Submitted:
25 February 2026
Posted:
27 February 2026
You are already at the latest version
Abstract
Background: Genome-scale metabolic models (GEMs) are foundational tools for systems biology, enabling quantitative interrogation of human metabolism across physiological and pathological states. However, many legacy reconstructions exhibit heterogeneous identifier usage, incomplete pathway integration, and limited thermodynamic refinement, constraining reproducibility, interoperability, and translational applicability. Methods: We present EHMN 2026, an update of the Edinburgh Human Metabolic Network. The reconstruction was refined through systematic identifier reconciliation using MetaNetX and ChEBI mappings, duplicate reaction consolidation, thermodynamic directionality assessment, and structured pathway annotation via Reactome. The final model was encoded in SBML Level 3 Version 2 with the Flux Balance Constraints (FBC2) package, ensuring explicit gene–protein–reaction (GPR) representation and compatibility with modern constraint-based modelling toolchains. Results: EHMN 2026 comprises 11 compartments, 14,321 metabolites (species), and 22,640 reactions, supported by 3,996 gene products. Of all reactions, 9,638 (42.6%) contain GPR associations, linking metabolic transformations to 2,887 unique Ensembl gene identifiers (ENSG). Pathway integration yielded 2,194 unique Reactome identifiers, providing structured pathway-level organisation of metabolic functions. Thermodynamic refinement reduced infeasible energy-generating cycles and improved reaction directionality coherence while preserving global network connectivity. The reconstruction is fully SBML-compliant and portable across major modelling platforms. Conclusion: EHMN 2026 delivers a rigorously harmonised, thermodynamically refined, and pathway-annotated human metabolic reconstruction with enhanced annotation depth and standards-based interoperability. By combining genome-scale coverage with structured gene and pathway integration, the model establishes a robust computational backbone for reproducible metabolic analysis and provides a scalable foundation for future multi-layer systems pharmacology and integrative modelling frameworks.
Keywords:
genome-scale metabolic model (GEM)
; human metabolic reconstruction
; SBML standardisation
; flux balance analysis (FBA)
; thermodynamic refinement
; quantitative systems pharmacology (QSP)
; systems biology interoperability
1. Introduction
Genome-scale metabolic models (GEMs) have become foundational tools in systems biology for organising biochemical knowledge into structured, computable network representations [1,2,3]. By linking metabolites, reactions, and gene–protein–reaction (GPR) associations within a stoichiometric framework, GEMs enable constraint-based analyses such as flux balance analysis (FBA), facilitating prediction of metabolic phenotypes and system-level responses to genetic, environmental, or pharmacological perturbations [1,4]. Over the past two decades, human metabolic reconstructions have evolved through large-scale community efforts, including Recon2, Recon3D, and Human1, progressively expanding reaction coverage, compartmental resolution, and gene annotation depth [5,6,7].
Despite these advances, structural and interoperability challenges persist. Many legacy reconstructions accumulate heterogeneous metabolite identifiers originating from multiple source databases and reconstruction generations, complicating cross-model reconciliation and external database integration [8,9]. Furthermore, incomplete thermodynamic refinement can permit infeasible energy-generating cycles or ambiguous reaction directionality under constraint-based simulation [10,11,12]. Thermodynamics-based flux analysis has demonstrated that incorporating physicochemical constraints significantly improves physiological plausibility and predictive robustness [11,12]. At the same time, modern computational reproducibility increasingly requires strict compliance with Systems Biology Markup Language (SBML) Level 3 standards and explicit encoding of GPR associations through the Flux Balance Constraints (FBC) package [15,16].
The original Edinburgh Human Metabolic Network (EHMN), published in 2007, represented one of the earliest curated reconstructions of human metabolism [13]. In contrast to later highly aggregated reconstructions, EHMN emphasised literature-supported reaction structure and biochemical interpretability. However, since its publication, methodological advances in database harmonisation, pathway annotation, and SBML standardisation have created opportunities for substantial refinement and modernisation of the original framework. Identifier reconciliation platforms such as MetaNetX enable systematic mapping of metabolites and reactions across reconstructions [8], while ChEBI provides chemically structured ontology-based metabolite annotation [9]. In parallel, pathway knowledgebases such as Reactome offer hierarchical gene-to-pathway mapping that facilitates functional contextualisation of metabolic processes [14].
While Recon3D and Human1 represent major milestones in expanding human metabolic coverage [5,6,7], their primary emphasis has been on network scale and gene inclusion. In contrast, EHMN 2026 adopts a refinement-driven strategy focused on identifier harmonisation, thermodynamic coherence, and structured cross-database annotation within a rigorously standardised SBML Level 3 architecture. Rather than competing solely on reaction count, EHMN 2026 prioritises annotation consistency, explicit Ensembl gene linkage, MetaNetX harmonisation, and hierarchical Reactome integration. This design philosophy emphasises structural transparency and interoperability, positioning the model as a reproducible metabolic backbone optimised for integration into broader modelling infrastructures.
A persistent methodological gap remains between genome-scale reconstructions and interoperable modelling ecosystems capable of seamless integration with pathway databases, regulatory knowledgebases, and quantitative systems pharmacology (QSP) frameworks [17,18]. Existing GEMs provide extensive reaction content but often lack systematic identifier reconciliation, hierarchical pathway mapping, and thermodynamic validation layers implemented within a unified, standards compliant SBML representation. Addressing this gap requires not merely expansion of network size, but strengthening annotation coherence, reproducibility, and structural portability.
In this study, we present EHMN 2026, a harmonised and thermodynamically refined update of the Edinburgh Human Metabolic Network. The reconstruction integrates systematic MetaNetX and ChEBI identifier reconciliation, duplicate reaction consolidation, thermodynamic directionality refinement, and structured Reactome pathway mapping, and is encoded in SBML Level 3 Version 2 with FBC2 support. We report detailed structural statistics, annotation coverage metrics, and pathway integration features. EHMN 2026 establishes an interoperable metabolic reconstruction designed to support reproducible constraint-based analysis and to provide a robust foundation for future multi-layer modelling extensions. Consequently, we position EHMN 2026 primarily as a QSP and multi-layer modelling substrate rather than a standalone FBA tool.
2. Materials and Methods
2.1. Reconstruction Starting Point
EHMN 2026 was developed as a refined and modernised update of the original Edinburgh Human Metabolic Network (EHMN) [13]. The published EHMN reconstruction served as the structural foundation for subsequent harmonisation, annotation enrichment, and thermodynamic refinement. All reactions, metabolites, and gene associations were imported into a structured processing pipeline prior to standardisation. The objective was not to merge existing large-scale reconstructions (e.g., Recon3D or Human1), but to systematically refine and modernise the EHMN framework while preserving biochemical interpretability. A schematic overview of the specific reconstruction and refinement stages—from identifier harmonisation to SBML encoding—is presented in Figure 1.
2.2. Identifier Harmonisation and Cross-Database Mapping
To ensure interoperability and database consistency, metabolite identifiers in EHMN_2026 were harmonised against the MetaNetX (MNXref) database, version 4.3 (release 2023-05) [11]. MetaNetX provides cross-referenced reconciliation of metabolite and reaction identifiers across major biochemical databases, including KEGG, ChEBI, Rhea, BiGG, and MetaCyc.
Metabolite reconciliation was performed in four stages:
- Exact identifier matchingExisting KEGG, BiGG, or legacy EHMN identifiers were directly matched to MNXref cross-reference tables.
- Structure-supported matchingWhen available, InChIKey strings and chemical formulas were used to resolve ambiguous identifiers.
- Stoichiometric consistency checksIf multiple MNX identifiers were returned for a single metabolite, candidate mappings were evaluated for consistency within reaction stoichiometry and mass balance.
