
Submitted:
18 April 2023
Posted:
18 April 2023
You are already at the latest version
Abstract
Nucleotide-binding domain like receptor protein 3 (NLRP3) inflammasome in the kidney and the heart is increasingly being suggested to play a key role in mediating inflammation. In the kidney, NLRP3 activation has been associated with the progression of diabetic kidney disease. In the heart, activation of the NLRP3 inflammasome has been related to enhanced release of interleukin-1β (IL-1β) and the subsequent induction of atherosclerosis and heart failure. Apart from their glucose-lowering effects, SGLT-2 Inhibitors have been documented to attenuate activation of the NLRP3, thus, resulting in the constellation of an anti-inflammatory milieu. In this review, we will focus on the interplay between SGLT-2 Inhibitors and the inflammasome in the kidney, the heart and the neurons in the context of diabetes mellitus and its complications.
Keywords:
SGLT-2 Inhibitors
; Inflammasome
; NLRP3
; reno-protective effects
; Cardio-protective effects
; neuro-protective effects.
Introduction
SGLT (sodium glucose co-transporters) inhibitors are transmembrane proteins that normally promote glucose reabsorption in the intestine and renal glomerulus through a sodium-dependent process that leads to concomitant sodium retention [1]. SGLT-1s are mainly expressed in the small intestine, in contrast to SGLT-2s, which are located mainly in the proximal renal tubule, specifically in the S1 and S2 segments, where they ensure the reabsorption of 90% of the glucose filtered into the renal glomerulus. The remaining 10% of the filtered glucose is reabsorbed with the help of SGLT-1 inhibitors, in the S3 segment of the renal tubule, where the available glucose in the lumen is in a smaller amount. Sodium-glucose co-transporter 2 (SGLT-2) inhibitors or gliflozins are a novel class of anti-diabetic drugs with a unique mechanism of action, as they promote the reduction of serum glucose, by inducing glycosuria in an insulin-independent manner. At the same time, they cause natriuresis, resulting in a reduction in blood pressure [2,3,4].
Phlorizin is the first natural SGLT1/2 inhibitor, first isolated in 1835, which belongs to the family of flavonoids. It was originally isolated from apple bark and was shown to cause glycosuria through non-selective action in SGLT-1 and SGLT-2 co-transporters, resulting in inhibition of intestinal and renal glucose reabsorption. Despite its beneficial effects on the complications of diabetes mellitus and the improvement of insulin resistance, it was eventually withdrawn, as it presented significant drawbacks, such as limited oral bioavailability, reduced intestinal absorption and rapid renal excretion [5,6,7]. Many years later, the scientific community in its quest to discover new therapeutic weapons in an effort to achieve glycemic control in diabetic patients, is bringing back gliflozins to the fore and creating newer synthetic SGLT inhibitors. Thus, in March 2013, the Food and Drug Administration (FDA) approved Canagliflozin, while a year later Dapagliflozin and Empagliflozin were also endorsed by the FDA [8]. Beyond their role in glycemic regulation, SGLT-2 inhibitors seem to have remarkable pleiotropic effects, with multiple reports highlighting their potential anti-inflammatory action. According to more recent data, SGLT-2 inhibitors are no longer considered as pure anti-diabetic drugs, as they have now received an indication for administration to patients with heart failure and chronic kidney disease, regardless of the presence of diabetes mellitus [8].
NLR Family Pyrin Domain Containing 3 (NLRP3 inflammasome) is a protein complex of the innate immune system, with major triggers for its activation, infection and cell damage. Structurally, it consists of a sensor, NLRP3, expressed in cells of the immune system and especially in macrophages, an adapter (PYCARD, PYD and CARD Domain Containing) and finally an effector, caspase-1, which in turn cleaves Gasdermin D (GSDMD). This results in the formation of a pore in the cell membrane, leading to the leakage of intracellular components, including the mediators of the inflammatory response IL-1β and IL-18. GSDMD is the final driver of pyroptosis, a newer form of programmed cell death, as the inflammatory response to pathogens. While NLRP3 activation plays a central role in host defense against invading pathogens, its deleterious activation with consequent activation of pyroptosis appears to contribute significantly to the pathogenesis of many metabolic diseases, such as type 2 diabetes mellitus and chronic kidney disease [9,10,11,12,13]. In this article, we review the potential effects of SGLT2 inhibitors in inflammation, the potential actions of SGLTs in inflammation, emphasizing on their effect on NLRP3 function.
Inflammasome's Mechanism of action
Inflammation is a protective immune response to harmful stimuli, and can be caused by both infectious and non-infectious factors. The triggering of inflammation starts with the recognition of PAMPs and DAMPs. PAMPs (pathogen-associated molecular patterns) originate from microbial invaders, while DAMPs (danger-related molecular patterns) originate from damaged cells and tissues [14,15]. Furthermore, PAMPs and DAMPs are recognized by special receptors, known as Pattern Recognition Receptors (PRRs), which are mostly expressed on cells of innate immunity, such as macrophages, monocytes and dendritic cells. They belong to various protein groups and are subdivided into two main categories based on their location in the cell. TLRs (Toll-like receptors) and CTLs (C-type lectin receptors) are found in the cytoplasmic membrane, while NLRs (NOD-like receptors) and RLRs (RIG-I-like receptors) are located in the cytoplasm. TLRs mostly recognize elements of pathogenic bacteria, such as bacterial DNA, lipopolysaccharides (LPS), peptidoglycan and teichoic acid, in contrast with CTLs, which recognize fungi components, with β-D-glucan being a typical recognition target. On the other hand, RLRs act as sensory receptors of viruses’ elements, such as the envelope membrane, and NLRs receptors recognize intermediate ingredients of cellular metabolism.
