
Submitted:
03 May 2024
Posted:
09 May 2024
You are already at the latest version
Abstract
Graphene oxide (GO) is considered as one of the promising adsorbents for the removal of various heavy metal contaminants in aquatic environment, due to its beneficial structural, chemical and electronic properties. Using the density functional theory (DFT) approach, we investigate the reactivity of a model GO nanoparticule (C30H14O15) toward neutral and charged lead (Pb) and cadmium (Cd) atoms. We found the model metal-free GO to have a high chemical reactivity. To study the adsorption of the metal atoms on the GO nanoparticle, we have used a single and doubly-adsorbed neutral (Pb0 and Cd0) and charged (Pb2+, Cd2+) atoms. After identified possible adsorption sites on the GO, we found that a single charged metal ion binds more strongly than a neutral atom of the same type. To determine the possibility of multiple adsorptions of GO nanoparticle, two metal atoms of the same species are co-adsorbed on its surface. Our calculations reveals a site-dependent adsorption energy such that when two atoms of the same specie are adsorbed at sites Si and Sj , the binding energy per atom depends on the whether one of the two atoms is adsorbed firstly on the Si or Sj sites. It is also found that the binding energy per atom for two co-adsorbed atoms of the same specie is less than the binding energy of a singly-adsorbed atom which suggests that atoms may become less likely to be adsorbed on the GO nanoparticle when their concentrations increase. This applies to both neutral and charged atoms. We adduce the origin of this observation to be interplay between metal-metal interaction on the one hand and GO-metal on the other, with the former resulting in a less binding for the charged adsorbed metals in particular, due to repulsive interaction between two positively charged ions. The frontier molecular orbitals analysis and the calculated global reactivity descriptors of the respective GO- metal complexes revealed that all the GO-metal complexes have a smaller HOMO-LUMO gap (HLG) relative to that of pristine metal-free GO nanoparticle. This suggests that although the GO-metal complexes are stable, they are less stable compared to metal-free GO nanoparticles. The negative values of the chemical potentials obtained for all the GO-metal complexes further confirms their stability. Our work calculates parameters that will be crucial to rational design of GO nanoparticles for Pb abd Cd ions contaminants removal, and may find application in water purification for example.
Keywords:
Graphene oxide
; Density Functional Theory
; Heavy metals
; Adsorption
1. Introduction
With the industrial revolution, the issues related to the management and control of mining waste have increased. These mining wastes, mainly heavy metals, constitute a real source of pollution to the ecosystem since they are not generally biodegradable [1]. Present in soils, waters and air, heavy metals can also be found in many uses such as paints, dyes, in some fertilizers, soaps as well in certain medicines [2]. Also, the bioaccumulation of metals such as Pb and Cb in the body is one of the causes of carcinogenic, cardiovascular and kidney diseases, and can lead to death when absorbed in high concentrations [2,3].
Recent researches have indicated that two-dimensional nanomaterials containing oxygen, epoxy, hydroxyl, carboxylic groups [4] have a great potential for the adsorption of heavy metals [5,6,7,8,9,10] . In this context, graphene oxide (GO) nanoparticles have shown a great potential due to its high adsorption capacity, large surface area and solubility. It can also serve as an electrode material for monitoring contaminants [11]. They are hydrophilic in nature and have large negative charge surfaces that help to effectively remove cationic impurities such as heavy metal cations and cationic dyes by electrostatic interactions. Furthermore, due to their unique physico-chemical characteristics, they have the potential of becoming excellent adsorbents [12]. Indeed, Weijun et al have shown that GO nanoparticles can be superior adsorbents for the removal of heavy metal ions from water [13]. They can also act as excellent catalysts or further hybridize with effective catalysts to convert harmful gases and organic species in wastewater [14,15].
Recently, many experimental works have been carried out in the field of wastewater treatment [9,16,17]. These include the use of treatment techniques to remove heavy metal ions from wastewater such as chemical precipitation, membrane separation, ion exchange, electrochemical treatment as well metal adsorption, etc. Out of all these techniques, metal adsorption has been found to show remarkable effectiveness. Besides, due to its low cost, it is considered as a relatively cheap and fast approach for wastewater treatment. Also, to our knowledge, only a few theoretical works have been reported on this topic [13,18,19,20].
In this study, we use DFT calculations to investigate the structural and electronic properties of the GO nanoparticle from Ref. [21], having chemical the formula CHO, to heavy metals adsorption. Specifically, we consider the structural and electronic response of the nanoparticles to Cd and Pb in their neutral gas-phase, i.e., Pb and Cd respectively, as well as in their cationic state, i.e., Pb and Cd. We determine the active sites on the GO nanoparticle surface and the binding mechanisms underpinning metal adsorption. Furthermore, we provide deep insight into electronic interactions between the GO substrate and the adsorbed metal. The rest of the paper is organized as follows : In section II, we present the computational details and atomic models use in the study. Fundamental structural and electronic properties of the GO nanoparticle are then presented in section IIIA which are followed by adsorption and binding mechanism of Cd and Pb on GO as well as underlying electronic interactions (section IIIB).
