expressed in adipose tissue and is involved in adipocyte metabolism and fatty acid metabolism [18,19]. In delving into the similar functional roles of SCD1 and SCD5, we found that SCD5 was able to partially or fully compensate for the function of SCD1 and that the differences in this catalytic effect depended on the specific cell type [20,21]. Bovine SCD genes are expressed predominantly in the mammary gland and adipose tissue. Gervais R et al. [22] identified the SCD5 gene in the mammary gland of dairy cows and noted that it is involved in regulating fatty acid metabolism in conjunction with SCD1.
Studies have shown that the SCD5 gene has a role involved in the control of cellular fat distribution, lipid synthesis, cell signaling, and cell growth and replication [23,24], but less research has been done on the mechanism of regulating fatty acid metabolism and fat deposition. Zhang HB et al. [25] investigated the correlation between IMF content and the expression of lipid metabolism-related genes in yaks, and found that the external spine could promote intramuscular fat deposition and increase the tenderness of muscle tissues by increasing the expression of the SCD5 gene, thus improving the quality of the meat, but the specific regulatory mechanism was not clear. Fang QH et al. [26] knocked out the SCD5 gene using the gene targeting knockout technology and found that the deletion of SCD5 significantly reduced the relative content of erucic acid. Erucic acid was able to regulate the differentiation of mesenchymal stem cells by inhibiting the transcriptional activity of PPARγ and significantly reduced the expression of genes related to adipocyte differentiation. Therefore, chronic intake of high erucic acid may affect lipid metabolism [27,28]. In this study, we screened differential genes between high intramuscular fat and low intramuscular fat content from the longissimus dorsi muscle samples of Xinjiang Brown Cattle, and one of the differentially upregulated genes was SCD5. It is hypothesized that SCD5 may have a role in fat metabolism and be able to regulate IMF deposition.
CPT1C belongs to the CPT1 family of transmembrane integrins and is associated with the transmembrane transport of mitochondrial outer membrane peptide chains [29]. It is involved in the regulation of fatty acid oxidative catabolism (FAO) action, which is the main source of cellular energy. In this study CPT1C gene belongs to the differentially upregulated genes, which were found to be involved in Fatty acid degradation, Fatty acid metabolism, Glucagon signaling pathway, and PPAR signaling pathway. Hada T et al. [30] overexpressed three CPT1 isoforms in COS7 cells and normalized the activity values according to CPT1 expression levels. CPT1C was found to have low catalytic activity, with only 2% of CPT1A-specific activity and 5% of CPT1B-specific activity. However, Sierra AY et al. [31] isolated FAO metabolizing enzyme activity in microsomal fractions after transfection of HEK293T and PC-12 cells using pIRES-CPT1c and compared it with FAO metabolizing enzyme activity in fractions transfected with empty pIRES vector. They found that there was a significant elevation of residual CPT1A activity in microsomes, while the inhibitory effect of malonyl coenzyme A on CPT1 was significantly reduced. This suggests that CPT1C may indirectly enhance the ability of FAO by binding to malonyl coenzyme A, thus promoting the oxidative catabolism metabolism of fatty acids.
Most of the current studies on CPT1C have focused on tumors, where it is widely involved in cancer cell differentiation as a member of a family of key rate-limiting enzymes for fatty acid oxidative metabolism [32]. By overexpressing CPT1C in the MCF-7 breast cancer cell line, some studies have found that this enhances the stress resistance of tumor cells to a low-oxygen and glucose-free environment and increases tumor cell ATP production. On the contrary, if CPT1C is interfered with, it will have an inhibitory effect on the growth and proliferation of tumor cells [33,34]. CPT1C has an anti-aging effect in tumors.Chen P et al. [35] found that CPT1C was able to reverse the senescence process of MRC-5 fibroblasts in proliferation and senescence studies of human embryonic lung MRC-5 fibroblasts. Further lipidomic analysis showed that gain of function of CPT1C resulted in reduced lipid accumulation and reversal of aberrant lipid metabolic reprogramming in MRC-5 advanced cells. By oil red O staining and Nile red fluorescence detection, they found that lipid accumulation was significantly reduced when CPT1C function was absent, contrary to previous observations. Therefore, in combination with the result that CPT1C belongs to the differentially upregulated genes in the longissimus dorsi muscle of Xinjiang Brown Cattle in the present study, it is hypothesized that it has a promotional role with intramuscular fat deposition.
As a key enzyme in glucose metabolism, the FBP2 gene is not only involved in regulating the expression of lipid metabolism-related enzyme genes, but also has the function of catalyzing the conversion of fructose 1,6-bisphosphate to fructose 6-phosphate. This conversion process is essential for the conversion of carbohydrate precursors such as lactate into glycogen synthesis. In addition, the FBP2 gene exhibits phosphatase activity and plays a regulatory role in gluconeogenesis [36,37]. In the present study, FBP2 belongs to the differentially downregulated genes in the AMPK signaling pathway, Fructose and mannose metabolism, Fructose and mannose metabolism, Carbon metabolism, Glycolysis/Glycogenesis, and Glucagon signaling pathways. Ectopic expression of FBP2 was found to activate the AMPK signaling pathway and inhibit the Akt-mTOR pathway, leading to inhibition of glucose metabolism [38]. Bakshi I et al. [39] proposed that elevated levels of FBP2 activity may promote inefficient cycling and increase metabolic sensitivity thereby elevating energy demand and leading to increased substrate oxidation. Specifically, enhanced FBP2 activity promotes glycolytic fluxes in mouse extensor digitorum longus (EDL muscle). In addition, overexpression of FBP2 leads to an increase in glucose oxidation in red EDL muscle while decreasing glucose oxidation in white EDL muscle, a process that may trigger differences in insulin-stimulated glucose uptake. In this study, the FBP2 gene was among the differentially down-regulated genes. The relatively low expression level of FBP2 in the muscles of the HIMF group compared with the LIMF group may be a potential reason for the more abundant intramuscular fat content in the HIMF group. Meanwhile, FBP2 also reduces unnecessary energy consumption during gluconeogenesis and helps to achieve an optimal state of glycolytic efficiency.
It was noted that the FBP2 gene can regulate lipid metabolism in the liver and blood by activating glucokinase to maintain hepatic metabolic homeostasis [40]. In a study by Rukkwamsuk T et al. [41], it was found that the decrease in FBP2 activity in cattle with fatty liver during 1-7 days postpartum led to a slowing down of the rate of gluconeogenesis which in turn triggered a delay in lipolysis, ultimately impairing the gluconeogenic capacity of the liver. In this study, FBP2 belonged to the differentially down-regulated genes in the longissimus dorsi muscle of Xinjiang Brown Cattle. It is hypothesized that this gene may play a role in delaying lipolysis in muscle tissue, thus promoting more abundant deposition of intramuscular fat in the LIMF group.
5. Conclusions
In summary, the present study showed that intramuscular fat content deposition differences also existed within the species in Xinjiang Brown Cattle. A total of 165 differentially expressed genes were screened by tissue transcriptome sequencing of meat samples with high and low intramuscular fat content in the longissimus dorsi muscle. The SCD5 and CPT1C genes among the differentially expressed genes are mainly enriched in Fatty acid metabolism and PPAR signaling pathways, which can promote intramuscular fat deposition indirectly or directly. The FBP2 gene among the differentially expressed genes is predominantly enriched in the Glycolysis/Glycogenesis and AMPK signaling pathways, which can affect lipid metabolism in fat through glucose metabolism. Therefore, the present study identified SCD5, CPT1C and FBP2 as differentially expressed genes closely related to lipid metabolism, a finding that can provide a valuable reference for subsequent in-depth investigation of key regulatory genes affecting the fat deposition process in cattle.
Author Contributions
Conceptualization, methodology, validation and writing-original draft preparation, Y.G. and L.Y.; Investigation and resources, L.Y. and K.Y.; Data curation, Y.W. and X.Z.; Visualization, M.Y. and Y. W.; Project administration and funding acquisition, W.S.; Conceptualization, writing—review and editing, and supervision, W.R. All authors have read and agreed to the published versi-on of the manuscript.