Intracellular (cytoplasmic) receptors (NLRs, RLRs) are part of a complex, called inflammasome, which is a key-parameter of non-specific immunity and acts as a "factory" for the production of pro-inflammatory cytokines (such as IL-1β), with pleiotropic effects on inflammation. The inflammasome is a polyprotein intracellular complex and consists of 3 parts; a protein-sensor, one or more protein-adaptors, which contain the caspase binding region, also known as the CARD region and pro-caspase-1. Caspases are a family of cysteine proteases that serve as main effectors during apoptosis to proteolytically dismantle most cellular structures. They act as specific molecules that are localized in the cytoplasm in the form of their inactive proenzymes, the pro-caspases. There are many types of inflammasome, although the best studied is the inflammasome 3 (NLRP3), which is the therapeutic target of SGLT-2 inhibitors. NLRP3 belongs to the NOD-like receptor family. In the case of NLRP3, the sensor protein is the NLRP3 receptor (LYR, NACHT and PYD domains), while the adaptor protein is ASC. Specifically, it contains a leucine-rich region (LRR), which recognizes and binds PAMPs or DAMPs, an intermediate nucleotide-binding domain (NACHT), which is responsible for polymerization and a pyrin domain (PYD), whose main role is the activation of caspase-1 through the CARD region [10,11,12,13,14,15].
Inflammasome formation and activation require two signals. The first one induces transcription and production of individual components, while the second one promotes polymerization and assembly into an active inflammasome. The first signal comes after the recognition and binding of PAMPs and DAMPs to the respective receptors of the innate immune cells. Binding of PAMPs and DAMPs triggers end-to-end intracellular signal transduction pathways resulting in the activation of the cytoplasmic transcription factor NF-κB (Nuclear Factor-κB). NF-κB, in turn, induces transcription of the genes encoding pro-IL-18 and pro-IL-1β cytokines, which polymerize and activate the NLRP3 receptor. Then, secondary signals, such as potassium efflux and the production of oxygen free radicals, contribute to the polymerization and the connection of the remaining parts (ACS and procaspase-1), resulting in the formation and activation of the inflammasome. As a result, the production of active caspase-1, leads to hydrolysis of pro-IL-18 and pro-IL-1β to IL-18 and IL-1β, which are finally secreted into the environment [10,11,12,13,14,15].
IL-18 and IL-1β further stimulate non-specific and specific immunity. Also, the production of IL-18 and IL-1β is associated with the induction of insulin resistance in tissues, which plays a role in the treatment of infections. This local insulin resistance develops in order to conserve glucose and free fatty acids, essential energy substrates for immune cells to fight pathogen invasion. Caspase-1, resulting from inflammasome activation, along with caspase-11, hydrolyzes gasdermin-D, resulting in lysis pore formation and as a consequence the death of the infected cell, a process known as autophagy. As a sequelae, a release of DAMPs is observed, which in turn can strengthen immunity, as they can activate the inflammasome of the neighboring cells. Overall, the inflammasome is a major regulator of non-specific immunity and the goal of its activation is both the destruction of the infectious invader and the limitation of the tissue damage, through the production of interleukins and the routing of autophagy [15,16].
SGLT-2 inhibitors' Anti-inflammatory effects
In addition to their effect on glycemic regulation, numerous studies have revealed the cardioprotective and nephroprotective effect of SGLT inhibitors. SGLT-2 inhibitors display remarkable pleiotropic actions, targeting multiple mechanisms that go beyond the context of glucose homeostasis, although these are not completely understood to date. The favorable effects of this class of agents on weight loss through glucose excretion, blood pressure lowering through induced natriuresis, and mitochondrial function and biogenesis make these drugs a first-line therapeutic option in patients with cardiovascular disease. In addition, gliflozins appear to exhibit remarkable effects on mechanisms such as pyroptosis, autophagy, and oxidative stress, a finding that highlights their multisystem efficacy and their potential to target mechanisms at multiple pathophysiological levels [15,16,17,18].
Recent studies have shown that gliflozins exerts an anti-inflammatory activity, with a potential therapeutic role in the treatment of diseases, such as obesity, atherosclerosis and non-alcoholic steatohepatitis. In the abovementioned disorders, inflammation is well known to play a central pathophysiological role. In its initial stages, inflammation is a protective mechanism of the immune system and is mediated by molecules, which aim to heal the damage, through the clearance of harmful agents. Progressively, the beneficial role of inflammation is exhausted, it becomes chronic and the adverse effects on the body are reflected by causing tissue damage, as a vicious cycle is created, which perpetuates and at the same time worsens this damage. Inflammation appears to be a factor that accelerates atherosclerosis in diabetic patients with type II diabetes mellitus (T2DM), and thus it is believed that interventions that reduce inflammation may achieve beneficial effects in this group of patients. Relatively recently, a significant number of studies have described the anti-inflammatory effect of SGLT inhibitors, based on different models of experimental approach [16,17,18,19].
Macrophages are immune cells that play a central role in the inflammatory process, as they contribute both to the defense against the invading microorganism and to the repair of damaged tissues. Traditionally, they are classified into two groups, the M1 subgroup (classically activated macrophages), which are stimulated by Th1 cytokines or LPS bacterial lipopolysaccharides and stimulate the production of proinflammatory cytokines, and the M2 subgroup (alternatively activated macrophages), which exert an anti-inflammatory effect after its activation by Th2 cytokines. Macrophage cells possess remarkable plasticity, which enables them to transform from one form to another, a process called polarization, in response to the respective stimulus of their microenvironment. SGLT inhibitors appear to modulate the conversion of macrophages from one form to another in such a way as to favorably alter the inflammatory response [20]. Existing studies mainly concern the three main SGLT-2 inhibitors, dapagliflozin, canagliflozin and empagliflozin.
The main mechanisms through which gliflozins interfere with macrophage cells concern their ability to promote the conversion of M1 macrophages to M2, a property that is maintained even in hyperglycemic conditions, while they inhibit the LPS-induced secretion of pro-inflammatory cytokines (IL and TNF-a). Furthermore, SGLT inhibitors increase the expression of macrophage-mediated regulators of immune and inflammatory responses, with notable action on the TLR4 receptor and NF-kB. Their aforementioned actions have as a final consequence the limitation of the clinical effects of inflammation, such as atherosclerosis and fibrosis, both in the cardiovascular system and in the liver and kidneys. In particular, empagliflozin reduces the infiltration of atheromatous plaque by macrophage cells, by inhibiting the proliferation of macrophages in the plaque, thus contributing to its regression. In the liver, in addition to reducing tissue infiltration by macrophages, it activates their autophagy, through the activation of the mammalian target of rapamycin (mTOR) target. Finally, SGLT-2 inhibitors exert a favorable effect on adipose tissue as well, suppressing obesity-induced macrophage accumulation, while reducing the amount of adipose tissue and the size of adipocytes [15,20].