2. Computational Details
All the results presented in this work have been obtained with the density function theory (DFT) as implemented in the Gaussian 16 package [22]. We used the B3LYP exchange-correlation functional with LANL2DZ basis set [23,24] and 6-31g for the calculation of GO vibrational frequencies. Figure 1(a) shows the atomic model of GO nanoparticle used in the study. It has the chemical formula CHO and is made up of 59 atoms which also contains carboxylic (−COOH), alcohol (−OH) and epoxy (−O−) chemical groups. [21]In fact, the GO nanoparticle has to be constructed in such a way that the overall charge is zero by passivating the residual dangling bonds with H atoms. To simulate the metal-adsorption behaviour of GO nanoparticle, a metal atom in a gas-phase is placed on its most nucleophilic sites, which are denoted as S and S. Each Pb or Cd atom is placed about 2.0 Å above the GO surface. Such a separating distance is sufficent for metal atom to interact with the GO nanoparticle. Furthermore, the placement of the metal atom at the nucleophilic S and S sites is informed by the fact that they have the highest electrophilic condensed Fukui functions [25,26,27,28] as will be shown in the later section of this paper. It is worthwhile to state at this point that the condensed Fukui functions at a particular site, measures its chemical reactivity to an external perturbation. Three different types of condensed Fukui functions for a particular atom or site k have been defined, depending upon the type of electron transfer,
where are the grosses electronic population of the atom or site k, calculated from the , and 0 charged molecule. It should be noted that, the greater the value of condensed Fukui function, the more reactive is the particular atomic site in the molecule [27].
Three separate models of metal-adsorbed GO have been considered. These are named as GO-M/S, GO-M/S and GO-2M/SS. In the model termed as GO-M/S, a single metal atom M is adsorbed on the S site (Figure 1(a)) while S is the adsorption site of M in the model GO-M/S (Figure 1(b)). In the case of GO-2M/SS, two metal atoms of the same type occupy the adsorption sites S and S (Figure 1(c)). The structural model of GO-2M/SS enables us to determine if metal atoms can be co-adsrbed at the two adsorption sites. To determine the minimium energy configuration for each of the three models, GO with neutral metal atoms of Pb and Cd as well as GO with metal cations of Pb and Cd, each attached at their respective sites were optimized. The chemical stability and reactivity were determined through the global reactivity descriptors, using the frontier molecular orbitals which are the HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) with the Koopmans theorem [29] :
where , , and are defined as the HOMO-LUMO gap (HLG), chemical potential, chemical hardness and global electrophilicity index, respectively. It has been demonstrated that, a high HOMO energy corresponds to a more reactive molecule in the reactions with electrophiles, while a low LUMO energy favorizes molecular reactions with nucleophiles [30,31]. Note that a large HOMO-LUMO gap implies high kinetic stability and low chemical reactivity because it is energetically unfavorable to add electrons to a high-lying LUMO or to extract electrons from a low-lying HOMO [28,32,33,34,35,36]. A negative chemical potential suggests that a chemical compound or an atomic system is stable and will not decompose spontaneously into its constituents elements [37,38]. The chemical hardness may be interpreted as a measure of resistance toward the deformation of electron cloud of a chemical system under small perturbations encountered during chemical process. The global electrophilicity index is related to the ability of an electrophile to acquire additional electronic charge and the resistance to exchange electronic charge with the environment [31,39,40].
Following the energy minimization process, we calculated the binding energies per metal atom on the metal-free GO as well as the GO-metal complexes as :
where and are the minimum energies of GO nanoparticles having n and numbers of adsorbed metal atoms respectively, while is the total energy of the isolated metal. In this convention, the positive value of means the adsorption process is favorable. Moreover, for a binding energy lower than 0.5 eV, the adsorbate (i.e, the metal atom) is generally considered to be physisorbed on the adsorbent. A higher binding energy indicates chemisorption [41]. Finally, to confirm that the optimized GO system is in the minimum energy configuration, which is chemically stable, we performed vibrational frequency calculations. It has to be noticed that, imaginary frequencies in the vibrational modes will indicate an unstable molecule.
3. Results and Discussion
3.1. Stability and Reactivity of GO Nanoparticle
The molecule CHO composed of 59 atoms has 174 normal modes of vibration. Depending on whether the vibration modes are given by the formula 3N-6 (N:number of atoms). The results of the frequency calculation have revealed no imaginary frequency, so we can deduce that our GO is stable see supplementary information. Also, we have found that the paramagnetic ground-state is the preferred magnetic state for the GO nanoparticle, a result which is consistent with previous experimental and theoretical investigations of GO nanoparticles [42]. Also, we have found that the paramagnetic ground-state is the preferred magnetic state for the GO nanoparticle, a result which is consistent with previous experimental and theoretical investigations of GO nanoparticles [42].
Figure 1.
(a) Graphene oxide nanoparticle (GO), (b) GO nanoparticle with the metal atom adsorbed at the S site, i.e., GO-M/S model, (c) GO nanoparticle with the metal atom adsorbed at the S site, i.e., GO-M/S model, (d) GO-2M/SS model where two metal atoms are co-adsorbed at the S and S sites. In each of the models, M = Pb, Cd, Pb and Cd. Carbon, hydrogen, oxygen and metal atoms are represented as gray, white, red and blue spheres.
Figure 1.
(a) Graphene oxide nanoparticle (GO), (b) GO nanoparticle with the metal atom adsorbed at the S site, i.e., GO-M/S model, (c) GO nanoparticle with the metal atom adsorbed at the S site, i.e., GO-M/S model, (d) GO-2M/SS model where two metal atoms are co-adsorbed at the S and S sites. In each of the models, M = Pb, Cd, Pb and Cd. Carbon, hydrogen, oxygen and metal atoms are represented as gray, white, red and blue spheres.