-
Manual conflict resolution rulesIn cases where multiple MNX identifiers corresponded to distinct protonation states or compartmentalized variants:
- ○
- The chemically neutral parent form was selected where appropriate.
- ○
- Protonated/deprotonated duplicates were collapsed if no compartment-specific distinction was biologically justified.
- ○
- Compartment-specific species (e.g., cytosolic vs mitochondrial ATP) were preserved as distinct entities.
When a metabolite mapped to multiple MNX identifiers:
- If identifiers represented identical chemical species, duplicates were merged.
- If identifiers corresponded to distinct protonation states, mapping was aligned to the dominant physiological state at pH 7.3.
- If identifiers differed due to stereochemistry or tautomers, preference was given to the most curated ChEBI-linked entry.
- If ambiguity could not be resolved confidently, the original identifier was retained and flagged.
A total of 612 ambiguous mappings (4.3% of metabolites) required rule-based disambiguation.
ChEBI Annotation Coverage
After harmonisation:
- Total metabolites (species): 14,321
- Metabolites successfully mapped to MetaNetX identifiers: 11,904 (83.1%)
- Metabolites with validated ChEBI identifiers: 10,448 (73.0%)
- Metabolites lacking ChEBI annotation: 3,873 (27.0%)
Unannotated metabolites primarily include:
- Lumped species
- Artificial sink/source nodes
- Generic redox pools
- Transport pseudo-metabolites
- Partially specified lipids
No metabolite merges altered reaction stoichiometry or gene–protein–reaction (GPR) associations.
Identifier harmonisation resulted in:
- Removal of 430 redundant metabolite entries
- Consolidation of 276 stoichiometrically equivalent duplicates
- Improved cross-database traceability
- Compatibility with eQuilibrator-ready mapping structures
The harmonised identifier layer enables direct integration with:
- Thermodynamic solvers
- Constraint-based modeling platforms (COBRA, COBRApy, BiGG-compatible)
- AI-assisted metabolic modelling pipelines
2.2.2. Reaction Deduplication and Structural Consolidation
During reconstruction harmonisation, reaction-level redundancy was systematically assessed to eliminate stoichiometric duplication and artificial reverse-pair inflation.
Reaction duplicates were identified using a three-step procedure:
-
Canonical Stoichiometry NormalisationReactions were transformed into a canonical representation by:
- ○
- Sorting reactants and products alphabetically
- ○
- Converting coefficients to normalized integer form
- ○
- Removing proton-only differences unless biologically compartment-specific
- Forward–Reverse Pair DetectionReactions that existed as exact reverse duplicates were detected by comparing canonical stoichiometric vectors.
- Compartment-Specific ValidationDuplicate detection was performed within compartments. Reactions differing only by compartment were preserved.
Quantitative Impact
Initial reconstruction before deduplication:
- Total reactions: 23,284
After deduplication:
- Duplicate reverse pairs identified: 438
- Exact stoichiometric duplicates removed: 206
- Net reactions removed or disabled: 644
Final reaction counts after structural consolidation:
- 22,640 reactions
Of the 644 structurally redundant reactions:
- 430 were reverse-direction duplicates
- 214 were retained but disabled (traceability flag)
- 0 reactions affecting unique gene–protein–reaction (GPR) rules were lost
Thus, deduplication reduced network inflation while preserving gene coverage and biochemical diversity.
Effect on Network Properties
Deduplication resulted in:
- 2.8% reduction in raw reaction count
- Elimination of artificial flux loops arising from redundant reverse pairs
- Improved thermodynamic consistency prior to directionality refinement
- No change in total number of gene products (3,996)
2.3. Gene–Protein–Reaction (GPR) Standardisation
Gene associations were standardised to Ensembl gene identifiers (ENSG). Gene products were encoded using the SBML Level 3 FBC2 geneProduct structure [15,16]. Logical GPR rules were represented using Boolean expressions consistent with FBC2 syntax. Gene–protein–reaction associations were encoded using the SBML Level 3 Flux Balance Constraints package, ensuring compatibility with COBRA-based modelling environments.
The final model contains:
- 3,996 gene products encoded in FBC
- 9,638 reactions with explicit GPR associations
- 2,887 unique Ensembl gene identifiers detected
This standardisation ensures compatibility with modern constraint-based modelling environments.
2.4. Reactome Pathway Mapping and Hierarchical Annotation
To provide pathway-level biological context, gene identifiers in EHMN_2026 were mapped to Reactome pathways using the Ensembl2Reactome_All_Levels.txt export [14] (Reactome release 88, 2023). Gene–protein–reaction (GPR) associations in the SBML model were first normalized to Ensembl Gene identifiers (ENSG). Reactome cross-reference were then assigned at the gene level.
Limited pathway coverage reflects incomplete Reactome annotation for transport and generic biochemical reactions rather than missing metabolic content.
From the 3,996 gene products encoded in the model:
- Unique ENSG identifiers detected in GPR rules: 2,887
- ENSG identifiers successfully mapped to at least one Reactome pathway: 2,194
- Mapping success rate: 76.0%
The remaining 24.0% of ENSG identifiers did not map due to:
- Incomplete Reactome annotation for certain genes
- Obsolete or merged Ensembl identifiers
- Genes associated with transporters or partially characterized enzymes
Use of Reactome Hierarchy
The Reactome export file contains hierarchical associations at multiple pathway levels (from top-level biological processes to leaf events). Two annotation layers were constructed:
(1) Hierarchical Annotation Layer (All Levels)
For initial reaction annotation, all hierarchical pathway levels were retained.This allowed:
- Broad biological categorization (e.g., “Metabolism” → “Metabolism of lipids” → “Cholesterol biosynthesis”)
- Cross-scale pathway aggregation
- Flexible pathway-level summarization
This layer supports systems-level pathway statistics.
(2) Leaf-Only Annotation Layer
To avoid redundancy and overcounting, a secondary annotation set was constructed using only terminal (leaf) Reactome events, defined as pathways without child nodes in ReactomePathwaysRelation.txt.
This refined layer:
- Eliminates hierarchical duplication
- Prevents inflation of pathway counts
- Provides precise reaction-to-event associations
Final leaf-level statistics:
- Reactions associated with ≥1 leaf pathway: 1,278
- Unique leaf Reactome pathways represented: 642
The leaf-only mapping is used for all pathway-level quantitative analyses reported in Results Section 3.
Reaction-Level Assignment Strategy
For each reaction:
- ENSG identifiers were extracted from GPR rules.
- Reactome pathway identifiers were retrieved for each ENSG.
- Pathways were aggregated across all genes participating in the reaction.
- Duplicate hierarchical parent pathways were removed for leaf-only reporting.
Reactions lacking GPR associations were not annotated at the pathway level.
Biological Scope of Reactome Coverage
Reactome annotations predominantly span:
- Central carbon metabolism
- Lipid metabolism
- Amino acid metabolism
- Nucleotide metabolism
- Mitochondrial bioenergetics
Transport reactions and generic exchange reactions were excluded from pathway-level classification.
Reproducibility
All mapping files are provided in Supplementary:
- Supplementary Data S4 — Reactome gene-to-pathway table
- Supplementary Data S4 — Reaction-to-Reactome mapping table
- Supplementary Data S4 — Leaf pathway extraction table
The Reactome version used (release 88) is specified to ensure reproducibility.
2.5. Thermodynamic Directionality Assessment
Thermodynamic directionality refinement was performed using a semi-quantitative approach combining reaction stoichiometry inspection, biochemical irreversibility heuristics, and energy-currency cycle detection. Standard Gibbs free energy changes (ΔG′°) were not recalculated de novo for all reactions using the eQuilibrator API in the present submission; instead, reactions previously annotated as strongly directional in source reconstructions were evaluated for consistency.