SGLT-2 and the heart
SGLT-2 inhibitors are glucose-sodium co-transporters and, thus, inhibition of their activity results in osmotic diuresis and natriuresis. The ensuing decrease in plasma volume has multiple benefits for the cardiovascular system, as it contributes to the reduction of stress on the cardiovascular wall, with a concomitant drop in blood pressure. Despite compensatory activation of the renin-angiotensin-aldosterone axis (RAAS), the reduction in plasma volume is long-lasting, likely due to continued glycosuria and the initial reduction in total body sodium content. In addition, the administration of an SGLT-2 inhibitor may enhance the action of diuretics in patients with heart failure, as it is believed that their action on the proximal tubule is not limited to SGLT-2 co-transporters, but may also inhibit the sodium-hydrogen exchanger (NHE3), which normally regulates the re-absorption of sodium bicarbonate. This protein is up-regulated in heart failure, with the main consequence being the reduced effect of diuretic therapy, through an increase in resistance to natriuresis. At the same time, it leads to cardiotoxicity, through changes in the intracellular space, with an increase in sodium and calcium. Inhibition of NHE in myocardial tissue, in a SGLT-2-independent manner, leads to limitation of hypertrophy and fibrosis in myocardial tissue [21].
According to Paulus and Tschope, systemic inflammation and oxidative stress play a key role in myocardial dysfunction. Inhibition of NHE1 appears to be the key point of empagliflozin's action, as it leads to a reduction of mitochondrial swelling, through inhibition of intracellular calcium entry and reduced release of ROS from myocardial cells. At the same time, the signaling pathway that serves the activation of AKT1/AKT2/Unit AKT318 is inhibited, preventing the stimulation of inducible NOS2 production, with the final result of reducing oxidative stress. The reduction in oxidative stress appears to be achieved through the inhibition of other signaling pathways, which normally serve the increased expression of NOS2 (NFAT1/NFAT2/NFAT3 pathway) and HDAC1 (histone deacetylase 1) by NF-κB downstream of AKT. Inactivation of NHE1 appears to show beneficial effects in myocardial extracellular matrix remodeling, cardiomyocyte stiffness and heart concentric hypertrophy, through multiple mechanisms. Inhibition of NHE1 also affects the function of the directly involved in HFpEF, NLRP3 inflammasome, and IL-1β, through inhibition of cathepsin B activation. In addition, the activation of TNF-α and ICAM1 molecules involved in the inflammatory process is inhibited, as the deactivation of NEH1 leads to the deactivation of their activators, specifically NFAT2 and NFAT3 [15,22,23].
One of the most characteristic side effects of SGLT2 inhibitors is the induction of euglycemic diabetic ketoacidosis. The observed increase in beta-hydroxybutyrate (β-OHB) levels can be fatal if not treated in time, but paradoxically can provide significant benefits. The above finding has been the subject of studies in recent years and according to existing evidence, it is attributed to the ability of β-OHB to inhibit oxidative stress, while at the same time can protect the function of mitochondria and exert an anti-inflammatory effect, as it is an endogenous NLRP3 Inflammasome inhibitor. The increase in β-OHB levels by the administration of SGLT-2 inhibitors seems to be an alternative source of energy for the myocyte in conditions of heart failure, changing in a favorable way the energy environment of the cell, while promoting a reduction in the size of the heart infarct in ischemia-reperfusion injury (I/R injury). The above observation is in agreement with the findings of Horton et al, according to whom, in the final stages of heart failure, the utilization of β-OHB by the heart muscle is significantly increased [24].
The inhibitory effect of β-OHB on the NLRP3 inflammasome has been shown in experimental models in both the heart and the kidney. In 2015, Youm et al, showed that the adoption of the ketogenic diet can bring beneficial anti-inflammatory effects, through the inhibition of the NLRP3 inflammasome, which results from the observed increase in β-OHB levels. The proposed mechanisms, through which elevated levels of β-OHB paradoxically exert a beneficial effect on heart muscle through inhibition of NLRP3 action, are several and emerge from recent data in experimental models [25]. Bae et al showed that increased β-OHB levels can activate AMPK and thereby reduce endoplasmic reticulum stress and the subsequent increase in NLRP3 levels [26]. In addition, attenuation of NLRP3 activity and subsequent prevention of mitochondrial dysfunction through increased levels of β-OHB in HFpEF can markedly slow the progression of the disease, as recently found by Deng et al [27]. It is worth noting that it is still unknown whether elevated blood levels of β-OHB have the same hemodynamic effects among patients with HFrEF or HFpEF, an issue that is expected to be clarified in future studies [27]. Figure 1 depicts the main mechanisms of action of SGLT-2 inhibitors and the inflammasome in the heart, the kidney and the central nervous system (CNS) [Figure 1].
A high-calorie diet appears to lead to NLRP3 activation in several organs, including the liver and kidney, with administration of the SGLT-2 inhibitor empagliflozin reducing this activation. In contrast, Liakos et al showed that hypercaloric diets do not show similar effects in myocardial tissue, as they do not lead to activation of the NLRP3 inflammasome in conditions of myocardial steatosis [28].
Recent data advocate that the cardioprotective effect of SGLT-2 inhibitors could be attributed to their ability to stimulate autophagy, a process of maintaining cellular homeostasis under stress conditions. According to what we know today, the protective mechanism of autophagy is disturbed in heart failure, in such a way, that its enhancement appears favorable in the progression of the disease. Through autophagy, the cell can remove waste and potentially toxic products of cellular stress, ensuring a more metabolically favorable environment. Autophagy can inhibit the action of NLRP3, as it removes endogenous activators of the inflammasome, such as dysfunctional mitochondria, which are a substrate for the occurrence of oxidative stress. Although the mechanism by which SGLT2 inhibitors stimulate autophagy is not entirely clear, it seems likely that they induce activation of adenosine monophosphate-activated protein kinase (AMPK), sirtuin-1 (SIRT1) and hypoxia-inducible factor (HIF-1α and HIF-2α). The activation of these molecules ensures a favorable metabolic balance for the myocardial cell, while at the same time it is related to the transcription of genes that ensure the oxygen supply to the myocyte, while it is hypothesized that the ketogenesis caused by SGL-2 inhibitors can itself induce the mechanism of autophagy, through its ability to activate the aforementioned molecules [29].