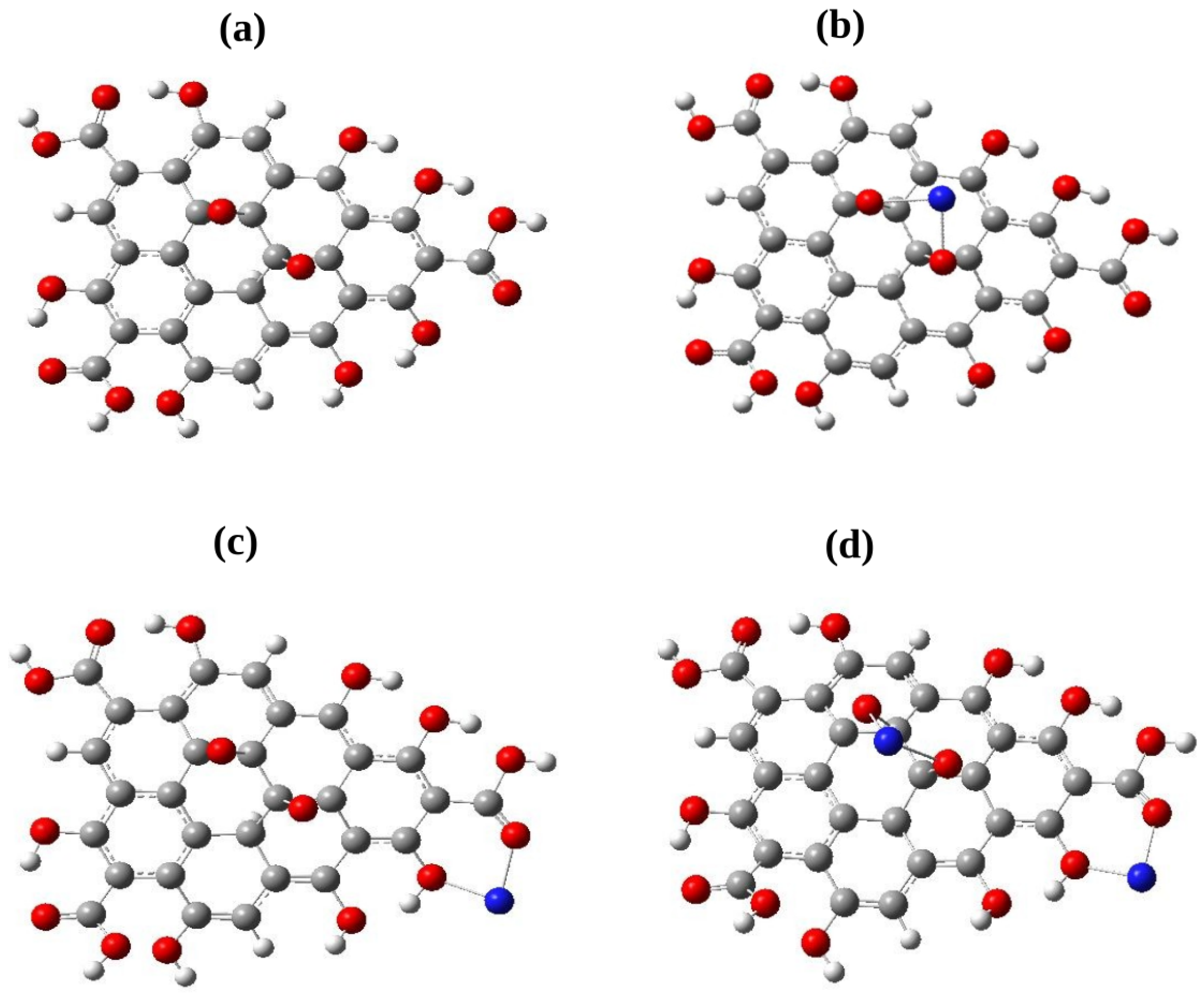
Table 2 shows our calculated values for the LUMO-HOMO gap (E), chemical potential (), chemical hardness (), and global electrophilicity index (). The HOMO−LUMO gap is found to be about eV. It should be noted that, chemical systems with large E are hard and much less polarizable compared to systems with small E). A HUMO-LUMO gap (HLG) greater than eV may be considered large [43]. Therefore, our model GO nanoparticle of relatively high E can be considered to have a high chemical stability and chemical hardness, and thus will be less polarizable. Its electron cloud will be resistive to small perturbations which will result in low chemical reactivity [31]. The chemical potentials () is found to be eV. This negative also suggests that the GO nanoparticle is stable and does not undergo decomposition into elements. The chemical hardness of about eV may is large enough to consider the GO nanoparticle relatively resistant toward the deformation of its electron clouds under small perturbations, which is further affirmed by the relatively high E, i.e., greater than eV. The global electrophilicity index of about eV, which is small enough to consider that the GO nanoparticle may be subject to electrophilic attacks.
In order to map out nucleophilic sites onto the GO nanoparticle, we plot the molecular electrostatic potential (MEP) isosurface. The plot is displayed in Figure 2. The figure shows variation in the electron density in the GO nanoparticle. The blue colored regions for example, show low electronic densities while white colored regions indicate medium electronic densities. The orange colored regions indicate high electron densities and may be considered as nucleophilic sites, and are thus potential sites for electrophilic attacks.
Also, Figure 2 clearly shows that the most prominent nucleophilic sites are mainly localized around epoxy groups (site S), and the vicinity of carboxylic and alcohol groups (sites S, S, S, S, S, S). For a better understanding of local reactivity properties, we calculated the condensed dual descriptors of atoms localized at the potential nucleophilic sites, defined as . The negative value of indicates the site is favoured for an electrophilic attack while the positive value indicates that the site may be favorable for a nucleophilic attack [25,26,27,28]. The calculated values of are presented in Table 1, where we have demonstrated the varation of Fukui function values in the molecule by chosing the corresponding values for the sites S (k = 1-7). Here, S (k = 1-6) have negative condensed dual descriptors while the site S has a positive condensed dual descriptor. The latter indicates that the electrophilicity of the site S prevails over its nucleophilicity, which may be explained by the proximity with a highly electrophilic site as shown by the MEP isosurface. Furthermore, the ranking of the electrophilic Fukui condensed functions of potential nucleophilic sites is as follows : . This clearly suggests that the site S has higher ability to attract electrophilic ions, followed by the site S. For this reason, the following section will be devoted to the adsorption behaviour of both sites S and S.
