
Submitted:
13 February 2026
Posted:
14 February 2026
You are already at the latest version
Abstract
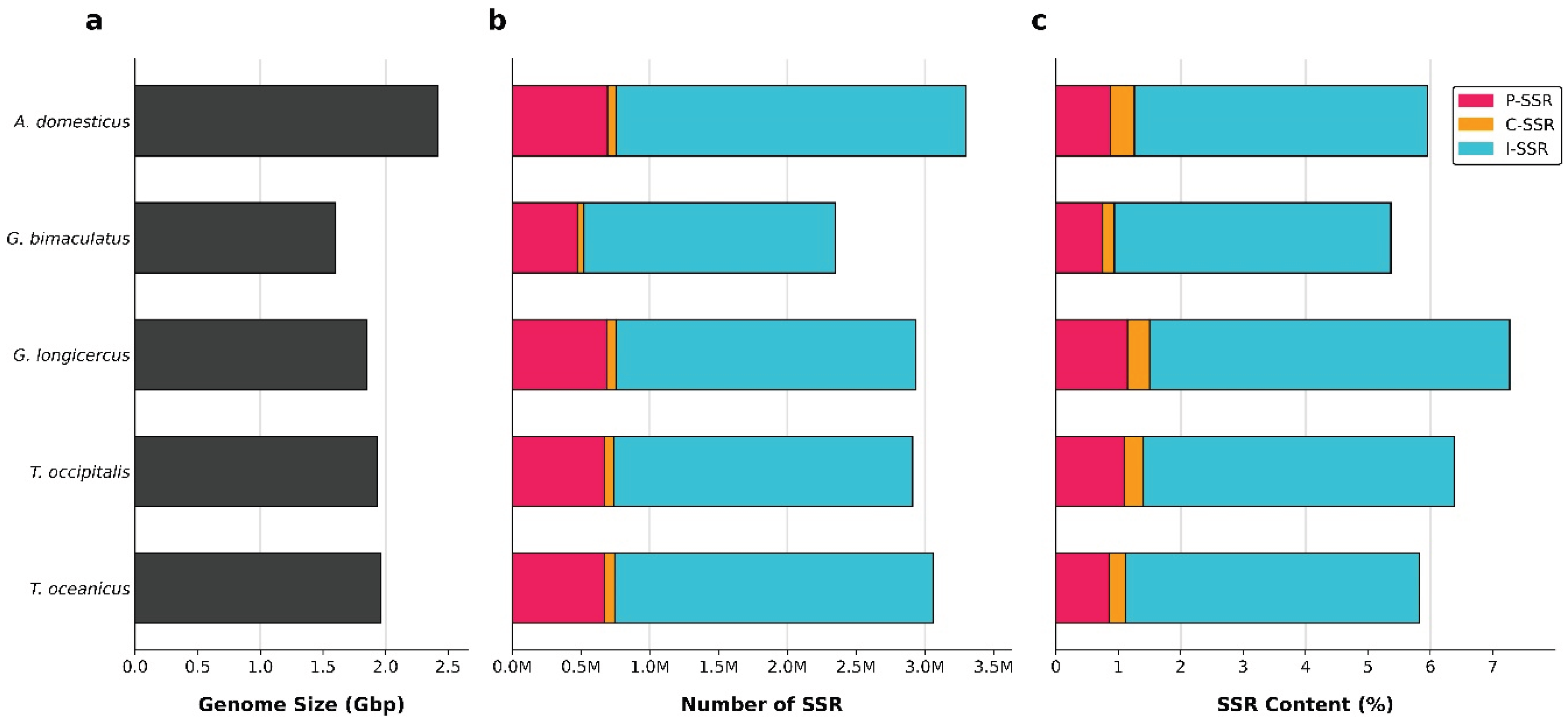
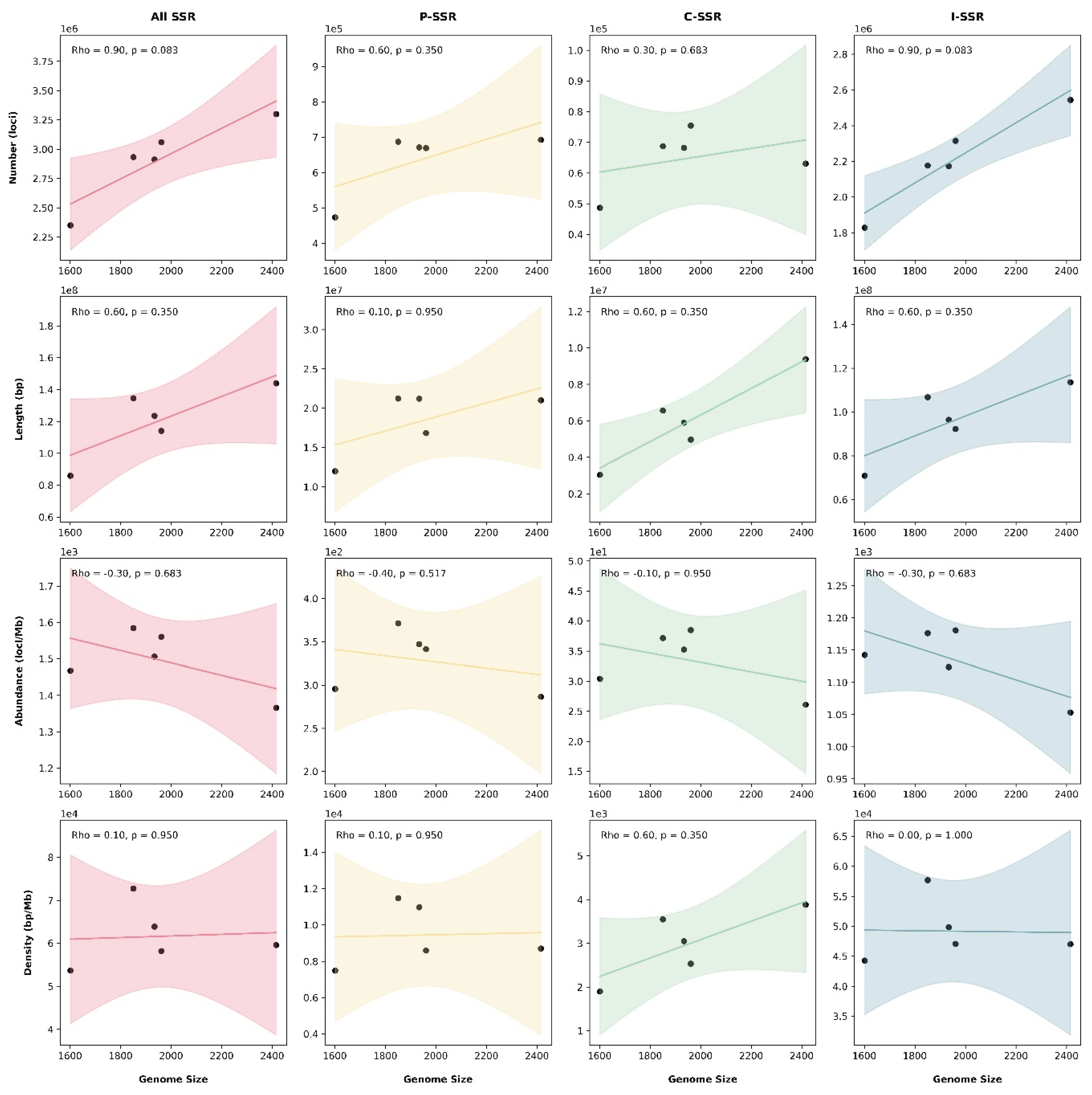
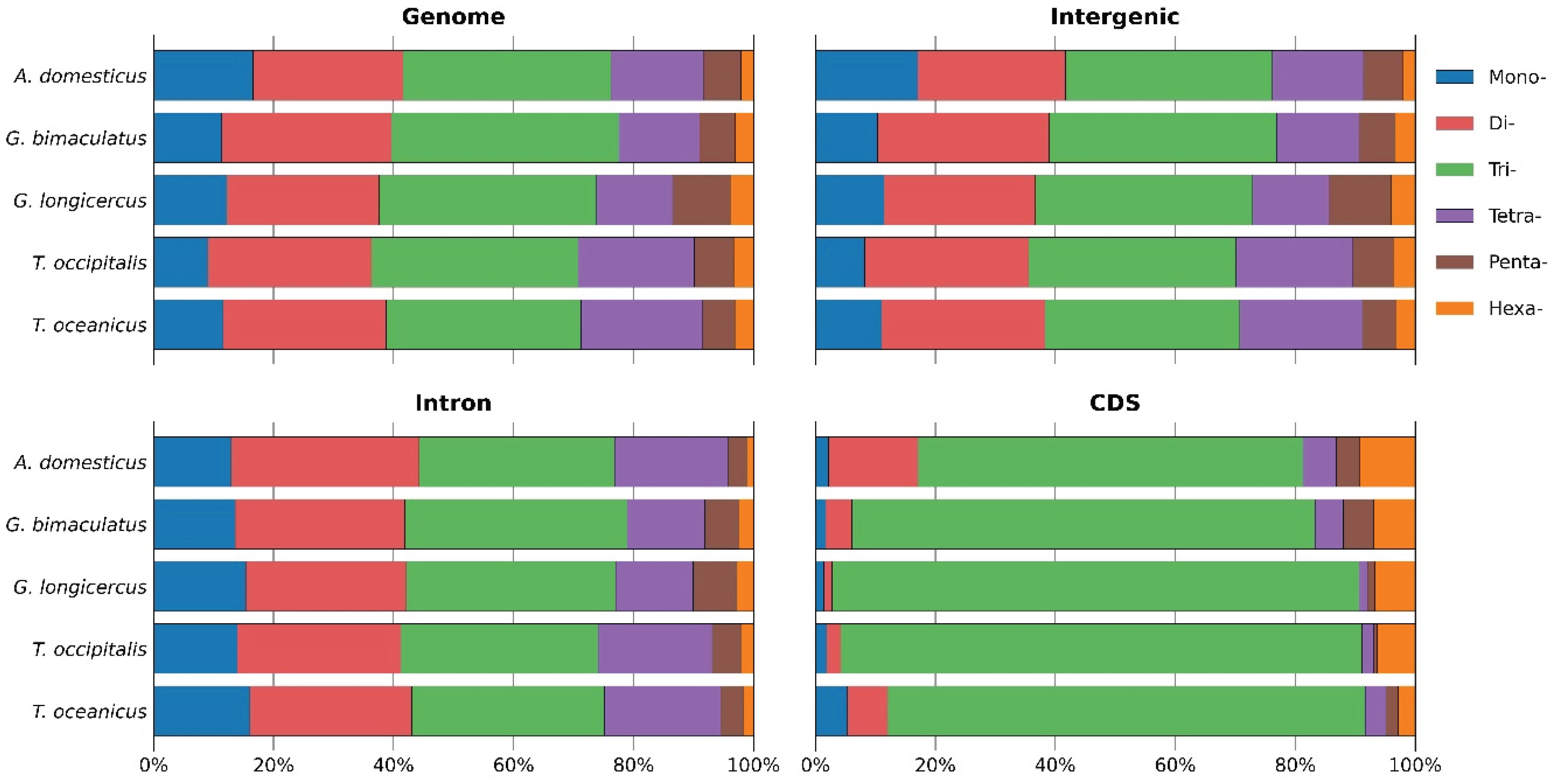
Microsatellites or simple sequence repeats (SSRs) are valuable markers for understanding genome structure, function, and evolution. However, their distribution and characteristics remain largely unexplored in cricket species. We conducted a genome-wide identification and analysis of SSRs (P-SSRs, C-SSRs, and I-SSRs) across five cricket genomes. The total number of SSRs ranged from 2,350,765 to 3,299,527, representing 5.37%–7.27% of the genomes. Abundance followed the pattern I-SSRs > P-SSRs > C-SSRs across genomic regions (genome, intergenic, intronic, and CDSs). The total SSR number showed a strong but statistically non-significant positive correlation with genome size, whereas SSR length, abundance, and density showed no correlation. Trinucleotide repeats were consistently the most common P-SSR type. The (AAT)n motif predominated in genome, intergenic, and intron regions, while (CCG)n was most frequent in CDSs. Consequently, AT-rich repeats dominated non-coding regions, whereas GC-rich repeats were enriched in CDSs. Coefficient of variation (CV) analysis of repeat copy numbers (RCN) revealed distinct trends in P-SSR distribution across regions and species. Functional annotation of CDSs containing P-SSRs indicated involvement in binding, signal transduction, and transcription. This study represents, to our knowledge, the first comprehensive family-level comparative analysis of SSRs in crickets, providing new insights into their genomic architecture.