The vast majority of the available experimental data show that the cardioprotective properties of SGLT-2 inhibitors are induced, in part, by the inhibition of the NLRP3 inflammasome. However, in 2021 Gordon et al, published the results of an experimental study, which supports that the administration of empagliflozin for the period of 8 weeks did not ultimately lead to the inhibition of NLRP3 or IL-1β levels, suggesting the redirection to other pathophysiological mechanisms that could potentially explain the cardioprotective effects of SGLT2 inhibitors [30].
The Inflammasome and Chronic Kidney Disease (CKD)
The inflammasome has been demonstrated to be involved in the progression of diabetic kidney disease (DKD) [21,23]. This association has been confirmed by the finding of increased levels of IL-1β and Interleukin 18 (IL-18) in the serum and renal tissue among patients with DKD [31,32]. Furthermore, the activation of the NLRP3 inflammasome has been documented in renal infiltrating macrophages, podocytes, tubular epithelial cells as well as in mesangial cells [33,34,35,36,37,38,39]. In addition, confirmatory results of the involvement of the inflammasome in the progression of DKD have come from studies in which Inhibition or knockout of NLRP3 have been performed. For example, the NLRP3-specific inhibitor MCC950 has been demonstrated to ameliorate renal damage as well as fibrosis in diabetic mice (db/db mice) [40,41]. Also, knockout of the NLRP3 by Wu et al had a significant reno-protective effect in STZ-induced diabetic mice [42]. Apart from the NLRP3 inflammasome, the NLRP1 and NLRC4 inflammasome have also been activated in the kidneys of DKD mice. Their activation has been related to increased urinary albumin excretion and renal damage as well [43,44]. Furthermore, Luan et al. have demonstrated that the NLRC5 gene deficiency is associated with reductions in renal inflammation and damage, while they documented a delay in the progression of renal fibrosis in STZ-induced diabetic mice [45]. Other inflammasomes, such as the NLRP2, NLRP6, NLRP10, NLRP12, and AIM2 are also suggested to be implicated in the pathogenesis of non-diabetic kidney diseases [47,48,49]. Valiño-Rivas et al showed that the NLRP6 deficiency led to increased ischemia-reperfusion induced acute kidney injury [50]. In particular, the AIM2 activation has been related to the development of lupus nephritis and hepatitis B associated glomerulonephritis [51,52]. These data are suggestive of a plausible role of the inflammasome in the pathogenesis of chronic kidney disease in general and not only of DKD.
Based upon the above mentioned data, the inflammasome is documented to be implicated in DKD or/and non-diabetic CKD. Therefore, the use of SGLT2 Inhibitors, which have been documented to ameliorate renal damage, is increasingly being recognized as a potential target of the inflammasome in the kidney. Interestingly, empagliflozin has been shown to reduce obesity-related kidney disease in animal models. More specifically, Ye et al have demonstrated that empagliflozin by inhibiting the NLRP3 inflammasome, has resulted in protection against obesity related kidney disease. The exact mechanisms of this protection are not fully elucidated. However, empagliflozin is postulated to affect genes, which play a crucial role in inhibition of the NLRP3 inflammasome [53]. In addition, Benetti et al have also documented the inhibitory effects of empagliflozin upon the NLRP3 inflammasome and the subsequent mitigation of inflammatory responses in DKD in a mouse model of high fat high sugar fed diet [54]. Regarding dapagliflozin, Birnbaum et al have demonstrated an inhibitory effect on the inflammasome in an animal model of T2DM, which resulted in attenuation of the renal damage [55]. Besides, only recently dapagliflozin has been shown to inhibit the NLRP3 inflammasome, thereby enhancing the anti-inflammatory and anti-fibrotic changes in DKD [56]. To our knowledge, there are no studies with other gliflozins until now, regarding other SGLT-2 inhibitors, the inhibition of the inflammasome and the reno-protective potential. Table 1 depicts major studies in animal models regarding glucose-lowering drugs and the inflammasome [Table 1].
The Inflammasome and Central Nervous System
There is a growing interest regarding SGLT2 inhibitors and their neuro-protective potential. SGLT2 inhibitors are lipid-soluble molecules, which have the capacity to cross the blood-brain barrier. A brain-to-serum ratio of the area under the curve has been reported to be 0.3 for canagliflozin and dapagliflozin and up to 0.5 for empagliflozin [57]. As SGLT1 and SGLT2 receptors are expressed in the brain, SGLT2 inhibitors are capable of directly affecting their target. Brain expression of SGLT2 is lower than SGLT1, and it occurs mainly in the microvessels of the blood-brain barrier as well as in the amygdala, hypothalamus, periaqueductal gray, and the dorsomedial medulla [58,59]. It is noteworthy that the brain locations, where SGLTs have been found, are suggested to be responsible for learning process, food intake, energy and glucose homeostasis. Moreover, these regions have been implicated in central cardiovascular and autonomic regulation as well [59,60]. It is possible that SGLT2 receptors also exert a cardioprotective effect through central mechanisms by directly influencing cardiovascular regulation and autonomic pathways, including the paraventricular nucleus of the hypothalamus, the nucleus of the solitary tract, and the periaqueductal gray [61].
NLRP3 inflammasome is activated not only by microorganisms, but also in cases of chronic inflammatory diseases, such as atherosclerosis and Alzheimer’s disease [62,63]. NLRP3 contributes to the inflammatory process in atherosclerosis, as its activation has been demonstrated in arterial walls, as a result of lipoprotein accumulation [64]. Indeed, in an animal model of atherosclerosis, the inhibition of the NLRP3 inflammasome by MCC950 led to a significant decrease in atherosclerotic lesions [65]. SGLT2 inhibitors are suggested to ameliorate atherosclerosis and cognitive dysfunction by inhibiting the NLRP3 inflammasome. Kim et al used empagliflozin or glimepiride, a sulfonylurea, among 61 patients with T2DM and high CVD risk and measured several metabolic parameters after 30 days of treatment [18]. They documented that empagliflozin resulted in a more significant reduction in the release of IL-1β and serum insulin levels together with increased serum levels of BHB, when compared to sulfonylureas. Furthermore, they performed an ex vivo study with human macrophages that confirmed the inhibitory effects of empagliflozin on the NLRP3 inflammasome [18].