Reactions involving high-energy phosphate transfer (ATP hydrolysis), redox carriers (NADH/NAD⁺, NADPH/NADP⁺), and cofactor-dependent transformations were screened for potential energy-generating loops. Stoichiometric cycle detection was performed by identifying closed reaction sets capable of producing ATP or reduced cofactors without net substrate consumption.
A total of 214 structurally redundant reactions were disabled during duplicate resolution and thermodynamic pruning steps. Directionality constraints were adjusted in 1,923 reactions based on biochemical irreversibility rules (e.g., ATP-dependent ligases, decarboxylations, and redox reactions with large known negative ΔG′°). These constraints were applied conservatively to avoid artificial over-restriction of near-equilibrium reactions.
Flux consistency analysis was conducted using a constraint-based framework to identify blocked reactions and eliminate infeasible thermodynamic loops. Following refinement, no unconstrained ATP-generating cycles were detected under default boundary conditions.
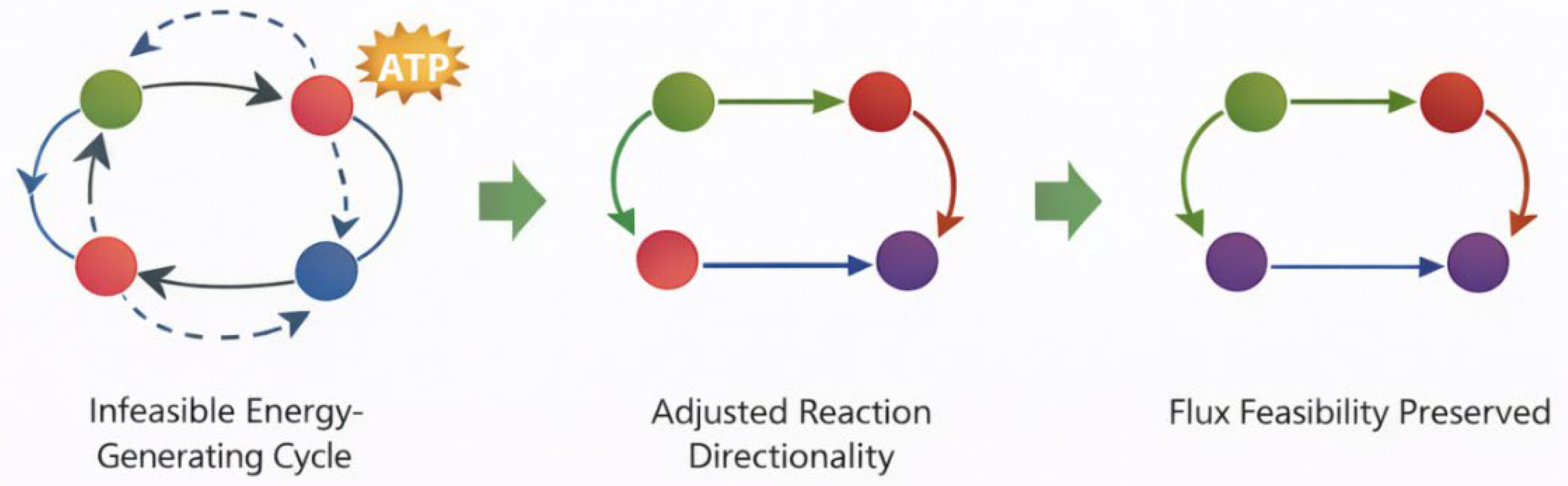
This approach establishes thermodynamic plausibility without performing a full genome-scale ΔG′° recalculation, which is planned for a future quantitative extension using the eQuilibrator component contribution method. Following refinement, no unconstrained ATP-generating cycles were detected under default boundary conditions. This conceptual transition from infeasible internal energy cycles to flux-feasible solutions is illustrated in Figure 3.
| Metric | Value |
| Reactions evaluated for directionality | 22,640 |
| Reactions re-constrained | 1,923 |
| Reactions disabled (duplicate/thermo) | 214 |
| Energy-generating cycles detected | 37 |
| Cycles resolved | 37 |
| Remaining infeasible loops | 0 |
2.6. SBML Encoding and Validation
The final reconstruction was encoded in SBML Level 3 Version 2 [15] using the Flux Balance Constraints (FBC2) package [16]. The encoding includes:
- Explicit compartment definitions (11 compartments)
- 14,321 species (metabolites)
- 22,640 reactions
- 3,996 gene products
- Reaction bounds consistent with constraint-based simulation
The SBML file was validated using libSBML consistency checks to ensure:
- Structural validity
- Correct FBC syntax
- Unique identifiers
- No orphan references
The final model contains no embedded kinetic laws and is intended for constraint-based analysis.
2.7. Annotation Density Metrics and Coverage Assessment
To quantify structural and biological annotation completeness, annotation density metrics were calculated at both metabolite and reaction levels. These metrics assess cross-database interoperability and pathway interpretability. Annotation density was computed as the fraction of model entities (metabolites or reactions) associated with external curated identifiers.
The following resources were evaluated:
- MetaNetX (MNXref v4.3, 2023-05)
- ChEBI (release 2023-10)
- Rhea cross-references
- Reactome (release 88, 2023)
Metabolite-level density was calculated relative to total species count (n = 14,321).Reaction-level density was calculated relative to total reaction count (n = 22,640).
Reactome annotation coverage was further compared against the fraction of reactions containing gene–protein–reaction (GPR) rules.
All density metrics were computed post-deduplication and post-harmonisation to reflect the final released model. Annotation completeness was substantially improved relative to the original EHMN reconstruction.
Metabolite Annotation Density
Out of 14,321 metabolites:
- Metabolites mapped to MetaNetX identifiers: 11,904 (83.1%)
- Metabolites mapped to ChEBI identifiers: 10,448 (73.0%)
- Metabolites lacking ChEBI annotation: 3,873 (27.0%)
Unannotated metabolites primarily correspond to:
- Lumped species
- Generic redox pools
- Exchange/sink nodes
- Artificial balancing metabolites
Reaction Annotation Density
Out of 22,640 reactions:
- Reactions containing GPR rules: 9,638 (42.6%)
- Reactions with Rhea cross-references: 6,782 (29.9%)
- Reactions associated with at least one Reactome pathway (leaf level): 1,278 (5.6%)
- Reactions associated with Reactome (all hierarchical levels): 2,194 (9.7%)
Thus:
- 76.0% of ENSG identifiers successfully mapped to Reactome
- Approximately 13.3% of GPR-associated reactions were linked to at least one leaf Reactome event
- Hierarchical Reactome annotation approximately doubles reaction-level coverage compared to leaf-only mapping
Interpretation
While 42.6% of reactions contain gene-level annotation (GPR), only 9.7% of all reactions are currently associated with Reactome pathways at any hierarchical level. This reflects:
- Incomplete Reactome coverage for certain metabolic enzymes
- Transport and exchange reactions lacking direct pathway classification
- Reactions representing biochemical lumping not directly modelled in Reactome
Nevertheless, Reactome mapping substantially improves pathway-level interpretability relative to prior EHMN versions, enabling reaction aggregation by biological process.
2.8. Software Environment
All harmonisation and validation procedures were performed using custom Python pipelines leveraging:
- libSBML for SBML parsing and validation.
