
Submitted:
30 January 2024
Posted:
31 January 2024
You are already at the latest version
Abstract
Duchenne muscular dystrophy (DMD) is a progressive neuromuscular disease caused by mutations to the dystrophin gene - resulting in deficiency of dystrophin protein, loss of myofiber integrity in skeletal and cardiac muscle, and eventual cell death and replacement with fibrotic tissue. Pathologic cardiac manifestations occur in nearly every DMD patient, with development of cardiomyopathy - the leading cause of death - inevitable by adulthood. As early cardiac abnormalities are difficult to detect, timely diagnosis and appropriate treatment modalities remain a challenge. There is no cure for DMD – treatment is aimed at delaying disorder progression and alleviating symptoms. A comprehensive understanding of the pathophysiological mechanisms is crucial to development of targeted treatments. While established hypotheses of underlying mechanisms include sarcolemmal weakening, upregulation of pro-inflammatory cytokines, and perturbed ion homeostasis, mitochondrial stress has recently come into focus as a potential key contributor. Several experimental compounds targeting the skeletal muscle pathology of DMD are in development, but effects of such agents on cardiac function remain unclear. Synergistic integration of small molecule- and gene-target-based drugs with metabolic, immune, or ion balance-enhancing compounds into a combinatorial therapy offers potential for treating dystrophin deficiency-induced cardiomyopathy, making it crucial to understand the underlying mechanisms driving the disorder.
Keywords:
Dystrophin
; mitochondria
; inflammation
; reactive oxygen species
; glucocorticoids
; cardiomyopathy
; neuromuscular disease
; therapeutic target
; treatment
; stress response
; ATP
; respiration
; calcium
; ion homeostasis
1. Introduction
1.1. Overview
Duchenne muscular dystrophy (DMD) is a devastating progressive neuromuscular disorder caused by X-linked recessive mutations to the dystrophin gene (DMD), located on chromosome Xp21 (Nigro et al., 1983; Jacobs et al., 1981). The mutation results in deficiency of functional dystrophin protein and subsequent loss of myofiber integrity in skeletal and cardiac muscle, thus leaving muscle fibers susceptible to contraction-induced damage (Petrof et al., 1993). As DMD progresses, muscle turnover and repair cannot keep pace with the constitutive cellular damage that arises, resulting in cell death and replacement with fibrotic and/or fatty tissue (reviewed in Wallace and McNally, 2009).
Phenotypically, dystrophin deficiency is characterized by progressive locomotor-skeletal, respiratory, and cardiac muscle weakness, ultimately resulting in loss of ambulation as well as respiratory and cardiac failure (McNally et al., 2015; Bach et al., 1987; Zambon et al., 2022; reviewed in Schultz et al., 2022). Pathologic cardiac manifestations occur in nearly every DMD patient, with development of cardiomyopathy inevitable by adulthood (Nigro et al., 1990; de Kermadec et al., 1994), and ultimately progressing to heart failure and mortality in 44-57% of patients (Szabo et al., 2021). Due to early pharmacological intervention and advancements in respiratory care for DMD patients, cardiomyopathy is now the leading cause of death in this patient population (Bushby and Connor, 2011; Passamano et al., 2012). While timely detection and management are essential to slow progression of dystrophin deficiency-induced cardiomyopathy, uncovering appropriate diagnostic and treatment modalities still remains a challenge due to the early cardiac abnormalities that are often difficult to detect and limitations in cardiac imaging techniques. Additionally, functional measurements in dystrophin deficiency-induced cardiomyopathy are confounded by skeletal muscle weakness given that typical heart failure symptoms, such as exercise limitations and dyspnea, are masked in this population (reviewed in McNally et al., 2015).
No cure exists for DMD—therefore, treatment is aimed at delaying disorder progression and alleviating symptoms. A comprehensive appreciation of the pathophysiological mechanisms that underlie cardiomyopathy is of the utmost importance to develop targeted treatments. It is worth noting that a single mechanism cannot explain the multifactorial pathogenesis of dystrophin deficiency-induced cardiomyopathy; rather, it is a result of several, interrelated mechanisms. While prevailing hypotheses of underlying mechanisms include sarcolemmal weakening, upregulation of pro-inflammatory cytokines, metabolic disruptions, and perturbed calcium homeostasis, mitochondrial dysfunction has come into focus as a potential key contributor (reviewed in Bellissimo et al., 2022; Schultz et al., 2022). Several experimental compounds targeted to the skeletal muscle pathology of dystrophin deficiency are in development, but the effect of agents on cardiac function remain unclear. Combinatory therapies are most likely the best approach for treating dystrophin deficiency-induced cardiomyopathy, again making it crucial to understand the underlying pathologic mechanisms driving the disorder.
1.2. Epidemiology of Duchenne muscular dystrophy (DMD)
DMD is the most common inherited muscular dystrophy diagnosed in childhood, with an incidence of approximately 1 in 5,000 newborn males (Crisafulli et al., 2020), although variations have been reported elsewhere (Ryder et al., 2017; Mah et al., 2014; Bladen et al., 2013). Since DMD is X-linked, it primarily affects males. Female carriers are typically asymptomatic, however, females with mild to severe symptoms have been reported (Florian et al., 2016). Approximately 25% and 59% of DMD patients develop cardiomyopathy by 6 and 10 years of age, respectively, with electrocardiogram (ECG) abnormalities and sinus tachycardia (ST) representing the primary early clinical manifestations (Nigro et al., 1990; Finsterer and Cripe, 2014; McNally et al., 2015; van Westering et al., 2015). By 18 years of age, cardiomyopathy occurs in 98% of patients and progresses to heart failure in 40% of patients (Nigro et al., 1983; Judge et al., 2011; Verhaart et al., 2011). Although limited, some studies have shown that female carriers present with indices of cardiac abnormalities, including cardiomyopathy, and similar to affected males, cardiac risk increases with age (Florian et al., 2016; Birnkrant et al., 2018). A higher mortality is linked to dystrophin deficiency-induced cardiomyopathy than to other cardiomyopathies (McNally et al., 2015).
1.3. Genetics
The DMD gene is the largest in the human genome (Koenig et al., 1987; Hoffman et al., 2020), spanning 2.4 Mb in the Xp21 region (Kamdar and Garry, 2016). It consists of 79 exons and has several internal promoters that produce an array of dystrophin isoforms expressed in striated and smooth muscles, brain, retina, and kidney (Kamdar and Garry, 2016). Thousands of mutations in the DMD gene have been found to cause dystrophin deficiency (Bladen et al., 2013).
Deletions account for over 70% of mutations and typically result in an altered reading frame that produces a premature stop codon (Bladen et al., 2015; Flanigan, 2017). Approximately 20% of dystrophin mutations are point mutations, small deletions, or insertions, whereas duplications make up 5-15% of mutations (Aartsma-Rus et al., 2006; Magri et al., 2011). Deletions and duplications tend to cluster in hotspot regions, located at exons 45-55 and 3-9 on the DMD gene (Nakamura et al., 2016). De novo mutations are responsible for one-third of DMD cases (Bladen et al., 2015; Laing, 1993). Mutations that disrupt the reading frame or introduce a premature stop codon cause the absence of dystrophin. Mutations that maintain the reading frame allow for truncated dystrophin to be produced resulting in the phenotype of Becker muscular dystrophy (BMD) (Aartsma-Rus and den Dunnen, 2019). Mutations can potentially confer cardioprotective properties against dilated cardiomyopathy (DCM) (exons 51-52) (Jefferies et al., 2005), perpetuate cardiomyopathy (exons 12, 14-17, 31-42, 45, 48-49, and 79) (reviewed in Yamamoto et al., 2018), or be neutral. As mutations can affect different exons on the DMD gene, it is unlikely that a single genetic therapy will benefit patients across the spectrum of mutations. Instead, there is substantial value in targeting secondary dysfunctions, such as mitochondrial stress, which are ubiquitously exhibited in most dystrophin-deficiency models.
1.4. Dystrophin and membrane instability
Dystrophin is a rod-shaped protein, located beneath the muscle fiber membrane, that acts as a molecular shock absorber by transmitting forces generated by sarcomere contraction to the extracellular matrix (ECM) (Le et al., 2018). Dystrophin consists of four major functional domains: an amino-terminal actin-binding domain, a large central rod domain, a carboxyl-terminal domain, and a cysteine-rich domain that binds dystroglycan (reviewed in Kaspar et al., 2009). Dystrophin is an integral part of the dystrophin-glycoprotein complex (DGC) that connects the intracellular cytoskeleton to the ECM (Ervasti and Campbell, 1993) and provides structural support to the sarcolemma. Aside from structural support, dystrophin also serves as a scaffold for proteins involved in various signaling cascades, including sarcolemmal ion channels, to be discussed in detail later (Allen et al., 2010). In cardiomyocytes, the Dp427 isoform of dystrophin is expressed and located at the cardiac sarcolemma as well as in the cardiac T-tubules (Masubuchi et al., 2013). DMD-induced cardiomyopathy appears to be predominantly attributed to the loss of the Dp427 isoform (Masubuchi et al., 2013).
The absence of dystrophin results in sarcolemmal membrane fragility and susceptibility to contraction-induced damage leading to micro-tears in the membrane (Kaspar et al., 2009). In one study examining dystrophin deficiency-induced membrane tears, it was determined through an Evans blue dye assay that left ventricular (LV) cardiomyocyte leakage was significantly increased in C57BL/10 mdx mice compared to age-matched control mice (Van Erp et al., 2010). Membrane fragility and tears are hypothesized to precipitate secondary pathophysiological mechanisms, such as excessive extracellular calcium influx and altered calcium homeostasis, that lead to myocyte degradation and necrosis (Allen et al., 2016; reviewed in Meyers and Townsend, 2019), which will be discussed in detail later. Furthermore, elevated serum cardiac troponin I (cTnI) (as a result of destruction to the myocardium) reflects the increased cellular permeability of the dystrophin-deficient heart (Matsumura et al., 2007). Use of membrane sealants, such as Poloxamer 188, have demonstrated stabilization of the sarcolemma and improvement in acute cardiac function in murine and canine dystrophin-deficiency models (Yasuda et al., 2005, Townsend et al., 2010).
In skeletal muscle, dystrophin mutations also lead to disorganized microtubule organization and oxidized actin which contributes to weakness and is thought to contribute to metabolic stress and other abnormalities (Ramos et al., 2020; Prins et al., 2009; Belanto et al., 2016; Nelson et al., 2018; Belanto et al., 2014; Olthoff et al., 2018).
1.5. Preclinical models of DMD
Animal models have been critical in delineating the pathophysiologic mechanisms of dystrophin deficiency, and for subsequently trialing therapeutic candidates. The C57BL/10 mdx mouse, a genetic and biochemical homolog of human DMD, is the most widely used animal model of dystrophin deficiency and has been invaluable in providing knowledge for therapeutic strategies (Coley et al., 2016). The C57BL/10 mdx mouse lacks full-length dystrophin due to a nonsense point mutation in exon 23 of the DMD gene (Coley et al., 2016). This model only exhibits a mild cardiomyopathy late in life that never fully advances into heart failure (Howard et al., 2021). Additionally, cardiac fibrosis and dysfunction present very late, if at all, in this model (Bulfield et al., 1984). Although the milder skeletal muscle phenotype and subtle nature of cardiac involvement limits translation to human disorder progression (reviewed in McNally et al., 2015), some features such as cardiomyopathy can be unmasked in C57BL/10 mdx mice via cardiac stressors, such as ischemia-reperfusion (IR) insults (Burelle et al., 2009; Ascah et al., 2011). Due to the ease of breeding and maintaining, with a long-life expectancy, the C57BL/10 mdx mouse models remain a common tool in dystrophin deficiency research (Shirokova et al., 2013). Other mouse models exist with a variety of backgrounds and mutations, but the majority result from the crossing of the C57BL/10 mdx mouse with different mutations to worsen the dystrophin deficiency phenotype (Swiderski and Lynch, 2021; Wong et al., 2020)). However, the natural history of cardiomyopathy in each model remains largely uncharacterized.
The DBA/2J-mdx (D2.mdx) and utrophin-dystrophin double knockout mice have drawn attention as representing better models of DMD-induced cardiomyopathy, including earlier onset cardiac deficits (Coley et al., 2016). In the D2.mdx mouse, cardiac fibrosis is noted at 18 weeks of age (Hayes et al., 2022), but the degree to which cardiac dysfunction occurs at later ages is not understood. Several mouse models for dystrophin deficiency have been developed, but none of the models completely recapitulates the phenotype and disorder progression seen in humans. However, more severe models have a reduced lifespan, are more difficult to breed, and carry an additional mutation not affected in humans, which causes difficulty in reliably extrapolating data to the human phenotype (Shirokova et al., 2013; Coley et al., 2016).
To bridge the gap between small rodent (mouse) DMD models and the human phenotype, several different rat DMD models have been investigated. To date, the majority of rat DMD models have been developed to genetically target the genomic region that spans exons 3-26 (Taglietti et al., 2022; Iyer et al., 2020; Larcher et al., 2014; Nakamura et al., 2014). This is a major limitation in the model given that a major mutation hotspot for the classical, progressive DMD phenotype is between exons 45-55 (Taglietti et al., 2022). For this reason, a new rat DMD model was generated with a deletion mutation in exon 52 on the rat DMD gene, known as the R-DMDdel52 rat (Taglietti et al., 2022). In comparison to the mdx mouse model, the R-DMDdel52 rat exhibits remodelling of the entire striated musculature, including both the heart and diaphragm, which culminates leading to premature lethality between 10-14 months of age (Taglietti et al., 2022).
The golden retriever muscular dystrophy (GRMD) model offers remarkable resemblance to the progressive locomotor, respiratory, and cardiac muscular phenotype observed in human DMD patients (Howell et al., 1997). Similar to humans, these canines are characterized by highly variable cardiomyopathy progression, making assessment of therapies challenging (Le Guiner et al., 2017; Felsburg, 2002). Unfortunately, high costs, limited supply, and difficulty in maintaining a colony preclude widespread use of GRMD models (Shirokova et al., 2013).
In order to more closely recapitulate the human DMD phenotype (in comparison to small rodent models), without limitations imposed by the financial burden of large mammals (e.g., GRMD), a DMD rabbit model was developed by cytoplasmic microinjections of Cas9 mRNA and single guide RNA (sgRNA) (Sui et al., 2018). The rabbit DMD model was developed by targeting exon 51 (commonly mutated in human DMD patients) to disrupt the open reading frame of DMD in rabbits, thereby generating DMD KO (knock-out) rabbits (Sui et al., 2018). This model exhibits many hallmark pathologies of DMD, including elevated serum creatine kinase, impaired mobility/ambulation, muscle necrosis and regeneration, and importantly, cardiomyopathic manifestations (increased cardiac fibrosis, impaired function, etc.) (Sui et al., 2018). Although further research is still required to comprehensively validate and characterize this model, the clear evidence of cardiomyopathy at 4 months of age (demonstrated by reduced left ventricular ejection fraction and fractioning shortening) paired with the close resemblance to its human DMD counterpart, makes this an attractive model for preclinical drug screening studies (Sui et al., 2018).
Induced pluripotent stem cells (iPSCs), another preclinical model for dystrophin deficiency, are reprogrammed from patient-specific somatic cells and carry the same genetic defects as DMD patients (Lin et al., 2015). These cells make it possible to obtain functional cardiomyocytes from DMD patients and offer an important complement to animal models in studying dystrophin deficiency (reviewed in Law et al., 2020). However, iPSCs do not manifest a fully mature phenotype, which may limit their utility in pre-clinical research (Svobodova et al., 2021).
1.6. Clinical manifestations of DMD
The earliest symptoms in DMD patients typically present between 1 to 5 years of age and includes waddling gait, Gower’s maneuver, difficulty climbing stairs, and frequent falls (Gardner-Medwin, 1980; Emery, 2002; Ryder et al., 2017). Calf hypertrophy tends to develop during early childhood due to localized accumulation of fatty and fibrotic tissue (Torriani et al., 2012). Motor skills deteriorate at 6 to 8 years of age when lordosis and scoliosis advance (Florczyk-Soluch et al., 2021). Scoliosis increases the risk of respiratory failure (Archer et al., 2016). Most DMD patients are wheelchair bound at approximately 13 years of age (Mercuri et al., 2019). Cognitive impairments are observed in roughly one third of all DMD patients (Banihani et al., 2015). Secondary to progressive muscle degradation and leakage of myofiber contents, elevated creatine kinase (CK) levels are detected early, with levels being about 10 times higher in newborns with DMD and approximately 50 to 100 times higher in children with DMD as compared to healthy age-matched boys (Birkrant et al., 2018; Mendell et al., 2012).
During disorder progression, cardiac symptoms advance from diastolic dysfunction to myocardial remodeling and fibrosis, leading to contractile weakness and subsequent DCM with decreased systolic function (Finsterer and Cripe, 2014; van Westering et al., 2015). Heart failure and arrhythmias develop gradually, increasing the risk of sudden cardiac death (Schultz et al., 2022). Largely attributable to advancements in respiratory interventions, such as assistive breathing devices (ventilators), the predominant cause of death has shifted from respiratory failure towards cardiomyopathy (reviewed in Passamano et al., 2012), and resultantly, patients may survive well into their third and fourth decades (Bushby et al., 2010; Wagner et al., 2007).
2. Cardiomyopathy in DMD: Functional and Histological Manifestations
As previously noted, typical cardiac symptoms of exercise intolerance and dyspnea are masked in DMD patients due to the influence of skeletal muscle weakness on these same parameters (McNally et al., 2015). Nonetheless, detection of early changes in the structure and function of the heart is crucial to initiating timely treatment in hopes of yielding better outcomes (Lee et al., 2021). Current guidelines recommend yearly cardiac screening at diagnosis (Birkrant et al., 2018).
2.1. Echocardiography
Echocardiography is recommended until at least 6 to 7 years of age, when the child can lie still without anesthesia, at which point cardiac magnetic resonance imaging (CMR) is recommended due to its ability to detect subtle cardiac changes prior to overt cardiac dysfunction (Birkrant et al., 2018, Lee et al., 2021). Transthoracic echocardiography (TTE) is a non-invasive, readily available diagnostic tool, but measurement of left ventricular ejection fraction (LVEF) using standard TTE rarely detects abnormal cardiac function in DMD patients within the first decade of life (Soslow et al., 2016; D’Amario et al., 2017). Two-dimensional fractional shortening (FS) and 5/6 area-length LVEF were found to be the most accurate and reproducible objective measures of left ventricular (LV) function using TTE in DMD patients (Soslow et al., 2016). However, TTE has been observed to underestimate LV function compared to CMR (Soslow et al., 2016; Buddhe et al., 2016). Myocardial performance index (MPI) and doppler tissue imaging (DTI) may be capable of detecting myocardial dysfunction prior to the development of systolic dysfunction (Spurney et al., 2011). MPI, an assessment of global heart function, has been observed to correlate with EF (LaCorte et al., 2003). However, in DMD patients, calculation of the MPI showed abnormalities in 79% of patients, whereas 40% of patients had an abnormal LVEF on TTE (Bahler et al., 2005). DTI, which does not require good resolution, can detect early changes in the development of cardiomyopathy through measures of myocardial tissue velocities and strain (Spurney et al., 2011). Decreased tissue velocities have been observed in asymptomatic patients as young as 8.8 years and were able to predict poor outcomes with 85% accuracy (Giatrakos et al., 2006). Reduction in peak systolic radial strain has been observed in the posterior wall of DMD patients (Mori et al., 2007), commonly seen in the outer portion of the wall (Frankel and Rosser, 1976), and in those with normal systolic function (Ogata et al., 2007). DTI-derived strain measurement is less reliable in DMD patients due to their skeletal deformities obscuring proper Doppler beam placement and thus speckle tracking echocardiography (STE) is preferred to measure 2D strain. STE can detect subclinical LV dysfunction prior to decrease in LVEF through measurement of myocardial strain (Adorisio et al., 2020; Amedro et al., 2019) and is able to assess segmental and global myocardial function in the longitudinal, radial, and circumferential displacements (Mondillo et al., 2011). Before the appearance of overt cardiomyopathy in patients with DMD, significant reduction in global LV STE strain has been reported (Amedro et al., 2019). Myocardial strain, as measured by STE, was observed to be abnormal in approximately 50% of DMD patients, even in those with normal EF, suggesting that strain may identify early cardiac involvement (Levy et al., 2016; Patrianakos et al., 2015). Lower global longitudinal strain (GLS) values were seen in DMD patients with a decrease of 0.34% per year according to age (Amedro et al., 2019). The magnitude of difference in strain was greatest in the inferolateral and anterolateral segments (Amedro et al., 2019; Bilchick et al., 2011). Studies have reported variable findings using STE, as longitudinal peak-systolic strain was reported to be more pronounced in the apical area and mid-anterior segment in one study (Taqatqa et al., 2016), while another study revealed more prominent changes in the basal lateral segments (Bilchick et al., 2011) with decreased peak-systolic strain (Cho et al., 2018). In addition, a non-controlled study found that circumferential STE strain correlated moderately well with CMR strain and reported a trend toward reduced STE strain in patients with late gadolinium enhancement, a sign of fibrosis on CMR (Amedro et al., 2019). This could represent a good cardiac assessment option in DMD children too young to undergo CMR without anesthesia.
2.2. Limitations of echocardiography
Patients with DMD have poor acoustic windows due to increased adiposity, altered body habitus, lung hyper-inflation, and limited mobility (Romfh and McNally, 2010; Power et al., 2017). These factors degrade echocardiographic image quality and negatively impact interpretation (Buddhe et al., 2016). As a result, in several studies, echocardiography has been deemed inadequate for detecting cardiac involvement in DMD patients in the first decade of life, and sometimes beyond (Hor et al., 2013; Buddhe et al., 2016). Additionally, TTE cannot perform tissue characterization to detect early myocardial fibrosis (Prakash et al., 2022) and is unable to accurately account for the regional distribution typically apparent in DMD-induced cardiomyopathy (Lee et al., 2021). It was observed that TTE misclassified 20% of DMD patients, and 37% of the myocardial segments were unable to be visualized compared to CMR (Soslow et al., 2016; Buddhe et al., 2016). Despite these limitations, more advanced imaging may be contraindicated and TTE still remains valuable in the assessment and monitoring of DMD patients.
2.3. Cardiac magnetic resonance imaging
CMR is a non-invasive imaging tool that provides accurate volumetric measurements, assessment of wall motion abnormalities, and tissue characterization without negative effects from body habitus or other similar external factors (Soslow et al., 2016). Advantages to this technique include its excellent reproducibility and operator independency (Mavrogeni et al., 2013). CMR has been proven efficient in detecting myocardial fibrosis in patients with DMD (Ogata et al., 2009; Viollet et al., 2012). CMR in DMD patients has demonstrated a better diagnostic yield for detecting preclinical features of cardiomyopathy and greater sensitivity for identifying subtle changes in cardiac function, wall motion, and structural abnormalities than TTE (Prakash et al., 2022). LVEF as derived from CMR was found to poorly correlate with LVEF derived from TTE, and CMR-derived LVEF was found to moderately correlate to TTE-derived FS (Soslow et al., 2014), even with adequate imaging quality (Buddhe et al., 2018). As such, CMR is now the preferred cardiac imaging technique for patients with DMD (Birkrant et al., 2018). Even in the absence of overt cardiomyopathy, CMR has identified a pattern of fibrosis in female carriers similar to that observed in DMD patients (Mavrogeni et al., 2013). Detection of LV wall motion abnormalities on CMR has demonstrated good predictive value for the presence of regional cardiac dysfunction (Gloss et al., 2016), whereas the transmural pattern often located at the inferolateral wall independently predicts adverse cardiac event in DMD patients with normal LVEF (Florian et al., 2014b).
Late gadolinium enhancement (LGE), a sensitive marker of myocardial fat and fibrosis, appears as a result of diminished contrast washout on CMR (Prakash et al., 2022). This tool can be used to detect subclinical cardiac disease prior to LV dysfunction and may predict adverse cardiac events in DMD patients (Florian et al., 2014b; Hor et al., 2013; Silva et al., 2017). In DMD-induced cardiomyopathy, LGE is subepicardial and absent in the subendocardium (Lee et al., 2021). LGE appears to correlate with age such that patients with LGE tend to be significantly older with decreased LVEF (Hor et al., 2013; Hor et al., 2009). In fact, the presence of LGE correlated with a 2.2% decline in LVEF per year (Tandon et al., 2015). LGE in DMD patients has also been linked to greater LV dilation and dysfunction, as well as higher incidence of arrhythmias (Menon et al., 2014). LGE was reported in 30% of patients with normal LVEF and in 84% of patients with abnormal LVEF (Hor et al., 2013). One study revealed that patients with normal function on TTE had LGE on CMR and that several segments with abnormal wall motion by CMR were not detected on TTE (Soslow et al., 2016). LGE has been observed in patients under 10 years (Buddhe et al., 2016; Spurney et al., 2014), predominantly in the inferoseptal and anterolateral segments, but also typically involves the basal inferolateral free wall (Prakash et al., 2022). In addition, LGE of the subepicardium of the lateral LV wall is commonly observed, with intramural septal LGE becoming more prevalent with disorder progression (Puchalski et al., 2009; Florian et al., 2014b). It appears that the presence of LGE can predict the severity of cardiomyopathy in DMD (Tandon et al., 2015) and that it correlates to higher risk of arrhythmias in DMD (Menon et al., 2014). LGE may guide therapeutic interventions as the most recent DMD treatment guidelines recommend initiating cardiac therapy when LGE presence is first observed (Lee et al., 2021).
Strain imaging quantifies regional tissue deformation, and its measurements are potential early markers of cardiac involvement in DMD (Gotte et al., 2006). CMR strain correlates more closely with CMR LVEF compared to TTE strain (Buddhe et al., 2016). A sensitive marker used to identify cardiac pathology is peak circumferential strain (εcc) at the mid-ventricular level, which can also assess effects of therapeutic interventions on cardiac function and measure the degree of cardiac pathology (Hor et al., 2009; Siddiqui et al., 2020; Hor et al., 2015; Ashford et al., 2005; Raman et al., 2015; Bilchick et al., 2011; Batra et al., 2022). This measure was able to detect cardiac pathology as early as 5 years of age and the findings of this study revealed the heterogeneity in cardiac progression in patients with DMD (Batra et al., 2022). Additionally, circumferential uniformity ratio estimate (CURE) was identified in this study to have potential as a tool in understanding cardiac pathology in the DMD population (Batra et al., 2022). Global εcc appears to be the most sensitive strain marker for subclinical myocardial changes in DMD (Panovsky et al., 2021; Siddiqui et al., 2020) with more prominent differences in anterolateral, inferolateral, and inferior segments (Siegel et al., 2017). Reduced myocardial strain has been observed in the presence of normal LVEF (Hor et al., 2009; Ashford et al., 2005; Hagenbuch et al., 2010). Specifically, decreased global circumferential and segmental strain as well as mitral annular plane systolic excursion (MAPSE) has been observed in DMD patients with normal LV function and absence of LGE (Panovsky et al., 2021, Ashford et al., 2005), Similarly, decreased LV myocardial peak circumferential strain was observed in DMD patients under 10 years of age and declined with age (Hor et al., 2009). In DMD patients who develop overt LV dysfunction, CMR strain was found to be significantly worse than in those who do not develop cardiomyopathy (Siddiqui et al., 2020).
In cases where contrast is contraindicated, such as patients with vascular access issues, renal insufficiency, or contrast allergies, non-contrast CMR techniques are important (Buddhe et al., 2016). CMR-feature tracking does not use contrast and has been able to differentiate DMD patients from controls (Siegel et al., 2018; Aikawa et al., 2018). Without contrast, εcc can still detect myocardial global and regional alterations (Hor et al., 2009; Hor et al., 2015; Ashford et al., 2005).
Other CMR tools are T1 and T2 mapping. T1 mapping, which measures diffuse myocardial fibrosis and extracellular volume expansion, can identify early myocardial fibrosis even in the absence of LGE (Olivieri et al., 2016). Both pre- and post-contrast T1 mapping can detect earlier and more subtle signs of cardiac dysfunction (Magrath et al., 2018). In contrast to LGE that demonstrates focal fibrosis, T1 mapping can identify diffuse fibrosis in cardiomyopathy (Haaf et al., 2016). T1 values are increased in DMD patients and thus may serve as early markers of myocardial fibrosis (Florian et al., 2014). T1 mapping is often unable to differentiate fibrosis from inflammation or fat infiltration which may limit its use in DMD patients (Roujol et al., 2014; Kellman et al., 2015).
T2 mapping may be able to identify fat infiltration, edema, and inflammation within the affected muscle (Magrath et al., 2018). In a study using T2 mapping in DMD boys, the full-width half-max, a measure of T2 heterogeneity, correlated well with reduced LVEF and circumferential strain (Wansapura et al., 2010).
2.4. Limitations of cardiac magnetic resonance imaging
Disadvantages to CMR include expense, limited availability, patient discomfort due to position and immobility, claustrophobia, length of study, and the need for sedation in some patients (Soslow et al. 2016). The presence of hardware, such as spinal rods to treat scoliosis, may distort the image, limiting the use of CMR in DMD patients (Shih et al., 2020). With repeated exposure, gadolinium has been reported to accumulate in organs and may lead to toxicity (Perazella, 2009; Olchowy et al., 2017; Gale et al., 2017). In addition, the breath-holding segment of this procedure has been a limitation for the DMD population, but advancements have decreased breath holding times and free-breathing sequences are sometimes an option (Kellman et al., 2009; Lee at al., 2021). Despite these limitations, CMR is a powerful imaging modality for monitoring the cardiac involvement in DMD patients and circumferential strain and LGE are sensitive tools that can be utilized to detect occult cardiomyopathy prior to LVEF decline (Lee et al., 2021).