3.2. Adsorption of Pb and Cd
To determined the stability of metal at the various sites, we calculated the binding energy {} of adsorbed metal on the sites S and S of GO nanoparticle using the relaxed minimum energy structures of GO-M complexes. The resulting values are presented in Table 2. For a single metal adsorption, the is positive in all the cases, which indicates that the adsorption of a single Cd or Pb as a neutral atom or in ionic form is stable. Also, to be noted from the table is that, the Pb adsorption at the S and S sites of GO nanoparticle leads to a chemisorption with eV and eV respectively. These are much stronger adsorption compared to Cd, which is also chemisorbed at the S site with a much lower binding energy of eV, but physisorbed at the S site with eV. Our results are in agreement with theoretical investigations conducted by Sara M. Elgengehi et al [20] in the case of graphene and GO as adsorbents for Cd and Pb heavy metals. They also found the same trend, namely, neutral Pb atom absorbs strongly than Cd.

Table 2.
HOMO-LUMO gap (), chemical potential (), chemical hardness () and global electrophilicity index () of GO nanoparticle and GO-M complexes (M = Pb, Cd). Mulliken charge transfers for the metal atom adsorbed at the site S {}. is the adsorption energy per metal atom adsorbed on the GO nanoparticle. Similarly, {} represents the binding energy per adsorbed metal atom when two atoms are co-adsorbed in such a way that the first atom is adsorbed at the S prior to the adsorption of a second atom at S site. In other words, a metal atom pre-exists at the S before the second atom is adsorbed S.
Table 2.
HOMO-LUMO gap (), chemical potential (), chemical hardness () and global electrophilicity index () of GO nanoparticle and GO-M complexes (M = Pb, Cd). Mulliken charge transfers for the metal atom adsorbed at the site S {}. is the adsorption energy per metal atom adsorbed on the GO nanoparticle. Similarly, {} represents the binding energy per adsorbed metal atom when two atoms are co-adsorbed in such a way that the first atom is adsorbed at the S prior to the adsorption of a second atom at S site. In other words, a metal atom pre-exists at the S before the second atom is adsorbed S.
| Site | Structure | (eV) | (eV) | (eV) | (eV) | (eV) | (eV) | |
| GO | − | − | − | |||||
| GO-Pb | ||||||||
| GO-Cd | ||||||||
| S | GO-Pb | |||||||
| GO-Cd | ||||||||
| GO-Pb | ||||||||
| S | GO-Cd | |||||||
| GO-Pb | ||||||||
| GO-Cd |
The aforementioned results can be related to Mulliken charge transfer between the GO nanoparticle and the adsorbed metal [44]. Table 2 presents different values of (where i is the site index), defined as the Muliken charge difference between the electronic charge on the metal before and after adosorption on the GO surface. According to our definition, a negative value of implies that the metal adsorbate loses charges to the GO surface while the positive value corresponds to a gain of electronic charges by the metal. We calculated a higher charge transfers from Pb to the GO nanoparticle ( and ) compared to those occurring from Cd ( and ), for the S and S sites respectively. Thus, neutral Pb at the S and S sites act as electron donors while Cd at the S acts as electron acceptor. Interestingly, Cd with a positive has the lowest binding energy of all the single neutral metal atoms considered. Furthermore, the difference in charge transfers between Pb and Cd can be understood through the difference between their ionization energies (IE) [45,46,47]. Neutral Pb and Cd has IE of eV and eV respectively [46]. Thus, it is easier to exract electronic charge from Pb compared to Cd due to the former’s lower IE. Hence, Pb transfers electrons more easily than Cd to GO nanoparticle. Indeed, similar trend in IE and charge transfer were obtained via first-principle investigations of adsorption properties of toxic heavy metals (i.e., Cd, Hg, Pb) on graphene quantum dots and infinite graphene structure [48].
We also calculated the harmonic vibration frequencies of both GO and GO-M with M being adsorbed on S and S sites. The results show that both GO and GO-M are stable, while the vibration frequencies rang from 25.8716 cm to 3694.89 cm for GO nanoparticle and 19.6641 cm to 3722.71 cm, 24.8106 cm to 3719.83 cm respectively for GO-Pb and GO-Cd concerning the adsorption on the site S, and from 15.9952 cm to 3709.68 cm and 15.0921 cm to 3711.59 cm for GO-Pb and GO-Cd concerning the site S (see supplementary information for more details).
The adsorption of a single cationic adsorbate Pb or Cd at the S and S sites leads to a stronger chemisorption compared to those of a single neutral atoms (see Table 2). The binding energies of a single Pb on the S and S sites are eV and eV respectively. Also, Cd is adsorbed with eV and eV at the S and S sites respectively. These binding energies are order of magnitude higher than those obtained for corresponding neutral Pb and Cd adsorptions as shown in the Table 2. In general, charged metal ions, irrespective of the adsorption sites, binds more strongly compared to neutral atom of the same type. This result is in agreement with previous findings by Shtepliuk et al [48].
Muliken charge analysis shows a positive charge transfer, i.e., positive , and thus electron transfer from the GO to the cations. Thus, each of Pb and Cd cations act as an electron acceptor from the GO nanoparticle. Therefore, positively charged ions reverse the direction of charge transfer compared to the charge-donating neutral atoms which is consistent with previously reported results by Shtepliuk et al [48].