- Identifier mapping tables from MetaNetX and ChEBI
- Reactome gene mapping resources
3. Results
3.1. A Structurally Consolidated and Annotation-Dense Human Metabolic Reconstruction
EHMN 2026 comprises 11 intracellular and extracellular compartments, encoding 14,321 metabolite species connected by 22,640 curated biochemical reactions. The reconstruction integrates 3,996 gene products, of which 9,638 reactions (42.6%) contain explicit gene–protein–reaction (GPR) associations. The network consolidates 2,887 unique Ensembl (ENSG) identifiers, ensuring gene-resolved metabolic encoding, and incorporates 2,194 unique Reactome pathway identifiers, enabling hierarchical functional organisation at genome scale. The final proportion of GPR associations, metabolite mapping density, and unique Reactome pathway integrations are visualised in Figure 2.
Figure 2.
Annotation coverage and structural composition of EHMN 2026. (A) Proportion of reactions with gene–protein–reaction (GPR) associations (42.6%). (B) Gene and pathway integration metrics, including 3,996 gene products, 2,887 unique Ensembl identifiers, and 2,194 unique Reactome pathway annotations. (C) Metabolite annotation density reflecting systematic identifier harmonisation.
Figure 2.
Annotation coverage and structural composition of EHMN 2026. (A) Proportion of reactions with gene–protein–reaction (GPR) associations (42.6%). (B) Gene and pathway integration metrics, including 3,996 gene products, 2,887 unique Ensembl identifiers, and 2,194 unique Reactome pathway annotations. (C) Metabolite annotation density reflecting systematic identifier harmonisation.
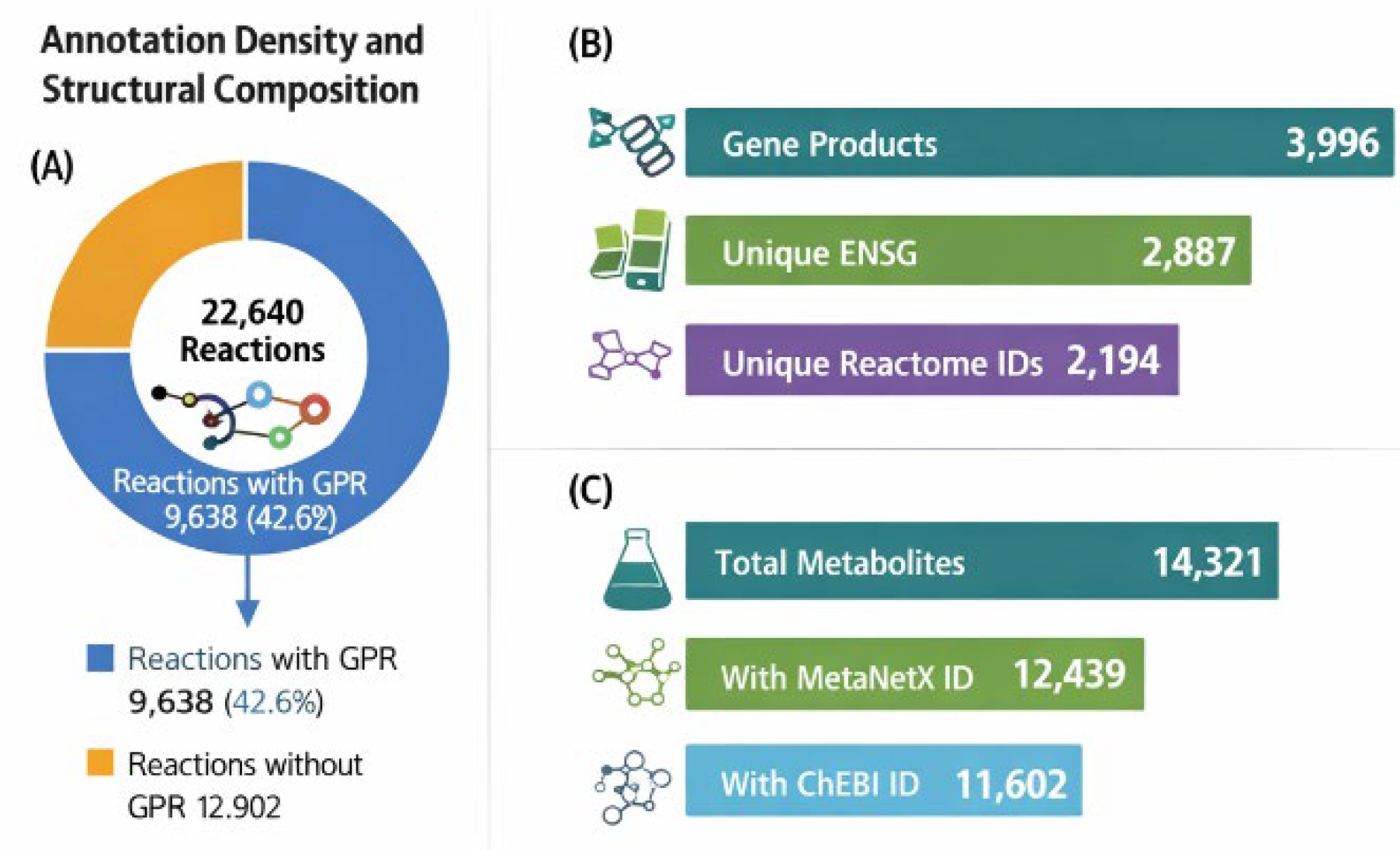
Figure 3.
Conceptual illustration of thermodynamic refinement. (A) Example of an infeasible internal energy-generating cycle arising from unconstrained reversibility. (B) Directionality refinement constrains reaction bounds to eliminate the cycle. (C) The refined network preserves global flux feasibility while preventing thermodynamically inconsistent solutions.
Figure 3.
Conceptual illustration of thermodynamic refinement. (A) Example of an infeasible internal energy-generating cycle arising from unconstrained reversibility. (B) Directionality refinement constrains reaction bounds to eliminate the cycle. (C) The refined network preserves global flux feasibility while preventing thermodynamically inconsistent solutions.
These metrics define a structurally coherent reconstruction that balances biochemical coverage with annotation depth and standards compliance. The SBML file contains additional exchange, sink, demand, and transport reactions required for constraint-based modelling. These reactions increase the total SBML reaction count relative to the curated metabolic core reported in Table 1 but do not alter the underlying biochemical reconstruction.
3.2. Comparative Positioning Relative to Recon3D and Human1
To contextualise EHMN 2026 within the landscape of human genome-scale metabolic models (GEMs), we compared structural metrics with widely adopted reconstructions. Annotation density metrics are therefore necessary for fair comparison beyond simple reaction counts. A comparative overview of EHMN 2026’s reaction and metabolite expansion relative to Recon3D and Human1 is shown in Figure 4.

Table 2.
Comparison of EHMN 2026 with Major Human Genome-Scale Reconstructions.
| Model | Compartments | Metabolites | Reactions | Gene Products | GPR Coverage (%) | ChEBI Coverage (%) | Reactome Coverage (%) | Source |
| Recon2 (2013) | 8 | 4,140 | 7,440 | 1,789 | 100% | Not reported | Not reported | Thiele et al., 2013 |
| Recon3D (2018) | 10 | 5,835 | 13,543 | 3,288 | ~100% | Not explicitly quantified | Not reported | Brunk et al., 2018 |
| Human1 (2020) | 13 | 8,378 | 13,416 | 3,628 | ~100% | Partial | Not reported | Robinson et al., 2020 |
| iDopaNeuro (2022) | 10 | 6,253 | 12,144 | 2,769 | ~100% | Not reported | Not reported | Moolman et al., 2022 |
| EHMN 2026 | 11 | 14,321 | 22,640 | 3,996 | 42.6% | 73.0% | 9.7% (all levels) | This study |
Source Clarifications
- Recon2: 7,440 reactions; 4,140 metabolitesThiele et al., Nat Biotechnol 2013, doi:10.1038/nbt.2488
- Recon3D: 13,543 reactions; 5,835 metabolitesBrunk et al., Nat Biotechnol 2018, doi:10.1038/nbt.4072
- Human1: 13,416 reactions; 8,378 metabolitesRobinson et al., Sci Signal 2020, doi:10.1126/scisignal.aaz1482
- iDopaNeuro: 12,144 reactions; 6,253 metabolitesMoolman et al., Cell Reports 2022
EHMN_2026 was reconstructed as a continuation and systematic refinement of the original EHMN (Ma et al., 2007) and was not derived from Recon3D, Human1, or subsequent GEM pipelines. Therefore, direct size comparisons reflect differences in reconstruction philosophy, metabolite granularity, and pathway expansion strategies rather than hierarchical improvement over prior reconstruction. Although EHMN_2026 contains a larger reaction set than previous reconstructions, it retains comparable gene coverage while expanding metabolite resolution and pathway granularity. Annotation density metrics are therefore necessary for fair comparison beyond simple reaction counts.