2.5. Hemodynamic biomarkers
Biomarkers indicative of DMD are suspected to be released into circulation as a result of sarcolemmal tearing, a phenomenon that that occurs in response to the mechanical stress of contraction caused by compromised membrane integrity arising from the dystrophin mutation. Cardiac troponin I (cTnI) and T (cTnT) are proteins that comprise the contractile unit of myocardial cells and are biomarkers for damage to the myocardium (Spurney et al., 2021). Serum cTnT can be elevated in the setting of neuromuscular disease and has been shown to correlate better with creatine kinase and myoglobin levels than with cTnI (Rittoo et al., 2014; Wens et al., 2016). Therefore, cTnI is more specific for myocardial injury in DMD as it is not expressed in skeletal muscle during regenerative processes (Schmid et al., 2018; Bodor et al., 1995). Cases of acutely elevated cTnI in DMD patients have been reported with abnormal ECGs, but no evidence of coronary artery disease (Hor et al., 2018; Abutaleb et al., 2018; Cinteza et al., 2017; Cucia et al., 1984; Politano et al., 2003; Schoeffler et al., 2008). Heterogeneity of cTnI levels have also been reported throughout the literature (Spurney et al., 2021). Significantly elevated levels of cTnI have been reported in DMD patients with mild LGE compared to those without LGE. However, cTnI levels were not elevated in patients with moderate-to-severe LGE. Since LGE is representative of cardiac fibrosis on CMR, this finding is likely due to increased cell death and fibrofatty tissue replacement in early disease and decreased enzyme leak at later stages of the disease when fibrofatty tissue has replaced most of the myocardium (Voleti et al., 2020). Elevated troponin levels have also been reported in asymptomatic patients prior to development of cardiac disease (Voleti et al., 2020), which may represent the necrosis and fibrosis resulting from subclinical or early cardiac remodeling. Decreased cTnI was observed in dystrophic dogs treated with membrane sealant, which was thought to ameliorate cardiac injury (Townsend et al., 2010). No current recommendations exist for routine monitoring of cTnI in DMD patient and therefore, only the symptomatic patients tend to undergo testing, making unbiased assessment of the distribution of troponin levels difficult (Spurney et al., 2021).
In addition to troponin, B-type natriuretic peptide (BNP) is a well-known serological marker of cardiac dysfunction; however, studies have revealed that BNP levels are inconsistent and thus are not appropriate measurements of cardiomyopathy in DMD patients (Dittrich et al., 2015). Despite this, lower BNP levels have been reported in patients with DMD-induced cardiomyopathy versus those with idiopathic cardiomyopathy (Demachi et al., 2004). Only a mild elevation of BNP levels is seen and typically only in the presence of severe cardiac dysfunction (Mohyuddin et al., 2007; van Bockel et al., 2009).
Other potential biomarkers and indicators include plasma alpha-ANP, CK-MB, and diastolic abnormalities. Elevation of plasma alpha-ANP levels may be an indication of poor prognosis in DMD patients as it is correlated with congestive heart failure and respiratory failure (Yanagisawa et al., 1990). Since CK-MB can be seen during skeletal muscle regeneration, this protein is not a good marker for cardiac involvement in DMD patients as it can also be indicative of damage to other muscles in the body (Wang et al., 2011). Furthermore, DCM and systolic dysfunction are preceded by diastolic dysfunction in DMD and therefore, diastolic abnormalities may serve as early indicators for early cardiac decompensation (Markham et al., 2006).
2.6. Systolic and diastolic dysfunction
Several studies have reported the presence of systolic and diastolic dysfunction in patients with DMD. It is hypothesized that the calcium dysregulation resulting from the absence of dystrophin presents phenotypically as sustained ion-driven myocyte contraction, which appears as underfilling of the LV and impaired relaxation prior to the development of DCM (Su et al., 2016; Markham et al., 2006). Su et al. (2016) described the heart in this phase as “tonic contraction”, a term describing the abnormally increased tone without ability to relax (Figure 1). Although the LV appears smaller due to its inability to relax, the mass is unaltered. These changes likely lead to a decrease in stroke volume which may be correlated with the resting tachycardia often present in DMD patients (Su et al., 2016). Furthermore, impaired relaxation (which may be attributed to increased myocardial fibrosis leading to stiffness of the ventricular walls), diminished LV cavity size at the end of diastole (a possible outcome of impaired calcium handling - to be discussed later), and thickened myocardium have been observed in DMD animal models (Greally et al., 2013; Townsend, 2011; Wagner et al., 2012; Su et al., 2016). Prior to the appearance of clinical symptoms and decreased LVEF in DMD patients, early changes in diastolic function were observed through DTI (Cho et al., 2018). Specifically, abnormal measures of impaired diastolic function included altered myocardial velocity E’ basal lateral and the mitral valve velocity/myocardial basal lateral E-wave ratio E/E’ (Mertens et al., 2008; Ryan et al., 2013). In a recent study utilizing CMR in DMD patients, LV atrioventricular plane displacement (LVAPD), a marker of systolic and diastolic dysfunction, was decreased in patients with DMD, indicating reduced atrioventricular (AV) plane displacement (Batra et al., 2022). Decreased AV displacement correlates to a reduction in contractility and relaxation of the heart, resulting in reduced diastolic filling pressure and diastolic volume (Batra et al., 2022). This study found that LV end-diastolic volume (LVEDV) was significantly lower than controls, an observation well supported by the literature (Amedro et al., 2019; Panovsky et al., 2021; Dual et al., 2021). Other significant findings in the study by Batra et al. (2022) included reduced LV end-diastolic index (LVEDI) and changes in LV mass and LV end-systolic volume (LVESV).
2.7. Cardiac fibrosis
Deterioration of cardiomyocytes observed during dystrophin deficiency-induced cardiomyopathy activates an inflammatory cascade that results in macrophages clearing damaged cells, and fibroblasts invading the compromised area to form fibrotic (collagenous) scar tissue. In DMD, a prolonged subclinical phase of myocardial fibrosis is typically observed, beginning early in life (Markham et al., 2006).
Death of cardiomyocytes and subsequent development of cardiac fibrosis initially present in the posterobasal segment of the LV (Goodwin and Muntoni, 2005; Finsterer and Stollberger, 2003) and extend to the outer third and ultimately the entire LV and septum (Perloff et al., 1967). The fibrotic lesions are more prominent in the subepicardium and spare the muscle fibers nearest to the chambers in GRMD (Schneider et al., 2022). Additionally, fibrosis of the papillary muscles and posterobasal area contributes to mitral valve regurgitation in DMD (Sanyal et al., 1980). Fibrosis is prevalent from a young age, as evidenced by LGE on CMR (Spurney, 2011; Mertens et al., 2008), where cardiac fibrosis was identified in 17% of patients under 10 years, 34% of patients between 10-15 years, and 59% of patients over 15 years (Hor et al., 2013). Fibrosis occurs prior to the onset of decreased systolic function, which has been demonstrated with LGE on CMR (Spurney, 2011; Mertens et al., 2008). Myocardial fibrotic changes typically have a heterogeneous distribution, according to evidence of regional wall motion abnormalities determined via imaging modalities (Buddhe et al., 2016). While widespread scarring leads to stretching and thinning of the myocardial walls, focal fibrosis increases the risk of sudden death (Magrath et al., 2018).
2.8. Dilated cardiomyopathy
As myocardial fibrosis increases with disorder progression, the fibrotic region of the heart gradually stretches and thins, losing its contractility and resulting in DCM (reviewed in Kaspar et al., 2009). This may involve dilation of the LV or both ventricles, although right ventricle (RV) function tends to be relatively preserved in the setting of improved respiratory interventions (Mehmood et al., 2015). Dystrophin deficiency-induced cardiomyopathy is different than other types of cardiomyopathy in that the LV dilatation is less prominent, while the prognosis is worse (Connuck et al., 2008). DCM typically occurs in the second decade of life in DMD patients, although it has been reported in children under six years of age (Nigro et al., 1990). Clinical signs of DCM in DMD patients include increased LV diameter and volume, reduced FS, decreased LVEF, and development of mitral valve regurgitation (Kaspar et al., 2009).
2.9. Arrhythmia
Frequent ECG changes detected in DMD after development of cardiac fibrosis consist of ST, short PR interval, inferolateral Q waves, increased R/S ratio in precordial leads with tall R waves, left atrial abnormality, and right axis deviation (Shih et al., 2020). In addition to dystrophin deficiency-induced cardiac fibrosis, calcium transients, and elevated reactive oxygen species (ROS), ECG abnormalities may be secondary to dysregulated sodium, calcium, and potassium channels; kinases; and nitric oxide synthase (nNOS) triggered by the absence of dystrophin (Adams et al., 2018; Koenig et al., 2018). Briefly, dystrophin is thought to play an important role in scaffolding voltage-dependent sarcolemmal ion channels in the heart via syntrophin binding, which is an adaptor protein that binds directly to two sites in dystrophin’s carboxyl-terminal region (Adams et al., 2018; Koenig et al., 2018). Dystrophic cardiomyocytes have demonstrated considerable sodium channel loss-of-function, while the activity of some potassium channels may also be reduced (Koenig et al., 2018). It is hypothesized that sarcolemmal ion channel abnormalities may occur prior to onset of cardiomyopathy in the dystrophic heart and may represent a primary effect of the DMD mutation (Koenig et al., 2018). QRS duration appears to increase with age, regardless of systolic function (Segawa et al., 2017). The onset of resting ST is typically seen in DMD patients by 5 years of age and conduction changes are observed by 10 years of age (Kaspar et al., 2009). Cellular mechanisms, including altered calcium homeostasis and elevated ROS, are hypothesized to cause arrhythmias in these patients and will be discussed in more detail in following sections. More serious arrhythmias, such as atrial fibrillation, AV block, ventricular tachycardia, and ventricular fibrillation, have been reported in the presence of advanced fibrosis (Corrado et al., 2002; Kaspar et al., 2009). One study estimated that approximately 44% of DMD patients had arrhythmias, including frequent atrial (3%) or ventricular premature contractions (22%), atrial (16%) or ventricular couplets (32%), supraventricular tachycardia (SVT) (9%), and ventricular tachycardia (VT) (13%) (Chiang et al. 2016). It has also been noted that DMD patients exhibit Wolff Parkinson White (WPW) patterns in their ECGs (Takami et al., 2008; reviewed in Fayssoil et al., 2017). Arrhythmias have been shown to be significantly correlated with decreased systolic function (Chiang et al., 2016; Villa et al., 2015) and an age older than 17 years was significantly associated with development of SVT or VT (Chiang et al., 2016). Sudden death does occur in DMD patients, but the percentage due to arrhythmias is unknown (Groh, 2012). Continuous Holter recordings have shown resting ST, loss of cardiac circadian rhythm, and heart rate variability (Santos et al., 2010). Although one study showed that ECG abnormalities frequently precede cardiac dysfunction by several years (Shah et al., 2010), no differences were noted on ECG of DMD patients with cardiomyopathy compared to those in the subclinical stages (Thrush et al., 2009). These inconsistencies suggest that ECG is not a useful diagnostic tool; however, periodic Holter monitoring is recommended in this population (Birkrant et al., 2018).
3. Mechanisms of Cardiomyopathy in DMD
3.1. Inflammatory signaling and immune response
Although inflammation is necessary for healing, it can also be damaging if it occurs aberrantly. Inflammatory and immune infiltrate are responsible for clearing damaged muscle cells and are often present prior to the onset of symptoms. The immune infiltrate primarily consists of macrophages and T cells in young (2 to 8 years of age) DMD patients (Spencer et al., 1997; reviewed in Evans et al., 2009). Since the heart has a limited capacity to regenerate, macrophages secrete chemokines, such as transforming growth factor beta (TGF-β), to activate resident fibroblasts and endothelial cells in response to injury, thus increasing the ratio of nascent myofibroblasts to quiescent fibroblasts. The fibroblasts are activated into collagen-secreting myofibroblasts and enhance ECM deposition at the injury site, forming fibrocollagenous scar tissue within the wall of the ventricular myocardium (reviewed in Kanisicak et al., 2016; Borthwick et al., 2013). Muscle biopsies from DMD patients demonstrated that TGF-β expression is associated with skeletal muscle fibrosis (Bernasconi et al., 1995). A high concentration of macrophages and T lymphocytes have been reported in pooled dystrophic muscles (quadriceps, hamstrings, and gastrocnemius) beginning early in disorder progression, suggesting that these cells play a crucial role in the pathology of dystrophic muscle (Spencer et al., 2001; reviewed in Evans et al., 2009). Sarcolemmal lesions in dystrophic skeletal muscles initiate calcium leakage and inflammatory cytokine upregulation; produce and release tumour necrosis factor alpha (TNF-α) and histamine; and contribute to mast cell degranulation and elevations to eosinophils (Gorospe et al., 1994; Cai et al., 2000; reviewed in Evans et al., 2009). A combination of these cytokines contributes to a proinflammatory environment and consequently promotes muscle necrosis (Evans et al., 2009). While inflammatory mechanisms have been discovered in dystrophic skeletal muscle, these pathways have not been extensively examined in the dystrophic heart, thus representing a future avenue of research.
Two main inflammatory pathways are involved in DMD: the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway and the nucleotide-binding oligomerization domain (NOD)-like receptor family pyrin domain containing 3 (NLRP3) pathway. NF-κB, a transcription factor that regulates the expression of chemokines and cytokines, may be activated by dystrophin deficiency-induced mechanical stretch. Increased activation of NF-κB is seen in dystrophic skeletal muscle (Kumar and Boriek, 2003; reviewed in Evans et al., 2009). Overexpression of chemokines is hypothesized to occur prior to initial disorder onset (defined as the initial mechanical damage that results from the absence of the membrane-stabilizing effects of dystrophin) and induce macrophage and T-cell infiltration (Wehling et al., 2001; reviewed in Evans et al., 2009). It has been previously determined that inhibition of NF-κB in utrophin/dystrophin-deficient mice improves cardiac contractile function (Delfin et al., 2011).
Before NLRP3 can be activated, it must be primed, which can be accomplished through activation of NF-κB and subsequent transcription of NLRP3 components and pro-inflammatory cytokines (reviewed in Reid and Alexander, 2021). Stressed mitochondria in dystrophin deficient models exhibit elevated mitochondrial hydrogen peroxide (mH2O2) release during attenuated oxidative phosphorylation (Hughes et al., 2019; Hughes et al., 2020; Ramos et al., 2020, Bellissimo et al., 2023), and it has been proposed that elevated ROS (Naik and Dixit, 2011) and possibly mitochondrial cardiolipin release (Iyer et al., 2013) can activate the NF-κB pathway through the NLRP3 inflammasome (reviewed in Elliot and Sutterwala, 2015). The degree to which such mitochondrial stress contributes to cellular degeneration and inflammation versus a reciprocal inflammation-induced mitochondrial stress requires extensive research.
3.2. Calcium handling dysregulation and cell death
In healthy cardiac muscle, contraction and relaxation are driven by the cycling of calcium between the sarcoplasmic reticulum (SR) and cytoplasm. Briefly, this process involves plasma membrane depolarization that subsequently activates the L-type calcium channels (LTCC), which allows calcium to flow into the cell and subsequently causes a larger calcium flux from the SR through the ryanodine receptor 2 (RyR2), resulting in muscle contraction. This process is termed calcium-induced calcium release (CICR). It is imperative to note that the relationship between muscular dystrophy-induced elevations to net intracellular influx of calcium and muscle-cell necrosis was first proposed 40 years ago (Wrogemann and Pena, 1976; reviewed in Zulian et al., 2016). It was hypothesized that this increased intracellular calcium concentration triggers a vicious cycle of downstream mitochondrial calcium overload and eventual insufficient ATP synthesis, which further perpetuates cytoplasmic calcium levels by impairing calcium pumps, leading to hypercontraction of muscle fibres and subsequent cell necrosis (Wrogemann and Pena 1976). Interestingly, Wrogemann and colleagues were among the first to determine that calcium chelators such as EDTA could ameliorate the depressed respiration in dystrophic mitochondria, at least from skeletal muscle, by lowering mitochondrial calcium concentrations (Wrogemann et al., 1973). Although this finding was not originally established in dystrophic cardiac tissue, it is still a seminal finding that highlights the importance of intracellular calcium balance in dystrophic tissue and how calcium handling can be targeted in pursuit of a therapy.
Multiple mechanisms are believed to contribute to calcium overload and disrupted calcium homeostasis. Increased calcium influx and resting levels of mitochondrial calcium have been observed in myocytes from C57BL/10 mdx mice (Viola et al., 2013). In addition to the extracellular calcium that leaks through the sarcolemmal membrane tears, ion channels and calcium handling proteins likely contribute to the excessive calcium found in dystrophic myocytes. Stretch-activated calcium influx differences, increased diastolic calcium levels, altered calcium transient kinetics, and changes in calcium-handling protein expression, activation, or post-translational modifications have been observed in cardiomyocytes of C57BL/10 mdx mice (Yasuda et al., 2005; Williams et al., 2007; reviewed in Vallejo-Illarramendi et al., 2014; reviewed in Meyers and Townsend, 2019) (Figure 2).
Excessive cytoplasmic calcium may activate calcium-dependent proteases, such as calpains, contributing to apoptosis (Feng et al., 2001). A study investigating single ventricular myocytes from C57BL/10 mdx proposed that calcium-activated calpains degrade troponin I, resulting in contractile dysfunction and decreased calcium sensitivity (Williams and Allen, 2007). A separate study assessing calpains in 4-week-old C57BL/10 mdx mice determined that total concentrations of calpains were elevated in necrotic hind limb muscle from dystrophic mice compared to controls, however, further research on the heart is required (Spencer et al., 1995).
Another possible source of calcium overload in dystrophic cardiomyocytes is through transient receptor potential channels (TRPC) and the LTCC Cav1.2 (Johnstone et al., 2017; Esposito et al., 2019; Koenig et al., 2018). TRPCs are hypothesized to engage in the augmented stretch-activated cation influx seen in C57BL/10 mdx cardiomyocytes and overexpression of these channels has been observed in dystrophic cardiomyocytes (Johnstone et al., 2017; Allen and Whitehead, 2010; Lorin et al., 2015; Allen et al., 2005). A decrease in excessive calcium was demonstrated in C57BL/10 mdx cardiomyocytes using inhibitors of TRPC, stretch activated channels and transient receptor potential vanilloid (TRPV2) (Williams and Allen, 2007; Lorin et al., 2015). Redox modifications of Cav1.2 alpha1 subunit that occur during periods of elevated oxidative stress leads to increased channel-mediated calcium influx (Cserne et al., 2017; Muralidharan et al., 2017; Koenig et al., 2018). Resultantly, ROS may partly explain the gain-of-function calcium channel abnormalities observed in dystrophic cardiomyocytes (Koenig et al., 2018). Indeed, communication between LTCC and mitochondria has proven to be crucial in healthy cells for metabolic function and this communication was disrupted in C57BL/10 mdx cardiomyocytes (Viola et al., 2009; Viola et al., 2014). Studies in mice lacking either dystrophin alone or dystrophin and utrophin demonstrated increased calcium influx through the LTCC in cardiomyocytes (Williams and Allen, 2007; Lorin et al., 2015). The LTCC pathway to calcium overload is also suspected to play a role in the calcium-dependent arrythmias seen in DMD (Koenig et al., 2014).
RyR2 and sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA2a) may enhance the release of store-operated calcium in cardiomyocytes of DMD patients (Andersson and Marks, 2010; Fauconnier et al., 2010; Bellinger et al., 2009). It has been observed that in C57BL/10 mdx hearts, RyR2 levels are 2 to 3 times greater compared to those of wild-type (WT) mice (Williams and Allen, 2007). Also, protein kinase A (PKA)- or calcium/calmodulin-dependent protein kinase II (CaMKII)-mediated hyperphosphorylation and S-nitrosylation of RyR2 have been demonstrated in dystrophic cardiomyocytes and lead to dissociation from the stabilizing-protein calstabin2 and subsequent increased release of calcium from the SR (Andersson and Marks, 2010; Fauconnier et al., 2010; Bellinger et al., 2009; Shirokova et al., 2013). This is supported by studies demonstrating that agents blocking RyR2 phosphorylation result in normalization of cytosolic calcium levels as well as improvement in cardiac pathology and arrythmias (Fauconnier et al., 2010; Bellinger et al., 2009; Voit et al., 2017; Rohman et al., 2003). SERCA2a activity is significantly reduced in the C57BL/10 mdx dystrophic hearts due to lower SERCA2a expression and impaired regulation by inhibitory proteins phospholamban (PLN) and sarcolipin (Williams and Allen, 2007; Voit et al., 2017; Rohman et al., 2003). Phosphorylation of PLN via PKA or CaMKII is mostly regulated by β-adrenergic signaling (reviewed in Kranias and Hajjar, 2012; MacLennan and Kranias, 2003). Inositol trisphosphate (IP3) receptors, which are calcium release channels on the SR, are activated by the downstream product of phospholipase C (PLC) (Johnstone et al., 2017). PLC inhibitors have demonstrated a normalization of intracellular calcium levels in C57BL/10 mdx cardiomyocytes to WT levels (Mijares et al., 2014).
Sodium ions can also affect mitochondrial-cytoplasmic calcium exchange through the Na+-Ca2+ exchanger (NCX), which drives calcium out of the mitochondria in exchange for sodium when cytosolic sodium begins to accumulate as a result of dystrophin deficiency-induced microtears. A significant increase of cytosolic sodium levels (Kyrychenko et al., 2015) and elevated levels of NCLX (Na+/Li+/Ca2+) have been observed in cardiomyocytes of C57BL/10 mdx mice (Dubinin et al., 2020b). To decrease intracellular sodium and calcium overload caused by pathologic reversal of the NCX, a potent and selective sodium-proton exchanger isoform 1 (NHE-1) inhibitor known as rimeporide has been in development (Porte-Thomé et al., 2015; Previtali et al., 2020). NHE-1 is a membrane transporter responsible for catalyzing the electroneutral counter transport of sodium and hydrogen ions through the plasma membrane (Porte-Thomé et al., 2015). Interestingly, when rimeporide was tested on mdx mice, protective effects against inflammation and accumulation of fibrosis were observed in the heart, but additional work in this field to identify potential side-effects and dose-response curves is still required (Porte-Thomé et al., 2015). Additionally, previous work in gastrocnemius and longissimus dorsi muscles from 5- to 6-month-old C57BL/10 mdx mice demonstrated elevations in Na+/K+ ATPase content (Dunn et al., 1995). Albeit not in the heart, this further suggests that sodium regulation is abnormal in dystrophic muscle, and potentially precedes cell death via abnormal regulation of cell volume (Dunn et al., 1995) (Figure 1). Collectively, these findings highlight opportunities for further investigation into the potential role of ion (e.g. Na+, Ca2+, Li+, K+, H+) dysregulation as a prospective driver of mitochondrial stress responses.
Cardiac mitochondria do not typically act as a significant dynamic buffer of cytosolic calcium in healthy hearts; however, prolonged elevations of intracellular calcium substantially enhance mitochondrial calcium uptake (Boyman et al., 2014; Williams et al., 2013). Recently, increased calcium uptake has been observed in the mitochondria of C57BL/10 mdx skeletal muscle likely due to enhanced expression of the channel-forming MCU subunit and reduced expression of dominant-negative MCUb subunit (Dubinin et al., 2020b). While the precise mechanisms regulating excess mitochondrial calcium uptake requires more investigation, current theories posit that calcium overload triggers cell death through mitochondrial permeability transition pore activity in conjunction with impaired oxidative phosphorylation and elevated ROS production as described below.
3.3. Mitochondrial permeability transition pore and apoptosis
Mitochondrial permeability transition pore (PTP) formation is an event that fuses the inner (IMM) and outer mitochondrial membrane (OMM) while permeabilizing the inner membrane. Recent evidence supports a model whereby ATP synthase components dimerize to form the mPTP in response to reactive oxygen species and excess calcium stress (Bernardi et al., 2022; Reviewed in Giorgio et al., 2018) which then leads to depolarization, a loss of ATP synthesis, and rapid mitochondrial calcium release to the cytosol through an event known as permeability transition (PT). Following subsequent release of cytochrome c, caspases 9/3 are activated which activate proteases that contribute to apoptosis (Budihardjo et al., 1999) (Figure 2).
Myotubes cultured from C57BL/10 mdx demonstrate ~10-fold higher calcium in the matrix compared to the cytosol (Robert et al., 2001). Calcium-induced PT was elevated following ischaemia reperfusion in young C57BL/10 mdx hearts and was accompanied by the release of proapoptotic factors (including cytochrome c into the cytosolic fraction) as well as enhanced activities of caspases 9/3 prior to the onset of cardiac dysfunction (Ascah et al., 2011; reviewed in Burelle et al., 2010) (Figure 2). In LV of 4-week-old D2.mdx mice, no differences in calcium-induced PT or caspases 9/3 activities were observed (Hughes et al., 2020) in contrast to increases in both measures seen in certain skeletal muscles (Hughes et al., 2019). As there were no reductions in ejection fraction at this young age, the study concluded that mitochondria do not demonstrate PT in LV in early-stage disease. A separate study conducted on 7-month-old C57BL/10 mdx mice using mitochondria isolated from whole hearts demonstrated a greater propensity for calcium-induced PT (Stevens et al., 2024). Collectively, these findings suggest that introducing physiological stressors such as ischaemia reperfusion or assessing more advanced disease states may be required to reveal an underlying mitochondrial propensity for PT, although the degree to which PT exists across specific regions of the heart is unknown. The contributions of PT to cardiomyopathy at specific stages of disease require further research.
It has been proposed that different analytical interpretations can be associated with the specific technique that is used to determine and quantify PT. Techniques include quantifying the concentration of calcium required to trigger its opening via bolus titrations in a permeabilized fiber system, versus the elapsed time that is required to trigger this opening in response to a single bolus of calcium. To this end, differing conclusions from studies have led to the belief that disease effects on time may not be mirrored by changes in total calcium uptake by PT (proposed and reviewed in Bellissimo et al., 2022). In one such study, both approaches were compared and demonstrated no change in total calcium uptake despite lower time for calcium to trigger PT in C57BL/10 mdx mice (Ascah et al., 2011). This finding implies that a greater propensity for calcium-induced PT may be due to greater velocities of calcium uptake independent of potential differences in total uptake. Such methodological considerations could be considered when assessing PT in relation to cardiomyopathy in mdx models.
Although this has not been studied extensively in the heart, pharmacological inhibitors of mPTP in skeletal muscle have shown promise by improving indices of muscle function in C57BL/10 mdx mice (reviewed in Bellissimo et al., 2022; Millay et al., 2008; De Luca et al., 2005). Whether this can be attributed to the inhibitory effects on PT itself or the indirect benefit of immunosuppression is still a contentious topic. This is important given that immunosuppressive glucocorticoids have been known to suppress PT in C57BL/10 mdx skeletal muscle (Dubinin et al., 2020a).
Overall, the specific importance and role of PT-induced cell death in dystrophic cardiomyopathy is still not fully understood and requires more research. Insight may be gained by considering the effects of mPTP inhibitors in skeletal muscle of mdx mouse models. For example, Debio 025 (d-MeAla3EtVal4-cyclosporin), an inhibitor of the mPTP regulator cyclophilin, showed beneficial roles in locomotor and respiratory muscles of 3-week-old mdx5Cv mice (Reutenauer et al., 2008; Wissing et al., 2010) but had no effect on echocardiograph assessments of heart function or fibrosis in 18-week-old D2.mdx mice (Hayes et al., 2022). Alisporivir, another cyclophilin inhibitor, is a cyclosporin A derivative that desensitizes the PTP without inhibiting calcineurin (Schiavone et al., 2017). Albeit not in the heart, treatment of alisporivir in primary cultures obtained from muscle biopsies of DMD patients demonstrated that this compound could restore the maximal respiratory capacity in dystrophic muscle cells without interfering with basal oxygen consumption, therefore restoring physiological respiratory reserve (Schiavone et al., 2017). In addition, treatment with alisporivir also recovered respiratory function, which matched improvements to muscle ultrastructure and survival in the sapje zebrafish model of DMD (Schiavone et al., 2017). TR001, which is a metabolically stable triazole analog and inhibitor of mPTP improved markers of motility 6 days post-fertilization when administered in sapje zebrafish (Stocco et al., 2021). In addition to improving muscle structure and function recovery, as well as mitochondrial respiration and survival in these zebrafish, TR001 also improved respiration in skeletal muscle-derived myoblasts and myotubes from DMD patients (Stocco et al., 2021). The promise and efficacy of mPTP inhibitor treatment modalities on dystrophic locomotor and respiratory muscle in various pre-clinical DMD models supports the need for these compounds to be tested on cardiac tissue and/or cardiomyocytes.