Keywords:
1. Introduction
2. Materials and Methods
2.1. Sources of Genomic Dataset
2.2. Microsatellites Identification, Classification, and Localization
2.3. Microsatellite Attributes Calculation
2.4. Functional Analysis of CDSs Containing P-SSRs
2.5. Statistical Analysis
3. Results
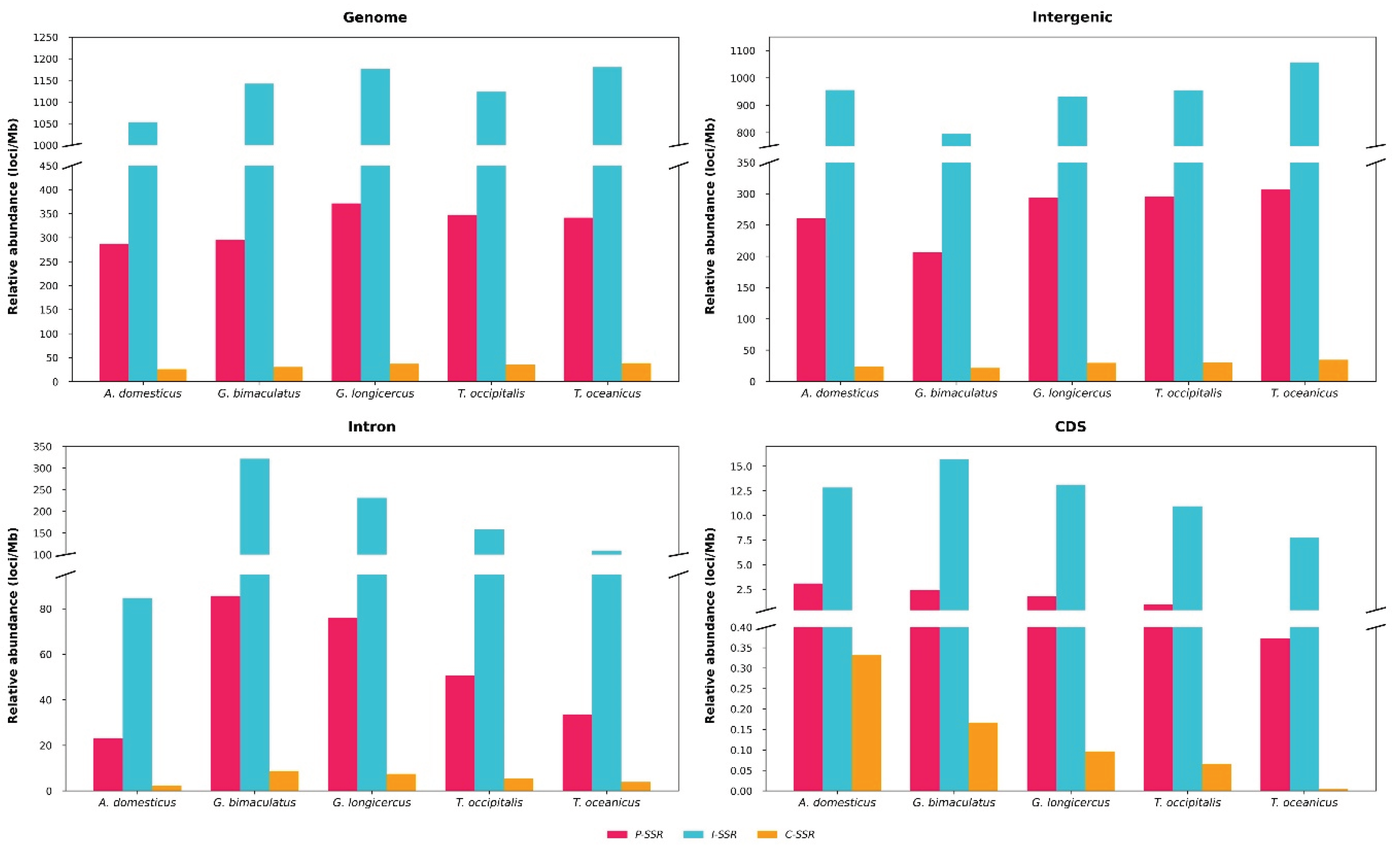
3.1. Global Characteristics of Microsatellites in Cricket Genomes
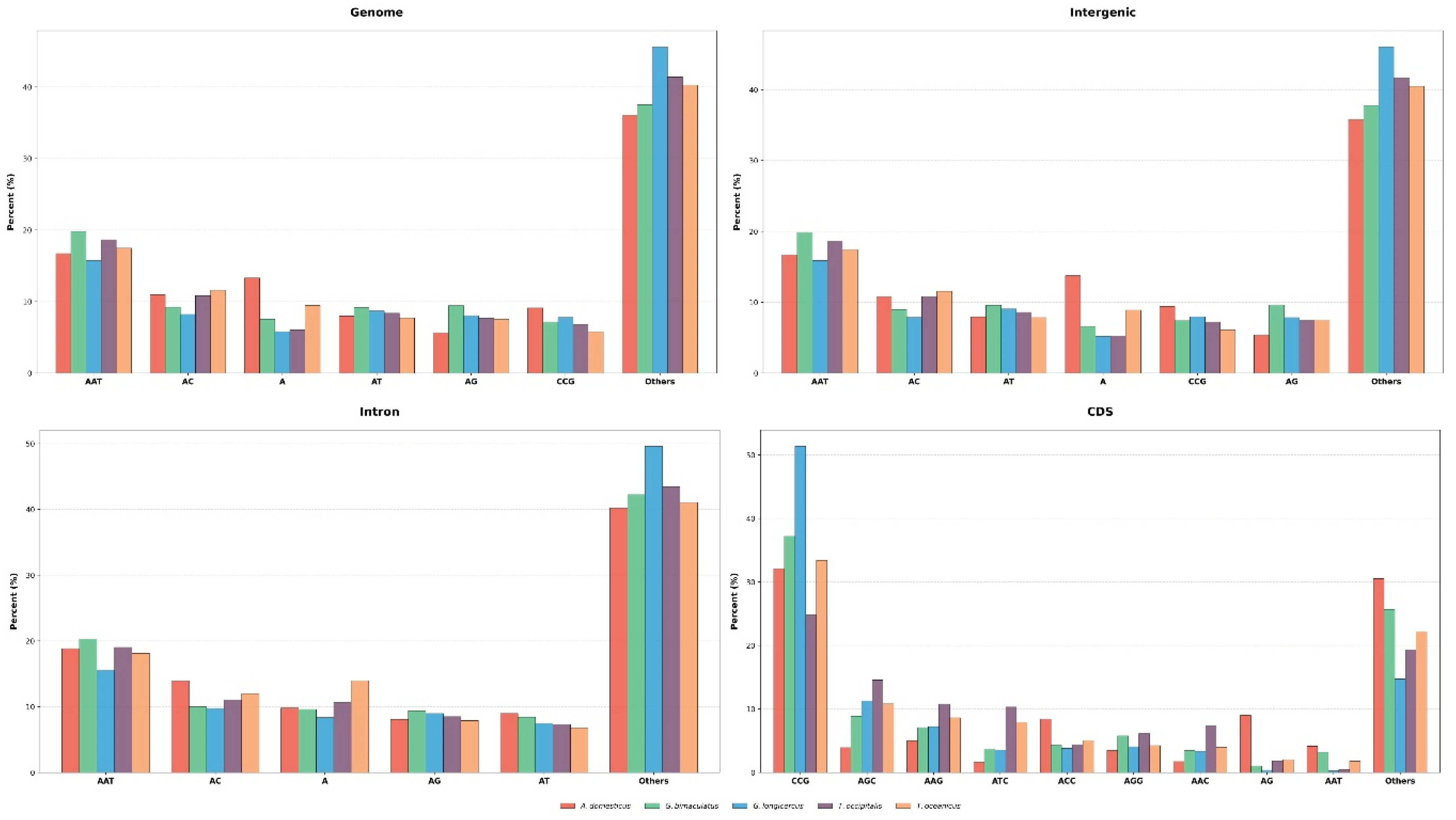
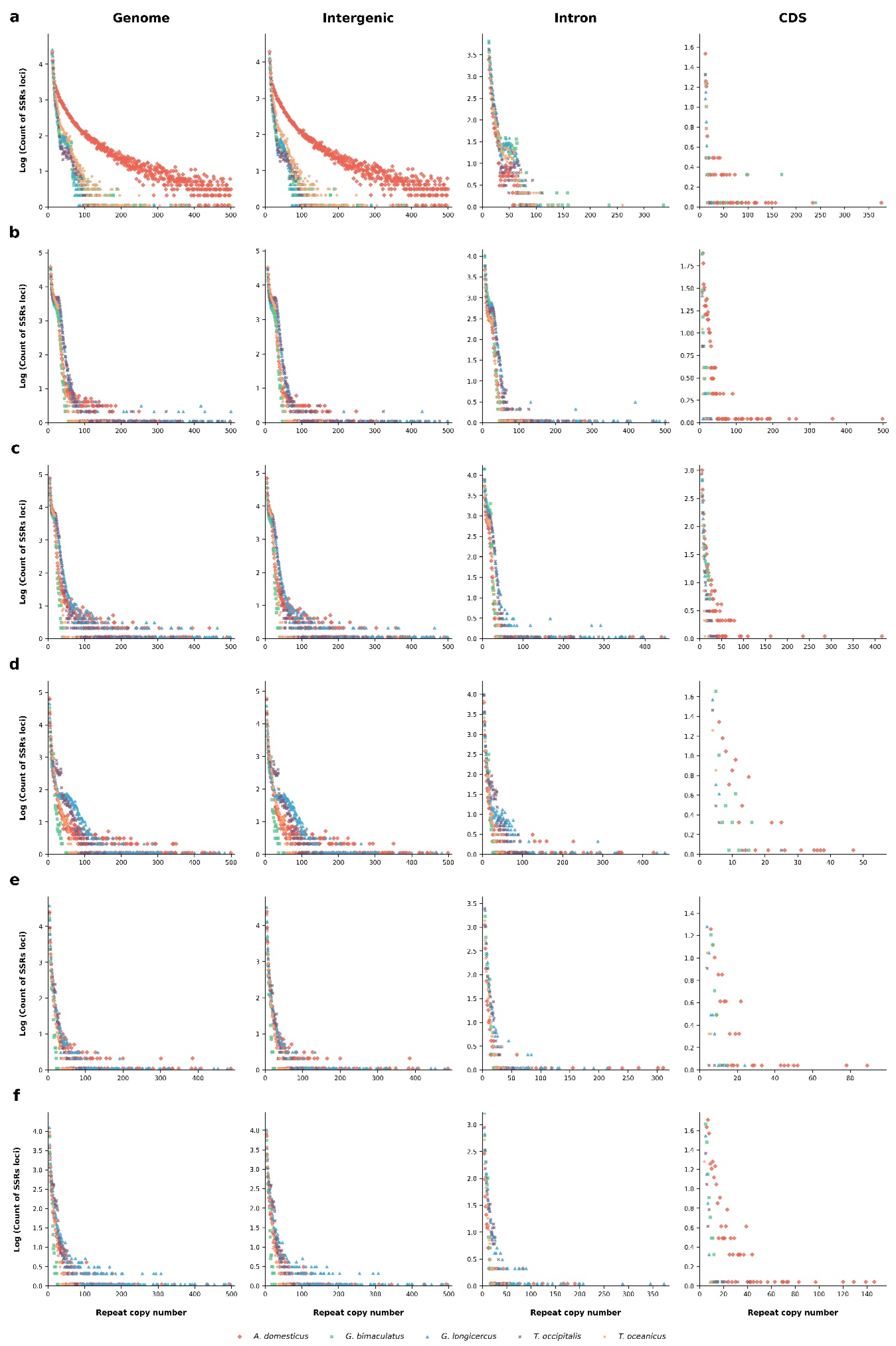
3.2. Distribution Patterns of P-SSRs in Cricket Genomes
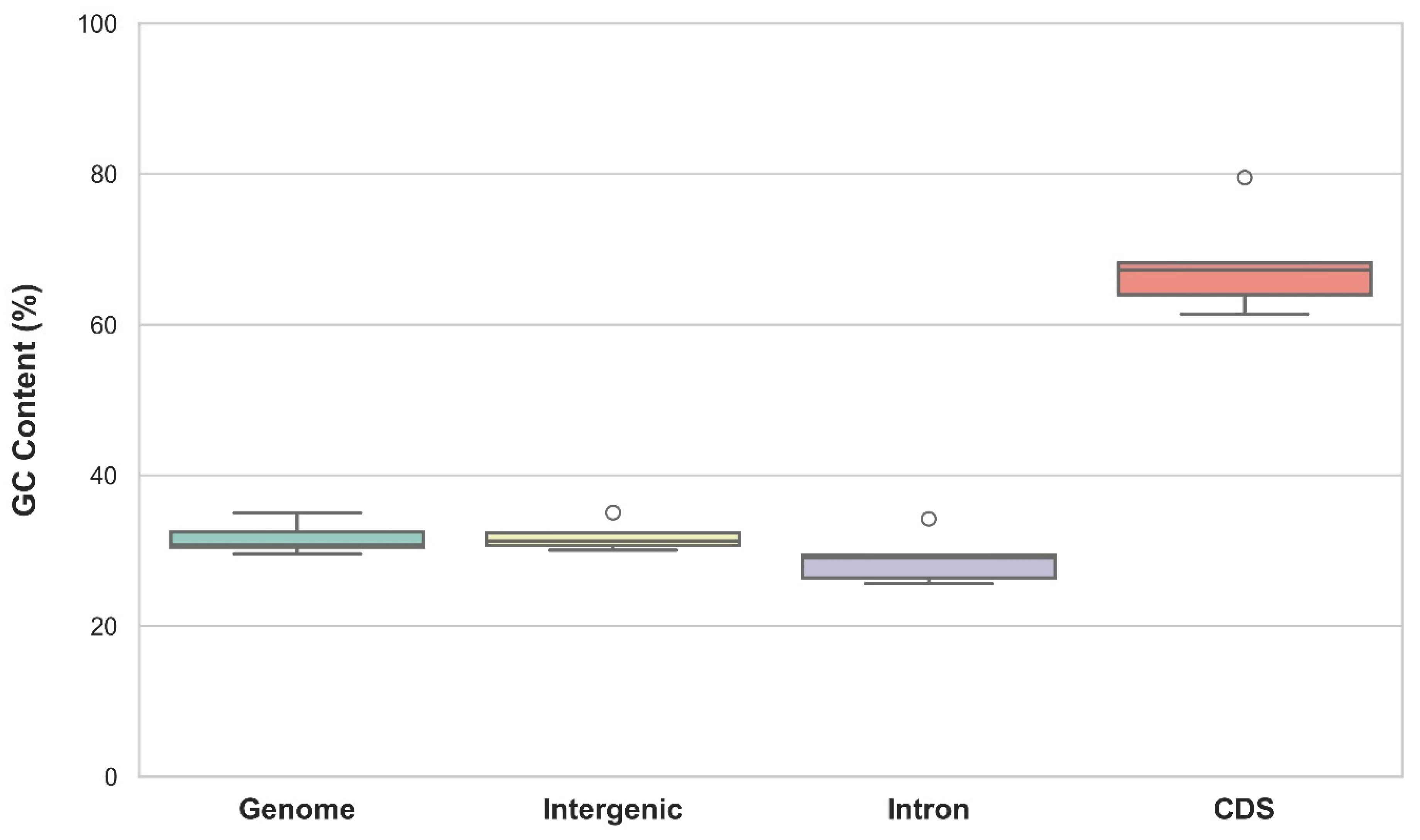
3.3. GC Content of P-SSRs in Different Genomic Regions of Cricket Genomes
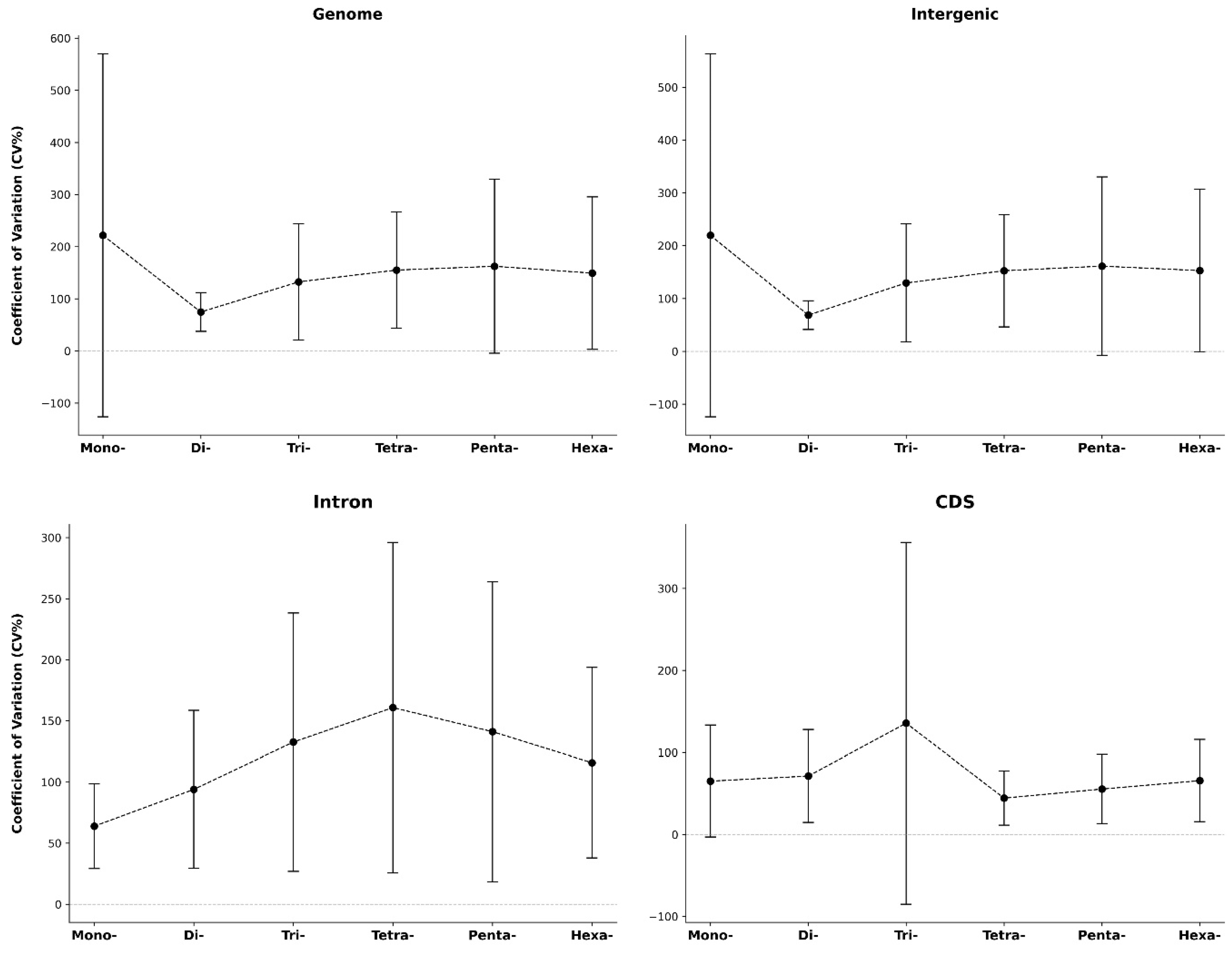
3.4. Analysis of the Coefficient of Variability (CV) of P-SSRs
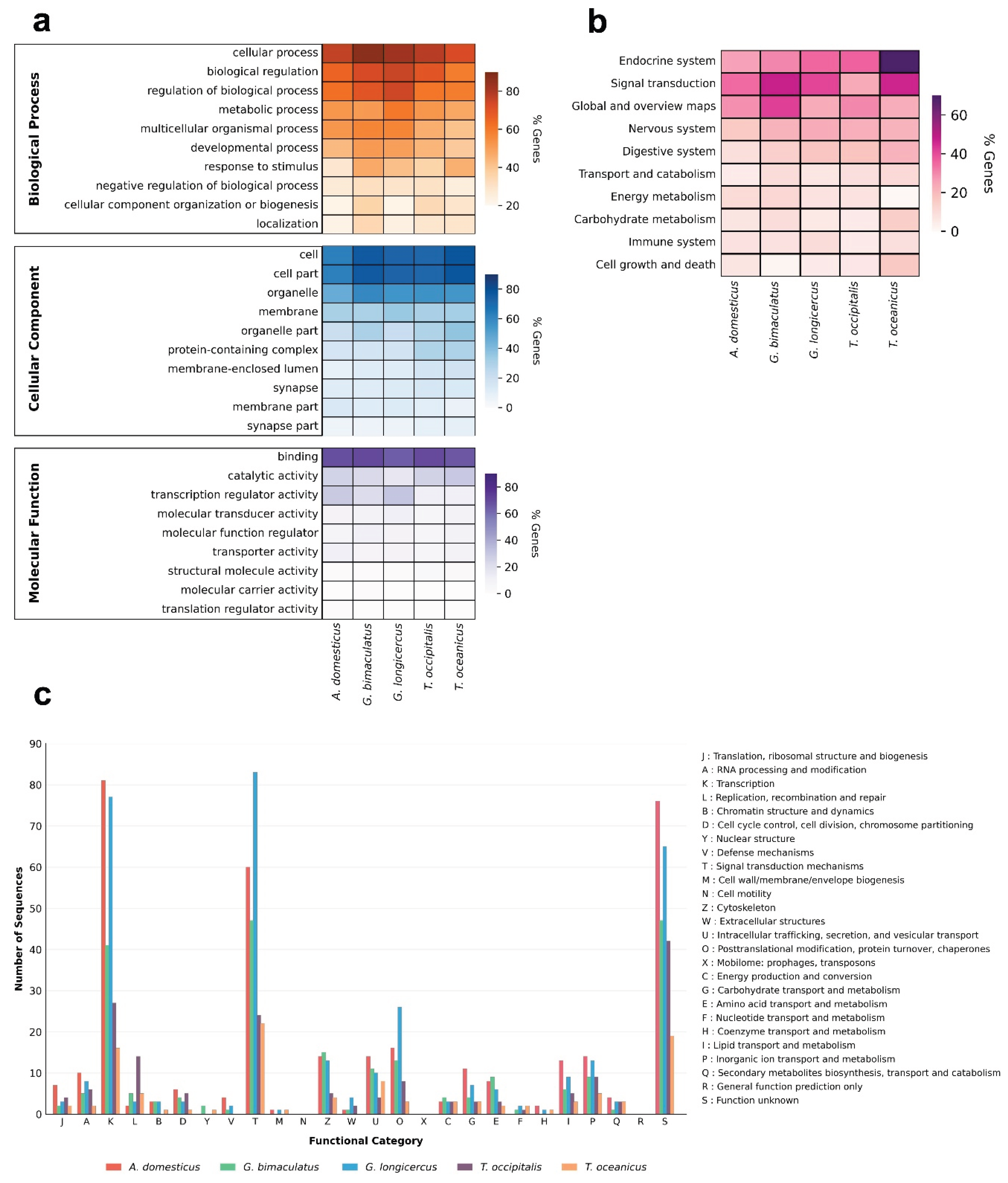
3.5. Functional Analysis of CDSs with P-SSRs in Cricket Genomes
4. Discussion
4.1. The Number of Microsatellites
4.2. The Characteristic of P-SSRs
4.3. Potential Function of CDSs Containing P-SSRs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tóth, G.; Gáspári, Z.; Jurka, J. Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res. 2000, 10, 967–981. [Google Scholar] [CrossRef]
- Bhargava, A.; Fuentes, F.F. Mutational Dynamics of Microsatellites. Mol. Biotechnol. 2010, 44, 250–266. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Hellemans, B.; Volckaert, F.A.M. Microsatellites and their genomic distribution, evolution, function and applications: A review with special reference to fish genetics. Aquaculture 2006, 255, 1–29. [Google Scholar] [CrossRef]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-C.; Korol, A.B.; Fahima, T.; Nevo, E. Microsatellites Within Genes: Structure, Function, and Evolution. Mol. Biol. Evol. 2004, 21, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, J.R.; Willemsen, R.; Oostra, B.A. Microsatellite repeat instability and neurological disease. Bioessays 2009, 31, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Gelsomino, F.; Barbolini, M.; Spallanzani, A.; Pugliese, G.; Cascinu, S. The evolving role of microsatellite instability in colorectal cancer: A review. Cancer Treat. Rev. 2016, 51, 19–26. [Google Scholar] [CrossRef]
- Kataoka, K.; Togawa, Y.; Sanno, R.; Asahi, T.; Yura, K. Dissecting cricket genomes for the advancement of entomology and entomophagy. Biophys. Rev. 2022, 14, 75–97. [Google Scholar] [CrossRef]
- Donoughe, S.; Extavour, C.G. Embryonic development of the cricket Gryllus bimaculatus. Dev. Biol. 2016, 411, 140–156. [Google Scholar] [CrossRef]