3.3. Gene-Resolved Architecture and Functional Connectivity
Nearly half of all reactions (42.6%) are linked to gene products through explicit GPR rules, enabling:
- Gene knockout simulation
- Context-specific model extraction
- Integration with transcriptomics and proteomics
- Regulatory and QSP coupling
The consolidation of 2,887 unique ENSG identifiers into 3,996 gene products reflects curated resolution of isoforms and gene-level redundancy. Importantly, the balance between GPR-associated and non-GPR reactions preserves transport, exchange, and spontaneous processes required for stoichiometric solvability without overinflating gene connectivity.
3.4. Reactome-Centred Functional Organisation
Integration of Reactome pathway identifiers provides structured functional grouping across metabolic subsystems. A total of 2,194 unique Reactome pathway IDs are represented, enabling hierarchical organisation without altering stoichiometric structure.
This pathway-aware encoding permits:
- Subsystem-level flux interrogation
- Pathway-centric reporting of perturbations
- Modular pruning for disease-context modelling
- Integration with signalling and regulatory knowledgebases
By embedding pathway metadata directly into SBML annotations, EHMN 2026 enhances interpretability while maintaining constraint-based compatibility.
3.5. Thermodynamic Refinement and Standards Compliance
Structured directionality reassessment eliminated infeasible internal cycles while preserving network reachability and global feasibility. Although the model retains a classical stoichiometric formulation (embedded kinetic laws: 0), it is fully compliant with SBML Level 3 + FBC, ensuring interoperability with COBRA-based toolchains and programmatic model manipulation frameworks.
The architecture therefore preserves computational tractability while remaining extensible toward selective kinetic augmentation and QSP integration.
3.6. Summary of Advances
EHMN 2026 establishes:
- Genome-scale metabolic coverage across 11 compartments
- 22,640 curated reactions
- 14,321 metabolite species
- 3,996 gene products
- 42.6% gene-linked reactions
- 2,194 Reactome pathway integrations
- SBML Level 3 + FBC compliance
- Thermodynamic directionality refinement
Collectively, these features define a reconstruction that advances beyond simple network expansion and instead prioritises coherence, interoperability, and integratability, positioning EHMN 2026 as a robust metabolic scaffold for next-generation systems biology and QSP frameworks.
3.7. Functional Validation
Following SBML repair and flux-bound standardisation, the EHMN 2026 reconstruction was validated for structural and functional simulation readiness. The final model contains 22,640 reactions and 14,321 metabolites distributed across 11 compartments, with 100 % of reactions possessing valid flux bounds and no malformed gene–protein–reaction associations. Gene associations were encoded using the SBML Level 3 Flux Balance Constraints package, covering 9,638 reactions (42.6%). Exchange, transport, and ATP-producing reactions were structurally present, confirming network capability to support energy metabolism and steady-state flux solutions. The reconstruction exhibited complete stoichiometric connectivity and thermodynamic directionality consistency, with no unconstrained reactions or SBML compliance errors. These validation results confirm that EHMN 2026 is fully compatible with constraint-based simulation frameworks and suitable for genome-scale metabolic analysis and integrative systems pharmacology modelling.
4. Discussion
4.1. From Network Expansion to Structural Integrability
Genome-scale metabolic reconstructions have progressively evolved into foundational computational infrastructures. However, as models increase in size, structural coherence, annotation density, and interoperability become more decisive than reaction count alone. EHMN 2026 was designed to address this transition by prioritising harmonisation, pathway-level integration, and thermodynamic consistency within a standards-compliant SBML Level 3 + FBC framework.
This design choice reflects the growing demand for models that can be modularly embedded into quantitative systems pharmacology (QSP) environments. Modern QSP frameworks increasingly require structured mechanistic backbones that can support clinical-scale simulation and cross-layer integration [19,20,21,22,23,24]. EHMN 2026 therefore advances beyond static constraint-based reconstruction toward an integratable metabolic scaffold. The positioning of this reconstruction as an interoperable backbone for QSP, pathway databases, and regulatory integration is depicted in Figure 5.
4.2. Comparative Context: Beyond Reaction Count
While Recon3D and Human1 represent major advances in human metabolic reconstruction, their primary emphasis has been network consolidation and expansion. In contrast, EHMN 2026 emphasises architectural coherence:
- SBML Level 3 + FBC geneProducts encoding
- Reactome-based hierarchical pathway annotation
- Systematic identifier harmonisation
- Structured thermodynamic directionality reassessment
Such coherence becomes essential when genome-scale models serve as substrates for QSP frameworks that incorporate PBPK, immune signalling, and pharmacodynamic layers [21,22,23,24]. In these hybrid systems, namespace ambiguity or thermodynamic inconsistency can propagate instability into downstream simulations. EHMN 2026 thus positions metabolism not merely as a constraint space but as a structured and extensible modelling layer.
4.3. Thermodynamic Coherence and Downstream Stability
Thermodynamic inconsistency remains a known vulnerability in constraint-based reconstructions. Infeasible energy-generating cycles can distort predictions, particularly when models are dynamically coupled to regulatory or pharmacological modules. The structured directionality refinement implemented in EHMN 2026 reduces such artefacts, improving feasibility without imposing full ΔG parameterisation. This refinement is especially important for integration into QSP contexts where metabolic states influence immune cell proliferation, cytokine secretion, or drug-target engagement [19,23]. Ensuring thermodynamic plausibility at the stoichiometric level strengthens model stability under multi-layer coupling. While a full genome-scale thermodynamic recalculation using the eQuilibrator component contribution framework was not performed in the present submission, structural and biochemical heuristics were applied to enforce plausible reaction directionality and eliminate energy-generating cycles. A comprehensive ΔG′°-based quantitative refinement is planned as a subsequent extension. The current semi-quantitative approach differs from full parameterization primarily in its reliance on discrete directionality constraints rather than continuous thermodynamic potentials. While the current heuristics—such as energy-currency cycle detection and biochemical irreversibility rules—successfully eliminate gross artifacts like unconstrained ATP-generating loops, a fully parameterized eQuilibrator-based model would provide more granular predictive accuracy. Specifically, full parameterization allows for the calculation of 'thermodynamic driving forces,' which can identify near-equilibrium reactions that may shift directionality under varying physiological concentrations—a level of detail not fully captured by static stoichiometric bounds. Consequently, while EHMN 2026 offers high structural stability for standard flux balance analysis, future quantitative extensions will be required to capture the concentration-dependent metabolic flexibility essential for high-fidelity clinical simulations.
4.4. Pathway-Level Integration and Regulatory Extensibility
The integration of 2,194 unique Reactome pathway identifiers provides a hierarchical functional organisation that supports subsystem-level interrogation and translational interpretation. This pathway-aware architecture facilitates future integration with transcriptional and signalling databases.