3.4. Oxidative phosphorylation and substrate catabolism
Mitochondrial ATP synthesis occurs predominantly through oxidative phosphorylation within the electron transport chain (ETC), a process vital for the energy-demanding heart. In healthy mitochondria, this process depends upon the governance of membrane potential across the IMM and is stimulated by temporary elevations to mitochondrial matrix calcium caused by contraction (Glancy et al., 2012; Nicholls and Ferguson, 2013). Early identification of mitochondrial deficits in muscular dystrophies using direct assessments of respirometry in isolated mitochondria in people with DMD were reported in 1967 albeit in skeletal muscle (Ionăsescu et al., 1967). Other reports around this time focused on skeletal muscle animal models of undefined muscular dystrophies (Wrogemann and Blanchaer 1967; Wrogemann and Blanchaer 1968; Wrogemann et al., 1970; Wrogemann et al., 1973) prior to the identification of the mdx mouse (Bulfield et al., 1984). Numerous factors need to be considered when assessing the regulation of mitochondrial oxidative phosphorylation in muscle, including phenotype severity (due to the progressive nature of dystrophin deficiency), and that not all mitochondrial protein markers uniformly change across a spectrum of muscle-types, models, or age ranges (reviewed in Bellissimo et al., 2023). This is important to note because mitochondrial protein content should not always be interpreted as being reflective of mitochondrial function (discussed in Bellissimo et al., 2023).
While no study has performed functional assessments of cardiac mitochondrial oxidative phosphorylation in people with DMD, one study found iPSC-derived cardiomyocytes from adults with DMD had significant reductions oxygen consumption coupled to ATP synthesis (Willi et al., 2022). Attenuations in the phosphocreatine (PCr) energy system indicated by lower PCr concentrations were also observed in these cells (Willi et al., 2022). In the C57BL/10 mdx mouse, studies have shown significant reductions in the ratio of cytoplasmic PCr to ATP ratio which reflects a compromised ability to maintain energy homeostasis prior to the development of cardiac fibrosis (Cui et al., 2015). This observation is consistent with many reports of attenuated mitochondrial respiration in dystrophin deficient heart (see Bellissimo et al., 2022for review).
Of interest, mitochondria can also export phosphocreatine as an alternative to ATP (reviewed in Schlattner et al., 2018; Guzun et al., 2012). Regulated by mitochondrial creatine kinase, this model posits that PCr is recycled back to ATP by local ATP-hydrolyzing proteins through cytoplasmic creatine kinases. As PCr diffuses at a rate several fold faster than ATP, with the creatine product returning to mitochondria ~2,000 fold faster than ADP, this fast phosphate shuttling mechanism may be advantageous to maintaining energy homeostasis in cells that can experience very high rates of demand for ATP, such as the heart (Meyer et al., 1984; reviewed in Schlattner et al., 2018; and Wallimann et al., 2011). One study found that this faster creatine-dependent phosphate shuttling mechanism was more attenuated than the direct ATP export system when assessed with respirometry in permeabilized muscle fibres from the LV of 4-week-old D2.mdx mice (Hughes et al., 2020) which has also been reported in skeletal muscle (Hughes et al., 2019; Bellissimo et al., 2023). Such ‘creatine insensitivity’ occurring early in the disease was suggested to be a potential unique mechanism preceding the eventual development of cardiomyopathy.
Additional work is required to address the fates of glucose and other substrates being utilized in dystrophic cardiac tissue. Whether reductions in mitochondrial oxidative phosphorylation and elevations to fat oxidation occur concurrently, and should be considered ‘mitochondrial dysfunctions’, are still questions remaining to be answered. For this reason, when assessing new therapeutic avenues, careful consideration should be given to designing experiments that consider the role of mitochondrial oxidative phosphorylation as part of an integrated perspective on substrate fates (reviewed in Bellissimo et al., 2022).
A variety of other metabolic changes occur in the dystrophic heart. For example, a shift in metabolism from fatty acid to carbohydrate oxidation was observed in vivo in C57BL/10 mdx hearts prior to cardiomyopathy, suggesting that altered mitochondrial metabolism is one of the first metabolic changes to occur (Khairallah et al., 2007). In ex vivo perfused C57BL/10 mdx hearts, a shift in substrate selection from long chain fatty acids (LCFA) to carbohydrate (glucose) oxidation in C57BL/10 mdx hearts was demonstrated by a ~30% lower oleate flux ratio, ~120% higher pyruvate decarboxylation flux ratio, and ~80% increased glycolysis rate (Khairallah et al., 2007). This shift is supported by evidence from cardiac positron emission tomography (PET) studies in DMD patients using 18F-deoxyglucose or a radioiodinated branched fatty acid (Krumenacker et al., 2001; Momse et al., 2001; Naruse et al., 2004; Perloff et al., 1984). On the note of carbohydrate oxidation, glucose transporter type 4 (GLUT4) is the main transporter of glucose in cardiomyocytes (Zorzano et al., 1997; Fischer et al., 1997) and glucose transport into muscle cells in response to insulin or contraction occurs via translocation of GLUT4 from the cytoplasm to the sarcolemma/T-tubules (Birnbaum, 1989; Fischer et al., 1997). GLUT4 was observed to be abnormally localized in the sarcolemmal membrane and a trend toward elevated GLUT4 expression was observed in skeletal muscle of GRMD, but not in the LV (Schneider et al., 2018). Primary cardiac insulin resistance was also observed in the LV of GRMD (Nikolaidis et al., 2004). Together, this data suggests that glucose/carbohydrate metabolism can potentially be used as a preclinical biomarker of the dystrophic phenotype in skeletal muscle of GRMD dogs, but whether this can be implemented in cardiac tissue remains to be determined (Schneider et al., 2018).
Interventions that correct the substrate imbalance may lead to improvements or recovery of the cardiac contractile dysfunction (reviewed in Glatz et al., 2020). Improved contractile performance was observed in C57BL/10 mdx hearts perfused with multiple substrates, including carbohydrates (glucose, lactate, and pyruvate) and a LCFA (oleate), in comparison to C57BL/10 mdx hearts with glucose as the sole substrate (Burelle et al., 2010; Danialou et al., 2001). Another potential explanation for substrate shift is the p38 mitogen-activated protein kinase (MAPK) pathway, which is involved through the nuclear receptor peroxisome proliferator-activated receptor (PPARα), a transcriptional regulator of fatty acid oxidation enzyme expression (Khairallah et al., 2007). A 2-fold decrease in p38 MAPK phosphorylation may account for the decrease in LCFA oxidation observed in the C57BL/10 mdx heart (Burelle et al., 2010). This emphasizes the importance of increased and balanced substrate supply to maintain adequate cellular energy in the C57BL/10 mdx heart.
It is also possible that certain metabolic stress responses are compensatory in nature. For instance, a shift from fat to carbohydrate oxidation could be beneficial given carbohydrate oxidation produces more adenosine triphosphate (ATP) for a given amount of oxygen consumption (Burkhoff et al., 1991; reviewed in Stanley et al., 2005). However, in DMD, competition for glucose, fatty acids, and proteins may exist for uses unrelated to bioenergetics, such as for membrane repair, as reviewed elsewhere (Bellissimo et al., 2023). Interestingly, greater levels of fatty acid biosynthesis enzymes that convert fatty acid constituents to acetyl-CoA for the tricarboxylic (TCA) cycle were observed in skeletal muscle from people with DMD (Capitanio et al., 2020). Despite this finding, decreases in TCA pool size and lower aconitase activity were found in C57BL/10 mdx hearts (Khairallah et al., 2007, Griffin et al., 2009) which could limit oxidative phosphorylation. Likewise, lower mitochondrial respiratory sensitivity to ADP, reduced mitochondrial creatine metabolism, and attenuated complex I activity were reported in LV of 4-week-old D2.mdx mice (Hughes et al., 2020). Some of these metabolic changes (Khairallah et al., 2007, Griffin et al., 2009, Hughes et al., 2020) preceded the development of overt cardiac dysfunction, suggesting metabolic stress could be an initial event in the disorder. This will be explored extensively in section 3.6.
The relationship of such mitochondrial reprogramming and/or deficiencies to impaired nitric oxide (NO)/cyclic guanosine monophosphate (cGMP) signaling is relatively understudied in DMD, but several reports supporting continued investigation in this area are warranted. NO/cGMP is believed to regulate nuclear gene expression supporting mitochondrial biogenesis (Nisoli et al, 2004). Increased expression of the atrial natriuretic factor (anf) gene, an activator of the NO/cGMP signaling pathway and marker of cardiac remodeling, was observed in C57BL/10 mdx hearts at 10-12 weeks of age (Khairallah et al., 2007). Increased levels of the alpha unit of soluble guanylate cyclase (sgcα1) gene were observed in hearts of 25-week-old mice, a gene which typically negatively correlates with NO/cGMP (Krumenacker et al., 2004). These results suggest that an increase in anf expression may compensate for a defective NO/cGMP pathway in young mice, but this adaptive mechanism appears to diminish with age (Khairallah et al., 2007; Krumenacker et al., 2004). However, despite lower nNOS protein content, a study examining skeletal muscle in C57BL/10 mdx reported that nitrate supplementation (an approach used to increase NO) did not rescue mitochondrial oxidative phosphorylation, increased mitochondrial peroxynitrite, and promoted muscle damage. Collectively, these data suggest that a defect in the NO/cGMP signaling pathway potentially contributes to the metabolic abnormalities in the heart (Khairallah et al., 2007; Burelle et al., 2010) but negative effects of nitrate supplementation in skeletal muscle identify an incomplete understanding of this pathway’s role in contributing to mitochondrial dysfunction in DMD.
Collectively, the regulation of mitochondrial substrate oxidation is altered in the heart during DMD. However, the degree to which such stress responses contribute to cardiomyopathy or serve as an intentional reprogramming to permit alternative fates of glucose or fatty acid substrates requires considerable attention given the latter possibility has rarely been considered (proposed in Bellissimo et al., 2022).
3.5. Reactive oxygen species (ROS) and mitochondrial H2O2 emission
Many studies measuring ROS in mdx models used relatively non-specific fluorophores that do not permit definitive conclusions regarding the precise subcellular origin or type of ROS assessed (reviewed in Bellissimo et al., 2022). In this regard, very few studies have used methodologies that isolated the source of ROS from mitochondria or other sources.
Increased pyruvate-stimulated, complex I-supported mH2O2 emission was observed during attenuated oxidative phosphorylation in permeabilized LV cardiac muscle fibres of dystrophin-deficient mice due to a reduced ability of ADP to suppress mH2O2, as normally occurs during oxidative phosphorylation (Nicholls and Ferguson, 2013). As creatine normally accelerates mitochondrial ADP/ATP cycling (reviewed in Schlattner et al., 2018), and has been shown to enhance ADP’s ability to attenuate mH2O2 (Meyer et al., 2006), the additional finding that mitochondrial ADP attenuation of mH2O2 was no longer sensitive in D2.mdx LV fibres suggests that mitochondrial creatine kinase may represent a unique mechanism of mitochondrial dysfunction in dystrophin deficient hearts (Hughes et al., 2020). The precise mechanisms by which this elevation in mH2O2/O2 may contribute to cardiac dysfunction remains unknown, particularly given there were no changes in the glutathione equilibrium (Hughes et al., 2020 or the ability of mitochondria to scavenge H2O2 in C57BL/10 mdx hearts (Ascah et al., 2011), but such elevations in mH2O2/O2 occurred during a time of apparent cardiac compensations as mice showed slight elevations in ejection fraction albeit before overt cardiac dysfunction (Hughes et al., 2020).
Similar observations were made in isolated mitochondria from mixed hearts (rather than a specific chamber) in the C57BL/10.mdx mouse also at 4 weeks of age (Dubinin et al., 2020b) as well as skeletal muscle of D2.mdx mice (Hughes et al., 2019; Ramos et al., 2020; Bellissimo et al., 2023). While various antioxidants that do not target mitochondria per se have been tested in C57BL/10 mdx mice for their potential cardioprotective properties, and have demonstrated discordant results (Gartz et al., 2022), to our knowledge, there are no studies assessing the effects of mitochondrial-targeted antioxidants on mitochondrial bioenergetics and cardiac function in mdx models.
The precise signaling mechanisms downstream of mH2O2 that may mediate cardiac dysfunction in dystrophin deficient hearts have not been explored extensively in mdx mice or DMD patients. Understanding how mitochondrial-derived ROS contributes to cardiac dysfunction with consideration of ROS-sensitive targets mediating cell membrane injury, metabolic dysfunction, calcium dysregulation, and cardiomyocyte degeneration could guide the development of ROS-specific targeted therapies.
The superoxide-generating enzyme Nicotinamide Adenine Dinucleotide Phosphate (NADPH) is a major source of non-mitochondrial ROS in skeletal and cardiac muscle (Prosser et al., 2011; Gonzalez et al., 2014), especially during mechanical stress and at early stages of disorder progression (Shirokova et al., 2013; Kyrychenko et al., 2015) (Figure 2). NOX2-mediated ROS generation is significantly increased in C57BL/10mdx hearts (Prosser et al., 2011; Khairallah et al., 2012; Williams and Allen, 2007; Gonzalez et al., 2014), which has been attributed to altered microtubule association with NOX2 (Khairallah et al., 2012; Prosser et al., 2013). Inhibiting NOX2-derived ROS (Jung et al., 2007; Kyrychenko et al., 2013) or using the non-specific ROS scavenger N-acetylcysteine (NAC) (Prosser et al., 2011; Williams and Allen, 2007) restored calcium homeostasis in dystrophin-deficient muscle. NOX2 inhibition with the drug apocynin improved single sarcomeric shortening in isolated cardiomyocytes (Gonzalez et al., 2014) while NAC improved fractional shortening (Williams and Allen, 2007). However, the therapeutic potential of NAC is uncertain given it also reduces muscle weights in C57BL10/mdx mice (Pinniger et al., 2017) although the degree to which this occurred because of attenuated superoxide is not clear.
3.6. Altered mitochondrial autophagy (mitophagy)
In healthy tissue, defective mitochondria can be removed through mitochondrial autophagy, also known as mitophagy, in a process intended to uphold quality control of the healthy mitochondrial pool. This process mitigates damage that may be caused by dysfunctional mitochondria (Reid and Alexander, 2021) to maintain energy metabolism stability (Li et al., 2020; reviewed in Eisner et al., 2017). Mitophagy is regulated by PTEN-induced kinase 1 (PINK1) and Parkinson juvenile disease protein 2, (PARKIN) (reviewed in Bellissimo et al., 2023). In a damaged mitochondrion, PINK1 is not degraded and is able to phosphorylate mitofusin 2 (Mfn2), which subsequently signals PARKIN to tag the mitochondria for degradation (Reid and Alexander, 2021; Kang et al., 2018). Important mitophagy-related genes, including PINK1, PARK2, and BNIP3 were decreased in DMD patients (dystrophic quadriceps) and dystrophin-deficient rodent models (12-month-old C57BL/10 mdx mice) (Luan et al., 2021; Kang et al., 2018). Improving mitophagy has shown promise in the diaphragm of C57BL/10 mdx mice (Pauly et al., 2012), but further research on the heart is required to elucidate if PINK/PARKIN pathways represent potential therapeutic targets.
Given that loss of PINK1 increases the vulnerability of the heart to IR-injury, it appears to possess some cardioprotective properties (Kang et al., 2018). A study by Kuno et al., (2018) revealed that the co-localization of LC3 dots with fragmented mitochondria was significantly increased in hearts of 22-week-old C57BL/10 mdx mice compared to healthy controls, indicating that the elimination of damaged mitochondria via mitophagy was impaired and subsequent accumulation of damaged mitochondria persisted. Other studies in skeletal muscle from C57BL/10 mdx mice and DMD patients demonstrated significantly reduced levels of autophagy (De Palma et al., 2012), suggesting that the suppression of autophagy may be muscle-type dependent. Further, mitophagy is a dynamic process and therefore, changes to protein content and gene expression of common mitophagic markers may not always be proportional to mitophagic activity and/or flux. It is essential to understand phenotype severity, muscle-type, and age when considering mitophagy as a therapeutic target. The majority of research to date investigating the role of mitophagy in dystrophin deficient models has been conducted in skeletal muscle, thus, advancements in cardiac muscle remain to be addressed.
3.7. Altered mitochondrial content and structure
iPSC-cardiomyocytes from DMD patients possess increased mitochondria with abnormal morphologies (Willi et al., 2022). Degeneration of mitochondrial structure (swelling and loss of cristae) has been identified in dystrophic cardiomyocytes from hearts of 1-month-old and 3-4-month-old mdx mice prior to disorder onset (Kyrychenko et al., 2015). Furthermore, a significant increase in structurally abnormal mitochondria were observed in hearts of 12-month and older C57BL/10 mdx mice after developing DCM (Kang et al., 2018) which has also been reported as early as 4 weeks of age in this model (Dubinin et al., 2020b). Various mitochondrial proteins contents have been assessed in mdx mouse models with divergent responses observed depending on the pathway, age and model suggesting that altered control of oxidative phosphorylation could occur through post-translational modifications that have yet to be fully identified, and that compensations in the content of certain pathways may occurs (Hughes et al., 2020; Ascah et al., 2011 as reviewed in Bellissimo et al., 2022).
4. Current interventional targets and approved non-genetic therapies for cardiomyopathy in DMD
No cure exists for DMD or its associated cardiac dysfunction, and therefore, most treatments are aimed at treating the symptoms and delaying the progression of dystrophic cardiomyopathy. Unfortunately, cardiac transplantation is generally contraindicated in DMD patients due to muscular weakness and respiratory insufficiency (Connuck et al., 2008). Left ventricular assist devices (LVAD), if implemented as a destination therapy rather than a bridge to transplantation, can be considered as a therapeutic option in DMD patients (Rose et al., 2001). While novel therapies have been in development to specifically address defects resulting from dystrophin deficiency, their effectiveness with respect to cardiac function has been limited due to the diverse range of genetic mutations in DMD and the complex risks of each prospective therapy (reviewed in Shah and Yokota 2023). For example, certain small molecule-based therapies used in practice or under development target secondary cellular dysfunctions that are common in most if not all DMD patients and therefore have potential for widespread use. Such therapies tend to focus on symptom management and delaying development of cardiomyopathy and heart failure. Other gene-based treatments focus on restoring dystrophin expression in a truncated or complete sequence must be customized to each unique mutation that occurs in DMD.
4.1. Glucocorticoid therapy
The most widely used intervention in DMD is corticosteroids, which have proven to reduce inflammation, prolong strength, and delay loss of ambulation by 1 to 3 years in DMD patients (Escolar et al., 2011). However, beneficial effects of chronic steroids in the heart are less established. Therapy most often involves daily dosing of either prednisone or deflazacort initiated around 2 to 5 years of age (Flanigan, 2017). Although it is unknown how steroids delay DMD progression, the following mechanisms are suspected to contribute: reduced cytokine production, activation of insulin-like growth factors, increased myoblast proliferation, decreased lymphocyte reaction, and upregulation of synergistic molecules (Angelini and Peterle, 2012). Cardioprotective mechanisms in boys with DMD may include cell membrane stabilization, decreased myocardial inflammation and fibrosis, and improvement in skeletal muscle function leading to secondary cardiac effects (Barber et al., 2013). Steroids also activate mineralocorticoid receptors which likely causes adverse effects, including reduced bone density, obesity, hypertension, adrenal insufficiency, and increased muscle catabolism (Hoffman et al., 2012; Bylo et al., 2020; Liu et al., 2013). This will be discussed in detail later. Recent studies have suggested that pulsed steroid regimens may limit the adverse effects while maintaining the benefits of daily steroid dosing in DMD patients (Connolly et al., 2019; Quattrocelli et al., 2019). Preclinical studies have demonstrated that steroids may worsen the progression of cardiomyopathy in the heart of C57BL/10 mdx mice with findings including decreased cardiac function, increased dilation, and increased cardiac fibrosis (Janssen et al., 2014; Bauer et al., 2009; Guerron et al., 2010), consistent with similar findings in the diaphragm (Howard et al., 2022). It should be noted that these studies used a more continuous method of drug delivery than is equivalent to the single daily dose therapies administered in DMD patients (Spurney et al., 2011).
Studies in DMD patients collectively revealed the protective benefits of steroid treatment in dystrophic hearts, including decreased fibrosis, preserved ventricular function, and better survival (Schram et al., 2013; Markham et al., 2005; Barber et al., 2013; Silversides et al., 2003; Raman et al., 2015; Spurney et al., 2011). When steroid therapy is initiated prior to the onset of cardiomyopathy, delayed development of cardiac dysfunction has been observed (Markham et al., 2008). In fact, a delay in the onset of cardiomyopathy by 4% for each year of steroid use has been observed in the setting of chronic steroid use (Barber et al., 2013). Data from a surveillance program demonstrated that longer duration of steroid use correlates with a greater improvement in LV function (Barber et al., 2013). Furthermore, the duration of steroid treatment has been proven to be inversely correlated with the incidence of cardiomyopathy in DMD patients (Barber et al., 2013). Long-term steroid treatment beyond loss of ambulation in DMD led to improvement in all-cause mortality secondary to improved cardiac outcome (Schram et al., 2013). Improvement in LVEF and FS was demonstrated in DMD patients treated with steroids (Markham et al., 2008; Houe et al., 2008). On a molecular level, a study in 7-week-old C57BL/10 mdx mice demonstrated that glucocorticoids prevented calcium-induced mPTP opening and restored ADP-stimulated respiration as the result of enhanced expression of Complex III, Complex IV, and IV protein content markers in skeletal muscle (Dubinin et al., 2020a), but further work on cardiac muscle is required in this area.
4.2. Angiotensin-inhibiting therapies
The goal of blocking the renin-angiotensin system is to improve the adverse remodeling that results from cardiomyocyte loss. Angiotensin II and angiotensin II type 1 receptor induce many harmful cardiac effects, including increased fibrosis, remodeling, ROS production, and cardiomyocyte death (Dikalov and Nazarewicz, 2013; Dasgupta and Zhang, 2011; Kawai et al., 2017). The two main angiotensin-inhibiting drug classes used in heart failure and DMD patients are angiotensin receptor blockers (ARBs) and angiotensin converting enzyme inhibitors (ACEIs). ACEIs prevent an enzyme from converting angiotensin I to angiotensin II, which decreases the activation of genes that enhance fibrosis and scarring of the myocardium (Greenberg et al., 1999), and block the action of angiotensin II, allowing veins and arteries to dilate.
Based on findings that the first clinical signs of cardiac involvement appear around 6 to 10 years of age, current guidelines advise that DMD patients begin an angiotensin-inhibiting agent by age 10 or earlier in the asymptomatic patient (Birnkrant et al., 2018). Since ACEIs are used as the first line of therapy for general heart failure and have been studied more extensively, these agents are typically the first recommended drug for the treatment of cardiac dysfunction in DMD patients (Kaspar et al., 2009). In fact, ACEIs were the first class of drugs to improve cardiac function in DMD patients in clinical trials (Duboc et al., 2005; Duboc et al., 2007). For example, the onset and progression of LV dysfunction was delayed, and survival rate improved in DMD patients when treated early with the ACEI perindopril (Duboc et al., 2005; Duboc et al., 2007). In addition, improvement in LV function was reported with use of ACEI alone or in combination with a beta-blocker in DMD patients (Viollet et al., 2012).
Despite being equally as effective and better tolerated than ACEIs (Allen et al., 2013), ARBs are an alternative treatment for cardiac dysfunction in DMD patients and typically reserved for patients who do not tolerate ACEIs (Birnkrant et al., 2018). In C57BL/10 mdx mice with pharmacologically induced myocardial injury, treatment with the ARB, Losartan, ameliorated cardiac injury (Meyers Heitzman, Krebsbach et al., 2019). Losartan reduced cardiac damage 2.8-fold in C57BL/10 mdx hearts without injury on WT hearts, but ACEI treatment had no effect on the myocardial injury (Meyers, Heitzman, and Townsend, 2019). This reveals that direct blockade of the angiotensin II type 1 receptor may be necessary in the dystrophic heart (Meyers, Heitzman, and Townsend, 2019).
4.3. Beta-adrenergic receptor blockers
The cardiac dysfunction in DMD is typically characterized by tachycardia (Perloff et al., 1967). Activation of β-adrenergic receptors enhances heart rate elevations and increases contractility through its action on the calcium transients in the cardiomyocyte (Wagner and Maier, 2015). Beta-blockers may limit these adverse effects through inhibition of β-receptor binding and subsequent catecholamine binding (Wagner and Maier, 2015). The latest guidelines recommend considering use of a beta-blocker at the onset of tachycardia or ventricular dysfunction (Birnkrant et al., 2018). Beta-blockers are considered a second-line candidate for cardiac-related therapy in DMD patients, typically administered in addition to an angiotensin-inhibiting agent (Viollet et al., 2012; reviewed in McNally et al., 2015). Its benefit in this patient population is unclear and not all beta-blockers have a similar efficacy in DMD (Schultz et al., 2022; reviewed in McNally et al., 2015). For example, administration of metoprolol to C57BL/10 mdx mice with early cardiomyopathy led to worsening right ventricular ejection fraction (RVEF) with no effect on myocardial calcium influx (Blain et al., 2013). Carvedilol administered for 6 months improved LVEF and decreased the incidence of ventricular tachycardia in DMD patients (Rhodes et al., 2008). Significantly higher survival rates and reduction in severe cardiac events were observed in DMD patients treated with carvedilol over 5 years (Matsumura et al., 2010). Furthermore, a prospective open study demonstrated no difference between subjects receiving ACEIs alone versus ACEIs with beta-blockers (Viollet et al., 2012). However, other trials have shown that this combination therapy improves LV function (Jefferies et al., 2005; Kajimoto et al., 2006), prevention of major cardiac events (Matsumura et al., 2010), and long-term survival (Ogata et al., 2009) in comparison to ACEIs alone. Beta-blockers administered to DMD human iPSC-derived cardiomyocytes decreased the incidence of arrhythmogenesis (Kamdar et al., 2020). Since beta-blockers are second-line agents, there is difficulty in determining their benefit in DMD patient as their use is often delayed and used in combination with a first-line therapy (Viollet et al., 2012; reviewed in McNally et al., 2015).
Systolic blood pressure is typically lower in DMD patients, which complicates the use of these and other standard heart failure medications (McNally et al., 2015). There is, however, a cross-sectional study which showed increased systolic and diastolic BP, and heart rate in adolescents with DMD in response to an active sitting test that promoted reduced autonomic modulation (Rodrigues et al., 2021). Of note, it was observed that infusion of the β-1 adrenergic receptor agonist dobutamine in 7-month-old C57BL/10 mdx Langendorff perfused hearts resulted in significantly lower peak systolic pressure, developed pressure, and dP/dtMAX compared to control mice (Stevens et al., 2024).
4.4. Mineralocorticoid receptor antagonists
Aldosterone activates the mineralocorticoid receptor (MR), resulting in cardiomyocyte death, hypertrophy, and fibrosis in DMD cardiomyopathy (Raman et al., 2015). MR antagonists, such as eplerenone and spironolactone, are treatments for heart failure with reduced LVEF and are used in some cases of DMD cardiomyopathy (Kamdar and Garry, 2016). Myeloid cells that infiltrate the heart in dystrophic mice express aldosterone synthase, which may increase the injury and subsequent remodeling (Chadwick et al., 2016). Corticosteroids, previously mentioned as common first-line interventions among DMD patients, activate MR as well as their target receptor (Heier et al., 2019). Administration of spironolactone and ACEIs together demonstrated a protective effect in mice models of DMD cardiomyopathy. However, this effect lessened with age and reiterated the importance of early initiation of treatment (Rafael-Fortney et al., 2011; Janssen et al., 2014). Spironolactone and eplerenone have been observed to be of equal efficacy in the DMD population (Raman et al., 2019). Clinical trials involving patients with preserved ejection fraction (EF) on an angiotensin-limiting drug prior to and during the trial revealed marked improvements in myocardial strain, EF, and chamber dilation with eplerenone treatment (Raman et al., 2015b). The open label extension of this trial demonstrated preservation of the myocardial circumferential strain over 36 months while being treated with an angiotensin-limiting drug and eplerenone (Raman et al., 2017). Vamorolone, an MR antagonist that matches the potency of eplerenone and possesses anti-inflammatory benefits similar to corticosteroids, was recently approved in the USA for DMD (Guglieri et al., 2022).
4.5. Gene-targeted therapies
Gene-targeted therapies for DMD aim to restore normal myocyte function through production of a functional dystrophin gene product (Shah and Yokota, 2023). Although gene therapy is an attractive technology, these complex therapies pose many challenges to achieving appropriate and lasting correction. While the impact of many of these therapies has been examined in skeletal muscle, assessing the benefits of these approaches on cardiac outcomes has proven difficult (reviewed in Shah and Yokota, 2023, and Meyers and Townsend, 2019). Restoring dystrophin in skeletal muscle without addressing the cardiomyopathy could lead to increased cardiac demand, exacerbate cardiomyopathy, and accelerate development of heart failure (reviewed in Meyers and Townsend, 2019; Townsend et al., 2008). Gene-targeted therapies, including stop codon readthrough, antisense oligonucleotides (AONs), viral gene therapy, and clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (Cas9) gene editing, have been thoroughly reviewed elsewhere (reviewed in Shah and Yokota 2023; and Meyers and Townsend, 2019) and will therefore only be briefly discussed here.
Stop codon readthrough therapy, such as Ataluren which is approved in Europe, involves binding to ribosomal RNA and bypassing the premature stop codon, typically resulting in partial dystrophin expression in skeletal muscle (Namgoog and Bertoni, 2016). However, this type of treatment would only be useful in about 10% of DMD cases (i.e. those with nonsense mutations) (Bladen et al., 2015) and studies have demonstrated no effect on cardiac function with Ataluren administration (Ebrahimi-Fakhari et al., 2018).