Indeed, in an animal model of diabetes mellitus and Alzheimer’s disease, empagliflozin was documented to improve both cerebral microvascular impairment and cognitive dysfunction, too [66]. More specifically, empagliflozin was shown to reduce thinning of the cortex, hemorrhages and microglia burden as well as senile plaques in this mouse model of T2DM and Alzheimer’s disease [67]. In Alzheimer’s disease, NLRP3 inflammasome has been postulated to interconnect systemic inflammation with neuro-inflammation by impairing the removal of amyloid-beta via the microglia [68]. This effect may be clinically significant, as, in another study, the inhibition of NLRP3 by OLT1177 significantly improved cognitive impairment in an animal model of Alzheimer’s disease [68]. Apart from animal models, Wu et al have very recently published an intriguing study in Diabetes Care. Among 106,903 individuals that were enrolled in this study, SGLT-2 inhibitors compared with DPP-4 inhibitors were associated with a decreased risk of dementia (14.2/1,000 person-years; aHR 0.80 [95% CI 0.71-0.89]). Regarding SGLT-2 inhibitors, dapagliflozin showed the lowest risk (aHR 0.67 [95% CI 0.53-0.84]), followed by empagliflozin (aHR 0.78 [95% CI 0.69-0.89]), whereas no relationship between canagliflozin and dementia was noted (aHR 0.96 [95% CI 0.80-1.16]) [69]. Therefore, nowadays, there is limited evidence for a truly neuroprotective role of SGLT-2 inhibitors in the context of cognitive impairment and especially diabetes. However, further large-scale studies are needed to support this notion and the study by Wu et al may be just the beginning.
Conclusion
The inflammasome, especially NLRP3 inflammasome, seems to play a crucial role in mediating inflammation among patients with diabetes and heart failure, CKD and cognitive impairment. SGLT-2 inhibitors, apart from their glucose-lowering effects are postulated to mitigate activation of the inflammasome and thus, create an anti-inflammatory milieu. As chronic low-grade inflammation is the cornerstone of diabetes, heart failure, CKD and cognitive impairment, it seems likely that SGLT-2 inhibitors may exert beneficial effects on the abovementioned clinical entities. In fact, SGLT-2 inhibitors have been demonstrated to exhibit their therapeutic potential among patients with heart failure and CKD, even without diabetes. Their utility regarding cognitive impairment is still under investigation. Nowadays, there is growing evidence for the pleiotropic effects of SGLT-2 inhibitors [70]. These pleiotropic effects are largely attributed to their attenuation of inflammasome activation. This attenuation as well as other mechanisms of action of this advantageous class of glucose-lowering drugs will be further elucidated in the 21st century. Overall, their utilization among patients with diabetes and heart failure, CKD or even cognitive impairment should be a fact, not a fiction.
Abbreviations
AMPK: Adenosine Monophosphate-activated Protein Kinase; CVD: β-HOB: beta hydroxybutyrate; Cardiovascular Disease; CKD: Chronic Kidney Disease; CNS: Central Nervous System; DAMPs: Danger related molecular patterns; DKD: Diabetic Kidney Disease; DPP4-Inhibitors: Dipeptidyl Peptidase 4 Inhibitors; FDA: Food and Drug Administration; GSDMD: Gasdermin D; HFpEF: Heart Failure with preserved Ejection Fraction; HFrEF: Heart Failure with reduced Ejection Fraction; HIF: Hypoxia Inducible Factor; IL: Interleukin; LPS: Lipopolysaccharide; mTOR: mammalian Target Of Rapamycin NLRP3: Nucleotide-binding domain like receptor protein 3; PAMPs: Pathogen associated molecular patterns; SIRT1: Sirtuin 1; SGLT: Sodium Glucose Co-transporters; NHE3: sodium-hydrogen exchanger; TLRs: Toll Like Receptors; T2DM: Type 2 Diabetes Mellitus; TNF-α: Tumor Necrosis Factor alpha.
References
- Caparrotta, T.M.; Greenhalgh, A.M.; Osinski, K.; Gifford, R.M.; Moser, S.; Wild, S.H.; Reynolds, R.M.; Webb, D.J.; Colhoun, H.M. Sodium–Glucose Co-Transporter 2 Inhibitors (SGLT2i) Exposure and Outcomes in Type 2 Diabetes: A Systematic Review of Population-Based Observational Studies. Diabetes Ther. 2021, 12, 991–1028. [Google Scholar] [CrossRef] [PubMed]
- Kang, A.; Jardine, M.J. SGLT2 inhibitors may offer benefit beyond diabetes. Nat. Rev. Nephrol. 2020, 17, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Cefalu, W.T.; Riddle, M.C. SGLT2 Inhibitors: The Latest “New Kids on the Block”! Diabetes Care 2015, 38, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Vallianou, N.G.; Christodoulatos, G.S.; Kounatidis, D.; Dalamaga, M. Sotagliflozin, a dual SGLT1 and SGLT2 inhibitor: In the heart of the problem. Metab. Open 2021, 10, 100089. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C. Analyse des Phloridzins. Eur. J. Org. Chem. 1835, 15, 178–178. [Google Scholar] [CrossRef]
- White, J.R. Apple Trees to Sodium Glucose Co-Transporter Inhibitors: A Review of SGLT2 Inhibition. Clin. Diabetes 2010, 28, 5–10. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Trigkidis, K.; Kazazis, C. Sodium glucose co-transporter 2 inhibitors and their nephroprotective potential. Clin. Nephrol. 2017, 87, 212–216. [Google Scholar] [CrossRef]
- Available online: www.uptodate.com (accessed on 15 April 2023).
- The Human Protein Atlas Single Cell Type—NLRP3. Available online: https://www.proteinatlas.org/ENSG00000162711-NLRP3 /single+cell+type (accessed on 12 April 2023).