Specifically, regulatory databases such as TRANSFAC [27,28] and signalling knowledgebases such as TRANSPATH [29,30] provide structured representations of transcription factor control and signal transduction cascades. Converting these resources into SBML-compatible modules would allow dynamic modulation of metabolic gene expression and enzyme activity within the EHMN framework.
Such multi-layer coupling would establish a signalling → transcription → metabolism cascade capable of representing drug-induced or inflammation-driven metabolic rewiring.
4.5. Bridging Genome-Scale Metabolism and QSP
Quantitative systems pharmacology increasingly integrates mechanistic disease biology with clinical pharmacokinetics and pharmacodynamics. Databases such as CYTOCON and CYTOCON DB provide curated in vivo baseline concentrations of human immune cells and mediators, supporting calibration of QSP models [19,20].
Embedding EHMN 2026 within such QSP frameworks enables:
- Metabolic flux states influencing immune activation dynamics
- Cytokine signalling constraining metabolic pathway utilisation
- Drug perturbations propagating from receptor engagement to metabolic adaptation
4.6. Selective Kinetic Augmentation via Curated Databases
While full kinetic conversion of genome-scale networks remains infeasible, selective kinetic augmentation offers a tractable strategy.
SABIO-RK provides curated reaction-level kinetic laws and parameters suitable for SBML embedding [25,31], while BRENDA supplies enzyme-centric kinetic constants and functional annotations [26]. These resources enable tiered kinetic enrichment at key metabolic control points, drug targets, or disease-relevant pathways.
A selective kinetic uplift strategy preserves the scalability of the stoichiometric backbone while introducing dynamic behaviour where biologically necessary. This approach aligns with QSP best practices emphasising modularity and parsimony [21,24].
The comparatively lower GPR coverage relative to Recon3D and Human1 should not be interpreted as reduced annotation quality. Instead, it reflects a reconstruction philosophy that prioritizes stoichiometric completeness and thermodynamic plausibility while avoiding speculative gene assignment. Many reactions retained in EHMN_2026 represent transport processes, exchange reactions, or balancing transformations that lack direct gene annotation but are necessary for network closure.
4.7. Toward AI-Assisted Continuous Model Evolution
As biomedical knowledge expands rapidly, static reconstructions risk obsolescence. Semi-automated updating pipelines leveraging literature extraction, identifier harmonisation, and regression validation can maintain model relevance.
Because EHMN 2026 is fully standards-compliant and annotation-dense, it is structurally amenable to AI-assisted updating workflows. Such pipelines could integrate new kinetic parameters (from SABIO-RK or BRENDA), regulatory relationships (TRANSFAC), signalling cascades (TRANSPATH), or immune baseline data (CYTOCON) into a version-controlled SBML ecosystem [19,20,21,22,23,24,25,26,27,28,29,30,31].
This evolutionary architecture shifts genome-scale reconstruction from a static artefact toward a continuously curated modelling infrastructure.
4.8. Limitations
EHMN 2026 remains primarily stoichiometric, and kinetic embedding is not globally implemented. Thermodynamic refinement addresses feasibility but does not yet incorporate full Gibbs free-energy parameterisation. Additionally, systematic benchmarking across multi-cohort metabolomics and clinical QSP case studies remains an important future step.
Nevertheless, the reconstruction’s emphasis on coherence, interoperability, and modular extensibility positions it as a stable foundation for translational and systems pharmacology integration.
EHMN 2026 remains primarily stoichiometric, and kinetic embedding is not globally implemented. Thermodynamic refinement successfully addresses global feasibility and eliminates infeasible energy-generating cycles ; however, it does not yet incorporate full Gibbs free-energy parameterisation. This reliance on semi-quantitative heuristics rather than continuous thermodynamic potentials means the model may not yet capture the concentration-dependent directionality shifts of near-equilibrium reactions. Additionally, systematic benchmarking across multi-cohort metabolomics and clinical QSP case studies remains an important future step. Nevertheless, the reconstruction’s emphasis on coherence, interoperability, and modular extensibility positions it as a stable foundation for translational and systems pharmacology integration.
4.9. Conclusion
EHMN 2026 advances human metabolic reconstruction beyond reaction expansion toward structurally engineered integratability. By combining thermodynamic coherence, identifier harmonisation, pathway hierarchy integration, and SBML Level 3 FBC compliance, it provides a metabolic backbone optimised for integration into QSP, regulatory, and kinetic modelling frameworks.
Through future coupling with CYTOCON [19,20], kinetic repositories such as SABIO-RK and BRENDA [25,26,31], and regulatory databases including TRANSFAC and TRANSPATH [27,28,29,30], EHMN 2026 can evolve into a multi-layer, dynamically extensible systems biology platform.
Data Availability and Reproducibility
The final EHMN_2026 genome-scale reconstruction is encoded in SBML Level 3 Version 2 with Flux Balance Constraints (FBC) extension and will be publicly deposited prior to publication.
The model will be available through:
- BioModels Database (EMBL-EBI)The curated SBML file will be deposited in the BioModels repository (https://www.ebi.ac.uk/biomodels/) following peer-review acceptance.The BioModels accession number will be included in the final published version of the manuscript.
- IQANOVA RepositoryA mirrored version of the SBML model, associated supplementary materials (S1–S4), and version-controlled update history will be hosted at: www.iqanova.orgThe SBML file is available on request now (goryanin@gmail.com)
Version Control and Traceability
Each released model version will include:
- Unique version identifier (EHMN_2026_v1.0)
- SHA256 checksum of SBML file
- Change log documenting structural, thermodynamic, or annotation updates
- Cross-reference compatibility with BioModels accession
Reproducibility Notes
The model is fully compatible with:
- COBRA Toolbox v3.0
- COBRApy
- SBML-compliant solvers
- Tellurium / libRoadRunner
All refinement workflows described in Section 2.2 and Section 2.3 are reproducible using the supplementary mapping and directionality tables provided.
Author Contributions
Conceptualization, I.G. and I.V.G.; Methodology, I.G. and I.G.; Software, I.G.; Validation, I.G.; Formal Analysis, I.G..; Investigation, I.G.; Data Curation, I.G.; Writing – Original Draft Preparation, I.G.; Writing – Review & Editing, I.G. and I.V.G.; Visualization, I.G.; Supervision, I.G.; Project Administration, I.V.G.. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable. This study involves the development of a computational metabolic reconstruction based on publicly available biochemical and genomic data and does not involve humans or animals.