Funding
Supported by the earmarked fund for XJARS (XJARS-11), Studies on the metabolic basis and molecular mechanism of the efficient synthesis of mammary casein in dairy cows (2022D01D10) and Tianchi talent program (2223RSTTCYC).
Informed Consent Statement
Not applicable.
Data Availability Statement
We confirm that our experimental data are accurate, which supports the results and conclusions of this study.
Acknowledgments
This study received guidance and assistance from Wanping Ren, Liang Yang, and Wei Shao etc. of Xinjiang Agricultural University in experimental design, and paper writing. We also thank Wen Jiang, Min Yang, Kangyu Yao, and others for their assistance with software and Methodology Writing. We sincerely thank them!
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Chen, Q.; Xu, L.; Zhang, M.; Zhang, T.; Yan, M.; Zhai, M.; Huang, X. Whole genome resequencing reveals the genetic contribution of Kazakh and Swiss Brown Cattle to a population of Xinjiang Brown Cattle. Gene 2022, 839, 146725. [Google Scholar] [CrossRef] [PubMed]
- Brewer, M.S.; Zhu, L.G.; McKeith, F.K. Marbling effects on quality characteristics of pork loin chops: consumer purchase intent, visual and sensory characteristics. Meat Sci. 2001, 59, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Pickworth, C.L.; Loerch, S.C.; Velleman, S.G.; Pate, J.L.; Poole, D.H.; Fluharty, F.L. Adipogenic differentiation state-specific gene expression as related to bovine carcass adiposity. J Anim Sci. 2011, 89, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Huang, Y.; Das, A.K.; Yang, Q.; Duarte, M.S.; Dodson, M.V.; Zhu, M.J. Meat Science and Muscle Biology Symposium: manipulating mesenchymal progenitor cell differentiation to optimize performance and carcass value of beef cattle. J Anim Sci. 2013, 91, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, J.; Wang, X.; Han, L.; Yang, Y.; Wang, Q.; Yu, Q. Metagenomic and transcriptomic analyses reveal the differences and associations between the gut microbiome and muscular genes in ang-us and Chinese Simmental Cattle. Front Microbiol 2022, 13, 815915. [Google Scholar] [CrossRef]
- Bonnet, M.; Faulconnier, Y.; Leroux, C.; Jurie, C.; Cassar-Malek, I.; Bauchart, D.; Boulesteix, P.; Pethick, D.; Hocquette, J.F.; Chilliard, Y. Glucose-6-phosphate dehydrogenase and leptin are related to marbling differences among Limousin and Angus or Japanese Black x Angus steers. J Anim Sci. 2007, 85, 2882–2894. [Google Scholar] [CrossRef] [PubMed]
- Pethick, W.D.; Harper, S.G.; Oddy, H.V. Growth, development and nutritional manipulation of marbling in cattle: a review. J AUST J EXP AGR. 2004, 44, 705–715. [Google Scholar] [CrossRef]
- Liu, S.; Huang, J.; Wang, X.; Ma, Y. Transcription factors regulate adipocyte differentiation in beef cattle. Anim Genet 2020, 51, 351–357. [Google Scholar] [CrossRef]
- Berton, M.P.; Fonseca, L.F.; Gimenez, D.F.; Utembergue, B.L.; Cesar, A.S.; Coutinho, L.L.; de Lemos, M.V.; Aboujaoude, C.; Pereira, A.S.; Silva, R.M.; et al. Gene expression profile of intramuscular muscle in Nellore cattle with extreme values of fatty acid. BMC Genomics 2016, 17, 972. [Google Scholar] [CrossRef]
- Cesar, A.S.; Regitano, L.C.; Koltes, J.E.; Fritz-Waters, E.R.; Lanna, D.P.; Gasparin, G.; Mourão, G.B.; Oliveira, P.S.; Reecy, J.M.; Coutinho, L.L. Putative regulatory factors associated with intramuscular fat content. PLoS One. 2015, 10, e0128350. [Google Scholar] [CrossRef]
- Essén-Gustavsson, B.; Karlsson, A.; Lundström, K.; Enfält, A.C. Intramuscular fat and muscle fibre lipid contents in halothane-gene-free pigs fed high or low protein diets and its relation to meat quality. Meat Sci. 1994, 38, 269–277. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008, 31, 91–123. [Google Scholar] [CrossRef]
- Keating, D.J. Mitochondrial dysfunction, oxidative stress, regulation of exocytosis and their relevance to neurodegenerative diseases. J Neurochem 2008, 104, 298–305. [Google Scholar] [CrossRef]
- Taniguchi, M.; Guan, L.L.; Zhang, B.; Dodson, M.V.; Okine, E.; Moore, S.S. Adipogenesis of bovine perimuscular preadipocytes. Biochem Biophys Res Commun. 2008, 366, 54–59. [Google Scholar] [CrossRef]
- Forest, C.; Tordjman, J.; Glorian, M.; Duplus, E.; Chauvet, G.; Quette, J.; Beale, E.G.; Antoine, B. Fatty acid recycling in adipocytes: a role for glyceroneogenesis and phosphoenolpyruvate carboxykinase. Biochem Soc Trans. 2003, 31, 1125–1129. [Google Scholar] [CrossRef]
- Chaves, V.E.; Frasson, D.; Kawashita, N.H. Several agents and pathways regulate lipolysis in adipocytes. Biochimie 2011, 93, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Jaubert, A.M.; Penot, G.; Niang, F.; Durant, S.; Forest, C. Rapid nitration of adipocyte phosphoenolpyruvate carboxykinase by leptin reduces glyceroneogenesis and induces fatty acid release. PLoS One. 2012, 7, e40650. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Yan, H.; Xia, M.; Chang, X.; Xu, X.; Wang, L.; Sun, X.; Lu, Y.; Bian, H.; Li, X.; et al. Metformin attenuates triglyceride accumulation in HepG2 cells through decreasing stearyl-coenzyme A desaturase 1 expression. Lipids Health Dis. 2018, 17, 114. [Google Scholar] [CrossRef] [PubMed]
- Salmani Izadi, M.; Naserian, A.A.; Nasiri, M.R.; Majidzadeh Heravi, R.; Valizadeh, R. Evaluation of SCD and FASN gene expression in Baluchi, Iran-Black, and Arman Sheep. Rep Biochem Mol Biol. 2016, 5, 33–39. [Google Scholar] [PubMed]
- Sinner, D.I.; Kim, G.J.; Henderson, G.C.; Igal, R.A. StearoylCoA desaturase-5: a novel regulator of neuronal cell proliferation and differentiation. PLoS One. 2012, 7, e39787. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Burhans, M.S.; Flowers, M.T.; Ntambi, J.M. Hepatic oleate regulates liver stress response partially through PGC-1α during high-carbohydrate feeding. J Hepatol 2016, 65, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Gervais, R.; McFadden, J.W.; Lengi, A.J.; Corl, B.A.; Chouinard, P.Y. Effects of intravenous infusion of trans-10, cis-12 18:2 on mammary lipid metabolism in lactating dairy cows. J Dairy Sci. 2009, 92, 5167–5177. [Google Scholar] [CrossRef]