- Cayre, M.; Scotto-Lomassese, S.; Malaterre, J.; Strambi, C.; Strambi, A. Understanding the Regulation and Function of Adult Neurogenesis: Contribution from an Insect Model, the House Cricket. Chem. Senses 2007, 32, 385–395. [Google Scholar] [CrossRef]
- Pascoal, S.; Cezard, T.; Eik-Nes, A.; Gharbi, K.; Majewska, J.; Payne, E.; Ritchie, Michael G.; Zuk, M.; Bailey, Nathan W. Rapid Convergent Evolution in Wild Crickets. Curr. Biol. 2014, 24, 1369–1374. [Google Scholar] [CrossRef]
- Moriyama, Y.; Sakamoto, T.; Karpova, S.G.; Matsumoto, A.; Noji, S.; Tomioka, K. RNA Interference of the Clock Gene period Disrupts Circadian Rhythms in the Cricket Gryllus bimaculatus. J. Biol. Rhythms 2008, 23, 308–318. [Google Scholar] [CrossRef]
- Mitchaothai, J.; Grabowski, N.T.; Lertpatarakomol, R.; Trairatapiwan, T.; Chhay, T.; Keo, S.; Lukkananukool, A. Production Performance and Nutrient Conversion Efficiency of Field Cricket (Gryllus bimaculatus) in Mass-Rearing Conditions. Animals 2022, 12, 2263. [Google Scholar] [CrossRef]
- Akiyama, D.; Kaewplik, T.; Fujisawa, T.; Kurosu, T.; Sasaki, Y. Crickets (Gryllus Bimaculatus) using food waste usefulness of self-selection feed design method through each growth stage. J. Insects Food Feed 2023, 10, 247–258. [Google Scholar] [CrossRef]
- Magara, H.J.O.; Niassy, S.; Ayieko, M.A.; Mukundamago, M.; Egonyu, J.P.; Tanga, C.M.; Kimathi, E.K.; Ongere, J.O.; Fiaboe, K.K.M.; Hugel, S.; et al. Edible Crickets (Orthoptera) Around the World: Distribution, Nutritional Value, and Other Benefits—A Review. Front. Nutr. 2021, 7. [Google Scholar] [CrossRef]
- Tang, C.; Yang, D.; Liao, H.; Sun, H.; Liu, C.; Wei, L.; Li, F. Edible insects as a food source: a review. Food Prod. Process. Nutr. 2019, 1, 8. [Google Scholar] [CrossRef]
- Sharma, P.C.; Grover, A.; Kahl, G. Mining microsatellites in eukaryotic genomes. Trends Biotechnol. 2007, 25, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Merkel, A.; Gemmell, N. Detecting short tandem repeats from genome data: opening the software black box. Briefings in Bioinformatics 2008, 9, 355–366. [Google Scholar] [CrossRef]
- Grover, A.; Aishwarya, V.; Sharma, P.C. Searching microsatellites in DNA sequences: approaches used and tools developed. Physiol. Mol. Biol. Plants 2012, 18, 11–19. [Google Scholar] [CrossRef]
- Srivastava, S.; Avvaru, A.K.; Sowpati, D.T.; Mishra, R.K. Patterns of microsatellite distribution across eukaryotic genomes. BMC Genomics 2019, 20, 153. [Google Scholar] [CrossRef]
- Song, X.; Yang, T.; Yan, X.; Zheng, F.; Xu, X.; Zhou, C. Comparison of microsatellite distribution patterns in twenty-nine beetle genomes. Gene 2020, 757, 144919. [Google Scholar] [CrossRef]