- Marcuzzi, A.; Melloni, E.; Zauli, G.; Romani, A.; Secchiero, P.; Maximova, N.; Rimondi, E. Autoinflammatory Diseases and Cytokine Storms—Imbalances of Innate and Adaptative Immunity. Int. J. Mol. Sci. 2021, 22, 11241. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Burdette, B.E.; Esparza, A.N.; Zhu, H.; Wang, S. Gasdermin D in pyroptosis. Acta Pharm. Sin. B 2021, 11, 2768–2782. [Google Scholar] [CrossRef]
- Lee, H.-M.; Kim, J.-J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.-K. Upregulated NLRP3 Inflammasome Activation in Patients with Type 2 Diabetes. Diabetes 2012, 62, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Toldo, S.; Marchetti, C.; Kron, J.; Van Tassell, B.W.; Dinarello, C.A. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ. Res. 2020, 126, 1260–1280. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Meng, X.-F.; Zhang, C. NLRP3 Inflammasome in Metabolic-Associated Kidney Diseases: An Update. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Feijóo-Bandín, S.; Aragón-Herrera, A.; Otero-Santiago, M.; Anido-Varela, L.; Moraña-Fernández, S.; Tarazón, E.; Roselló-Lletí, E.; Portolés, M.; Gualillo, O.; González-Juanatey, J.R.; et al. Role of Sodium-Glucose Co-Transporter 2 Inhibitors in the Regulation of Inflammatory Processes in Animal Models. Int. J. Mol. Sci. 2022, 23, 5634. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, A.; Akter, A.; Hossain, S.; Sarker, T.; Safa, S.A.; Mustafa, Q.G.; Muhammad, S.A.; Munir, F. Role of NLRP3 inflammasome in liver disease. J. Dig. Dis. 2020, 21, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, S.-G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Benetti, E.; Mastrocola, R.; Vitarelli, G.; Cutrin, J.C.; Nigro, D.; Chiazza, F.; Mayoux, E.; Collino, M.; Fantozzi, R. Empagliflozin Protects against Diet-Induced NLRP-3 Inflammasome Activation and Lipid Accumulation. Experiment 2016, 359, 45–53. [Google Scholar] [CrossRef]
- Tsigalou, C.; Vallianou, N.; Dalamaga, M. Autoantibody Production in Obesity: Is There Evidence for a Link Between Obesity and Autoimmunity? Curr. Obes. Rep. 2020, 9, 245–254. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Packer, M.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Masood, H.; Che, R.; Zhang, A. Inflammasomes in the Pathophysiology of Kidney Diseases. Kidney Dis. 2015, 1, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. J. Clin. Investig. 2019, 4, e124079. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.-D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.R.; Kim, D.H.; Park, M.H.; Lee, B.; Kim, M.J.; Lee, E.K.; Chung, K.W.; Kim, S.M.; Im, D.S.; Chung, H.Y. β-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget 2016, 7, 66444–66454. [Google Scholar] [CrossRef]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Liakos, A.; Tsapas, A.; Bekiari, E. Some glucose-lowering drugs reduce risk for major adverse cardiac events. Ann. Intern. Med. 2020, 173, JC9–JC10. [Google Scholar] [CrossRef] [PubMed]
- Nasiri-Ansari, N.; Nikolopoulou, C.; Papoutsi, K.; Kyrou, I.; Mantzoros, C.S.; Kyriakopoulos, G.; Chatzigeorgiou, A.; Kalotychou, V.; Randeva, M.S.; Chatha, K.; et al. Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE(-/-) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. Int. J. Mol. Sci. 2021, 22, 818. [Google Scholar] [CrossRef]
- Gordon, M.; Meagher, P.; Connelly, K.A. Effect of Empagliflozin and Liraglutide on the Nucleotide-Binding and Oligomerization Domain-Like Receptor Family Pyrin Domain-Containing 3 Inflammasome in a Rodent Model of Type 2 Diabetes Mellitus. Can. J. Diabetes 2020, 45, 553–556. [Google Scholar] [CrossRef]
- Uzu, T.; Yokoyama, H.; Itoh, H.; Koya, D.; Nakagawa, A.; Nishizawa, M.; Maegawa, H.; Yokomaku, Y.; Araki, S.-I.; Abiko, A.; et al. Elevated serum levels of interleukin-18 in patients with overt diabetic nephropathy: effects of miglitol. Clin. Exp. Nephrol. 2010, 15, 58–63. [Google Scholar] [CrossRef]