- Igal, R.A.; Sinner, D.I. Stearoyl-CoA desaturase 5 (SCD5), a Δ-9 fatty acyl desaturase in search of a function. Biochim Biophys Acta Mol Cell Biol Lipids. 2021, 1866, 158840. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Xiao, W.; Qin, X.; Cai, G.; Chen, H.; Hua, Z.; Cheng, C.; Li, X.; Hua, W.; Xiao, H.; et al. Myostatin regulates fatty acid desaturation and fat deposition through MEF2C/miR222/SCD5 cascade in pigs. Commun Biol. 2020, 3, 612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.B.; Guan, J.Q.; Zhou, X.J.; Liao, X.P.; Guo, D.S.; Luo, X.L. Correlation Analysis of Intramuscular Fat Content and Fat Metabolism Related Gene Expression in Different Muscle Tissues of Yaks. Chinese Journal of Animal Science 2020, 56, 73–77. (In China) [Google Scholar] [CrossRef]
- Fang, Q.H.; Bai, W.Z.; Li, Z.M.; Chen, H.B.; Bi, Z.T. Studies on the effect of pig SCD5 gene deletion on fatty acid composition. Chinese Journal of Animal Science 2023, 59, 224–231. (In China) [Google Scholar] [CrossRef]
- Chen, X.; Shang, L.; Deng, S.; Li, P.; Chen, K.; Gao, T.; Zhang, X.; Chen, Z.; Zeng, J. Peroxisomal oxidation of erucic acid suppresses mitochondrial fatty acid oxidation by stimulating malonyl-CoA formation in the rat liver. J Biol Chem. 2020, 295, 10168–10179. [Google Scholar] [CrossRef]
- Takahashi, A.; Dohi, H.; Egashira, Y.; Hirai, S. Erucic acid derived from rosemary regulates differentiation of mesenchymal stem cells into osteoblasts/adipocytes via suppression of peroxisome proliferator-activated receptor γ transcriptional activity. Phytother Res. 2020, 34, 1358–1366. [Google Scholar] [CrossRef]
- Roa-Mansergas, X.; Fadó, R.; Atari, M.; Mir, J.F.; Muley, H.; Serra, D.; Casals, N. CPT1C promotes human mesenchymal stem cells survival under glucose deprivation through the modulation of autophagy. Sci Rep. 2018, 8, 6997. [Google Scholar] [CrossRef]
- Hada, T.; Yamamoto, T.; Yamamoto, A.; Ohkura, K.; Yamazaki, N.; Takiguchi, Y.; Shinohara, Y. Comparison of the catalytic activities of three isozymes of carnitine palmitoyltransferase 1 expressed in COS7 cells. Appl Biochem Biotechnol. 2014, 172, 1486–1496. [Google Scholar] [CrossRef]
- Sierra, A.Y.; Gratacós, E.; Carrasco, P.; Clotet, J.; Ureña, J.; Serra, D.; Asins, G.; Hegardt, F.G.; Casals, N. CPT1c is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity. J Biol Chem. 2008, 283, 6878–6885. [Google Scholar] [CrossRef]
- Pucci, S.; Zonetti, M.J.; Fisco, T.; Polidoro, C.; Bocchinfuso, G.; Palleschi, A.; Novelli, G.; Spagnoli, L.G.; Mazzarelli, P. Carnitine palmitoyl transferase-1A (CPT1A): a new tumor specific target in human breast cancer. Oncotarget 2016, 7, 19982–19996. [Google Scholar] [CrossRef] [PubMed]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef]
- Wang, R.; Cheng, Y.; Su, D.; Gong, B.; He, X.; Zhou, X.; Pang, Z.; Cheng, L.; Chen, Y.; Yao, Z. Cpt1c regulated by AMPK promotes papillary thyroid carcinomas cells survival under metabolic stress conditions. J Cancer. 2017, 8, 3675–3681. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhang, Q.; Zhang, H.; Gao, Y.; Zhou, Y.; Chen, Y.; Guan, L.; Jiao, T.; Zhao, Y.; Huang, M.; et al. Carnitine palmitoyltransferase 1C reverses cellular senescence of MRC-5 fibroblasts via regulating lipid accumulation and mitochondrial function. J Cell Physiol. 2021, 236, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Khan, S.A.; Peng, L.J.; Lange, A.J. Roles for fructose-2,6-bisphosphate in the control of fuel metabolism: beyond its allosteric effects on glycolytic and gluconeogenic enzymes. Adv Enzyme Regul. 2006, 46, 72–88. [Google Scholar] [CrossRef]
- Dzugaj, A. Localization and regulation of muscle fructose-1,6-bisphosphatase, the key enzyme of glyconeogenesis. Adv Enzyme Regul. 2006, 46, 51–71. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, J.; Xu, H.; Xing, R.; Pan, Y.; Li, W.; Cui, J.; Zhang, H.; Lu, Y. Decreased fructose-1,6-bisphosphatase-2 expression promotes glycolysis and growth in gastric cancer cells. Mol Cancer. 2013, 12, 110. [Google Scholar] [CrossRef]
- Bakshi, I.; Suryana, E.; Small, L.; Quek, L.E.; Brandon, A.E.; Turner, N.; Cooney, G.J. Fructose bisphosphatase 2 overexpression increases glucose uptake in skeletal muscle. J Endocrinol. 2018, 237, 101–111. [Google Scholar] [CrossRef]
- Agius, L. Hormonal and Metabolite Regulation of Hepatic Glucokinase. Annu Rev Nutr. 2016, 36, 389–415. [Google Scholar] [CrossRef]
- Rukkwamsuk, T.; Wensing, T.; Geelen, M.J. Effect of fatty liver on hepatic gluconeogenesis in periparturient dairy cows. J Dairy Sci. 1999, 82, 500–505. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Morphological observation on the longissimus dorsi muscle of Xinjiang Brown Cattle . (A) Five samples with low intramuscular fat content were selected for paraffin sectioning from the LIMF group; (B) five samples with high intramuscular fat content were selected for paraffin sectioning from the HIMF group. Hematoxylin eosin staining was used, the nucleus was dyed to blue, muscle fibers were dyed to red, fat tissue was white, the adipose tissue is white. Magnification: 100×.
Figure 1.
Morphological observation on the longissimus dorsi muscle of Xinjiang Brown Cattle . (A) Five samples with low intramuscular fat content were selected for paraffin sectioning from the LIMF group; (B) five samples with high intramuscular fat content were selected for paraffin sectioning from the HIMF group. Hematoxylin eosin staining was used, the nucleus was dyed to blue, muscle fibers were dyed to red, fat tissue was white, the adipose tissue is white. Magnification: 100×.
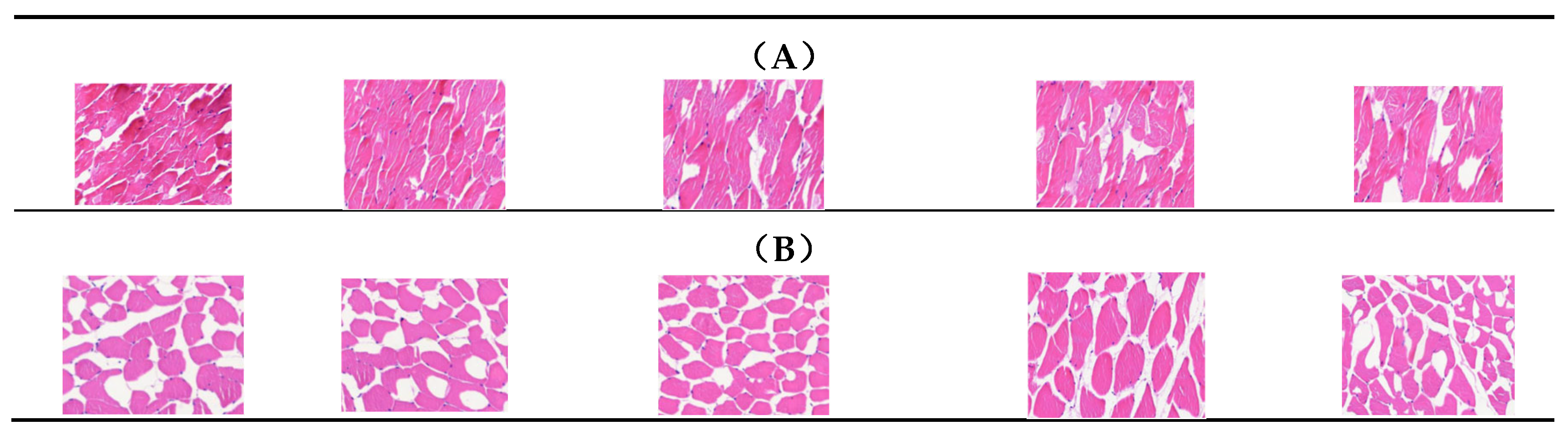
Figure 2.