In turn, AONs, which can restore the reading frame to generate a truncated yet functional protein (Piga et al., 2019), are useful in the case of deletions. Although approximately 80% of DMD patients have mutations that may be amenable to this approach, AONs can only target one exon at a time. In addition, while AONs were observed to restore dystrophin expression in skeletal muscles in preclinical studies, negligible effects on dystrophin levels were demonstrated in the heart (Lu et al., 2005; Yokota et al., 2009). Peptide-conjugated phosphorodiamidate morpholino oligomer (PPMO) delivery of AONs, which facilitates the delivery of AONs across the plasma membrane, has demonstrated substantial expression of dystrophin in the hearts of DMD animal models (Yin et al., 2008). The AONs Eteplirsen, Golodirsen, Viltolarsen, and Casimersen are FDA-approved based on modest levels of dystrophin restoration rather than on functional outcomes (Duan et al., 2021; Johnston et al., 2021).
Viral gene therapy in DMD patients aims to replace or repair defective dystrophin with micro-dystrophin, which is one-third the size of the full-length dystrophin protein (Chamberlain and Chamberlain, 2017; Hakim et al., 2018). The adeno-associated virus (AAV) is currently the only gene therapy vector that has been considered due to the vector’s capability for increasing dystrophin in cardiac and skeletal muscles (Hakim et al., 2018; Duan, 2016). Elevidys, the first and only FDA-approved gene therapy for DMD, uses a MHCK7 promoter and has demonstrated expression of micro-dystrophin in both skeletal and cardiac muscle in animal models (Potter et al., 2021). The MHCK7 promoter has been shown to have higher cardiac expression in mice (Potter et al.,2021; Salva et al., 2007). This is due to the inclusion of the alpha myosin heavy chain enhancer. However, to our knowledge, there is no data demonstrating that the MHCK7 promoter is expressed in human ventricles and thus the efficacy of this therapy has not been fully established. Human atria do express the MHCK7 promoter, but it is unknown how this expression pattern of micro-dystrophin affects conductivity and cardiac function. Multiple micro-dystrophin constructs are also in development, including the Pfizer vector, which employs a skeletal muscle promoter, and the CK8 promoter in the Solid Biosciences construct (Duan, 2018; Salva et al., 2007; Hakim et al., 2017). The impact of both Elevidys and the constructs in development on cardiac muscle is still unknown clinically and some findings suggest that higher doses may have potentially toxic implications (Duan et al., 2018; Hart et al., 2022).
CRISPR-Cas9 gene editing aims to restore muscle physiology by producing truncated dystrophin constructs (Johnston et al., 2021) and involves a guide RNA (gRNA) that targets a genomic sequence as well as an endonuclease that produces a double-stranded break in the DNA at targeted sites (Ran et al., 2013). The subsequently activated DNA repair pathways include homology-directed repair (HDR) and nonhomologous end-joining (NHEJ), which is used to restore the reading frame in non-dividing cells, such as cardiomyocytes (Duan et al., 2021). Several studies showed significant expression of dystrophin and improvement of cardiac pathology in mouse and GRMD models treated using CRISPR-Cas9 technology with a cardiac-expressing promoter (Nelson et al., 2016; Amoasii et al., 2018; Amoasii et al., 2017; Bengtsson et al., 2017; El Refaey et al., 2017). This correction persisted long-term (18 months) in mice, suggesting that permanent gene repair could be possible with this approach (Hakim et al., 2018; Xu et al., 2019; Nelson et al., 2019). Although exciting, gene editing for DMD is in the preclinical stage and challenges must be overcome to apply this technology to humans.
4.6. Other therapies
Tamoxifen, which is a first-generation selective estrogen receptor modulator (SERM), demonstrates potent antiestrogenic activity on the mammary gland and is generally used for the prevention and treatment of breast cancer (Dorchies et al., 2013). In a study orally administering Tamoxifen on dystrophic mdx5Cv mice starting at 3 weeks of age for 15 months at 10 mg/kg/day, remarkable improvements to cardiac structure were noted, with cardiac fibrosis concurrently being diminished by ~50%, compared to WT mice (Dorchies et al. 2013).
5. Chamber-specific cardiomyopathy in DMD
To date, the majority of research in the hearts of DMD patients and dystrophin-deficient animal models has been conducted specifically in the LV or, more commonly, in the unspecified ‘whole heart’. Emerging data now suggests that dystrophic cardiomyopathy may affect the heart heterogeneously, with differing data on fibrosis, calcification, function (e.g. echocardiography, hemodynamics, etc.), and other related parameters observed across ventricles.
5.1. Human Duchenne muscular dystrophy
A study by Mehmood et al., which was the first to use CMR to define RV systolic function and size in DMD patients, determined that in DMD patients with reduced LVEF, the RVEF was still relatively unchanged (Mehmood et al., 2016; Bosser et al., 2004). The relative preservation of RV function is thought to be attributed to the advancements that have been made in respiratory care, including improvements to ventilatory support. Furthermore, in a study by Bosser et al., normal resting RVEF (>45%) was observed in 95% of DMD patients, whereas only 79% of patients demonstrated a normal LVEF (50%) (Bosser et al. 2004). This has largely led to the inaccurate belief that the RV is generally less impacted, and therefore less crucial to the outcomes of cardiomyopathy in DMD patients.
Despite these findings, other studies have indicated that RV insults do persist in DMD. Electrical right ventricular hypertrophy (RVH) is frequent in DMD patients, reaching 37% without correlating to LV dysfunction (Takami et al., 2008; reviewed in Fayssoil et al., 2017). Dual et al. demonstrated that pre-contrast RV-T1 (via CMR), which indicates presence of diffuse myocardial fibrosis, is elevated in boys with DMD and that this is negatively correlated with RVEF, compared to age- and sex-matched healthy boys (Dual et al., 2021). This finding is particularly important because boys with DMD develop respiratory impairments prior to heart failure, and the respiratory dysfunction and resulting hypoxia are predictors of increased afterload as seen through the RV due to constriction of pulmonary arteries (Dual et al., 2021). This study also demonstrated abnormal RV function in DMD-affected boys, which was indicated by significantly decreased tricuspid annular excursion (TAE) (a measure of RV longitudinal contraction) and RV mass (Dual et al., 2021). Additionally, the group examined the role of the septum in DMD-induced cardiomyopathy. Given that no differences in pre-contrast septal T1 were determined when compared between groups, it was concluded that when classifying RV mass and BMI as covariates, RV-T1 best predicts DMD status relative to the septum (Dual et al., 2021). It was thus determined that in DMD patients, the septum remains relatively unchanged as cardiomyopathy worsens (Dual et al. 2021). The study by Dual et al. (2021) also reported higher RV-T1 compared to LV-T1 in both healthy patients and those with DMD. This chamber-specific elevation is thought to be attributed to naturally increased collagen content of the RV wall.
Further exploring the RV in DMD, a study by Subramanian et al. investigated the relationship between pulmonary function and RV functional parameters in young patients with DMD (Subramaniam et al., 2015). They determined that abnormal pulmonary function was correlated with reduced RV stroke volume and preserved LV systolic function. Work comparing the LV to the RV has demonstrated that young DMD patients with preserved EF and normal FS present with subclinical regional myocardial dysfunction (Mertens et al., 2008). This work established that during the earlier stages of disorder progression, function of the interventricular septum and RV free wall were better preserved than the LV, as indicated by longitudinal measurements.
It has been theorized that RV lesions in DMD patients are more difficult to assess with LGE (which is commonly used to assess LV fibrosis) because the RV has a smaller mass, potentially smaller-sized lesions, and a more complex geometry (Meyers and Townsend, 2015). Also, it has been hypothesized that sympathetic activation or hypoxia-induced constriction of the pulmonary artery leads to elevated afterload, resulting in the increased RV (Meyers and Townsend, 2015). Refer to the cardiac fibrosis section above for more detail.
5.2. Dystrophin-deficient rodent models
While much of what is known about RV functional parameters has been learned from human DMD patients, there are several known studies that examined RV function in mdx mice, including one which tested the effects of early beta-blocker (metoprolol) use on chamber-specific cardiomyopathy (Blain et al., 2013; Stuckey et al., 2012). Blain et al. compared ventricular function in 24-week-old C57BL/10 mdx mice without beta-blocker treatment and determined that there was general ventricular hypertrophy, reduced LV stroke volume with a small LV cavity size, and normal LVEF, despite reduced RVEF compared to WTs (Blain et al., 2013). This study concluded that in the untreated, aged C57BL/10 mdx mouse model, RV dysfunction potentially preceded LV dysfunction (Blain et al., 2013; Stuckey et al., 2012). Based on their findings, the authors cautioned against early beta-blocker usage due to the lasting effects they may have on the RV. It has been postulated that the apparent improvements in LV function and deterioration in RV function may be linked through a mechanism whereby untreated hearts have a relatively low stroke volume, which may protect the RV from increased volume overload. Given that beta-blockers improve cardiac output and stroke volume to normal levels, greater dilatation of the RV can result from the increased flow to the right heart (Blain et al., 2013). In a separate study, Hayes et al. (2022) demonstrated that 18-week-old D2.mdx hearts exhibit thick epicardial fibrotic-calcinosis layers that are limited and restricted to the RV, but such changes are not detectable in the LV or septum (Meyers and Townsend, 2015). While systolic dysfunction is evident in both ventricles of the D2.mdx heart, it was determined that RV function was slightly better than LV despite the elevated fibrosis (Meyers and Townsend, 2015). Together, this data suggests that RV damage potentially precedes onset of significant cardiac complication (Meyers and Townsend, 2015). Hemodynamic output is similar between ventricles in both the D2.mdx mouse model and in DMD patients (Meyers and Townsend, 2015). Stuckey et al. (2012) determined that C57BL/10 mdx mice exhibited normal RV resting function at 1 month of age, but this was noted to be abnormal beginning at 3 months of age, elevated RV-ESV and decreased RVEF. These responses were noted to occur prior to LV dysfunction, with LVEF being reduced at a later time point (1 year of age).
5.3. Limitations and future directions of chamber-specific cardiomyopathy
Historically, the role of the RV in dystrophin deficiency-induced cardiomyopathy has been underappreciated in place of the larger, more physiologically demanding LV. For this reason, literature comparing pathology across ventricles is limited. Although, to date, some work has been conducted to elucidate RV function and its role among the respiratory impairments associated with dystrophin deficiency, further research is required to investigate parameters such as hemodynamics and echocardiography in young dystrophin deficient rodent models (mice aged >24 weeks). Additionally, there is an abundance of conflicting data with regards to ventricle-specific disorder progression due to the use of different dystrophin-deficient models and time points. Future research should seek to track time-course progression of ventricular abnormalities in an established dystrophin-deficient rodent model with cardiomyopathy (mdx mice) to determine the order in which these pathologies arise.
Furthermore, researchers should consider conducting four-chamber histopathological assessments of the heart, which can provide valuable insight towards early- and late-stage cardiomyopathy (at least in dystrophin deficient rodent models due to logistical limitations encountered in human DMD models). Exploring these parameters across chambers can help guide therapy development beyond just LV impairments, such that lung and diaphragm abnormalities can be addressed concurrently using RV and atrial data. To date, most papers omit collection of this data due to restrictions brought on by the complex geometry of the RV, coupled with the extremely small free wall size, which is a limitation that can be overcome by repeating experiments to obtain multiple phases for various measures. Future work in both human and dystrophin-deficient rodent models should seek to explore chamber-specific cardiomyopathy holistically- from histopathological and functional assessments to molecular and metabolic underpinnings of the disorder using a time-course experimental design.
6. Mitochondria as a potential therapeutic target
6.1. Targeting calcium handling
Given mitochondrial dysfunctions are thought to arise, in part, by elevated calcium uptake, preventing mitochondrial dysfunction could be achieved by improving cytoplasmic handling itself. For example, due to the hypersensitivity to excitation-contraction coupling in cardiomyopathy of DMD patients, targeting RyR2 may offer therapeutic benefit. PKA phosphorylation of RyR2 appears to contribute to increased calcium release from the SR, and therefore, PKA could be a potential therapeutic target (Figure 1). In dystrophic mice, inhibition of PKA-mediated phosphorylation of RyR2 reduced SR calcium leak and in turn prevented cardiomyopathy (Sarma et al., 2010). Oxidative stress can lead to the nitrosylation of RyR2 and subsequent disassociation of calstabin2 from RyR2, increasing SR calcium leak (Fauconnier et al., 2010). Treatment of C57BL/10 mdx mice with either NAC to inhibit RyR2 nitrosylation, or the RyR2 stabilizer Rycal, prevented depletion of calstabin2 and subsequent SR calcium leak, aberrant depolarization in cardiomyocytes, and arrhythmias (Fauconnier et al., 2010).
The stretch-sensitive channel TRPV2 is another calcium channel target to consider, as increased TRPV2 has been documented in the cytoplasmic membrane of mdx cardiac and skeletal cells and the sarcolemmal membrane of DMD patients (Iwata et al., 2013). Treatment of DMD patients with the antiallergy drug Tranilast, which has anti-TRPV2 activity, resulted in reduction of heart failure biomarkers (Matsmura et al., 2018).
Enhancing SR calcium uptake through overexpression of SERCA or targeting its inhibitors can be considered in restoring calcium homeostasis. In C57BL/10 mdx mice 12 months of age, administration of AVV9-SERCA2a gene therapy significantly improved cardiac electrophysiology (Shin et al., 2011), whereas in 3-month-old mdx mice a similar therapy ameliorated DCM for at least 18 months (Wasala et al., 2020). Decreasing sarcolipin expression in mdx mice restored cardiac SERCA function and calcium cycling, thus preventing cardiomyopathy. Decreased LV internal diameter in diastole as well as decreased fibrotic and necrotic tissue likely contributed to the improved cardiac function (Voit et al., 2017). While targeting sarcolipin may be a promising approach, cardiac function worsened in phospholamban (PLN) knockout C57BL/10 mdx mice (Law et al., 2018).
6.2. Mitochondrial calcium overload
As described previously, frequent PT activity may contribute to calcium-dependent mitochondrial dysfunction and therefore, prevention of this opening could be beneficial. As discussed in Section 3, inhibition of a key regulator of mPTP, cyclophilin, may impede opening of the pore and subsequent mitochondrial dysfunction (Millay et al., 2008; Reutenauer et al., 2008).
6.3. Targeting cellular/mitochondrial antioxidant systems
Since cardiac mH2O2 is elevated early in DMD (Hughes et al., 2020; Dubinin et al., 2020b), mitochondrial-targeted antioxidant therapies could provide therapeutic benefit in DMD patients. Several general antioxidants, including MitoQ and SkQ1, are conjugated with the lipophilic cation triphenylphosphonin (TTP) to target the mitochondria (reviewed in Murphy, 2008). The small ‘SS’ peptides are cell permeable agents that bind to cardiolipin on the IMM and can preserve oxidative phosphorylation, attenuate mH2O2, and prevent oxidative damage (Szeto and Birk, 2014; Zhao et al., 2003; Birk et al., 2013). Furthermore, SS peptides can inhibit the dissociation of cytochrome c from cardiolipin, hindering the activation of cell death pathways (Zhao et al., 2004) (Figure 3). To our knowledge, no publication has investigated the effects of any mitochondrial-ROS lowering compound in models of DMD.
6.4. Cardiolipin and membrane stability
Cardiolipin, a phospholipid enriched with unsaturated fatty acids located in the IMM, is essential to maintain mitochondrial structure and function. Through its binding to key modulators of energy exchange and subsequent proteolipid complex formation, cardiolipin plays a key role in mitochondrial bioenergetics (Schlattner et al., 2018) (Figure 3). This complex can regulate energy exchange, reduce mitochondrial ROS generation, and prevent opening of mPTP (Schlattner et al., 2018). In the presence of oxidative stress, cardiolipin and the key regulator mtCK, lose their binding capacity, leading to dissociation of the proteolipid complex (Koufen et al., 1999; Dolder et al., 2001) (Figure 3). Since mH2O2 generation is elevated in dystrophin deficient heart (Hughes et al., 2020; Dubinin et al., 2020b), a cardiolipin targeting agent, such as elamipretide, may be appropriate for targeting impaired mitochondrial bioenergetics. Although data on cardiolipin physiology in mdx is limited, the altered mitochondrial cristae structure reported in section 3.7 serves as a foundation for continuing the investigation on cardiolipin in various dystrophic models.
Albeit not published in a dystrophin deficient model, SS-31, or elamipretide, is an agent that has demonstrated improvements to mitochondrial stress including reduction of mitochondrial ROS across several pathologies (Siegel et al., 2013; Szeto and Schiller, 2011; Szeto, 2014). Elamipretide has also shown evidence of direct cardioprotective mechanisms including amelioration of apoptosis and fibrosis in preclinical models of heart failure (Dai et al., 2011; Eirin et al., 2014) (Figure 3).
Idebenone, a synthetic analogue of Coenzyme Q10 with electron shuttling activity, can alter respiratory chain activity (Zs-Nagy, 1990; Gillis et al., 1994), although this may require very high micromolar doses (Suno and Nagaoka, 1984; Suno and Nagaoka, 1989). Long-term idebenone therapy improved cardiac diastolic dysfunction, reduced cardiac inflammation and fibrosis, improved running performance, and limited mortality from cardiac pump failure induced by dobutamine stress testing in vivo in mdx mice (Buyse et al., 2009). Early clinical trials in patients with DMD revealed improved cardiac and respiratory function with idebenone treatment (Buyse et al., 2011; Buyse et al., 2015; Buyse et al., 2017; McDonald et al., 2016). Specifically, a trend for increased peak systolic radial strain in the LV was seen in these patients (Buyse et al., 2011). However, no measures of mitochondrial bioenergetics were performed to verify that the drug was acting through a mitochondrial mechanism. As reviewed elsewhere, idebenone has a variety of effects on other organelles which may not involve antioxidant properties of the compound (Gueven et al., 2021). This suggests that the beneficial effects in some studies may not be due to alterations in mitochondrial functions despite claims to this effect. These factors might be consistent with a subsequent clinical trial in people with DMD that was unsuccessful (NCT02814019). As such, no study to date has definitively tested the potential of mitochondrial-targeted therapies on cardiac dysfunction in pre-clinical models of dystrophin deficiency or in clinical studies.
Mitochondria are hypothesized to modulate phosphorodiamidate morpholino oligomer (PMO) efficacy by mediating membrane repair (Brown et al., 2022). In fact, mitochondria are known to hone to the site of sarcolemmal injury as well as contribute to the production of membrane phospholipids (Brown et al., 2022). However, PMO uptake by the cells requires ATP (Brown et al., 2022). Elamipretide localizes to the IMM where it binds to cardiolipin to improve membrane stability, enhance ATP synthesis, optimize cristae architecture, and attenuate ROS generation (Szeto and Birk, 2014). This agent has also been observed to improve mitochondrial ultrastructure (Allen et al., 2020) and restore cellular bioenergetics (Obi et al., 2022), which are known to be altered in dystrophin deficient models. In a preclinical study, elamipretide significantly increased dystrophin protein expression evoked by an exon-skipping PMO in mdx mice (Brown et al., 2022). When elamipretide was administered alone, trends to reduction of inflammation were observed (Brown et al., 2022). These data suggest that a potential synergy exists between elamipretide and exon-skipping PMOs which may represent a beneficial treatment strategy in patients with DMD (Brown et al., 2022). Given many of these mitochondrial abnormalities exist in cardiac tissue from mdx models, there is sufficient rationale to investigate whether such compounds preserve mitochondrial bioenergetics and treat cardiomyopathy in DMD.
7. Summary and future directions
Since emerging therapies are thus far unable to completely restore the DMD gene, there remains a need to develop additional therapies treating secondary contributors to cardiomyopathy. While curative approaches customized to individual mutations are actively pursued, developing new combinatorial therapies targeting a variety of cellular and physiological stressors could complement existing conventional approaches in a manner that benefits the majority of people with DMD. In this regard, the collective evidence warrants the development of new approaches targeting partial dystrophin restoration, inflammation, calcium and other ion dysregulation, membrane tearing, oxidative stress, cytoskeletal disorganization, metabolic stress and other cellular dysfunctions. Many of these stressors are related to mitochondrial-specific stress responses linked to disrupted energy homeostasis, redox balance and calcium-induced cell death. The degree to which each mitochondrial stress response contributes to cardiomyopathy in DMD requires careful consideration of the mechanisms of specific mitochondrial targeting compounds, the age and type of animal model used in pre-clinical investigations, and cardiac chamber-specific dysfunctions. The overview provided in this article serves as a basis for guiding experimental design in a way that captures the specific mechanisms by which mitochondria contribute to cardiac dysfunction and histopathology in DMD.
Author Contributions
SG and CP contributed equally to the preparation of the manuscript while all authors contributed to writing and editing specific sections.
Funding
Writing and editing assistance were funded by Stealth BioTherapeutics. CP has previously received research funding from the Sponsor. All authors declare independence from the Sponsor.
Acknowledgments
Writing and editing assistance, including preparation of a draft manuscript under the direction and guidance of the authors, incorporating author feedback, and manuscript submission, was provided by Jamie L Dermatis, DPM, and James A. Shiffer, RPh (Write On Time Medical Communications, LLC, NJ).
Competing interests
The authors declare no conflicts of interest.
References
- Aartsma-Rus A,Van Deutekom JCT, Fokkema IF, Van Ommen GJB & den Dunnen JT Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144.
- Aartsma-Rus A & den Dunnen, JT. Phenotype predictions for exon deletions/duplications: A user guide for professionals and clinicians using Becker and Duchenne muscular dystrophy as examples. Hum Mutat 2019, 40, 1630–1633. [Google Scholar]
- Abutaleb ARA, McNally EM, Khan SS, Anderson AS, Carr JC & Wilcox JE. Myocarditis in Duchenne muscular dystrophy after changing steroids. JAMA Cardiol 2018, 3, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Adorisio R, Mencarelli E, Cantarutti N, Calvieri C, Amato L, Cicenia M, Silvetti M, D’Amico A, Grandinetti M, Drago F & Amodeo A. Duchenne dilated cardiomyopathy: cardiac management from prevention to advanced cardiovascular therapies. J Clin Med 2020, 9, 3186. [Google Scholar] [CrossRef] [PubMed]
- Aikawa T, Takeda A, Oyama-Manabe N, Naya M, Yamazawa H, Koyanagawa K, Ito YM & Anzai T. Progressive left ventricular dysfunction and myocardial fibrosis in Duchenne and Becker muscular dystrophy: a longitudinal cardiovascular magnetic resonance study. Pediatr Cardiol 2018, 40, 384–392. [Google Scholar]
- Aliev M, Guzun R, Karu-Varikmaa M, Kaambre T, Wallimann T & Saks V. Molecular system bioenergics of the heart: experimental studies of metabolic compartmentation and energy fluxes versus computer modeling. Int J Mol Sci 2011, 12, 9296–9331. [Google Scholar] [CrossRef] [PubMed]
- Allen DG, Whitehead NP & Yeung EW. Mechanisms of stretch-induced muscle damage in normal and dystrophic muscle: role of ionic changes. J Physiol 2005, 567, 723–735. [Google Scholar] [CrossRef]
- Allen DG, Gervasio OL, Yeung EW & Whitehead NP. Calcium and the damage pathways in muscular dystrophy. Can J Physiol Pharmacol 2010, 88, 83–91. [Google Scholar]
- Allen DG & Whitehead, NP. Duchennz muscular dystrophy—What causes the increased membrane permeability in skeletal muscle. Int J Biochem Cell Biol 2010, 43, 290–294. [Google Scholar]
- Allen DG, Whitehead NP, & Froehner SC. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol Rev 2016, 96, 253–305. [Google Scholar] [CrossRef]
- 11. Allen HD, Flanigan KM, Thrush PT, Dvorchik I, Yin H, Canter C, Connolly AM, Parrish M, McDonald CM, Braunlin E, Colan SD, Day J, Darras B & Mendell JR. A randomized, double-blind trial of Lisinopril and Losartan for the treatment of cardiomyopathy in Duchenne muscular dystrophy. PLoS Curr, 2013; 5.
- Allen ME, Pennington ER, Perry JB, Dadoo S, Makrecka-Kuka M, Dambrova M, Moukdar F, Patel HD, Han X, Kidd GK, Benson EK, Raisch TB, Poelzing S, Brown D A & Shaikh SR. The cardiolipin-binding peptide elamipretide mitigates fragmentation of cristae networks following cardiac ischemia reperfusion in rats. Commun Biol 2020, 3, 389. [Google Scholar]
- Amedro P, Vincenti M, De La Villeon G, Lavastre K, Barrea C, Guillaumont S, Bredy C, Gamon L, Meli AC, Cazorla O, Fauconnier J; Meyer P; Rivier F, Adda J, Mura T & Lacampagne A. Speckle-tracking echocardiography in children with Duchenne muscular dystrophy: a prospective multicenter controlled cross-sectional study. J Am Soc Echocardiogr 2019, 32, 412–422. [Google Scholar] [CrossRef]
- Amoasii L, Li H, Sanchez-Ortiz E, Caballero D, Harron R, Massey C, Shelton J, Piercy R & Olson EN. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef]
- Amoasii L, Long C, Li H, Mireault AA, Shelton JM, Sanchez-Ortiz E, McAnally JR, Bhattacharyya S, Schmidt F, Grimm D, Hauschka SD, Bassel-Duby R & Olson EN. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci Transl Med 2017, 9, 1–11. [Google Scholar]
- Andersson DC & Marks, AR. Fixing ryanodine receptor Ca2+ leak—A novel therapeutic strategy for contractile failure in heart and skeletal muscle. Drug Discov Today Dis Mech 2010, 7, e151–e157. [Google Scholar]
- Angelini C & Peterle, E. Old and new therapeutic developments in steroid treatment in Duchenne muscular dystrophy. Acta Myol 2012, 31, 9–15. [Google Scholar]
- Archer JE, Gardner AC, Roper HP, Chikermane AA & Tatman AJ. Duchenne muscular dystrophy: the management of scoliosis. J Spine Surg 2016, 2, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Ascah A, Khairallah M, Daussin F, Bourcier-Lucas C, Godin R, Allen BG, Petrof BJ, Des Rosiers C & Burelle Y. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am J Physiol Heart Circ Physiol 2011, 300, H144–H153. [Google Scholar] [CrossRef] [PubMed]
- Ashford MW, Liu W, Lin SJ, Abraszewski P, Caruthers SD, Connolly AM, Yu X & Wickline SA. Occult cardiac contractile dysfunction in dystrophin-deficient children revealed by cardiac magnetic resonance strain imaging. Circulation 2005, 112, 2462–2467. [Google Scholar] [CrossRef]
- Bach JR, O’Brien J, Krotenberg R & Alba AS. Management of end stage respiratory failure in Duchenne muscular dystrophy. Muscle Nerve 1987, 10, 177–182. [Google Scholar] [CrossRef]
- Bahler RC, Mohyuddin T, Finkelhor RS & Jacobs IB. Contribution of Doppler tissue imaging and myocardial performance index to assessment of left ventricular function in patients with Duchenne’s muscular dystrophy. J Am Soc Echocardiogr 2005, 18, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Banihani R, Smile S, Yoon G, Dupuis A, Mosleh M, Snider A & McAdam L. Cognitive and neurobehavioral profile in boys with Duchenne muscular dystrophy. J Child Neurol 2015, 30, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Barber BJ, Andrews JG, Lu Z, West NA, Meaney FJ, Price ET, Gray A, Sheehan DW, Pandya S, Yang M & Cunniff C. Oral corticosteroids and onset of cardiomyopathy in Duchenne muscular dystrophy. J Pediatr 2013, 163, 1080–1084e1. [Google Scholar] [CrossRef]
- Batra A, Barnard AM, Lott DJ, Willcocks RJ, Forbes SC, Chakraborty S, Daniels MJ, Arbogast J, Triplett W, Henricson EK, Dayan JG, Schmalfuss C, Sweeney L, Byrne BJ, McDonald CM, Vandenborne K & Walter GA. Longitudinal changes in cardiac function in Duchenne muscular dystrophy population as measured by magnetic resonance imaging. BMC Cardiovasc Disord 2022, 22, 260. [Google Scholar]
- Bauer R, Straub V, Blain A, Bushby K & MacGowan GA. Contrasting effects of steroids and angiotensin-converting-enzyme inhibitors in a mouse model of dystrophin-deficient cardiomyopathy. Eur J Heart Fail 2009, 11, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Belanto JJ, Mader TL, Eckhoff MD, Strandjord DM, Banks GB, Gardner MK, Lowe DA, & Ervasti JM. Microtubule binding distinguishes dystrophin from utrophin. Proc Natl Acad Sci USA 2014, 111, 5723–5728. [Google Scholar] [CrossRef] [PubMed]
- Belanto JJ, Olthoff JT, Mader TL, Chamberlain CM, Nelson DM, McCourt PM, Talsness DM, Gundersen GG, Lowe DA, & Ervasti JM. Independent variability of microtubule perturbations associated with dystrophinopathy. Hum Mol Genet 2016, 25, 4951–4961. [Google Scholar]
- Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, Matecki S, Lacampagne A & Marks AR. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med 2009, 15, 325–330. [Google Scholar] [CrossRef]
- Bellissimo CA, Garibotti MC & Perry CGR. Mitochondria stress responses in Duchenne muscular dystrophy: metabolic dysfunction or adaptive reprogramming? Am J Physiol Cell Physiol 2022, 323, C718–C730. [Google Scholar] [CrossRef]
- Bellissimo CA, Delfinis LJ, Hughes MC, Turnbull PC, Gandhi S, DiBenedetto SN, Rahman FA, Tadi P, Amaral CA, Dehghani A, Cobley JN, Quadrilatero J, Schlattner U & Perry CGR. Mitochondrial creatine sensitivity is lost in the D2mdx model of Duchenne muscular dystrophy and rescued by the mitochondrial-enhancing compound. Olesoxime Am J Physiol Cell Physiol 2023, 324, C1141–C1157.