- Ding, S.; Wang, S.; He, K.; Jiang, M.; Li, F. Large-scale analysis reveals that the genome features of simple sequence repeats are generally conserved at the family level in insects. BMC Genomics 2017, 18, 848. [Google Scholar] [CrossRef]
- Liu, W.; Xu, Y.; Li, Z.; Fan, J.; Yang, Y. Genome-wide mining of microsatellites in king cobra (Ophiophagus hannah) and cross-species development of tetranucleotide SSR markers in Chinese cobra (Naja atra). Mol. Biol. Rep. 2019, 46, 6087–6098. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, S.; Chen, Y.; Zhang, S.; Zhao, Q.; Li, M.; Gao, Y.; Yang, L.; Bennetzen, J.L. Comparative genome-wide characterization leading to simple sequence repeat marker development for Nicotiana. BMC Genomics 2018, 19, 500. [Google Scholar] [CrossRef]
- Feng, K.; Zhou, C.; Wang, L.; Zhang, C.; Yang, Z.; Hu, Z.; Yue, B.; Wu, Y. Comprehensive Comparative Analysis Sheds Light on the Patterns of Microsatellite Distribution across Birds Based on the Chromosome-Level Genomes. Animals 2023, 13, 655. [Google Scholar] [CrossRef]
- Huang, J.; Li, W.; Jian, Z.; Yue, B.; Yan, Y. Genome-wide distribution and organization of microsatellites in six species of birds. Biochem. Syst. Ecol. 2016, 67, 95–102. [Google Scholar] [CrossRef]
- Zhong, H.; Shao, X.; Cao, J.; Huang, J.; Wang, J.; Yang, N.; Yuan, B. Comparison of the Distribution Patterns of Microsatellites Across the Genomes of Reptiles. Ecol. Evol. 2024, 14, e70458. [Google Scholar] [CrossRef]
- Song, X.; Yang, T.; Zhang, X.; Yuan, Y.; Yan, X.; Wei, Y.; Zhang, J.; Zhou, C. Comparison of the Microsatellite Distribution Patterns in the Genomes of Euarchontoglires at the Taxonomic Level. Front. Genet. 2021, 12. [Google Scholar] [CrossRef]
- Du, L.; Zhang, C.; Liu, Q.; Zhang, X.; Yue, B. Krait: an ultrafast tool for genome-wide survey of microsatellites and primer design. Bioinformatics 2017, 34, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Li, F.; Wen, Q.; Price, M.; Yang, N.; Yue, B. Characterization of microsatellites in the endangered snow leopard based on the chromosome-level genome. Mammal Res. 2021, 66, 385–398. [Google Scholar] [CrossRef]
- Jiang, Q.; Li, Q.; Yu, H.; Kong, L. Genome-Wide Analysis of Simple Sequence Repeats in Marine Animals—a Comparative Approach. Mar. Biotechnol. 2014, 16, 604–619. [Google Scholar] [CrossRef]
- Jurka, J.; Pethiyagoda, C. Simple repetitive DNA sequences from primates: compilation and analysis. J. Mol. Evol. 1995, 40, 120–126. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Peng, Z.; Sun, H.; Yue, X.; Lou, Y.; Dong, L.; Wang, L.; Gao, Z. Developing genome-wide microsatellite markers of bamboo and their applications on molecular marker assisted taxonomy for accessions in the genus Phyllostachys. Sci. Rep. 2015, 5, 8018. [Google Scholar] [CrossRef]