Quantitative analysis. (A)Sample Co-Expression Venn Plot; (B)Plot of results of principal component analysis (coordinates first principal component, vertical second principal component); (C)Box plot of gene expression distribution of samples (horizontal coordinates were sample names, vertical coordinates were log2(FPKM+1)); (D)Heat map of inter-sample correlations (horizontal and vertical coordinates were squared correlation coefficients for each sample).
Figure 2.
Quantitative analysis. (A)Sample Co-Expression Venn Plot; (B)Plot of results of principal component analysis (coordinates first principal component, vertical second principal component); (C)Box plot of gene expression distribution of samples (horizontal coordinates were sample names, vertical coordinates were log2(FPKM+1)); (D)Heat map of inter-sample correlations (horizontal and vertical coordinates were squared correlation coefficients for each sample).
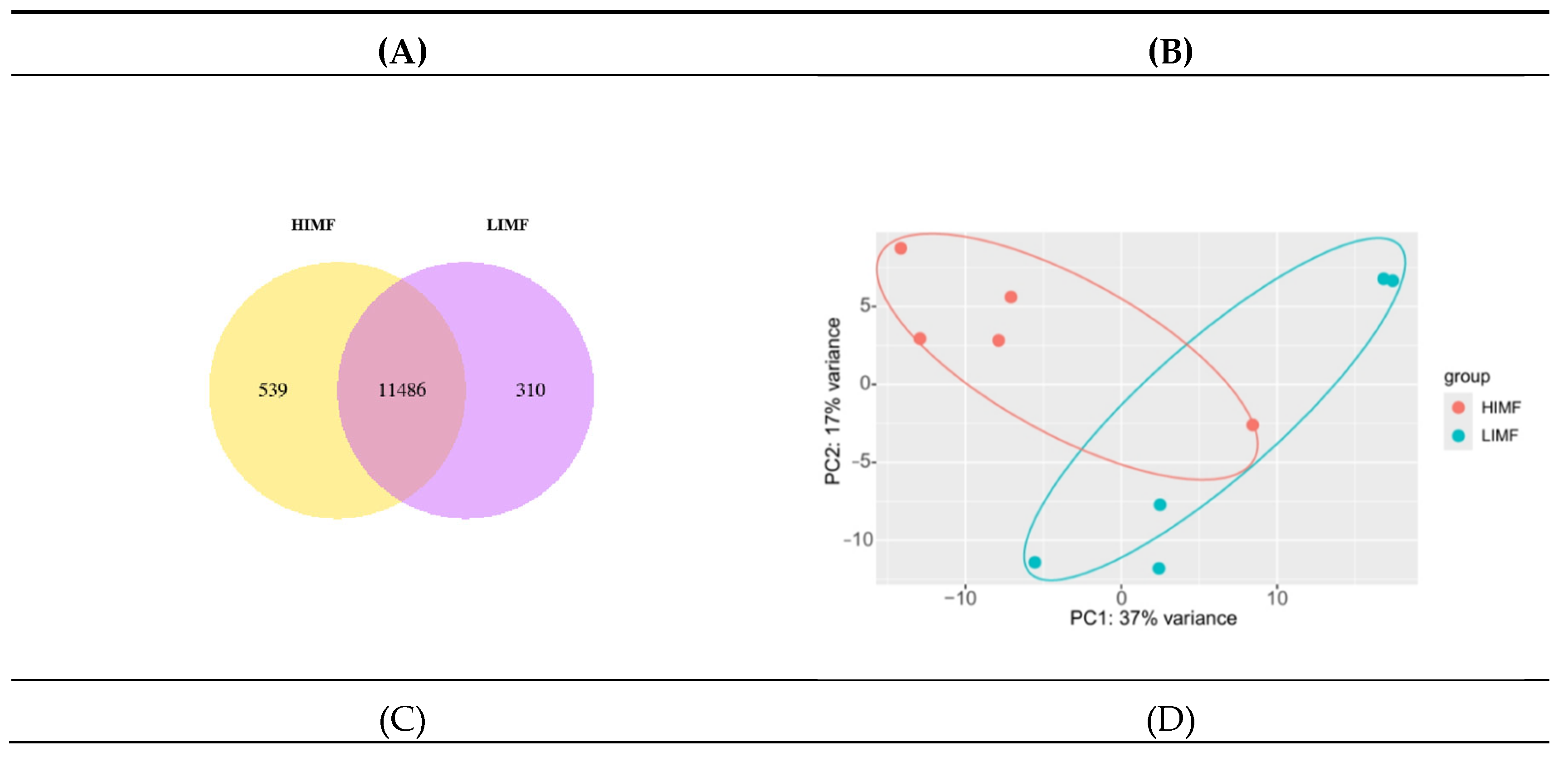
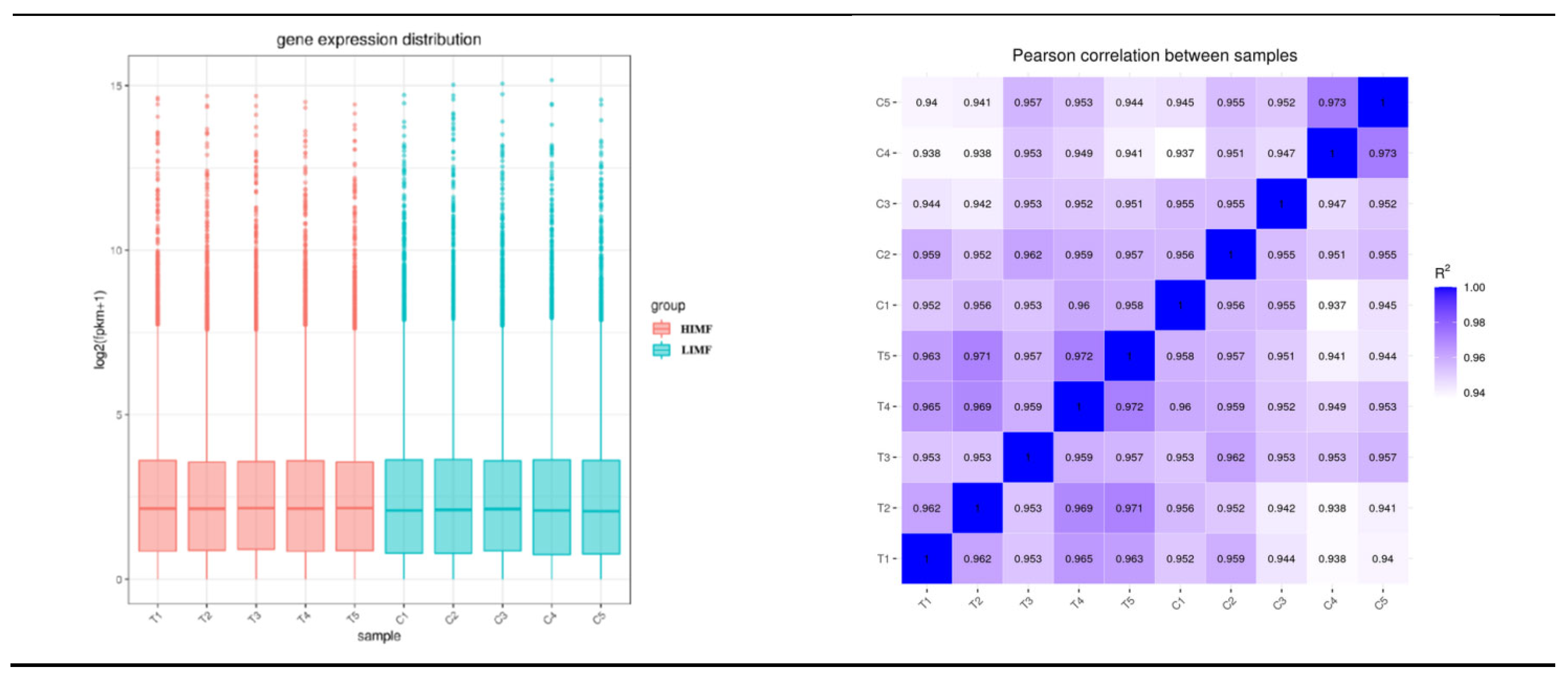
Figure 3.