- Bengtsson NE, Hall JK, Odom GL, Phelps MP, Andrus CR, Hawkins RD, Hauschka SD, Chamberlain JR & Chamberlain JS. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun 2017, 8, 1–9. [Google Scholar]
- Bernardi P & von Stockum, S. The permeability transition pore as a Ca(2þ) release channel: new answers to an old question. Cell Calcium 2012, 52, 22–27. [Google Scholar]
- Bernardi P, Carraro M & Lippe G. The mitochondrial permeability transition: recent progress and open questions. FEBS J 2022, 289, 7051–7074. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi P, Torchiana E, Confalonieri P, Brugnoni R, Barresi R, Mora M, Cornelio F, Morandi L & Mantegazza R. Expression of transforming growth factor-beta 1 in dystrophic patient muscles correlates with fibrosis: pathogenetic role of a fibrogenic cytokine. J Clin Invest 1995, 96, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, Seshan SV, Pardee JD, & Szeto HH. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol 2013, 24, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Bilchick KC, Salerno M, Plitt D, Dori Y, Crawford TO, Drachman D & Thompson WR. Prevalence and distribution of regional scar in dysfunctional myocardial segments in Duchenne muscular dystrophy. J Cardiovasc Magn Reson 2011, 13, 20. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, MJ. Identification of a novel gene encoding an insulin-responsive glucose transporter protein. Cell 1989, 57, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, Case LE, Cripe L, Hadjiyannakis S, Olson AK, Sheehan DW, Bolen J, Weber DR, Ward LM & DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol 2018, 17, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Bladen CL, Rafferty K, Straub V, Monges S, Moresco A, Dawkins H, Roy A, Chamova T, Guergueltcheva V, Korngut L, Campbell C, Dai Y, Barišić N, Kos T, Brabec P, Rahbek J, Lahdetie J, Tuffery-Giraud S, Claustres M,… Lochmüller H. The TREAT-NMD Duchenne 22 muscular dystrophy registries: conception, design, and utilization by industry and academia. Hum Mutat 2013, 34, 1449–1457. [Google Scholar] [CrossRef]
- Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, Dawkins H, Lamont L, Roy AJ, Chamova T, Guergueltcheva V, Chan S, Korngut L, Campbell C, Dai Y, Wang J, Barišić N, Brabec P, Lahdetie J, Walter MC,… Lochmüller H. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Blain A, Greally E, Laval S, Blamire A, Straub V & MacGowan GA. Beta-blockers, left and right ventricular function, and in vivo calcium influx in muscular dystrophy cardiomyopathy. PLOS One 2013, 8, e57260. [Google Scholar]
- Bodor GS, Porterfield D, Voss EM, Smith S & Apple FS. Cardiac troponin-I is not expressed in fetal and healthy or diseased adult human skeletal muscle tissue. Clin Chem 1995, 41, 1710–1715. [Google Scholar] [CrossRef]
- Borthwick LA, Wynn TA & Fisher AJ. Cytokine mediated tissue fibrosis. Biochim Biophys Acta 2013, 1832, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Bosser G, Lucron H, Lethor JP, Burger G, Beltramo F, Marie PY & Marçon F. Evidence of early impairments in both right and left ventricular inotropic reserves in children with Duchenne’s muscular dystrophy. Am J Cardiol 2004, 93, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Boyman L, Chikando AC, Williams GS, Khairallah RJ, Kettlewell S, Ward CW, Smith GL, Kao JP & Lederer WJ. Calcium movement in cardiac mitochondria. Biophys J 2014, 107, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Brown DA, Vining A, Mackinnon A, Elustondo P, McKenna M & Nagaraju K (2022). The Mitochondria-targeting Peptide Elamipretide Potentiates Dystrophin Expression Induced by an Exon-skipping Morpholino in the mdx Mouse Model. Proceedings of Muscular Dystrophy Association (MDA) Clinical & Scientific Conference; Muscular Dystrophy Association: Nashville, TN, USA.
- Buddhe S, Lewin M, Olson A, Ferguson M & Soriano BD. Comparison of left ventricular function assessment between echocardiography and MRI in Duchenne muscular dystrophy. Pediatr Radiol 2016, 46, 1399–1408. [Google Scholar] [CrossRef]
- Buddhe S, Cripe L; Friedland-Little J, Kertesz N, Eghtesady P, Finder J, Hor K, Judge DP, Kinnett K, McNally EM, Raman S, Thompson WR, Wagner KR & Olson AK. Cardiac management of the patient with Duchenne muscular. dystrophy Pediatrics 2018, 142, S72–S81.
- Budihardjo I, Oliver H, Lutter M, Luo X & Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol 1999, 15, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Bulfield G, Siller WG & Wight PA. Moore KJ X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci U S A 1984, 81, 1189–1192. [Google Scholar] [CrossRef]
- Burelle Y, Khairallah M, Ascah A, Allen BG, Deschepper CF, Petrof BJ & Des Rosiers C. Alterations in mitochondrial function as a harbinger of cardiomyopathy: lessons from the dystrophic heart. J Mol Cell Cardiol 2010, 48, 310–321. [Google Scholar] [CrossRef]
- Burkhoff D, Weiss RG, Schulman SP, Kalil-Filho R, Wannenburg T & Gerstenblith G. Influence of metabolic substrate on rat heart function and metabolism at different coronary flows. Am J Physiol 1991, 261, H741–H750. [Google Scholar]
- Bushby K & Connor, E. Clinical outcome measures for trials in Duchenne muscular dystrophy: report from International Working Group meetings. Clin Investig 2011, 1, 1217–1235. [Google Scholar]
- Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J & Constantin C. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010, 9, 77–93. [Google Scholar] [CrossRef]
- Buyse GM, Goemans N, van den Hauwe M, Thijs D, de Groot IJ, Schara U, Ceulemans B, Meier T & Mertens L. Idebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: Results from a 12 month, double-blind, randomized placebo-controlled trial. Neuromuscul Disord 2011, 21, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Buyse GM, Van der Mieren G, Erb M, D’Hooge J, Herijgers P, Verbeken E, Jara A, Van Den Bergh A, Mertens L, Courdier-Fruh I, Barzaghi P & Meier T. Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: cardiac protection and improved exercise performance. Eur Heart J 2009, 30, 116–124. [Google Scholar]
- Buyse GM, Voit T, Schara U, Straathof CS, D’Angelo MG, Bernert G, Cuisset JM, Finkel RS, Goemans N, McDonald CM, Rummey C & Meier T. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): a double-blind randomised placebo-controlled phase 3 trial. Lancet 2015, 385, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Buyse GM, Voit T, Schara U, Straathof CS, D’Angelo MG, Bernert G, Cuisset JM, Finkel RS, Goemans N, Rummey C, Leinonen MS, Mayer OH, Spagnolo P, Meier T & McDonald CM. Treatment effect of idebenone on inspiratory function in patients with Duchenne muscular dystrophy. Pediatr Pulmonol 2017, 52, 508–515. [Google Scholar] [CrossRef]
- Bylo M, Farewell R, Coppenrath VA & Yogaratnam D. A review of deflazacort for patients with Duchenne muscular dystrophy. Ann Pharmacother 2020, 54, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Cai J, Yang J & Jones D. Mitochondrial control of apoptosis: the role of cytochrome c. Biochim Biophys Acta 1998, 1366, 139–149. [Google Scholar] [CrossRef]
- Cai B, Spencer MJ, Nakamura G, Tseng-Ong L & Tidball JG. Eosinophilia of dystrophin-deficient muscle is promoted by perforin-mediated cytotoxicity by T cell effectors. Am J Pathol 2000, 156, 1789–1796. [Google Scholar] [CrossRef]
- Capitanio D, Moriggi M, Torretta E, Barbacini P, De Palma S, Vigano A, Lochmüller H, Muntoni F, Ferlini A, Mora M & Gelfi C. Comparative proteomic analyses of Duchenne muscular dystrophy and Becker muscular dystrophy muscles: changes contributing to preserve muscle function in Becker muscular dystrophy patients. J Cachexia Sarcopenia Muscle 2020, 11, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Chadwick JA, Swager SA, Lowe J, Welc SS, Tidball JG, Gomez-Sanchez CE, Gomez-Sanchez EP & Rafael-Fortney JA. Myeloid cells are capable of synthesizing aldosterone to exacerbate damage in muscular dystrophy. Hum Mol Genet 2016, 25, 5167–5177. [Google Scholar]
- Chamberlain JR & Chamberlain, JS. Progress toward gene therapy for Duchenne muscular dystrophy. Mol Ther 2017, 25, 1125–1131. [Google Scholar] [PubMed]
- Chemello F, Wang Z, Li H, McAnally JR, Liu N, Bassel-Duby R & Olson EN. Degenerative and regenerative pathways underlying Duchenne muscular dystrophy revealed by single-nucleus RNA sequencing. Proc Natl Acad Sci U S A 2020, 117, 29691–29701. [Google Scholar] [CrossRef] [PubMed]
- Chiang DY, Allen HD, Kim JJ, Valdes SO, Wang Y, Pignatelli RH, Lotze TE & Miyake CY. Relation of cardiac dysfunction to rhythm abnormalities in patients with Duchenne or Becker muscular dystrophies. Am J Cardiol 2016, 117, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- CHMP Assessment Report for Sovrima; European Medicines Agency: London, UK, 2008. Available at: https://wwwemaeuropaeu/en/documents/assessment-report/sovrima-epar-public-assessment-report_enpdf.
- Cho MJ, Lee JW, Lee J & Shin YB. Evaluation of early left ventricular dysfunction in patients with Duchenne muscular dystrophy using two-dimensional speckle tracking echocardiography and tissue Doppler imaging. Pediatr Cardiol 2018, 39, 1614–1619. [Google Scholar] [CrossRef] [PubMed]
- Cinteza E, Stoicescu C, Butoianu N, Balgradean M, Nicolescu A & Angrés M. Acut myocardial injury in a child with Duchenne muscular dystrophy: pulse steroid therapy? Maedica 2017, 12, 180–183. [Google Scholar]
- Coley WD, Bogdanik L, Vila MC, Yu Q, Van Der Meulen JH, Rayavarapu S, Novak JS, Nearing M, Quinn JL, Saunders A, Dolan C, Andrews W, Lammert C, Austin A, Partridge TA, Cox GA, Lutz C & Nagaraju K. Effect of genetic background on the dystrophic phenotype in mdx mice. Hum Mol Genet 2016, 25, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Connolly AM Zaidman CM, Golumbek PT, Cradock MM, Flanigan KM, Kuntz NL, Finkel RS, McDonald CM, Iannaccone ST, Anand P, Siener CA, Florence JM, Lowes LP, Alfano LN, Johnson LB, Nicorici A, Nelson LL, Mendell JR & MDA DMD Clinical Research Network. Twice-weekly glucocorticosteroids in infants and young boys with Duchenne muscular dystrophy. Muscle Nerve 2019, 59, 650–657. [Google Scholar] [CrossRef]
- Connuck DM, Sleeper LA, Colan SD, Cox GF, Towbin JA, Lowe AM, Wilkinson JD, Orav EJ, Cuniberti L, Salbert BA & Lipshultz SE. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: a comparative study from the Pediatric Cardiomyopathy Registry. Am Heart J 2008, 155, 998–1005. [Google Scholar] [CrossRef]
- Corrado G, Lissoni A, Beretta S, Terenghi L, Tadeo G, Foglia-Manzillo G, Tagliagambe LM, Spata M & Santarone M. Prognostic value of electrocardiograms, ventricular late potentials, ventricular arrhythmias, and left ventricular systolic dysfunction in patients with Duchenne muscular dystrophy. Am J Cardiol 2002, 89, 838–841. [Google Scholar] [CrossRef] [PubMed]
- Crisafulli S, Sultana J, Fontana A, Salvo F, Messina S & Trifirò G. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet J Rare Dis 2020, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Cserne Szappanos H, Muralidharan P, Ingley E, Petereit J, Millar AH & Hool LC. Identification of a novel cAMP dependent protein kinase, A phosphorylation site on the human cardiac calcium channel. Sci Rep 2017, 7, 15118. [Google Scholar] [CrossRef] [PubMed]
- Cuccia C, Benedini G, Leonzi O, Pagnoni N & Giubbini R. [Acut myocardial infarction in a 13-year-old boy with Duchenne’s disease.] G Ital Cardiol 1984, 14, 817–820.
- Cui W, Jang A, Zhang P, Thompson B, Townsend D, Metzger JM & Zhang J. Early detection of myocardial bioenergetic deficits: a 94 tesla complete non invasive 31P MR spectroscopy study in mice with muscular dystrophy. PLOS One 2015, 10, e0135000. [Google Scholar]
- D’Amario D, Amodeo A, Adorisio R, Tiziano FD, Leone AM, Perri G, Bruno P, Massetti M, Ferlini A, Pane M, Niccoli G, Porto I, D’Angelo GA, Borovac JA, Mercuri E & Crea F. A current approach to heart failure in Duchenne muscular dystrophy. Heart 2017, 103, 1770–1779. [Google Scholar] [CrossRef]
- Dai DF, Chen T, Szeto H, Nieves-Cintrón M, Kutyavin V, Santana LF & Rabinovitch PS. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol 2011, 58, 73–82. [Google Scholar] [CrossRef]
- Danialou G, Comtois AS, Dudley R, Karpati G, Vincent G, Des Rosiers C & Petrof BJ. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J 2001, 15, 1655–1657. [Google Scholar] [CrossRef]
- Dasgupta C & Zhang, L. Angiotensin II receptors and drug discovery in cardiovascular disease. Drug Discov Today 2011, 16, 22–34. [Google Scholar]
- de Kermadec JM, Bécane HM, Chénard A, Tertrain F & Weiss Y. Prevalence of left ventricular systolic dysfunction in Duchenne muscular dystrophy: an echocardiographic study. Am Heart J 1994, 127, 618–623. [Google Scholar] [CrossRef]
- De Luca A, Nico B, Liantonio A, Didonna MP, Fraysse B, Pierno S, Burdi R, Mangieri D, Rolland JF, Camerino C, Zallone A, Confalonieri P, Andreetta F, Arnoldi E, Courdier-Fruh I, Magyar JP, Frigeri A, Pisoni M, Svelto M & Conte Camerino D. A multidisciplinary evaluation of the effectiveness of cyclosporine A in dystrophic mdx mice. Am J Pathol 2005, 166, 477–489. [Google Scholar] [CrossRef] [PubMed]
- De Palma C, Morisi F, Cheli S, Pambianco S, Cappello V, Vezzoli M, Rovere-Querini P, Moggio M, Ripolone M, Francolini M, Sandri M & Clementi E. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis 2012, 3, e418. [Google Scholar] [CrossRef] [PubMed]
- Delfin DA, Xu Y, Peterson JM, Guttridge DC, Rafael-Fortney JA & Janssen PM. Improvement of cardiac contractile function by peptide-based inhibition of NF-kappaB in the utrophin/dystrophin-deficient murine model of muscular dystrophy. J Transl Med 2011, 9, 68. [Google Scholar] [CrossRef]
- Demachi J, Kagaya Y, Watanabe J, Sakuma M, Ikeda J, Kakuta Y, Motoyoshi I, Kohnosu T, Sakuma H, Shimazaki S, Sakai H, Kimpara T, Takahashi T, Omura K, Okada M, Saito H & Shirato K. Characteristics of the increase in plasma brain natriuretic peptide level in left ventricular systolic dysfunction, associated with muscular dystrophy in comparison with idiopathic dilated cardiomyopathy. Neuromuscul Disord 2004, 14, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Dikalov SI & Nazarewicz, RR. Angiotensin II-induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal 2013, 19, 1085–1094. [Google Scholar]
- Dittrich S, Tuerk M, Haaker G, Greim V, Buchholz A, Burkhardt B, Fujak A, Trollmann R, Schmid A & Schroeder R. Cardiomyopathy in Duchenne muscular dystrophy: current value of clinical, electrophysiological and imaging findings in children and teenagers. Klin Padiatr 2015, 227, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Dolder M, Wendt S & Wallimann T. Mitochondrial creatine kinase in contact sites: interaction with porin and adenine nucleotide translocase, role in permeability transition and sensitivity to oxidative damage. Neurosignals 2001, 10, 93–111. [Google Scholar] [CrossRef]
- Dorchies OM, Reutenauer-Patte J, Dahmane E, Ismail HM, Petermann O, Patthey-Vuadens O, Comyn SA, Gayi E, Piacenza T, Handa RJ, Décosterd LA, & Ruegg UT. The Anticancer Drug Tamoxifen Counteracts the Pathology in a Mouse Model of Duchenne Muscular Dystrophy. Am. J. Pathol. 2013, 182, 485–504. [Google Scholar] [CrossRef]
- Dual SA, Maforo NG, McElhinney DB, Prosper A, Wu HH, Maskatia S, Renella P, Halnon N & Ennis DB. Right ventricular function and T1-mapping in boys with Duchenne muscular dystrophy. J Magn Reson Imaging 2021, 54, 1503–1513. [Google Scholar] [CrossRef]
- Duan, D. Systemic delivery of adeno-associated viral vectors. Curr Opin Virol 2016, 21, 16–25. [Google Scholar] [CrossRef]
- Duan, D. Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol Ther 2018, 26, 2337–2356. [Google Scholar] [CrossRef] [PubMed]
- Duan D, Goemans N, Takeda S, Mercuri E & Aartsma-Rus A. Duchenne muscular dystrophy. Nat Rev Dis Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Dubinin MV, Talanov EY, Tenkov KS, Starinets VS, Belosludtseva NV & Belosludtsev KN. The effect of deflazacort treatment on the functioning of skeletal muscle mitochondria in Duchenne muscular dystrophy. Int J Mol Sci 2020, 21, 8763. [Google Scholar] [CrossRef] [PubMed]
- Dubinin MV, Talanov EY, Tenkov KS, Starinets VS, Mikheeva IB, & Belosludtsev KN. Transport of Ca2+ and Ca2+-dependent permeability transition in heart. Biochim. Biophys. Acta 2020, 1861, 148250. [Google Scholar]
- mitochondria in the early stages of Duchenne muscular dystrophy.
- Duboc D, Meune C, Lerebours G, Devaux JY, Vaksmann G & Bécane HM. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol 2005, 45, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Duboc D, Meune C, Pierre B, Wahbi K, Eymard B, Toutain A, Berard C, Vaksmann G, Weber S & Bécane HM. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am Heart J 2007, 154, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Dunn JF, Burton KA & Dauncey MJ. Ouabain sensitive Na+ K+-ATPase content is elevated in mdx mice: Implications for the regulation of ions in dystrophic muscle. J Neurol Sci 1995, 133, 11–5. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari D, Dillmann U, Flotats-Bastardas M, Poryo M, Abdul-Khaliq H, Shamdeen MG, Mischo B, Zemlin M & Meyer S. Off-label use of ataluren in four non-ambulatory patients with nonsense mutation Duchenne muscular dystrophy: effects on cardiac and pulmonary function and muscle strength. Front Pediatr 2018, 6, 316. [Google Scholar] [CrossRef] [PubMed]
- Eirin A, Ebrahimi B, Zhang X, Zhu XY, Woollard JR, He Q, Textor SC, Lerman A & Lerman L O. Mitochondrial protection restores renal function in swine atherosclerotic renovascular disease. Cardiovasc Res 2014, 103, 461–472. [Google Scholar] [CrossRef]
- Eisner DA, Caldwell JL, Kistamás K & Trafford AW. Calcium and excitation-contraction coupling in the heart. Circ Res 2017, 121, 181–195. [Google Scholar] [CrossRef]
- El Refaey M, Xu L, Gao Y, Canan BD, Adesanya TMA, Warner SC, Akagi K, Symer DE, Mohler PJ, Ma J; Janssen PML & Han R. In Vivo Genome Editing Restores Dystrophin Expression and Cardiac Function in Dystrophic. Mice Circ Res 2017, 121, 923–929.
- Elliott EI & Sutterwala, FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev 2015, 265, 35–52. [Google Scholar]
- Emery AEH. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Ervasti JM & Campbell, KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol 1993, 122, 809–823. [Google Scholar]
- Escolar DM, Hache LP, Clemens PR, Cnaan A, McDonald CM, Viswanathan V, Kornberg AJ, Bertorini TE, Nevo Y, Lotze T, Pestronk A, Ryan MM, Monasterio E, Day JW, Zimmerman A, Arrieta A, Henricson E, Mayhew J, Florence J,… Connolly AM. Randomized, blinded trial of weekend vs daily prednisone in Duchenne muscular dystrophy. Neurology 2011, 77, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Esposito G & Carsana, A. Metabolic alterations in cardiomyocytes of patients with Duchenne and Becker muscular dystrophies. J Clin Med 2019, 8, 2151. [Google Scholar]
- Evans NP, Misyak SA, Robertson JL, Bassaganya-Riera J & Grange RW. Immune-mediated mechanisms potentially regulate the disease time-course of Duchenne muscular dystrophy and provide targets for therapeutic intervention. PM R 2009, 1, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Fauconnier J, Thireau J, Reiken S, Cassan C, Richard S, Matecki S, Marks AR & Lacampagne A. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 2010, 107, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Fayssoil A, Abasse S & Silverston K. Cardiac involvement classification and therapeutic management in patients with Duchenne muscular dystrophy. J Neuromuscul Dis 2017, 4, 17–23. [Google Scholar] [CrossRef]
- Felsburg, PJ. Overview of immune system development in the dog: comparison with humans. Hum Exp Toxicol 2002, 21, 487–492. [Google Scholar] [CrossRef]
- Feng J, Schaus BJ, Fallavollita JA, Lee TC & Canty JM. Preload induces troponin I degradation independently of myocardial ischemia. Circulation 2001, 103, 2035–2037. [Google Scholar] [CrossRef]
- Finsterer J, Cripe L. Treatment of dystrophin cardiomyopathies. Nat Rev Cardiol 2014, 11, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Finsterer J, Stöllberger C. The heart in human dystrophinopathies. Cardiology 2003, 99, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Fischer Y, Thomas J, Sevilla L, Muñoz P, Becker C, Holman G, Kozka IJ, Palacín M, Testar X, Kammermeier H & Zorzano A. Insulin-induced recruitment of glucose transporter 4 (GLUT4) and GLUT1 in isolated rat cardiac myocytes: evidence of the existence of different intracellular GLUT4 vesicle populations. J Biol Chem 1997, 272, 7085–7092. [Google Scholar] [CrossRef]
- Flanigan KM (2017). Duchenne and Becker muscular dystrophies. In Swaiman’s Pediatric Neurology, 6th edn, ed. Elsevier Inc, pp. e2482–e2492. Edinburgh, UK.
- Florczyk-Soluch U, Polak K & Dulak J. The multifaceted view of heart problem in Duchenne muscular dystrophy. Cell Mol Life Sci 2021, 78, 5447–5468. [Google Scholar] [CrossRef] [PubMed]
- Florian A, Ludwig A, Rösch S, Yildiz H, Sechtem U & Yilmaz A. Myocardial fibrosis imaging based on T1- mapping and extracellular volume fraction (ECV) measurement in muscular dystrophy patients: diagnostic value compared with conventional late gadolinium enhancement (LGE) imaging. Eur Heart J Cardiovasc Imaging 2014, 15, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Florian A-R, Ludwig A, Engelen M, Waltenberger J, Rösch S, Sechtem U & Yilmaz A. Left ventricular systolic function and the pattern of late-gadolinium-enhancement independently and additively predict adverse cardiac events in muscular dystrophy patients. J Cardiovasc Magn Reson 2014, 16, 81. [Google Scholar] [CrossRef]
- Florian A, Rösch S, Bietenbeck M, Engelen M, Stypmann J, Waltenberger J, Sechtem U & Yilmaz A. Cardiac involvement in female Duchenne and Becker muscular dystrophy carriers in comparison to their first-degree male relatives: a comparative cardiovascular magnetic resonance study. Eur Heart J Cardiovasc Imaging 2016, 17, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Frankel KA & Rosser, RJ. The pathology of the heart in progressive muscular dystrophy: epimyocardial fibrosis. Hum Pathol 1976, 7, 375–386. [Google Scholar]
- Gale EM, Caravan P, Rao AG, McDonald RJ, Winfeld M, Fleck RJ & Gee MS. Gadolinium-based contrast agents in pediatric magnetic resonance imaging. Pediatr Radiol 2017, 47, 507–521. [Google Scholar] [CrossRef]
- Gartz M, Haberman M, Prom MJ, Beatka MJ, Strande JL & Lawlor MW. A Long-Term Study Evaluating the Effects of Nicorandil Treatment on Duchenne Muscular Dystrophy-Associated Cardiomyopathy in mdx Mice. J Cardiovasc Pharmacol Ther 2022, 27, 10742484221088655. [Google Scholar]
- Gardner-Medwin, D. Clinical features and classification of the muscular dystrophies. Br Med Bull 1980, 36, 109–15. [Google Scholar] [CrossRef]
- Giatrakos N, Kinali M, Stephens D, Dawson D, Muntoni F & Nihoyannopoulos P. Cardiac tissue velocities and strain rate in the early detection of myocardial dysfunction of asymptomatic boys with Duchenne’s muscular dystrophy: relationship to clinical outcome. Heart 2006, 92, 840–842. [Google Scholar]
- Gillis JC, Benfield P & McTavish D. Idebenone. Drugs & Aging 1994, 5, 133–152. [Google Scholar]
- Giorgio V, Guo L, Bassot C, Petronilli V, & Bernardi P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 2018, 70, 56–63. [Google Scholar] [CrossRef]
- Glancy B & Balaban, RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–7293. [Google Scholar]
- Glatz JFC, Nabben M, Young ME, Schulze PC, Taegtmeyer H & Luiken JJFP. Re-balancing cellular energy substrate metabolism to mend the failing heart. Biochimica et Biophysica Acta: Molecular Basis of Disease 2020, 1866, 165579. [Google Scholar]
- Gloss D, Moxley RT III; Ashwal S & Oskoui M. Practice guideline update summary: corticosteroid treatment of Duchenne muscular dystrophy: report of the guideline development Subcommittee of the American Academy of Neurology. Neurology 2016, 86, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez DR, Treuer AV, Lamirault G, Mayo V, Cao Y, Dulce RA & Hare JM. NADPH oxidase-2 inhibition restores contractility and intracellular calcium handling and reduces arrhythmogenicity in dystrophic cardiomyopathy. Am J Physiol Heart Circ Physiol 2014, 307, H710–H721. [Google Scholar] [CrossRef]
- Goodwin FC & Muntoni, F. Cardiac involvement in muscular dystrophies: molecular mechanisms. Muscle & Nerve, 2005; 32, 577–588. [Google Scholar]
- Gorospe JR, Tharp M, Demitsu T & Hoffman EP. Dystrophin-deficient myofibers are vulnerable to mast cell granule-induced necrosis. Neuromusc Disord 1994, 4, 325–333. [Google Scholar] [CrossRef]
- Götte MJ, Germans T, Rüssel IK, Zwanenburg JJ, Marcus JT, van Rossum AC & van Veldhuisen DJ. Myocardial strain and torsion quantified by cardiovascular magnetic resonance tissue tagging: studies in normal and impaired left ventricular function. J Am Coll Cardiol 2006, 48, 2002–2011. [Google Scholar] [CrossRef] [PubMed]
- Greally E, Davison BJ, Blain A, Laval S, Blamire A, Straub V & MacGowan GA. Heterogeneous abnormalities of in vivo left ventricular calcium influx and function in mouse models of muscular dystrophy cardiomyopathy. J Cardiovasc Magn Reson 2013, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, BH. Role of angiotensin receptor blockers in heart failure: not yet resolved. Circulation 1999, 100, 1032–1034. [Google Scholar] [CrossRef]
- Griffin JL & Des Rosiers, C. Applications of metabolomics and proteomics to the mdx mouse model of Duchenne muscular dystrophy: lessons from downstream of the transcriptome. Genome Med 2009, 1, 32. [Google Scholar]
- Groh, WJ. Arrhythmias in the muscular dystrophies. Heart Rhythm 2012, 9, 1890–1895. [Google Scholar] [CrossRef] [PubMed]
- Guerron AD, Rawat R, Sali A, Spurney CF, Pistilli E, Cha HJ Pandey GS, Gernapudi R, Francia D, Farajian V, Escolar DM, Bossi L, Becker M, Zerr P, de la Porte S, Gordish-Dressman H, Partridge T, Hoffman EP & Nagaraju K. Functional and molecular effects of arginine butyrate and prednisone on muscle and heart in the mdx mouse model of Duchenne muscular dystrophy. PLOS One 2010, 5, e11220. [Google Scholar]
- Gueven N, Ravishankar P, Eri R & Rybalka E. Idebenone: When an antioxidant is not an antioxidant. Redox Biol 2021, 38, 101812. [Google Scholar]
- Guglieri M, Clemens PR, Perlman SJ, Smith EC, Horrocks I, Finkel RS, Mah JK, Deconinck N, Goemans N, Haberlova J, Straub V, Mengle-Gaw LJ, Schwartz BD, Harper AD, Shieh PB, De Waele L, Castro D, Yang ML, Ryan MM, McDonald CM, Tulinius M, Webster R, McMillan HJ, Kuntz NL, Rao VK, Baranello G, Spinty S, Childs A-M, Sbrocchi AM, Selby KA, Monduy M, Nevo Y, Vilchez-Padilla JJ, Nascimento-Osorio A, Niks EH, de Groot IJM, Katsalouli M, James MK, van den Anker J, Damsker JM, Ahmet A, Ward LM, Jaros M, Shale P, Dang UJ, & Hoffman EP. Efficacy and Safety of Vamorolone vs Placebo and Prednisone Among Boys With Duchenne Muscular Dystrophy: A Randomized Clinical Trial. JAMA Neurol 2022, 79, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Guzun R, Gonzalez-Granillo M, Karu-Varikmaa M, Grichine A, Usson Y, Kaambre T, Guerrero-Roesch K, Kuznetsov A, Schlattner U & Saks V. Regulation of respiration in muscle cells in vivo by VDAC through interaction with the cytoskeleton and MtCK within Mitochondrial Interactosome. Biochim Biophys Acta 2012, 1818, 1545–1554. [Google Scholar] [CrossRef]
- Haaf P, Garg P, Messroghli DR, Broadbent DA, Greenwood JP & Plein S. Cardiac T1 mapping and extracellular volume (ECV) in clinical practice: a comprehensive review. J Cardiovasc Magn Reson 2016, 18, 89. [Google Scholar] [CrossRef]
- Hagenbuch SC, Gottliebson WM, Wansapura J, Mazur W, Fleck R, Benson DW & Hor KN. Detection of progressive cardiac dysfunction by serial evaluation of circumferential strain in patients with Duchenne muscular dystrophy. Am J Cardiol 2010, 105, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Hakim CH, Wasala NB, Nelson CE, Wasala LP, Yue Y, Louderman JA, Lessa TB, Dai A, Zhang K, Jenkins GJ, Nance ME, Pan X, Kodippili K, Yang NN, Chen SJ, Gersbach CA & Duan D. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight 2018, 3, 1–13. [Google Scholar]
- Hakim CH, Wasala NB, Pan X, Kodippili K, Yue Y, Zhang K, Yao G, Haffner B, Duan SX, Ramos J, Schneider JS, Yang NN, Chamberlain JS & Duan D. A five-repeat micro-dystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J-mdx model of Duchenne muscular dystrophy. Mol Ther Methods Clin Dev 2017, 6, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Hart CC, il Lee Y, Xie J & Gao G, Hammers DW, Sweeney L (2022). Potential limitations of micro-dystrophin gene therapy for Duchenne muscular dystrophy bioRxiv DOI: 1002510519. [CrossRef]
- Hayes HM, Angerosa J, Piers AT, White JD, Koleff J, Thurgood M, Moody J, Cheung MM, & Pepe S. Preserved Left Ventricular Function despite Myocardial Fibrosis and Myopathy in the Dystrophin-Deficient D2.B10-Dmdmdx/J Mouse. Oxid Med Cell Longev, 2022; 5362115.