- Qi, W.-H.; Jiang, X.-M.; Yan, C.-C.; Zhang, W.-Q.; Xiao, G.-S.; Yue, B.-S.; Zhou, C.-Q. Distribution patterns and variation analysis of simple sequence repeats in different genomic regions of bovid genomes. Sci. Rep. 2018, 8, 14407. [Google Scholar] [CrossRef] [PubMed]
- The Galaxy Community. The Galaxy platform for accessible, reproducible, and collaborative data analyses: 2024 update. Nucleic Acids Res. 2024, 52, W83–W94. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; et al. WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Sainudiin, R.; Durrett, R.T.; Aquadro, C.F.; Nielsen, R. Microsatellite mutation models: insights from a comparison of humans and chimpanzees. Genetics 2004, 168, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Bagshaw, A.T.M. Functional Mechanisms of Microsatellite DNA in Eukaryotic Genomes. Genome Biol. Evol. 2017, 9, 2428–2443. [Google Scholar] [CrossRef]
- Qi, W.-H.; Jiang, X.-M.; Du, L.-M.; Xiao, G.-S.; Hu, T.-Z.; Yue, B.-S.; Quan, Q.-M. Genome-wide survey and analysis of microsatellite sequences in bovid species. PLoS One 2015, 10, e0133667. [Google Scholar] [CrossRef]
- Manee, M.M.; Algarni, A.T.; Alharbi, S.N.; Al-Shomrani, B.M.; Ibrahim, M.A.; Binghadir, S.A.; Al-Fageeh, M.B. Genome-wide characterization and analysis of microsatellite sequences in camelid species. Mammal Res. 2020, 65, 359–373. [Google Scholar] [CrossRef]
- Liu, S.; Hou, W.; Sun, T.; Xu, Y.; Li, P.; Yue, B.; Fan, Z.; Li, J. Genome-wide mining and comparative analysis of microsatellites in three macaque species. Mol. Genet. Genomics 2017, 292, 537–550. [Google Scholar] [CrossRef]
- Zuo, B.; Nneji, L.M.; Sun, Y.-B. Comparative genomics reveals insights into anuran genome size evolution. BMC Genomics 2023, 24, 379. [Google Scholar] [CrossRef]
- Blommaert, J.; Riss, S.; Hecox-Lea, B.; Mark Welch, D.B.; Stelzer, C.P. Small, but surprisingly repetitive genomes: transposon expansion and not polyploidy has driven a doubling in genome size in a metazoan species complex. BMC Genomics 2019, 20, 466. [Google Scholar] [CrossRef]
- Wang, X.T.; Zhang, Y.J.; Qiao, L.; Chen, B. Comparative analyses of simple sequence repeats (SSRs) in 23 mosquito species genomes: identification, characterization and distribution (Diptera: Culicidae). Insect Sci. 2019, 26, 607–619. [Google Scholar] [CrossRef]
- Xu, Y.; Li, W.; Hu, Z.; Zeng, T.; Shen, Y.; Liu, S.; Zhang, X.; Li, J.; Yue, B. Genome-wide mining of perfect microsatellites and tetranucleotide orthologous microsatellites estimates in six primate species. Gene 2018, 643, 124–132. [Google Scholar] [CrossRef]