Map of differentially expressed genes. (A)Histogram of the number of differential genes counted in the differential comparison combinations; (B)Differential gene volcano map (blue dashed lines indicate threshold lines for differential gene screening criteria; (C)Heatmap of clustering of differentially expressed genes (the redder the color the higher the expression, the greener the color the lower the expression).
Figure 3.
Map of differentially expressed genes. (A)Histogram of the number of differential genes counted in the differential comparison combinations; (B)Differential gene volcano map (blue dashed lines indicate threshold lines for differential gene screening criteria; (C)Heatmap of clustering of differentially expressed genes (the redder the color the higher the expression, the greener the color the lower the expression).
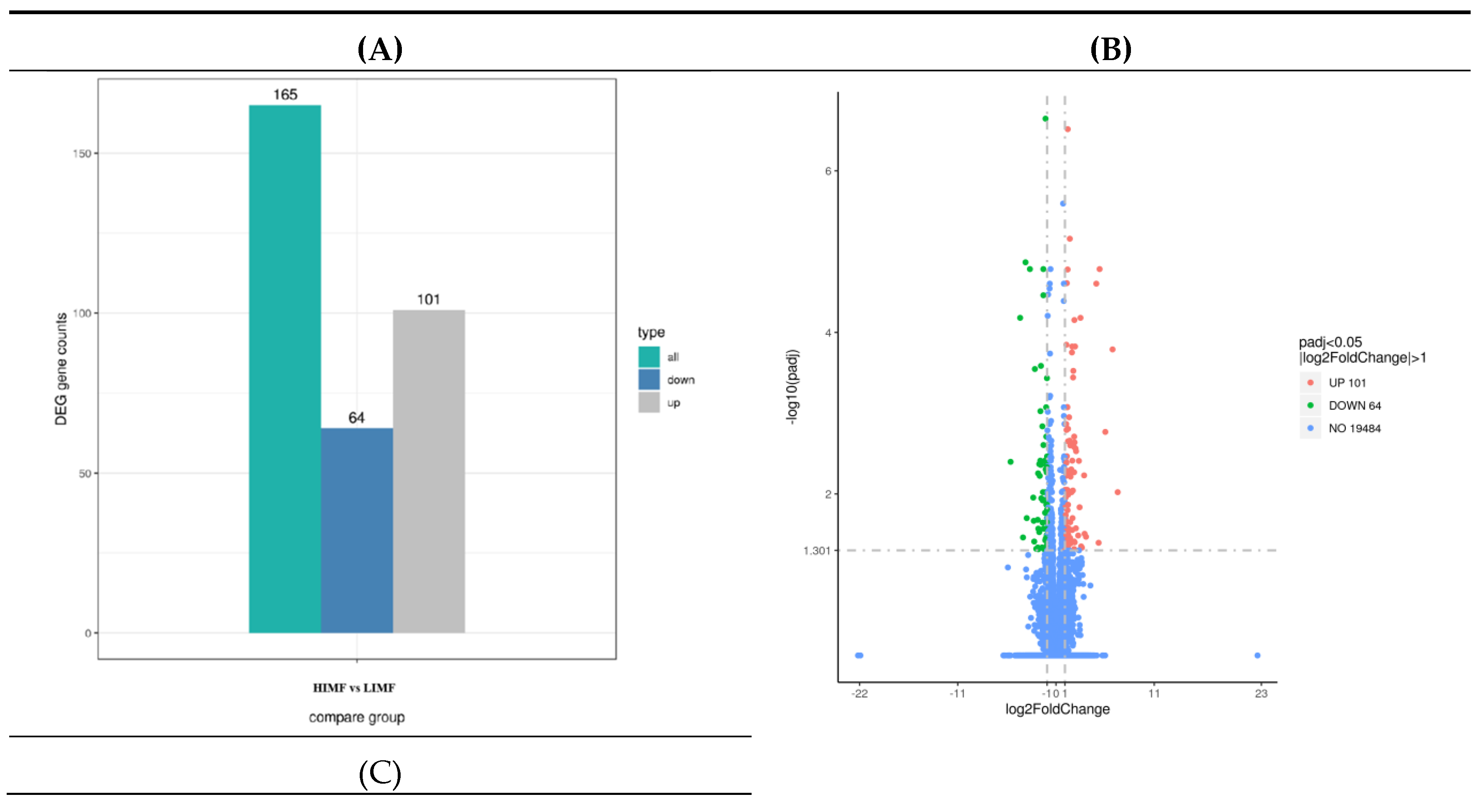
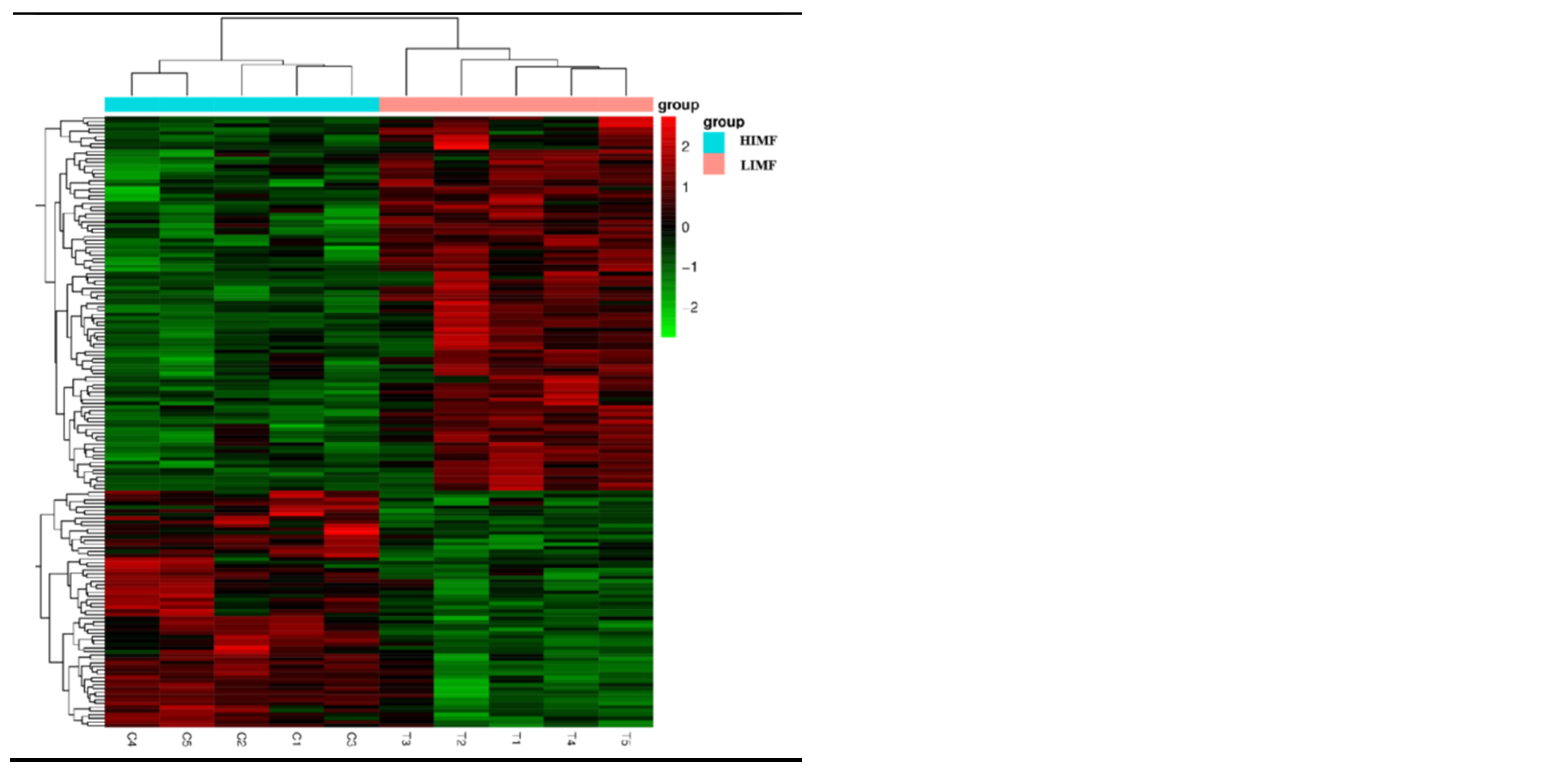
Figure 4.