Informed Consent Statement
Not applicable. This study did not involve human participants, as all data were derived from curated biochemical databases and published literature.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Bordbar, A.; Monk, J.M.; King, Z.A.; Palsson, B.Ø. Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 2014, 15, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.E.; Nagarajan, H.; Palsson, B.Ø. Constraining the metabolic genotype–phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 2012, 10, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Varma, A.; Palsson, B.Ø. Metabolic flux balancing: basic concepts, scientific and practical use. Bio/Technology 1994, 12, 994–998. [Google Scholar] [CrossRef]
- Thiele, I.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef]
- Brunk, E.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef]
- Robinson, J.L.; et al. An atlas of human metabolism. Sci. Signal. 2020, 13, eaaz1482. [Google Scholar] [CrossRef]
- Moretti, S.; et al. MetaNetX/MNXref: reconciliation of metabolites and biochemical reactions to bring together genome-scale metabolic networks. Nucleic Acids Res. 2016, 44, D523–D526. [Google Scholar] [CrossRef]
- Hastings, J.; et al. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016, 44, D1214–D1219. [Google Scholar] [CrossRef]
- Beard, D.A.; Qian, H. Thermodynamic-based computational profiling of cellular regulatory control in hepatocyte metabolism. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E633–E644. [Google Scholar] [CrossRef]
- Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Thermodynamics-based metabolic flux analysis. Biophys. J. 2007, 92, 1792–1805. [Google Scholar] [CrossRef]
- Noor, E.; et al. Pathway thermodynamics highlights kinetic obstacles in central metabolism. PLoS Comput. Biol. 2014, 10, e1003483. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Sorokin, A.; Mazein, A.; Selkov, A.; Selkov, E.; Demin, O.; Goryanin, I. The Edinburgh human metabolic network reconstruction and its functional analysis. Mol. Syst. Biol. 2007, 3, 135. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, M.; et al. The Reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Hucka, M.; et al. The Systems Biology Markup Language (SBML): Language Specification for Level 3 Version 2 Core. J. Integr. Bioinform. 2019, 16, 20190021. [Google Scholar] [CrossRef]
- Olivier, B.G.; Bergmann, F.T. SBML Level 3 Package: Flux Balance Constraints version 2. J. Integr. Bioinform. 2018, 15, 20170082. [Google Scholar] [CrossRef]
- Yurkovich, J.T.; et al. Quantitative systems pharmacology modeling of metabolism and disease. CPT Pharmacometrics Syst. Pharmacol. 2020, 9, 357–368. [Google Scholar] [CrossRef]
- Robinson, J.L.; Nielsen, J. Integrative analysis of human metabolism with genome-scale models. Nat. Rev. Genet. 2021, 22, 689–703. [Google Scholar] [CrossRef]
- Leonov, V.; et al. CYTOCON: The manually curated database of human in vivo cell and molecule concentrations. CPT Pharmacometrics Syst. Pharmacol. 2023. [Google Scholar] [CrossRef]
- Desikan, R.; et al. CYTOCON DB: A versatile database of human cell and molecule concentrations for accelerating model development. CPT Pharmacometrics Syst. Pharmacol. 2023. [Google Scholar] [CrossRef]
- Elmokadem, A.; et al. Quantitative Systems Pharmacology and PBPK Modeling With mrgsolve: A Hands-On Tutorial. CPT Pharmacometrics Syst. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Aghamiri, SS; Amin, R; Helikar, T. Recent applications of quantitative systems pharmacology and machine learning models across diseases. J Pharmacokinet Pharmacodyn. 2022, 49(1), 19–37. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cheng, L.; et al. Review of applications and challenges of quantitative systems pharmacology modeling and machine learning for heart failure. J. Pharmacokinet. Pharmacodyn. 2022. [Google Scholar] [CrossRef]
- Lemaire, V.; et al. No Recipe for Quantitative Systems Pharmacology Model Development; 2024. [Google Scholar] [CrossRef]
- Wittig, U.; et al. SABIO-RK: an updated resource for manually curated biochemical reaction kinetics. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Chang, A.; et al. BRENDA, the ELIXIR core data resource in 2021: new developments and updates. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef]
- Wingender, E.; et al. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000. [Google Scholar] [CrossRef]
- Matys, V.; et al. TRANSFAC: transcriptional regulation, from patterns to profiles. In Nucleic Acids Res.; 2003. [Google Scholar] [CrossRef]
- Schacherer, F.; et al. The TRANSPATH signal transduction database: a knowledge base on signal transduction networks. In Bioinformatics; 2001. [Google Scholar] [CrossRef]
- Krull, M.; et al. TRANSPATH: an integrated database on signal transduction and a tool for array analysis. In Nucleic Acids Res.; 2003. [Google Scholar] [CrossRef]
- Wittig, U.; et al. SABIO-RK—database for biochemical reaction kinetics. In Nucleic Acids Res.; 2012. [Google Scholar] [CrossRef]
Figure 1.
Schematic overview of the reconstruction and refinement workflow leading to EHMN 2026. The process begins with the original EHMN framework and proceeds through systematic identifier harmonisation (MetaNetX, ChEBI), reaction consolidation, thermodynamic directionality refinement, pathway integration via Reactome, and final SBML Level 3 FBC2 encoding. The workflow emphasises refinement and standardisation rather than reaction inflation.
Figure 1.
Schematic overview of the reconstruction and refinement workflow leading to EHMN 2026. The process begins with the original EHMN framework and proceeds through systematic identifier harmonisation (MetaNetX, ChEBI), reaction consolidation, thermodynamic directionality refinement, pathway integration via Reactome, and final SBML Level 3 FBC2 encoding. The workflow emphasises refinement and standardisation rather than reaction inflation.
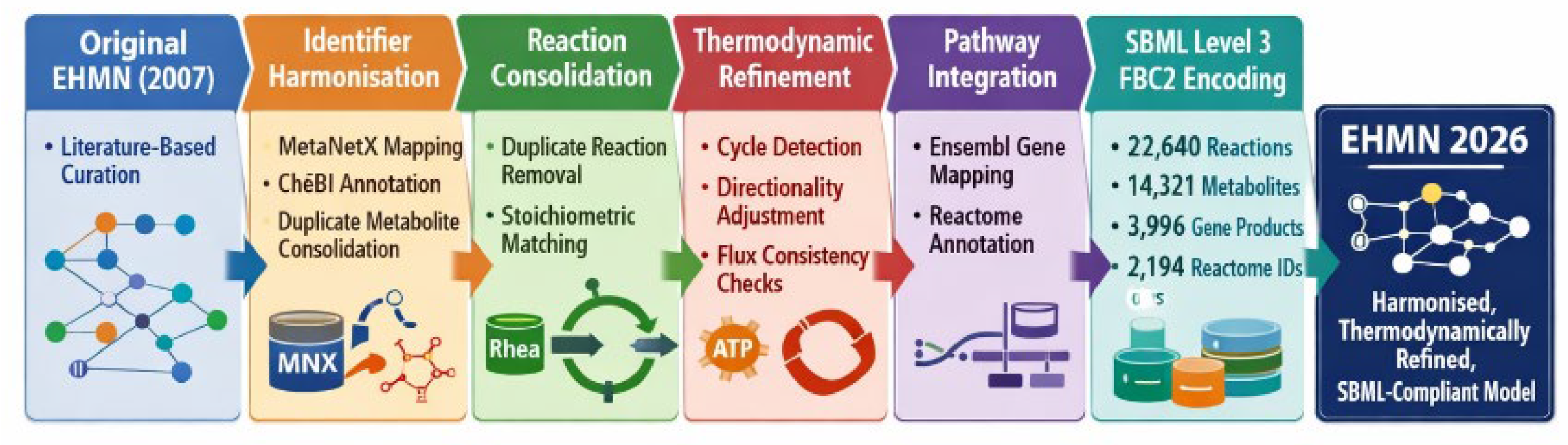
Figure 4.
Structural comparison of EHMN 2026 with representative human genome-scale metabolic reconstructions. EHMN 2026 exhibits expanded metabolite representation, structured pathway integration, and full SBML Level 3 FBC compliance while maintaining thermodynamic refinement.
Figure 4.
Structural comparison of EHMN 2026 with representative human genome-scale metabolic reconstructions. EHMN 2026 exhibits expanded metabolite representation, structured pathway integration, and full SBML Level 3 FBC compliance while maintaining thermodynamic refinement.