- Heier CR, Yu Q, Fiorillo AA, Tully CB, Tucker A, Mazala DA, Uaesoontrachoon K, Srinivassane S, Damsker JM, Hoffman EP, Nagaraju K & Spurney CF. Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy. Life Sci Alliance 2019, 2, e201800186. [Google Scholar] [CrossRef] [PubMed]
- Hoffman EP, Reeves E, Damsker J, Nagaraju K, McCall JM, Connor EM & Bushby K. Novel approaches to corticosteroid treatment in Duchenne muscular dystrophy. Phys Med Rehabil Clin N Am 2012, 23, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, EP. The discovery of dystrophin, the protein product of the Duchenne muscular dystrophy gene. FEBS J 2020, 287, 3879–3887. [Google Scholar] [CrossRef] [PubMed]
- Hor KN, Mah ML, Johnston P, Cripe TP & Cripe LH. Advances in the diagnosis and management of cardiomyopathy in Duchenne muscular dystrophy. Neuromuscul Disord 2018, 28, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Hor KN, Kissoon N, Mazur W, Gupta R, Ittenbach RF, Al-Khalidi HR, Cripe LH, Raman SV, Puchalski MD, Gottliebson WM & Benson DW. Regional circumferential strain is a biomarker for disease severity in Duchenne muscular dystrophy heart disease: A cross-sectional study. Pediatr Cardiol 2015, 36, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Hor KN, Taylor MD, Al-Khalidi HR, LH, Raman SV, Jefferies JL, O’Donnell R, Benson DW & Mazur W. Prevalence and distribution of late gadolinium enhancement in a large population of patients with Duchenne muscular dystrophy: Effect of age and left ventricular systolic function. J Cardiovasc Magn Reson 2013, 15, 107. [Google Scholar] [CrossRef]
- Hor KN, Wansapura J, Markham LW, Mazur W, Cripe LH, Fleck R, Benson DW & Gottliebson WM. Circumferential strain analysis identifies strata of cardiomyopathy in Duchenne muscular dystrophy: A cardiac magnetic resonance tagging study. J Am Coll Cardiol 2009, 53, 1204–1210. [Google Scholar] [CrossRef]
- Houde S, Filiatrault M, Fournier A, Dubé J, D'Arcy S, Bérubé D, Brousseau Y, Lapierre G & Vanasse M. Deflazacort use in Duchenne muscular dystrophy: An 8-year follow-up. Pediatr Neurol 2008, 38, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Howard ZM, Dorn LE, Lowe J, Gertzen MD, Ciccone P, Rastogi N, Odom GL, Accornero F, Chamberlain JS & Rafael-Fortney JA. Micro-dystrophin gene therapy prevents heart failure in an improved Duchenne muscular dystrophy cardiomyopathy mouse model. JCI Insight 2021, 6, e146511. [Google Scholar] [CrossRef] [PubMed]
- Howell JM, Fletcher S, Kakulas BA, O’Hara M, Lochmuller H & Karpati G. Use of the dog model for Duchenne muscular dystrophy in gene therapy trials. Neuromuscul Disord 1997, 7, 325–328. [Google Scholar] [CrossRef]
- Hughes MC, Ramos SV, Turnbull PC, Edgett BA, Huber JS, Polidovitch N, Schlattner U, Backx PH, Simpson JA & Perry CGR. Impairments in left ventricular mitochondrial bioenergetics precede overt cardiac dysfunction and remodelling in Duchenne muscular dystrophy. J Physiol 2020, 598, 1377–1392. [Google Scholar] [CrossRef] [PubMed]
- Hughes MC, Ramos SV, Turnbull PC, Rebalka IA, Cao A, Monaco CMF, Varah NE, Edgett BA, Huber JS, Tadi P, Delfinis LJ, Schlattner U, Simpson JA, Hawke TJ & Perry CGR. Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2O2 emission during impaired oxidative phosphorylation. J Cachexia Sarcopenia Muscle 2019, 10, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Iwata Y, Ohtake H, Suzuki O, Matsuda J, Komamura K & Wakabayashi S. Blockade of sarcolemmal TRPV2 accumulation inhibits progression of dilated cardiomyopathy. Cardiovasc Res 2013, 99, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Ionăsescu V, Luca N, & Vuia O. Respiratory control and oxidative phosphorylation in the dystrophic muscle. Acta Neurol Scand 1967, 43, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL & Sutterwala FS. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Jacobs PA, Hunt PA, Mayer M & Bart RD. Duchenne muscular dystrophy (DMD) in a female with an X/autosome translocation: further evidence that the DMD locus is at Xp21. Am J Hum Genet 1981, 33, 513–518. [Google Scholar]
- Janssen PML, Murray JD, Schill KE, Rastogi N, Schultz EJ, Tran T, Raman SV & Rafael-Fortney JA. Prednisolone attenuates improvement of cardiac and skeletal contractile function and histopathology by lisinopril and spironolactone in the mdx mouse model of Duchenne muscular dystrophy. PLOS One 2014, 9, e88360. [Google Scholar]
- Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, Neish SR, Smith EO & Towbin J. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation 2005, 112, 2799–2804. [Google Scholar] [CrossRef]
- Johnston JR & McNally, EM. Genetic correction strategies for Duchenne muscular dystrophy and their impact on the heart. Prog Pediatr Cardiol 2021, 63, 101460. [Google Scholar]
- Johnstone VP, Viola HM & Hool LC. Dystrophic cardiomyopathy—potential role of calcium in pathogenesis, treatment and novel therapies. Genes (Basel) 2017, 8, 108. [Google Scholar] [CrossRef]
- Judge DP, Kass DA, Thompson WR & Wagner KR. Pathophysiology and therapy of cardiac dysfunction in Duchenne muscular dystrophy. Am J Cardiovasc Drugs 2011, 11, 287–294. [Google Scholar] [CrossRef]
- Jung C, Martins AS, Niggli E & Shirokova N. Dystrophic cardiomyopathy: amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovas Res 2007, 77, 766–773. [Google Scholar]
- Kajimoto H, Ishigaki K, Okumura K, Tomimatsu H, Nakazawa M, Saito K, Osawa M & Nakanishi T. Beta-blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Circulation 2006, 70, 991–994. [Google Scholar] [CrossRef]
- Kamdar F, Das S, Gong W, Kamdar AK, Meyers TA, Shah P, Ervasti JM, Townsend D, Kamp TJ, Wu JC, Garry MG, Zhang J & Garry DJ. Stem cell-derived cardiomyocytes and beta-adrenergic receptor blockade in Duchenne muscular dystrophy cardiomyopathy. J Am Coll Cardiol 2020, 75, 1159–1174. [Google Scholar] [CrossRef] [PubMed]
- Kamdar F & Garry, DJ. Dystrophin-deficient cardiomyopathy. J Am Coll Cardiol 2016, 67, 2533–2546. [Google Scholar]
- Kang C, Badr MA, Kyrychenko V, Eskelinen EL & Shirokova N. Deficit in PINK1/PARKIN-mediated mitochondrial autophagy at late stages of dystrophic cardiomyopathy. Cardiovasc Res 2018, 114, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, Lin SCJ, Aronow BJ, Tallquist MD & Molkentin JD. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun 2016, 7, 12260. [Google Scholar] [CrossRef] [PubMed]
- Kaspar RW, Allen HD & Montanaro F. Current understanding and management of dilated cardiomyopathy in Duchenne and Becker muscular dystrophy. J Am Acad Nurse Pract 2009, 21, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Kawai T, Forrester SJ, O’Brien S, Baggett A, Rizzo V & Eguchi S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol Res 2017, 125, 4–13. [Google Scholar] [CrossRef]
- Kellman P, Chefd’hotel C, Lorenz CH, Mancini C, Arai AE & McVeigh ER. High spatial and temporal resolution cardiac cine MRI from retrospective reconstruction of data acquired in real time using motion correction and resorting. Magn Reson Med 2009, 62, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Kellman P, Bandettini WP, Mancini C, Hammer-Hansen S, Hansen MS & Arai AE. Characterization of myocardial T1-mapping bias caused by intramyocardial fat in inversion recovery and saturation recovery techniques. J Cardiovasc Magn Reson 2015, 17, 1–11. [Google Scholar]
- Khairallah RJ, Shi G, Sbrana F, Prosser BL, Borroto C, Mazaitis MJ, Hoffman EP, Mahurkar A, Sachs F, Sun Y, Chen YW, Raiteri R, Lederer WJ, Dorsey SG & Ward CW. Microtubules underlie dysfunction in Duchenne muscular dystrophy. Sci Signal 2012, 5, ra56. [Google Scholar]
- Khairallah M; Khairallah R, Young ME, Dyck JR, Petrof BJ & Des Rosiers C. Metabolic and signaling alterations in dystrophin-deficient hearts precede overt cardiomyopathy. J Mol Cell Cardiol 2007, 43, 119–129. [Google Scholar] [CrossRef]
- Koenig X, Rubi L, Obermair GJ, Cervenka R, Dang XB, Lukacs P, Kummer S, Bittner RE, Kubista H, Todt H & Hilber K. Enhanced currents through L-type calcium channels in cardiomyocytes disturb the electrophysiology of the dystrophic heart. Am J Physiol Heart Circ Physiol 2014, 306, H564–H573. [Google Scholar] [CrossRef]
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C & Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Koufen P, Rück A, Brdiczka D, Wendt S, Wallimann T & Stark G. Free radical-induced inactivation of creatine kinase: influence on the octameric and dimeric states of the mitochondrial enzyme (Mib-CK). Biochem J 1999, 344, 413–417. [Google Scholar] [CrossRef]
- Kranias EG & Hajjar, RJ. Modulation of cardiac contractility by the phopholamban/SERCA2a regulatome. Circ Res 2012, 110, 1646. [Google Scholar]
- Krumenacker JS, Hanafy KA & Murad F. Regulation of nitric oxide and soluble guanylyl cyclase. Brain Res Bull 2004, 62, 505–515. [Google Scholar] [CrossRef]
- Krumenacker JS, Hyder SM & Murad F. Estradiol rapidly inhibits soluble guanylyl cyclase expression in rat uterus. Proc Natl Acad Sci U S A 2001, 98, 717–722. [Google Scholar] [CrossRef]
- Kumar A & Boriek, AM. Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: a possible role in Duchenne muscular dystrophy. FASEB Jo 2003, 17, 386–396. [Google Scholar]
- Kuno A, Hosoda R, Sebori R, Hayashi T, Sakuragi H, Tanabe M & Horio Y. Resveratrol ameliorates mitophagy disturbance and improves cardiac pathophysiology of dystrophin-deficient mdx mice. Sci Rep 2018, 8, 15555. [Google Scholar] [CrossRef]
- Kyrychenko V, Poláková E, Janíček R & Shirokova N. Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium 2015, 58, 186–195. [Google Scholar] [CrossRef]
- Kyrychenko S, Poláková E, Kang C, Pocsai K, Ullrich ND, Niggli E & Shirokova N. Hierarchical accumulation of RyR post-translational modifications drives disease progression in dystrophic cardiomyopathy. Cardiovasc Res 2013, 97, 666–675. [Google Scholar] [CrossRef]
- LaCorte JC, Cabreriza SE, Rabkin DG, Printz BF, Coku L, Weinberg A, Gersony WM & Spotnitz HM. Correlation of the Tei index with invasive measurements of ventricular function in a porcine model. J Am Soc Echocardiogr 2003, 16, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Laing NG, Siddique T, Bartlett R, Yamaoka LH, Hung WY, Pericak-Vance MA & Roses AD. Duchenne muscular dystrophy: detection of deletion carriers by spectrophotometric densitometry. Clin Genet 1989, 35, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Larcher T, Lafoux A, Tesson L, Remy S, Thepenier V, François V, Le Guiner C, Goubin H, Dutilleul M, Guigand L, Toumaniantz G, De Cian A, Boix C, Renaud JB, Cherel Y, Giovannangeli C, Concordet JP, Anegon I & Huchet C. Characterization of dystrophin deficient rats: a new model for Duchenne muscular dystrophy. PLoS One 2014, 9, e110371. [Google Scholar]
- Law ML, Prins KW, Olander ME & Metzger JM. Exacerbation of dystrophic cardiomyopathy by phospholamban deficiency mediated chronically increased cardiac Ca(2+) cycling in vivo. Am J Physiol Heart Circ Physiol 2018, 315, H1544–H1552. [Google Scholar] [CrossRef]
- Law ML, Cohen H, Martin AA, Angulski ABB & Metzger JM. Dysregulation of calcium handling in Duchenne muscular dystrophy-associated dilated cardiomyopathy: mechanisms and experimental therapeutic strategies. J Clin Med 2020, 9, 520. [Google Scholar] [CrossRef]
- Lu QL, Rabinowitz A, Chen YC, Yokota T, Yin H, Alter J, Jadoon A, Bou-Gharios G & Partridge T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc Natl Acad Sci U S A 2005, 102, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Le S, Yu M, Hovan L, Zhao Z, Ervasti J & Yan J. Dystrophin as a molecular shock absorber. ACS Nano 2018, 12, 12140–12148. [Google Scholar] [CrossRef] [PubMed]
- Le Guiner C Servais L, Montus M, Larcher T, Fraysse B, Moullec S, Allais M, François V, Dutilleul M, Malerba A, Koo T, Thibaut JL, Matot B, Devaux M, Le Duff J, Deschamps JY, Barthelemy I, Blot S, Testault I,… Dickson G. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat Commun 2017, 8, 16105. [Google Scholar] [CrossRef] [PubMed]
- Lee S, Lee M & Hor KN. The role of imaging in characterizing the cardiac natural history of Duchenne muscular dystrophy. Pediatr Pulmonol 2021, 56, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Lemasters JJ, Theruvath TP, Zhong Z & Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta 2009, 1787, 1395–1401. [Google Scholar] [CrossRef]
- Levy PT, Machefsky A, Sanchez AA, Patel MD, Rogal S, Fowler S, Yaeger L, Hardi A, Holland MR, Hamvas A & Singh GK. Reference ranges of left ventricular strain measures by two-dimensional speckle-tracking echocardiography in children: a systematic review and meta-analysis. J Am Soc Echocardiogr 2016, 29, 209–225e6. [Google Scholar] [CrossRef]
- Li X, Zhang W, Cao Q, Wang Z, Zhao M, Xu L & Zhuang Q. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov 2020, 6, 80. [Google Scholar] [CrossRef]
- Lin B, Li Y, Han L, Kaplan AD, Ao Y, Kalra S, Bett GC, Rasmusson RL, Denning C & Yang L. Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with Duchenne muscular dystrophy. Dis Model Mech 2015, 8, 457–466. [Google Scholar] [CrossRef]
- Liu D, Ahmet A, Ward L, Krishnamoorthy P, Mandelcorn ED, Leigh R, Brown JP, Cohen A & Kim H. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin Immunol 2013, 9, 30. [Google Scholar] [CrossRef]
- Lorin C, Vögeli I & Niggli E. Dystrophic cardiomyopathy: role of TRPV2 channels in stretch-induced cell damage. Cardiovasc Res 2015, 106, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Luan P, D’Amico D, Andreux PA, Laurila PP, Wohlwend M, Li H, Imamura de Lima T, Place N, Rinsch C, Zanou N & Auwerx J. Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy. Sci Transl Med 2021, 13, eabb0319. [Google Scholar] [CrossRef] [PubMed]
- MacLennan D & Kranias, E. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol 2003, 4, 566–577. [Google Scholar]
- Magrath P, Maforo N, Renella P, Nelson SF, Halnon N & Ennis DB. Cardiac MRI biomarkers for Duchenne muscular dystrophy. Biomark Med 2018, 12, 1271–1289. [Google Scholar] [CrossRef] [PubMed]
- Magri F, Govoni A, D’Angelo MG, Del Bo R, Ghezzi S, Sandra G, Turconi AC, Sciacco M, Ciscato P, Bordoni A, Tedeschi S, Fortunato F, Lucchini V, Bonato S, Lamperti C, Coviello D, Torrente Y, Corti S, Moggio M,… Comi, GP. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol 2011, 258, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T & Jette N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord 2014, 24, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Markham LW, Spicer RL, Khoury PR, Wong BL, Mathews KD & Cripe LH. Steroid therapy and cardiac function in Duchenne muscular dystrophy. Pediatr Cardiol 2005, 26, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Markham LW, Kinnett K, Wong BL, Benson DW & Cripe LH. Corticosteroid treatment retards development of ventricular dysfunction in Duchenne muscular dystrophy. Neuromuscul Disord 2008, 2008, 18–365. [Google Scholar]
- Markham LW, Michelfelder EC, Border WL, Khoury PR, Spicer RL, Wong BL, Benson DW & Cripe LH. Abnormalities of diastolic function precede dilated cardiomyopathy associated with Duchenne muscular dystrophy. J Am Soc Echocardiogr 2006, 19, 865–871. [Google Scholar] [CrossRef]
- Masubuchi N, Shidoh Y, Kondo S, Takatoh J & Hanaoka K. Subcellular localization of dystrophin isoforms in cardiomyocytes and phenotypic analysis of dystrophin-deficient mice reveal cardiac myopathy is predominantly caused by a deficiency in full-length dystrophin. Exp Anim 2013, 62, 211–217. [Google Scholar] [CrossRef]
- Matsumura T, Saito T, Fujimura H & Shinno S. Cardiac troponin I for accurate evaluation of cardiac status in myopathic patients. Brain Dev 2007, 29, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Matsumura T, Tamura T, Kuru S, Kikuchi Y & Kawai M. Carvedilol can prevent cardiac events in Duchenne muscular dystrophy. Intern Med 2010, 49, 1357–1363. [Google Scholar] [CrossRef] [PubMed]
- Mavrogeni S, Bratis K, Papavasiliou A, Skouteli E, Karanasios E, Georgakopoulos D, Kolovou G & Papadopoulos G. CMR detects subclinical cardiomyopathy in mother-carriers of Duchenne and Becker muscular dystrophy. JACC Cardiovasc Imaging 2013, 6, 26–28. [Google Scholar]
- McDonald CM, Meier T, Voit T, Schara U, Straathof CS, D’Angelo MG, Bernert G, Cuisset JM, Finkel RS, Goemans N, Rummey C, Leinonen M, Spagnolo P, Buyse GM & DELOS Study Group. Idebenone reduces respiratory complications in patients with Duchenne muscular dystrophy. Neuromuscul Disord 2016, 26, 473–480. [Google Scholar] [CrossRef]
- McNally EM, Kaltman JR, Benson DW, Canter CE, Cripe LH, Duan D, Finder JD, Hoffman EP, Judge DP, Kertesz N, Kinnett K, Kirsch R, Metzger JM, Pearson GD, Rafael-Fortney JA, Raman SV, CF, Targum SL, Wagner KR & Markham LW. Contemporary cardiac issues in Duchenne muscular dystrophy. Circulation 2015, 131, 1590–1598. [Google Scholar] [CrossRef]
- Mehmood M, Hor KN, Al-Khalidi HR, Benson DW, Jefferies JL, Taylor MD, Egnaczyk GF, Raman SV, Basu SK, Cripe LH, Germann J & Mazur W. Comparison of right and left ventricular function and size in Duchenne muscular dystrophy. Eur J Radiol 2015, 84, 1938–1942. [Google Scholar] [CrossRef]
- Mehmood M, Ambach SA, Taylor MD, Jefferies JL, Raman SV, Taylor RJ, Sawani H, Mathew J, Mazur W, Hor KN & Al-Khalidi HR. Relationship of right ventricular size and function with respiratory status in Duchenne muscular dystrophy. Pediatr Cardiol 2016, 37, 878–883. [Google Scholar] [CrossRef]
- Mendell JR, Shilling C, Leslie ND, Flanigan KM, al-Dahhak R, Gastier-Foster J; Kneile K, Dunn DM, Duval B, Aoyagi A, Hamil C, Mahmoud M, Roush K, Bird L, Rankin C, Lilly H, Street N, Chandrasekar R & Weiss RB. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol 2012, 71, 304–313. [Google Scholar] [CrossRef]
- Menon SC, Etheridge SP, Liesemer KN, Williams RV, Bardsley T, Heywood MC & Puchalski MD. Predictive value of myocardial delayed enhancement in Duchenne muscular dystrophy. Pediatr Cardiol 2014, 35, 1279–1285. [Google Scholar] [CrossRef]
- Mercuri E, Bonnemann CG & Muntoni F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Mertens L, Ganame J, Claus P, Goemans N, Thijs D, Eyskens B, Van Laere D, Bijnens B, D’hooge J, Sutherland GR & Buyse G. Early regional myocardial dysfunction in young patients with Duchenne muscular dystrophy. J Am Soc Echocardiogr 2008, 21, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Meyer LE, Machado LB, Santiago APSA, da-Silva WS, Felice FGD, Holub O, Oliveira MF, & Galina A. Mitochondrial creatine kinase activity prevents reactive oxygen species generation: antioxidant role of mitochondrial kinase-dependent ADP re-cycling activity. J Biol Chem 2006, 281, 37361–37371. [Google Scholar] [CrossRef] [PubMed]
- Meyer RA & Sweeney HL, Kushmerick MJ. A simple analysis of the “phosphocreatine shuttle”. Am J Physiol 1984, 246, C365–C377. [Google Scholar] [CrossRef] [PubMed]
- Meyers TA & Townsend, D. Early right ventricular fibrosis and reduction in biventricular cardiac reserve in the dystrophin-deficient mdx heart. Am J Physiol Heart Circ Physiol 2015, 308, H303–H315. [Google Scholar]
- Meyers TA & Townsend, D. Cardiac pathophysiology and the future of cardiac therapies in Duchenne muscular dystrophy. Int J Mol Sci 2019, 20, E4098. [Google Scholar]
- Meyers TA, Heitzman JA & Townsend D. 2019. Acute myocardial injury in mdx hearts ameliorated by ARB but not ACE inhibitor treatment. bioRxiv. [CrossRef]
- Meyers TA, Heitzman JA, Krebsbach A, Aufdembrink LM, Hughes R, Bartolomucci A & Townsend D. Acute AT1R blockade prevents isoproterenol-induced injury in mdx hearts. J Mol Cell Cardiol 2019, 128, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Mijares A, Altamirano F, Kolster J, Adams JA & Lopez JR. Age-dependent changes in diastolic Ca2+ and Na+ concentrations in dystrophic cardiomyopathy: role of Ca2+ entry and IP3. Biochem Biophys Res Commun 2014, 452, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, Sweeney HL, Robbins J & Molkentin JD. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Mohyuddin T, Jacobs IB & Bahler RC. B-type natriuretic peptide and cardiac dysfunction in Duchenne muscular dystrophy. Int J Cardiol 2007, 119, 389–391. [Google Scholar] [CrossRef]
- Momose M, Iguchi N, Imamura K, Usui H, Ueda T, Miyamoto K & Inaba S. Depressed myocardial fatty acid metabolism in patients with muscular dystrophy. Neuromuscul Disord 2001, 11, 464–469. [Google Scholar] [CrossRef]
- Mondillo S, Galderisi M, Mele D, Cameli M, Lomoriello VS, Zacà V, Ballo P, D’Andrea A, Muraru D, Losi M, Agricola E, D’Errico A, Buralli S, Sciomer S, Nistri S, Badano L & Echocardiography Study Group of the Italian Society of Cardiology (Rome, Italy). Speckle-tracking echocardiography: a new technique for assessing myocardial function. J Ultrasound Med 2011, 30, 71–83. [Google Scholar] [CrossRef]
- Mori K, Hayabuchi Y, Inoue M, Suzuki M, Sakata M, Nakagawa R, Kagami S, Tatara K, Hirayama Y & Abe Y. Myocardial strain imaging for early detection of cardiac involvement in patients with Duchenne’s progressive muscular dystrophy. Echocardiography 2007, 24, 598–608. [Google Scholar] [CrossRef]
- Muralidharan P, Cserne Szappanos H, Ingley E & Hool LC. The cardiac L-type calcium channel alpha subunit is a target for direct redox modification during oxidative stress-the role of cysteine residues in the alpha interacting domain. Clin Exp Pharmacol Physiol 2017, 44, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Murphy, MP. Targeting lipophilic cations to mitochondria. Biochim Biophys Acta 2008, 1777, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Naik E & Dixit, VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med 2011, 208, 417–420. [Google Scholar]
- Nakamura K, Fujii W Tsuboi M, Tanihata J, Teramoto N, Takeuchi S, Naito K, Yamanouchi K & Nishihara M. Generation of muscular dystrophy model rats with a CRISPR/Cas system. Sci Rep 2014, 4, 5635. [Google Scholar] [CrossRef]
- Nakamura A, Fueki N, Shiba N, Motoki H, Miyazaki D, Nishizawa H, Echigoya Y, Yokota T, Aoki Y & Takeda S. Deletion of exons 3-9 encompassing a mutational hot spot in the DMD gene presents an asymptomatic phenotype, indicating a target region for multiexon skipping therapy. J Exp Med 2016, 61, 663–667. [Google Scholar]
- Namgoog JH & Bertoni, C. Clinical potential of ataluren in the treatment of Duchenne muscular dystrophy. J Exp Med 2016, 6, 37–48. [Google Scholar]
- Naruse H, Miyagi J, Arii T, Ohyanagi M, Iwasaki T & Jinnai K. The relationship between clinical stage, prognosis and myocardial damage in patients with Duchenne-type muscular dystrophy: five-year follow-up study. Ann Nucl Med 2004, 18, 203–208. [Google Scholar] [CrossRef]
- Nelson CE, Wu Y, Gemberling MP, Oliver ML, Waller MA, Bohning JD, Robinson-Hamm JN, Bulaklak K, Castellanos Rivera RM, Collier JH, Asokan A & Gersbach CA. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat Med 2019, 25, 427–432. [Google Scholar] [CrossRef]
- Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, Madhavan S, Pan X, Ran FA, Yan WX, Asokan A, Zhang F, Duan D & Gersbach CA. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Nelson DM, Lindsay A, Judge LM, Duan D, Chamberlain JS, Lowe DA, & Ervasti JM. Variable rescue of microtubule and physiological phenotypes in mdx muscle expressing different miniaturized dystrophins. Hum Mol Genet 2018, 27, 2090–2100. [Google Scholar] [CrossRef] [PubMed]
- Nicholls DG & Ferguson SJ (2013). Bioenergetics 4. Elsevier: p. 434.