We also investigated the adsorption of two neutral or charged Pb or Cd atoms on the S and S sites. Only co-adsorption of atoms of the same species are considered. Thus, we examined two models of adsorption which are the following : (i) The first metal atom pre-exists at the the S site while the second atom adsorption occurs on the site S. Here, the binding energy of doubly-adsorbed Pb or Cd is termed {}, where indicates that metal atom pre-exists at site S before the adsorption of another atom of the same specie occurs at S ; (ii) The second adsorption occurs on the S site while the first one pre-exists on the S site. In this case, the binding energy of doubly-adsorbed Pb or Cd is termed { where indicate that metal atom pre-exists at site S before the adsorption of another atom occurs at S. These binding energies per atom () are presented in the last column of Table 2. If these are compared to the binding energies of a singly adsorbed metal atoms (), it is clear that the atoms becomes less binded to the GO nanoparticle with increasing number of adsorbed atoms. As an example, for a single Pb adsorption, eV where for a doubly adsorbed atom of the same specie, eV. The same trend can be observed for all the metals, neutral or charged. In the case of Pb ion, the binding energy per atom becomes negative (i.e, an unstable configuration or desorption) when the two charged ions are are co-adsorbed. Another observation from Table 2 is that the magnitude of is not the same as . In all cases (for neutral and cations atoms adsorption) is larger than . For example, eV for Pb while eV for the same atom specie. Similar trend is observed for all the atoms as show in Table 2. Thus, when a metal atom is initially adsorbed at the S site, it attracts and binds more strongly an atom of the same specie at the S site. In this case, the co-adsorption is thus energetically more favoured. However, when a metal atom is initally adsorbed at the S site, the second atom adsorbed on the S site experiences less binding. In actual fact, the co-adsorption may not be energetically favoured (that is, an unstable co-adsorption or desorption) as shown in the case of Pb where is positive , i.e., 1.31 eV whereas is negative, i.e., eV. Thus, the lattice site of a pre-exisitng or an adsorbed metal atom on GO has strong influence on subsequent binding or attraction of another atom of the same specie.
Furthermore, in order to gain deeper inisght into the electronic origin of stability of adsorbed metal atoms on the GO nanoparticle, we performed frontier molecular orbitals analysis and calculated the global reactivity descriptors of the respective complexes (see Table 2). We found that the HLG of all GO-M complexes are smaller than that of pristine metal-free GO nanoparticle, which suggests that when GO adsorbs metal, the GO-M complexes are still energetically stable, although less stable compared to metal-free GO. Also, the lower HLG of GO-M complexes indicates that they are chemically soft, more polarized and have higher chemical reactivity compared to the GO nanoparticle. However, we found the chemical potentials to be negative for all the metal adsorption on both the S and S sites, suggesting that the resulting complexes are stable and may not spontaneously decompose into separate metal ions and GO nanoparticle. Also, the chemical hardness of GO-metal complexes are smaller relative to that of pure GO, which indicates that GO-metal complexes are less resistant toward the deformation of their electron clouds under small perturbations. Furthermore, metal adsorption on GO leads to a higher global electrophilicity indexes compared to that of metal-free GO nanoparticule. This indicates the reduction of nucleophilic character of GO nanoparticle following metal adsorptions.
Based on the analyses of the binding energy and frontier molecular orbitals, we conclude that in general, multiple adsoprption of metal atoms on GO is favoured, i.e., preponderance of postive binding energy for the co-adsorbed atom. However, as the number or concentration of adsorbed atom increases, the metal atom becomes less binded to the GO. Such a behaviour, in the case of charged ions in particular may be rationalized as due to repulsive interaction between atoms of the same charge type, i.e., positive charge Cd and Pb ions. Indeed, Shtepliuk postulated that the competition between metal-metal interaction on the one hand and that of GO-metal on the other [48] is the origin of less binding experienced by multiple metal atoms when adsorbed on the GO nanoparticle. This important observation will be useful to evaluate the adsorption and retention of ions capability of GO nanoparticle as a medium to sequestre heavy metal cations, in particular, those investigated in this work.
4. Summary and Conclusions
Density-functional theory (DFT) calculations were employed to investigate the reactivity and interaction of a graphene oxide (GO) nanoparticule with neutral metal atoms Pb and Cd as well as charged ions Pb and Cd ions. Binding energy and global reactivity descriptors such as HOMO-LUMO gap (HLG) chemical potential (), chemical hardness () and global electrophilicity index () were used to characterize the interaction and reactivity of the metal atoms with the GO nanoparticle. We demonstrated that our model GO nanoparticle is stable and has high chemical reactivity. We identified seven potential nucleophilic sites on the GO nanoparticle, however only two of these were selected due to their greater occurences, to study the adsorption of metal atoms. Using a single and doubly-adsorbed neutral and charged metal atoms on GO as our model of GO-metal complexes, we found that the adsorption characteristics of charged metal ions, i.e., Pb and Cd differs significantly from those of neutral atoms Pb and Cd. Firstly, we found that charged metal ions bind more strongly than a neutral atoms of the same specie for single adsorptions. Also, we investigated the adsorption and binding capability of GO nanoparticles by considering model systems consisting of two Pb or Cd in a neutral or charged atoms. Only co-adsorption of atoms of the same type have been considered. We found site-dependency of the adsorption energy. Specifically, the binding energy of one atom at a nucleophilic site S when one other atom of the same specie is adsorbed firstly at another site S, differs significantly to the binding energy of the same atom when the adsorption order on the two sites is reversed. Furthermore, it was observed that the binding energy per atom for two co-adsorbed atoms of the same specie is less than the binding energy of a singly-adsorbed atom. This may suggests that with increasing number or concentration of the adsorbed atoms, they become less likey to be adsorbed on the GO surface. The observed behaviour may be ascribed to the metal-metal interaction in one hand and GO-metal on the other. The former, in particular, may results in less binding for the charged metal system due to repulsive interaction between two positively charged ions. Furthermore, we performed frontier molecular orbitals analysis and calculated the global reactivity descriptors of the respective GO-metal complexes in order to gain deeper inisght into the electronic origin of their stability. Our analyses revealed that all the GO-metal complexes have smaller HLG relative to that of pristine metal-free GO nanoparticle, which indicates that although the GO-metal complexes are satble, they are less stable compared to metal-free GO nanoparticles. Furthermore, we found the chemical potentials to be negative for all the studied GO-metal complexes, which confirms their stability. Our work provides a basis for rational design of GO for harmful metal ions capturing which may be important in water purification when these are present as water pollutants.