GO enrichment analysis. The color from red to purple represents the magnitude of the significance of the enrichment, and the size of the dots represents the number of enriched entries.
Figure 4.
GO enrichment analysis. The color from red to purple represents the magnitude of the significance of the enrichment, and the size of the dots represents the number of enriched entries.
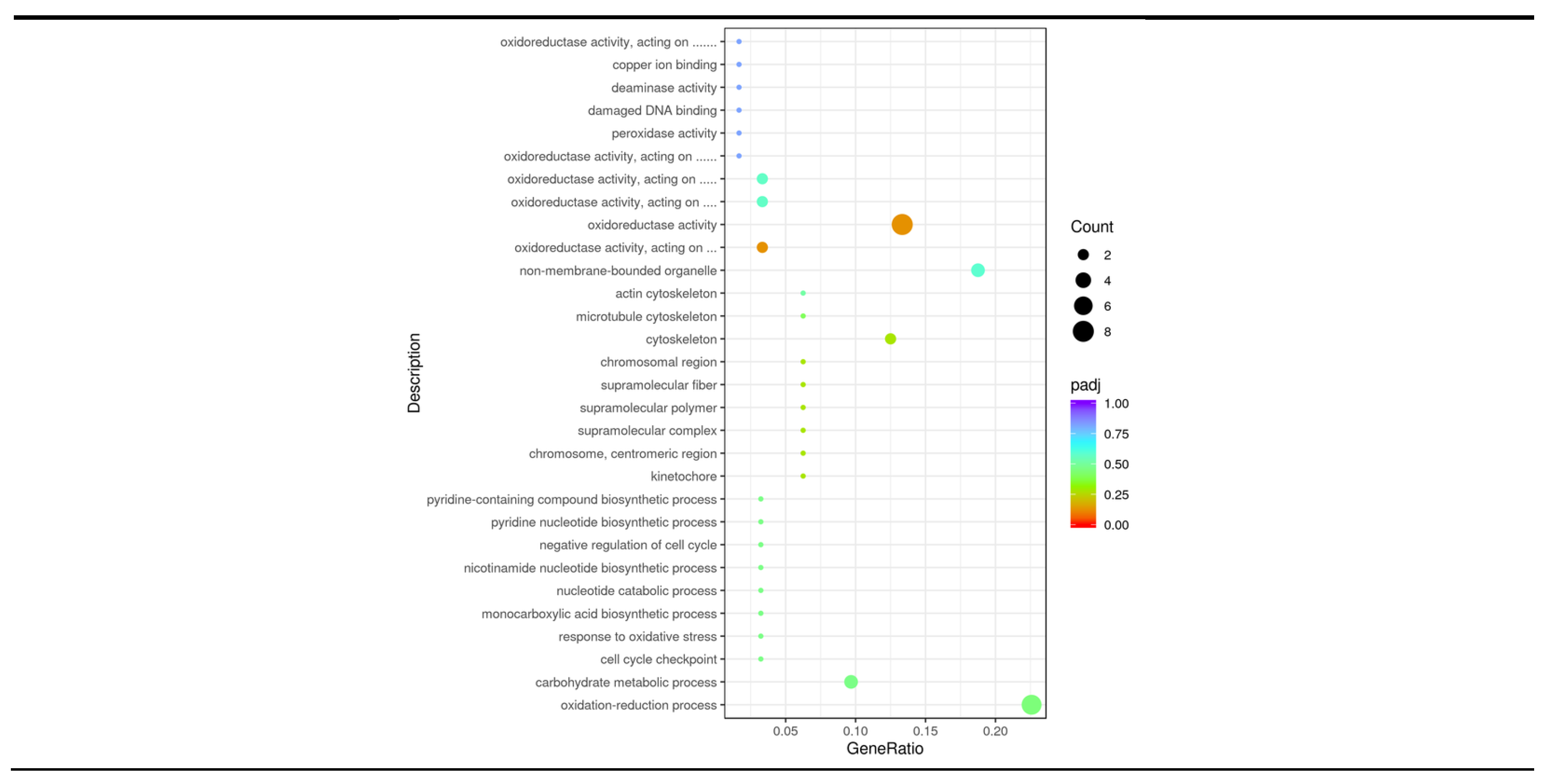
Figure 5.
KEGG enrichment analysis. The KEGG pathway is annotated as the ratio of the number of differential genes to the total number of differential genes on the horizontal coordinate, and the KEGG pathway is annotated on the vertical coordinate.
Figure 5.
KEGG enrichment analysis. The KEGG pathway is annotated as the ratio of the number of differential genes to the total number of differential genes on the horizontal coordinate, and the KEGG pathway is annotated on the vertical coordinate.
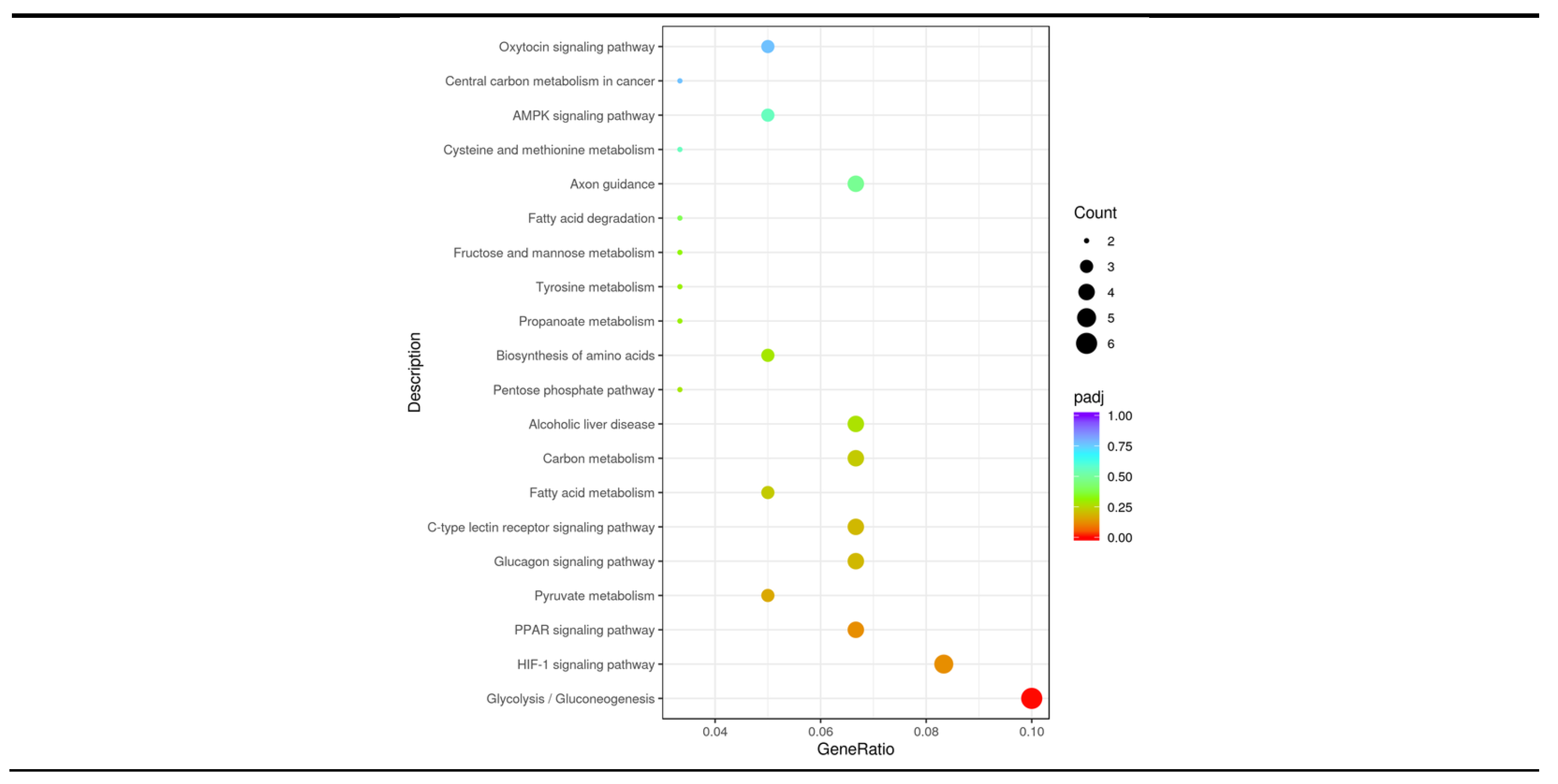
Figure 6.