- Ma, Z. Genome-wide characterization of perfect microsatellites in yak (Bos grunniens). Genetica 2015, 143, 515–520. [Google Scholar] [CrossRef]
- Qi, W.H.; Lu, T.; Zheng, C.L.; Jiang, X.M.; Jie, H.; Zhang, X.Y.; Yue, B.S.; Zhao, G.J. Distribution patterns of microsatellites and development of its marker in different genomic regions of forest musk deer genome based on high throughput sequencing. Aging (Albany NY) 2020, 12, 4445–4462. [Google Scholar] [CrossRef] [PubMed]
- Metzgar, D.; Bytof, J.; Wills, C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10, 72–80. [Google Scholar]
- Katti, M.V.; Ranjekar, P.K.; Gupta, V.S. Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol. Biol. Evol. 2001, 18, 1161–1167. [Google Scholar] [CrossRef]
- Mayer, C.; Leese, F.; Tollrian, R. Genome-wide analysis of tandem repeats in Daphnia pulex - a comparative approach. BMC Genomics 2010, 11, 277. [Google Scholar] [CrossRef]
- Umumararungu, T.; Gahamanyi, N.; Mukiza, J.; Habarurema, G.; Katandula, J.; Rugamba, A.; Kagisha, V. Proline, a unique amino acid whose polymer, polyproline II helix, and its analogues are involved in many biological processes: a review. Amino Acids 2024, 56, 50. [Google Scholar] [CrossRef] [PubMed]
- Lynch, V.J.; Wagner, G.P. Cooption of polyalanine tract into a repressor domain in the mammalian transcription factor HoxA11. J. Exp. Zool. B Mol. Dev. Evol. 2023, 340, 486–495. [Google Scholar] [CrossRef]
- Pirone, L.; Caldinelli, L.; Di Lascio, S.; Di Girolamo, R.; Di Gaetano, S.; Fornasari, D.; Pollegioni, L.; Benfante, R.; Pedone, E. Molecular insights into the role of the polyalanine region in mediating PHOX2B aggregation. FEBS J. 2019, 286, 2505–2521. [Google Scholar] [CrossRef] [PubMed]
- Reddy Chichili, V.P.; Kumar, V.; Sivaraman, J. Linkers in the structural biology of protein–protein interactions. Protein Sci. 2013, 22, 153–167. [Google Scholar] [CrossRef]
- Paloni, M.; Bussi, G.; Barducci, A. Arginine multivalency stabilizes protein/RNA condensates. Protein Sci. 2021, 30, 1418–1426. [Google Scholar] [CrossRef]
- Qi, W.H.; Yan, C.C.; Li, W.J.; Jiang, X.M.; Li, G.Z.; Zhang, X.Y.; Hu, T.Z.; Li, J.; Yue, B.S. Distinct patterns of simple sequence repeats and GC distribution in intragenic and intergenic regions of primate genomes. Aging (Albany NY) 2016, 8, 2635–2654. [Google Scholar] [CrossRef]
- Kaessmann, H.; Vinckenbosch, N.; Long, M. RNA-based gene duplication: mechanistic and evolutionary insights. Nat. Rev. Genet. 2009, 10, 19–31. [Google Scholar] [CrossRef]