- Lei, Y.; Devarapu, S.K.; Motrapu, M.; Cohen, C.D.; Lindenmeyer, M.T.; Moll, S.; Kumar, S.V.; Anders, H.-J. Interleukin-1β Inhibition for Chronic Kidney Disease in Obese Mice With Type 2 Diabetes. Front. Immunol. 2019, 10, 1223. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, X.; Zhang, X.; Lu, D.; Guo, R. Electro-Acupuncture Protects Diabetic Nephropathy-Induced Inflammation Through Suppression of NLRP3 Inflammasome in Renal Macrophage Isolation. Endocrine, Metab. Immune Disord. - Drug Targets 2021, 21, 2075–2083. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Meng, X.-F.; Zhang, C. Inflammasome activation in podocytes: a new mechanism of glomerular diseases. Inflamm. Res. 2020, 69, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Yang, Z.; Zhang, C.; Shi, Y.; Han, W.; Song, S.; Mu, L.; Du, C.; Shi, Y. Inhibition of NLRP3 inflammasome ameliorates podocyte damage by suppressing lipid accumulation in diabetic nephropathy. Metabolism 2021, 118, 154748. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Wu, W.; Tang, A.; Luo, N.; Tan, Y. lncRNA GAS5/miR-452-5p Reduces Oxidative Stress and Pyroptosis of High-Glucose-Stimulated Renal Tubular Cells. Diabetes, Metab. Syndr. Obesity: Targets Ther. 2019, 12, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Peng, R.; Zhang, L.-Y.; Sun, Y.; Peng, H.-M.; Liu, H.-D.; Yu, L.-J.; Li, A.-L.; Zhang, Y.-J.; Jiang, W.-H.; et al. LincRNA-Gm4419 knockdown ameliorates NF-κB/NLRP3 inflammasome-mediated inflammation in diabetic nephropathy. Cell Death Dis. 2017, 8, e2583. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.; Hsu, Y.; Shih, Y.; Chang, P.; Lin, C. Glomerular mesangial cell and podocyte injuries in diabetic nephropathy. Nephrology 2018, 23, 32–37. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, X.; Li, L.; Ma, T.; Shi, M.; Yang, Y.; Fan, Q. A small molecule inhibitor MCC950 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3 inflammasome activation. Diabetes, Metab. Syndr. Obesity: Targets Ther. 2019, 12, 1297–1309. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Fan, J.; Zhang, X.; Luan, J.; Bian, Q.; Ding, T.; Wang, Y.; Wang, Z.; Song, P.; et al. Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of NLRP3 inflammasome activation. Cell Death Dis. 2017, 8, e2937. [Google Scholar] [CrossRef]
- Wu, M.; Han, W.; Song, S.; Du, Y.; Liu, C.; Chen, N.; Wu, H.; Shi, Y.; Duan, H. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol. Cell. Endocrinol. 2018, 478, 115–125. [Google Scholar] [CrossRef]
- Soares JL, S.; Fernandes, F.P.; Patente, T.A.; Monteiro, M.B.; Parisi, M.C.; Giannella-Neto, D.; Corrêa-Giannella, M.L.; Pontillo, A. Gain-of-function Variants in NLRP1 Protect against the Development of Diabetic Kidney Disease: NLRP1 Inflammasome Role in Metabolic Stress Sensing? Clin. Immunol. 2018, 187, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Kolb, R.; Pandey, G.; Li, W.; Sun, L.; Liu, F.; Sutterwala, F.S.; Liu, Y.; Zhang, W. Involvement of the NLRC4-Inflammasome in Diabetic Nephropathy. PLOS ONE 2016, 11, e0164135. [Google Scholar] [CrossRef] [PubMed]
- Luan, P.; Zhuang, J.; Zou, J.; Li, H.; Shuai, P.; Xu, X.; Zhao, Y.; Kou, W.; Ji, S.; Peng, A.; et al. NLRC5 deficiency ameliorates diabetic nephropathy through alleviating inflammation. FASEB J. 2018, 32, 1070–1084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, L.; Shi, X.; Yang, L.; Hua, F.; Ma, J.; Zhu, W.; Liu, X.; Xuan, R.; Shen, Y.; et al. Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway. Aging 2020, 12, 24270–24287. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.; Avila-Ferrufino, A.; Skuginna, V.; Susanti, H.E.; Anders, H.-J. Quantitative expression of RIG-like helicase, NOD-like receptor and inflammasome-related mRNAs in humans and mice. Int. Immunol. 2010, 22, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Komada, T.; Chung, H.; Lau, A.; Platnich, J.M.; Beck, P.L.; Benediktsson, H.; Duff, H.J.; Jenne, C.N.; Muruve, D.A. Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD. J. Am. Soc. Nephrol. 2018, 29, 1165–1181. [Google Scholar] [CrossRef] [PubMed]
- Komada, T.; Muruve, D.A. The role of inflammasomes in kidney disease. Nat. Rev. Nephrol. 2019, 15, 501–520. [Google Scholar] [CrossRef] [PubMed]
- Valiño-Rivas, L.; Cuarental, L.; Nuñez, G.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Loss of NLRP6 expression increases the severity of acute kidney injury. Nephrol. Dial. Transplant. 2019, 35, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cai, Y.; Xu, W.; Yin, Z.; Gao, X.; Xiong, S. AIM2 Facilitates the Apoptotic DNA-induced Systemic Lupus Erythematosus via Arbitrating Macrophage Functional Maturation. J. Clin. Immunol. 2013, 33, 925–937. [Google Scholar] [CrossRef]