RT-qPCR verification of RNA-Seq results.
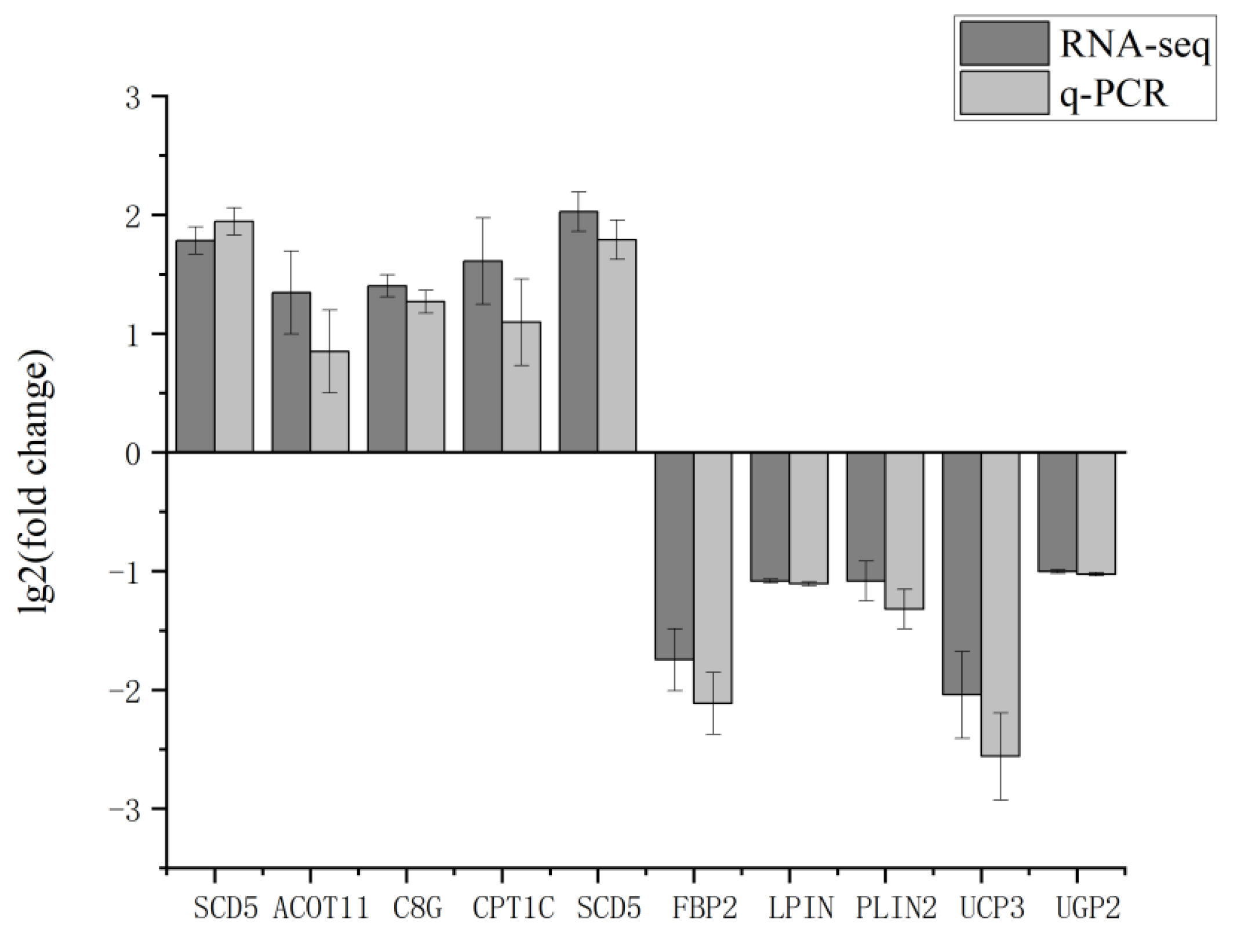

Table 1.
Primer sequences for RT-qPCR.
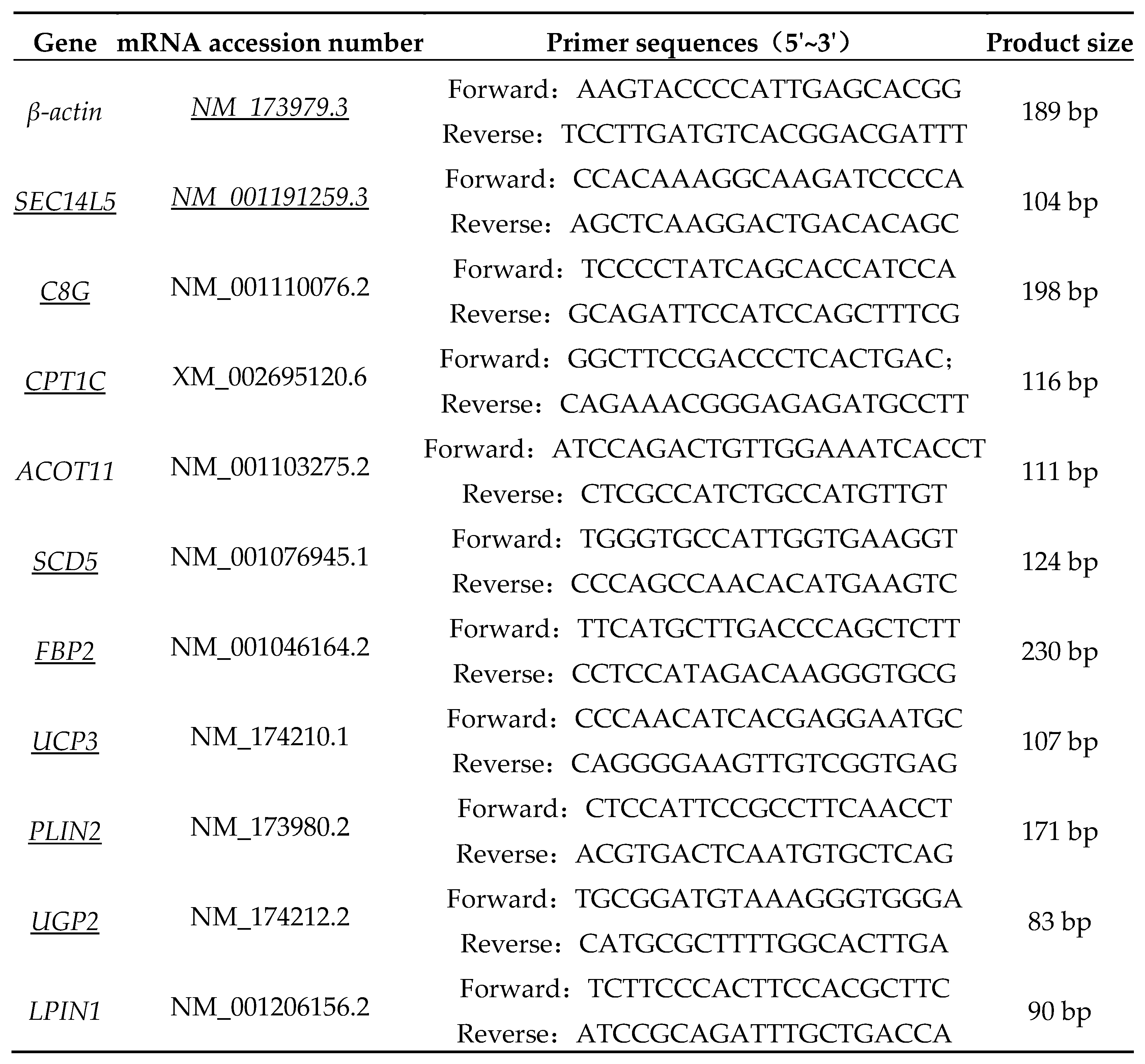
Table 2.