Acknowledgments
The computational resources are provided by the Center for High Performance Computing (CHPC)
in South Africa through the MATS0988 project. In particular, we are thankful to Dr. Anton Lopis
(CHPC) for his technical assistance. One of the authors, ATR, is grateful to the National Research
Foundation/Departement of Science and Technology (NRF/DST, South Africa) for their supports.
Also, ATR acknowledges that part of this workis based on the research supported by the NRF(Grant
number: 141942).
References
- Yang, S.; Chen, C.; Chen, Y.; Li, J.; Wang, D.; Wang, X.; Hu, W. Competitive adsorption of PbII, NiII, and SrII ions on graphene oxides: a combined experimental and theoretical study. ChemPlusChem 2015, 80, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Jalilian, R.; Jauregui, L.A.; Lopez, G.; Tian, J.; Roecker, C.; Yazdanpanah, M.M.; Cohn, R.W.; Jovanovic, I.; Chen, Y.P. Scanning gate microscopy on graphene: charge inhomogeneity and extrinsic doping. Nanotechnology 2011, 22, 295705. [Google Scholar] [CrossRef] [PubMed]
- Kabata-Pendias, A.; Mukherjee, A.B. Humans; Springer, 2007.
- Boukhvalov, D.W.; Katsnelson, M.I. Modeling of graphite oxide. Journal of the American Chemical Society 2008, 130, 10697–10701. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Y.; Pang, H.; Yu, S.; Ai, Y.; Ma, X.; Song, G.; Hayat, T.; Alsaedi, A.; Wang, X. Effect of graphene oxide surface modification on the elimination of Co (II) from aqueous solutions. Chemical Engineering Journal 2018, 344, 380–390. [Google Scholar] [CrossRef]
- Kong, Q.; Wei, J.; Hu, Y.; Wei, C. Fabrication of terminal amino hyperbranched polymer modified graphene oxide and its prominent adsorption performance towards Cr (VI). Journal of hazardous materials 2019, 363, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Wei, C.; Preis, S.; Hu, Y.; Wang, F. Facile preparation of nitrogen and sulfur co-doped graphene-based aerogel for simultaneous removal of Cd2+ and organic dyes. Environmental Science and Pollution Research 2018, 25, 21164–21175. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wang, X.; Yao, W.; Wang, J.; Ji, Y.; Ai, Y.; Alsaedi, A.; Hayat, T.; Wang, X. Macroscopic, spectroscopic, and theoretical investigation for the interaction of phenol and naphthol on reduced graphene oxide. Environmental Science & Technology 2017, 51, 3278–3286. [Google Scholar]
- Kong, Q.; Preis, S.; Li, L.; Luo, P.; Wei, C.; Li, Z.; Hu, Y.; Wei, C. Relations between metal ion characteristics and adsorption performance of graphene oxide: A comprehensive experimental and theoretical study. Separation and Purification Technology 2020, 232, 115956. [Google Scholar] [CrossRef]
- Chen, D.; Feng, H.; Li, J. Graphene oxide: preparation, functionalization, and electrochemical applications. Chemical reviews 2012, 112, 6027–6053. [Google Scholar] [CrossRef]
- Zhao, D.; Zhang, L.; Siebold, D.; DeArmond, D.; Alvarez, N.T.; Shanov, V.N.; Heineman, W.R. Electrochemical studies of three dimensional graphene foam as an electrode material. Electroanalysis 2017, 29, 1506–1512. [Google Scholar] [CrossRef]
- Gandhi, M.R.; Vasudevan, S.; Shibayama, A.; Yamada, M. Graphene and graphene-based composites: A rising star in water purification-a comprehensive overview. ChemistrySelect 2016, 1, 4358–4385. [Google Scholar] [CrossRef]
- Peng, W.; Li, H.; Liu, Y.; Song, S. A review on heavy metal ions adsorption from water by graphene oxide and its composites. Journal of Molecular Liquids 2017, 230, 496–504. [Google Scholar] [CrossRef]
- Zuo, Y.; Xu, J.; Zhu, X.; Duan, X.; Lu, L.; Yu, Y. Graphene-derived nanomaterials as recognition elements for electrochemical determination of heavy metal ions: a review. Microchimica Acta 2019, 186, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dimiev, A.M.; Eigler, S. Graphene oxide: fundamentals and applications; John Wiley & Sons, 2016.
- Kong, Q.; Shi, X.; Ma, W.; Zhang, F.; Yu, T.; Zhao, F.; Zhao, D.; Wei, C. Strategies to improve the adsorption properties of graphene-based adsorbent towards heavy metal ions and their compound pollutants: A review. Journal of Hazardous Materials 2021, 415, 125690. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Shi, X.; Ma, W.; Zhang, F.; Yu, T.; Zhao, F.; Zhao, D.; Wei, C. Strategies to improve the adsorption properties of graphene-based adsorbent towards heavy metal ions and their compound pollutants: A review. Journal of Hazardous Materials 2021, 415, 125690. [Google Scholar] [CrossRef] [PubMed]
- Sitko, R.; Turek, E.; Zawisza, B.; Malicka, E.; Talik, E.; Heimann, J.; Gagor, A.; Feist, B.; Wrzalik, R. Adsorption of divalent metal ions from aqueous solutions using graphene oxide. Dalton transactions 2013, 42, 5682–5689. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, Y.; Zhang, L.; Huang, H.; Hu, J.; Shah, S.M.; Su, X. Adsorption and removal of tetracycline antibiotics from aqueous solution by graphene oxide. Journal of colloid and interface science 2012, 368, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Elgengehi, S.M.; El-Taher, S.; Ibrahim, M.A.; Desmarais, J.K.; El-Kelany, K.E. Graphene and graphene oxide as adsorbents for cadmium and lead heavy metals: A theoretical investigation. Applied Surface Science 2020, 507, 145038. [Google Scholar] [CrossRef]
- Hattab, A.H.; Tapabashi, N.O.; Khalil, N.J. Density Functional Theory calculations for Graphene Oxide, Zinc Oxide and Graphene oxide/zinc oxide composite structure 2023.