- Zhen, J.; Zhang, L.; Pan, J.; Ma, S.; Yu, X.; Li, X.; Chen, S.; Du, W. AIM2 Mediates Inflammation-Associated Renal Damage in Hepatitis B Virus-Associated Glomerulonephritis by Regulating Caspase-1, IL-1β, and IL-18. Mediat. Inflamm. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Ye, T.; Zhang, J.; Wu, D.; Shi, J.; Kuang, Z.; Ma, Y.; Xu, Q.; Chen, B.; Kan, C.; Sun, X.; et al. Empagliflozin Attenuates Obesity-Related Kidney Dysfunction and NLRP3 Inflammasome Activity Through the HO-1–Adiponectin Axis. Front. Endocrinol. 2022, 13, 907984. [Google Scholar] [CrossRef] [PubMed]
- Benetti, E.; Mastrocola, R.; Vitarelli, G.; Cutrin, J.C.; Nigro, D.; Chiazza, F.; Mayoux, E.; Collino, M.; Fantozzi, R. Empagliflozin Protects against Diet-Induced NLRP-3 Inflammasome Activation and Lipid Accumulation. Experiment 2016, 359, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, Y.; Bajaj, M.; Yang, H.-C.; Ye, Y. Combined SGLT2 and DPP4 Inhibition Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Nephropathy in Mice with Type 2 Diabetes. Cardiovasc. Drugs Ther. 2018, 32, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Shi, C.; Lv, Y.; Wang, L.; Luo, J.; Jiang, L.; Yang, J.; Zhou, Y. SGLT2 inhibitor counteracts NLRP3 inflammasome via tubular metabolite itaconate in fibrosis kidney. FASEB J. 2021, 36, e22078. [Google Scholar] [CrossRef] [PubMed]
- Tahara, A.; Takasu, T.; Yokono, M.; Imamura, M.; Kurosaki, E. Characterization and comparison of sodium-glucose cotransporter 2 inhibitors in pharmacokinetics, pharmacodynamics, and pharmacologic effects. J. Pharmacol. Sci. 2016, 130, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.K.; DeSilva, S.; Abbruscato, T.J. The Role of Glucose Transporters in Brain Disease: Diabetes and Alzheimer's Disease. Int. J. Mol. Sci. 2012, 13, 12629–12655. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in the brain in health and disease. Pflugers Arch. Eur. J. Physiol. 2020, 472, 1299–1343. [Google Scholar] [CrossRef]
- Enerson, B.E.; Drewes, L.R. The Rat Blood—Brain Barrier Transcriptome. Br. J. Pharmacol. 2005, 26, 959–973. [Google Scholar] [CrossRef]
- Nguyen, T.; Wen, S.; Gong, M.; Yuan, X.; Xu, D.; Wang, C.; Jin, J.; Zhou, L. Dapagliflozin Activates Neurons in the Central Nervous System and Regulates Cardiovascular Activity by Inhibiting SGLT-2 in Mice. Diabetes, Metab. Syndr. Obesity: Targets Ther. 2020, 13, 2781–2799. [Google Scholar] [CrossRef]
- Gaur, A.; Pal, G.K.; Ananthanarayanan, P.H.; Pal, P. Role of Ventromedial hypothalamus in high fat diet induced obesity in male rats: Association with lipid profile, thyroid profile and insulin resistance. Ann. Neurosci. 2014, 21, 104–107. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Fu, J. Novel Insights Into the NLRP3 Inflammasome in Atherosclerosis. J. Am. Hear. Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slütter, B.; Foks, A.C.; Bot, I.; Kuiper, J.; T, W.; et al. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E–Deficient Mice—Brief Report. Arter. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Hierro-Bujalance, C.; Infante-Garcia, C.; del Marco, A.; Herrera, M.; Carranza-Naval, M.J.; Suarez, J.; Alves-Martinez, P.; Lubian-Lopez, S.; Garcia-Alloza, M. Empagliflozin reduces vascular damage and cognitive impairment in a mixed murine model of Alzheimer’s disease and type 2 diabetes. Alzheimer's Res. Ther. 2020, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance through NLRP 3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Iskander, C.; Wang, C.; Xiong, L.Y.; Shah, B.R.; Edwards, J.D.; Kapral, M.K.; Herrmann, N.; Lanctôt, K.L.; Masellis, M.; et al. Association of Sodium–Glucose Cotransporter 2 Inhibitors With Time to Dementia: A Population-Based Cohort Study. Diabetes Care 2022, 46, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Vallianou, N.G.; Geladari, E.; Kazazis, C.E. SGLT-2 inhibitors: Their pleiotropic properties. Diabetes Metab. Syndr. Clin. Res. Rev. 2017, 11, 311–315. [Google Scholar] [CrossRef]
- Ye, Y.; Bajaj, M.; Yang, H.-C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef]
- Birnbaum, Y.; Tran, D.; Bajaj, M.; Ye, Y. DPP-4 inhibition by linagliptin prevents cardiac dysfunction and inflammation by targeting the Nlrp3/ASC inflammasome. Basic Res. Cardiol. 2019, 114, 35. [Google Scholar] [CrossRef]
Figure 1.
The main mechanisms of action of SGLT-2 Inhibitors and the Inflammasome.
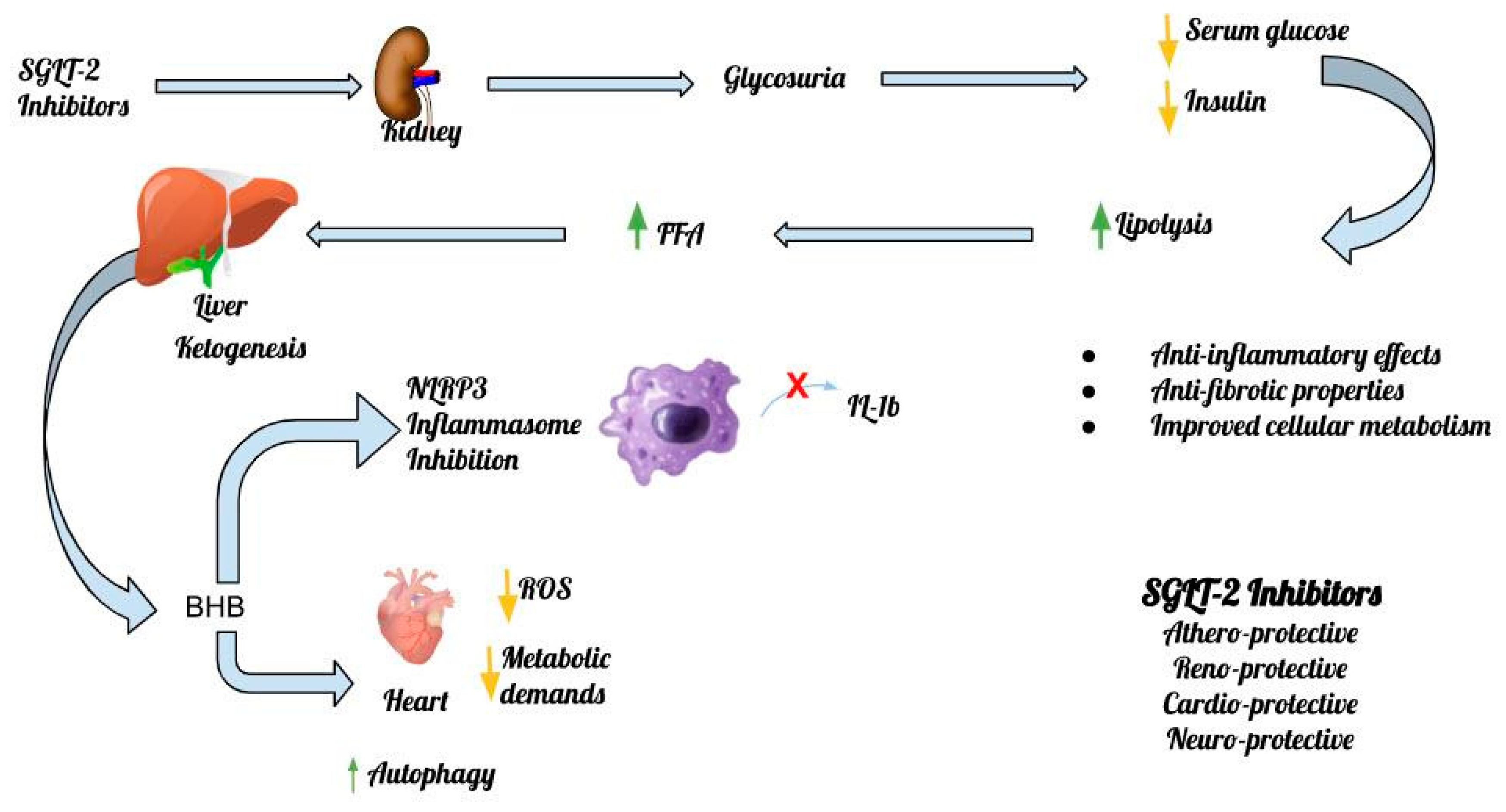

Table 1.
List of main studies in animal models associating SGLT-2 Inhibitors with the NLRP3 Inflammasome.
Table 1.
List of main studies in animal models associating SGLT-2 Inhibitors with the NLRP3 Inflammasome.



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