IMF content of the longissimus dorsi muscle in Xinjiang Brown Cattle.
| Item1 | IMF content (%) | Minimum (%) | Maximu (%) | p-value |
|---|---|---|---|---|
| HIMF | 12.379±0.046A | 5.418 | 21.177 | 0.0001 |
| LIMF | 2.523±0.003B | 1.996 | 3.086 |
1HIMF: set of samples with high intramuscular fat content, LIMF: set of samples with low intramuscular fat content. Results were expressed as mean ± standard deviation, n=21. AB Values in the same column with different superscripts are extremely different (P < 0.01).
Table 3.
Summary of sample sequencing data quality.
| Sample | Raw reads | Raw bases | Clean reads | Clean bases | Error rate | Q20 | Q30 | GC pct |
|---|---|---|---|---|---|---|---|---|
| T1 | 43246440 | 6.49G | 42439552 | 6.37G | 0.03 | 97.81 | 93.85 | 51.02 |
| T2 | 43426076 | 6.51G | 42377682 | 6.36G | 0.03 | 97.87 | 94.04 | 52.11 |
| T3 | 41992442 | 6.3G | 41202940 | 6.18G | 0.02 | 98.03 | 94.47 | 52.4 |
| T4 | 41644788 | 6.25G | 40126880 | 6.02G | 0.02 | 98.02 | 94.46 | 52.64 |
| T5 | 48098022 | 7.21G | 46682976 | 7.0G | 0.03 | 97.9 | 94.16 | 51.8 |
| C1 | 42494264 | 6.37G | 41037266 | 6.16G | 0.03 | 97.9 | 94.1 | 54.01 |
| C2 | 43497090 | 6.52G | 42302406 | 6.35G | 0.02 | 98.03 | 94.44 | 51.74 |
| C3 | 46125528 | 6.92G | 43760976 | 6.56G | 0.02 | 97.95 | 94.24 | 52.16 |
| C4 | 48924916 | 7.34G | 47173470 | 7.08G | 0.02 | 98.08 | 94.58 | 50.79 |
| C5 | 47427100 | 7.11G | 45659038 | 6.85G | 0.02 | 98.07 | 94.53 | 51.84 |
T1, T2, T3, T4, and T5 in the table are samples from the HIMF group; C1, C2, C3, C4, and C5 are samples from the LIMF group. The same as below.
Table 4.
Statistics on the comparison of the samples with the reference genome.
| Sample | Total reads | Total map | Map rate | Unique map | Unique map rate | Multi map | Multi map rate |
|---|---|---|---|---|---|---|---|
| T1 | 42439552 | 40217936 | 94.77% | 39141933 | 92.23% | 1076003 | 2.54% |
| T2 | 42377682 | 40157154 | 94.76% | 38944182 | 91.9% | 1212972 | 2.86% |
| T3 | 41202940 | 39045874 | 94.76% | 37817486 | 91.78% | 1228388 | 2.98% |
| T4 | 40126880 | 38521569 | 96.0% | 37350423 | 93.08% | 1171146 | 2.92% |
| T5 | 46682976 | 44779474 | 95.92% | 43397543 | 92.96% | 1381931 | 2.96% |
| C1 | 41037266 | 39309911 | 95.79% | 38015521 | 92.64% | 1294390 | 3.15% |
| C2 | 42302406 | 40332401 | 95.34% | 39075620 | 92.37% | 1256781 | 2.97% |
| C3 | 43760976 | 40739851 | 93.10% | 39403210 | 90.04% | 1336641 | 3.05% |
| C4 | 47173470 | 44649391 | 94.65% | 43182346 | 91.54% | 1467045 | 3.11% |
| C5 | 45659038 | 43293569 | 94.82% | 41735269 | 91.41% | 1558300 | 3.41% |
Table 5.
KEGG enrichment pathway and differentially expressed genes.
| KEGG id | KEGG description | P-value | Upregulated genes | Downregulated genes |
|---|---|---|---|---|
| bta04152 | AMPK signaling pathway | 0.0710 | FBP2; SCD5 | CPT1C |
| bta04921 | Oxytocin signaling pathway | 0.1093 | RGS2; PTGS2 | NFATC3 |
| bta05230 | Central carbon metabolism in cancer | 0.1063 | ENSBTAG00000032217; LDHB | / |
| bta04360 | Axon guidance | 0.0550 | RND1 | SSH2; UNC5A; NFATC3 |
| bta00270 | Cysteine and methionine metabolism | 0.0682 | ENSBTAG00000032217; LDHB | / |
| bta00071 | Fatty acid degradation | 0.0417 | CPT1C | ENSBTAG00000052243 |
| bta00051 | Fructose and mannose metabolism | 0.0312 | / | FBP2; ALDOA |
| bta00350 | Tyrosine metabolism | 0.0280 | ENSBTAG00000046264 | TYRP1 |
| bta00640 | Propanoate metabolism | 0.0264 | ENSBTAG00000032217; LDHB | / |
| bta01230 | Biosynthesis of amino acids | 0.0222 | / | ALDOA; ENSBTAG00000018554 |
| bta00030 | Pentose phosphate pathway | 0.0205 | / | FBP2; ALDOA |
| bta04936 | Alcoholic liver disease | 0.0175 | SCD5; CPT1C | LPIN1; ENSBTAG00000052243 |
| bta01200 | Carbon metabolism | 0.0129 | / | FBP2; ALDOA; ENSBTAG00000018554 |
| bta01212 | Fatty acid metabolism | 0.0118 | SCD5;CPT1C | ENSBTAG00000052243 |
| bta04625 | C-type lectin receptor signaling pathway | 0.0083 | EGR3; PTGS2 | CCL22; NFATC3 |
| bta04922 | Glucagon signaling pathway | 0.0072 | ENSBTAG00000032217;LDHB;CPT1C | FBP2 |
| bta00620 | Pyruvate metabolism | 0.0047 | ACOT11; ENSBTAG00000032217;LDHB | / |
| bta03320 | PPAR signaling pathway | 0.0026 | SCD5; CPT1C | ENSBTAG00000052243; PLIN2 |
| bta04066 | HIF-1 signaling pathway | 0.0018 | ENSBTAG00000032217; LDHB | ALDOA; ENSBTAG00000018554 |
| bta00010 | Glycolysis/Gluconeogenesis | <0.0001 | ENSBTAG00000032217; LDHB | FBP2; ALDOA; ENSBTAG00000018554 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