- Nigro G, Comi LI, Limongelli FM, Giugliano MAM, Politano L, Petretta V, Passamano L & Stefanelli S. Prospective study of X-linked progressive muscular dystrophy in campania. Muscle & Nerve 1983, 6, 253–262. [Google Scholar]
- Nigro G, Comi LI, Politano L &, Bain RJI. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Nikolaidis LA, Sturzu A, Stolarski C, Elahi D, Shen YT & Shannon RP. The development of myocardial insulin resistance in conscious dogs with advanced dilated cardiomyopathy. Cardiovasc Res 2004, 61, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Nisoli E, Falcone S, Tonello C, & Clementi E. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 16507–16512. [Google Scholar] [CrossRef]
- Obi C, Smith AT, Hughes GJ & Adeboye AA. Targeting mitochondrial dysfunction with elamipretide. Heart Fail Rev 2022, 27, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Ogata H, Ishikawa Y, Ishikawa Y & Minami R. Beneficial effects of beta-blockers and angiotensin-converting enzyme inhibitors in Duchenne muscular dystrophy. J Cardiol 2009, 53, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Ogata H, Nakatani S, Ishikawa Y, Negishi A, Kobayashi M & Minami R. Myocardial strain changes in Duchenne muscular dystrophy without overt cardiomyopathy. Int J Cardiol 2007, 115, 190–195. [Google Scholar] [CrossRef]
- Olchowy C, Cebulski K, Łasecki M, Chaber R, Olchowy A, Kałwak K & Zaleska-Dorobisz U. The presence of the gadolinium-based contrast agent depositions in the brain and symptoms of gadolinium neurotoxicity: a systematic review. PLOS One 2017, 12, e0171704. [Google Scholar]
- Olivieri LJ, Kellman P, McCarter RJ, Cross RR, Hansen MS & Spurney CF. Native T1 values identify myocardial changes and stratify disease severity in patients with Duchenne muscular dystrophy. J Cardiovasc Magn Reson 2016, 18, 72. [Google Scholar] [CrossRef] [PubMed]
- Olthoff JT, Lindsay A, Abo-Zahrah R, Baltgalvis KA, Patrinostro X, Belanto JJ, Yu D-Y, Perrin BJ, Garry DJ, Rodney GG, Lowe DA, & Ervasti JM. Loss of peroxiredoxin-2 exacerbates eccentric contraction-induced force loss in dystrophin-deficient muscle. Nat Commun 2018, 9, 5104. [Google Scholar] [CrossRef] [PubMed]
- Panovský R, Pešl M, Máchal J, Holeček T, Feitová V, Juříková L, Masárová L, Pešlová E, Opatřil L, Mojica-Pisciotti ML & Kincl V. Quantitative assessment of left ventricular longitudinal function and myocardial deformation in Duchenne muscular dystrophy patients. Orphanet J Rare Dis 2021, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Passamano L, Taglia A, Palladino A, Viggiano E, D’Ambrosio P, Scutifero M, Cecio MR, Torre V, De Luca F, Picillo E, Paciello O, Piluso G, Nigro G & Politano L. Improvement of survival in Duchenne muscular dystrophy: retrospective analysis of 835 patients. Acta Myol 2012, 31, 121–125. [Google Scholar]
- Patrianakos AP, Zacharaki A, Kalogerakis A, Solidakis G, Parthenakis F & Vardas P. Two-dimensiona global and segmental longitudinal strain: are the results from software in different high-end ultrasound systems comparable? Echo Res Pract 2015, 2, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Pauly M, Daussin F, Burelle Y, Li T, Godin R, Fauconnier J, Koechlin-Ramonatxo C, Hugon G, Lacampagne A, Coisy-Quivy M, Liang F, Hussain S, Matecki S & Petrof BJ. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J Pathol 2012, 181, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Perazella, MA. Current status of gadolinium toxicity in patients with kidney disease. Clin J Am Soc Nephrol 2009, 4, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Perloff JK, Roberts WC, de Leon AC & O’Doherty D. The distinctive electrocardiogram of Duchenne’s progressive muscular dystrophy. Am J Med 1967, 42, 179–188. [Google Scholar] [CrossRef]
- Perloff JK, Henze E & Schelbert HR. Alterations in regional myocardial metabolism, perfusion, and wall motion in Duchenne muscular dystrophy studied by radionuclide imaging. Circulation 1984, 69, 33–42. [Google Scholar] [CrossRef]
- Petrof BJ, Shrager JB, Stedman HH, Kelly AM & Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A 1993, 90, 3710–3714. [Google Scholar] [CrossRef]
- Piga D, Salani S, Magri F, Brusa R, Mauri E, Comi GP, Bresolin N & Corti S. Human induced pluripotent stem cell models for the study and treatment of Duchenne and Becker muscular dystrophies. Ther Adv Neurol Disord 2019, 12, 175628641983347. [Google Scholar] [CrossRef] [PubMed]
- Pinniger GJ, Terrill JR, Assan EB, Grounds MD, & Arthur PG. Pre-clinical evaluation of N-acetylcysteine reveals side effects in the mdx mouse model of Duchenne muscular dystrophy. J Physiol 2017, 595, 7093–7107. [Google Scholar] [CrossRef] [PubMed]
- Politano L, Palladino A, Petretta VR, Mansi L, Passamano L, Nigro G, Comi LI & Nigro G. ST-segmen displacement in Duchenne muscular dystrophy: myocardial necrosis or apoptosis? Acta myol 2003, 22, 5–10. [Google Scholar]
- Porte-Thomé F, Nagaraju K, Yu Q, Tatem K, Bkaily G, Scholz W, Slade A, Bot N & Kant C. Development of Rimeporide, a sodium-hydrogen exchanger (NHE-1) inhibitor, for patients with Duchenne muscular dystrophy. Neuromuscul Disord 2015, 25, S259–S260. [Google Scholar] [CrossRef]
- Potter RA, Griffin DA, Heller KN, Peterson EL, Clark EK, Mendell JR & Rodino-Klapac LR. Dose-escalation study of systemically delivered rAAVrh74MHCK7 micro-dystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther 2021, 32, 375–389. [Google Scholar] [CrossRef]
- Power A, Poonja S, Disler D, Myers K, Patton DJ, Mah JK, Fine NM & Greenway SC. Echocardiographic image quality deteriorates with age in children and young adults with Duchenne muscular dystrophy. Front Cardiovasc Med 2017, 4, 82. [Google Scholar] [CrossRef]
- Prakash N, Suthar R, Sihag BK, Debi U, Kumar RM & Sankhyan N. Cardiac MRI and echocardiography for early diagnosis of cardiomyopathy among boys with Duchenne muscular dystrophy: a cross-sectional study. Front Pediatr 2022, 10, 818608. [Google Scholar] [CrossRef] [PubMed]
- Previtali SC, Gidaro T, Díaz-Manera J, Zambon A, Carnesecchi S, Roux-Lombard P, Spitali P, Signorelli M, Szigyarto CA, Johansson C, Gray J, Labolle D, Porte Thomé F, Pitchforth J, Domingos J & Muntoni F. Rimeporide as a first- in-class NHE-1 inhibitor: Results of a phase Ib trial in young patients with Duchenne Muscular Dystrophy. Pharmacol Res 2020, 159, 104999. [Google Scholar] [CrossRef] [PubMed]
- Prins KW, Humston JL, Mehta A, Tate V, Ralston E, & Ervasti JM. Dystrophin is a microtubule-associated protein. J Cell Biol 2009, 186, 363–369. [Google Scholar] [CrossRef]
- Prosser BL, Ward CW & Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef]
- Prosser BL, Khairallah RJ, Ziman AP, Ward CW & Lederer WJ. X-ROS signaling in the heart and skeletal muscle: stretch-dependent local ROS regulates [Ca2+]i. J Mol Cell Cardiol 2013, 58, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Puchalski MD, Williams RV, Askovich B, Sower CT, Hor KH, Su JT, Pack N, Dibella E & Gottliebson WM. Lat gadolinium enhancement: precursor to cardiomyopathy in Duchenne muscular dystrophy? Int J Cardiovasc Imaging 2009, 25, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Quattrocelli M, Zelikovich AS, Jiang Z, Peek CB, Demonbreun AR, Kuntz NL, Barish GD, Haldar SM & Bass J, McNally EM. Pulsed glucocorticoids enhance dystrophic muscle performance through epigenetic-metabolic reprogramming. JCI Insight 2019, 4, e132402. [Google Scholar] [CrossRef] [PubMed]
- Rafael-Fortney JA, Chimanji NS, Schill KE, Martin CD, Murray JD, Ganguly R, Stangland JE, Tran T, Xu Y, Canan BD, Mays TA, Delfín DA, Janssen PML & Raman SV. Early treatment with lisinopril and spironolactone preserves cardiac and skeletal muscle in Duchenne muscular dystrophy mice. Circulation 2011, 124, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Raman SV, Hor KN, Mazur W, Cardona A, He X, Halnon N, Markham L, Soslow JH, Puchalski MD, Auerbach SR, Truong U, Smart S, McCarthy B, Saeed IM, Statland JM, Kissel JT & Cripe LH. Stabilization of early Duchenne cardiomyopathy with aldosterone inhibition: results of the multicenter AIDMD trial. J Am Heart Assoc 2019, 8, e013501. [Google Scholar] [CrossRef] [PubMed]
- Raman SV & Cripe, LH. Glucocorticoi therapy for Duchenne cardiomyopathy: a Hobson’s choice. J Am Heart Assoc 2015, 4, 1–3. [Google Scholar]
- 287. Raman SV, Hor KN, Mazur W, Halnon NJ, Kissel JT, He X, Tran T, Smart S, McCarthy B, Taylor MD, Jefferies JL, Rafael-Fortney JA, Lowe J, Roble SL & Cripe LH. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2015, 14, 153–161.
- Raman SV, Hor KN, Mazur W, He X, Kissel JT, Smart S, McCarthy B, Roble SL & Cripe LH. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: results of a two-year open-label extension trial. Orphanet J Rare Dis 2017, 12, 1–5. [Google Scholar]
- Ramos SV, Hughes MC, Delfinis LJ, Bellissimo CA & Perry CGR. Mitochondrial bioenergetic dysfunction in the D2mdx model of Duchenne muscular dystrophy is associated with microtubule disorganization in skeletal muscle. PLOS One 2020, 15, e0237138. [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA & Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Reid AL & Alexander, MS. The interplay of mitophagy and inflammation in Duchenne muscular dystrophy. Life (Basel) 2021, 11, 648. [Google Scholar]
- Reutenauer J, Dorchies OM, Patthey-Vuadens O, Vuagniaux G, & Ruegg UT. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br. J. Pharmacol. 2008, 155, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Rhodes J, Margossian R, Darras BT, Colan SD, Jenkins KJ, Geva T & Powell AJ. Safety and efficacy of carvedilol therapy for patients with dilated cardiomyopathy secondary to muscular dystrophy. Pediatr Cardiol 2008, 29, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Rittoo D, Jones A, Lecky B & Neithercut D. Elevation of cardiac troponin T, but not cardiac troponin I, in patients with neuromuscular diseases: implications for the diagnosis of myocardial infarction. J Am Coll Cardiol 2014, 63, 2411–2420. [Google Scholar] [CrossRef]
- Robert V, Massimino ML, Tosello V, Marsault R, Cantini M, Sorrentino V & Pozzan T. Alteration in calcium handling at the subcellular level in mdx myotubes. J Biol Chem 2001, 276, 4647–4651. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues MV, Stoco-Oliveira MC, Silva TDD, Ferreira C, Valente HB, Vanzella LM & Vanderlei LCM. Autonomic modulation at rest and in response to postural change in adolescents with Duchenne muscular dystrophy: a cross-sectional study. Arq Neuropsiquiatr 2021, 79, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Rohman MS, Emoto N, Takeshima Y, Yokoyama M & Matsuo M. Decreased mAKAP, ryanodine receptor, and SERCA2a gene expression in mdx hearts. Biochem Biophys Res Commun 2003, 310, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Romfh A & McNally, EM. Cardiac assessment in Duchenne and Becker muscular dystrophies. Curr Heart Fail Rep 2010, 7, 212–218. [Google Scholar]
- Roujol S, Weingärtner S, Foppa M, Chow K, Kawaji K, Ngo LH, Kellman P, Manning WJ, Thompson R & Nezafat R. Accuracy, precision, and reproducibility of four T1 mapping sequences: a head-to-head comparison of MOLLI, ShMOLLI, SASHA, and SAPPHIRE. Radiology 2014, 272, 683–689. [Google Scholar] [CrossRef]
- Ryan TD, Taylor MD, Mazur W, Cripe LH, Pratt J, King EC, Lao K, Grenier MA, Jefferies JL, Benson DW & Hor KN. Abnormal circumferential strain is present in young Duchenne muscular dystrophy patients. Pediatr Cardiol 2013, 34, 1159–1165. [Google Scholar] [CrossRef]
- Rybalka E, Timpani CA, Cooke MB, Williams AD & Hayes A. Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLOS One 2014, 9, e115763. [Google Scholar]
- Ryder S, Leadley RM, Armstrong N, Westwood M, de Kock S, Butt T, Jain M & Kleijnen J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis 2017, 12, 79. [Google Scholar] [CrossRef]
- Salva MZ, Himeda CL, Tai PWL, Nishiuchi E, Gregorevic P, Allen JM, Finn EE, Nguyen QG, Blankinship MJ, Meuse L, Chamberlain JS & Hauschkaet SD. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol Ther 2007, 15, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Santos MA, Costa Fde A, Travessa AF, Bombig MTN, Fonseca FH, Filho BL, Mussi A, de Souza D, de Oliveira A & Povoa R. Duchenne muscular dystrophy: electrocardiographic analysis of 131 patients. Arq Bras Cardiol 2010, 94, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Sanyal SK, Johnson WW, Dische MR, Pitner SE & Beard C. Dystrophic degeneration of papillary muscle and ventricular myocardium: a basis for mitral valve prolapse in Duchenne’s muscular dystrophy. Circulation 1980, 62, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Sarma S, Li N, van Oort RJ, Reynolds C, Skapura DG & Wehrens XHT. Genetic inhibition of PKA phosphorylation of RyR2 prevents dystrophic cardiomyopathy. Proc Natl Acad Sci U S A 2010, 107, 13165–13170. [Google Scholar] [CrossRef] [PubMed]
- Schiavone M, Zulian A, Menazza S, Petronilli V, Argenton F, Merlini L, Sabatelli P, & Bernardi P. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Schlattner U, Kay L & Tokarska-Schlattner M. Mitochondrial Proteolipid Complexes of Creatine Kinase. Subcell Biochem 2018, 87, 365–408. [Google Scholar]
- Schmid J, Liesinger L, Birner-Gruenberger R, Stojakovic T, Scharnagl H, Dieplinger B, Asslaber M, Radl R, Beer M, Polacin M, Mair J, Szolar D, Berghold A, Quasthoff S, Binder JS & Rainer PP. Elevated cardiac troponin T in patients with skeletal myopathies. J Am Coll Cardiol 2018, 71, 1540–1549. [Google Scholar] [CrossRef]
- Schneider SM, Sridhar V, Bettis AK, Heath-Barnett H, Balog-Alvarez CJ, Guo LJ, Johnson R, Jaques S, Vitha S, Glowcwski AC, Kornegay JN & Nghiem PP. Glucose metabolism as a pre-clinical biomarker for the golden retriever model of Duchenne muscular dystrophy. Mol Imaging Biol 2018, 20, 780–788. [Google Scholar] [CrossRef]
- Schneider SM, Sansom GT, Guo LJ, Furuya S, Weeks BR & Kornegay JN. Natural history of histopathologic changes in cardiomyopathy of golden retriever muscular dystrophy. Front Vet Sci 2022, 8, 759585. [Google Scholar] [CrossRef] [PubMed]
- Schoeffler M, Wallet F & Robert MO. Elévation de troponine I coronarographie normale chez un patient porteur d’une myopathie de Duchenne [Increased troponin I level in a Duchenne muscular dystrophy patient with normal coronarography]. Ann Fr Anesth Reanim 2008, 27, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Schram G, Fournier A, Leduc H, Dahdah N, Therien J, Vanasse M & Khairy P. All-cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J Am Coll Cardiol 2013, 61, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Schultz TI, Raucci FJ Jr & Salloum FN. Cardiovascular disease in Duchenne muscular dystrophy: overview and insight into novel therapeutic targets. JACC Basic Transl Sci 2022, 7, 608–625. [Google Scholar] [CrossRef] [PubMed]
- Segawa K, Komaki H, Mori-Yoshimura M, Oya Y, Kimura K, Tachimori H, Kato N, Sasaki M & Takahashi Y. Cardiac conduction disturbances and aging in patients with Duchenne muscular dystrophy. Medicine (Baltimore) 2017, 96, e8335. [Google Scholar] [CrossRef] [PubMed]
- Shah AM, Jefferies JL, Rossano JW, Decker JA, Cannon BC & Kim JJ. Electrocardiographic abnormalities and arrhythmias are strongly associated with the development of cardiomyopathy in muscular dystrophy. Heart Rhythm 2010, 7, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Shah MNA & Yokota, Y. Cardiac therapies for Duchenne muscular dystrophy. Ther Adv Neurol Disord 2023, 16, 17562864231182934. [Google Scholar]
- Shih JA, Folch A & Wong BL. Duchenne muscular dystrophy: the heart of the matter. Curr Heart Fail Rep 2020, 17, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Shin JH, Bostick B, Yue Y, Hajjar R & Duan D. SERCA2a gene transfer improves electrocardiographic performance in aged mdx mice. J Transl Med 2011, 9, 132. [Google Scholar] [CrossRef]
- Shirokova N & Niggli, E. Cardiac phenotype of Duchenne muscular dystrophy: insights from cellular studies. J Mol Cell Cardiol 2013, 58, 217–224. [Google Scholar]
- Siddiqui S, Alsaied T, Henson SE, Gandhi J, Patel P, Khoury P, Villa C, Ryan TD, Wittekind SG, Lang SM & Taylor MD. Left ventricular magnetic resonance imaging strain predicts the onset of Duchenne muscular dystrophy-associated cardiomyopathy. Circ Cardiovasc Imaging 2020, 13, e011526. [Google Scholar] [CrossRef] [PubMed]
- Siegel B, Olivieri L, Gordish-Dressman H & Spurney C. Myocardial strain using cardiac MR feature tracking and speckle tracking echocardiography in Duchenne muscular dystrophy patients. Pediatr Cardiol 2017, 39, 478–483. [Google Scholar]
- Siegel MP, Kruse SE, Percival JM, Goh J, White CC, Hopkins HC, Kavanagh TJ, Szeto HH, Rabinovitch PS & Marcinek DJ. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 2013, 12, 763–771. [Google Scholar] [CrossRef]
- Silva MC, Magalhães TA, Meira ZMA, Rassi CHRE, de Souza Andrade AC, Gutierrez PS, Azevedo CF, Gurgel-Giannetti J, Vainzof M, Zatz M, Kalil-Filho R & Rochitte CE. Myocardial fibrosis progression in Duchenne and Becker muscular dystrophy: a randomized clinical trial. JAMA Cardiol 2017, 2, 190–199. [Google Scholar] [CrossRef]
- Silversides CK, Webb GD, Harris VA & Biggar DW. Effects of deflazacort on left ventricular function in patients with Duchenne muscular dystrophy. A J Cardiol 2003, 91, 769–772. [Google Scholar] [CrossRef]
- Soslow JH, Xu M, Slaughter JC, Stanley M, Crum K, Markham LW & Parra DA. Evaluation of echocardiographic measures of left ventricular function in patients with Duchenne muscular dystrophy: assessment of reproducibility and comparison to cardiac magnetic resonance imaging. J Am Soc Echocardiogr 2016, 29, 983–991. [Google Scholar] [CrossRef]
- Spencer MJ, Croall DE & Tidball JG. Calpains are activated in necrotic fibers from mdx dystrophic mice. J Biol Chem 1995, 270, 10909–10914. [Google Scholar] [CrossRef] [PubMed]
- Spencer MJ, Walsh CM, Dorshkind KA, Rodriguez EM & Tidball JG. Myonuclear apoptosis in dystrophic mdx muscle occurs by perforin-mediated cytotoxicity. J Clin Invest 1997, 99, 2745–2751. [Google Scholar] [CrossRef]
- Spencer MJ, Montecino-Rodriguez E, Dorshkind K & Tidball JG. Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin Immunol 2001, 98, 235–243. [Google Scholar] [CrossRef]
- Spurney C, Shimizu R, Morgenroth LP, Kolski H, Gordish-Dressman H, Clemens PR & CINRG Investigators. Cooperative International Neuromuscular Research Group Duchenne Natural History Study demonstrates insufficient diagnosis and treatment of cardiomyopathy in Duchenne muscular dystrophy. Muscle & Nerve 2014, 50, 250–256. [Google Scholar]
- Spurney CF, Ascheim D, Charnas L, Cripe L, Hor K, King N, Kinnett K, McNally EM, Sauer JM, Sweeney L, Villa C & Markham LW. Current state of cardiac troponin testing in Duchenne muscular dystrophy cardiomyopathy: review and recommendations from the Parent Project Muscular Dystrophy expert panel. Open Heart 2021, 8, e001592. [Google Scholar] [CrossRef] [PubMed]
- Spurney, CF. Cardiomyopathy of Duchenne muscular dystrophy: current understanding and future directions. Muscle & Nerve 2011, 44, 8–19. [Google Scholar]
- Stevens JA, Dobratz TC, Fischer KD, Palmer A, Bourdage K, Wong AJ, Chapoy-Villanueva H, Garry DJ, Liu JC, Kay MW, Kuzmiak-Glancy S, & Townsend D. Mechanisms of reduced myocardial energetics of the dystrophic heart. Am J Physiol Heart Circ Physiol 2024, 326, H396–H407. [Google Scholar] [CrossRef]
- Stocco A, Smolina N, Sabatelli P, Sileikyte J, Artusi E, Mouly V, Cohen M, Forte M, Schiavone M, & Bernardi P. Treatment with a triazole inhibitor of the mitochondrial permeability transition pore fully corrects the pathology of sapje zebrafish lacking dystrophin. Pharmacol. Res. 2021, 165, 105421. [Google Scholar] [CrossRef] [PubMed]
- Stanley WC, Recchia FA & Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Stuckey DJ, Carr CA, Camelliti P, Tyler DJ, Davies KE & Clarke K. In vivo MRI characterization of progressive cardiac dysfunction in the mdx mouse model of muscular dystrophy. PLOS One 2012, 7, e28569. [Google Scholar]
- Su JA, Ramos-Platt L & Menteer J. Left ventricular tonic contraction as a novel biomarker of cardiomyopathy in Duchenne muscular dystrophy. Pediatr Cardiol 2016, 37, 678–685. [Google Scholar] [CrossRef]
- Subramanian S, Hor KN, Mazur W, Smart S, Tran T, Halnon N, Taylor MD, Cripe L & Raman SV. Effect of pulmonary function on right heart function in Duchenne muscular dystrophy patients. J Cardiovasc Magn Reson 2015, 17, P279. [Google Scholar] [CrossRef]
- Sui T, Lau YS, Liu D, Liu T, Xu L, Gao Y, Lai L, Li Z & Han R. A novel rabbit model of Duchenne muscular dystrophy generated by CRISPR/Cas9. Dis Model Mech 2018, 11, dmm032201. [Google Scholar] [CrossRef]
- Suno M, Nagaoka A. Inhibition of lipid peroxidation by a novel compound (CV-2619) in brain mitochondria and mode of action of the inhibition. Biochem Biophys Res Commun 1984, 125, 1046–52. [Google Scholar] [CrossRef]
- Suno M, Nagaoka A. Inhibition of lipid peroxidation by idebenone in brain mitochondria in the presence of succinate. Arch Gerontol Geriatr 1989, 8, 291–7. [Google Scholar] [CrossRef] [PubMed]
- Svobodova B, Jelinkova S, Pesl M, Beckerová D, Lacampagne A, Meli AC & Rotrekl V. Cellular pathology of the human heart in Duchenne muscular dystrophy (DMD): lessons learned from in vitro modeling. Pflügers Archiv 2021, 473, 1099–1115. [Google Scholar] [CrossRef] [PubMed]
- Swiderski K & Lynch, GS. Murine models of Duchenne muscular dystrophy: is there a best model? Am J Physiol 2021, 321, C409–C412. [Google Scholar]
- Szabo SM, Salhany RM, Deighton A, Harwood M, Mah J & Gooch KL. The clinical course of Duchenne muscular dystrophy in the corticosteroid treatment era: a systematic literature review. Orphanet J Rare Dis 2021, 16, 237. [Google Scholar] [CrossRef] [PubMed]
- Szeto HH & Schiller. PW Novel therapies targeting inner mitochondrial membrane--from discovery to clinical development. Pharm Res 2011, 28, 2669–2679. [Google Scholar] [CrossRef] [PubMed]
- Szeto, HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [PubMed]
- Szeto HH & Birk, AV. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther 2014, 96, 672–683. [Google Scholar]
- Taglietti V, Kefi K, Bronisz-Budzyńska I, Mirciloglu B, Rodrigues M, Cardone N, Coulpier F, Periou B, Gentil C, Goddard M, Authier FJ, Pietri-Rouxel F, Malfatti E, Lafuste P, Tiret L & Relaix F. Duchenne muscular dystrophy trajectory in R-DMDdel52 preclinical rat model identifies COMP as biomarker of fibrosis. Acta Neuropathol Commun 2022, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Takami Y, Takeshima Y, Awano H, Okizuka Y, Yagi M & Matsuo M. High incidence of electrocardiogram abnormalities in young patients with Duchenne muscular dystrophy. Pediatr Neurol 2008, 39, 399–403. [Google Scholar] [CrossRef]
- Tandon A, Villa CR, Hor KN, Jefferies JL, Gao Z, Towbin JA, Wong BL, Mazur W, Fleck RJ, Sticka JJ, Benson DW & Taylor MD. Myocardial fibrosis burden predicts left ventricular ejection fraction and is associated with age and steroid treatment duration in Duchenne muscular dystrophy. J Am Heart Assoc 2015, 4, 4. [Google Scholar]
- Taqatqa A, Bokowski J, Al-Kubaisi M, Khalil A, Miranda C, Alaksham H, Fughhi I, Kenny D & Diab KA. The use of speckle tracking echocardiography for early detection of myocardial dysfunction in patients with Duchenne muscular dystrophy. Pediatr Cardiol 2016, 37, 1422–1428. [Google Scholar] [CrossRef]
- Thrush PT, Allen HD, Viollet L & Mendell JR. Re-examination of the electrocardiogram in boys with Duchenne muscular dystrophy and correlation with its dilated cardiomyopathy. Am J Cardiol 2009, 103, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Torriani M, Townsend E, Thomas BJ, Bredella MA, Ghomi RH & Tseng BS. Lower leg muscle involvement in Duchenne muscular dystrophy: an MR imaging and spectroscopy study. Skeletal Radiol 2012, 41, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Townsend DW, Turner I, Yasuda S, Martindale J, Davis J, Shillingford M, Kornegay JN & Metzger JM. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest 2010, 120, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Townsend D, Yasuda S, McNally E & Metzger JM. Distinct pathophysiological mechanisms of cardiomyopathy in hearts lacking dystrophin or the sarcoglycan complex. FASEB J 2011, 25, 3106–3114. [Google Scholar] [CrossRef] [PubMed]
- Townsend D, Yasuda S, Li S, Chamberlain JS & Metzger JM. Emergent dilated cardiomyopathy caused by targeted repair of dystrophic skeletal muscle. Mol Ther 2008, 16, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Illarramendi A, Toral-Ojeda I, Aldanondo G & López de Munain A. Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev Mol Med 2014, 16, e16. [Google Scholar] [CrossRef] [PubMed]
- van Bockel EA, Lind JS, Zijlstra JG, Wijkstra PJ, Meijer PM, van den Berg MP, Slart RHJA, Aarts LPHJ & Tulleken JE. Cardiac assessment of patients with late stage Duchenne muscular dystrophy. Neth Heart J 2009, 17, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Van Erp C, Loch D, Laws N, Trebbin A & Hoey AJ. Timeline of cardiac dystrophy in 3– 18-month-old MDX mice. Muscle & Nerve 2010, 42, 504–513. [Google Scholar]
- van Westering TL, Betts CA & Wood MJ. Current understanding of molecular pathology and treatment of cardiomyopathy in Duchenne muscular dystrophy. Molecules, 2015, 20, 8823–8855. [Google Scholar] [CrossRef]
- Verhaert D, Richards K, Rafael-Fortney JA & Raman SV. Cardiac involvement in patients with muscular dystrophies: magnetic resonance imaging phenotype and genotypic considerations. Circ Cardiovasc Imaging 2011, 4, 67–76. [Google Scholar] [CrossRef]
- Villa CR, Czosek RJ, Ahmed H, Khoury PR, Anderson JB, Knilans TK, Jefferies JL, Wong B & Spar DS. Ambulatory monitoring and arrhythmic outcomes in pediatric and adolescent patients with Duchenne muscular dystrophy. J Am Heart Assoc 2015, 5, e002620. [Google Scholar]
- Viola HM, Davies SMK, Filipovska A & Hool LC. L-type Ca2+ channel contributes to alterations in mitochondrial calcium handling in the mdx ventricular myocyte. Am J Physiol Heart Circ Physiol 2013, 304, H767–H775. [Google Scholar] [CrossRef] [PubMed]
- Viola HM, Arthur PG & Hool LC. Evidence for regulation of mitochondrial function by the L-type Ca2+ channel in ventricular myocytes. J Mol Cell Cardiol 2009, 46, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Viola HM, Adams AM, Davies SMK, Fletcher S, Filipovska A & Hool LC. Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc Natl Acad Sci U S A 2014, 111, E2905–E2914. [Google Scholar]
- Viollet L, Thrush PT, Flanigan KM, Mendell JR & Allen HD. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in Duchenne muscular dystrophy. Am J Cardiol 2012, 110, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Voit A, Patel V, Pachon R, Shah V, Bakhutma M, Kohlbrenner E, McArdle JJ, Dell’Italia LJ, Mendell JR, Xie LH, Hajjar RJ, Duan D, Fraidenraich D & Babu GJ. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat Commun 2017, 8, 1068. [Google Scholar] [CrossRef] [PubMed]
- Voleti S, Olivieri L, Hamann K, Gordish-Dressman H & Spurney C. Troponin I levels correlate with cardiac MR LGE and native T1 values in Duchenne muscular dystrophy cardiomyopathy and identify early disease progression. Pediatr Cardiol 2020, 41, 1–7. [Google Scholar]
- Wagner S, Knipp S, Weber C, Hein S, Schinkel S, Walther A, Bekeredjian R, Müller OJ & Friedrich O. The heart in Duchenne muscular dystrophy: early detection of contractile performance alteration. J Cell Mol Med 2012, 16, 3028–3036. [Google Scholar] [CrossRef]
- Wagner KR, Lechtzin N & Judge DP. Current treatment of adult Duchenne muscular dystrophy. Biochi Biophys Acta 2007, 1772, 229–237. [Google Scholar] [CrossRef]
- Wagner S, Maier LS & Bers DM. Role of sodium and calcium dysregulation in tachyarrhythmias in sudden cardiac death. Circ Res 2015, 16, 1956–1970. [Google Scholar]
- Wallimann T, Tokarska-Schlattner M & Schlattner U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011, 40, 1271–1296. [Google Scholar] [CrossRef] [PubMed]
- Wang H, Liu S, Xing YL & Chen R. [The limitation of MB isoenzyme of creatine kinase mass in assess myocardial injury with muscular disease]. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue [Chinese Critical Care Medicine] 2011, 23, 723–726. [Google Scholar]
- Wansapura JP, Hor KN, Mazur W, Fleck R, Hagenbuch S, Benson DW & Gottliebson WM. Left ventricular T2 distribution in Duchenne muscular dystrophy. Cardiovasc Magn Reson 2010, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Wasala NB, Yue Y, Lostal W, Wasala LP, Niranjan N, Hajjar RJ, Babu GJ & Duan D. Single SERCA2a therapy ameliorated dilated cardiomyopathy for 18 months in a mouse model of Duchenne muscular dystrophy. Mol Ther 2020, 28, 845–854. [Google Scholar] [CrossRef]
- Wehling M, Spencer MJ & Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol 2001, 155, 123–131. [Google Scholar] [CrossRef]
- Wens SCA, Schaaf GJ, Michels M, Kruijshaar ME, van Gestel TJM, in 't Groen S, Pijnenburg J, Dekkers DHW, Demmers JAA, Verdijk LB, Brusse E, van Schaik RHN, van der Ploeg AT, van Doorn PA & Pim Pijnappel WWM. Elevated plasma cardiac troponin T levels caused by skeletal muscle damage in Pompe disease. Circ Cardiovasc Genet 2016, 9, 6–13. [Google Scholar] [CrossRef] [PubMed]
- 378. Wissing ER, Millay DP, Vuagniaux G, & Molkentin JD. Debio-025 is more effective than prednisone in reducing muscular pathology in mdx mice. Neuromuscul Disord, 20, 753–760.