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; others. Gaussian 16 Revision C. 01. 2016; Gaussian Inc. Wallingford CT 2016, 421. [Google Scholar]
- Albertsen, J.; Knudsen, J.; Roy-Poulsen, N.; Vistisen, L. Meteorites and thermodynamic equilibrium in fcc iron-nickel alloys (25-50% Ni). Physica Scripta 1980, 22, 171. [Google Scholar] [CrossRef]
- Lee, H.; Spanos, G.; Shiflet, G.; Aaronson, H. Mechanisms of the bainite (non-lamellar eutectoid) reaction and a fundamental distinction between the bainite and pearlite (lamellar eutectoid) reactions. Acta Metallurgica 1988, 36, 1129–1140. [Google Scholar] [CrossRef]
- Sarkar, U.; Roy, D.; Chattaraj, P.; Parthasarathi, R.; Padmanabhan, J.; Subramanian, V. A conceptual DFT approach towards analysing toxicity. Journal of Chemical Sciences 2005, 117, 599–612. [Google Scholar] [CrossRef]
- Frau, J.; Muñoz, F.; Glossman-Mitnik, D. A molecular electron density theory study of the chemical reactivity of cis-and trans-resveratrol. Molecules 2016, 21, 1650. [Google Scholar] [CrossRef] [PubMed]
- Chandrakumar, K.; Pal, S. The concept of density functional theory based descriptors and its relation with the reactivity of molecular systems: A semi-quantitative study. International Journal of Molecular Sciences 2002, 3, 324–337. [Google Scholar] [CrossRef]
- Armaković, S.; Armaković, S.J.; Abramović, B.F. Theoretical investigation of loratadine reactivity in order to understand its degradation properties: DFT and MD study. Journal of molecular modeling 2016, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, T. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den einzelnen Elektronen eines Atoms. physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Fukui, K. Role of frontier orbitals in chemical reactions. science 1982, 218, 747–754. [Google Scholar] [CrossRef]
- Choudhary, V.; Bhatt, A.; Dash, D.; Sharma, N. DFT calculations on molecular structures, HOMO–LUMO study, reactivity descriptors and spectral analyses of newly synthesized diorganotin (IV) 2-chloridophenylacetohydroxamate complexes. Journal of computational chemistry 2019, 40, 2354–2363. [Google Scholar] [CrossRef] [PubMed]
- Aihara, J.i. Reduced HOMO- LUMO gap as an index of kinetic stability for polycyclic aromatic hydrocarbons. The Journal of Physical Chemistry A 1999, 103, 7487–7495. [Google Scholar] [CrossRef]
- Aihara, J.i. Weighted HOMO-LUMO energy separation as an index of kinetic stability for fullerenes. Theoretical Chemistry Accounts 1999, 102, 134–138. [Google Scholar] [CrossRef]
- Yoshida, M.; Aihara, J.i. Validity of the weighted HOMO–LUMO energy separation as an index of kinetic stability for fullerenes with up to 120 carbon atoms. Physical Chemistry Chemical Physics 1999, 1, 227–230. [Google Scholar] [CrossRef]
- Parr, R.G.; Zhou, Z. Absolute hardness: unifying concept for identifying shells and subshells in nuclei, atoms, molecules, and metallic clusters. Accounts of chemical research 1993, 26, 256–258. [Google Scholar] [CrossRef]
- Kosar, B.; Albayrak, C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino) methyl] phenol. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2011, 78, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Li, S.; Priest, C.; Wang, T.; Wu, G.; Li, Q. Effective approaches for designing stable M–Nx/C oxygen-reduction catalysts for proton-exchange-membrane fuel cells. Advanced materials 2022, 34, 2200595. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.; Di Santo, A.; Arias, J.M.; Gil, D.M.; Altabef, A.B. Ab-initio and DFT calculations on molecular structure, NBO, HOMO–LUMO study and a new vibrational analysis of 4-(Dimethylamino) Benzaldehyde. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2015, 136, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.K.; Roy, D.R. Update 1 of: electrophilicity index. Chemical reviews 2007, 107, PR46–PR74. [Google Scholar] [CrossRef]
- Parr, R.; Szentpaly, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 121 1922. [Google Scholar] [CrossRef]
- Leenaerts, O.; Partoens, B.; Peeters, F. Adsorption of Molecules on Graphene. Graphene Chemistry: Theoretical Perspectives, 2013. [Google Scholar]
- Zhang, X.; Li, G.; Li, Q.; Shaikh, M.; Li, Z. The pure paramagnetism in graphene oxide. Results in Physics 2021, 26, 104407. [Google Scholar] [CrossRef]
- Perepichka, D.F.; Bryce, M.R. Molecules with exceptionally small HOMO-LUMO gaps. Angewandte Chemie International Edition 2005, 44. [Google Scholar] [CrossRef] [PubMed]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. The Journal of chemical physics 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Legesse, M.; El Mellouhi, F.; Bentria, E.T.; Madjet, M.E.; Fisher, T.S.; Kais, S.; Alharbi, F.H. Reduced work function of graphene by metal adatoms. Applied Surface Science 2017, 394, 98–107. [Google Scholar] [CrossRef]
- Levels, N.G. Ionization Energies for the Neutral Atoms. Data File on the Site http://physics. nist. gov/PhysRefData/IonEnergy/tblNew. html.