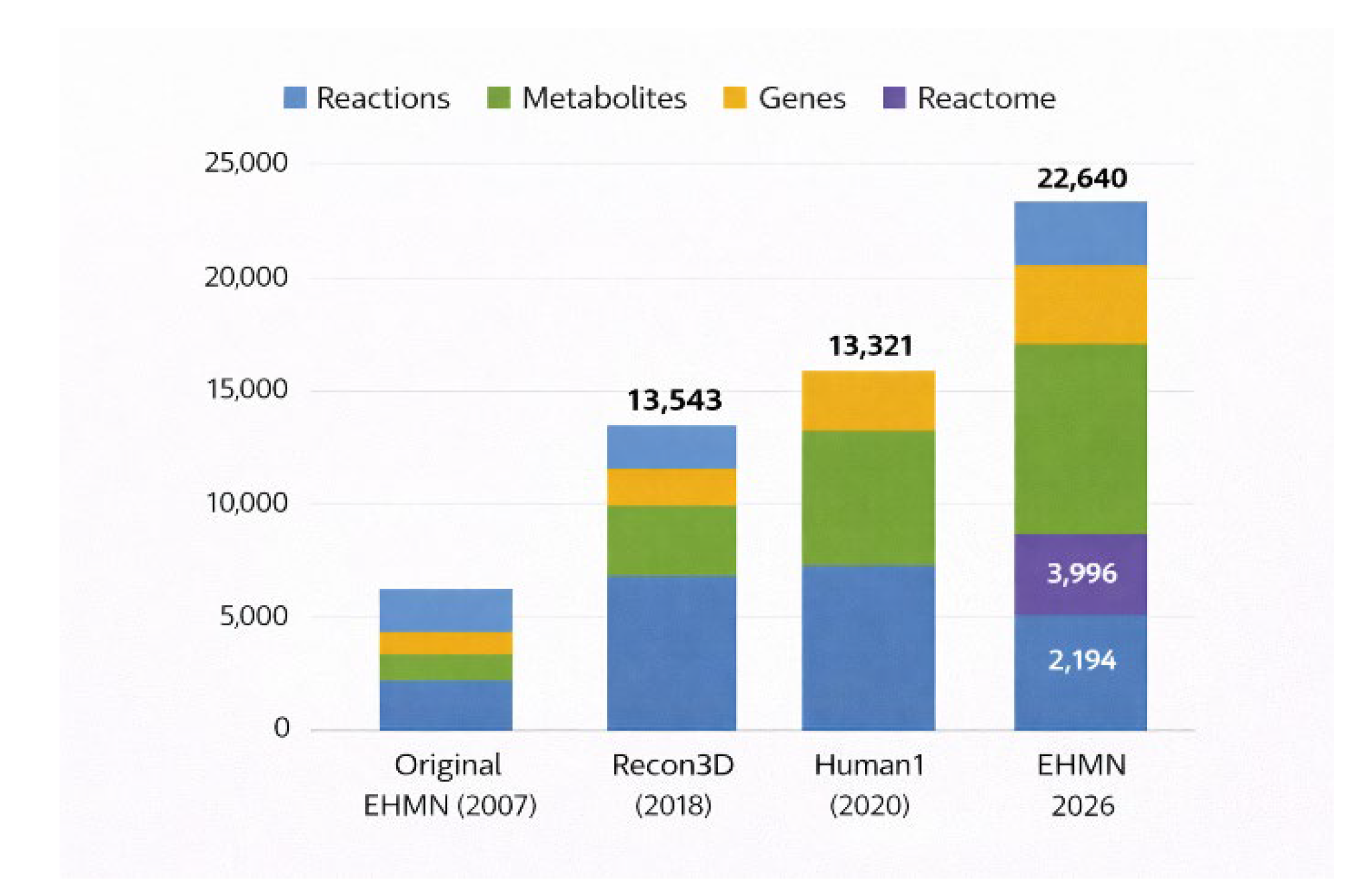
Figure 5.
Positioning EHMN 2026 as an Interoperable Metabolic Backbone.
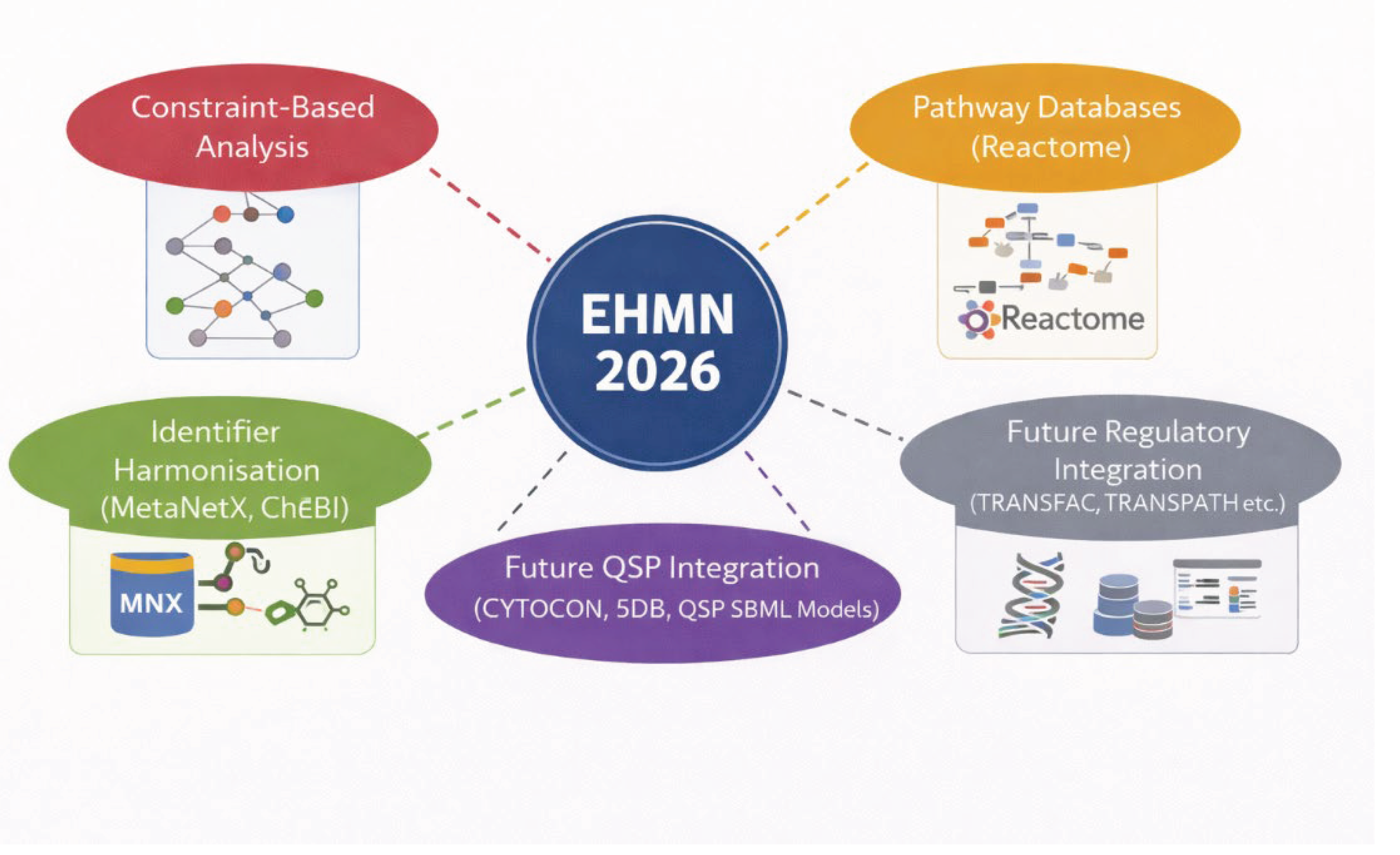
Table 1.
Core structural metrics of EHMN 2026.
| Feature | Value |
| Compartments | 11 |
| Metabolites | 14,321 |
| Reactions | 22,640 |
| Gene products | 3,996 |
| Reactions with GPR | 9,638 (42.6%) |
| Unique ENSG | 2,887 |
| Unique Reactome IDs | 2,194 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