- Willi L, Abramovich I, Fernandez-Garcia J, Agranovich B, Shulman M, Milman H, Baskin P, Eisen B, Michele DE, Arad M, Binah O & Gottlieb E. Bioenergetic and metabolic impairments in induced pluripotent stem cell-derived cardiomyocytes generated from Duchenne muscular dystrophy patients. Int J Mol Sci 2022, 23, 9808. [Google Scholar] [CrossRef] [PubMed]
- Williams GS, Boyman L, Chikando AC, Khairallah RJ & Lederer WJ. Mitochondrial calcium uptake. Proc Natl Acad Sci U S A 2013, 110, 10479–10486. [Google Scholar] [CrossRef]
- Williams IA & Allen, DG. Intracellular calcium handling in ventricular myocytes from mdx mice. Am J Physiol Heart Circ Physiol 2007, 292, H846–H855. [Google Scholar]
- Wong TWY, Ahmed A, Yang G, Maino E, Steiman S, Hyatt E, Chan P, Lindsay K, Wong N, Golebiowski D, Schneider J, Delgado-Olguin P, Ivakine EA, & Cohn, RD. A novel mouse model of Duchenne muscular dystrophy carrying a multi-exonic Dmd deletion exhibits progressive muscular dystrophy and early-onset cardiomyopathy. Dis Model Mech 2020, 13, dmm045369. [Google Scholar] [CrossRef] [PubMed]
- Wrogemann K, & Blanchaer, MC. Oxidative phosphorylation by muscle mitochondria of dystrophic mice. Can. J. Biochem 1967, 45, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Wrogemann K, & Blanchaer, MC. Respiration and oxidative phosphorylation by muscle and heart mitochondria of hamsters with hereditary myocardiopathy and polymyopathy. Can. J. Biochem 1968, 46, 323–329. [Google Scholar] [CrossRef] [PubMed]
- 385. Wrogemann K, Blanchaer, MC, & Jacobson, BE. A calcium-associated magnesium-responsive defect of respiration and oxidative phosphorylation by skeletal muscle mitochondria of BIO 14.6 dystrophic hamsters. Life Sci, 1970; II9, 1167–1173.
- Wrogemann K, Jacobson, BE, & Blanchaer, MC. On the Mechanism of a Calcium-Associated Defect of Oxidative Phosphorylation in Progressive Muscular Dystrophy. Arch. Biochem. Biophys. 1973, 159, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Wrogemann K, & Pena, SDJ. Mitochondrial calcium overload: a general mechanism for cell-necrosis in muscle diseases. The Lancet 1976, 307, P672–P674. [Google Scholar] [CrossRef]
- Xu L, Lau YS, Gao Y, Li H & Han R. Life-long AAV-mediated CRISPR genome editing in dystrophic heart improves cardiomyopathy without causing serious lesions in mdx mice. Mol Ther 2019, 27, 1–8. [Google Scholar]
- Yamamoto T, Awano H, Zhang Z, Sakuma M, Kitaaki S, Matsumoto M, Nagai M, Sato I, Imanishi T, Hayashi N, Matsuo M, Iijima K & Saegusa J. Cardiac dysfunction in Duchenne muscular dystrophy is less frequent in patients with mutations in the dystrophin Dp116 coding region than in other regions. Circ Genom Precis Med 2018, 11, e001782. [Google Scholar]
- Yanagisawa A, Yokota N, Miyagawa M, Kawamura J, Ishihara T, Aoyagi T & Ishikawa K. Plasma levels of atrial natriuretic peptide in patients with Duchenne’s progressive muscular dystrophy. Am Heart J 1990, 120, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Yasuda S, Townsend D, Michele DE, Favre EG, Day SM & Metzger JM. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 2005, 436, 1025–1029. [Google Scholar] [CrossRef]
- Yin H, Moulton HM, Seow Y, Boyd C, Boutilier J, Iverson P & Wood MJA. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum Mol Genet 2008, 17, 3909–3918. [Google Scholar] [CrossRef]
- Yokota T, Lu QL, Partridge T, Kobayashi M, Nakamura A, Takeda S & Hoffman E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol 2009, 65, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Zambon AA, Ayyar Gupta V, Ridout D, Manzur AY, Baranello G, Trucco F, Muntoni F & UK Northstar Clinical Network. Peak functional ability and age at loss of ambulation in Duchenne muscular dystrophy. Dev Med Child Neurol 2022, 64, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW & Szeto HH. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 2004, 279, 34682–34690. [Google Scholar] [CrossRef] [PubMed]
- Zhao K, Luo G, Zhao G-M, Schiller PW, & Szeto HH. Transcellular transport of a highly polar 3+ net charge opioid tetrapeptide. J Pharmacol Exp Ther 2003, 304, 425–32. [Google Scholar] [CrossRef] [PubMed]
- Zorzano A, Sevilla L, Tomàs E, Gumà A, Palacín M & Fischer Y. GLUT4 trafficking in cardiac and skeletal muscle: isolation and characterization of distinct intracellular GLUT4-containing vesicle populations. Biochem Soc Trans 1997, 25, 968–974. [Google Scholar] [CrossRef]
- Zs-Nagy, I. Chemistry toxicology, pharmacology and pharmacokinetics of idebenone: a review. Arch Gerontol Geriatr 1990, 11, 177–186. [Google Scholar] [CrossRef]
- 399. Zulian A, Schiavone M, Giorgio V, & Bernardi P. Forty years later: Mitochondria as therapeutic targets in muscle diseases. Pharmacol Res, 2016; 113 (Pt A), 563–573.
Figure 1.
Proposed mechanism demonstrating how dystrophin deficiency-induced cardiac mitochondrial stress contributes to diastolic dysfunction. As an outcome of mitochondrial stress, left ventricular [ATP]:[ADP] is decreased, while mitochondrial reactive oxygen species (mH2O2) emission is elevated (Hughes et al., 2020; Dubinin et al., 2020b). Yellow question marks denote pathways where ATP insufficiency and elevated mH2O2 provision may precede impaired muscle cross-bridge relaxation, with independent hypotheses broadly reflecting extensive previous literature. Pathways include Na+/K+ ATPases along the t-tubule (which require ATP to maintain action potential charge balance and membrane excitability) (Dunn et al., 1995); ATP-dependent SERCA (which requires ATP re-uptake of excess cytosolic Ca2+ to allow for cardiac relaxation) (Williams and Allen, 2007; Voit et al., 2017; Rohman et al., 2003); blocked myosin detachment (which requires ATP to drive filament sliding during muscle cross-bridge cycling); and ROS-induced RyR2 oxidative stress (which may aberrantly release Ca2+ from its SERCA storage site) (Fauconnier et al., 2010; Bellinger et al., 2009; Voit et al., 2017; Rohman et al., 2003). Potentially as an outcome of elevated mitochondrial stress, and consequent decreased [ATP]:[ADP], impaired relaxation and the corresponding stiffer heart muscles are hypothesized to precede diastolic dysfunction exhibited in various late-stage dystrophic models. Created on BioRender.com.
Figure 1.
Proposed mechanism demonstrating how dystrophin deficiency-induced cardiac mitochondrial stress contributes to diastolic dysfunction. As an outcome of mitochondrial stress, left ventricular [ATP]:[ADP] is decreased, while mitochondrial reactive oxygen species (mH2O2) emission is elevated (Hughes et al., 2020; Dubinin et al., 2020b). Yellow question marks denote pathways where ATP insufficiency and elevated mH2O2 provision may precede impaired muscle cross-bridge relaxation, with independent hypotheses broadly reflecting extensive previous literature. Pathways include Na+/K+ ATPases along the t-tubule (which require ATP to maintain action potential charge balance and membrane excitability) (Dunn et al., 1995); ATP-dependent SERCA (which requires ATP re-uptake of excess cytosolic Ca2+ to allow for cardiac relaxation) (Williams and Allen, 2007; Voit et al., 2017; Rohman et al., 2003); blocked myosin detachment (which requires ATP to drive filament sliding during muscle cross-bridge cycling); and ROS-induced RyR2 oxidative stress (which may aberrantly release Ca2+ from its SERCA storage site) (Fauconnier et al., 2010; Bellinger et al., 2009; Voit et al., 2017; Rohman et al., 2003). Potentially as an outcome of elevated mitochondrial stress, and consequent decreased [ATP]:[ADP], impaired relaxation and the corresponding stiffer heart muscles are hypothesized to precede diastolic dysfunction exhibited in various late-stage dystrophic models. Created on BioRender.com.
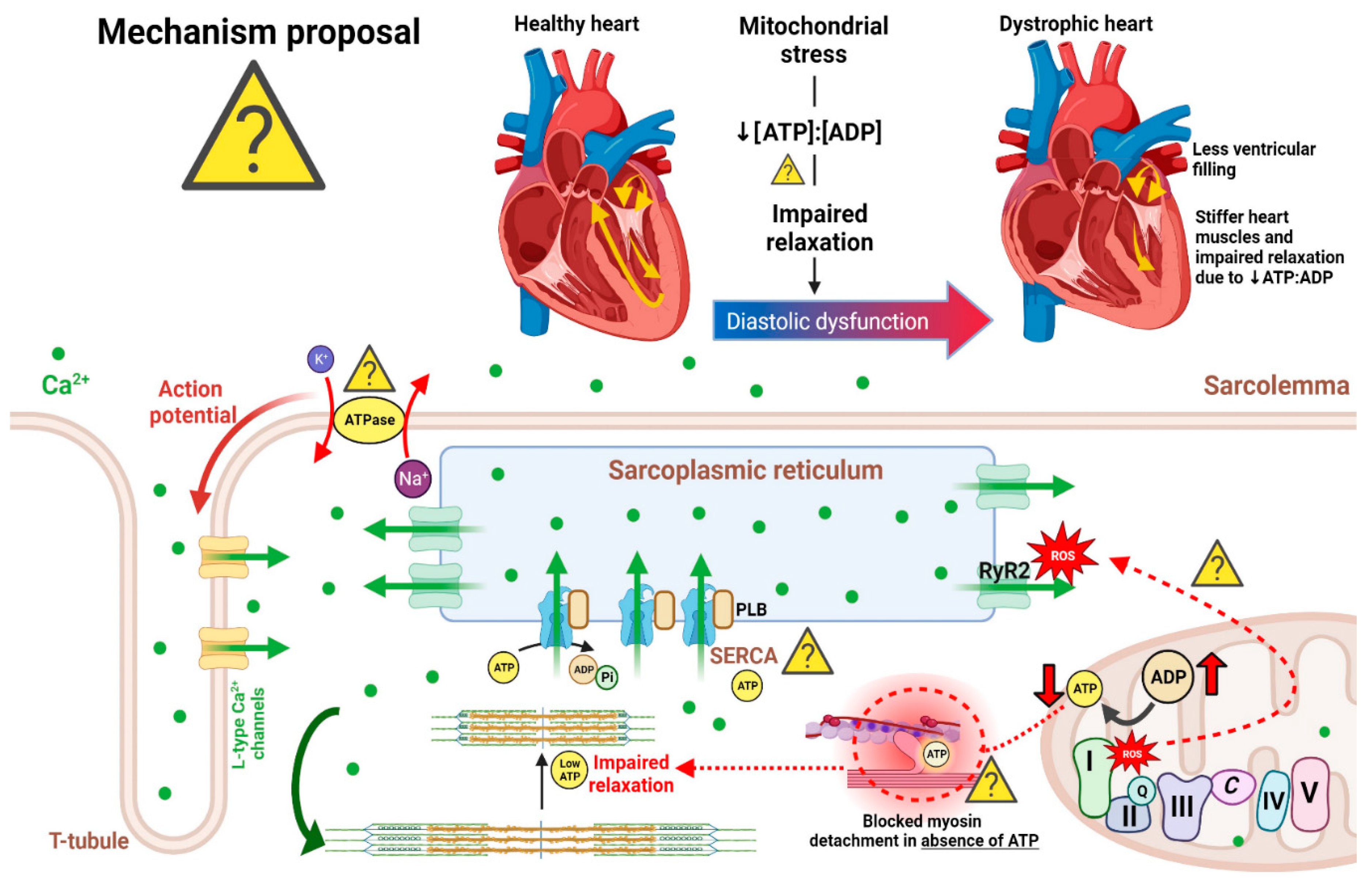
Figure 2.
Role of pathological Ca2+ overload in perpetuating cardiomyocyte death, and mPTP as a prospective therapeutic target for attenuating cardiomyopathic symptoms in DMD. Dystrophin deficiency-induced sarcolemmal tears lead to rapid cytosolic Ca2+ influx via stretch-activated channels (SACs) along the membrane (Yasuda et al., 2005; Williams et al., 2007; reviewed in Vallejo-Illarramendi et al., 2014). NOX2-derived oxidative damage to SACs further damages these channels, thus forcing additional Ca2+ to enter the cytosol (Allen and Whitehead, 2010). During cytosolic Ca2+ overload, the mitochondrion takes up excess Ca2+ via voltage-dependent anion channel (VDAC) and mitochondrial calcium uniporter (MCU) channels along the OMM and IMM, respectively. Subsequent mitochondrial Ca2+ overload results in mPTP opening, which expels cytochrome c into the cytosol (Burelle et al., 2010). It is also hypothesized that complex I and complex III (yellow question mark denotes insufficient data)-derived reactive oxygen species (ROS) damage stimulates mPTP opening, however, further research is required to characterize the presence of complex III-derived ROS in mdx models (Burelle et al., 2010; Hughes et al., 2020). It is also presently unknown if mitochondrial ROS directly triggers cardiomyocyte death in dystrophic hearts (denoted by yellow question mark). Cytochrome c release from the OMM interacts with the apoptosome-containing adaptor Apaf-1 and other initiator caspases (9/3) (Ascah et al., 2011; Budihardjo et al., 1999), which induce cardiomyocyte death downstream. In healthy physiological systems, regulated control of cardiomyocyte death is required for remodeling events following contraction-induced damage; however, in pathological states, excessive cardiomyocyte death leads to irreversible damage and fibro-fatty tissue replacement, also known as cardiomyopathy. Created on BioRender.com.
Figure 2.
Role of pathological Ca2+ overload in perpetuating cardiomyocyte death, and mPTP as a prospective therapeutic target for attenuating cardiomyopathic symptoms in DMD. Dystrophin deficiency-induced sarcolemmal tears lead to rapid cytosolic Ca2+ influx via stretch-activated channels (SACs) along the membrane (Yasuda et al., 2005; Williams et al., 2007; reviewed in Vallejo-Illarramendi et al., 2014). NOX2-derived oxidative damage to SACs further damages these channels, thus forcing additional Ca2+ to enter the cytosol (Allen and Whitehead, 2010). During cytosolic Ca2+ overload, the mitochondrion takes up excess Ca2+ via voltage-dependent anion channel (VDAC) and mitochondrial calcium uniporter (MCU) channels along the OMM and IMM, respectively. Subsequent mitochondrial Ca2+ overload results in mPTP opening, which expels cytochrome c into the cytosol (Burelle et al., 2010). It is also hypothesized that complex I and complex III (yellow question mark denotes insufficient data)-derived reactive oxygen species (ROS) damage stimulates mPTP opening, however, further research is required to characterize the presence of complex III-derived ROS in mdx models (Burelle et al., 2010; Hughes et al., 2020). It is also presently unknown if mitochondrial ROS directly triggers cardiomyocyte death in dystrophic hearts (denoted by yellow question mark). Cytochrome c release from the OMM interacts with the apoptosome-containing adaptor Apaf-1 and other initiator caspases (9/3) (Ascah et al., 2011; Budihardjo et al., 1999), which induce cardiomyocyte death downstream. In healthy physiological systems, regulated control of cardiomyocyte death is required for remodeling events following contraction-induced damage; however, in pathological states, excessive cardiomyocyte death leads to irreversible damage and fibro-fatty tissue replacement, also known as cardiomyopathy. Created on BioRender.com.
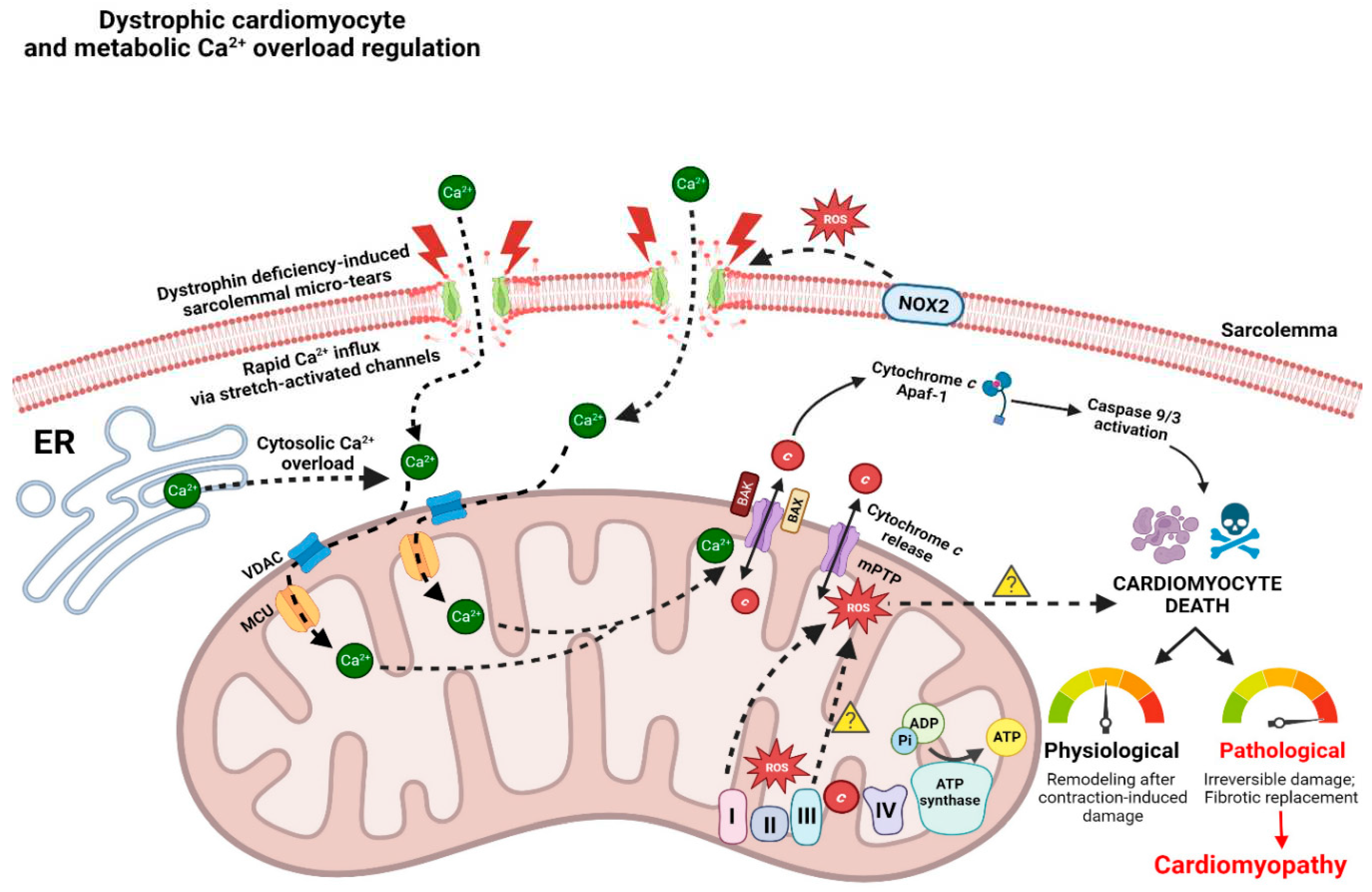
Figure 3.
Cardiolipin as a prospective therapeutic target for treating mitochondrial stress in dystrophic cardiac tissue. Since cardiolipin is highly redox-sensitive (attributed to its high structural content of unsaturated fatty acids), and mitochondrial reactive oxygen species (mH2O2) provision is elevated in dystrophic cardiac tissue (Hughes et al., 2020), targeting cardiolipin may be beneficial for stabilizing mitochondrial membranes. (Top panel) Herein, we propose that when cardiolipin is oxidized, electron transport chain supercomplexes are unevenly distributed and mitochondrial membranes are abnormally linear, thus resulting in elevated mH2O2 production and reductions to [ATP]:[ADP] (Koufen et al. 1999; Dolder et al. 2001). Additionally, cytochrome c is released into the cytosolic fraction, thus impairing its association with mitochondrial creatine kinase (mtCK) (Schlattner et al. 2018; Koufen et al. 1999; Dolder et al. 2001). Although elevations to cytochrome c in the cytosolic fraction of dystrophic hearts have been previously characterized (Burelle et al., 2010), the yellow question mark denotes a lack of causal data linking this elevation to oxidized cardiolipin. The release of cytochrome c from the IMM, and eventually OMM (yellow question mark once again denotes a lack of data linking this to oxidized cardiolipin), may also lead to pathological cytosolic cytochrome c accumulation, thus perpetuating apoptotic events (Burelle et al. 2010). (Bottom panel) In the presence of a cardiolipin-stabilizing agent such as elamipretide, electron transport chain (ETC) supercomplexes can be tethered to each other in a less linear mitochondrial membrane, while cytochrome c remains tethered to the ETC and mtCK. This results in lower mH2O2 emissions and a higher [ATP]:[ADP] ratio, akin to healthy physiological systems. Created on BioRender.com.
Figure 3.
Cardiolipin as a prospective therapeutic target for treating mitochondrial stress in dystrophic cardiac tissue. Since cardiolipin is highly redox-sensitive (attributed to its high structural content of unsaturated fatty acids), and mitochondrial reactive oxygen species (mH2O2) provision is elevated in dystrophic cardiac tissue (Hughes et al., 2020), targeting cardiolipin may be beneficial for stabilizing mitochondrial membranes. (Top panel) Herein, we propose that when cardiolipin is oxidized, electron transport chain supercomplexes are unevenly distributed and mitochondrial membranes are abnormally linear, thus resulting in elevated mH2O2 production and reductions to [ATP]:[ADP] (Koufen et al. 1999; Dolder et al. 2001). Additionally, cytochrome c is released into the cytosolic fraction, thus impairing its association with mitochondrial creatine kinase (mtCK) (Schlattner et al. 2018; Koufen et al. 1999; Dolder et al. 2001). Although elevations to cytochrome c in the cytosolic fraction of dystrophic hearts have been previously characterized (Burelle et al., 2010), the yellow question mark denotes a lack of causal data linking this elevation to oxidized cardiolipin. The release of cytochrome c from the IMM, and eventually OMM (yellow question mark once again denotes a lack of data linking this to oxidized cardiolipin), may also lead to pathological cytosolic cytochrome c accumulation, thus perpetuating apoptotic events (Burelle et al. 2010). (Bottom panel) In the presence of a cardiolipin-stabilizing agent such as elamipretide, electron transport chain (ETC) supercomplexes can be tethered to each other in a less linear mitochondrial membrane, while cytochrome c remains tethered to the ETC and mtCK. This results in lower mH2O2 emissions and a higher [ATP]:[ADP] ratio, akin to healthy physiological systems. Created on BioRender.com.
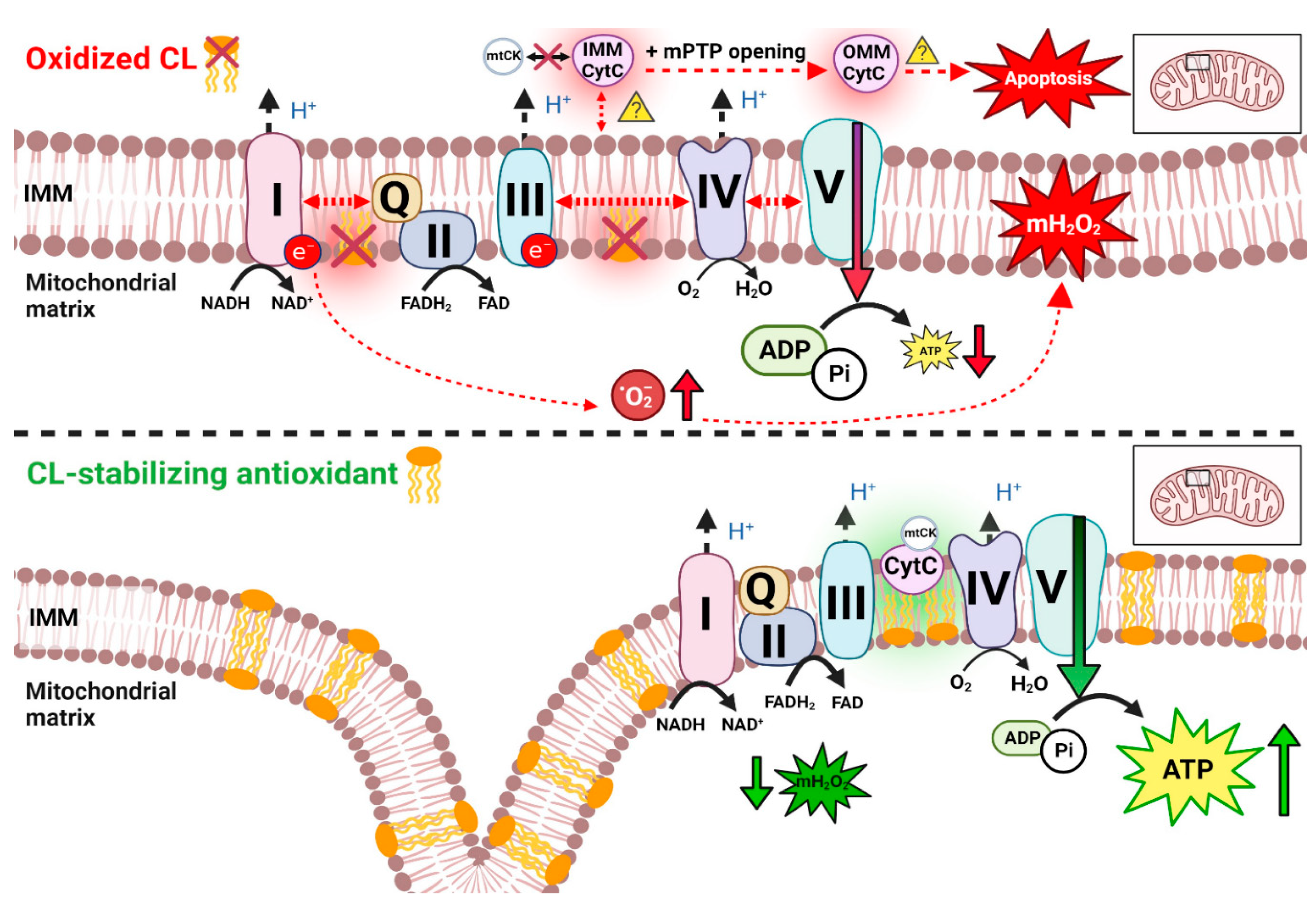
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