- Lide, D.R. CRC handbook of chemistry and physics; Vol. 85, CRC press, 2004.
- Shtepliuk, I.; Caffrey, N.M.; Iakimov, T.; Khranovskyy, V.; Abrikosov, I.A.; Yakimova, R. On the interaction of toxic Heavy Metals (Cd, Hg, Pb) with graphene quantum dots and infinite graphene. Scientific reports 2017, 7, 3934. [Google Scholar] [CrossRef]
Figure 2.
Molecular electrostatic potential (MEP) isosurfaces of the GO nanoparticle. The blue color indicates low electronic density, the white color indicates medium electronic density, and the orange color indicates high electron density region. S, S, S, S, S, S and S are the most prominent sites for electrophilic attacks.
Figure 2.
Molecular electrostatic potential (MEP) isosurfaces of the GO nanoparticle. The blue color indicates low electronic density, the white color indicates medium electronic density, and the orange color indicates high electron density region. S, S, S, S, S, S and S are the most prominent sites for electrophilic attacks.
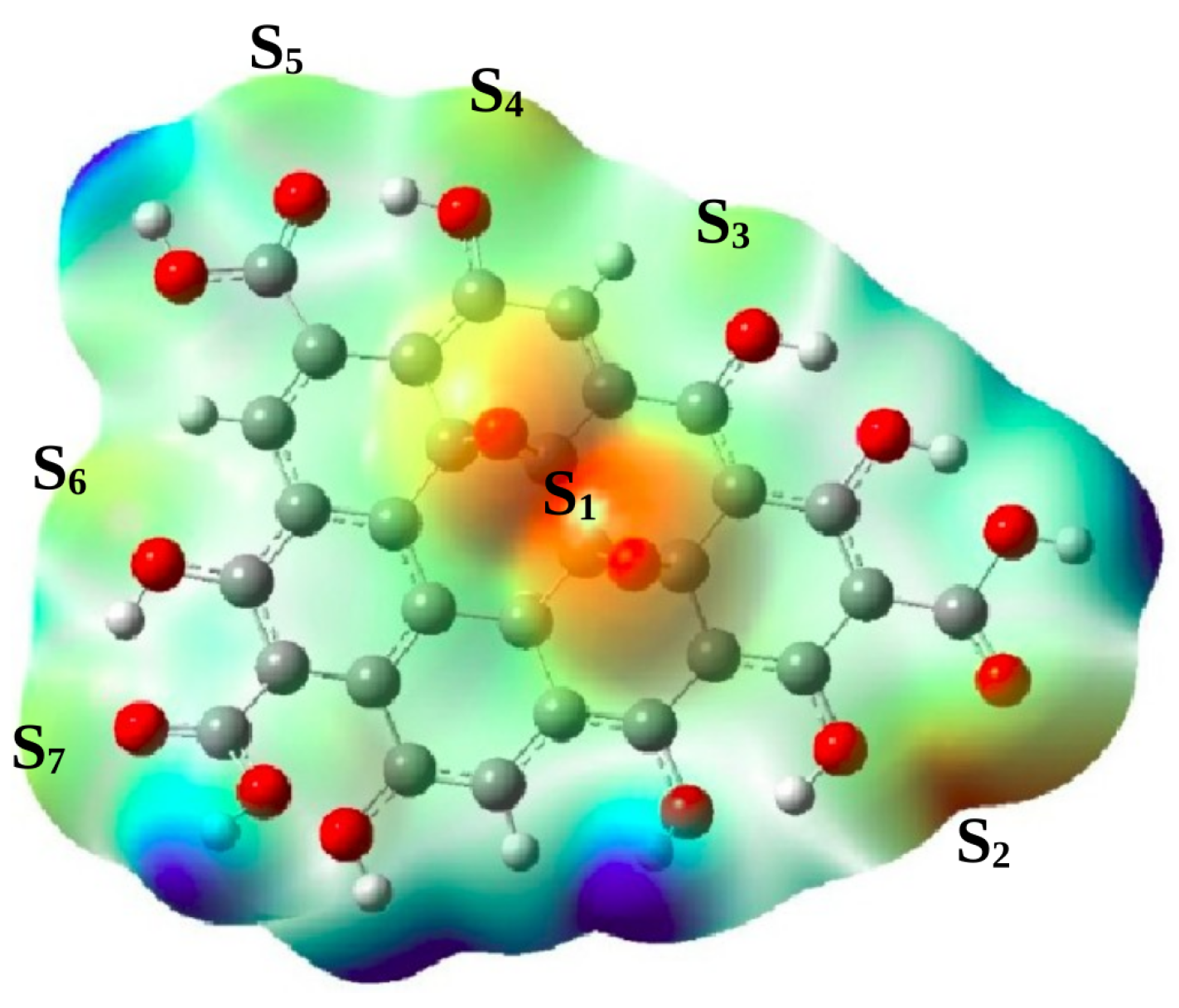
Table 1.
Nucleophilic and electrophilic condensed Fukui functions, and the condensed dual descriptor for different adsorption sites onto the free GO nanoparticle.
Table 1.
Nucleophilic and electrophilic condensed Fukui functions, and the condensed dual descriptor for different adsorption sites onto the free GO nanoparticle.
| Structure | Site | |||
| S | ||||
| S | ||||
| S | ||||
| GO | S | |||
| S | ||||
| S | ||||
| S |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