- Grandi, F.C.; An, W. Non-LTR retrotransposons and microsatellites. Mob. Genet. Elements 2013, 3, e25674. [Google Scholar] [CrossRef]
- Pavlícek, A.; Paces, J.; Elleder, D.; Hejnar, J. Processed pseudogenes of human endogenous retroviruses generated by LINEs: their integration, stability, and distribution. Genome Res. 2002, 12, 391–399. [Google Scholar] [CrossRef]
- Schorderet, D.F.; Gartler, S.M. Analysis of CpG suppression in methylated and nonmethylated species. Proc. Natl. Acad. Sci. U. S. A 1992, 89, 957–961. [Google Scholar] [CrossRef]
- Fotsing, S.F.; Margoliash, J.; Wang, C.; Saini, S.; Yanicky, R.; Shleizer-Burko, S.; Goren, A.; Gymrek, M. The impact of short tandem repeat variation on gene expression. Nat. Genet. 2019, 51, 1652–1659. [Google Scholar] [CrossRef]
- Zhou, H.; Qiang, G.; Xia, Y.; Tan, J.; Fu, Q.; Luo, K.; Meng, X.; Chen, B.; Chen, M.; Sui, J.; et al. Copy Number Variations in Short Tandem Repeats Modulate Growth Traits in Penaeid Shrimp Through Neighboring Gene Regulation. Animals 2025, 15, 262. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Luo, C.; Xiang, G.; Huang, J.; Shao, L.; Huang, H.; Fan, S. Genome-Wide Microsatellites in Acanthopagrus latus: Development, Distribution, Characterization, and Polymorphism. Animals 2024, 14, 3709. [Google Scholar] [CrossRef] [PubMed]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O'Donovan, C.; Apweiler, R. QuickGO: a web-based tool for Gene Ontology searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Vera Alvarez, R.; Karamycheva, S.; Makarova, K.S.; Wolf, Yuri I.; Landsman, D.; Koonin, E.V. COG database update 2024. Nucleic Acids Res. 2024, 53, D356–D363. [Google Scholar] [CrossRef]
- Gemayel, R.; Vinces, M.D.; Legendre, M.; Verstrepen, K.J. Variable Tandem Repeats Accelerate Evolution of Coding and Regulatory Sequences. Annu. Rev. Genet. 2010, 44, 445–477. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.E.; Todd, P.K. Native functions of short tandem repeats. eLife 2023, 12, e84043. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, L.A.; Hennessy, J.M.; Peixoto, A.A.; Rosato, E.; Parkinson, H.; Costa, R.; Kyriacou, C.P. Natural Variation in a Drosophila Clock Gene and Temperature Compensation. Science 1997, 278, 2117–2120. [Google Scholar] [CrossRef] [PubMed]
- Verbiest, M.; Maksimov, M.; Jin, Y.; Anisimova, M.; Gymrek, M.; Bilgin Sonay, T. Mutation and selection processes regulating short tandem repeats give rise to genetic and phenotypic diversity across species. J. Evol. Biol. 2023, 36, 321–336. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



